

Abstract
Many organisms localize mRNAs to specific subcellular destinations to spatially and temporally control gene expression. Recent studies have demonstrated that the majority of the transcriptome is localized to a nonrandom position in cells and embryos. One approach to identify localized mRNAs is to biochemically purify a cellular structure of interest and to identify all associated transcripts. Using recently developed high-throughput sequencing technologies it is now straightforward to identify all RNAs associated with a subcellular structure. To facilitate transcript identification it is necessary to work with an organism with a fully sequenced genome. One attractive system for the biochemical purification of subcellular structures are egg extracts produced from the frog Xenopus laevis. However, X. laevis currently does not have a fully sequenced genome, which hampers transcript identification. In this article we describe a method to produce egg extracts from a related frog, X. tropicalis, that has a fully sequenced genome. We provide details for microtubule polymerization, purification and transcript isolation. While this article describes a specific method for identification of microtubule-associated transcripts, we believe that it will be easily applied to other subcellular structures and will provide a powerful method for identification of localized RNAs.
Keywords: Molecular Biology, Issue 76, Genetics, Developmental Biology, Biochemistry, Bioengineering, Cellular Biology, RNA, Messenger, Stored, RNA Processing, Post-Transcriptional, Xenopus, microtubules, egg extract, purification, RNA localization, mRNA, Xenopus tropicalis, eggs, animal model
Introduction
Spatial and temporal control of gene expression is important for all cells, and is especially important for the control of early embryonic pattering1. Spatial control of gene expression is achieved through the active localization of mRNAs to specific destinations within cells or embryos. In many very large cell types, (e.g. oocytes, embryos, and neurons) mRNA localization is used to restrict protein expression to the site of action of the coded protein. Since a localized mRNA can catalyze many rounds of protein production it is more efficient to localize an mRNA than to localize individual protein molecules. Localized mRNAs are typically translationally repressed until they reach their destination, which serves to further limit the localization of the coded protein2. In addition to the many well-documented cases of RNA localization to control embryonic patterning, several studies have documented mRNAs that are localized to the site of action of the encoded protein. Prominent examples include localization of the β-actin3 and Arp2/34 mRNAs to the leading edge of motile fibroblasts and localization of the mRNAs for many mitotic regulators to meiotic and mitotic spindles5-7.
Many of the classic examples of localized mRNAs were identified through genetic screens for maternal effect mutations and were later determined to encode localized RNAs. However, recent genome-wide studies have begun to provide broader insight into the scope of localized RNAs. A recent in situ hybridization screen in Drosophila embryos demonstrated that ~70% of all mRNAs have a specific localization, including many novel destinations8. Purification of pseudopodia from mouse fibroblasts identified a diverse group of localized mRNAs9. Work from our group using biochemical purification of microtubules from meiotic Xenopus egg extracts identified hundreds of mRNAs that copurify with the spindle5,7. Our work showed that the majority of microtubule-localized mRNAs encode proteins that function in control of mitosis, supporting the idea that mRNAs are localized to the site of action of the coded protein. Furthermore, the ability to detect mRNA enrichment in a subcellular fraction by biochemical purification highlights the power of this approach for identification of localized mRNAs.
Most localized RNAs use active transport on the cytoskeleton, either actin or microtubules, to achieve transport to their final destination10. To gain a better understanding of the extent and types of RNAs that are localized to specific destinations using a biochemical approach it is necessary to have an in vitro system that can recapitulate cytoskeletal processes. One of the premier systems for studying cytoskeletal biology is egg extracts produced from unfertilized eggs from the frog Xenopus laevis. X. laevis egg extracts have been used for decades to study a wide array of cytoskeletal processes and have contributed much to our understanding of the mechanisms and molecules that control cytoskeletal assembly and dynamics11. Furthermore, X. laevis egg extracts are amenable to large-scale purifications of microtubules and associated proteins12,13 and there are well-designed methods for the production of various types of egg extracts14-16. However, for genomic studies there are several drawbacks to the use of X. laevis as a model system.
For decades Xenopus laevis frogs have been a powerful system for the study of developmental and cell biology, owing to the large oocyte size and robust external development17. Furthermore, the development of egg extract systems that can recapitulate many cellular processes in a test tube has made this frog a powerful experimental model. However, Xenopus laevis has been hampered by the lack of a complete genome sequence, which has been slowed by the allotetraploid nature of the genome.In contrast, a closely related species, Xenopus tropicalis, has a diploid genome that was sequenced in 201018. While X. tropicalis is not as experimentally tractable as X. laevis17the availability of a sequenced genome makes it an attractive model system to perform genome wide analyses.
In this report we describe a method to make meiosis II-, cytostatic factor-arrested extracts (CSF) from X. tropicalis19. We then describe a simple method to purify microtubules and associated RNAs from this extract. The RNAs can then be converted into libraries amenable to sequencing using recently developed high throughput sequencing technologies. Once the libraries are sequenced they can be aligned to the genome of the frog to identify specific mRNAs that are enriched in the microtubule sample compared to total extract. This provides a powerful method to detect microtubule-targeted mRNA localization on a genome-wide scale. In addition to being able to detect localized mRNAs, the use of high-throughput sequencing and a sequenced genome offer the possibility of discovering novel transcripts that are not currently present in public database annotations.
Protocol
1. Generation of X. tropicalis Eggs
All Xenopus tropicalis frogs are ordered from NASCO. Our frogs are housed in an Aquatic Habitats recirculating water system kept at 27 °C. There are many options for water systems for care of X. tropicalis. Some good general information on this frog species can be found on the web sites of the Harland and Grainger labs (http://tropicalis.berkeley.edu/home/, http://www.faculty.virginia.edu/xtropicalis/). Our frogs are maintained in tankwater consisting of (0.4 g Ciclid Lake Salts, 0.6 g marine salt, 0.625 g NaHCO3 per liter of water, pH 7.0)20. This recipe results in a conductivity of ~1800 μS, which is a high salinity for X. tropicalis. However, we have found that our frogs thrive in this environment and oocyte quality is improved. Alternative tankwater recipes can be found above at the resources listed for general X. tropicalis care.
Frogs are injected with human Chorionic Gonadotropin (hCG) on three successive days to stimulate egg laying: First, prepare two concentrations of hCG solution. Resuspend 10,000 U of lyophilized hCG powder in 10 ml sterile, deionized H2O for a final concentration of 1,000 U/ml. Then, dilute 1 ml of 1,000 U/ml hCG solution in 9 ml H2O for a final concentration of 100 U/ml. Store both solutions at 4 °C.
On day 1, prepare 4-6 frogs for egg laying by injecting with hCG between 2:00-3:00 PM. Inject each frog in the dorsal lymph sac near the cloaca with 0.2 ml 100 U/ml hCG solution. Having the frogs fast during the subsequent two injections will minimize the amount of frog waste present during egg laying, but is optional.
On day 2, inject the same frogs with 0.2 ml 100 U/ml hCG solution between 2:00-3:00 PM.
On day 3, inject the same frogs with 0.2 ml 1,000 U/ml hCG solution, between 7:00-10:00 AM. Set up frogs to lay eggs: fill a 6-quart plastic bucket with fresh tankwater, add frogs and place in the dark at 25 °C. After this injection, egg laying will begin after 4 hr and will be complete by 7 hr. Frogs should lay eggs in an environment that is maintained at a minimum of 25 °C.
Make extract solutions and have equipment ready immediately before collecting eggs.
20X MMR: 100 mM HEPES, pH 7.8; 2 mM EDTA pH 7.8; 2 M NaCl; 40 mM KCl; 20 mM MgCl2; 40 mM CaCl2. Autoclave and store at room temperature. Prepare 1 L of 1X MMR just prior to extract preparation.
10X XB: 100 mM HEPES, pH7.7; 10 mM MgCl2; 1 mM CaCl2; 1 M KCl; 500 mM sucrose. Autoclave and store at 4 °C. Prepare 1 L of 1X XB just prior to extract preparation. Dejelly solution: Prepare 250 ml 3% cysteine solution in deionized H2O and pH to 7.8-8.0 with 10 N NaOH. Prepare just prior to extract preparation.
CSF-XB: take 200 ml of 1X XB and add 2 ml 0.5 M EGTA pH 7.7 and 200 μl 1 M MgCl2. Prepare just prior to extract preparation.
CSF-XB+: take 50 ml of CSF-XB and add 50 μl of LPC (10 mg/ml each stock of Leupeptin, Pepstatin, and Chymostatin in DMSO). Add 50 μl Cytochalasin D (10 mg/ml in DMSO). Prepare just prior to extract preparation.
Prepare a 0.2% gelatin solution in deionized H2O, microwave to dissolve and filter sterilize. Store at room temperature.
Reserve 2 Beckman 2 x ½ inch Ultracentrifuge tubes.
Prepare two 15 ml glass round-bottomed centrifuge tubes with 0.5 ml of H2O in each to cushion the ultracentrifuge tube.
Make fire-polished glass Pasteur pipettes. Snap the end off of 5 ¾ inch glass pipettes to expose a broad opening, and expose to flame to smooth the new exposed pipette tip.
Prepare a 500 ml glass beaker for storing eggs by swirling a 0.2% gelatin solution around to coat the walls of the beaker. Discard gelatin solution from beaker after use.
Collect eggs from the plastic bucket used for laying 6-7 hr after the third injection on day 3. If desired, gently squeeze each frog once to get any remaining eggs. Wash eggs once with fresh tankwater and transfer to the 500 ml glass beaker coated with 0.2% gelatin solution.
2. Preparation of Extract from X. tropicalis Eggs
All steps of extract preparation can be performed at room temperature, approximately 25 °C. Throughout the washes, it is important to keep the eggs submerged under liquid so that they remain wet. Exposure to air can cause the eggs to escape cell cycle arrest or lyse.
Decant as much tankwater as possible while reserving enough liquid to keep the eggs wet. Tilt the beaker containing eggs to the side and add ~300 ml 1X MMR slowly to the wall of the beaker, so that physical agitation of the eggs is minimized. Let eggs settle, then decant off supernatant containing debris. X. tropicalis eggs are stringy at this step, so removal of activated eggs is done after dejellying. Repeat for a total of three 1X MMR washes.
Dejelly the eggs. Decant off as much MMR as possible and add half of the dejelly solution. Swirl continuously for approximately 5 min. Dissolving jelly coats will be visible in the supernatant after a couple of minutes. Decant off and add the remaining dejelly solution. Continue to swirl continuously until eggs pack very tightly and all orient with their vegetal pole (the pole with white pigment) toward the bottom of the dish. Quickly decant off as much dejelly solution as possible. Once the eggs are dejellied they are very sensitive to mechanical manipulations.
Carefully add XB to the eggs. In the first XB wash, remove eggs that have escaped CSF arrest by removing lysed, puffy, white, and pseudocleavage eggs. Activated X. tropicalis eggs tend to settle in the top center, so use a plastic transfer pipette to pull these out. Also remove pieces of skin and frog waste. Wash eggs a total of three times with ~ 300 ml 1X XB solution, gently swirling eggs between washes and allowing them to settle on the bottom of the beaker. As before, decant as much of each wash solution as possible while keeping eggs wet.
Wash eggs twice with CSF-XB and decant.
Add CSF-XB+ to eggs. Using a gelatin-treated fire-polished Pasteur pipette, transfer eggs to Ultra-centrifuge tubes with CSF-XB+, taking care not to expose the eggs to air. Place inside the 15 ml glass centrifuge tubes with the water cushion.
Spin eggs in a clinical centrifuge at 200 x g for 1 min, increase speed to 800 x g and spin for 30 sec.
Use an aspirator to remove as much buffer as possible from eggs. They should be almost dry on top. Quickly move eggs to a Sorvall RC-6 centrifuge equipped with a HB-6 rotor (or equivalent) and spin 17,000 x g for 15 min at 20 °C.
Remove the yellow cytoplasmic layer between the pigment and lipid layers using an 18 gauge needle attached to a 1 ml syringe. Puncture the side of the tube and pull the syringe barrel slowly to obtain the cytoplasmic extract layer. Avoid pigment granules as much as possible.
Transfer cytoplasm to new ultracentrifugation tube. It is normal for the extract to appear slightly cloudy at this step. Place inside the 15 ml glass centrifuge tube with water cushion. Spin again 17,000 x g for 10 min at 20 °C. Repeat extraction with 18 gauge needle.
Transfer cytoplasm to a 1.5 ml microfuge tube. Estimate the extract volume and dilute Cytochalasin D and LPC 1:1,000 into the extract. Mix well with a 1 ml pipette tip, pipetting up and down many times without introduction of air bubbles. A typical yield from a healthy frog colony is approximately 300-500 μl of extract/frog. To preserve maximum activity, it is necessary to store the extract and perform experimental manipulations at room temperature (20-25 °C).
3. Purification Taxol-stabilized Microtubules from X. tropicalis Extract
Add Taxol to a 100-200 μl aliquot of extract at a final concentration of 10 μM and incubate at room temperature for 30 min. For control reactions, treat an equivalent volume of extract with the microtubule-destablilzing drug Nocodazole (10 μM). Reserve 100 μl of untreated extract for analysis.
Dilute the drug-treated extract with 10 volumes BRB-80 (80 mM PIPES pH 6.8, 1 mM MgCl2, 1 mM EGTA) + 30% glycerol. Assemble 14 ml round-bottom polypropylene tubes containing 10 ml of BRB-80 + 60% glycerol cushion. Using a wide bore pipette tip, layer the drug-treated extract reaction gently on top of the BRB-80 + 60% glycerol cushion. Centrifuge for 10 min at 17,000 x g at 20 °C in a Sorvall RC-6 centrifuge equipped with a HB-6 rotor (or equivalent) and tube adapters.
Aspirate the supernatant containing unsedimented extract material, and wash the interface twice with deionized H2O. Aspirate the remaining cushion volume slowly, taking care not to disturb the gel-like pellet containing microtubules, microtubule-associated proteins, and microtubule-associated RNAs in the Taxol-treated sample. The Nocodazole-treated sample does not contain visible material. Resuspend the pellet in 1 ml TRIzol and proceed with the manufacturer's instructions for isolating RNA. Untreated extract (up to 100 μl) can be resuspended directly in 1 ml TRIzol.
There are now commercially available kits for preparing transcriptome libraries suitable for RNA-seq. These may be purchased through http://www.illumina.com/ and http://www.454.com/.
Representative Results
To identify X. tropicalis transcripts associated with microtubules, we prepare a cytosolic extract from unfertilized eggs arrested in metaphase of meiosis II (CSF). Treatment of this extract with taxol allows the formation of stable microtubules that can be purified by sedimentation through a glycerol cushion (Figure 1A). Coomassie gel analysis confirms that α/β-tubulin sediments in a taxol-dependent manner, and represents the major protein species recovered in these preparations (Figure 1B). Lower levels of other proteins are also present in the taxol pellet, but not in preparations treated with the microtubule depolymerizing drug nocodazole, indicating that proteins in the taxol fraction specifically associate with microtubules (MAPs).
An Agilent Bioanalyzer is used to examine general RNA composition in all X. tropicalis extract fractions (Figure 1C). Both rRNA and tRNA species are present in CSF extract and the microtubule-containing taxol pellet, consistent with previous findings that translation occurs on microtubules and spindles in X. laevis egg extract5,21. A line trace of the gel projection reveals the mRNA signal is markedly lower in the microtubule-containing taxol pellet, most notably in the region migrating above 28S rRNA, indicating that a subset of mRNAs cosediment with microtubules in X. tropicalis. RNA isolated in this manner is suitable for RNA-seq experiments using commercially available reagents.
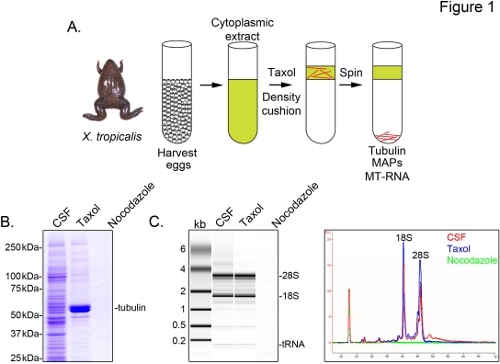 Figure 1. Purification of MT-RNA for RNA-seq. (A) Purification scheme to isolate MT-RNA. Eggs are harvested from female X. tropicalis frogs. After preparation of a cytoplasmic extract, taxol is added to induce microtubule polymerization. Microtubules and MT-RNA are purified by sedimentation through a glycerol cushion. (B) Coomassie gel analysis of proteins isolated using the scheme described in (A). Total CSF extract compared to proteins sedimented in the presence of taxol or nocodazole. (C) Bioanalyzer gel analysis of RNA isolated using the scheme described in (A). RNA isolated from CSF extract compared to RNA sedimented in the presence of taxol or nocodazole. Both the gel projection and the line traces are shown.Reprinted with permission from Sharp, et al., (2011). Click here to view larger image.
Figure 1. Purification of MT-RNA for RNA-seq. (A) Purification scheme to isolate MT-RNA. Eggs are harvested from female X. tropicalis frogs. After preparation of a cytoplasmic extract, taxol is added to induce microtubule polymerization. Microtubules and MT-RNA are purified by sedimentation through a glycerol cushion. (B) Coomassie gel analysis of proteins isolated using the scheme described in (A). Total CSF extract compared to proteins sedimented in the presence of taxol or nocodazole. (C) Bioanalyzer gel analysis of RNA isolated using the scheme described in (A). RNA isolated from CSF extract compared to RNA sedimented in the presence of taxol or nocodazole. Both the gel projection and the line traces are shown.Reprinted with permission from Sharp, et al., (2011). Click here to view larger image.
Discussion
In this report we have described a simple method to produce CSF-arrested egg extracts from X. tropicalis19and use this extract to study microtubule-associated RNAs7. The basic procedure for producing CSF-arrested egg extracts from X. tropicalis is the same as used for X. laevis with a few key differences. One of the most challenging aspects to working with X. tropicalis frogs is obtaining enough high quality eggs to make an extract with microtubule nucleation or spindle assembly activity comparable to X. laevis egg extracts. To achieve optimal egg laying conditions while preventing slippage from meiosis II cell cycle arrest, the interval between hormone injections for X. tropicalis is shorter than that used for X. laevis, and the timing from the third hCG injection to the beginning of egg laying is also much shorter. With X. laevis the timing from the hCG injection to the beginning of egg laying is such that it is convenient and efficient for eggs to be laid overnight into buffer. However, because of the shorter time between hCG injection and egg laying with X. tropicalis it is frequently necessary to manually express the eggs from frogs. Another significant difference between making egg extract from the two different frogs is the dejellying step. With X. laevis the eggs are so large that it is easy to determine when the jelly coat has dissolved by observing how closely the eggs are spaced in the beaker. As the dejellying reaction commences, the eggs begin to pack more densely. However, X. tropicalis eggs are much smaller and it can be quite difficult to determine when the jelly coat has dissolved by egg packing density alone. We have found that the most reliable method to determine when the jelly coat has dissolved is to monitor the orientation of the animal (black) and vegetal (white) poles. When all the vegetal poles orient toward the bottom of the beaker the jelly coat has been removed enough to proceed with the extract. Finally, whereas X. laevis egg extract can be stored at cool temperatures (4-12 °C) we have observed that it is critical to maintain X. tropicalis egg extract at room temperature (20-25 °C) during preparation and experimental manipulations to preserve biochemical activity. Because of the differences in ease of use we prefer to use X. laevis frogs for the production of egg extract. However, for experiments that require or are facilitated by an organism with a sequenced genome, X. tropicalis is an excellent alternative system.
The method that we have described in this report uses taxol as a microtubule-stabilizing agent to induce microtubule polymerization. We chose this method because taxol is a robust microtubule-stabilizing agent that facilitates the large-scale isolation of purified microtubules. The method that we described could likely be improved by comparing the proteins and RNAs associated with microtubules using alternative microtubule polymerization methods. Alternatives could include polymerization using GTP-induced polymerization (a classic technique),22 or using Ran-GTP as a microtubule polymerizer to mimic the microtubules induced by chromatin-driven spindle assembly23. Finally, use of purified sperm nuclei to induce microtubule polymerization would be the closest mimic to the types of microtubules that are nucleated during mitosis (centrosome, chromatin, and kinetochore mediated). Drawbacks to these alternative sources of microtubule nucleation are that the nucleating agents are not as readily available as taxol and they do not nucleate or stabilize microtubules as efficiently as taxol. Therefore, each of these methods would be more difficult to use for large-scale purifications. The advantage of comparing multiple different types of microtubule nucleators is that it could be possible to identify proteins and/or RNAs that are specific to each pathway of microtubule nucleation.
The method that we have described here takes advantage of cytoplasmic extracts of amphibians. However, this approach could be extended to the use of extract system from other organisms. Mitotic extracts have been described from synchronized human tissue culture cells24 that faithfully recapitulate many aspects of microtubule assembly. We have successfully used these extracts to identify microtubule-associated RNAs from HeLa cells5. Similar microtubule purification schemes have been described for many different organisms25,26, although the microtubule associated RNAs have not been examined. The approach described here could be used with any organism that can produce a concentrated cytoplasmic extract capable of nucleating microtubules.
Finally, although the approach that we describe here discusses the purification of microtubules and associated proteins and RNAs, this approach could be generalized to other subcellular structures. While most localized mRNAs have not been identified using biochemical methods the recent advances in DNA and RNA sequencing technologies make this approach an attractive method to identify localized RNAs. In this approach any subcellular or sub-embryo structure of interest could be isolated or purified. Then the associated proteins and RNAs can be identified on a genome wide scale. RNAs can then be compared to the RNA content of the total cell or embryo to identify enriched localized RNAs. This approach could be used with whole eggs (animal and vegetal separation, similar to the approach that identified the first localized RNAs in Xenopus27), actin associated RNAs, ER-associated RNAs, mitochondria-associated RNAs, or to any subcellular structure that can be purified with associated RNAs intact. Based on our work on microtubule-associated RNA we predict that this would be an excellent method to discover new proteins that function at a given location. Furthermore, identification of the location and extent of all localized RNAs will provide insight into how cells and embryos use mRNA localization to control gene expression.
Disclosures
No conflicts of interest declared.
References
- Martin KC, Ephrussi A. mRNA localization: gene expression in the spatial dimension. Cell. 2009;136:719–730. doi: 10.1016/j.cell.2009.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse F, Ephrussi A. Translational control of localized mRNAs: restricting protein synthesis in space and time. Nat. Rev. Mol. Cell Biol. 2008;9:971–980. doi: 10.1038/nrm2548. [DOI] [PubMed] [Google Scholar]
- Lawrence JB, Singer RH. Intracellular localization of messenger RNAs for cytoskeletal proteins. Cell. 1986;45:407–415. doi: 10.1016/0092-8674(86)90326-0. [DOI] [PubMed] [Google Scholar]
- Mingle LA, et al. Localization of all seven messenger RNAs for the actin-polymerization nucleator Arp2/3 complex in the protrusions of fibroblasts. J. Cell Sci. 2005;118:2425–2433. doi: 10.1242/jcs.02371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower MD, Feric E, Weis K, Heald R. Genome-wide analysis demonstrates conserved localization of messenger RNAs to mitotic microtubules. J. Cell Biol. 2007;179:1365–1373. doi: 10.1083/jcb.200705163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliscovich C, Peset I, Vernos I, Mendez R. Spindle-localized CPE-mediated translation controls meiotic chromosome segregation. Nat. Cell Biol. 2008;10:858–865. doi: 10.1038/ncb1746. [DOI] [PubMed] [Google Scholar]
- Sharp JA, Plant JJ, Ohsumi TK, Borowsky M, Blower MD. Functional analysis of the microtubule-interacting transcriptome. Mol. Biol Cell. 2011;22:4312–4323. doi: 10.1091/mbc.E11-07-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuyer E, et al. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell. 2007;131:174–187. doi: 10.1016/j.cell.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Mili S, Moissoglu K, Macara IG. Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature. 2008;453:115–119. doi: 10.1038/nature06888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Bullock SL. Subcellular mRNA localization in animal cells and why it matters. Science. 2009;326:1212–1216. doi: 10.1126/science.1176488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Murray A, Mitchison TJ, Walczak CE. The use of Xenopus egg extracts to study mitotic spindle assembly and function in vitro. Methods Cell Biol. 1999;61:385–412. doi: 10.1016/s0091-679x(08)61991-3. [DOI] [PubMed] [Google Scholar]
- Gache V, Waridel P, Luche S, Shevchenko A, Popov AV. Purification and mass spectrometry identification of microtubule-binding proteins from Xenopus egg extracts. Methods Mol. Med. 2007;137:29–43. doi: 10.1007/978-1-59745-442-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gache V, et al. Xenopus meiotic microtubule-associated interactome. PLoS One. 2010;5:e9248. doi: 10.1371/journal.pone.0009248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross MK, Powers M. Obtaining eggs from Xenopus laevis females. J. Vis. Exp. 2008. p. e890. [DOI] [PMC free article] [PubMed]
- Cross MK, Powers M. Preparation and fractionation of Xenopus laevis egg extracts. J. Vis. Exp. 2008. p. e891. [DOI] [PMC free article] [PubMed]
- Cross M, Powers M. In vitro nuclear assembly using fractionated Xenopus egg extracts. J. Vis. Exp. 2008. p. e908. [DOI] [PMC free article] [PubMed]
- Harland RM, Grainger RM. Xenopus research: metamorphosed by genetics and genomics. Trends Genet. 2011;27:507–515. doi: 10.1016/j.tig.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten U. The genome of the Western clawed frog Xenopus tropicalis. Science. 2010;328:633–636. doi: 10.1126/science.1183670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KS. Xenopus tropicalis egg extracts provide insight into scaling of the mitotic spindle. J. Cell Biol. 2007;176:765–770. doi: 10.1083/jcb.200610043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey EW, Sanders GE. Effect of water hardness on oocyte quality and embryo development in the African clawed frog (Xenopus laevis) Comp. Med. 2004;54:170–175. [PubMed] [Google Scholar]
- Groisman I, et al. CPEB, maskin, and cyclin B1 mRNA at the mitotic apparatus: implications for local translational control of cell division. Cell. 2000;103:435–447. doi: 10.1016/s0092-8674(00)00135-5. [DOI] [PubMed] [Google Scholar]
- Budde PP, Desai A, Heald R. Analysis of microtubule polymerization in vitro and during the cell cycle in Xenopus egg extracts. Methods. 2006;38:29–34. doi: 10.1016/j.ymeth.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Kalab P, Pu RT, Dasso M. The ran GTPase regulates mitotic spindle assembly. Curr. Biol. 1999;9:481–484. doi: 10.1016/s0960-9822(99)80213-9. [DOI] [PubMed] [Google Scholar]
- Gaglio T, Saredi A, Compton DA. NuMA is required for the organization of microtubules into aster-like mitotic arrays. J. Cell Biol. 1995;131:693–708. doi: 10.1083/jcb.131.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JR, et al. A microtubule interactome: complexes with roles in cell cycle and mitosis. PLoS Biol. 2008;6:e98. doi: 10.1371/journal.pbio.0060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suprenant KA, Tempero LB, Hammer LE. Association of ribosomes with in vitro assembled microtubules. Cell Motil. Cytoskeleton. 1002;14:401–415. doi: 10.1002/cm.970140310. [DOI] [PubMed] [Google Scholar]
- Rebagliati MR, Weeks DL, Harvey RP, Melton DA. Identification and cloning of localized maternal RNAs from Xenopus eggs. Cell. 1985;42:769–777. doi: 10.1016/0092-8674(85)90273-9. [DOI] [PubMed] [Google Scholar]