

Abstract
Genomic approaches to characterizing bacterial communities are revealing significant differences in diversity and composition between environments. But bacterial distributions have not been mapped at a global scale. Although current community surveys are way too sparse to map global diversity patterns directly, there is now sufficient data to fit accurate models of how bacterial distributions vary across different environments and to make global scale maps from these models. We apply this approach to map the global distributions of bacteria in marine surface waters. Our spatially and temporally explicit predictions suggest that bacterial diversity peaks in temperate latitudes across the world's oceans. These global peaks are seasonal, occurring 6 months apart in the two hemispheres, in the boreal and austral winters. This pattern is quite different from the tropical, seasonally consistent diversity patterns observed for most macroorganisms. However, like other marine organisms, surface water bacteria are particularly diverse in regions of high human environmental impacts on the oceans. Our maps provide the first picture of bacterial distributions at a global scale and suggest important differences between the diversity patterns of bacteria compared with other organisms.
Keywords: bacteria, marine, species distribution model, niche model, diversity gradient, range map
Introduction
The rich tradition of mapping macroorganism diversity patterns and ranges (Lamarck and Candolle, 1805) has been crucial for understanding the evolutionary and ecological processes that shape contemporary biodiversity. If similar distribution maps could be constructed for bacteria, they would clarify the mechanisms structuring bacterial communities and the processes shaping global diversity. This knowledge would improve our understanding of the biogeochemical cycles and ecosystem services for which bacteria are critical components (Martiny et al., 2006). Bacterial diversity maps may also be useful for informing ecosystem-level conservation and management decisions (Richardson and Whittaker, 2010).
Unfortunately, there have been impediments to mapping bacterial distributions, including most notably the geographic sparsity of microbial community surveys. The situation is improving due to genomic approaches to characterizing microbial assemblages via high-throughput sequencing of phylogenetically informative marker genes such as the small-subunit ribosomal RNA gene (rDNA; International Census of Marine Microbes, 2011) and entire metagenomes (Rusch et al., 2007; Fierer et al., 2012). However, for the foreseeable future, data are likely to be too sparse to directly map bacterial distributions from observations.
We propose that statistical modeling, if carefully applied, can be used to predict bacterial diversity and the ranges of bacterial taxonomic groups based on the environmental conditions at the sampling sites, where we do have data about microbial communities. The idea is to learn associations between environmental variables and microbial distributions using observed data and then apply the learned model to estimate what bacteria might be present at the many locations where we have environmental data but no microbial survey. An established method for this type of analysis is species distribution modeling (SDM). SDM has been a fundamental tool for predicting the diversity patterns of macroorganisms (Franklin and Miller, 2009). Although previously applied to bacterial communities at a regional scale (Larsen et al., 2012), SDM has not been used to infer global bacterial distributions.
We used SDM to map diversity of bacteria in marine surface waters on a global scale. Our models employ publicly available environmental data and a database of rDNA sequences, which we complied from a variety of marine sampling studies from around the world. Marine bacteria are well suited to SDM because of their strong environmental sorting (Tamames et al., 2010) and potentially low-dispersal limitation (Hubert et al., 2009; Caporaso et al., 2012). The marine surface water environment is also particularly amenable to SDM, because its physical properties are well characterized; high-resolution global rasters are freely available for a large number of environmental variables, specific to many times of year. This environmental data allowed us to make spatially and temporally explicit diversity predictions with low estimated error and to avoid extrapolating far beyond our observed data.
We generated and mapped global predictions of bacterial diversity in marine surface waters for each month of the year. These maps uncovered several novel patterns. First, marine bacterial diversity peaks globally in temperate latitudes in winter, extending previous studies that found local temperate peaks of diversity in winter (Ghiglione and Murray, 2012; Gilbert et al., 2012). These high latitude, seasonally dependent diversity peaks contrast with the tropical, seasonally consistent diversity peaks observed for most marine and terrestrial macroorganisms (Hillebrand, 2004). In addition, global hotspots of bacterial diversity occur in marine surface waters with high levels of human impact (Halpern et al., 2008). These findings contribute to the expanding foundation of microbial biogeography by generating predictions that can be tested through future hypothesis-driven research in specific ecosystems across the world's oceans.
Materials and methods
To create our diversity maps, we used SDM methodology. This approach generates maps by regressing observations of diversity on environmental conditions and then projecting the regression into geographic space (Franklin and Miller, 2009). Specifically, we employ an assemble-first SDM approach, wherein richness of operational taxonomic units (OTUs) is modeled directly as a function of environmental conditions and then projected (Ferrier and Guisan, 2006). We applied this approach to rDNA sequence data from the MICROBIS project (International census of marine microbes, 2011) and validated our predictions using several independent data sets (Supplementary Table S1).
Data
Constructing and implementing an SDM model requires local measurements of community composition and rasters of environmental data. For measurements of community composition, we assembled a database of rDNA data from 377 marine samples from 164 distinct locations with depth ⩽150 m (Supplementary Figure S1). We excluded samples from vents, anoxic water, sediment and fresh water. Data came from four sources (Supplementary Table S1); we used MICROBIS for our primary analysis and the other sources for model selection and validation. Three data sources contributed 16S sequences, and Fuhrman et al. (2008) contributed ARISA data (Supplementary Table S1). Although ARISA data are generated from the intergenic spacer region between the 16S and 23S ribosomal genes, we employ the term ‘rDNA' data for ease of communication. OTUs for the ARISA data were from reference Fuhrman et al. (2008). For all non-ARISA data, to define OTUs we implemented reference-based classification (Supplementary Methods) and also used de novo clustering of sequences into OTUs from the original publications.
For the rasters of environmental data, from 45 environmental variables mapped at a 0.5° latitude/longitude resolution across the world ocean, we selected 21 variables (starred in Supplementary Table S2) that correlated with diversity, were not highly correlated with each other (Supplementary Table S3), and had multivariate environmental similarity surface (Elith et al., 2010) scores >−20 for 99.5% of the world ocean (Supplementary Table S4). Incorporation of multivariate environmental similarity surface scores ensured that models could be projected into geographic space with minimal extrapolation (Elith et al., 2010). Many of the rasters were depth- and month-specific, although these were often less predictive than their averaged counterparts.
Model fitting
To construct SDMs, we fit models using the MICROBIS data, and performed extensive variable selection and validation analyses using all four data sets (see below and Supplementary Table S1). We constructed SDMs with linear or nonlinear models, rarefaction depths of 4266 or 150 rDNA sequences per sample (with more than 4266 sequences, many samples would have to be excluded), and sequences classified using de novo clustering or reference-based classification (Supplementary Methods). Regardless of the methodology, the resulting maps showed temperate diversity peaks in the winter (Figure 1, Supplementary Figures S2–5). Thus, we focus on a linear model at a rarefaction depth of 4266 sequences, with de novo sequence classification. To estimate ranges of individual taxa, we used SDMs with a logistic regression model (Franklin and Miller, 2009). Data used for model fitting are available in Supplementary File 3.
Figure 1.

Maps of predicted global marine bacterial diversity. Color scale shows relative richness of marine surface waters as predicted by SDM. Samples were rarefied to 4266 rDNA sequences to enable accurate estimation of relative richness patterns on a global scale from data sets with different sequencing depths. True richness is expected to exceed estimated values. (a) In December, OTU richness peaks in temperate and higher latitudes in the Northern Hemisphere. (b) In June, OTU richness peaks in temperate latitudes in the Southern Hemisphere. Predicted richness during the spring and fall is intermediate, with roughly globally uniform richness near the equinoxes (movie available in Supplementary File 2). Predicted richness patterns remain qualitatively the same regardless of the taxonomic classification method (Supplementary Figure S2), modeling method (Supplementary Figure S3), choice of environmental predictors (Supplementary Figure S4) and sequencing depth (Supplementary Figure S5). Error rates for the predictions are generally low, as indicated by 95% confidence intervals on the marginal plots (right panels, shaded gray) and maps of standard errors (Supplementary Figure S6). Grayed regions on the maps are areas where environmental raster data and, hence, predictions are unavailable. Richness estimates in most regions are interpolated rather than extrapolated (Supplementary Figure S7).
We performed 15 analyses, labeled Analyses I–XV, to check the robustness of the diversity maps and to model the distributions of different taxa and groups of taxa (Supplementary Tables S1 and S5). For Analyses I–XI, we log-transformed richness and Shannon diversity.
Robustness analyses
Analyses I–V checked the robustness of overall diversity patterns that we report. Analysis I used a linear model, with OTUs identified using de novo clustering, and a rarefaction depth of 4266 sequences. Analysis II checked whether the patterns are affected by the classification method. It was the same as Analysis I, but used OTUs identified by the Ribosomal Database Project (RDP) classifier, a reference-based procedure. We ran the RDP classifier with and without a 50% bootstrap threshold. Using the bootstrap threshold introduces significant bias to the data set, because sequences with high similarity to known bacterial genera are not evenly distributed across latitudes. Without a bootstrap threshold, the relative diversity patterns of RDP classified genera are very similar to those from de novo OTUs (Supplementary Figure S2). Anaylsis III checked for effects of rarefaction depth. It was the same as Analysis I, but used a rarefaction depth of 150 sequences rather than 4266 sequences. Analysis IV checked whether using a linear model affected our results. It implemented a nonlinear, multiple adaptive regression splines model (MARS) in lieu of the linear model, but was otherwise like Analysis I. Analysis V checked whether our patterns were dependent on the diversity metric used. It was the same as Analysis I, but used Shannon diversity instead of OTU richness. The results of all five analyses were qualitatively alike (Figure 1, Supplementary Figures S2–5), so in the main text we focus on the results from Analysis I.
Additional diversity maps
Analyses VI–XI mapped the distribution of richness of OTUs within certain phyla. Analyses XII–XV mapped the distributions of select genera of marine bacteria.
Model selection
For all analyses, we fit models using just the MICROBIS data, and used all four data sets for model selection and validation. Specifically, for Analysis I, using the MICROBIS data we evaluated all linear models with subsets of zero to eight predictors (that is, environmental variables) to determine which environmental variables to include. Among the models with each number of predictors that had variance inflation factors less than 5, we chose the one having the best predictive power as measured by leave-one-out cross-validation (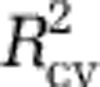 ; equivalent to PRESS) for further consideration (Supplementary Table S6). We also examined models with the lowest Akaike information criterion and Bayesian information criterion scores. In general, the latter models coincided with the models with the best
; equivalent to PRESS) for further consideration (Supplementary Table S6). We also examined models with the lowest Akaike information criterion and Bayesian information criterion scores. In general, the latter models coincided with the models with the best 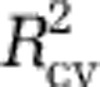 . To choose among the best models with zero to eight predictors, one approach would have been to choose the model with the overall best
. To choose among the best models with zero to eight predictors, one approach would have been to choose the model with the overall best 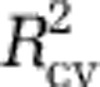 , Akaike information criterion or Bayesian information criterion (Supplementary Figures S8A–C). However, these criteria suggested models that were overfit: the resulting maps had obvious artifacts, and the models had poor predictive power with the independent data sets (that is, the Pommier et al., 2007; Fuhrman et al., 2008; and GOS data; Supplementary Table S1). Thus, we evaluated the predictive power of several models with high values of
, Akaike information criterion or Bayesian information criterion (Supplementary Figures S8A–C). However, these criteria suggested models that were overfit: the resulting maps had obvious artifacts, and the models had poor predictive power with the independent data sets (that is, the Pommier et al., 2007; Fuhrman et al., 2008; and GOS data; Supplementary Table S1). Thus, we evaluated the predictive power of several models with high values of 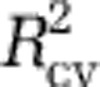 on the independent data sets: we fit the models with the MICROBIS data and calculated the proportion of variability in the diversity of the independent data sets that was predicted by each, hereafter referred to as
on the independent data sets: we fit the models with the MICROBIS data and calculated the proportion of variability in the diversity of the independent data sets that was predicted by each, hereafter referred to as 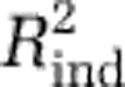 . For all independent data sets, the model with three predictors had the best
. For all independent data sets, the model with three predictors had the best 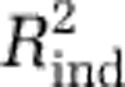 (Supplementary Figure S8D—F), and maps created using it lacked artifacts. Moreover, although the three-predictor model had slightly lower
(Supplementary Figure S8D—F), and maps created using it lacked artifacts. Moreover, although the three-predictor model had slightly lower 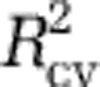 than possible, the difference was negligible (Supplementary Figure S8A). Based on these considerations, we proceeded with the three-predictor model indicated by the independent data sets. We also experimented with other model selection schemes (for example, backward model selection with Akaike information criterion) and other sets of predictors, including time-lagged variables. Regardless of the specific model selection algorithm used, so long as overfitting was controlled, our main result of high temperate diversity in the winter was clearly evident from resulting SDMs (for example, Supplementary Figure S4).
than possible, the difference was negligible (Supplementary Figure S8A). Based on these considerations, we proceeded with the three-predictor model indicated by the independent data sets. We also experimented with other model selection schemes (for example, backward model selection with Akaike information criterion) and other sets of predictors, including time-lagged variables. Regardless of the specific model selection algorithm used, so long as overfitting was controlled, our main result of high temperate diversity in the winter was clearly evident from resulting SDMs (for example, Supplementary Figure S4).
For the other analyses of diversity with linear models (Analyses II and IV–XI), we followed a procedure analogous to that used in Analysis I, selecting models by a combination of cross validation and independent data. In some analyses (for example, diversity maps within phyla (Analyses VI–XI)), not all independent data sets could be used because they lacked relevant diversity data. In these cases, we used just the independent data sets that were applicable. If no independent data sets were applicable or predictive (all 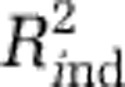 <0.1), we chose the model with the number of predictors such that adding another predictor would increase
<0.1), we chose the model with the number of predictors such that adding another predictor would increase 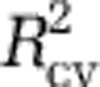 less than 2.5%. Applying this criterion to cases where independent data were available, indicated models close to those that were indicated by the independent data.
less than 2.5%. Applying this criterion to cases where independent data were available, indicated models close to those that were indicated by the independent data.
To check whether the linearity of the model affected results, we used a MARS model (Analysis III; Friedman, 1991; Elith et al., 2006). To fit the MARS model, we offered the environmental variables previously found to be predictive, and used a maximum interaction degree of 1 with a forward stepping threshold of 0.001. Both the linear and non-linear models yielded qualitatively similar diversity maps and had similar predictive power (Supplementary Figure S9 vs S10), as measured by 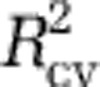 . In addition, regression diagnostics and correlation plots indicated that the linear model was justified, so we focus on the results from the linear model in the text.
. In addition, regression diagnostics and correlation plots indicated that the linear model was justified, so we focus on the results from the linear model in the text.
To model the relative abundance of individual genera (Analyses XII–XV), we fit logistic regression models (Franklin and Miller, 2009). For these analyses, we used the MICROBIS sequences classified by RDP (Supplementary Table S1). To perform model selection and speed calculations, we used logit-transformed data and a linear model, with the 2.5% 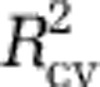 criterion described above, as independent data were unavailable. Upon selection of a model, we fit a logistic regression model with untransformed data. Logistic regression models were justified: we could find little evidence for nonlinearity or lack of fit.
criterion described above, as independent data were unavailable. Upon selection of a model, we fit a logistic regression model with untransformed data. Logistic regression models were justified: we could find little evidence for nonlinearity or lack of fit.
For all selected models, we examined plots of observed values vs predicted values from leave-one-out cross-validation (Supplementary Figures S9–15). These plots generally indicated good to excellent predictive power. Data used for model selection are given in Supplementary Figures S6 and S16–S28. Selected models and those that performed best for alternative numbers of predictors are listed in Supplementary Tables S6 and S7.
Latitudinal diversity gradients
From the diversity maps generated from Analyses I–V, we generated plots of mean predicted diversity vs latitude (Figure 1, Supplementary Figures S2, S3, S5 and S29). We quantified the uncertainty associated with the diversity predictions by calculating 95% confidence intervals based on the estimated standard errors of the regression coefficients.
Results and discussion
Our diversity predictions reveal two remarkable patterns: a reverse latitudinal diversity gradient and an extreme seasonality to that gradient. Specifically, maps of predicted bacterial diversity indicate the greatest richness in temperate latitudes in the winter. We predict that diversity peaks at latitudes 30° north and south, with consistently greater richness predictions at higher latitudes compared with lower ones (Figure 1). These patterns are robust: they are evident regardless of the method used to classify rDNA sequences into OTUs (Supplementary Figure S2), SDM to infer the patterns (Supplementary Figure S3), exact set of environmental predictors in the SDM (Supplementary Figure S4) and rarefaction depth (Supplementary Figure S5), and they are evident in scatter plots of the raw data (Supplementary Figure S30). The patterns are also statistically significant (Figure 1 marginal plots). These results are consistent with results reporting seasonal fluctuations of diversity in three temperate and high-latitude locales (Ghiglione and Murray, 2012; Gilbert et al., 2012) and show that seasonal fluctuations in fact dominate the pattern of global bacterial diversity.
The seasonal, temperate peaks in marine bacteria diversity contrast with marine (Tittensor et al., 2010) and terrestrial (Hillebrand, 2004) macroorganism diversity, which typically peaks in the tropics and does not reverse on a seasonal basis (Hillebrand, 2004). Until now, global marine bacterial diversity was thought to follow the same pattern as macroorganisms, that of seasonally consistent and high tropical diversity (Pommier et al., 2007; Fuhrman et al., 2008). The differences between our results and the reported patterns almost certainly stem from differences when samples were collected. In previous bacterial studies, temperate locations were sampled year-round, but latitude and day length were confounded because all of the samples from high latitudes were collected during the summer (Pommier et al., 2007; Fuhrman et al., 2008). In contrast, the MICROBIS data set we used to train our SDM includes samples collected from high latitudes in the winter (Supplementary Figure S31). Summer diversity in temperate and polar oceans is low. Hence, the bias of previous studies toward sampling high latitudes in summer likely resulted in the appearance of higher tropical diversity. This sampling bias underscores the importance of time series data from many seasons at individual sampling locations, as demonstrated by previous studies (Ghiglione and Murray, 2012; Gilbert et al., 2012). Importantly, the SDMs we constructed predict the diversity observed in the samples from previous studies (Supplementary Figure S32), despite major differences in data collection methodologies (Supplementary Table S1). Incorporating samples collected in the winter and controlling for sampling date with SDMs reveal that diversity actually peaks globally in the temperate latitudes in the winter.
In our SDMs, the three strongest predictors of bacterial richness are proximity to the thermocline (sensu reference Montegut et al. (2004)), daylength and phosphate concentration (Supplementary Table S6 and Supplementary Figure S8). Specifically, we find a strong positive correlation between richness and distance from the thermocline (Spearman r=0.364, P-value<0.0001, variables log-transformed). This positive correlation is largely responsible for the predicted seasonal, temperate diversity peaks, as the thermocline reaches its greatest depth at temperate latitudes and in the winter (Montegut et al., 2004). Daylength (Spearman r=−0.638, P-value<0.0001, richness log-transformed) also contributes to the seasonality in the maps. Daylength strongly correlates with richness of marine bacteria in temperate regions, with short photoperiods being associated with high richness (Ghiglione and Murray, 2012; Gilbert et al., 2012). In addition, although phosphate predicts diversity (Spearman r=−0.517, P-value<0.0001, richness log-transformed), other nutrients, such as iron (dust) and nitrate, lack predictive power, suggesting a relatively small role of nutrient limitation in determining global diversity patterns. Naturally, although thermocline proximity, daylength and phosphate concentration predict diversity, other closely related variables might be the causal factors.
The seasonal shifts in local bacterial diversity in temperate latitudes result from shifts in the relative abundance of OTUs, with most OTUs always present, albeit at low abundance during summer (Caporaso et al., 2012). Consistent with this finding, we find that Shannon diversity, which measures the evenness of the distribution of individuals among OTUs, is low in temperate latitudes in summer (Supplementary Figure S29). Thus, in summer, our maps indicate that at a global scale temperate communities are dominated by a few OTUs with high relative abundance. To further examine relative abundance patterns, we generated range maps for select genera of marine bacteria by fitting SDMs of relative abundance (Figure 2, Supplementary Methods). In all seasons, the Cyanobacteria genera Prochlorococcus and Synechococcus show high (25%) relative abundance in tropical and subtropical waters but not elsewhere, consistent with previous reports (Partensky et al., 1999; Wietz et al., 2010). This high relative abundance may contribute to the low Shannon diversity at low latitudes (Supplementary Figure S29). By contrast, Pelagibacter is widely distributed, but shows pronounced winter peaks in relative abundance in the Arctic and Antarctic. In agreement with the notion of summertime blooms depressing Shannon diversity at high latitudes, Polaribacter is abundant in Arctic and Antarctic oceans during the summer, but has low relative abundance during the summer. Other genera, such as Sphingopyxis, show more spatially heterogeneous distributions.
Figure 2.

Range maps of representative genera. Each map shows the probability that a randomly selected rDNA sequence belongs to a particular genus (that is, relative abundance). Primarily autotrophic genera, such as Prochlorococcus and Synechococcus (grouped together by sequence classifier used here; see Methods Summary and Supplementary Methods), occur in high abundance in the tropics and mid-latitudes. Other genera show seasonally variable, high relative abundance (Pelagibacter) or summertime blooms (Polaribacter). The distributions of other taxa follow more complex patterns (Sphingopyxis). Figure S33 presents color versions of these maps.
Based on the variability of ranges among genera, we generated separate diversity maps for dominant phyla of bacteria to determine whether diversity patterns for marine bacteria are consistent across phyla (Figure 3 and Supplementary Figure S34). We find different phylum-specific diversity patterns, potentially reflecting the large range of functional diversity encompassed by bacteria. Alphaproteobacteria richness patterns are qualitatively similar to the aggregate richness pattern of all bacteria, whereas those of other phyla are unique. For example, Gammaproteobacteria richness is highest at very high latitudes, whereas richness within the Cyanobacteria peaks in tropical latitudes. Consistent with the latter pattern, light availability is implicated in the evolution of distinct Procholorococcus ecotypes (Johnson et al., 2006).
Figure 3.

Patterns of OTU richness within bacterial phyla. Columns show maps for different phyla; rows show maps for different seasons. Within the Cyanobacteria, richness peaks primarily at low latitudes, as might be expected for primarily autotrophic taxa. Within the Alphaproteobacteria, richness is distributed similarly to all bacteria taken together. Among Actinobacteria, richness follows primary productivity. Gammaproteobacteria show high polar diversity in the winter. Patterns within other representative phyla are shown in Supplementary Figure S34. Figure S35 presents color versions of these maps.
To demonstrate how our diversity predictions can be used to generate hypotheses for future research, we used our maps to identify bacterial diversity hotspots (Orme et al., 2005), defined here as the 10% of ocean surface with the greatest OTU richness (Tittensor et al., 2010). Most hotspots are centered along 30° latitude north and south, extending up to 15° northward and southward. We compared the global distribution of diversity hotspots to publicly available maps of various human environmental impacts (Halpern et al., 2008) and with previously published marine macroorganism diversity maps (Tittensor et al., 2010). Within hotspots of marine bacterial diversity, total human impacts are disproportionately high (Figures 4a and b). Individual human impacts, such as sea surface temperature anomalies (Supplementary Figure S36), ocean acidification (Supplementary Figure S37) and ultraviolet radiation (Supplementary Figure S38), show similar trends but are not significantly associated with diversity hotspots. Human impacts on the oceans are also disproportionately high in hotspots of pelagic macroorganism diversity (Tittensor et al., 2010). However, bacterial and macroorganism diversity hotspots are situated primarily in different locations (Figures 4c and d). Human impacts often have negative effects on marine macroorganism diversity (Tittensor et al., 2010), but it is uncertain whether similar negative effects occur for bacteria. With the role of bacteria in global biogeochemical cycling and their importance for bioprospecting (Kirchman, 2008), the effects of human impacts on global marine bacterial biodiversity require further investigation.
Figure 4.

Bacterial diversity hotspots. (a) Hotspots of marine bacterial richness overlaid on a map of human impacts to the oceans (Halpern et al., 2008). Hotspots are outlined with black borders, and are defined as the 10% of ocean surface with the greatest diversity in December and June (primarily in the Northern and Southern hemispheres, respectively). (b) The distribution of human impacts across the entire ocean and within December and June diversity hotspots. December and June diversity hotspots have disproportionately high levels of human impacts. (c) Hotspots of marine bacterial richness overlaid on a map of marine macroorgnism diversity (Tittensor et al., 2010) and (d) the distribution of macroorganism richness across the entire ocean and within hotspots. Macroorganism diversity is disproportionately low within bacterial hotspots.
The SDM framework provides a foundation for further investigations of bacterial distributions. Our diversity predictions and range maps with supporting data are publicly available through an interactive web-portal at http://docpollard.org/marine_diversity. The training data for this study are globally distributed and largely cover the range of environmental conditions for which we make diversity predictions, as quantified by multivariate environmental similarity surface plots (Supplementary Figure S7). Our ability to make accurate predictions from this relatively small and unevenly distributed data set is supported by generally low error estimates (gray regions in marginal distribution curves in the right panels of Figure 1) and high agreement between our predictions and observed diversity in an external data set (Pommier et al., 2007, ∼90%). Nonetheless, our predictions are less accurate for other external data sets and in some geographic regions. It is therefore essential to update and validate our predictions as data from additional marine sampling surveys are publicly released. Our maps should therefore be viewed as material for generating hypotheses about individual ecosystems that can be tested through focused studies. In addition, as sequence and environmental data become available, SDM can be directly applied to deeper ocean environments, and global atmospheric and terrestrial ecosystems. Mapping the distributions of microbial functions (for example, by performing SDM analysis on metabolically important genes from metagenomic studies) will complement emerging efforts to investigate the geographic distribution of bacterial metabolism (Raes et al., 2011).
There is a great need to understand diversity hotspots as well as seasonal and meridional patterns of diversity in marine microbes (Barton et al., 2010; Giovannoni and Vergin, 2012). Our analyses predict that marine bacterial diversity follows a highly significant seasonal fluctuation across the globe with a reversed latitudinal gradient, unlike that seen for macroorganisms. Indeed, although some pelagic marine macroorganisms show temperate peaks in diversity (Tittensor et al., 2010), these differ considerably from the patterns reported here and they do not reverse on a seasonal basis. The bacteria differ in many respects from macroorganisms. They have more rapid generation times, allowing them to adapt more quickly to changing environments and potentially to be more widespread (Poole et al., 2003), they can form dormant, resistant stages that allow them to travel unscathed through inhospitable environments (Hubert et al., 2009), and they experience their environment at smaller scales than macroorganisms, so regions that contain niches for only a few macroorganisms may contain niches for many microorganisms (Whitaker and Banfield, 2006). Understanding how such traits affect the global distributions of bacteria may be key in understanding the processes shaping the evolution and maintenance of global biodiversity. The present study is an important starting point for these investigations.
Acknowledgments
S Riesenfeld, B Dorazio, J Eisen, JC Thrash, JA Gilbert, N Fierer, G Howard and VL Sharpton provided valuable insight and discussion. This research was funded by Gordon and Betty Moore Foundation Grant #1660, institutional funding from the J David Gladstone Institutes, and the Omidyar Fellowship at the Santa Fe Institute.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Barton AD, Dutkiewicz S, Flierl G, Bragg J, Follows MJ. Patterns of diversity in marine phytoplankton. Science. 2010;327:1509–1511. doi: 10.1126/science.1184961. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Paszkiewicz K, Field D, Knight R, Gilbert JA. The western english channel contains a persistent microbial seed bank. ISME J. 2012;6:1089–1093. doi: 10.1038/ismej.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elith J, Graham CH, Anderson RP, Dudik M, Ferrier S, Guisan A, et al. Novel methods improve prediction of species distributions from occurrence data. Ecography. 2006;29:129–151. [Google Scholar]
- Elith J, Kearney M, Phillips S. The art of modelling range-shifting species. Method Ecol Evol. 2010;1:330–342. [Google Scholar]
- Ferrier S, Guisan A. Spatial modelling of biodiversity at the community level. J Appl Ecol. 2006;43:393–404. [Google Scholar]
- Fierer N, Leff JW, Adams BJ, Nielsend UN, Bates ST, Lauber CL, et al. Crossbiome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci USA. 2012;109:21390–21395. doi: 10.1073/pnas.1215210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin J, Miller JA. Mapping species distributions: Spatial Inference and Prediction. Cambridge University Press: Cambridge, UK; 2009. [Google Scholar]
- Friedman JH. Multivariate adaptive regression splines. Ann Stat. 1991;19:1–141. doi: 10.1177/096228029500400303. [DOI] [PubMed] [Google Scholar]
- Fuhrman JA, Steele JA, Hewson I, Schwalbach MS, Brown MV, Green JL, et al. A latitudinal diversity gradient in planktonic marine bacteria. Proc Natl Acad Sci USA. 2008;105:7774–7778. doi: 10.1073/pnas.0803070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglione JF, Murray AE. Pronounced summer to winter differences and higher wintertime richness in coastal antarctic marine bacterioplankton. Environ Microbiol. 2012;14:617–629. doi: 10.1111/j.1462-2920.2011.02601.x. [DOI] [PubMed] [Google Scholar]
- Gilbert JA, Steele JA, Caporaso JG, Steinbruck L, Reeder J, Temperton B, et al. Defining seasonal marine microbial community dynamics. ISME J. 2012;6:298–308. doi: 10.1038/ismej.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni SJ, Vergin KL. Seasonality in ocean microbial communities. Science. 2012;335:671–676. doi: 10.1126/science.1198078. [DOI] [PubMed] [Google Scholar]
- Halpern BS, Walbridge S, Selkoe KA, Kappel CV, Micheli F, D'Agrosa C, et al. A global map of human impact on marine ecosystems. Science. 2008;319:948–952. doi: 10.1126/science.1149345. [DOI] [PubMed] [Google Scholar]
- Hillebrand H. On the generality of the latitudinal diversity gradient. Am Nat. 2004;163:192–211. doi: 10.1086/381004. [DOI] [PubMed] [Google Scholar]
- Hubert C, Loy A, Nickel M, Arnosti C, Baranyi C, Bruchert V, et al. A constant flux of diverse thermophilic bacteria into the cold arctic seabed. Science. 2009;325:1541–1544. doi: 10.1126/science.1174012. [DOI] [PubMed] [Google Scholar]
- International census of marine microbes 2011 ). http://icomm.mbl.edu .
- Johnson ZI, Zinser ER, Coe A, McNulty NP, Woodward EMS, Chisholm SW. Niche partitioning among prochlorococcus ecotypes along ocean-scale environmental gradients. Science. 2006;311:1737–1740. doi: 10.1126/science.1118052. [DOI] [PubMed] [Google Scholar]
- Kirchman DL.2008Microbial Ecology of the Oceans2nd ednWiley [Google Scholar]
- Lamarck JBPA, Candolle AP.1805Flore Francaise3rd ednDesray Publishers [Google Scholar]
- Larsen PE, Field D, Gilbert JA. Predicting bacterial community assemblages using an artificial neural network approach. Nat Methods. 2012;9:621–625. doi: 10.1038/nmeth.1975. [DOI] [PubMed] [Google Scholar]
- Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL, et al. Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol. 2006;4:102–112. doi: 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- Montegut CB, Madec G, Fischer AS, Lazar A. Mixed layer depth over the global ocean: an examination of profile data and a profile-based climatology. J Geophys Res. 2004;109:003. [Google Scholar]
- Orme CDL, Davies RG, Burgess M, Eigenbrod F, Pickup N, Olson VA, et al. Global hotspots of species richness are not congruent with endemism or threat. Nature. 2005;436:1016–1019. doi: 10.1038/nature03850. [DOI] [PubMed] [Google Scholar]
- Partensky F, Hess W, Vaulot D. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol Mol Biol R. 1999;63:106–127. doi: 10.1128/mmbr.63.1.106-127.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier T, Canback B, Riemann L, Bostrom KH, Simu K, Lundberg P, et al. Global patterns of diversity and community structure in marine bacterioplankton. Mol Ecol. 2007;16:867–880. doi: 10.1111/j.1365-294X.2006.03189.x. [DOI] [PubMed] [Google Scholar]
- Poole AM, Phillips MJ, Penny D. Prokaryote and eukaryote evolvability. Biosystems. 2003;69:163–185. doi: 10.1016/s0303-2647(02)00131-4. [DOI] [PubMed] [Google Scholar]
- Raes J, Letunic I, Yamada T, Jensen LJ, Bork P. Toward molecular trait-based ecology through integration of biogeochemical, geographical and metagenomic data. Mol Syst Biol. 2011;7:1–9. doi: 10.1038/msb.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DM, Whittaker RJ. Conservation biogeography – foundations, concepts and challenges. Divers Distrib. 2010;16:313–320. [Google Scholar]
- Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, et al. The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamames T, Abellan JJ, Pignatelli M, Camacho A, Moya A. Environmental distribution of prokaryotic taxa. BMC Microbiol. 2010;10:85. doi: 10.1186/1471-2180-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tittensor DP, Mora C, Jetz W, Lotze HK, Ricard D, Berghe EV, et al. Global patterns and predictors of marine biodiversity across taxa. Nature. 2010;466:1098–1101. doi: 10.1038/nature09329. [DOI] [PubMed] [Google Scholar]
- Whitaker RJ, Banfield JF. Population genomics in natural microbial communities. Trends Ecol Evol. 2006;21:508–516. doi: 10.1016/j.tree.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Wietz M, Gram L, Jorgensen B, Schramm A. Latitudinal patterns in the abundance of major marine bacterioplankton groups. Aquat Microb Ecol. 2010;61:179–189. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.