

Abstract
Processing of Okazaki fragments to complete lagging-strand DNA synthesis requires coordination among several proteins. RNA primers and DNA synthesised by DNA polymerase α are displaced by DNA polymerase δ to create bifurcated nucleic acid structures known as 5′-flaps. These 5′-flaps are removed by Flap Endonuclease 1 (FEN), a structure-specific nuclease whose divalent metal-ion-dependent phosphodiesterase activity cleaves 5′-flaps with exquisite specificity. FENs are paradigms for the 5′ nuclease superfamily, whose members perform a wide variety of roles in nucleic acid metabolism using a similar nuclease core domain that displays common biochemical properties and structural features. A detailed review of FEN structure is undertaken to show how DNA substrate recognition occurs and how FEN achieves cleavage at a single phosphate diester. A proposed double nucleotide unpairing trap (DoNUT) is discussed with regards to FEN and has relevance to the wider 5′-nuclease superfamily. The homotrimeric proliferating cell nuclear antigen protein (PCNA) coordinates the actions of DNA polymerase, FEN and DNA ligase by facilitating the hand-off intermediates between each protein during Okazaki fragment maturation to maximise through-put and minimise consequences of intermediates being released into the wider cellular environment. FEN has numerous partner proteins that modulate and control its action during DNA replication and is also controlled by several post-translational modification events, all acting in concert to maintain precise and appropriate cleavage of Okazaki fragment intermediates during DNA replication.
Keywords: Okazaki fragment maturation, Lagging-strand DNA replication, Double nucleotide unpairing, Structure-specific nuclease, Disorder-order transition
16.1 Introduction
Unlike leading-strand DNA replication, lagging-strand DNA is synthesised discontinuously as the replication fork moves in the opposite direction to the polymerase. As the replication fork progresses, newly exposed DNA on the lagging strand is continuously primed by primase/pol α then extended by pol δ with the assistance of PCNA. These segments of DNA on the lagging-strand are known as Okazaki fragments, and it is estimated that human DNA replication generates ∼50 million per cell cycle (Burgers 2009). To form a continuous piece of DNA, the RNA primer, and possibly the DNA laid down by pol α, must be removed, with DNA being subsequently ligated to complete Okazaki fragment maturation (Fig. 16.1a). Initial experiments aimed at reconstituting the DNA replication machinery in vitro using fractionated nuclear extracts identified a maturation factor (MF1) necessary for the completion of lagging-strand DNA replication (Waga et al. 1994). This maturation factor was later renamed Flap Endonuclease due to the enzyme's preference to cleave bifurcated DNA structures with displaced 5′-single-stranded DNA flaps (Fig. 16.1b–d) (Harrington and Lieber 1994). Flap endonucleases (FENs), which are present across all domains of life, are divalent metal ion-dependent nucleases, whose phosphodiesterase activity enhances the hydrolysis rate of targeted phosphodiester bonds at least ∼1017 fold (Tomlinson et al. 2010). FENs possess a single active site that can perform both endo-and exonucleolytic cleavages. Furthermore, FENs are considered prototypical members of the 5′-nuclease superfamily, which includes enzymes with diverse DNA processing activities such as EXO1, XPG and GEN1 (Finger and Shen 2010; Tomlinson et al. 2010; Grasby et al. 2011), as well as the 5′-exoribonucleases Xrn1 and Rat1 (Solinger et al. 1999; Yang 2010).
Fig. 16.1.
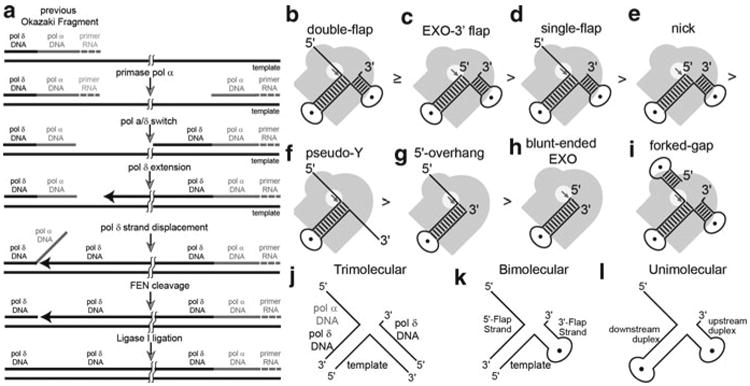
The role of FENs in lagging-strand DNA replication and various FEN substrates in vitro. (a) Simplified diagram of the Okazaki fragment maturation (b–i) Various activities that can be observed with FENs and model substrates in vitro. The grey image represents the protein with the active site highlighted by the white circle to show how these activities are achieved. (b–h) The strength of each activity is indicated by the order they are placed as illustrated by the symbols between each. (i) Example of a forked-gap substrate. (j–l) Examples of double flap constructs used in biochemical studies. Illustrations are labelled to (j) show how in vivo Okazaki fragments correspond to in vitro substrates and (k) each region or (l) component of the substrates are referred to as herein
Current paradigms of eukaryotic DNA replication are based mainly on studies in yeast (Burgers 2009). FENs were once thought to be solely responsible for cleavage of flaps of any length in vivo, but studies identified two additional proteins involved in yeast Okazaki fragment maturation – Pif1 and Dna2 (Budd and Campbell 1997; Budd et al. 2006). Pif1, a member of the IB helicase superfamily (Bochman et al. 2010), has been shown to increase the efficiency of pol δ strand displacement synthesis, thereby resulting in long (>30 nucleotides) 5′ ssDNA flaps. Exposure of 30 or more nucleotides of ssDNA recruits Replication Protein A (RPA; ssDNA binding protein), resulting in a 5′-flap-ssDNA-RPA complex that is refractory to FEN1 cleavage. To remove these RPA coated flaps, the RPA-activated Dna2 nuclease/helicase is recruited to imprecisely cleave the long flap, thereby creating a short flap that is then processed by FEN1 (Kang et al. 2010). The relevance of this yeast “two-step” Okazaki fragment maturation model to mammalian systems remains unclear. Nonetheless, both short and long flap pathways require the ability of FEN to cleave with exquisite precision to create a product that is a substrate for DNA ligase. Primers that are incorrectly processed or not removed by FEN would create gaps or overlaps, respectively, resulting in genomic instability, as seen in studies of budding and fission yeast lacking Rad27/Rad2 (FEN1 homologues in Saccharomyces cerevisiae and Schizosaccharomyces pombe, respectively) (Johnson et al. 1995; Reagan et al. 1995), which display severe mutator phenotypes (Liu et al. 2004; Navarro et al. 2007).
The importance of FEN is further highlighted in higher eukaryotes, where homozygous deletion of the fen1 gene (fen1−/−) is embryonically lethal in mice (Larsen et al. 2003), indicating that FEN1 is absolutely essential in mammals. FEN1 is expressed in all proliferative tissues in humans, including cancers (Kim et al. 2000; Warbrick et al. 1998). FEN1 expression levels in normal tissue are correlated with proliferative capacity. In addition, FEN1 over-expression in human cancers has been linked to tumour aggressiveness (Finger and Shen 2010); for this reason, the chemotherapeutic potential of FEN1 inhibitors has been investigated (Tumey et al. 2004, 2005). Furthermore, mutations that decrease expression levels or alter FEN1 biochemical properties predispose humans and mice to cancers (Kucherlapati et al. 2002; Larsen et al. 2008; Zheng et al. 2007b). Thus, a paradox of FEN activity emerges: optimal FEN1 activity is essential to prevent cancer, but overabundance or impaired function of FEN1 can promote cancer by increasing the efficiency of DNA replication and repair or reducing fidelity of DNA replication respectively.
An area of controversy regarding FENs is how the enzyme is able to precisely cleave at a single phosphate diester to create a ligatable nick based solely upon the structure instead of the sequence of the DNA. Early studies established that prokaryotic and eukaryotic FENs recognize the structure of bifurcated 5′-flaps rather than sequence (Harrington and Lieber 1995; Lyamichev et al. 1993), but controversy as to how this was accomplished quickly emerged. Dahlberg and co-workers suggested that specificity was achieved by threading the 5′-flap through a hole in the FEN protein. Subsequent biochemical studies on mammalian FENs supported this proposal and suggested that these enzymes thread the 5′ flap through a hole in the protein until it encounters the dsDNA, whereupon cleavage occurs (Murante et al. 1995). Structural studies with bacteriophage T5 FEN (T5FEN) revealed a helical archway above the active site whose dimensions could only accommodate ssDNA (Ceska et al. 1996), lending support to threading models. Further biochemical and structural work subsequently suggested instead that the helical arch was actually used to clamp onto 5′-flap ssDNA at the 5′-terminus and to then track along the flap until dsDNA was encountered and subsequently cleaved (Bornarth et al. 1999; Chapados et al. 2004). Alternatively, studies using the E. coli FEN homologue led Joyce and colleagues to suggest that FENs initially recognize the dsDNA portion of 5′-flap substrates and then, thread the 5′-flap DNA (Xu et al. 2001). More recent evidence from several groups suggests that the latter model is a better mechanistic description of eukaryotic FENs (Finger et al. 2009; Gloor et al. 2010; Hohl et al. 2007; Stewart et al. 2009). Furthermore, X-ray crystallographic studies of enzyme-substrate and enzyme-product complexes of human FEN1 (hFEN1) have shed light on how FENs identify their substrate and select the scissile phosphate (Tsutakawa et al. 2011). Here, we review both biochemical and structural aspects of FEN1 that give rise to a structure-specific nuclease with exquisite scissile phosphate diester selectivity, and then, discuss how this protein is assisted and regulated in vivo by sub-cellular localization, protein interaction partners, and post-translational modification.
16.2 Biochemical Activity
FENs cleave a large range of substrates in vitro with a 5′ to 3′ polarity both endo-and exo-nucleolytically (Fig. 16.1b–i) (Nazarkina et al. 2008; Shen et al. 2005). The divalent metal ion-dependent phosphodiesterase activity of FENs exclusively generates 5′-phosphate monoester and 3′-hydroxyl products (Pickering et al. 1999). However, the catalytic efficiency on each type of substrate structure varies greatly. In addition to DNA replication, FENs have also been implicated in other DNA metabolic pathways due to the ability to observe activities with certain substrates (Zheng et al. 2011b).
Eukaryotic and archaeal FENs prefer substrates with two dsDNA regions (Fig. 16.1b–e), but substrates having only a single dsDNA region of at least 12 base pairs can be cleaved as well, albeit weakly (Fig. 16.1f–i). Single 5′-flap substrates (Fig. 16.1d) were initially thought to be the preferred substrate of FENs (Harrington and Lieber 1995; Lyamichev et al. 1999), but 5′-flap and EXO substrates also having a single nucleotide 3′-flap (i.e., a double-flap substrate) were later shown to be the preferred substrates for eukaryotic FENs (Fig. 16.1b,c) for three reasons. Substrates bearing 3′-flaps have lower apparent KDs (Friedrich-Heineken and Hubscher 2004; Kao et al. 2002), are cleaved with greater efficiency than their single-flap cognates, and are cleaved exclusively at the phosphate diester between the first and second nucleotide of the downstream duplex. Importantly, this increased precision results in all dsDNA product being ligatable (Fig. 16.2a) (Finger et al. 2009; Kao et al. 2002). With single-flap substrates, cleavage is less efficient and predominantly occurs at the dsDNA-ssDNA flap junction, but to a lesser extent also one nucleotide into the downstream duplex (Fig. 16.2b) (Finger et al. 2009; Kao et al. 2002). The minor cleavage product results in a 1-nt gap that would require post-replication repair mechanisms to fill in and close the gap (Chapados et al. 2004; Finger and Shen 2010). Lower organism FENs, such as those from the T4 and T5 bacteriophages, do not possess a 3′-flap binding pocket (Friedrich-Heineken and Hubscher 2004; Shen et al. 1998). Thus, FENs from higher organisms have evolved such a feature to ensure that cleavage results in immediately ligatable nicks, avoiding the need for initiation of DNA repair mechanisms.
Fig. 16.2.

The importance of the 3′-flap in vitro and in vivo. Reactions on (a) double flaps result in all dsDNA product being a ligatable nick, whereas (b) single flaps produce a minor product that is a 1 nt gap. Examples of static (i.e., single conformation) double-(c) and single-(d) flaps that are commonly used in vitro. (e) Flaps generated in replication are potentially migrating flaps (equilibrating flaps) and can theoretically form multiple structures. Those labelled here represent the two conformations important for polymerase and FENs. (f) Diagram of the FEN reaction pathway on double-flap substrates deduced from studies of FENs to date. Note this is only a model, and there may be more steps in the reaction pathway than illustrated. The FEN enzyme (E) binds its substrate (S) to form an enzyme-substrate complex ([ES]). To be able to cleave the substrate, the protein, DNA, or both have to change conformation to create a cleavage competent complex ([ES]″). Upon cleavage, ssDNA (P) and dsDNA (Q) products are created. The ssDNA (P) likely dissociates form the complex immediately upon cleavage resulting in the enzyme-dsDNA product complex ([EQ]). The dissociation of [EQ] results in nicked dsDNA and enzyme turnover. The rates (k) associated with each steps are listed above or below the corresponding arrow: kon – bimolecular association (i.e., diffusion), koff – dissociation, kCC – conformational change, kRCC – reverse conformational change, kchem – chemical catalysis, krelease – product dissociation, kPA – product association. Note, kPA can be ignored when measuring initial rates of reaction. The macroscopic rate constants commonly measured kinetically are above or below a bracket that encompasses the rates that can in fluence the measured parameter. Because reactions with FEN have intermediates after initial [ES] formation, the KM is an overall dissociation constant for all enzyme-bound species ([ES] + [ES]″ + [EQ]). The turnover number (kcat) in WT FENs is mainly a reflection of the slowest step (enzyme product release), but can be affected by other first order rates in the reaction pathway. The second order rate constant (kcat/KM) for WT is mainly a measure of diffusion (kon), but mutants of FENs can sometimes change and represent anyone or some combination of steps within the bracket. The rate measured under single turnover conditions can measure any rates after initial [ES] complex formation and before [EQ] release and is a measure of some physical limitation such as conformational change in WT FENs
The substrates used in vitro are commonly designed to exclusively form a 5′-flap of a known length, with or without a single nt 3′-flap. These types of flap substrates are referred to as static double-flap and single-flap substrates, respectively (Fig. 16.2c,d), and are prepared either as tri-, bi-, or uni-molecular constructs (Fig. 16.1j). The ‘template strand’ oligonucleotide corresponds to the lagging-strand template in vivo. The strand that base pairs with the template to form the upstream duplex region is equivalent to the nascently synthesized pol δ DNA, whereas the strand that anneals to form the downstream dsDNA corresponds to pol α and/or pol δ synthesized DNA of the previous Okazaki fragment depending on the extent of strand displacement synthesis (Fig. 16.1j–l) and assuming that RNaseH has already removed all but the last nucleotide of the RNA primer (Chon et al. 2009; Mesiet-Cladiere et al. 2007; Qiu et al. 1999). In vivo pol α and pol δ use the same template, so the sequences they synthesise should be identical. Thus, the 5′-flap structures generated in vivo are ‘equilibrating’ double-flap substrates (Liu et al. 2004); as such, overlapping sequences can form multiple structures of varying 5′ and 3′ flap lengths by a mechanism analogous to Holliday junction migration (Fig. 16.2e). Model ‘equilibrating’ double flap structures are cleaved at a single phosphate diester in a manner analogous to the cleavage of a static double-flap substrate (i.e., one nucleotide into the downstream duplex) (Kao et al. 2002). The 3′-flap in the static and equilibrating double-flap substrates corresponds to the last nucleotide added by pol δ during strand displacement synthesis (Figs. 16.1a and 16.2e). Thus, FENs have evolved to recognize the last nucleotide added by pol δ and to identify the scissile phosphate accordingly. In addition to increased specificity, the 3′-flap augments “enzyme commitment” to the forward reaction by increasing first-order rates of reaction after initial enzyme substrate complex formation (Fig. 16.2f) (i.e., kCC ≫ koff) (Finger et al. 2009). In fact, the catalytic efficiency of FEN1 on a static double flap substrate approaches enzyme:substrate association rates in solution. Therefore, FEN reactions with static double flap substrates may be diffusion controlled under conditions whereby substrate is limiting ([E] < [S] < KM; kcat/KM conditions), implying that the enzyme has reached catalytic perfection (Sengerova et al. 2010).
Under saturating multiple turnover (MT) conditions ([S]> > KM > > [E]), the enzyme is rate limited by enzyme product release as single turnover (ST) rates of hydrolysis are faster than MT rates (Fig. 16.2f) (Finger et al. 2009; Williams et al. 2007). On double-flap substrates, FENs produce two products (denoted P and Q), which with a double-flap substrate would be nicked dsDNA product (Q) and a small ssDNA fragment (P). Product inhibition studies have deduced that the ssDNA product (P) is instantaneously released after cleavage or released much faster than the dsDNA product, whereas the dsDNA product (Q) is retained (Finger et al. 2009). Thus, release of the dsDNA product is rate limiting in vitro under MT conditions (Fig. 16.2f). The fact that FENs hold onto the dsDNA product is similar to the observation that many DNA metabolic enzymes chaperone their potentially toxic repair intermediates (Parikh et al. 1999). Furthermore, in FEN1-product DNA crystals, only electron density for the hFEN1-dsDNA product (Q) and not for the ssDNA flap (P) was observed, consistent with FEN retaining only the dsDNA product after cleavage (Tsutakawa et al. 2011). Thus, FENs release the 5′-flaps ssDNA product, but retain the dsDNA product, which is the substrate for the next step of Okazaki fragment maturation (i.e., ligation).
Because kcat/KM conditions are likely diffusion controlled and saturating MT conditions are rate-limited by enzyme product release (Fig. 16.2f), MT measurements of reaction of FENs are probably not physiologically relevant, as these physical limitations likely do not exist in the cell (Berg and von Hippel 1985) due to sequestration of FENs to replication forks (Beattie and Bell 2011) and potential PCNA-mediated handoff (see Chap. 15, this volume) of replication intermediates between processing proteins (i.e., pol δ and Ligase I, respectively) (Chapados et al. 2004; Tsutakawa et al. 2011). A more physiologically relevant measurement of FEN activity is single-turnover experiments where [S] < KM < <[E]. These experiments measure the rates of reaction after enzyme-substrate complex formation and before enzyme product release (Fig. 16.2c). Despite knowing the rate of reaction of the rate-limiting step under ST conditions, it is difficult to know exactly what is being measured (e.g., kchem or a physical step in the reaction cycle like kCC/kRCC.) So far, studies of T5FEN have shown that a physical limitation (e.g., protein, DNA or concerted protein/DNA conformational change (kCC)) is rate limiting under ST conditions (Fig. 16.2f) (Sengerova et al. 2010). This is likely to be the case for eukaryotic FENs in vitro as well.
16.3 FEN Structure and Substrate Recognition
16.3.1 Free Protein
Crystal structures of FENs lacking substrate have been solved from a wide variety of organisms (Ceska et al. 1996; Chapados et al. 2004; Hos field et al. 1998b; Hwang et al. 1998; Mase et al. 2009; Sakurai et al. 2005, 2008). These have revealed a common architecture among FENs. The nuclease core domains of FENs fold into an α/β structure known as a SAM or PIN fold (Fig. 16.3a). Moreover, a mixed twisted β-sheet of (usually) seven-strands is sandwiched between two α-helical regions and forms a saddle-like structure. Two sites for binding dsDNA are on either side of the β-sheet. Protruding from the saddle-like structure is the region called the helical arch or helical clamp that in some crystal structures is observed as a disordered, flexible loop, and in others is structured as two α helices poised above the active site. The active site of FENs are highly conserved and in eukaryotic and archaeal FENs, and is characterized by the presence of seven acidic residues (Lieber 1997; Shen et al. 1998) (Fig. 16.3a) that sequester the divalent metal ions requisite for catalysis. Consistent with a two metal ion mechanism for phosphate diester hydrolysis (Yang et al. 2006), these acidic residues in the hFEN1 substrate-free structure hold the divalent metal ion <4 A apart; however, the occupancy in the crystal structures varies for the metals (Sakurai et al. 2005). An unusual feature of archaeal and eukaryotic FENs is that their N-termini are structured and lie close to the active site. If electron density for the N-terminus can be seen, the sequence starts with Gly2, and thus, N-terminal aminopeptidases have removed the initiator methionine during translation (Bradshaw et al. 1998), at least when expressed as a recombinant protein in E. coli. The observation of structured N-termini near the active sites of FENs explains why cloning FENs with N-terminal affinity tags results in proteins that are insoluble (i.e., GST tagged protein) (Zheng and Shen, unpublished data) or have negligible activity (i.e., His-tags that are cleaved off leaving additional N-terminal residues) (Grasby and Tainer, unpublished data) (Mase et al. 2009). This is the reason for the exclusive use of C-terminal affinity tags for recombinant expression.
Fig. 16.3.

Comparison of DNA-free hFEN1 and DNA-bound hFEN1. Structures of (a) hFEN1 without DNA and (b) hFEN1-product dsDNA with domain maps to highlight the ordering of the helical gateway and cap. Filled circles and triangles represent α-helices and β-sheets, respectively, and are numbered accordingly. The open circle represents a single helical turn of 4 amino acids. The approximate locations of the seven acidic residues for divalent metal sequestration and of the three amino acid residues responsible for coordination of the K+ ion are represented by filled red stars and purple circle, respectively. Structures of the enzyme-product (c) and enzymesubstrate (d) complexes with the protein illustrated as ribbon diagrams with translucent surface representations. The DNA template, 5′-flap and 3′-flap strand are shown in brown, yellow and purple, respectively
16.3.2 Protein-Product and Protein-Substrate Complexes
Comparison of the crystal structures of human FEN1 with product and substrate DNA revealed an extraordinarily sophisticated dsDNA binding and ssDNA incision mechanism (Tsutakawa et al. 2011). The proposed model allows FEN to distinguish 5′-flap structures from 3′-flap structures and ssDNA and is biologically elegant in eliminating non-FEN substrates from inadvertent incision. First, we will discuss structural elements common to both the enzyme-substrate and enzyme-product complexes with respect to the template and 3′-flap strand interactions (Fig. 16.1l). Then, we will discuss the differences that exist between the two complexes at the extreme 5′-end of the 5′-flap strand.
The protein-substrate and protein-product complexes show that most of the binding surface is to the two dsDNA regions. The upstream dsDNA is bent at approximately a 100° angle at a single phosphate diester relative to the downstream dsDNA. Helix 2 of FEN is wedged between the basepairs formed at either side of the two-way dsDNA junction (Fig. 16.3c, d). Thus, an initial recognition mechanism is binding dsDNA containing a junction that can easily bend at a single phosphate diester. Recognition of the substrate is not mediated through the 5′ flap DNA strand as would be expected according to the original threading/clamping and tracking models, but via interactions with the template DNA strand. Moreover, approximately half of the protein:DNA interface is directed to the template strand (Fig. 16.4a–d), which corresponds to the parental strand of DNA in replication (Fig. 16.1j). Part of this interaction is mediated by a potassium ion that that is coordinated to amino acid peptide carbonyls from a helix-two-turn-helix motif (H2tH), a side chain hydroxyl of a serine residue, and a phosphate diester from the template DNA (Fig. 16.3b–d, 16.4a–f). In addition, there are contacts to the 5′-flap strand near its 3′-terminus (Fig. 16.4c–d). Another facet to the dsDNA binding is that the protein-dsDNA interaction surface is not contiguous. The downstream portion of the template strand departs from the surface of the protein after the H2tH motif and then, returns to the surface of the protein just before the bend in the DNA. The template strand enters a groove where the DNA bends sharply, giving rise to the 100° bend at the two-way dsDNA junction. The arc that is formed by the template strand (template arc) as it leaves and then, returns to the protein surface is used to deliver the 5′-flap strand to the proximity of the active site (Fig. 16.4a, b). Moreover, as the downstream primer strand follows the template strand, it is the 5′-terminus of the downstream flap strand that is directed towards the active site. When delivered to the proximity of the active site, helix 2 and helix 4 residues contact the 5′-terminal portion of the 5′-flap strand. However, the way in which these residues interact with the 5′-terminal residues of the downstream primer differs between the enzyme substrate and enzyme product complexes, and will be dealt with in the next two sections (Tsutakawa et al. 2011).
Fig. 16.4.

FENs effectively utilize the helical properties of DNA to deliver the scissile phosphate to the active site. Due to the spacing of the dsDNA binding regions and the bend induced on the template strand of the two-way DNA junction, the template arc directs the 5′-flap strand towards the active in both the enzyme product (a) and enzyme substrate (b) complexes. The DNA strands and translucent surface representation of the protein are coloured as in Fig. 16.3. The two trivalent samarium ions (Sm3+) and the K+ ions are represented as cyan and purple spheres, respectively. Looking at the DNA alone and representing sites of protein contact to DNA by spheres, the product (c) and substrate (d) DNA are almost the same except for important changes near the scissile phosphate. Note the lack of direct contacts in the template arc region. Focussing on the downstream duplex region, comparison of the product (e) and substrate (f) DNA near the active site shows that the two nucleotides have unpaired for the scissile phosphate to interact with the catalytically important metal ions. In addition, the Gly2 N-terminus (G2NT) interacts initially with the scissile phosphate diester in the enzyme substrate complex (f), but interacts with the phosphate diester 3′ to the scissile phosphate in the enzyme-product complex (e). (g) View of the active site (coloured as above) from the back of the helical gateway and helical cap to highlight some of the catalytically important residues. (h) View of the complete hFEN1 active site that includes the seven highly conserved carboxylates, two basic residues from helix four (Lys93 and Arg100), and an aromatic stacking partner (Tyr40) from helix two. (i) View of the four active site acidic residues (D86, E160, D179, and D181) and product complex unpaired base phosphate monoester directly coordinating the Sm3+ metals (grey dashed lines)
Despite being base paired to the upstream nucleotides of the template, the only interactions seen with the 3′-flap strand are localized to its last three nucleotides. The rest of the upstream primer strand does not contribute to binding and passively exits from hFEN1 by following the template DNA (Fig. 16.4c, d). Most of the interactions are to the one nucleotide 3′-flap. The 3′ flap binding pocket is constructed by ten amino acid residues, with most of the protein interaction with the DNA to the sugar-phosphate backbone and the 3′-hydroxyl group. None of the interactions are base-specific, as would be expected for a structure rather than sequence specific nuclease. The 3′-flap is bound unpaired, as would be needed to obtain a ligatable product from an equilibrating flap (Fig. 16.2a). The cleft formed by the 3′-flap binding site physically blocks anything larger than a one nucleotide 3′-flap being accommodated, consistent with biochemical experiments with substrates where the 3′-flap sugar moiety was modified (Kao et al. 2002). In conjunction with the 3′-flap binding pocket, a conserved group of acidic residues (denoted as the acid block) in the proximity of the 3′-flap binding pocket and presumably is present to prevent DNA moving beyond this point. Both the 3′-flap binding pocket and the acid block are features that are present only in FENs of the 5′-nuclease superfamily, suggesting this feature has evolved to enhance FEN function. To a lesser extent, there are also interactions between residues of helix 3 and the penultimate nucleotide of the 3′-flap strand. These residues are part of the hydrophobic wedge that stabilizes the bent conformation of the two-way dsDNA junction by interacting with the face of the last base pair created by the 3′-flap strand and template. In the free protein structures (Sakurai et al. 2005), the residues involved in 3′-flap binding are disordered (Fig. 16.3a), suggesting the 3′-flap pocket orders upon recognition and binding of this nucleotide. The only structural feature of the 3′-flap that distinguishes it from all other nucleotides in the upstream primer strand is the presence of a 3′-hydroxyl. Thus, it is not surprising that the 3′-hydroxyl is used a key recognition feature by FEN.
16.3.3 Protein-Product Complex 5′-Strand Interactions
Crystallization of hFEN1 in complex with a four nucleotide ‘quasi-equilibrating’ double flap substrate resulted in an enzyme product complex where the dsDNA was still in complex with the protein, consistent with the dsDNA product (Q) being a competitive inhibitor (Fig. 16.2f) (Finger et al. 2009). Unlike structures of hFEN1 in the absence of DNA, the residues of the helical arch and the top of helix two are ordered in a gate-like conformation (Fig. 16.4g). Helix two and most of helix four form the posts of the gateway, whereas the upper portion of helix 4 and all of helix 5 sits atop the two posts, and has been referred to as the helical ‘cap’ (Fig. 16.3c,d). This nomenclature diverges from the previous arch or archway, which does not include helix two. The helical gateway is 13 Å at its most narrow and is located over the active site at the base of the arch. This region of the protein would not be able to order if dsDNA is in the gateway. Although FENs can cleave flaps containing dsDNA (Fig. 16.1i), a sufficient number of single-stranded nucleotides must be present between the dsDNA in the flap and the two way-dsDNA junction to allow the helical gateway to order around the ssDNA portion of the flap and to bring critical catalytic residues into the active site. The helical cap sterically limits passage through the gateway to substrates with free 5′ terminus (i.e., no bubbles, bulges, etc.) (Tsutakawa et al. 2011). Interestingly, the helical gateway is strongly conserved in the 5′-nuclease superfamily. The cap, on the other hand, is conserved only in FEN1 and EXO1. GEN1 and XPG, which cleave Holliday junctions and DNA bubbles, respectively, do not appear to have a cap (Orans et al. 2011; Tsutakawa et al. 2011).
In the product complex, the nucleotide just to the 3′-side of the cleaved phosphate diester is unpaired, with the 5′-phosphate monoester product interacting with the two divalent metal ions in the active site. Comparison of the product and substrate complexes (Fig. 16.4c–f) shows that two downstream dsDNA nucleotides 5′ and 3′ of the scissile phosphate must unpair to enter the active site. Tyr40 of helix two stacks against the 3′-face of the base of the unpaired nucleotide, thereby stabilizing the unpaired state (Fig. 16.4g). Two residues of helix four, Lys93 and Arg100, interact with the terminal phosphate suggesting that these residues are also important in stabilizing the unpaired state. These conserved residues were also identified in a screen of toxic yeast rad27 mutants that had a dominant negative effect on cell viability and growth (Storici et al. 2002). In DNA-free FEN structures where the helical cap and gateway are disordered, Lys93 and Arg100 (or their equivalent homologues) are not near the active site (Hos field et al. 1998b; Hwang et al. 1998; Sakurai et al. 2005). Furthermore, evidence from studies of T5FEN suggests that Lys83, the equivalent of hFEN1 Lys93, acts as an electrophilic catalyst (Sengerova et al. 2010). Once flipped, the N-terminus of Gly2 contacts the phosphate diester of the next nucleotide in the 5′-flap strand. The trajectory of the phosphate backbone through the active site of the enzyme product complex suggests that the substrate 5′-flap would traverse the helical gateway under the helical cap (Tsutakawa et al. 2011) (Fig. 16.4e). This strongly suggests that a bind-then-thread mechanism of action. However, this is still being debated (Patel et al. 2012).
16.3.4 Protein-Substrate Complex 5′-Flap Strand Interactions
Overall, the protein-substrate and protein-product complexes share similar features in binding, except for differences in the location of some downstream/flap strand primer nucleotides and amino acid residues involved in the interaction with these two residues (Fig. 16.4d). Unlike the enzyme-product complex, the downstream 5′-flap strand of the enzyme substrate complex is base paired to the template strand, and the scissile phosphate is in the proximity of the active site, but not in contact with the critical active site metals or amino acid residues. Despite not being in the active site, the protein-substrate complexes show that binding of the dsDNA alone in this bent, non-contiguous manner results in the scissile phosphate being placed within 5–8 Å of the active site in enzyme substrate complexes. Moreover, the binding of a two-way dsDNA junction in this orientation is important to place the scissile phosphate in the proximity of the active site. By allowing the structural features of the placement of the scissile phosphate diester to be mainly with the template strand and 3′-flap rather than the 5′-flap strand itself may prevent inadvertent incision of ssDNA in the 5′-flap, creating products that would require DNA repair processes to correct the imprecise cleavage. The lack of interaction with the 5′-flap strand allows cleavage of substrates containing RNA or DNA in the 5′-flap or near the site of incision, which is consistent with the fact that deletion of RNase H in yeast does not result in a significant mutator phenotype (Qiu et al. 1999). The spacing between dsDNA binding regions of the protein formed by the potassium ion/H2tH and at the DNA bend, which are approximately one helical turn apart, is also critical to exclude incision of 3′-flaps. Using models, the spacing between dsDNA binding sites is too wide for positioning a 5′-flap near the active site. FENs, therefore, elegantly take advantage of the helical properties of two-way DNA junctions to position the scissile phosphate diester close to the active site and to distinguish 5′-flap and 3′-flap structures (Tsutakawa et al. 2011)
As noted above, one-fourth of the protein-DNA interactions are made by the helical gateway of the protein and the nucleotides at the base of the 5′-flap around the scissile phosphate. In the product complex, there is little direct interaction between the protein and the downstream primer strand between the gateway and the potassium ion binding regions (Fig. 16.4a–d). Tyr40 in helix 2 stacks with the 5′-face of the base of the terminal 5′-flap strand nucleotide, whereas another helix 2 residue, Ile44, contacts the 5′-face of the complimentary nucleotide of the template strand. In addition, the scissile phosphate in the enzyme substrate complex is in contact with the N-terminus (Gly2) (Fig. 16.4d), instead of the phosphate diester 3′ to the scissile phosphate in the enzyme-product complex (Fig. 16.4c). In the paired substrate complex, the terminal base pair of the downstream dsDNA makes only one of two possible H-bonds due to a pronounced base pair opening and stagger towards the major groove. Although the DNA helical parameters of the upstream duplex conform to B-DNA, the helical parameters of the downstream duplex differ. The dsDNA in contact with the K+ ion and the H2tH motif conforms to B-DNA (Fig. 16.3c,d), but the six base pairs of the downstream dsDNA nearest to the active site deviate from B-DNA parameters and become more like A-DNA (Fig. 16.4d). In this region, the nucleotides of the 5′-flap strand DNA are less stacked (Tsutakawa et al. 2011).
16.3.5 Bind-Then-Thread or Bind-Then-Clamp
Despite this advance in our understanding of FEN substrate recognition, it is still unknown whether the 5′-flap portion of the substrates is threaded through the arch consistent with the bind-then-thread model (Xu et al. 2001), or if the flaps are instead clamped by the helical arch (Chapados et al. 2004; Orans et al. 2011). Evidence for the bind-then-thread model comes from the structure of T4 FEN in complex with a pseudo-Y substrate, which suggests that the 5′-flap traverses the archway (Devos et al. 2007). However, in this structure, a portion of the helical arch is not observed and the 5′-flap is not in the active site due to the absence of the requisite divalent metals. The bind-then-thread model would be consistent with the detrimental effects on incision activity with substrates where the 5′-flap has been modified with streptavidin as passage through the helical arch would be blocked by the addition of a molecule as large as tetrameric streptavidin (Gloor et al. 2010; Murante et al. 1995). However, because the structured helical arch is not large enough to accommodate dsDNA, the ability to cleave a fold-back flap substrate is said to be more consistent with the bind-then-clamp model (Finger et al. 2009). To counter this, the helical arch region in several DNA-free FEN structures from several archaea and eukaryotes has been observed to be unstructured (Fig. 16.3a) (Hosfield et al. 1998a; Hwang et al. 1998; Mase et al. 2009; Sakurai et al. 2005). An unstructured arch is theoretically large enough to accommodate double stranded DNA, and could then reorder around the ssDNA portion of the fold-back flap structure (Tsutakawa et al. 2011). Thus, a bind-then-thread model, whereby substrates containing dsDNA in the flap are threaded through an unstructured arch with subsequent ordering after threading, is plausible (Patel et al. 2012).
16.3.6 Scissile Phosphate Placement: The DoubleNucleotide Unpairing Trap (DoNUT)
A disorder-to-order transition of the helical arch (gateway and cap) induced by DNA binding is suggested by comparing DNA-bound hFEN1 structures with DNA-free hFEN1 structures that had previously been solved in complex with PCNA, where three hFEN1 molecules were bound to one PCNA homotrimer with each in a unique crystallographic environment (Sakurai et al. 2005) (Fig. 16.5a,b). In all three DNA-free structures, the helical gateway and cap, as well as the hydrophobic wedge of helix two were disordered (Fig. 16.3a). Furthermore, the catalytically critical residues in helix four (Lys93 and Arg100) and helix two (Tyr40) were not correctly positioned. Thus, we proposed that the binding of the 3′-flap likely orders the hydrophobic wedge, which in turn leads to the ordering of the helical gateway and cap. When ordered, Lys93 and Arg100 are poised in the active site (Fig. 16.4g,h). Because ordering has occurred without unpairing in the fully base-paired substrate complexes, we propose that unpairing occurs after the ordering of the helical gateway. Although base flipping can occur via movement through the major or minor groove (Bouvier and Grubmuller 2007), the dinucleotide unpairing inferred by comparing enzyme-substrate and enzyme-product complexes would likely occur via the major groove, because as the minor groove pathway is blocked by the hydrophobic wedge of helix two. This is consistent with the pronounced base pair opening and stagger towards the major groove in the enzyme-substrate complex (Tsutakawa et al. 2011)
Fig. 16.5.

How FENs work with other proteins. (a) Structure of homotrimeric hPCNA in complex with three subunits of hFEN1. The nuclease core domain is coloured as in Fig. 16.3. PCNA subunits are shown as combined ribbon and transparent surface representation. The portion of the extended C-terminus of hFEN1 that was observed in the crystal (residues 336–356) and the regions important for interaction in PCNA are shown in yellow and magenta, respectively. (b) Turning the structure 90° shows that the protein is positioned on one face of the protein. (c) Closer view of the βA-αA-βB motif with schematic illustration below it coloured as above. Alpha helices are represented as cylinders and arrows indicate the position of the β-sheets. (d) Model of hFEN1 interacting with PCNA and DNA simultaneously and coloured as above. (e) Comparison of the hFEN1 (blue) DNA (coloured as above) structure with the pol β-DNA (Purple) structures shows that the only regions available for FENs to initially contact in a handoff model is the downstream dsDNA binding region. (f) Comparison of the DBD domain of ligase (green) and its known interaction site (minor groove) in comparison to hFEN1 (blue) DNA product complex (coloured as above). The groove necessary for DBD interaction is accessible. Below is a translucent surface representation of the ligase DBD showing a steric clash between the DNA and the helical cap of hFEN1. This steric interference may be the initial manner in which ligase facilitates hFEN1 release of its product
But what drives the unpairing of the DNA? It has been shown by von Hippel and co-workers that nucleotides at ssDNA-dsDNA junctions are much more prone to spontaneous base flipping than nucleotides farther away from the ssDNA-dsDNA junction (Jose et al. 2009). Therefore, FENs may take advantage of this and simply capture the unpaired state. Comparison of the helical parameters of the enzyme-substrate and enzyme-product complexes shows that some of the distortions from B-DNA parameters in the downstream dsDNA of the enzyme-substrate complex are alleviated in the unpaired state of the enzyme-product complex (i.e., returns to more B-DNA like parameters), thereby suggesting that binding energy may also be used to promote the dinucleotide unpairing (Fersht 1999). Regardless of whether the unpairing process is passive or is actively promoted by the enzyme, the ability of FEN to capture the unpaired state is clearly demonstrated in the product complex (Fig. 16.4c–h).
16.3.7 Cleavage of the Scissile Phosphate Diester: Active Site Structure
In the enzyme-product crystal structure, the active site of hFEN1 contains two samarium (Sm3+) cations, which directly interact with the terminal phosphate of the product complex (Fig. 16.4h,i). The Sm3+ ions are directly coordinated to FEN by four of the seven carboxylate residues that are invariantly conserved in the 5′-nuclease superfamily active site and occupy the same sites that Mg2+ is known to bind in the DNA-free hFEN1 protein structures (Sakurai et al. 2005). Asp86, Glu160, Asp179, and Asp181 make inner sphere contacts with the metal ions, with Glu160 acting as the crucial bridging residue between the two metals. The other three conserved acidic residues in addition to a phenolic hydroxide from a conserved tyrosine (Tyr234) make outer sphere contacts via water molecules. Using a model of a ‘cleavage-competent’ enzyme-substrate complex based upon the enzyme-product complex, we have proposed that the attacking water activated by the two divalent metals hydrolyze the scissile phosphate diester bond in a manner similar to a mechanism proposed for EndoIV (Garcin et al. 2008; Ivanov et al. 2007). However, others have proposed a more traditional two divalent ion mechanism for cleavage, where the nucleophilic water is activated by one of the two metal ions (Orans et al. 2011). More work is necessary to distinguish which mechanism accurately describes hFEN1 catalysis and to identify the mechanism responsible for activating the attacking nucleophile (Yang et al. 2006). Nevertheless, there is consensus that phosphate diester hydrolysis requires two ions, in accordance with functional data (Syson et al. 2008)
16.4 Regulation of FEN1 Activity
The activity of FEN must be tightly controlled and coordinat with other components of the DNA replication machinery. FEN has several protein-protein interaction partners that work with FENs to achieve efficient and faithful copying of the DNA. FEN is also the target of several post-translational modifications, adding a further level of control (Zheng et al. 2011b). FEN interacts with its partner proteins mainly through its C-terminal extension (Guo et al. 2008a). This C-terminus has been implicated in DNA binding in vitro (Friedrich-Heineken et al. 2003; Stucki et al. 2001), but may in fact be an artefact in vitro due to its role of this region in mediating protein-protein interactions in vivo, which would interfere with the ability to bind DNA (see Sect. 16.4.1). The C-terminus is likely disordered in the absence of partner proteins; a disorder to order transition upon partner presence may allow for multiple, simultaneous protein-protein interactions. Furthermore, FENs are sequestered where necessary (e.g., nucleus and mitochondria), and evidence also suggests that even subcellular as well as suborganellar location are regulated (Zheng et al. 2011b)
16.4.1 Protein-Protein Interactions
16.4.1.1 PCNA
Proliferating cell nuclear antigen (PCNA) is the major protein involved in coordination of FEN recruitment and activity in the processing of Okazaki fragments. PCNA is a trimeric ‘sliding clamp’ protein, localised at the sites of Okazaki fragment maturation (see Chap. 15, this volume). PCNA binds and coordinates the action of pol δ, FEN and DNA ligase I (Beattie and Bell 2011; Burgers 2009). Each subunit of the homotrimeric PCNA likely interacts with a particular protein. In support of this notion, archaeal PCNA is a heterotrimer, with each member of the trimer having specificity for either pol δ, FEN or ligase, suggesting a precise architecture in the maturation of Okazaki fragments (Dionne et al. 2003). How a eukaryotic cell could ensure that each subunit of the homotrimeric PCNA is loaded with a single pol δ, FEN1, and ligase is unknown.
PCNA has been shown to interact with FEN1 in the absence of DNA (Wu et al. 1996) through a PCNA binding motif QXX(I/L/M)XXF(F/Y) (Frank et al. 2001). Structural data also shows that FENs utilize amino acid residues Thr336 to Leu356 in their C-terminus to form two β-zippers (β A and βB) separated by a small α helix (αA) to interact with PCNA (Fig. 16.5a–c) (Chapados et al. 2004; Sakurai et al. 2005). Disruption of FEN and PCNA interactions by mutating Phe343 and Phe344 of FEN to alanine (FFAA) results in loss of binding as determined using non-equilibrium binding assays, but does not affect FEN nuclease activity in vitro or display gross phenotypes in yeast (Frank et al. 2001) However, a homozygous FFAA mutation in mice results in newborn lethality and other phenotypes indicative of poor replicative capacity (Zheng et al. 2007a). Furthermore, heterozygous FFAA mice (i.e., FFAA/WT fen1) also show defects in Okazaki fragment maturation and an increased rate of aneuploidy-associated cancers (Zheng et al. 2011a).
In the hFEN1-PCNA co-crystal structure, the various conformations of three hFEN1s were a result of different torsion angles among four amino acid residues of hFEN1 (aa residues 333-336) (Sakurai et al. 2005) (Fig. 16.5a,b). This ‘hinge region’ (Fig. 16.5c) is conserved among eukaryotic FENs. Using the recent hFEN1-product structure in combination with one of the conformations in the FEN-PCNA structure, a model of how FEN and PCNA would work together was generated (Fig. 16.5d). Although upstream dsDNA need only be as short as 6 nts when studying FENs in vitro, the upstream dsDNA for a PCNA/FEN complex would need to be 6 nts plus ∼15 base pairs to allow PCNA-DNA binding as well.
PCNA has been shown to stimulate in vitro FEN activity on static single-flap substrates (Frank et al. 2001; Li et al. 1995; Sakurai et al. 2005). Furthermore, kinetic analyses have shown that this PCNA stimulation occurs by facilitating FEN-DNA complex formation (i.e., decreases KM rather than increasing kcat (Hutton et al. 2009; Tom et al. 2000)). Because kcat for FEN1 is a measure of enzyme-product release under MT conditions (Finger et al. 2009; Williams et al. 2007), it is interesting that the addition of PCNA increases the affinity for substrate but does not affect the rate of enzyme-product release. Obviously more work on this is necessary to determine how PCNA can selectively enhance substrate binding without slowing enzyme-product release.
16.4.1.2 RecQ Helicase Family Interactions
The RecQ helicase family members, WRN (Werner syndrome ATP-dependent helicase) and BLM (Bloom syndrome protein), are also key modulators of FEN activity. WRN co-localises with FEN at stalled replication forks (Sharma et al. 2004), unwinding Holliday junctions and stimulating FEN cleavage in a structure-dependent manner. WRN has also been shown to stimulate both FEN cleavage on single-flap, EXO, and forked-gapped substrates (Sharma et al. 2004; Zheng et al. 2005). FEN-WRN interactions are mediated by a 144 residue domain of WRN that has homology to RecQ helicase, interacting with the terminal 18 residues of FEN's C-terminal domain (Brosh et al. 2001; Guo et al. 2008a; Sharma et al. 2005). These residues are disordered in the (hFEN1) 3-PCNA complex (Fig. 16.5c). WRN, unlike PCNA, purportedly stimulates FEN by increasing its turnover of DNA substrates directly (Brosh et al. 2002). The WRN/BLM binding site is adjacent to the PCNA binding site and should allow direct co-ordination of activities by these partner proteins (Sharma et al. 2005). Both WRN and BLM over-expression can rescue mutants lacking Dna2, indicating their ability to stimulate FEN (Imamura and Campbell 2003; Sharma et al. 2004). WRN is also purportedly required for nucleolar localisation of FEN (Guo et al. 2008b).
16.4.2 Post-translational Modifications
Phosphorylation, methylation and acetylation also play a key role in modulating FEN activity and localisation (Zheng et al. 2011b). FEN is phosphorylated at Ser187 in late S-phase by the cyclin-dependent kinases Cdk1 and Cdk2, in partnership with cyclin A (Cdk1) or cyclin E (Cdk2). Phosphorylation in vitro results in a decrease in nuclease activity, but not DNA binding (Henneke et al. 2003). Phosphorylated FEN is unable to associate with PCNA and possibly also abrogates interactions with other proteins like WRN (Zheng et al. 2011b). Interestingly, Ser187 is buried in both DNA–free and DNA–bound structures of FEN (Sakurai et al. 2005; Tsutakawa et al. 2011). Thus, for kinases to phosphorylate FENs, a conformational change will likely be necessary. PRMT5 methylates Arg192 (Guo et al. 2010), a residue whose side chain contacts DNA in the enzyme-product complex. Methylation prevents phosphorylation, and has the opposite effect; addition of a methyl group facilitates interaction with PCNA (Zheng et al. 2011b). Acetylation occurs on multiple lysine residues of FEN both in vitro and in vivo (Choudhary et al. 2009; Hasan et al. 2001). Acetylation in vitro of four lysines in the extreme C-terminus of the protein results in a decrease in PCNA independent nuclease activity (Hasan et al. 2001). It is speculated that acetylation could lead to blocking of the short flap pathway by retarding FEN activity, leading to Okazaki fragment maturation by the long flap method involving Dna2 (Balakrishnan et al. 2009). The roles of Lys80 and Lys267 acetylation, which were discovered in a genome-wide screen for acetylated proteins (Choudhary et al. 2009), remain unclear. Lys80 is solvent exposed and interacts with PCNA residues in the hFEN1-PCNA co-crystal structure (Sakurai et al. 2005); therefore, its modification could in fluence PCNA interaction as well as other protein interaction partners. Lys267 interacts with the 3′-terminus of the 5′-flap strand (Tsutakawa et al. 2011); therefore, the addition of an acetyl group could regulate downstream dsDNA binding (Zheng et al. 2011b).
16.5 Handoff of DNA Intermediates
Although physical limitations such as diffusion and enzyme-product release sometimes limit reaction rates in vitro, these limitations may not occur in vivo. To increase the efficiency of metabolic processes, cells have overcome physical limitations by sequestering the requisite proteins to their sites of action and channelling or handing-off intermediates from protein to protein in the metabolic pathway, best exemplified by the multifunctional enzyme tryptophan synthase (Yanofsky 1989). This channelling is important to cellular function as it prevents the release of potentially reactive intermediates into solution and prevents the establishment of enzyme-substrate equilibrium (Ovádi 1991). An analogy to DNA metabolic pathways like Okazaki fragment maturation can be hypothesized, but rather than channelling a small metabolite from one active site to another, the metabolic intermediate (i.e., DNA) is passed from protein to protein. Although DNA nicks or flaps may not necessarily be reactive intermediates, release of these species in vivo during Okazaki fragment maturation could result in competition between ligation versus initiation of DNA repair pathways. Although DNA repair pathways can usually faithfully replicate the DNA, these processes are not perfect (e.g., unequal sister chromatid exchange in trinucleotide repeat regions resulting in contractions and expansions). Thus, an attractive mechanism for the processing of Okazaki fragment maturation is for intermediates to be efficiently passed between each protein in the pathway with PCNA coordinating the overall process.
Although there is no direct biochemical evidence yet for this handoff model, support for it does come from structural studies conducted on proteins involved in the pathway. Currently, the best available polymerase structure in complex with downstream and upstream dsDNA is a structure of pol β (Krahn et al. 2004). Although pol β is not involved in DNA replication, it is involved in an analogous process of long-patch base excision repair (LP-BER), (Robertson et al. 2009). Inspection of the pol β-DNA complex shows that the upstream and downstream dsDNA regions are bent ∼100° and that the base of the 5′-flap and 3′-flap and upstream dsDNA duplex are in direct contact with the polymerase (Krahn et al. 2004), and thus, would sterically clash with FENs (Fig. 16.5e). The only region exposed for FENs to initially bind is the downstream dsDNA region. To access the 5′-and 3′-flaps, FENs would first interact with the downstream dsDNA and would then displace the polymerase (Tsutakawa et al. 2011). Because flaps in vivo can potentially migrate (Fig. 16.2e), the handoff of the substrate in this manner would prevent flaps from forming structures that would need to be extensively remodelled before cleavage, thereby ensuring efficient Okazaki fragment maturation. Still, at some point in this hypothetical transition between the polymerase and FEN, the one nucleotide 3′-flap must bind the single nucleotide binding pocket of FEN (Fig. 16.2e).
The structure of DNA ligase I in complex with DNA shows that the enzyme encircles the nicked DNA using three domains: the DNA binding domain (DBD), adenylation domain (AdD) and OB domain (OB) (see Chap. 17, this volume). It is known that the DBD is essential for interaction with DNA substrates (Tomkinson et al. 2006). The DBD interacts with the minor grooves on either side of the nick, whereas the OB fold interacts with the major groove (Pascal et al. 2004). Inspection of the hFEN1-product complex shows that the two minor grooves for DBD interaction are exposed (Fig. 16.5f). However, the product DNA is bent in this structure unlike in the ligase I–DNA structure, where DNA is not bent in order to align the termini for ligation. Despite this, the fact that the grooves necessary for DBD interaction are exposed in the hFEN1-product complex provides further compelling evidence for the handoff model, and suggests that hFEN1 and the DBD of ligase I could bind DNA simultaneously.
In summary, consideration of the buried interfaces of the pol β, hFEN1 and ligase I in complex with DNA shows that the choreography of handoff would be dictated by the exposed regions of dsDNA. As such, it is not surprising that FENs have a tendency to retain the product that is the substrate of the next step of the DNA metabolic pathway and simply release the other upon cleavage. Such a handoff or ‘passing of the baton’ concept (Parikh et al. 1999; Wilson and Kunkel 2000) is a simple but efficient way to envisage how the efficiency of Okazaki fragment maturation can be increased beyond the physical limitations of in vitro studies. The proteins necessary for the process are sequestered to the site of action by PCNA, and the intermediates could be passed between one another (Beattie and Bell 2011; Burgers 2009). Although FENs show a remarkable non-sequence specific ability to identify their substrate while preventing inadvertent activity on other DNA structures, further work to determine how FEN interacts with its partner proteins is likely to be as elegant as the structural and biochemical work to date that has dissected what we already know about this remarkable enzyme.
Acknowledgments
This article was supported by the European Union 7th Research Framework Programme (FP7)-Marie Curie International Incoming Fellowship Project No. 254386 (LDF), National Cancer Institute grants RO1CA081967, P01 CA092584 (JAT), BBSRC grant BBF0147321 (JAG), and RO1CA073764 (BS).
Contributor Information
L. David Finger, Email: d.finger@sheffield.ac.uk, Department of Chemistry, Centre for Chemical Biology, Krebs Institute, University of Sheffield, Sheffield S3 7HF, UK.
John M. Atack, Email: j.m.atack@sheffield.ac.uk, Department of Chemistry, Centre for Chemical Biology, Krebs Institute, University of Sheffield, Sheffield S3 7HF, UK.
Susan Tsutakawa, Email: setsutakawa@lbl.gov, Life Sciences Division, Lawrence Berkeley National, Laboratory, Berkeley, CA 94720, USA.
Scott Classen, Email: sclassen@lbl.gov, Physical Biosciences Division, The Scripps Research, Institute, La Jolla, CA 92037, USA.
John Tainer, Email: jat@scripps.edu, Life Sciences Division, Lawrence Berkeley, National Laboratory, Berkeley, CA 94720, USA, Department of Molecular Biology, The Scripps Research Institute, La Jolla, CA 92037, USA, Skaggs Institute for Chemical Biology, La Jolla, CA 92037, USA.
Jane Grasby, Email: j.a.grasby@sheffield.ac.uk, Department of Chemistry, Centre for Chemical Biology, Krebs Institute, University of Sheffield, Sheffield S3 7HF, UK.
Binghui Shen, Email: bshen@coh.org, Division of Radiation Biology, City of Hope National Medical Center and Beckman Research Institute, Duarte, CA 91010, USA, College of Life Sciences, Zhejiang University, Hangzhou 310058, China.
References
- Balakrishnan L, Stewart J, Polaczek P, Campbell JL, Bambara RA. Acetylation of Dna2 endonuclease/helicase and flap endonuclease 1 by p300 promotes DNA stability by creating long flap intermediates. J Biol Chem. 2009;285:4398–4404. doi: 10.1074/jbc.M109.086397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie TR, Bell SD. The role of the DNA sliding clamp in Okazaki fragment maturation in archaea and eukaryotes. Biochem Soc Trans. 2011;39:70–76. doi: 10.1042/BST0390070. [DOI] [PubMed] [Google Scholar]
- Berg OG, von Hippel PH. Diffusion-controlled macromolecular interactions. Annu Rev Biophys Biophys Chem. 1985;14:131–160. doi: 10.1146/annurev.bb.14.060185.001023. [DOI] [PubMed] [Google Scholar]
- Bochman ML, Sabouri N, Zakian VA. Unwinding the functions of the Pif1 family helicases. DNA Repair. 2010;9:237–249. doi: 10.1016/j.dnarep.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornarth CJ, Ranalli TA, Henricksen LA, Wahl AF, Bambara RA. Effect of flap modifications on human FEN1 cleavage. Biochemistry. 1999;38:13347–13354. doi: 10.1021/bi991321u. [DOI] [PubMed] [Google Scholar]
- Bouvier B, Grubmuller H. A molecular dynamics study of slow base flipping in DNA using conformational flooding. Biophys J. 2007;93:770–786. doi: 10.1529/biophysj.106.091751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw RA, Brickey WW, Walker KW. N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem Sci. 1998;23:263–267. doi: 10.1016/s0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- Brosh RM, Jr, von Kobbe C, Sommers JA, Karmakar P, Opresko PL, Piotrowski J, Dianova I, Dianov GL, Bohr VA. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J. 2001;20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh RM, Jr, Driscoll HC, Dianov GL, Sommers JA. Biochemical characterization of the WRN-FEN-1 functional interaction. Biochemistry. 2002;41:12204–12216. doi: 10.1021/bi026031j. [DOI] [PubMed] [Google Scholar]
- Budd ME, Campbell JL. A yeast replicative helicase, Dna2 helicase, interacts with yeast FEN-1 nuclease in carrying out its essential function. Mol Cell Biol. 1997;17:2136–2142. doi: 10.1128/mcb.17.4.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd ME, Reis CC, Smith S, Myung K, Campbell JL. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase delta. Mol Cell Biol. 2006;26:2490–2500. doi: 10.1128/MCB.26.7.2490-2500.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–4045. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceska TA, Sayers JR, Stier G, Suck D. A helical arch allowing single-stranded DNA to thread through T5 5′-exonuclease. Nature. 1996;382:90–93. doi: 10.1038/382090a0. [DOI] [PubMed] [Google Scholar]
- Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- Chon HG, Vassilev A, DePamphilis ML, Zhao YM, Zhang JM, Burgers PM, Crouch RJ, Cerritelli SM. Contributions of the two accessory subunits, RNASEH2B and RNASEH2C, to the activity and properties of the human RNase H2 complex. Nucleic Acids Res. 2009;37:96–110. doi: 10.1093/nar/gkn913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Devos JM, Tomanicek SJ, Jones CE, Nossal NG, Mueser TC. Crystal structure of bacteriophage T4 5′ nuclease in complex with a branched DNA reveals how flap endonuclease-1 family nucleases bind their substrates. J Biol Chem. 2007;282:31713–31724. doi: 10.1074/jbc.M703209200. [DOI] [PubMed] [Google Scholar]
- Dionne I, Robinson NP, McGeoch AT, Marsh VL, Reddish A, Bell SD. DNA replication in the hyperthermophilic archaeon Sulfolobus solfataricus. Biochem Soc Trans. 2003;31:674–676. doi: 10.1042/bst0310674. [DOI] [PubMed] [Google Scholar]
- Fersht A. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. W.H. Freeman and Company; New York: 1999. [Google Scholar]
- Finger LD, Shen B. FEN1 (flap endonuclease 1) Atlas Genet Cytogenet Oncol Haematol. 2010 (URL: http://www.atlasgeneticsoncology.org/Genes/FEN1ID40543ch11q12.html)
- Finger L, Blanchard M, Theimer C, Sengerova B, Singh P, Chavez V, Liu F, Grasby J, Shen B. The 3′-flap pocket of human flap endonuclease 1 is critical for substrate binding and catalysis. J Biol Chem. 2009;284:22184–22194. doi: 10.1074/jbc.M109.015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank G, Qiu J, Zheng L, Shen B. Stimulation of eukaryotic flap endonuclease-1 activities by proliferating cell nuclear antigen (PCNA) is independent of its in vitro interaction via a consensus PCNA binding region. J Biol Chem. 2001;276:36295–36302. doi: 10.1074/jbc.M103397200. [DOI] [PubMed] [Google Scholar]
- Friedrich-Heineken E, Henneke G, Ferrari E, Hubscher U. The acetylatable lysines of human Fen1 are important for endo-and exonuclease activities. J Mol Biol. 2003;328:73–84. doi: 10.1016/s0022-2836(03)00270-5. [DOI] [PubMed] [Google Scholar]
- Friedrich-Heineken E, Hubscher U. The Fen1 extrahelical 3′-flap pocket is conserved from archaea to human and regulates DNA substrate specificity. Nucleic Acids Res. 2004;32:2520–2528. doi: 10.1093/nar/gkh576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcin ED, Hosfield DJ, Desai SA, Haas BJ, Bjoras M, Cunningham RP, Tainer JA. DNA apurinic-apyrimidinic site binding and excision by endonuclease IV. Nat Struct Mol Biol. 2008;15:515–522. doi: 10.1038/nsmb.1414. [DOI] [PubMed] [Google Scholar]
- Gloor JW, Balakrishnan L, Bambara RA. Flap endonuclease 1 mechanism analysis indicates flap base binding prior to threading. J Biol Chem. 2010;285:34922–34931. doi: 10.1074/jbc.M110.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasby JA, Finger LD, Tsutakawa SE, Atack JM, Tainer JA. Unpairing and gating: sequence-independent substrate recognition by FEN superfamily nucleases. Trends Biochem Sci. 2011;37:74–84. doi: 10.1016/j.tibs.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Chavez V, Singh P, Finger LD, Hang H, Hegde ML, Shen B. Comprehensive mapping of the C-terminus of flap endonuclease-1 reveals distinct interaction sites for five proteins that represent different DNA replication and repair pathways. J Mol Biol. 2008a;377:679–690. doi: 10.1016/j.jmb.2007.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Qian L, Liu R, Dai H, Zhou M, Zheng L, Shen B. Nucleolar localization and dynamic roles of flap endonuclease 1 in ribosomal DNA replication and damage repair. Mol Cell Biol. 2008b;28:4310–4319. doi: 10.1128/MCB.00200-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Zheng L, Xu H, Dai H, Zhou M, Pascua MR, Chen QM, Shen B. Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat Chem Biol. 2010;6:766–773. doi: 10.1038/nchembio.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington JJ, Lieber MR. Functional domains within FEN-1 and RAD2 define a family of structure-specific endonucleases: implications for nucleotide excision repair. Genes Dev. 1994;8:1344–1355. doi: 10.1101/gad.8.11.1344. [DOI] [PubMed] [Google Scholar]
- Harrington JJ, Lieber MR. DNA structural elements required for FEN-1 binding. J Biol Chem. 1995;270:4503–4508. doi: 10.1074/jbc.270.9.4503. [DOI] [PubMed] [Google Scholar]
- Hasan S, Stucki M, Hassa PO, Imhof R, Gehrig P, Hunziker P, Hubscher U, Hottiger MO. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol Cell. 2001;7:1221–1231. doi: 10.1016/s1097-2765(01)00272-6. [DOI] [PubMed] [Google Scholar]
- Henneke G, Koundrioukoff S, Hubscher U. Phosphorylation of human Fen1 by cyclin-dependent kinase modulates its role in replication fork regulation. Oncogene. 2003;22:4301–4313. doi: 10.1038/sj.onc.1206606. [DOI] [PubMed] [Google Scholar]
- Hohl M, Dunand-Sauthier I, Staresincic L, Jaquier-Gubler P, Thorel F, Modesti M, Clarkson SG, Scharer OD. Domain swapping between FEN-1 and XPG defines regions in XPG that mediate nucleotide excision repair activity and substrate specificity. Nucleic Acids Res. 2007;35:3053–3063. doi: 10.1093/nar/gkm092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosfield DJ, Frank G, Weng Y, Tainer JA, Shen B. Newly discovered archaebacterial flap endonucleases show a structure-specific mechanism for DNA substrate binding and catalysis resembling human flap endonuclease-1. J Biol Chem. 1998a;273:27154–27161. doi: 10.1074/jbc.273.42.27154. [DOI] [PubMed] [Google Scholar]
- Hosfield DJ, Mol CD, Shen B, Tainer JA. Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: coupling DNA and PCNA binding to FEN-1 activity. Cell. 1998b;95:135–146. doi: 10.1016/s0092-8674(00)81789-4. [DOI] [PubMed] [Google Scholar]
- Hutton RD, Craggs TD, White MF, Penedo JC. PCNA and XPF cooperate to distort DNA substrates. Nucleic Acids Res. 2009;38:1664–1675. doi: 10.1093/nar/gkp1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang KY, Baek K, Kim HY, Cho Y. The crystal structure of flap endonuclease-1 from Methanococcus jannaschii. Nat Struct Biol. 1998;5:707–713. doi: 10.1038/1406. [DOI] [PubMed] [Google Scholar]
- Imamura O, Campbell JL. The human Bloom syndrome gene suppresses the DNA replication and repair defects of yeast dna2 mutants. Proc Natl Acad Sci USA. 2003;100:8193–8198. doi: 10.1073/pnas.1431624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I, Tainer JA, McCammon JA. Unraveling the three-metal-ion catalytic mechanism of the DNA repair enzyme endonuclease IV. Proc Natl Acad Sci USA. 2007;104:1465–1470. doi: 10.1073/pnas.0603468104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Kovvali GK, Prakash L, Prakash S. Requirement of the yeast RTH1 5′ to 3′ exonuclease for the stability of simple repetitive DNA. Science. 1995;269:238–240. doi: 10.1126/science.7618086. [DOI] [PubMed] [Google Scholar]
- Jose D, Datta K, Johnson NP, von Hippel PH. Spectroscopic studies of position-specific DNA “breathing” fluctuations at replication forks and primer-template junctions. Proc Natl Acad Sci USA. 2009;106:4231–4236. doi: 10.1073/pnas.0900803106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YH, Lee CH, Seo YS. Dna2 on the road to Okazaki fragment processing and genome stability in eukaryotes. Crit Rev Biochem Mol Biol. 2010;45:71–96. doi: 10.3109/10409230903578593. [DOI] [PubMed] [Google Scholar]
- Kao HI, Henricksen LA, Liu Y, Bambara RA. Cleavage specificity of Saccharomyces cerevisiae flap endonuclease 1 suggests a double flap structure as the cellular substrate. J Biol Chem. 2002;277:14379–14389. doi: 10.1074/jbc.M110662200. [DOI] [PubMed] [Google Scholar]
- Kim IS, Lee MY, Lee IH, Shin SL, Lee SY. Gene expression of flap endonuclease-1 during cell proliferation and differentiation. Biochim Biophys Acta. 2000;1496:333–340. doi: 10.1016/s0167-4889(00)00029-x. [DOI] [PubMed] [Google Scholar]
- Krahn JM, Beard WA, Wilson SH. Structural insights into DNA polymerase beta deterrents for misincorporation support an induced-fit mechanism for fidelity. Structure. 2004;12:1823–1832. doi: 10.1016/j.str.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Kucherlapati M, Yang K, Kuraguchi M, Zhao J, Lia M, Heyer J, Kane MF, Fan K, Russell R, Brown AM, et al. Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc Natl Acad Sci USA. 2002;99:9924–9929. doi: 10.1073/pnas.152321699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen E, Gran C, Saether BE, Seeberg E, Klungland A. Proliferation failure and gamma radiation sensitivity of Fen1 null mutant mice at the blastocyst stage. Mol Cell Biol. 2003;23:5346–5353. doi: 10.1128/MCB.23.15.5346-5353.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen E, Kleppa L, Meza TJ, Meza-Zepeda LA, Rada C, Castellanos CG, Lien GF, Nesse GJ, Neuberger MS, Laerdahl JK, et al. Early-onset lymphoma and extensive embryonic apoptosis in two domain-specific Fen1 mice mutants. Cancer Res. 2008;68:4571–4579. doi: 10.1158/0008-5472.CAN-08-0168. [DOI] [PubMed] [Google Scholar]
- Li X, Li J, Harrington J, Lieber MR, Burgers PM. Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J Biol Chem. 1995;270:22109–22112. doi: 10.1074/jbc.270.38.22109. [DOI] [PubMed] [Google Scholar]
- Lieber MR. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. Bioessays. 1997;19:233–240. doi: 10.1002/bies.950190309. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- Lyamichev V, Brow MA, Dahlberg JE. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science. 1993;260:778–783. doi: 10.1126/science.7683443. [DOI] [PubMed] [Google Scholar]
- Lyamichev V, Brow MA, Varvel VE, Dahlberg JE. Comparison of the 5′ nuclease activities of taq DNA polymerase and its isolated nuclease domain. Proc Natl Acad Sci USA. 1999;96:6143–6148. doi: 10.1073/pnas.96.11.6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mase T, Kubota K, Miyazono K, Kawarabayasi Y, Tanokura M. Crystallization and preliminary X-ray analysis of flap endonuclease 1 (FEN1) from Desulfurococcus amylolyticus. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2009;65:923–925. doi: 10.1107/S1744309109031248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesiet-Cladiere L, Norais C, Kuhn J, Briffotaux J, Sloostra JW, Ferrari E, Hubscher U, Flament D, Myllykallio H. A novel proteomic approach identifies new interaction partners for proliferating cell nuclear antigen. J Mol Biol. 2007;372:1137–1148. doi: 10.1016/j.jmb.2007.06.056. [DOI] [PubMed] [Google Scholar]
- Murante RS, Rust L, Bambara RA. Calf 5′ to 3′ exo/endonuclease must slide from a 5′ end of the substrate to perform structure-specific cleavage. J Biol Chem. 1995;270:30377–30383. doi: 10.1074/jbc.270.51.30377. [DOI] [PubMed] [Google Scholar]
- Navarro MS, Bi L, Bailis AM. A mutant allele of the transcription factor IIH helicase gene, RAD3, promotes loss of heterozygosity in response to a DNA replication defect in Saccharomyces cerevisiae. Genetics. 2007;176:1391–1402. doi: 10.1534/genetics.107.073056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarkina JK, Lavrik OI, Khodyreva SN. Flap endonuclease-1 and its role in the processes of DNA metabolism in eucaryotic cells. Mol Biol (Mosk) 2008;42:405–421. [PubMed] [Google Scholar]
- Orans J, McSweeney EA, Iyer RR, Hast MA, Hellinga HW, Modrich P, Beese LS. Structures of human exonuclease 1 DNA complexes suggest a uni fied mechanism for nuclease family. Cell. 2011;145:212–223. doi: 10.1016/j.cell.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovádi J. Physiological significance of metabolic channelling. J Theor Biol. 1991;152:1–22. [PubMed] [Google Scholar]
- Parikh SS, Mol CD, Hosfield DJ, Tainer JA. Envisioning the molecular choreography of DNA base excision repair. Curr Opin Struct Biol. 1999;9:37–47. doi: 10.1016/s0959-440x(99)80006-2. [DOI] [PubMed] [Google Scholar]
- Pascal JM, O'Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432:473–478. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- Patel N, Atack JM, Finger LD, Exell J, Thompson P, Tsutakawa SE, Tainer JA, Williams DM, Grasby JA. Flap endonucleases pass 5′-flaps through a flexible arch using a disorder-thread-order mechanism to confer specificity for free 5′-ends. Nucl Acids Res. 2012 doi: 10.1093/nar/gks051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering TJ, Garforth SJ, Thorpe SJ, Sayers JR, Grasby JA. A single cleavage assay for T5 5′→ 3′ exonuclease: determination of the catalytic parameters for wild-type and mutant proteins. Nucleic Acids Res. 1999;27:730–735. doi: 10.1093/nar/27.3.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Qian Y, Frank P, Wintersberger U, Shen B. Saccharomyces cerevisiae RNase H(35) functions in RNA primer removal during lagging-strand DNA synthesis, most efficiently in cooperation with Rad27 nuclease. Mol Cell Biol. 1999;19:8361–8371. doi: 10.1128/mcb.19.12.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan MS, Pittenger C, Siede W, Friedberg EC. Characterization of a mutant strain of Saccharomyces cerevisiae with a deletion of the RAD27 gene, a structural homolog of the RAD2 nucleotide excision repair gene. J Bacteriol. 1995;177:364–371. doi: 10.1128/jb.177.2.364-371.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: base excision repair: the long and short of it. Cell Mol Life Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai S, Kitano K, Yamaguchi H, Hamada K, Okada K, Fukuda K, Uchida M, Ohtsuka E, Morioka H, Hakoshima T. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 2005;24:683–693. doi: 10.1038/sj.emboj.7600519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai S, Kitano K, Morioka H, Hakoshima T. Crystallization and preliminary crystallographic analysis of the catalytic domain of human flap endonuclease 1 in complex with a nicked DNA product: use of a DPCS kit for efficient protein-DNA complex crystallization. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:39–43. doi: 10.1107/S1744309107065372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengerova B, Tomlinson C, Atack JM, Williams R, Sayers JR, Williams NH, Grasby JA. Bronsted analysis and rate-limiting steps for the T5 flap endonuclease catalyzed hydrolysis of exonucleolytic substrates. Biochemistry. 2010;49:8085–8093. doi: 10.1021/bi100895j. [DOI] [PubMed] [Google Scholar]
- Sharma S, Otterlei M, Sommers JA, Driscoll HC, Dianov GL, Kao HI, Bambara RA, Brosh RM., Jr WRN helicase and FEN-1 form a complex upon replication arrest and together process branchmigrating DNA structures associated with the replication fork. Mol Biol Cell. 2004;15:734–750. doi: 10.1091/mbc.E03-08-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Sommers JA, Gary RK, Friedrich-Heineken E, Hubscher U, Brosh RM., Jr The interaction site of Flap Endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 2005;33:6769–6781. doi: 10.1093/nar/gki1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Qiu J, Hos field D, Tainer JA. Flap endonuclease homologs in archaebacteria exist as independent proteins. Trends Biochem Sci. 1998;23:171–173. doi: 10.1016/s0968-0004(98)01199-2. [DOI] [PubMed] [Google Scholar]
- Shen B, Singh P, Liu R, Qiu J, Zheng L, Finger LD, Alas S. Multiple but dissectible functions of FEN-1 nucleases in nucleic acid processing, genome stability and diseases. Bioessays. 2005;27:717–729. doi: 10.1002/bies.20255. [DOI] [PubMed] [Google Scholar]
- Solinger JA, Pascolini D, Heyer WD. Active-site mutations in the Xrn1p exoribonuclease of Saccharomyces cerevisiae reveal a specific role in meiosis. Mol Cell Biol. 1999;19:5930–5942. doi: 10.1128/mcb.19.9.5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JA, Campbell JL, Bambara RA. Significance of the dissociation of Dna2 by flap endonuclease 1 to Okazaki fragment processing in Saccharomyces cerevisiae. J Biol Chem. 2009;284:8283–8291. doi: 10.1074/jbc.M809189200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Henneke G, Ferrari E, Gordenin DA, Hubscher U, Resnick MA. The flexible loop of human FEN1 endonuclease is required for flap cleavage during DNA replication and repair. EMBO J. 2002;21:5930–5942. doi: 10.1093/emboj/cdf587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucki M, Jonsson ZO, Hubscher U. In eukaryotic flap endonuclease 1, the C terminus is essential for substrate binding. J Biol Chem. 2001;276:7843–7849. doi: 10.1074/jbc.M008829200. [DOI] [PubMed] [Google Scholar]
- Syson K, Tomlinson C, Chapados BR, Sayers JR, Tainer JA, Williams NH, Grasby JA. Three metal ions participate in the reaction catalyzed by T5 flap endonuclease. J Biol Chem. 2008;283:28741–28746. doi: 10.1074/jbc.M801264200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom S, Henricksen LA, Bambara RA. Mechanism whereby proliferating cell nuclear antigen stimulates flap endonuclease 1. J Biol Chem. 2000;275:10498–10505. doi: 10.1074/jbc.275.14.10498. [DOI] [PubMed] [Google Scholar]
- Tomkinson AE, Vijayakumar S, Pascal JM, Ellenberger T. DNA ligases: structure, reaction mechanism, and function. Chem Rev. 2006;106:687–699. doi: 10.1021/cr040498d. [DOI] [PubMed] [Google Scholar]
- Tomlinson CG, Atack JM, Chapados B, Tainer JA, Grasby JA. Substrate recognition and catalysis by flap endonucleases and related enzymes. Biochem Soc Trans. 2010;38:433–437. doi: 10.1042/BST0380433. [DOI] [PubMed] [Google Scholar]
- Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G, Tomlinson CG, Thompson P, Sarker AH, Shen B, et al. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell. 2011;145:198–211. doi: 10.1016/j.cell.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumey LN, Huck B, Gleason E, Wang J, Silver D, Brunden K, Boozer S, Rundlett S, Sherf B, Murphy S, et al. The identification and optimization of 2,4-diketobutyric acids as flap endonuclease 1 inhibitors. Bioorg Med Chem Lett. 2004;14:4915–4918. doi: 10.1016/j.bmcl.2004.07.028. [DOI] [PubMed] [Google Scholar]
- Tumey LN, Bom D, Huck B, Gleason E, Wang J, Silver D, Brunden K, Boozer S, Rundlett S, Sherf B, et al. The identification and optimization of a N-hydroxy urea series of flap endonuclease 1 inhibitors. Bioorg Med Chem Lett. 2005;15:277–281. doi: 10.1016/j.bmcl.2004.10.086. [DOI] [PubMed] [Google Scholar]
- Waga S, Bauer G, Stillman B. Reconstitution of complete SV40 DNA replication with purified replication factors. J Biol Chem. 1994;269:10923–10934. [PubMed] [Google Scholar]
- Warbrick E, Coates PJ, Hall PA. Fen1 expression: a novel marker for cell proliferation. J Pathol. 1998;186:319–324. doi: 10.1002/(SICI)1096-9896(1998110)186:3<319::AID-PATH184>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Williams R, Sengerova B, Osborne S, Syson K, Ault S, Kilgour A, Chapados BR, Tainer JA, Sayers JR, Grasby JA. Comparison of the catalytic parameters and reaction specificities of a phage and an archaeal flap endonuclease. J Mol Biol. 2007;371:34–48. doi: 10.1016/j.jmb.2007.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nat Struct Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- Wu X, Li J, Li X, Hsieh CL, Burgers PM, Lieber MR. Processing of branched DNA intermediates by a complex of human FEN-1 and PCNA. Nucleic Acids Res. 1996;24:2036–2043. doi: 10.1093/nar/24.11.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Potapova O, Leschziner AE, Grindley ND, Joyce CM. Contacts between the 5′ nuclease of DNA polymerase I and its DNA substrate. J Biol Chem. 2001;276:30167–30177. doi: 10.1074/jbc.M100985200. [DOI] [PubMed] [Google Scholar]
- Yang W. Nucleases: diversity of structure, function and mechanism. Q Rev Biophys. 2010;44:1–93. doi: 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Lee JY, Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Yanofsky C. A second reaction catalyzed by the tryptophan synthetase of Escherichia coli. 1959. Biochim Biophys Acta. 1989;1000:137–145. [PubMed] [Google Scholar]
- Zheng L, Zhou M, Chai Q, Parrish J, Xue D, Patrick SM, Turchi JJ, Yannone SM, Chen D, Shen B. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005;6:83–89. doi: 10.1038/sj.embor.7400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Dai H, Qiu J, Huang Q, Shen B. Disruption of the FEN-1/PCNA interaction results in DNA replication defects, pulmonary hypoplasia, pancytopenia, and newborn lethality in mice. Mol Cell Biol. 2007a;27:3176–3186. doi: 10.1128/MCB.01652-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Dai H, Zhou M, Li M, Singh P, Qiu J, Tsark W, Huang Q, Kernstine K, Zhang X, et al. Fen1 mutations result in autoimmunity, chronic in flammation and cancers. Nat Med. 2007b;13:812–819. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- Zheng L, Dai H, Hegde ML, Zhou M, Guo Z, Wu X, Wu J, Su L, Zhong X, Mitra S, et al. Fen1 mutations that specifically disrupt its interaction with PCNA cause aneuploidy-associated cancer. Cell Res. 2011a;21:1052–1067. doi: 10.1038/cr.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Jia J, Finger LD, Guo Z, Zer C, Shen B. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 2011b;39:781–794. doi: 10.1093/nar/gkq884. [DOI] [PMC free article] [PubMed] [Google Scholar]