

Abstract
MgADP inhibition, which is considered as a part of the regulatory system of ATP synthase, is a well-known process common to all F1-ATPases, a soluble component of ATP synthase. The entrapment of inhibitory MgADP at catalytic sites terminates catalysis. Regulation by the ε subunit is a common mechanism among F1-ATPases from bacteria and plants. The relationship between these two forms of regulatory mechanisms is obscure because it is difficult to distinguish which is active at a particular moment. Here, using F1-ATPase from Bacillus subtilis (BF1), which is strongly affected by MgADP inhibition, we can distinguish MgADP inhibition from regulation by the ε subunit. The ε subunit did not inhibit but activated BF1. We conclude that the ε subunit relieves BF1 from MgADP inhibition.
Introduction
FoF1-ATPase/synthase (FoF1) catalyzes ATP synthesis from ADP and inorganic phosphate coupled with the flow of H+ driven by the electrochemical gradient of H+ across cellular membranes. FoF1 consists of a water-soluble ATP-driven F1 motor (F1-ATPase) connected to a membrane-embedded H+-driven Fo motor to couple ATP synthesis/hydrolysis and H+ flow via a unique rotary mechanism [1–4]. F1-ATPase comprises α3, β3, γ, δ and ε subunits and its hydrolysis of one ATP molecule drives a discrete 120° rotation of the γε subunits relative to the others [5,6]. In FoF1, rotation of the rotor subunits of F1 (γ and ε) is transferred to the c subunit-ring of Fo to couple ATP synthesis/hydrolysis and flow of H+.
The smallest subunit, ε, is an endogenous inhibitor of the ATPase activity of bacterial and chloroplast F1-ATPases and is believed to contribute to the regulation ATP synthase [7–10]. The mechanism of inhibition by the ε subunit (ε inhibition) varies among species. For example, when F1-ATPase is separated from Fo, the ε subunit works as a dissociative inhibitor in Escherichia coli (EF1) and plant chloroplasts (CF1). The ε subunit inhibits ATPase activity, and the enzyme is reactivated when it dissociates from F1-ATPase, and the addition of excess ε subunits restores inhibition. In contrast, the ε subunit of F1-ATPase from thermophilic Bacillus PS3 (TF1) does not dissociate from the TF1 complex, and the addition of excess ε subunits does not significantly inhibit activity [10]. Rather, the ε subunit controls the activation state of the enzyme by changing its conformation. Because the dissociation of the ε subunit may not occur within the ATP synthase holo-complex, the ε subunit of EF1 or CF1 may also work as a regulator in intact ATP synthase. When in the extended conformation, the C-terminal domain of the ε subunit elongates into the cavity of the α3β3 ring and inhibits ATPase activity [11–16]. Upon activation, the C-terminal α helices of the ε subunit are expelled from the α3β3 ring and the ε subunit takes a folded-state conformation in which the C-terminal α helices are folded into a helix-turn-helix conformation, and ATPase activity is not inhibited [17]. We recently demonstrated that, in the case of TFoF1, the coupling between ATPase activity and flow of H+ is altered when the ε subunit does not bind ATP [18].
F1-ATPase is most commonly regulated by MgADP inhibition [19–21], which affects all known ATP synthases, and it is caused by the entrapment of MgADP at the catalytic site(s). The recovery from MgADP inhibition is accelerated when ATP binds to non-catalytic sites [22–26]. MgADP inhibition can be observed as pauses of the rotation of the γ subunit [27]. The pause angle of the γ subunit during MgADP inhibition is the same as that of the catalytic dwell (80° from the ATP-binding dwell), which is also the same as that during ε inhibition [28–30]. From this and other results, some investigators have proposed that ε inhibition is caused by the stabilization of MgADP inhibition [28,29,31]. Conversely, ε inhibition is prominent even in the presence of the detergent, lauryl dimethyl amine oxide (LDAO) (see supplemental figures of ref[14].), which is known to reduce MgADP inhibition [32]. Further, MgADP inhibition occurs even in the absence of the ε subunit. We demonstrated that the ε subunit greatly reduces the affinity of catalytic sites for MgATP and MgADP [10,33], which counteracts MgADP inhibition rather than stabilizing it. We have shown that the ε subunit relieved MgADP inhibition of a mutant TF1 unable to bind nucleotides to non-catalytic sites although at low levels [30]. Sekiya et al. reported that the ε subunit does not significantly influence MgADP inhibition of E. coli F1-ATPase [34]. Konno et al. proposed the existence of different origins of MgADP inhibition and ε inhibition in cyanobacterial F1-ATPase [35]. This discrepancy may be explained by concurrent and indistinguishable MgADP inhibition and ε inhibition.
Although the FoF1-ATP synthase from Bacillus subtilis has been studied for decades [36–38],, to the best of our knowledge, no detailed kinetic analysis of the purified enzyme has been reported, particularly regarding ε inhibition or MgADP inhibition. To address this question, in the present study, we purified F1-ATPase from B. subtilis (BF1) and carried out detailed analyses of the relationship between MgADP inhibition and the function of the ε subunit. Because the activity of BF1 is strongly affected by MgADP inhibition, we were able to examine the effect of the ε subunit on MgADP inhibition in detail. The results clearly indicate that regulation by the ε subunit is not only distinct from MgADP inhibition but their effects counteract each other.
Materials and Methods
Construction of a Plasmid to Express the α3β3γ Complex of BF1
KOD-Plus DNA DNA polymerase (Toyobo) was used for PCR reactions. The region containing the genes encoding the α, γ, and β subunits of BF1 was amplified by genomic PCR by using two primers as follows: 5′-CCGAATTCATATGAGCATCAAAGCTGAAGAGATTAGCACGC-3′ contains EcoRI and NdeI sites. The initiation codon of the α subunit (GTG) was replaced with ATG; 5′-GGCTCGAGCTGCAGTTAAACTTCTACACCCATTTCTTTTGCTTTC-3′ contains PstI and XhoI sites and the termination codon for the β subunit. B. subtilis genomic DNA was used as template. The PCR product was cloned into the EcoRV site of the pZero2.1 vector (Invitrogen) to produce pZero-BF1. The initiation codon of the γ subunit was converted from TTG to ATG and the SD sequence of the γ subunit was converted from AAGG to AAGGAGG, as reported for the expression system of TF1 [39] using overlap-extension PCR [40,41] with the four primers as follows: The mutagenic primers, 5′-AGAGAAAAGGAGGTGAAATCCATGGCCTCATTACG-3′ and 5′-AATGAGGCCATGGATTTCACCTCCTTTTCTCTTC-3′ contain an NcoI site in addition to the modifications described above; flanking primers were 5′-GCTGTCCTTGCTTCTTCGCCGTCCGC-3′ and 5′-TCTTGTGTGATGGCTGCTTGGCGAG-3′. The resulting 1.6-kbp fragment, containing segments of the genes encoding the γ and α subunits, was cloned into the EcoRV site of pZero2.1. A 1-kbp BglII fragment containing the initiation codon for the γ subunit was transferred to the cognate site of pZero-BF1 in the correct orientation to generate pZero-BF1ATG. The full-length genes encoding the α, γ, and β subunits were excised from pZero-BF1ATG with NdeI and XhoI and cloned into the respective sites of the pET16b expression vector (Novagen), generating pET16b-BF1 in which a His10-tag was introduced at the N-terminus of the α subunit. However, we were unable to express or purify the α3β3γ complex of BF1 from this construct as most of the α subunits were expressed as monomers. To introduce the His6-tag at the N-terminus of the β subunit, overlap-extension PCR was carried out using the four primers as follows: The mutagenic primers containing the His6-tag were 5′-CGATGCATCATCATCATCATCACATGAAGAAAGGACGCGTTAGCCAGG-3′ and 5′-CTTCATGTGATGATGATGATGATGCATCGCTATCCCTCCTGACAAAATC-3′. The flanking primers were 5′-CAGTTCGGTTTTACGGAGTGCTTATC-3′ and 5′-GCGCCGGGTCAGTGTAGTCATCG-3′. The resulting 1.6-kbp fragment, which contained the region around the initiation codon for the β subunit, was cloned into the EcoRV site of pZero2.1 to generate pZero2.1-βhis. To remove the His10-tag at the N-terminus of the α subunit, the NdeI/XhoI-digested fragment of pET16b-BF1, which contains the genes for the α, γ, and β subunits, was transferred to the respective sites of pET21a (Novagen) to produce pET21-BF1nohis. Then, to introduce the His6-tag into the β subunit in pET21-BF1nohis, pZero2.1-βhis was digested with BseRI/BssSI. A 0.8-kbp fragment, which contained the N-terminus of the β subunit was isolated and ligated to a 7.2-kbp fragment of pET21-BF1nohis-digested with BseRI/DraIII, and a 1.3-kbp fragment, which contained most of the β subunit gene of pET21-BF1nohis, which was digested with BssSI/DraIII, to obtain pET21-BF1. The final product, pET21-BF1, contained the following modifications of the original genes as follows: the His6-tag was introduced at the N-terminus of the β subunit, the initiation codon of the γ subunit was replaced with ATG, the SD sequence of the γ subunit was modified, an NdeI site was introduced at the 5′-terminus of the α subunit gene, and an NcoI site was introduced at the 5′-terminus of the γ subunit gene.
Construction of plasmids to express mutant proteins
The plasmid expressing the mutant (γS3C) α3β3γ complex of BF1 was constructed as follows. The sequence of the genes encoding the entire γ subunit and part of the β subunit were amplified by PCR from pET21-BF1 with the primers as follows. The mutagenic primer was 5′-AAATCCATGGCCTGTTTACGCGATATTAAG-3′, which contains a γSer3 to Cys substitution, and the other primer was 5′-ATGTAAGGAGCAAGCAAATCAACAAC-3′. The resulting 1.3-kbp fragment was introduced into the EcoRV site of pZero2.1 to produce pZero-γS3C. Then, pZero-γS3C was digested with NcoI/SalI, and a 1-kbp fragment containing the γS3C region was recovered and ligated to a 7.4-kbp fragment of pET21-BF1 digested with MunI/SalI, and a 0.9-kbp fragment of MunI/NcoI-digested pET21-BF1, which contained a segment of the gene encoding the α subunit, was ligated to obtain pET21-BF1 (γS3C). The plasmid expressing a mutant BF1 ε subunit (133C, where a Cys was introduced at the C-terminus) was constructed using the following primers for PCR as follows: 5′-GCCGGCGAAGCTTAACATTTCCCTGCTAC-3′, which contains 133C at the C-terminus of the BF1 ε subunit and a HindIII site, and 5′-GAAATTAATACGACTCACTATAGG-3′, which corresponds to the upstream sequence of the gene encoding the BF1 ε subunit (T7 promoter). The expression plasmid for WT BF1 ε [42] was used as the template. The resulting DNA fragment was cloned into the EcoRV site of pZero2.1; the resulting plasmid was digested with NdeI/HindIII, and the DNA fragment was transferred to the respective cognate sites of the pET21b expression vector to produce pET21-BF1ε(133C).
Protein purification
WT or mutant (γS3C) α3β3γ complexes of BF1 were prepared as follows: E. coli BL21(DE3) was transformed with pET21-BF1 and grown in 1-L LB medium containing 100 mg/L ampicillin and 10 µM IPTG at 25° C for 24–36 h with vigorous shaking at 250 rpm in a 3-L baffled flask. Typically, approximately 6 g wet cells was produced. Cells were suspended in buffer A (20 mM Tris-H2SO4 (pH 7.5), 300 mM K2SO4, and 30 mM imidazole) to 0.1–0.2 g cells/ml and disrupted using a French Press. The rest of the procedures was carried out at 25° C. Cell debris was removed by centrifugation at 2,000 × g for 15 min at 25° C. The supernatant was diluted with the same volume of buffer A and applied to a 5 ml HisTrapFF crude column (GE Healthcare Life Sciences) equilibrated with buffer A at a flow rate at 2 ml/min. The column was washed with buffer A until the absorbance at 280 nm plateaued. The adsorbed proteins were eluted with buffer B (buffer A containing 500 mM imidazole) and collected. Fractions were purified using a gel-filtration column (Superdex 200 10/300 GL; GE Healthcare Life Sciences) equilibrated with buffer C (50 mM Tris-H2SO4 (pH 7.5) and 50 mM K2SO4), eluted at 0.5 ml/min, monitored at 280 nm. The peak fractions containing α3β3γ complex were pooled, adjusted to 65% saturated ammonium sulfate, and stored in suspension at 4° C. Approximately 15 mg of α3β3γ complex was obtained from a 1-L culture. Purified α3β3γ complex did not contain bound nucleotides (<0.06 mol/mol) as measured by HPLC [43]. The α3β3γ complex was collected by centrifugation and dissolved in 50 mM Tris-H2SO4 (pH 7.5) and 50 mM K2SO4.
The WT ε subunit of BF1 was purified as described previously [42], and the mutant ε133C subunit was purified as follows. Approximately 3 g of BL21(DE3)/pET21-BF1-ε(133C) cultivated as described previously [42], was suspended to ~0.2 g of wet cells/ml in buffer D (50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1 mM DTT, and protease inhibitor cocktail (Roche Diagnostics)) and then disrupted twice using a French Press. The cell lysate was centrifuged at 3,000 × g for 10 min at 4° C to remove cell debris, and the supernatant was centrifuged at 180,000 × g for 1 h at 4° C. The rest of the procedures was carried out at 25° C. The supernatant was applied to a DEAE Toyopearl column (40 ml, Tosoh) equilibrated with buffer D. The flow-through fractions containing the ε133C subunit were collected and solid ammonium sulfate was added to 65% saturation. The precipitate was stored at 4° C. The protein was collected by centrifugation at 6,000 × g for 15 min at 4° C and dissolved in 30 mL of buffer D containing 10% saturated ammonium sulfate and applied to a butyl Toyopearl column (20 mL; Tosoh) equilibrated and washed with the same buffer. The ε133C subunit was eluted with buffer D at a flow rate at ~3 ml/min and fractions containing the ε133C subunit were pooled, and solid ammonium sulfate was added to 65% saturation and stored at 4° C. Approximately 40 mg of ε133C was obtained from a 1-L culture. The ε133C subunit was collected for analysis by centrifugation and dissolved in 50 mM Tris-H2SO4 (pH 7.5) and 50 mM K2SO4.
ATPase assay
ATPase activity was measured spectrophotometrically with an ATP-regenerating system coupled to NADH oxidation at 25 °C [44]. The assay mixture (1.5 ml) consisted of 50 mM Tris-H2SO4 (pH 7.5), 50 mM K2SO4, 2 mM phosphoenolpyruvate, 2 mM MgSO4, 0.2 mM NADH, 50 µg/ml pyruvate kinase, 50 µg/ml lactate dehydrogenase, and the indicated concentration of ATP-Mg (equimolar mixture of ATP and MgSO4) was transferred to a glass cuvette. Absorbance at 340 nm was measured using a V-550 spectrophotometer (JASCO) at 0.5 or 1-s intervals. The α3β3γ complex with or without ε subunit was added 2 min after starting the measurements. The mixture was stirred with a magnetic stirrer for 5 s before and after the addition of α3β3γ complex. The rate of ATP hydrolysis was determined from the rate of NADH oxidation. The final concentration of α3β3γ complex was 30 nM when measuring ATPase activity in the absence of lauryldimethylamine oxide (LDAO). Typically, 15 µl of 3 µM α3β3γ complex solution was added to 1.5 ml of the assay mixture. When ATPase activity was measured in the presence of LDAO, the final concentration of α3β3γ complex was reduced to 3 nM. In that case, 0.1 mg/ml bovine serum albumin (BSA) was included in stock α3β3γ complex solution (450 nM) to avoid the adsorption of α3β3γ complex on the plastic tube. Ten microliters of α3β3γ complex solution (450 nM) was added to 1.5 ml of the assay mixture without LDAO. Then, LDAO (final concentration 0.1%) was added and the solution was stirred continuously. When the ATPase activity of α3β3γε complexes was measured, the ε subunit was included in the α3β3γ complex stock solution at a 1:10 (3 µM α3β3γ complex and 30 µM ε in the absence of LDAO) to 1:100 (450 nM α3β3γ complex and 45 µM ε in the presence of LDAO) molar ratio. Reaction rates were determined at 2–7 s (initial) and 12–13 min (steady-state) after adding BF1. The reaction rate in the presence of LDAO was determined 100–150 s after the addition of LDAO.
Preincubation with MgADP
The effect of preincubation with MgADP was determined as follows: BF1 (10 µM α3β3γ complex ± 100 µM ε) in 50 mM Tris-H2SO4 (pH 7.5), 50 mM K2SO4, and 4 mM MgSO4 was mixed with an equal volume of 2× MgADP (equimolar mixture of ADP and MgSO4) and incubated for more than 10 min at 25° C (Mg2+ concentration was in 2 mM excess ADP). Nine microliters of the mixture was added to 1.5 ml of ATPase assay mixture containing 2 mM MgATP (30 nM α3β3γ complex ± 300 nM ε). The initial rate (2–4 s after the addition of BF1) was determined in this experiment.
Crosslinking γ and ε subunits
Crosslinking of the γ subunit to the extended conformation of the ε subunit in α3β3γS3Cε133C was performed as follows. Ammonium sulfate suspensions of α3β3γS3C complex and ε133C were centrifuged individually at 20,000 × g for 15 min at 4° C. Each precipitate was dissolved in 50 mM Tris-H2SO4 (pH 7.5) and 50 mM K2SO4, and 10 mM DTT was added and incubated for 10 min at 25° C. The α3β3γS3C and ε133C were mixed at a 1: 10 molar ratio and incubated for 15 min at 25° C. Excess ε133C was removed by ultrafiltration with a centrifugal concentrator (Amicon Ultra, 100-kDa cutoff). The sample was concentrated to approximately 10-fold and ultrafiltration was repeated 3 times after the addition of the same buffer to the original volume. The sample (1 mg/ml) was incubated with or without 4 mM MgATP for 10 min at 25° C; the solution was divided into two tubes, and an equal volume of 100 µM CuCl2 or the buffer was added. After 1-h incubation at 25° C, 10 mM EDTA was added to terminate the reaction. After 10 min, 0.1% SDS and 15 mM N-ethyl maleimide were added. The samples were analyzed using non-reducing SDS-PAGE (12% acrylamide). Part of the sample without ATP and with CuCl2 was saved after the addition of 10 mM EDTA for the ATPase assay. A combination of WT α3β3γ complex and ε133C served as the control. During the ATPase assays, 50 mM DTT was added to reduce crosslinking between the γ and ε subunits at the time indicated in the figure.
Other methods
Protein concentrations were determined by the method of Bradford [45] using BSA as a standard. DNA sequences for all of the recombinant proteins were confirmed using an ABI 3130xl Genetic Analyzer (Applied Biosystems). Non-reducing PAGE was performed according the method of Laemmli [46]. Chemicals were of the highest grade available. Kinetic data analyses were performed using Spectra Manager (JASCO) and OriginPro 8.5 and 9.0 (OriginLab), and the kinetic parameters are expressed with standard errors.
Results
ATPase activity of BF1 and the effect of ε subunit
Typical time courses of ATP hydrolysis by α3β3γ and α3β3γε complexes of BF1 are shown in Figure 1. At ATP concentrations ≥ 20 µM, very large initial inactivation was observed, irrespective of the presence of the ε subunit. At ATP concentrations > 50 µM, the inactivation was rapid enough to achieve constant, steady-state ATPase activity within the measurement (13 min), and there were no significant differences between α3β3γ and α3β3γε at ATP concentrations > 200 µM (Figure 1A, B). At lower ATP concentrations, the rate of inactivation slowed and did not reach the steady state (Figure 1C). Under these conditions, the ATPase activity of α3β3γε (lower traces in Figure 1) was higher than that of α3β3γ. Inactivation was diminished at lower ATP concentrations (Figure 1D). Reaction rates determined at 2-7 s and 12–13 min as a function of ATP concentration are shown in Figure 2. The steady-state ATPase activity of BF1 exhibited a decrease between 10 and 100 µM ATP possibly due in part to slow inactivation that did not reach the steady-state at low ATP concentrations. The value of k cat (1.83 s-1 for α3β3γ and 1.80 s-1 for α3β3γε) for steady-state ATPase activity is very low compared with the F1-ATPases from other sources, for example, TF1 ~60 s-1 and EF1 ~75 s-1 at 25° C) [39,47]. The ATPase activity increased more than 100-fold by LDAO, which is known to relieve MgADP inhibition (Figure 2). Because the initial rate of ATP hydrolysis reached only about 80 s-1 (Figure 2), and 200 mM Pi, which is known to reduce MgADP inhibition [48], activated BF1 to only ~10-fold (data not shown), the effect of LDAO may not be entirely related to MgADP inhibition. Nevertheless, these findings indicate that the ATPase activity of α3β3γ and α3β3γε complexes of BF1 was highly suppressed by MgADP inhibition. Judging from the activation ratio by LDAO, the degree of MgADP inhibition is low at low ATP concentrations. This could account for the triphasic dependence on ATP concentration dependence of ATPase activity in the absence of LDAO in part. In the presence of LDAO, the concentration-dependence on ATP of ATPase activity followed simple sum of two Michaelis–Menten equations (Figure 2).
Figure 1. Time-course of ATP hydrolysis by BF1 with or without the ε subunit.

In each panel, the upper and lower traces represent α3β3γ and α3β3γε, respectively. The final concentration of α3β3γ or α3β3γε complex of BF1 was 30 nM. The ATP concentrations are indicated in the figure. The α3β3γ or α3β3γε complex of BF1 was added at the time indicated by the arrowheads. The vertical and horizontal bars denote 0.2 absorbance units and 200 s, respectively.
Figure 2. Dependence of BF1 ATPase activity on ATP concentration.

The ATPase activities of initial (closed diamonds; α3β3γ, and open diamonds; α3β3γε), steady-state (closed squares; α3β3γ, and open squares; α3β3γε) and in the presence of LDAO (closed circles; α3β3γ, and open circles; ATPase activities of α3β3γε) at each ATP concentration was calculated from the velocities at 2-7 s, 12–13 min after the start of the reaction, and 100–150 s after the addition of LDAO, respectively. Error bars represent standard errors. The solid lines were fitted to a single (initial and steady-state) or sum of two (in the presence of LDAO) Michaelis–Menten equation(s). Only data from 200 µM and the above concentrations of ATP were used to fit the steady-state rates of α3β3γ and α3β3γε. Data from 1 µM (and 2µM, in the case of α3β3γ) were not used to fit the initial rate. The K M and the associated k cat values are 12.7 ± 0.9 µM, 56.2 ± 0.9 s-1 (α3β3γ, initial); 13.8 ± 0.9 µM, 72.3 ± 1.3 s-1 (α3β3γε, initial); 296 ± 25 µM, 1.92 ± 0.06 s-1 (α3β3γ, steady-state); 209 ± 18 µM, 1.87 ± 0.04 s-1 (α3β3γε, steady-state); 16.0 ± 1.9 µM, 68.8 ± 10.9 s-1 and 184 ± 32 µM, 199 ± 10 s-1 (α3β3γ, +LDAO); and 18.7 ± 3.4 µM, 80.1 ± 19.2 s-1 and 138 ± 18 µM, 272 ± 18 s-1 (α3β3γε, +LDAO).
The ε subunit affected the ATPase activity of BF1 only at low concentrations of ATP (Figures 1 and 2). Surprisingly, no inhibitory effect of the ε subunit was observed, and activation by the ε subunit occurred at ATP concentrations <50 µM (Figure 1C, D, lower trace). The dissociation of ε subunit from α3β3γε complex may not account for the equvialent activities of α3β3γ and α3β3γε at high ATP concentrations, because the α3β3γε complex could be isolated by gel-filtration HPLC (Superdex 200 10/300GL) even in the presence of ATP and/or LDAO (data not shown). Further, the addition of up to 30 µM ε subunit to the ATPase assay mixture did not significantly affect steady-state ATPase activity at 2 mM ATP (data not shown). In the presence of LDAO, the ATPase activities of α3β3γ and α3β3γε were essentially the same at all ATP concentrations, although the k cat value of α3β3γε (352 s-1) was slightly higher than that of α3β3γε (268 s-1) (Figure 2). The initial rates of ATP hydrolysis were also not significantly different (Figure 2). Therefore, the inhibition by the ε subunit of BF1 might be very weak, if any.
Preincubation with MgADP
When α3β3γ was preincubated with MgADP, the initial ATPase activity was significantly inhibited (Figure 3, closed circles), as reported previously for other F1-ATPases [26,48]. Incubation of 5 µM α3β3γ with 1:1 and 1:2 MgADP resulted in about 50% and 70% inhibition, respectively. In the presence of the ε subunit (open circles), the ATPase activity of α3β3γε was inhibited only marginally compared with that of α3β3γ; incubation with 1:1 and 1:2 MgADP resulted in about 20% and 35% inhibition, respectively. Reduced inhibition by the preincubating α3β3γε with MgADP might be due to the suppression of MgADP binding by the ε subunit in its extended conformation.
Figure 3. Effect of preincubation with MgADP.

The α3β3γ or α3β3γε (5 µM) was incubated with the indicated concentrations of MgADP for more than 10 min at 25° C. Residual ATPase activity was measured in the presence of 2 mM ATP. The initial rate (2–4 s after the start of the reaction) was measured, and the values relative to the control without incubation with MgADP (82.9 ± 5.4 s-1 and 88.6 ± 3.6 s-1 for α3β3γ and α3β3γε, respectively) are plotted. Closed and open circles represent α3β3γ and α3β3γε, respectively. Error bars represent standard errors.
Catalytic properties of mutant BF1 with its ε subunit fixed in the extended conformation
The activation by the ε subunit was investigated in more detail by examining a mutant α3β3γε complex of BF1, in which the N-terminus of the γ subunit and C-terminus of the ε subunit can be crosslinked via engineering in Cys residues to fix the ε subunit in the extended conformation. Thus, a mutant equivalent to that of TF1 [13] was prepared. The endogenous Cys residues in the α subunit did not react with the introduced Cys residues in the γ or ε subunits. To determine whether the apparent absence of ε inhibition in BF1 resulted from the inability of the ε subunit to assume an extended conformation, the presence of γ-ε crosslink formation was determined in the presence or absence of ATP. The γ and ε bands disappeared and a band corresponding to the γ-ε crosslink product appeared only in the absence of ATP (Figures 4, and S2). The distance between the Cα of the residues corresponding to the introduced Cys residues in a recently reported EF1 structure is 12.9 Å [16]. Although this is a little bit long to form a disulfide bridge, the formation of a disulfide bridge within the mutant α3β3γS3Cε133C complex of BF1 indicates that the crosslinked structure may reflect the physiological conformation within the range of thermal fluctuation. In the presence of ATP, a dimer of the ε subunit was formed, indicating that ε changed its conformation from the extended to the intermediate or folded-state in which the C-terminal Cys was accessible on the surface of the molecule. We conclude from these results that the absence of ε inhibition was not caused by the absence of its extended conformation. However, because there were no significant differences in the initial activities of WT α3β3γ and α3β3γε complexes, the extended conformation of the ε subunit may readily change upon addition of ATP.
Figure 4. Non-reducing SDS-PAGE analysis of mutant α3β3γS3Cε133C.

The α3β3 γ WT ε 133C (WT) or α3β3γS3Cε133C (S3C) were incubated for 1 h at 25°C with combinations of 2 mM ATP and 50 μM CuCl2 as indicated at the top of the figure. After the incubation, the samples were subjected to non-reducing SDS-PAGE (12% acrylamide). Bands derived from γ and ε subunits are marked by arrowheads.
We next determined the ATPase activity of the crosslinked mutant α3β3γS3Cε133C. In the absence of LDAO, the activity of the mutant was significantly higher than that of WT, even at 2 mM ATP (Figure 5A). When crosslinking was reduced by the addition of 50 mM DTT, the activity gradually decreased to the same level as WT. The addition of LDAO after this reduction resulted in activation to the WT level. When LDAO was added before reduction, activation of ATPase activity was undetectable (Figure 5B). The subsequent addition of DTT resulted in full activation.
Figure 5. Kinetics of ATP hydrolysis by mutant α3β3γS3Cε133C.
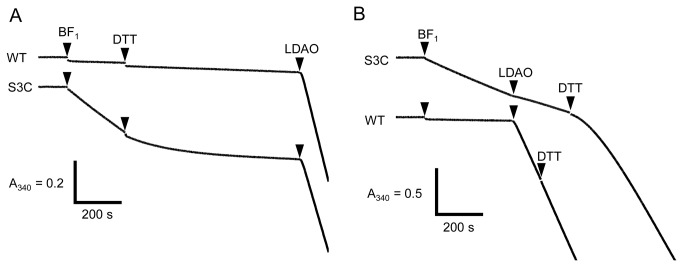
ATPase activities of α3β3γS3Cε133C (S3C), and α3β3γWTε133C (WT) at 2 mM ATP were determined. The α3β3γε complex of was added to 3 nM at the times indicated by the first arrowheads. (A) DTT (50 mM) and LDAO (0.1%) were added at the times indicated by the second and third arrowheads, respectively. (B) The order of addition of DTT and LDAO was reversed.
Discussion
ATPase activity of BF1 is strongly suppressed by MgADP inhibition
BF1 showed high initial ATPase activity, rapid inactivation, and very low steady-state ATPase activity. LDAO dramatically activated the steady-state ATPase activity of BF1 (Figure 2). The initial ATPase activity was >20-fold higher than the steady-state ATPase activity, 200 mM Pi also activated steady-state ATPase activity by ~10-fold (data not shown), and preincubation with MgADP greatly suppressed the initial ATPase activity (Figure 3). These suggest that the inactivation might be due to strong MgADP inhibition, which could mean that the B. subtilis ATP synthase functions as an ATP synthase that does not hydrolyze ATP, because MgADP inhibition does not inhibit ATP synthesis activity [49].
No inhibition by the ε subunit
The ε subunit did not significantly inhibit the ATPase activity of BF1 but activated at low concentrations of ATP presumably due to the suppression of MgADP inhibition. Further, the ε subunit, fixed in the extended conformation, did not inhibit the mutant enzyme. The ε subunit only inhibited the activity of the extended-state fixed mutant α3β3γS3Cε133C complex of BF1 in the presence of LDAO (Figure 5B, S3C after addition of LDAO). In this case, DTT activated the enzyme, indicating that the activity before the addition of DTT was actually suppressed by the extended-state ε subunit. We conclude, therefore, that due to the strong MgADP inhibition, ε inhibition is not evident, because the relief from MgADP inhibition by the ε subunit is more prominent.
Counteraction of MgADP inhibition and ε subunit
As discussed above, the ε subunit suppressed MgADP inhibition. In the absence of LDAO, the mutant α3β3γS3Cε133C complex of BF1 with an extended-state fixed ε subunit showed considerably higher ATPase activity than the WT, even at 2 mM ATP. LDAO did not activate the ATPase activity (Figure 5B). Thus, even before the addition of LDAO, the extended-state fixed α3β3γS3Cε133C complex of BF1 might be less inhibited by MgADP inhibition. This conclusion is further strengthened by the results when the WT BF1 was preincubated with MgADP (Figure 3). In the absence of the ε subunit, preincubation with MgADP suppressed the ATPase activity of α3β3γ proportionally at a α3β3γ: MgADP ratio of 1:2, suggesting that binding of MgADP is strong and binding of one or two MgADP is enough to induce MgADP inhibition of α3β3γ complex. In contrast, greater than 60% of the activity was retained in the presence of 1:2 MgADP and the ε subunit, indicating that binding of MgADP to α3β3γε was highly suppressed by the ε subunit. This agrees well with our previous observation that the ε subunit of TF1 significantly suppresses the binding of MgADP [33]. LDAO did not activate the extended-fixed α3β3γS3Cε133C complex of BF1 (Figure 5B), indicating that the extended-state ε subunit reduced MgADP inhibition. Considering all of these results, we conclude that ε inhibition is not due to the stabilization of MgADP inhibition [28,29,31], but due to an essentially different and counteracting mechanism. We believe, therefore, that these properties must be common among various F1-ATPases, despite the differences in the mechanisms of ε inhibition. It should be noted, however, the ε subunit did not protect mutant α3β3γS3Cε133C complex from MgADP inhibition by the preincubation with ADP (Figure S1). These apparent contradictory results may be due to the different catalytic site affinity for nucleotides between WT and the mutant (γS3C) α3β3γ complexes, and/or different mode of the action of MgADP during preincubation and ATPase turnover etc. Further experiments, for example, measurement of nucleotide binding to the catalytic sites with WT and mutant α3β3γ with and without ε subunit will give us a clue to resolve the differences between WT and the mutant in the MgADP preincubation experiment.
Significance of regulation by the ε subunit and MgADP inhibition in vivo
The results presented here indicate that the ATPase activity of BF1 is very low under normal conditions due to strong MgADP inhibition. Because B. subtilis lives in an aerobic environment and its ATP synthase is primarily used to synthesize ATP but not to hydrolyze ATP, as is the case for bacteria such as E. coli that can grow anaerobically. The ε subunit may not act as an inhibitor of the ATPase activity of B. subtilis ATP synthase. In contrast, its ability to attenuate MgADP inhibition may be its primary role in the regulatory system. Experiments using B. subtilis with mutant FoF1 to address these questions are underway in our laboratory. Elucidation of the balance and the interplay of these two regulatory systems in different bacteria may be required to understand the regulation of bacterial ATP synthases.
Supporting Information
Effect of preincubation with MgADP on α3β3γS3Cε133C.
The α3β3γS3C, DTT-treated α3β3γS3Cε133C and CuCl2-treated α3β3γS3Cε133C (5 µM) were subjected to the same experiment as shown in Figure 3. Closed circles, open circles and open squares represent α3β3γS3C, DTT-treated α3β3γS3Cε133C and CuCl2-treated α3β3γS3Cε133C, respectively. Error bars represent standard errors.
(TIF)
Enlargement of the part of Figure 4.
The part of Figure 4 is enlarged to visualize γ-ε crosslinked band clearer. Only the region around α, β and γ-ε from α3β3γS3Cε133C (S3C) treated with or without CuCl2 in the absence of ATP is shown.
(TIF)
Acknowledgments
We thank Professor F. Kawamura at Rikkyo University for providing us with B. subtilis genomic DNA and members of Y. Kato-Yamada’s laboratory for their help and critical discussions.
Funding Statement
This work was supported in parts by Grants-in-Aid for Scientific Research for Young Scientists (B) (No. 23770157), the Strategic Research Foundation Grant-aided Project for Private Universities (No. S1201003) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and Rikkyo University Special Fund for Research fro Rikkyo University (to Y. K.-Y.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Boyer PD (1997) The ATP synthase: a splendid molecular machine. Annu Rev Biochem 66: 717–749. doi:10.1146/annurev.biochem.66.1.717. PubMed: 9242922. [DOI] [PubMed] [Google Scholar]
- 2. Kinosita K Jr, Yasuda R, Noji H (2000) F1-ATPase: a highly efficient rotary ATP machine. Essays Biochem 35: 3–18. PubMed: 12471886. [DOI] [PubMed] [Google Scholar]
- 3. Yoshida M, Muneyuki E, Hisabori T (2001) ATP synthase: a marvellous rotary engine of the cell. Nat Rev Mol Cell Biol 2: 669–677. doi:10.1038/35088558. PubMed: 11533724. [DOI] [PubMed] [Google Scholar]
- 4. Senior AE, Nadanaciva S, Weber J (2002) The molecular mechanism of ATP synthesis by F1Fo-ATP synthase. Biochim Biophys Acta 1553: 188–211. doi:10.1016/S0005-2728(02)00185-8. PubMed: 11997128. [DOI] [PubMed] [Google Scholar]
- 5. Noji H, Yasuda R, Yoshida M, Kinosita K Jr (1997) Direct observation of the rotation of F1-ATPase. Nature 386: 299–302. doi:10.1038/386299a0. PubMed: 9069291. [DOI] [PubMed] [Google Scholar]
- 6. Yasuda R, Noji H, Kinosita K Jr, Yoshida M (1998) F1-ATPase is a highly efficient molecular motor that rotates with discrete 120 degree steps. Cell 93: 1117–1124. doi:10.1016/S0092-8674(00)81456-7. PubMed: 9657145. [DOI] [PubMed] [Google Scholar]
- 7. Smith JB, Sternweis PC, Heppel LA (1975) Partial purification of active δ and ε subunits of the membrane ATPase from Escherichia coli. J Supramol Struct 3: 248–255. doi:10.1002/jss.400030307. PubMed: 127087. [DOI] [PubMed] [Google Scholar]
- 8. Laget PP, Smith JB (1979) Inhibitory properties of endogenous subunit ε in the Escherichia coli F1 ATPase. Arch Biochem Biophys 197: 83–89. doi:10.1016/0003-9861(79)90222-4. PubMed: 161698. [DOI] [PubMed] [Google Scholar]
- 9. Ort DR, Oxborough K (1992) In situ regulation of chloroplast coupling factor activity. Annu Rev Plant Physiol Plant Mol Biol 43: 269–291. doi:10.1146/annurev.pp.43.060192.001413. [Google Scholar]
- 10. Kato Y, Matsui T, Tanaka N, Muneyuki E, Hisabori T et al. (1997) Thermophilic F1-ATPase is activated without dissociation of an endogenous inhibitor, ε subunit. J Biol Chem 272: 24906–24912. doi:10.1074/jbc.272.40.24906. PubMed: 9312092. [DOI] [PubMed] [Google Scholar]
- 11. Rodgers AJW, Wilce MCJ (2000) Structure of the γ-ε complex of ATP synthase. Nat Struct Biol 7: 1051–1054. doi:10.1038/80975. PubMed: 11062562. [DOI] [PubMed] [Google Scholar]
- 12. Tsunoda SP, Rodgers AJW, Aggeler R, Wilce MCJ, Yoshida M et al. (2001) Large conformational changes of the ε subunit in the bacterial F1Fo ATP synthase provide a ratchet action to regulate this rotary motor enzyme. Proc Natl Acad Sci U S A 98: 6560–6564. doi:10.1073/pnas.111128098. PubMed: 11381110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suzuki T, Murakami T, Iino R, Suzuki J, Ono S et al. (2003) FoF1-ATPase/synthase is geared to the synthesis mode by conformational rearrangement of ε subunit in response to proton motive force and ADP/ATP balance. J Biol Chem 278: 46840–46846. doi:10.1074/jbc.M307165200. PubMed: 12881515. [DOI] [PubMed] [Google Scholar]
- 14. Iino R, Murakami T, Iizuka S, Kato-Yamada Y, Suzuki T et al. (2005) Real time monitoring of conformational dynamics of the ε subunit in F1-ATPase. J Biol Chem 280: 40130–40134. doi:10.1074/jbc.M506160200. PubMed: 16203732. [DOI] [PubMed] [Google Scholar]
- 15. Feniouk BA, Kato-Yamada Y, Yoshida M, Suzuki T (2010) Conformational transitions of subunit ε in ATP synthase from thermophilic Bacillus PS3. Biophys J 98: 434–442. doi:10.1016/j.bpj.2009.12.2357. PubMed: 20141757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cingolani G, Duncan TM (2011) Structure of the ATP synthase catalytic complex (F1) from Escherichia coli in an autoinhibited conformation. Nat Struct Mol Biol 18: 701–707. doi:10.1038/nsmb.2058. PubMed: 21602818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kato S, Yoshida M, Kato-Yamada Y (2007) Role of the ε subunit of thermophilic F1-ATPase as a sensor for ATP. J Biol Chem 282: 37618–37623. doi:10.1074/jbc.M707509200. PubMed: 17933866. [DOI] [PubMed] [Google Scholar]
- 18. Kadoya F, Kato S, Watanabe K, Kato-Yamada Y (2011) ATP binding to the ε subunit of thermophilic ATP synthase is crucial for efficient coupling of ATPase and H+ pump activities. Biochem J 437: 135–140. doi:10.1042/BJ20110443. PubMed: 21510843. [DOI] [PubMed] [Google Scholar]
- 19. Vasilyeva EA, Minkov IB, Fitin AF, Vinogradov AD (1982) Kinetic mechanism of mitochondrial adenosine triphosphatase. ADP-specific inhibition as revealed by the steady-state kinetics. Biochem J 202: 9–14. PubMed: 6211173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vasilyeva EA, Minkov IB, Fitin AF, Vinogradov AD (1982) Kinetic mechanism of mitochondrial adenosine triphosphatase. Inhibition by azide and activation by sulphite. Biochem J 202: 15–23. PubMed: 6211171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou J-M, Xue ZX, Du ZY, Melese T, Boyer PD (1988) Relationship of tightly bound ADP and ATP to control and catalysis by chloroplast ATP synthase. Biochemistry 27: 5129–5135. doi:10.1021/bi00414a027. PubMed: 2901855. [DOI] [PubMed] [Google Scholar]
- 22. Milgrom YM, Ehler LL, Boyer PD (1991) The characteristics and effect on catalysis of nucleotide binding to noncatalytic sites of chloroplast F1-ATPase. J Biol Chem 266: 11551–11558. PubMed: 1828802. [PubMed] [Google Scholar]
- 23. Milgrom YM, Cross RL (1993) Nucleotide binding sites on beef heart mitochondrial F1-ATPase. Cooperative interactions between sites and specificity of noncatalytic sites. J Biol Chem 268: 23179–23185. PubMed: 8226836. [PubMed] [Google Scholar]
- 24. Hyndman DJ, Milgrom YM, Bramhall EA, Cross RL (1994) Nucleotide-binding sites on Escherichia coli F1-ATPase, Specificity of noncatalytic sites and inhibition at a catalytic sites by MgADP. J Biol Chem 269: 28871–28877. PubMed: 7961847. [PubMed] [Google Scholar]
- 25. Jault J-M, Matsui T, Jault FM, Kaibara C, Muneyuki E et al. (1995) The α3β3γ complex of the F1-ATPase from thermophilic Bacillus. PS 3 containing the αD261N substitution fails to dissociate inhibitory MgADP from a catalytic site when ATP binds to noncatalytic sites. Biochemistry 34: 16412–16418. [DOI] [PubMed] [Google Scholar]
- 26. Matsui T, Muneyuki E, Honda M, Allison WS, Dou C et al. (1997) Catalytic activity of the α3β3γ complex of F1-ATPase without noncatalytic nucleotide binding site. J Biol Chem 272: 8215–8221. doi:10.1074/jbc.272.13.8215. PubMed: 9079639. [DOI] [PubMed] [Google Scholar]
- 27. Hirono-Hara Y, Noji H, Nishiura M, Muneyuki E, Hara KY et al. (2001) Pause and rotation of F1-ATPase during catalysis. Proc Natl Acad Sci U S A 98: 13649–13654. doi:10.1073/pnas.241365698. PubMed: 11707579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Konno H, Murakami-Fuse T, Fujii F, Koyama F, Ueoka-Nakanishi H et al. (2006) The regulator of the F1 motor: inhibition of rotation of cyanobacterial F1-ATPase by the ε subunit. EMBO J 25: 4596–4604. doi:10.1038/sj.emboj.7601348. PubMed: 16977308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsumuraya M, Furuike S, Adachi K, Kinosita K Jr, Yoshida M (2009) Effect of ε subunit on the rotation of thermophilic Bacillus F1-ATPase. FEBS Lett 583: 1121–1126. doi:10.1016/j.febslet.2009.02.038. PubMed: 19265694. [DOI] [PubMed] [Google Scholar]
- 30. Haruyama T, Hirono-Hara Y, Kato-Yamada Y (2010) Inhibition of thermophilic F1-ATPase by the ε subunit takes different path from the ADP-Mg inhibition. Biophysics 6: 59–65. doi:10.2142/biophysics.6.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feniouk BA, Suzuki T, Yoshida M (2007) Regulatory interplay between proton motive force, ADP, phosphate, and subunit ε in bacterial ATP synthase. J Biol Chem 282: 764–772. PubMed: 17092944. [DOI] [PubMed] [Google Scholar]
- 32. Jault J-M, Dou C, Grodsky NB, Matsui T, Yoshida M et al. (1996) The α3β3γ subcomplex of the F1-ATPase from the thermophilic Bacillus. PS 3 with the βT165S substitution does not entrap inhibitory MgADP in a catalytic site during turnover. J. Biol. Chem. 271: 28818-28824. [DOI] [PubMed] [Google Scholar]
- 33. Yasuno T, Muneyuki E, Yoshida M, Kato-Yamada Y (2009) Modulation of nucleotide binding to the catalytic sites of thermophilic F1-ATPase by the ε subunit: implication for the role of the ε subunit in ATP synthesis. Biochem Biophys Res Commun 390: 230–234. doi:10.1016/j.bbrc.2009.09.092. PubMed: 19785990. [DOI] [PubMed] [Google Scholar]
- 34. Sekiya M, Hosokawa H, Nakanishi-Matsui M, Al-Shawi MK, Nakamoto RK et al. (2010) Single molecule behavior of inhibited and active states of Escherichia coli ATP synthase F1 rotation. J Biol Chem 285: 42058-42067. doi:10.1074/jbc.M110.176701. PubMed: 20974856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Konno H, Isu A, Kim Y, Murakami-Fuse T, Sugano Y et al. (2011) Characterization of the relationship between ADP- and ε-induced inhibition in cyanobacterial F1-ATPase. J Biol Chem 286: 13423-13429. doi:10.1074/jbc.M110.155986. PubMed: 21345803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hicks DB, Krulwich TA (1987) Purification and characterization of the F1 ATPase from Bacillus subtilis and its uncoupler-resistant mutant derivatives. J Bacteriol 169: 4743–4749. PubMed: 2888751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hicks DB, Cohen DM, Krulwich TA (1994) Reconstitution of energy-linked activities of the solubilized F1Fo ATP synthase from Bacillus subtilis. J Bacteriol 176: 4192–4195. PubMed: 8021203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Santana M, Ionescu MS, Vertes A, Longin R, Kunst F et al. (1994) Bacillus subtilis FoF1 ATPase: DNA sequence of the atp operon and characterization of atp mutants. J Bacteriol 176: 6802–6811. PubMed: 7961438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Matsui T, Yoshida M (1995) Expression of the wild-type and the Cys-/Trp-less α3β3γ complex of thermophilic F1-ATPase in Escherichia coli. Biochim Biophys Acta 1231: 139–146. doi:10.1016/0005-2728(95)00070-Y. PubMed: 7662694. [DOI] [PubMed] [Google Scholar]
- 40. Higuchi R, Krummel B, Saiki RK (1988) A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res 16: 7351–7367. doi:10.1093/nar/16.15.7351. PubMed: 3045756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene (Amst.)77: 51–59. [DOI] [PubMed]
- 42. Kato-Yamada Y, Yoshida M (2003) Isolated ε subunit of thermophilic F1-ATPase binds ATP. J Biol Chem 278: 36013–36016. doi:10.1074/jbc.M306140200. PubMed: 12837747. [DOI] [PubMed] [Google Scholar]
- 43. Hisabori T, Muneyuki E, Odaka M, Yokoyama K, Mochizuki K et al. (1992) Single site hydrolysis of 2′, 3′-O-(2,4,6-trinitrophenyl)-ATP by the F1-ATPase from thermophilic bacterium. PS 3 is accelerated by the chase-addition of excess ATP. J. Biol. Chem. 267: 4551–4556 [PubMed]
- 44. Stiggall DL, Galante YM, Hatefi Y (1979) Preparation and properties of complex V. Methods Enzymol 55: 308–315. doi:10.1016/0076-6879(79)55036-8. PubMed: 156835. [DOI] [PubMed] [Google Scholar]
- 45. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254. doi:10.1016/0003-2697(76)90527-3. PubMed: 942051. [DOI] [PubMed] [Google Scholar]
- 46. Laemmli UK. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685. doi:10.1038/227680a0. PubMed: 5432063. [DOI] [PubMed] [Google Scholar]
- 47. Kato Y, Sasayama T, Muneyuki E, Yoshida M (1995) Analysis of time-dependent change of Escherichia coli F1-ATPase activity and its relationship with apparent negative Cooperativity. Biochim Biophys Acta 1231: 275–281. doi:10.1016/0005-2728(95)00087-Y. PubMed: 7578215. [DOI] [PubMed] [Google Scholar]
- 48. Bald D, Muneyuki E, Amano T, Kruip J, Hisabori T et al. (1999) The noncatalytic site-deficient α3β3γ subcomplex and FoF1-ATP synthase can continuously catalyse ATP hydrolysis when Pi is present. Eur J Biochem 262: 563–568. doi:10.1046/j.1432-1327.1999.00410.x. PubMed: 10336643. [DOI] [PubMed] [Google Scholar]
- 49. Bald D, Amano T, Muneyuki E, Pitard B, Rigaud JL et al. (1998) ATP synthesis by FoF1-ATP synthase independent of noncatalytic nucleotide binding sites and insensitive to azide inhibition. J Biol Chem 273: 865–870. doi:10.1074/jbc.273.2.865. PubMed: 9422743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of preincubation with MgADP on α3β3γS3Cε133C.
The α3β3γS3C, DTT-treated α3β3γS3Cε133C and CuCl2-treated α3β3γS3Cε133C (5 µM) were subjected to the same experiment as shown in Figure 3. Closed circles, open circles and open squares represent α3β3γS3C, DTT-treated α3β3γS3Cε133C and CuCl2-treated α3β3γS3Cε133C, respectively. Error bars represent standard errors.
(TIF)
Enlargement of the part of Figure 4.
The part of Figure 4 is enlarged to visualize γ-ε crosslinked band clearer. Only the region around α, β and γ-ε from α3β3γS3Cε133C (S3C) treated with or without CuCl2 in the absence of ATP is shown.
(TIF)