

ABSTRACT
Vibrio cholerae, a Gram-negative bacterium, infects humans and causes cholera, a severe disease characterized by vomiting and diarrhea. These symptoms are primarily caused by cholera toxin (CT), whose production by V. cholerae is tightly regulated by the virulence cascade. In this study, we designed and carried out a high-throughput chemical genetic screen to identify inhibitors of the virulence cascade. We identified three compounds, which we named toxtazin A and toxtazin B and Bʹ, representing two novel classes of toxT transcription inhibitors. All three compounds reduce production of both CT and the toxin-coregulated pilus (TCP), an important colonization factor. We present evidence that toxtazin A works at the level of the toxT promoter and that toxtazins B and Bʹ work at the level of the tcpP promoter. Treatment with toxtazin B results in a 100-fold reduction in colonization in an infant mouse model of infection, though toxtazin A did not reduce colonization at the concentrations tested. These results add to the growing body of literature indicating that small-molecule inhibitors of virulence genes could be developed to treat infections, as alternatives to antibiotics become increasingly needed.
IMPORTANCE
V. cholerae caused more than 580,000 infections worldwide in 2011 alone (WHO, Wkly. Epidemiol. Rec. 87:289-304, 2012). Cholera is treated with an oral rehydration therapy consisting of water, glucose, and electrolytes. However, as V. cholerae is transmitted via contaminated water, treatment can be difficult for communities whose water source is contaminated. In this study, we address the need for new therapeutic approaches by targeting the production of the main virulence factor, cholera toxin (CT). The high-throughput screen presented here led to the identification of two novel classes of inhibitors of the virulence cascade in V. cholerae, toxtazin A and toxtazins B and Bʹ. We demonstrate that (i) small-molecule inhibitors of virulence gene production can be identified in a high-throughput screen, (ii) targeting virulence gene production is an effective therapeutic strategy, and (iii) small-molecule inhibitors can uncover unknown layers of gene regulation, even in well-studied regulatory cascades.
Introduction
Vibrio cholerae, a Gram-negative pathogen, colonizes the human intestine and causes cholera, an acute disease characterized by vomiting, profuse watery diarrhea, and severe dehydration. The symptoms are caused by the secreted cholera toxin (CT) (1), which binds and enters intestinal epithelial cells and increases cyclic AMP (cAMP) production. This leads to a decrease in sodium uptake and a concomitant increase in chloride extrusion into the lumen of the intestine, resulting in water secretion and thus causing diarrhea and dehydration (2).
Because CT is the major virulence factor produced by V. cholerae, much research has gone into understanding how its expression is regulated. Epidemic strains of V. cholerae are divided into two biotypes, classical and El Tor, both of which regulate the virulence cascade via the master virulence regulator, ToxT (see Fig. 6). Transcription of the ctxAB operon, which encodes the two CT subunits, and the tcp operon, which contains the genes for the toxin-coregulated pilus (TCP), is activated by ToxT (3, 4). Transcription of toxT is activated by a protein complex comprised of four inner membrane proteins, ToxRS and TcpPH (5). The transcription of tcpPH is activated by two transcription activators, AphA and AphB, which respond to cell density, anaerobiosis, and other factors (6–8).
Currently, cholera is treated with oral rehydration therapy (ORT), which restores fluids to the patient and allows the immune system to clear the infection (9, 10). Antibiotics are sometimes administered as a second line of treatment, as they can reduce vomiting and diarrheal volume by 50% and shorten illness duration by 50%. However, antibiotics are not effective alone because patients are still at risk of severe dehydration caused by CT (9, 11). Because of this, other treatment modalities could improve the treatment of cholera, and this is an area of active research (12–14). Previous work identified an inhibitor called virstatin, which inhibits ToxT dimerization and thereby alters its activity. Further experiments using virstatin revealed that ToxT dimerization affects its activity at various promoters (15). When ToxT was crystallized, it was bound to a sixteen-carbon fatty acid, cis-palmitoleate. This and other similar fatty acid ligands were shown to hold ToxT in a closed conformation, inhibiting its ability to bind and activate the tcp and ctx promoters (16).
Antivirulence drugs are becoming an increasingly popular strategy in combating diseases. Unlike antibiotics, antivirulence drugs aim to disarm a pathogen by eliminating its virulence potential and allowing the immune system to clear the infection, providing several advantages. First, targeting virulence genes imposes a weaker selective pressure than targeting growth, decreasing the potential for the emergence of resistant strains (17, 18). In fact, under some conditions, spontaneous nontoxigenic mutants of V. cholerae outcompete the pathogenic wild-type parental strains (19). Additionally, targeting pathogens with antivirulence drugs has minimal effects on the endogenous microbiota, diminishing the risk of dysbiosis caused by antibiotics, which can lead to acute and chronic intestinal problems (20, 21).
In addition to the potential therapeutic advantages of antivirulence compounds, small molecules have significant value as molecular probes for investigating the basic biology governing virulence. Because such compounds do not kill bacteria, they can be used experimentally to probe virulence traits without genetic manipulation of the organism. Compounds used as molecular probes act quickly and are often reversible, and their effects can be enhanced or diminished simply by changing the concentration.
In this study, we performed a high-throughput screen to identify small molecules that inhibit the expression of toxT. We identified three compounds, which we named toxtazin A and toxtazin B and Bʹ; the latter two are structural analogs of each other. All three compounds decrease CT and TCP levels. We determined that toxtazin A does not affect the protein or transcript levels of the ToxR, TcpP, AphA, or AphB, which are encoded by the genes upstream of toxT in the virulence cascade, indicating that it prevents toxT transcription. Toxtazins B and Bʹ both inhibit virulence by decreasing TcpP protein and tcpP transcript levels but not AphA or AphB protein or transcript levels, indicating that these compounds function by inhibiting tcpP transcriptional activation.
RESULTS
A GFP-based high-throughput screen identifies two novel classes of toxT expression inhibitors.
To identify small-molecule inhibitors of toxT transcription that do not affect general growth, a reporter strain was constructed consisting of wild-type V. cholerae harboring a plasmid on which the toxT promoter drives the expression of green fluorescent protein (GFP). Culturing the toxT::gfp reporter strain NB39 under toxin-inducing conditions resulted in high numbers of fluorescence intensity units in a wild-type background and relatively low numbers of fluorescence intensity units in the isogenic ΔtoxR strain NB40 (Fig. 1A). Molecules that decrease GFP expression in a wild-type cell could inhibit any event in the virulence cascade prior to toxT transcription.
FIG 1 .
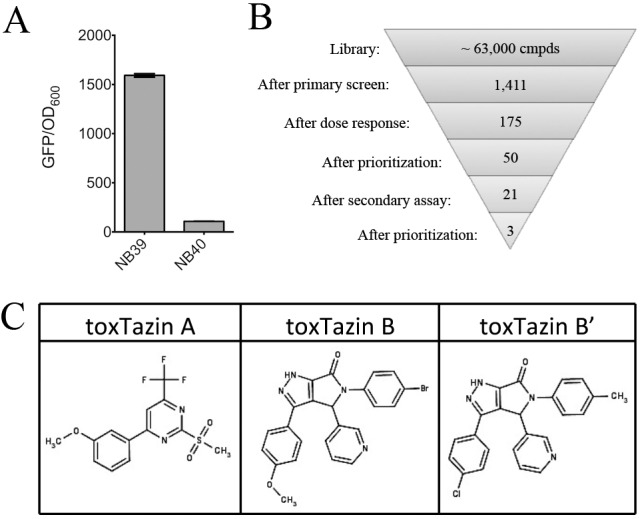
Identification of inhibitors of the virulence cascade in V. cholerae. (A) The wild-type reporter strain NB39 has a high GFP/OD600 ratio when grown overnight under toxin-inducing conditions, while the isogenic ΔtoxR strain NB39 does not. (B) Funnel figure illustrating the triage process used in the screen. (C) Structures of the toxtazins.
Using NB39, approximately 63,000 diverse compounds were screened at the University of Michigan Center for Chemical Genomics. A compound was scored as active if it reduced GFP fluorescence to below six standard deviations from the mean for the untreated controls, without decreasing growth (measured as changes in optical density at 600 nm [OD600]) >10% relative to the untreated controls, indicating that the compound is not toxic at that dose. Of the 1,411 compounds that met these requirements, 175 exhibited dose-dependent inhibition of GFP fluorescence (Fig. 1B). We prioritized these based on potency and purchased the top 50 compounds for further characterization. We measured the levels of CT produced in cultures treated with the top 50 compounds by ELISA and selected the three compounds that resulted in the lowest level of toxin production for further characterization. We named these compounds toxtazins A, B, and Bʹ, and their chemical structures are shown in Fig. 1C.
Toxtazin A, B, and Bʹ inhibit the virulence cascade.
To determine an optimal concentration for our experiments, cultures of NB39 were inoculated overnight under toxin-inducing conditions, with various concentrations of compounds or the equivalent volume of dimethyl sulfoxide (DMSO). Addition of compounds had significant and dose-dependent effects on GFP fluorescence (Fig. 2A), and the half-maximal effective concentrations (EC50) of toxtazins A, B, and Bʹ were 24.5 µM, 2.7 µM, and 7.2 µM, respectively. All three compounds had a statistically significant effect on growth at concentrations of 25 µM and higher (Fig. 2B) and a statistically significant effect on GFP fluorescence at concentrations of 5 µM and higher. Thus, subsequent in vitro experiments were performed at 10 µM unless otherwise indicated.
FIG 2 .

Toxtazin A, B, and Bʹ inhibit the virulence cascade in V. cholerae. (A and B) GFP expression (A) and terminal OD600 (B) after 16 h in cultures grown overnight under toxin-inducing conditions with 0.16 µM to 50 µM of compounds. (C) CT expression levels in wild-type cultures grown overnight under various toxin-inducing conditions in the presence or absence of 10 µM compounds. The dotted line indicates the limit of detection. An isogenic ΔtoxR strains served as a control. (D) Western blot of O395 cultures treated with 10 µM compounds overnight in LB pH 6.5 at 30°C, using a TcpA antibody. (E) CT levels in cultures of RA286 grown overnight under toxin-inducing conditions in the presence or absence of 10 µM compounds. Error bars represent the standard deviations for three biological replicates. The dotted line indicates the limit of detection.
If these compounds indeed inhibit toxT expression as opposed to altering GFP function, compound treatment should lead to decreased expression of ToxT-activated genes, such as those encoding CT and TCP. Additionally, for these compounds to have therapeutic potential, they should work in both the classical and El Tor biotypes of V. cholerae and should work under various toxin-inducing growth conditions. CT levels were determined in both classical and El Tor biotypes under various in vitro conditions known to induce the ToxT system for each of the two biotypes. For the classical strain O395, cultures were incubated overnight with 10 µM concentrations of each compound either in LB (pH 6.5) at 30°C or in minimal medium supplemented with amino acids asparagine, glutamate, arginine, and serine (NERS). For the El Tor strain E7946, cultures were incubated with a 10 µM concentration of each compound under AKI conditions (Fig. 2C). Toxtazins A, B, and Bʹ significantly inhibited CT production in both biotypes and under all three tested growth conditions, validating the target of the compounds as toxT transcription and ruling out trivial effects, such as inhibition of GFP activity. These results also indicate that the compounds are not biotype- or condition-specific inhibitors, though toxtazin B is more effective against the classical biotype. We note that toxtazin A (but not toxtazin B or Bʹ) completely inhibited growth in M9+NERS (data not shown), indicating that toxtazin A may affect a pathway required for growth under these conditions and that toxtazin A and B/Bʹ work by different mechanisms.
O395 cultures grown overnight in LB (pH 6.5) at 30°C in the presence or absence of compounds were also analyzed for TcpA expression by immunoblot (Fig. 2D). All three compounds decreased TcpA levels relative to the DMSO-treated control, supporting the conclusion that these compounds affect the virulence cascade by altering toxT expression.
To confirm that the compounds work on the cascade prior to toxT expression, we tested the effects of the toxtazins on bacteria expressing toxT ectopically, under the control of an IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible promoter. We predicted that if the compounds alter the cascade of gene regulation that leads to toxT expression, then removing toxT control from that cascade would confer resistance to the effects of the compounds. Strain RA286, which is a wild-type V. cholerae classical strain with a plasmid-encoded IPTG-inducible allele of toxT, was cultured overnight under toxin-inducing conditions in either DMSO, toxtazin A, or toxtazin B, and toxT was induced with 100 µM IPTG. The resulting supernatants were analyzed by enzyme-linked immunosorbent assay (ELISA) to quantify CT levels (Fig. 2E). Toxtazins A and B both led to decreased CT expression in bacteria cultured without IPTG, while CT levels were restored in bacteria cultured with IPTG to induce toxT transcription, indicating that both toxtazins A and B act upstream of toxT in the virulence cascade. We noted that overexpression of ToxT in the DMSO control led to a slight decrease in CT levels. While the mechanism for this is unclear, we have observed that overexpressing components of this regulatory cascade (including ToxR and TcpP) can have a slight inhibitory effect on CT, perhaps due to altered stoichiometry of activator complexes required for gene expression. This observation notwithstanding, the fact that ectopic ToxT expression restores CT expression in the presence of the toxtazins indicates that they inhibit the virulence cascade prior to toxT transcription.
Toxtazin B, but not toxtazin A, decreases V. cholerae colonization in an infant mouse model.
Host colonization by V. cholerae is dependent on TCP expression. Based on our in vitro findings that the toxtazins decrease TcpA levels, we hypothesized that they could decrease colonization loads of V. cholerae in a mouse model of colonization. To test toxtazin A activity in vivo, 4- to 6-day old mice were orogastrically inoculated with 106 bacteria and either toxtazin A or DMSO and received a booster of compound 3 h postinfection. Because not all the compound administered will reach the desired location within the mouse (i.e., the small intestine), and because some of the compound may be metabolized or otherwise degraded within the mouse, we used higher concentrations of compounds in these experiments. To ensure that any observed decreases in colonization are due to inhibition of TcpA and not to toxicity against V. cholerae, 3-ml LB cultures were started using the same inoculum, boosted with compound 3 h later, and then cultured overnight. Administration of either 20 µg, 40 µg, or 60 µg toxtazin A to mice inoculated with V. cholerae did not decrease colonization levels relative to those in the DMSO-treated mice (Fig. 3A). While toxtazin A did not reduce colonization even at the highest level tested, that concentration was toxic to V. cholerae grown in vitro (Fig. 3B). We note that cultures grown in sublethal concentrations of toxtazin A routinely grow better in LB, and this was also seen in the in vitro experiment at 20 µg and 40 µg (Fig. 3B). The mouse data indicated that toxtazin A was not toxic in vivo at 60 µg, but higher doses were not tested because it would be impossible to distinguish whether a decrease in colonization resulted from the ability of toxtazin A to inhibit ToxT activity or from its antibacterial activity. The lack of in vivo killing by toxtazin A at the 60- µg dose may reflect poor bioavailability of the compound in the infant mouse.
FIG 3 .
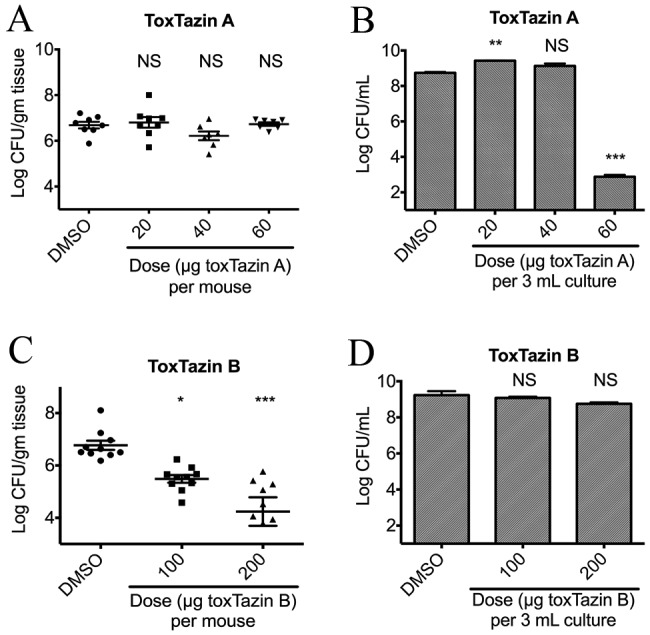
In vivo
efficacy of toxtazins A and B. Asterisks denote the statistical significance of a group relative to the DMSO control. NS, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. (A) Number of V. cholerae organisms recovered from mice orogastrically inoculated with 106 wild-type V. cholerae organisms and treated with either DMSO or 0 µg, 20 µg, 40 µg, or 60 µg toxtazin A. (B) Number of V. cholerae organisms recovered from 3-ml cultures grown overnight with the same inocula and boosters as the mice. (C) Number of V. cholerae recovered from mice orogastrically inoculated as described for panel A and treated with DMSO or 100 µg or 200 µg toxtazin B. (D) Number of V. cholerae organisms recovered from 3-ml cultures grown overnight with the same inocula and boosters as the mice.
Because toxtazin B and Bʹ are structural analogs and have behaved similarly in all assays thus far, and to reduce the number of animals used, we focused on toxtazin B in the following experiments. The same in vivo experiment performed with toxtazin A was performed with toxtazin B. Administration of either 100 µg or 200 µg toxtazin B to mice inoculated with V. cholerae decreased colonization levels approximately 100-fold relative to those in the DMSO-treated mice (Fig. 3C). This effect was not due to toxtazin B toxicity against V. cholerae, since the 3-ml in vitro cultures treated with toxtazin B had no growth defect compared to the DMSO-treated cultures (Fig. 3D). These results suggest that toxtazin B inhibits expression of the TcpA colonization determinant both in vitro and in vivo.
The toxtazins do not affect ToxR protein levels or activity.
To determine where in the regulatory cascade leading to toxT expression each compound works, a targeted approach was used. We first tested whether the compounds altered levels or activity of ToxR, one of the direct transcription activators of toxT. ToxR protein levels were unaffected by toxtazins A, B, and Bʹ in cultures grown overnight under toxin-inducing conditions (Fig. 4A). ToxR activity was investigated by measuring levels of OmpU and OmpT, outer membrane proteins whose transcription is activated or repressed, respectively, by ToxR. Inhibiting ToxR activity would decrease OmpU levels and elevate OmpT levels, similar to a toxR mutant. Cell lysates from overnight cultures grown with or without compounds under toxin-inducing conditions were subjected to SDS-PAGE and stained with Coomassie blue to visualize OmpU and OmpT (Fig. 4B). Toxtazin-treated cultures have the same OmpU/OmpT profile as the DMSO-treated wild-type culture, indicating that ToxR activity is not affected by toxtazin A, B, or Bʹ.
FIG 4 .
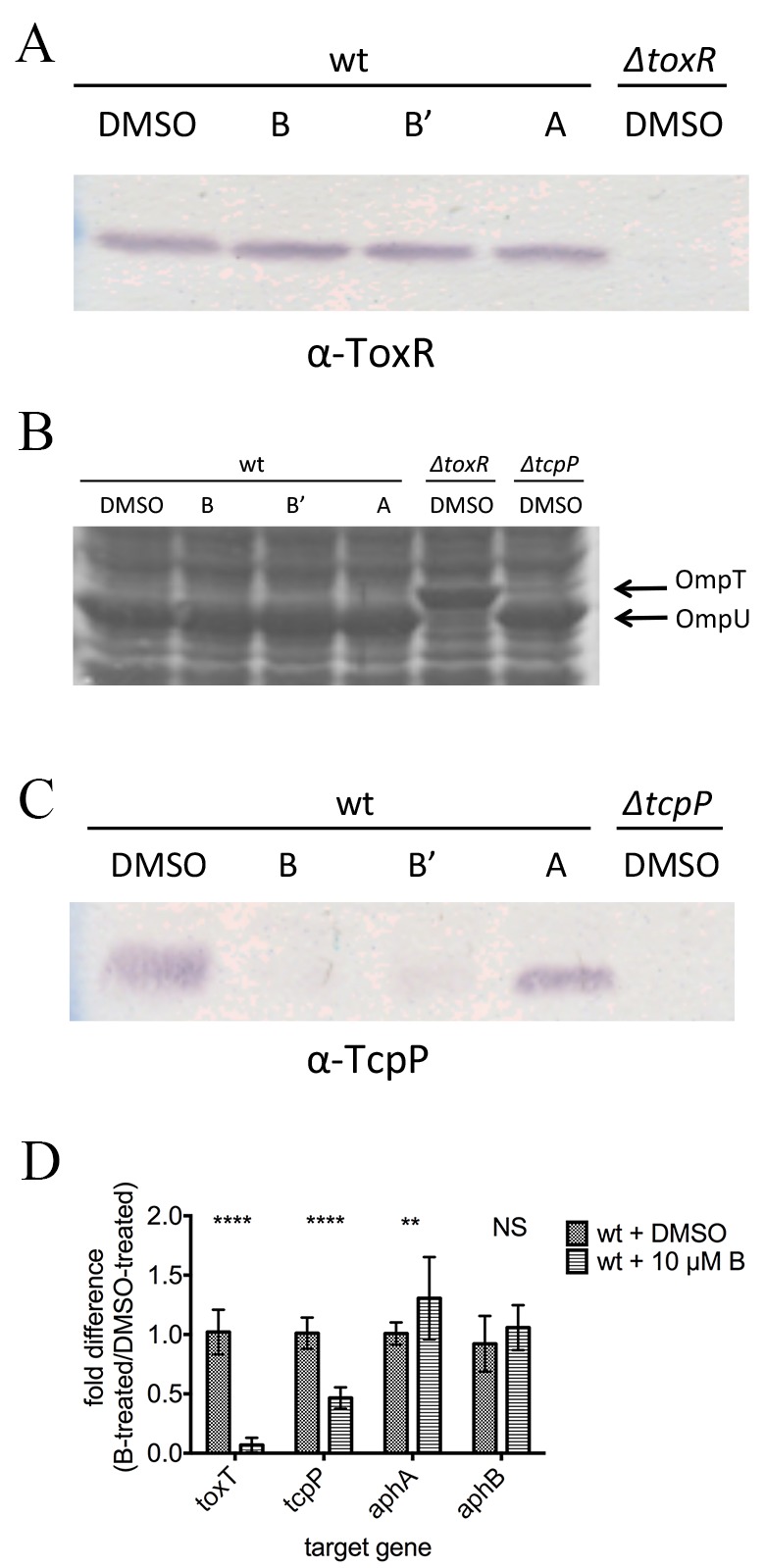
Effects of toxtazin A and B on the virulence cascade. (A) ToxR Western blot of cultures grown overnight under toxin-inducing conditions with or without 10 µM concentrations of compounds. (B) Coomassie stain showing OmpU and OmpT levels in cultures grown as described for panel A. (C) TcpP Western blot of cultures grown as described for panel A. (D) Transcript levels of toxT, tcpP, aphA, and aphB were determined for cultures grown as described for panel A. Significance was calculated by two-way ANOVA. NS, not significant; *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
TcpP protein levels are differentially affected by toxtazin A and B/Bʹ.
Next, we analyzed the effect on levels of TcpP, the other major transcription activator that regulates toxT. Cultures were grown overnight under toxin-inducing conditions in the presence or absence of 10 µM toxtazin A, B, or Bʹ, and cell lysates were analyzed by Western blotting with antiserum directed against TcpP. Toxtazin A did not alter TcpP protein levels; however, toxtazins B and Bʹ caused significant decreases in TcpP protein levels relative to those in the DMSO control (Fig. 4C). These results confirm that toxtazin A and B/Bʹ have different targets and mechanisms of action for inhibiting toxT transcription in V. cholerae. Toxtazin B and Bʹ inhibit TcpP expression, while toxtazin A affects toxT expression without altering TcpP levels.
Toxtazin B reduces transcript levels of toxT and tcpP but not of aphA or aphB.
Because toxtazin B and Bʹ behave similarly, we again focused on toxtazin B for the next set of experiments. To determine where in the TcpP branch of the regulatory cascade toxtazin B acts, qRT-PCR was used to quantify the levels of toxT, tcpP, aphA, and aphB transcripts in cultures treated overnight with 10 µM toxtazin B, incubated under toxin-inducing conditions (Fig. 4D). All results were normalized to 16S rRNA and are reported as the ratio of the mRNA levels in toxtazin B-treated cultures to those in DMSO-treated cultures. As expected, toxT expression was reduced by a factor of 10 in cultures treated with toxtazin B relative to the DMSO control. While the compound did not affect aphA or aphB transcript levels, tcpP transcript levels were decreased 50% compared to the DMSO controls, consistent with the decrease observed in TcpP protein levels. These data indicate that toxtazin B targets tcpP transcription.
Neither toxtazin A nor B affects AphA or AphB levels.
Having determined that aphA and aphB mRNA levels are not altered by toxtazin B, we wanted to determine whether their protein levels might be affected. Cultures grown overnight under toxin-inducing conditions in the presence or absence of 10 µM toxtazin A or B were analyzed by Western blotting (Fig. 5). No decrease in AphA or AphB protein levels was observed in the presence of toxtazin A or B. Toxtazin A consistently caused an increase in AphA protein levels, but it is unlikely that this accounts for the mechanism by which this compound leads to diminished toxT transcription. In any case, our results indicate that decreased tcpP transcription caused by toxtazin B is not due to an effect of the compound on AphA or AphB protein levels, pointing instead to the possibility that the compound may inhibit the activity of one of these proteins. Supporting this conclusion, ectopic expression of AphA or AphB did not make cells resistant to toxtazin B (see Fig. S1 in the supplemental material). As predicted, AphA and AphB levels were unaltered by growth in the presence of toxtazin A (Fig. 5).
FIG 5 .
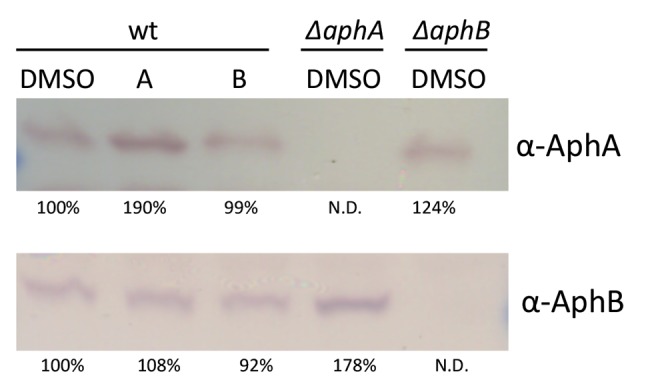
Toxtazins A and B do not affect AphA or AphB protein levels. Western blot for AphA and AphB in samples grown overnight under toxin-inducing conditions in the presence or absence of 10 µM concentrations of compounds. Band intensities were quantified with ImageJ and normalized to the wild-type DMSO-treated samples.
DISCUSSION
Despite the use of antibiotics and oral rehydration therapy (ORT), V. cholerae remains a major public health concern in much of the developing world. The number of yearly cholera cases has steadily increased since 2007, and the number of fatalities has increased in the past year (22). New treatment therapies that target toxin production and colonization by V. cholerae, either alone or in combination with current therapies, could be very useful in reducing the global health burden caused by this pathogen.
In this study, a bacterial-cell-based, high-throughput screen was used to identify and characterize three small molecules, toxtazin A, B, and Bʹ. All three reduce the virulence potential of V. cholerae in vitro, and toxtazin B also reduced colonization in vivo. We show that all three compounds reduce CT production in both the classical and El Tor biotypes and reduce TcpA levels relative to a DMSO control.
The two classes of compounds identified here work by different mechanisms. Toxtazin A does not alter TcpP or ToxR protein levels, suggesting that it affects toxT transcription via a different mechanism. We have not ruled out the possibility that toxtazin A inhibits ToxR or TcpP function, perhaps keeping one of them from physically binding to its site on the toxT promoter. Both ToxR and TcpP may sense environmental signals and transmit the information to the cytoplasm by activation of toxT transcription. Environmental signals such as temperature, pH, osmolarity, bile, oxygen, and amino acids have all been shown to affect toxT activation in vitro (8, 23–25). In fact, when ToxT was crystalized, a cis-palmitoleate was discovered in a solvent-inaccessible binding pocket. Oleic acid, another C-9 monosaturated fatty acid, may be the natural ligand for this binding pocket, as it also strongly regulates ToxT activity and is more abundant in bile than cis-palmitoleic acid. Other natural compounds have been reported to influence expression of the virulence cascade in V. cholerae (26, 27). The stressed physiological state of the cell grown with toxtazin A revealed by proteomic analysis (data not shown) and the growth-inhibitory phenotype we observed in minimal medium containing NERS (Fig. 2C) led us to propose that toxtazin A induces a nonpermissive physiological state in the cell which feeds back to shut off toxT transcription, and we are currently exploring this hypothesis. We investigated the potential for the toxtazins to act as general redox-active compounds by measuring the activity of alkaline phosphatase, which requires disulfide bond formation. Tested both in vitro and in cells treated with the toxtazins (data not shown), alkaline phosphatase activity was unaffected, suggesting that the toxtazins do not affect the general redox state of the cell. Furthermore, the compounds do not affect the bicinchoninic acid (BCA) protein assay, which does not work in the presence of a reducing agent, indicating that, at the concentrations used in our experiments, these compounds do not act as general reducing agents.
The mechanism of toxtazin B/Bʹ inhibition of virulence is clearer. Toxtazin B and Bʹ decrease TcpP but not ToxR protein levels, and they decrease the levels of toxT and tcpP transcripts but not those of aphA or aphB, nor do they decrease AphA or AphB protein levels. We noticed that toxtazin A consistently caused an increase in AphA protein levels, but it is unlikely that this accounts for the diminished toxT transcription levels. Taken together, the data suggest that toxtazin B inhibits the virulence cascade at the level of the tcpP promoter---downstream of AphA and AphB protein production but upstream of tcpP transcription. Supporting this, ectopic expression of either AphA or AphB does not restore CT production (Fig. 5) in the presence of toxtazin B.
Toxtazin B inhibits virulence gene expression in the classical biotype more strongly than in the El Tor biotype. It is notable that tcpP transcription, the level at which our data suggest this compound works, is regulated differently in the two biotypes. A single A-to-G base pair difference in the tcpP promoters of the two biotypes disrupts the dyad symmetry of the AphB binding motif (28–30); thus, AphB binds 10 times more strongly to the classical tcpP promoter than to the El Tor tcpP promoter (31, 32). We speculate that the natural differences in tcpP regulation in classical versus El Tor may be responsible for the different effectiveness of toxtazin B seen in the two biotypes in Fig. 2C, but further study must be done to confirm this.
The tcpP promoter is a highly regulated feature of the complex regulatory cascade controlling toxT transcription, assimilating multiple signals, including pH, osmolarity, cAMP levels, and phosphate levels. For example, when cells are grown at the nonpermissive pH, a protein called PepA partially inhibits tcpP transcription in the classical biotype (33). The tcpP promoter is also negatively regulated by the cAMP-CRP complex, whose binding site at the promoter overlaps the binding sites of AphA and AphB (30). Finally, the tcpP promoter is negatively regulated by PhoB, which binds at a site distinct from both the AphA and AphB binding sites (34). Toxtazin B may inhibit tcpP expression by altering the binding properties of any these proteins, or of the AphA and/or AphB proteins, at the tcpP promoter. Future work will aim to determine the precise mechanism by which toxtazin B inhibits gene expression required for colonization and pathogenicity.
In this study, the activity of the toxtazins in vivo was also determined using the infant mouse model of colonization. The ability of V. cholerae to colonize and replicate in this mammalian host is in large part dependent on expression of TCP, the major subunit of which is TcpA; wild-type strains are significantly more competitive than tcpA mutants in vivo (35, 36). Our in vitro results (Fig. 2B) demonstrate that toxtazin B leads to reduced TcpA levels, which we propose as the main reason for reduced colonization caused by toxtazin B. We note that a tcpA mutant colonizes more poorly than a toxtazin B-treated wild type, and we take this to mean that the local compound concentration (that is, the amount of toxtazin B that actually reaches V. cholerae in the gut) is lower than what would be required for complete inhibition. Pharmacodynamic and pharmacokinetic studies might optimize the ability of toxtazin B to inhibit colonization.
Unlike toxtazin B, toxtazin A did not decrease colonization level of V. cholerae at the concentrations tested. Perhaps this compound does not inhibit colonization because it does not reduce TcpA levels as efficiently as toxtazin B, or perhaps not enough compound reached the bacteria in the gut. Structure-activity relationship (SAR) studies to identify an analog with increased potency and lower toxicity would be useful in future in vivo studies and for determining whether toxtazin A decreases colonization.
In summary, we identified two novel classes of toxT transcription inhibitors, toxtazin A, B, and Bʹ. They are potent inhibitors of V. cholerae virulence gene expression and function at distinct points in the virulence regulatory cascade (Fig. 6). Toxtazin A inhibits by preventing toxT transcription late in the regulatory cascade, evidenced by the fact that ToxR and TcpP protein levels are equal to those of DMSO-treated cultures and that mRNA levels of tcpP, aphA, and aphB are not affected. Toxtazin B works by altering tcpP transcription, shown by a decrease in both transcript and protein levels of tcpP but no decrease in the protein or transcript levels of AphA or AphB relative to the DMSO-treated controls. Toxtazin B also reduces colonization of V. cholerae in an infant mice model. We are currently working on better characterizing the mechanism of action of the toxtazins to gain deeper insight into the requirements of V. cholerae pathogenesis. In addition to providing new chemical probes for richer study of the virulence cascade in V. cholerae, the toxtazins add further proof of principle that potent small-molecule inhibitors can be discovered by high throughput screening and can be used both as molecular probes for basic research and as a starting point for therapeutic development.
FIG 6 .
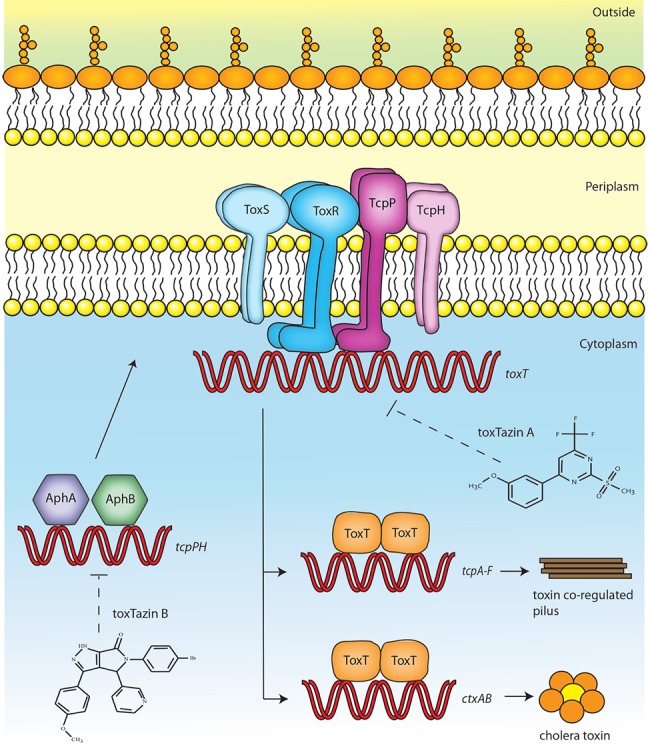
Model of the virulence cascade in V. cholerae and targets of the toxtazins. The virulence cascade in V. cholerae is tightly regulated. AphA and AphB activate transcription of tcpPH. TcpPH form an inner membrane complex with ToxR and ToxS to activate transcription of toxT. ToxT activates the transcription of tcpA-F, which encode the toxin coregulated pilus, and ctxAB, which encode the cholera toxin subunits. Based on our experiments, we propose that toxtazin A inhibits toxT transcription, while toxtazin inhibits tcpPH transcription.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and chemical inhibitors.
The strains and plasmids used in this study are listed in Table 1. Unless otherwise noted, the V. cholerae classical biotype strain O395 was used in these studies. Strains were maintained at −80°C in Luria-Bertani broth (LB) containing 20% glycerol. Overnight cultures were grown in LB medium at 37°C. Growth of the O395 classical strain under toxin-inducing conditions consists of subculturing an overnight culture 1:100 in LB pH 6.5 and growing at 30°C with shaking for 16 to 18 h, or as indicated above. Growth in minimal medium consists of subculturing an overnight culture 1:100 in M9 minimal medium containing NERS (M9 salts, 2 mM MgSO4, 4% glycerol, 0.1 mM CaCl2, and 5 mM [each] asparagine, glutamic acid, arginine, and serine) and growing overnight at 37°C with shaking (37). Growth under AKI conditions consists of diluting overnight cultures of the El Tor strain E7946 1:1,000 in 10 ml of AKI medium, growing them anaerobically for 4 h statically at 37°C, pouring the 10-ml cultures into 250-ml flasks, and growing them, with shaking, for an additional 4 h or longer (38, 39).
TABLE 1 .
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype and phenotype | Source |
|---|---|---|
| V. cholerae strains | ||
| O395 | Classical Ogawa, Smr | Laboratory collection |
| E7946 | El tor, Smr | Laboratory collection |
| RA25 | O395 ΔtoxR | 3 |
| RA6 | E7946 ΔtoxR | This work |
| NB39 | O395 + ptoxT-GFP | Laboratory collection |
| NB40 | O395 ΔtoxR + ptoxT-GFP | Laboratory collection |
| RA179 | O395 ΔtoxT | 43 |
| RA67 | O395 ΔtcpP | 44 |
| RA305 | O395 ΔaphA | This work |
| RA282 | O395 ΔaphB | This work |
| RA286 | O395 + pMT5 | 45 |
| RA289 | O395 + pMMB66EH-aphA | This work |
| RA290 | O395 + pMMB66EH-aphB | This work |
| RA306 | O395 ΔaphA + pMMB66EH | This work |
| RA307 | O395 ΔaphA + pMMB66EH- aphA |
This work |
| RA308 | O395 ΔaphB + pMMB66EH | This work |
| RA309 | O395 ΔaphB + pMMB66EH- aphB |
This work |
| Plasmids | ||
| ptoxT-GFP | pBH6119-toxTpro-gfp | This work |
| pMT5 | pMMB66HE-toxT | 45 |
| pWM91-ΔaphA | pWM91-ΔaphA | 46 |
| pWM91-ΔaphB | pWM91-ΔaphB | 46 |
| pMMB66EH- aphA |
pMMB66EH-aphA (O395) | This work |
| pMMB66EH- aphB |
pMMB66EH-aphB (O395) | This work |
Toxtazin A [IUPAC: 2-methanesulfonyl-4-(3-methoxyphenyl)-6-(trifluoromethyl)pyrimidine], toxtazin B [IUPAC: 5-(4-bromophenyl)-3-(4-methoxyphenyl)-4-(pyridine-3-yl)-1H,4H,5H,6H-pyrrolo[3,4-c] pyrazol-6-one], and toxtazin Bʹ [IUPAC: 3-(4-chlorophenyl)-5-(4-methylphenyl)-4-(pyridine-3-yl)-1H,4H,5H,6H-pyrrolo[3,4-c] pyrazol-6-one] were obtained from ChemDiv (San Diego, CA). Compound stocks were made to either 20 mM or 100 mM in DMSO and stored in the dark at −20°C. Cultures were treated with 10 µM unless otherwise specified, and the controls always received an equal volume of DMSO. Streptomycin was used at 100 µg/ml, ampicillin at 100 µg/ml, and X-Gal at 40 µg/ml.
The screening plasmid ptoxT-gfp was created by digesting pTLI2 (40) with EcoRI and BamHI to generate the −172 to +45 toxT promoter fragment. This fragment was ligated into the promoterless GFP reporter vector pBH6119 (41). The reporter plasmid was electroporated into wild-type O395 and an isogenic ΔtoxR mutant to generate the screening strains.
The E7946ΔtoxR strain was constructed using the pKAS32-ΔtoxR suicide plasmid as done previously (5) using wild-type E7946 as the recipient.
The clean deletions of aphA and aphB in O395 were generated by mating wild-type O395 cells with SM10λpir cells containing either pWM91-ΔaphA or pWM91-ΔaphB (kindly provided by Jun Zhu) and selecting for sucrose-resistant colonies. Mutants were confirmed by DNA sequencing.
The pMMB66EH-AphA and AphB constructs were constructed by amplifying the AphA sequence using primers RAP186 (GCAACGAATTCATGTCATTACCA) and RAP 187 (GTCAAGCTTTTATGCCATCGC) and the AphB sequence using primers RAP218 (GCC GAATTCTTGCAACATAATGTGTCAGA) and RAP219 (CCGAAGCTTTTATTGCAGGTGGTAGCC) from O395. The resulting products were digested with EcoRI and HindIII and ligated into pMMB66EH. The resulting constructs (Table 1) were verified by DNA sequencing.
IC50 and EC50 determination.
The concentration of compound that inhibits toxT-GFP activity to 50% of the DMSO-treated control (EC50) was measured by inoculating 2 ml of LB with a 1:100 dilution of an overnight culture and monitoring GFP expression under toxin-inducing conditions with 0.16 µM to 50 µM concentrations of compounds. The OD600 of these cultures was also monitored to determine the IC50, the concentration of compound that inhibits growth to 50% of that of the DMSO-treated control. EC50 and IC50 values were calculated in GraphPad Prism using the 4-parameter model (variable slope).
High-throughput screening for small-molecule inhibitors of toxT-gfp expression.
The screening strain used (NB39) is an O395 classical strain harboring a plasmid with the toxT promoter driving expression of GFPmut3. An isogenic O395 ΔtoxR strain with the toxT-GFP reporter (NB40) served as a control in the screen.
The primary screen, secondary screen, and dose-response studies were carried out at the Center for Chemical Genomics (University of Michigan), where approximately 63,000 compounds and 11,000 natural products were tested. Overnight cultures of NB39 were diluted to a final OD600 of 0.02 in LB (pH 6.5) containing streptomycin and ampicillin and transferred in a 40-µl volume to wells of a black, clear-bottom 384-well microtiter plate (Falcon 35-3948). The screening plates received 10 µM concentrations of compounds, which were pin transferred from stock plates. Each test plate contained positive (strain NB40 with DMSO) and negative (strain NB39 with DMSO) controls, and all compounds were tested in at least duplicate. The plates were incubated at 30°C for 16 to 18 h statically, after which the OD600 and GFP fluorescence (excitation λ = 385 nm, emission λ = 425 nm) were read for each well.
A compound was considered active if it met two criteria: (i) it caused a decrease in GFP expression of more than six standard deviations from the value for the DMSO control, and (ii) the OD600 of cultures grown with the compound was within 10% of the value for the DMSO control. A total of 1,411 compounds met both criteria and were retested for their effects on growth and toxT-gfp expression and for their dose responsiveness. Of the 175 compounds that retained activity and displayed dose-dependent activity, the 50 most potent (a low GFP/OD600 value) were ordered from ChemDiv and similarly retested for effects on growth and toxT transcription and for effects on CT production. Of the 21 compounds that caused significantly reduced CT levels, we focused on toxtazins A, B, and Bʹ because of their potency (Fig. 1C).
Detection of cholera toxin by ELISA.
Cultures of V. cholerae were grown under toxin-inducing conditions for 16 to 18 h with 10 µM compound or DMSO. GM1 ganglioside enzyme-linked immunosorbent CT assays were performed as previously described (42) on equal volumes of the resulting supernatants. CT expression values were normalized to the OD600 and are the averages for samples grown in at least duplicate.
Western blot analysis of TcpA, ToxR, TcpP, AphB, and AphA.
Cells were cultured under toxin-inducing conditions in the presence or absence of 10 µM compounds. Cell extracts were subjected to SDS-PAGE, transferred to a nitrocellulose membrane (GE Water and Processes Technologies, Feasterville-Trevose, PA), probed with the appropriate antibody, and visualized with alkaline phosphatase. The TcpA antibody was used at a 1:10,000 dilution, the ToxR antibody at a 1:1,000 dilution, and the TcpP antibody at a 1:500 dilution. The AphB antibody was kindly provided by Jun Zhu. The AphA antibody (kindly provided by Karen Skorupski) was used at a 1:10,000 dilution. Band densities were determined with the ImageJ software (http://rsb.info.nih.gov/ij/) and normalized to the wild-type DMSO-treated samples.
qRT-PCR analysis of mRNA expression.
Cells were cultured under toxin-inducing conditions in the presence or absence of 10 µM toxtazin B. RNA was harvested with Trizol (Invitrogen, Carlsbad, CA) according to the manufacturer’s directions, and DNA was removed using Turbo DNase (Ambion, Austin, TX). The qRT-PCR experiments were performed using the QuantiTect SYBR green RT-PCR kit (Qiagen, Valencia, CA) according the manufacturer’s manual. The qRT-PCR primers are shown in Table 2. Expression levels were normalized to 16S rRNA, and fold change was calculated using the 2−ΔΔCT method described in Applied Biosystems user bulletin no. 2 (P/N 4303859). Results are the averages for three biological replicates with three replicates each. Significance was determined by two-way analysis of variance (ANOVA).
TABLE 2 .
Primers used for qRT-PCR analysis
| Target | Forward primer | Reverse primer |
|---|---|---|
| toxT |
CAGCGATTTTCTTTGA CTTC |
CTCTGAAACCATTTAC CACTTC |
| tcpP |
GCTTTGCTACCTGTGA TTTGGTGG |
CCCGGTAACCTTGCTA AATCTCGT |
| aphA |
GAAGGCAAACCAGAT CGCAAGGTT |
AGACGGTAAGGTTCTG CCGATTGT |
| aphB |
TCAAATGGCAACTGA CCAACAGCC |
AGCTCCAATCCGACAG CACTTGTA |
| 16S rRNA |
GCATAACCTCGCAAGA GCAAAGCA |
TGTCTCAGTTCCAGTGT GGCTGAT |
Infant mouse colonization assays.
Four- to six-day-old CD1 mice (Charles River, Wilmington, MA) were orogastrically inoculated with a 30-µl bolus containing 106 CFU of V. cholerae O395, Cremaphor EL (20% final volume to solubilize compounds), and either DMSO or compound (doses are given above). An additional 30-µl bolus lacking bacteria was delivered to each mouse 3 h postinoculation, and the mice were incubated at 30°C. Mice were euthanized 18 to 24 h after inoculation, and the intestines were isolated, weighed, and homogenized in phosphate-buffered saline (PBS). Homogenates were serially diluted and plated on LB agar containing X-Gal and streptomycin to determine the number of CFU recovered. CFUs were normalized to the weight of the intestines and to the exact CFU of the initial inoculum. Significance was determined using one-way ANOVA.
SUPPLEMENTAL MATERIAL
Ectopic expression of AphA and AphB in toxtazin B-treated cells. (A) CT levels in a strain expressing AphA ectopically, grown overnight under toxin-inducing conditions in the presence or absence of 10 µM compound and in the presence or absence of 1 mM IPTG. (B) CT levels in a strain expressing AphB ectopically, grown as described for panel A. In both panels, error bars represent the standard deviations for three biological replicates. The dotted line denotes the limit of detection. Download
ACKNOWLEDGMENTS
We thank Jun Zhu for the AphB antibody and the pWM91-ΔaphA and pWM91-ΔaphB constructs used in this study, Karen Skorupski for the AphA antibody, Eric Krukonis for the construction of the E7946 ΔtoxR strain, and Nancy Beck for the construction of the reporter strains NB39 and NB40.
Footnotes
Citation Anthouard R, DiRita VJ. 2013. Small-molecule inhibitors of toxT expression in Vibrio cholerae. mBio 4(4):e00403-13. doi:10.1128/mBio.00403-13.
REFERENCES
- 1. Angelichio MJ, Spector J, Waldor MK, Camilli A. 1999. Vibrio cholerae intestinal population dynamics in the suckling mouse model of infection. Infect. Immun. 67:3733–3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanchez J, Holmgren J. 2011. Cholera toxin—a foe & a friend. Indian J. Med. Res. 133:153–163 [PMC free article] [PubMed] [Google Scholar]
- 3. Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc. Natl. Acad. Sci. U. S. A. 84:2833–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DiRita VJ, Parsot C, Jander G, Mekalanos JJ. 1991. Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 88:5403–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krukonis ES, Yu RR, Dirita VJ. 2000. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol. Microbiol. 38:67–84 [DOI] [PubMed] [Google Scholar]
- 6. Skorupski K, Taylor RK. 1999. A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol. Microbiol. 31:763–771 [DOI] [PubMed] [Google Scholar]
- 7. Kovacikova G, Skorupski K. 1999. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J. Bacteriol. 181:4250–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu Z, Yang M, Peterfreund GL, Tsou AM, Selamoglu N, Daldal F, Zhong Z, Kan B, Zhu J. 2011. Vibrio cholerae anaerobic induction of virulence gene expression is controlled by thiol-based switches of virulence regulator AphB. Proc. Natl. Acad. Sci. U. S. A. 108:810–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guerrant RL, Carneiro-Filho BA, Dillingham RA. 2003. Cholera, diarrhea, and oral rehydration therapy: triumph and indictment. Clin. Infect. Dis. 37:398–405 [DOI] [PubMed] [Google Scholar]
- 10. Hirschhorn N, Kinzie JL, Sachar DB, Northrup RS, Taylor JO, Ahmad SZ, Phillips RA. 1968. Decrease in net stool output in cholera during intestinal perfusion with glucose-containing solutions. N Engl J. Med. 279:176–181 [DOI] [PubMed] [Google Scholar]
- 11. Ayre J. 1849. Cholera: its nature, symptoms, and treatment. Stringer & Townsend, New York, NY. [Google Scholar]
- 12. Yamasaki S, Asakura M, Neogi SB, Hinenoya A, Iwaoka E, Aoki S. 2011. Inhibition of virulence potential of Vibrio cholerae by natural compounds. Indian J. Med. Res. 133:232–239 [PMC free article] [PubMed] [Google Scholar]
- 13. Fazil MH, Singh DV. 2011. Vibrio cholerae infection, novel drug targets and phage therapy. Future Microbiol. 6:1199–1208 [DOI] [PubMed] [Google Scholar]
- 14. Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ. 2005. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310:670–674 [DOI] [PubMed] [Google Scholar]
- 15. Shakhnovich EA, Hung DT, Pierson E, Lee K, Mekalanos JJ. 2007. Virstatin inhibits dimerization of the transcriptional activator ToxT. Proc. Natl. Acad. Sci. U. S. A. 104:2372–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lowden MJ, Skorupski K, Pellegrini M, Chiorazzo MG, Taylor RK, Kull FJ. 2010. Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc. Natl. Acad. Sci. U. S. A. 107:2860–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clatworthy AE, Pierson E, Hung DT. 2007. Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol. 3:541–548 [DOI] [PubMed] [Google Scholar]
- 18. Maeda T, García-Contreras R, Pu M, Sheng L, Garcia LR, Tomás M, Wood TK. 2012. Quorum quenching quandary: resistance to antivirulence compounds. ISME J. 6:493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paul K, Ghosh A, Sengupta N, Chowdhury R. 2004. Competitive growth advantage of nontoxigenic mutants in the stationary phase in archival cultures of pathogenic Vibrio cholerae strains. Infect. Immun. 72:5478–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McFarland LV. 2008. Antibiotic-associated diarrhea: epidemiology, trends and treatment. Future Microbiol. 3:563–578 [DOI] [PubMed] [Google Scholar]
- 21. Sekirov I, Russell SL, Antunes LC, Finlay BB. 2010. Gut microbiota in health and disease. Physiol. Rev. 90:859–904 [DOI] [PubMed] [Google Scholar]
- 22. WHO 2012. Cholera, 2011. Wkly. Epidemiol. Rec. 87::289–304 [PubMed] [Google Scholar]
- 23. Gardel CL, Mekalanos JJ. 1994. Regulation of cholera toxin by temperature, pH, and osmolarity. Methods Enzymol. 235:517–526 [DOI] [PubMed] [Google Scholar]
- 24. Hung DT, Mekalanos JJ. 2005. Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT-independent manner. Proc. Natl. Acad. Sci. U. S. A. 102:3028–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J. 2013. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc. Natl. Acad. Sci. U. S. A. 110:2348–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abuaita BH, Withey JH. 2009. Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect. Immun. 77:4111–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thomas S, Williams SG, Manning PA. 1995. Regulation of tcp genes in classical and El Tor strains of Vibrio cholerae O1. Gene 166:43–48 [DOI] [PubMed] [Google Scholar]
- 29. Ogierman MA, Voss E, Meaney C, Faast R, Attridge SR, Manning PA. 1996. Comparison of the promoter proximal regions of the toxin-co-regulated tcp gene cluster in classical and El Tor strains of Vibrio cholerae O1. Gene 170:9–16 [DOI] [PubMed] [Google Scholar]
- 30. Kovacikova G, Skorupski K. 2001. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol. Microbiol. 41:393–407 [DOI] [PubMed] [Google Scholar]
- 31. Kovacikova G, Skorupski K. 2000. Differential activation of the tcpPH promoter by AphB determines biotype specificity of virulence gene expression in Vibrio cholerae. J. Bacteriol. 182:3228–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kovacikova G, Skorupski K. 2002. Binding site requirements of the virulence gene regulator AphB: differential affinities for the Vibrio cholerae classical and El Tor tcpPH promoters. Mol. Microbiol. 44:533–547 [DOI] [PubMed] [Google Scholar]
- 33. Behari J, Stagon L, Calderwood SB. 2001. pepA, a gene mediating pH regulation of virulence genes in Vibrio cholerae. J. Bacteriol. 183:178–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pratt JT, Ismail AM, Camilli A. 2010. PhoB regulates both environmental and virulence gene expression in Vibrio cholerae. Mol. Microbiol. 77:1595–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merrell DS, Hava DL, Camilli A. 2002. Identification of novel factors involved in colonization and acid tolerance of Vibrio cholerae. Mol. Microbiol. 43:1471–1491 [DOI] [PubMed] [Google Scholar]
- 36. Chiang SL, Mekalanos JJ. 1998. Use of signature-tagged transposon mutagenesis to identify Vibrio cholerae genes critical for colonization. Mol. Microbiol. 27:797–805 [DOI] [PubMed] [Google Scholar]
- 37. Tischler AD, Camilli A. 2005. Cyclic diguanylate regulates Vibrio cholerae virulence gene expression. Infect. Immun. 73:5873–5882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Iwanaga M, Yamamoto K, Higa N, Ichinose Y, Nakasone N, Tanabe M. 1986. Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 el Tor. Microbiol. Immunol. 30:1075–1083 [DOI] [PubMed] [Google Scholar]
- 39. Iwanaga M, Kuyyakanond T. 1987. Large production of cholera toxin by Vibrio cholerae O1 in yeast extract peptone water. J. Clin. Microbiol. 25:2314–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Higgins DE, DiRita VJ. 1994. Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae. Mol. Microbiol. 14:17–29 [DOI] [PubMed] [Google Scholar]
- 41. Hammer BK, Swanson MS. 1999. Co-ordination of Legionella pneumophila virulence with entry into stationary phase by ppGpp. Mol. Microbiol. 33:721–731 [DOI] [PubMed] [Google Scholar]
- 42. Crawford JA, Krukonis ES, DiRita VJ. 2003. Membrane localization of the ToxR winged-helix domain is required for TcpP-mediated virulence gene activation in Vibrio cholerae. Mol. Microbiol. 47:1459–1473 [DOI] [PubMed] [Google Scholar]
- 43. Champion GA, Neely MN, Brennan MA, DiRita VJ. 1997. A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Mol. Microbiol. 23:323–331 [DOI] [PubMed] [Google Scholar]
- 44. Yu RR, DiRita VJ. 1999. Analysis of an autoregulatory loop controlling ToxT, cholera toxin, and toxin-coregulated pilus production in Vibrio cholerae. J. Bacteriol. 181:2584–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DiRita VJ, Neely M, Taylor RK, Bruss PM. 1996. Differential expression of the ToxR regulon in classical and E1 Tor biotypes of Vibrio cholerae is due to biotype-specific control over toxT expression. Proc. Natl. Acad. Sci. U. S. A. 93:7991–7995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu X, Stern AM, Liu Z, Kan B, Zhu J. 2010. Virulence regulator AphB enhances toxR transcription in Vibrio cholerae. BMC Microbiol. 10:3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ectopic expression of AphA and AphB in toxtazin B-treated cells. (A) CT levels in a strain expressing AphA ectopically, grown overnight under toxin-inducing conditions in the presence or absence of 10 µM compound and in the presence or absence of 1 mM IPTG. (B) CT levels in a strain expressing AphB ectopically, grown as described for panel A. In both panels, error bars represent the standard deviations for three biological replicates. The dotted line denotes the limit of detection. Download