

Abstract
Aminoacyl-tRNA synthetases are multi-domain enzymes that catalyze covalent attachment of amino acids to their cognate tRNA. Cross-talk between functional domains is a prerequisite for this process. In the present study, we investigate the molecular mechanism of site-to-site communication in Escherichia coli prolyl-tRNA synthetase (Ec ProRS). Earlier studies have demonstrated that evolutionarily conserved/co-evolved residues that are engaged in correlated motion are critical for the propagation of functional conformational changes from one site to another in modular proteins. Here, molecular simulation and bioinformatics-based analysis was performed to identify dynamically coupled and evolutionarily constrained residues that form contiguous pathways of residue-residue interactions between the aminoacylation and editing domains of Ec ProRS. The results of this study suggest that multiple pathways exist between these two domains to maintain the dynamic coupling essential for enzyme function. Moreover, residues in these interaction networks are generally highly conserved. Site-directed changes of on-pathway residues have a significant impact on enzyme function and dynamics suggesting that any perturbation along these pathways disrupts the native residue-residue interactions that are required for effective communication between the two functional domains. Free energy analysis revealed that communication between residues within a pathway, as well as cross-talk between pathways are important to coordinate functions of different domains of Ec ProRS for efficient catalysis.
Class II prolyl-tRNA synthetases (ProRSs) catalyze covalent attachment of proline to tRNAPro in a two-step reaction:
| (i) |
| (ii) |
ProRSs are modular proteins and are divided into two evolutionarily distinct groups based on sequence alignment and structural architecture (1, 2). The “prokaryotic-like” ProRSs contain an insertion domain (INS) between motifs 2 and 3 of the catalytic domain, whereas “eukaryotic-like” ProRSs have C- and/or N-terminal extension domains (3). ProRSs from all three kingdoms of life have been shown to misactivate noncognate alanine and cysteine, resulting in mischarged tRNAPro(4, 5). Many aminoacyl-tRNA synthetases (AARSs) have evolved proofreading capabilities to correct mistakes in amino acid activation (pre-transfer editing) and charging (post-transfer editing) (6). It has been shown that a domain (INS) of approximately 180 residues inserted within the catalytic core of prokaryotic-like ProRSs (Fig. 1) is a post-transfer editing active site that hydrolyzes specifically mischarged Ala-tRNAPro(4, 5, 7, 8). In contrast, Cys-tRNAPro is hydrolyzed by an INS homolog known as YbaK, which is encoded as a single-domain protein by many bacteria (9, 10). Unlike prokaryotic-like ProRSs, most eukaryotic-like ProRSs do not possess the INS domain but in some cases encode free-standing editing domain homologs. Some lower eukaryotic ProRSs encode an N-terminal domain that displays weak homology with bacterial INS. This domain exhibits post-transfer editing activity against Ala-tRNAPro in Plasmodium falciparum(11) but is a defunct editing domain in Saccharomyces cerevisiae(12).
Figure 1.
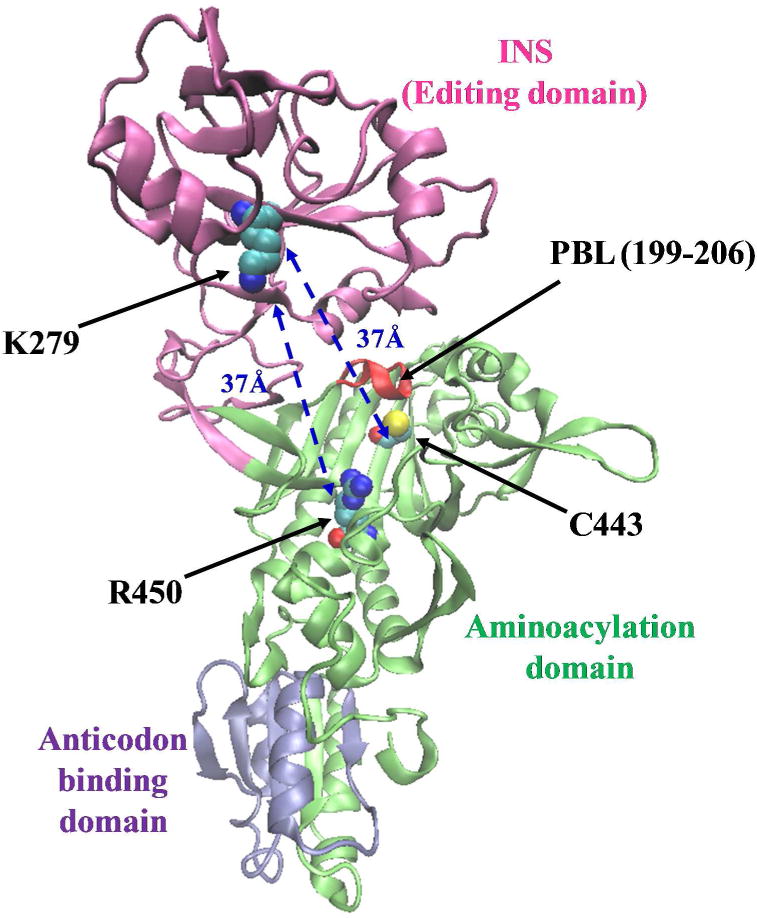
Ribbon representation of the 3-dimensional structural model of the monomeric form of Ec ProRS. The homology model was derived from the X-ray crystal structure of Ef ProRS (15). The Cα – Cα distances between the starting residues (C443/R450) and the end residue (K279) are shown.
Bacterial ProRSs are modular enzymes and efficient catalysis and editing requires effective communication between distant domains. Escherichia coli (Ec) ProRS, a representative member of the prokaryotic-like group, contains three distinct domains (Fig. 1). The aminoacylation domain (motifs 1, 2 and 3; residues 64–81, 128–164, and 435–465, respectively) catalyzes the activation of proline and the aminoacylation of tRNAPro, as well as pre-transfer editing (13); the anticodon binding domain (residues 506–570) is critical for recognition of cognate tRNA; the INS (residues 224–407, located between motifs 2 and 3 of the catalytic domain) is the post-transfer editing active site (7, 8). The aminoacylation domain and the INS of Ec ProRS have been observed to depend on each other in terms of their individual catalytic activities. For example, mutation of a highly conserved aspartate (D350) in INS to alanine resulted in reduced aminoacylation activity (7). Previous studies have also indicated that covalent connectivity between domains is a prerequisite for efficient aminoacylation and editing functions by ProRSs and other AARS systems (14). Deletion of the INS of Ec ProRS (ΔINS) has a severe impact on catalysis; the amino acid activation efficiency of the deletion variant was reduced 1200-fold (13). The addition of a separately cloned and purified INS in trans failed to stimulate the amino acid activation efficiency of the ΔINS construct (Hati and Musier-Forsyth, unpublished data). The requirement of covalent connectivity between domains for efficient function of Ec ProRS suggests the existence of inter-domain communication in this enzyme. Moreover, structural studies revealed that the catalytically important proline-binding loop (PBL) of bacterial ProRSs undergoes a conformational change from “open” to “closed” state upon substrate binding (15). A recent study showed that any perturbation in the surrounding structural elements has significant impact on PBL dynamics (16). The pathway by which substrate induced conformational change propagates from the activation center to the distant protein segments that modulate PBL dynamics and conformational change is unknown.
For multi-domain proteins like Ec ProRS, domain-domain communication is achieved by coupled domain dynamics (17–20). Recent MD simulation results revealed that the INS of Ec ProRS is engaged in coupled motion with structural elements of the catalytic domain (16). The collective dynamics of the PBL is altered by the deletion of the INS or point mutation at the INS-aminoacylation domain junction (16). To understand the molecular mechanism by which different structural elements of Ec ProRS coordinate their function, it is important to identify networks defined by key residue-residue interactions that promote coupled-domain dynamics in this enzyme.
In the present study, a bioinformatics-based computational method, statistical-thermal coupling analysis (STCA), was employed to trace pathways of site-to-site communication in Ec ProRS. Previous application of this method revealed that dynamically coupled and evolutionarily constrained residues are important to maintain coupled-domain dynamics in Thermus thermophilus leucyl-tRNA synthetase (Tt LeuRS) (20). Here STCA was used to identify the residue-residue interaction networks between the INS and aminoacylation domains of Ec ProRS. In addition, mutational and kinetic studies, as well as thermal fluctuation analyses, were performed to validate the predicted networks of residue-residue interactions. Taken together, the present study demonstrates that a modular protein like Ec ProRS employs multiple pathways of residue-residue interactions to communicate between distant functional sites. Moreover, networks of these pathways involve residues that are evolutionarily constrained and engaged in correlated motion.
MATERIALS AND METHODS
General Strategy
Based on reported experimental and structural data, the starting and ending points of interaction networks between the aminoacylation domain and the INS were selected; these active sites are > 30 Å apart (Fig. 1). The single cysteine residue (C443), which is important for amino acid activation, was chosen as one of the starting points within the amino acid activation site. The other starting residue is R450, found to be important for substrate binding (15) (Fig. 1). The end point was chosen to be K279 in the INS, which has been shown to be required for editing function (8) and is located 37 Å from C443 and R450 (Fig. 1). STCA was carried out in three discrete steps (Scheme 1). First, statistical coupling analysis (SCA) (21, 22) was performed to identify conserved and co-evolved residues in the ProRS family. In a parallel study, the collective motions of various domains of the protein were studied by performing a long-timescale molecular dynamics (MD) simulation of Ec ProRS. In the second step, the evolutionary dependence of the coupled-domain dynamics was explored. In this step, results of the SCA and MD simulation were computationally integrated (Scheme 1). This analysis identified a subset of residues that are not only dynamically coupled but also evolutionarily constrained. Next, distinct networks of interacting residues between the two distant sites were identified from this subset of residues using Dijkstra’s algorithm (23). Finally, the role of these selected residues in site-to-site communication was probed experimentally by conducting site-directed mutagenesis and kinetic studies. In addition, in silico mutations were performed and their impact on protein dynamics was examined by comparing root-mean-square (RMS) fluctuations of the WT and mutated variants (16).
Scheme 1.
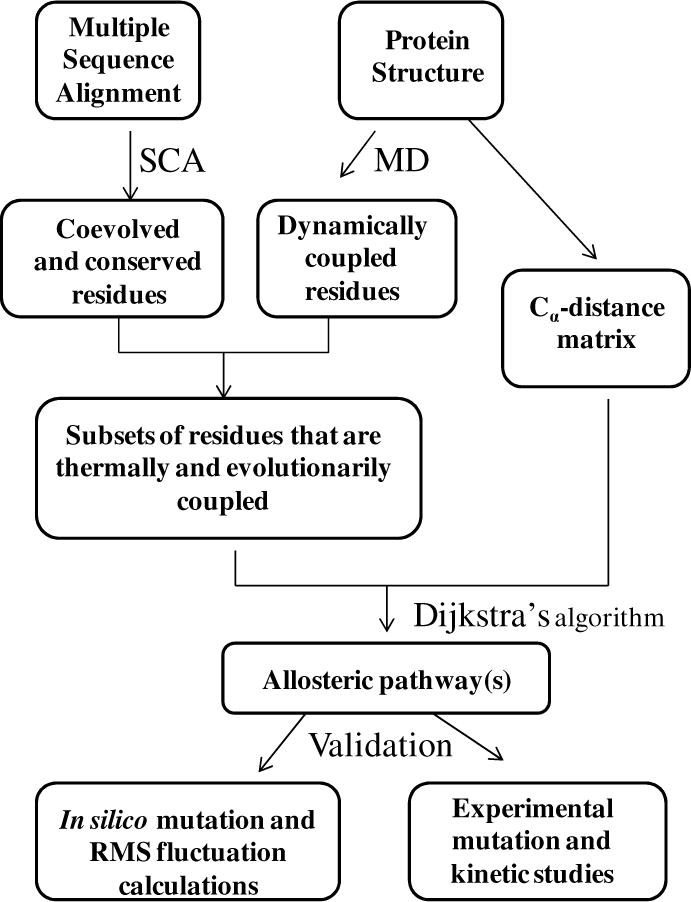
Proteins were visualized using VMD software (24). SCA was carried out using a MATLAB script obtained from the Ranganathan lab (http://systems.swmed.edu/rr_lab/sca.html). The MD plots, SCA plots, and all data processing were carried out using MATLAB R2006b (The MathWorks Inc., Natick, MA).
COMPUTATIONAL METHODS
Molecular Dynamics Simulations
MD simulations were carried out using the three-dimensional homology model of monomeric Ec ProRS (residues 1–567) (provided by S. Cusack). The model was generated using the crystal structure of Enterococcus faecalis ProRS (15), which is 48% identical to the Ec enzyme. Mutant proteins were built using the Mutator plug-in of VMD (24). Simulations were performed in water (TIP3P model (25)) with substrate-free enzymes using the all-atom CHARMM22 force field (26) within the NAMD package (27). All simulations were carried out with a 500 ps equilibration step followed by a 25 ns production MD run. The details of the MD simulation protocol were as described previously (16). To evaluate the statistical significance of the MD simulation analysis, three replicates were generated for each protein system, as described in the protein simulation studies by Roy and Laughton (28).
The correlated motions between residue pairs of distant structural elements were studied by principal component analysis (PCA, also known as essential dynamics analysis) of collective motions (29), as described earlier (16, 30). PCA is a procedure by which the low-frequency (high-amplitude) collective motions of a biomolecule, which are often more relevant for its functions, are extracted from a MD simulation trajectory (31). This method has been described in detail in our previous study with this enzyme (16). Briefly, a covariance matrix of the Cα atoms was generated using the simulated MD data set. The diagonalization of this covariance matrix produces eigenvectors and eigenvalues representing the direction and magnitude of the collective motion of the whole protein or protein segments of interest.
In the present study, the last 20 ns of the MD simulation data were used to generate principal components of atomic (backbone Cα atoms) fluctuations using Carma (32). The first three principal components were used to perform PCA-based cluster analysis, which produced a new trajectory of conformations representing the predominant conformational fluctuations. Finally, these conformations were used to generate dynamic cross-correlation matrix C, in which the ij-th element, Cij, represents the cross-correlation coefficient between fluctuations of residues at sites i and j during the simulation:
| (1) |
The atomic (Cα) displacements of residues i and j are represented by xi and xj, respectively; the angular brackets represent ensemble averages, and σxi and σxj represent the standard deviations of these displacements.
RMS fluctuations of Cα atoms averaged over three replicate simulations were also obtained for the WT and mutants. In these calculations, the last 20 ns of MD simulation data were used, each comprising an ensemble of 200,000 conformations. PCA was also carried out for the WT and mutant proteins in order to compare the effect of mutations on coupled dynamics. For each of these protein systems, an ensemble of 600,000 conformations, obtained by combining three replicate trajectories, was used to perform PCA. The first three clusters representing the predominant conformational fluctuations were considered in this study. In addition, the collective dynamics of the catalytically important PBL of the WT and various mutants were also analyzed following the method described earlier (16, 30).
Statistical Coupling Analysis
SCA is based on the assumption that the coupling of two sites in a protein, whether for structural or functional reasons should cause those two sites to coevolve (21, 22). SCA was carried out using the standard protocol, details of which are published (21, 22, 33) and are also available online at http://www.hhmi.swmed.edu/Labs/rr/sca.html. In the present work, a multiple-sequence alignment of 492 protein sequences of the ProRS family was generated using BLAST (34). Only ProRS sequences that bear significant sequence identity (> 67%) with Ec ProRS were included in this study. The conservation constant and the coupling constant were obtained using standard procedures described previously (20).
Statistical Thermal Coupling Analysis: Integration of Evolutionary and Dynamic Information
In our earlier study on Tt LeuRS, we observed that coupled-domain motions are facilitated by networks of thermally and evolutionarily constrained residues (20). To identify residue clusters that are important for maintaining coupled-domain dynamics, we extracted a subset of residues that are simultaneously coupled through evolution and dynamics (Scheme 1). The motional coupling information from MD was integrated with the evolutionary conservation and coevolution dataset obtained from the SCA study.
The conserved and co-evolved residues were treated separately. The conserved and dynamically coupled residues were chosen by selecting only those residues that exhibit significant conservation ( , values range between 0 and 1.0), as well as motional coupling [Cij ≥ 0.8, Cij values range between −1.0 (anticorrelated motion) and 1.0 (correlated motion)] with each other. Based on recent studies of the significance of correlated motion in long-range communication, only positive correlations were considered (35, 36). The value of Cij was set to ≥ 0.8 in order to obtain residues that are engaged in strong correlated motions.
To select the co-evolved and dynamically coupled residues, the dynamic cross-correlation matrix (C matrix) was truncated by including only those columns that are present in the normalized SCA-derived G matrix. Next, the coevolutionary dynamic coupling, CDC, matrix was created by multiplying each ij-th element of the G matrix with the corresponding element of the truncated C matrix:
| (5) |
The CDC matrix therefore contains the covariance information of residue pairs that are coevolved as well as dynamically coupled. In the present study, the co-evolved and dynamically correlated residues are extracted by choosing only those residues for which CDCij ≥ 0.4
Identification of Interaction Networks Using Dijkstra’s Algorithm
From the shortlisted residues (Scheme 1), residue-residue interaction networks between C443/R450 (aminoacylation domain) and K279 (INS) of Ec ProRS were identified using Dijkstra’s algorithm (23). In this method, each of the Cα atoms of the protein backbone represents a node. The connectivity between two adjacent nodes was described by a binary connection matrix P of inter-residue (Cα – Cα) contacts. The Cα – Cα distance matrix, D was computed from the Cartesian coordinates of all Cα atoms of the protein. Based on a Cα – Cα cutoff distance , Pij is equal to 1 if and zero otherwise. The interaction networks (paths) between two functional sites were identified and listed in terms of a “cost”, which is equal to the sum of all Cα – Cα distances between adjacent residues in a given path.
EXPERIMENTAL METHODS
Materials
All amino acids (Sigma) were of highest quality (>99% pure) and used without any further purification. [γ-32P]-ATP and [32P]-PPi were from Perkin Elmer. Primers for site-directed mutagenesis and PCR were from Integrated DNA Technologies.
Enzyme Preparation
Overexpression and purification of histidine-tagged WT and mutant Ec ProRS were performed as described previously (37, 38). Plasmids encoding D198A, E234A, H302A, N305A, G412A, F415A, H302A/G412A, N305A/G412A and E218A/N305A Ec ProRS were generated by site-directed mutagenesis of pCS-M1S (38) using primers listed in Table S1. Results of mutagenesis were confirmed by DNA sequencing (University of Wisconsin, Biotechnology Center-Madison). Protein expression was induced in Ec SG13009 (pREP4) competent cells with 0.1 mM isopropyl β-D-thiogalactoside for 4 h at 37°C. Histidine-tagged proteins were purified using a Talon cobalt affinity resin, and the desired protein was eluted with 100 mM imidazole. Protein concentrations were determined initially by the Bio-Rad assay (Bio-Rad Laboratories) followed by active-site titration (39).
RNA Preparation
Ec tRNAPro was transcribed using T7 RNA polymerase from BstN1-linearized plasmid as described (40) and purified by denaturing 12% polyacrylamide gel electrophoresis.
Enzyme Assays
The ATP-PPi exchange assay was performed at 37 °C according to the published method (41). The concentrations of proline ranged from 0.05–2 mM. The enzyme concentrations used were 10–40 nM for proline activation. Kinetic parameters were determined from Lineweaver-Burk plots and represent the average of at least three determinations.
ATP hydrolysis reactions to monitor pre-transfer editing were carried out as described previously (13) using 500 mM alanine and 4 μM ProRS.
Aminoacylation assays were performed at room temperature in a reaction mixture containing 50 mM HEPES (pH 7.5), 20 mM KCl, 25 mM MgCl2, 0.1 mg/ml bovine serum albumin, 20 mM 2-mercaptoethanol, and 4 mM ATP. Cognate aminoacylation reactions also contained 23 μM [3H]proline, 0.5 μM tRNAPro, and 100 nM Ec ProRS. When preparing charged tRNAPro to be used in deacylation assays, the same reaction mixture was used, and [14C]alanine was used to acylate G1:C72/U70 tRNAPro (8 μM) by Ec alanyl-tRNA synthetase (8 μM). After incubation for 1.5 hours, 1% acetic acid was used to quench the reaction and the mischarged tRNA was purified by repeated phenol:CHCl3 extraction (5:1 solution, pH 4.5), followed by ethanol precipitation.
Deacylation assays were carried out at room temperature in reactions containing 50 mM HEPES (pH 7.5), 5 mM MgCl2, 1 μM G1:C72/U70 [14C] Ala-tRNAPro, and were initiated with 0.5 μM Ec ProRS. A buffer only background curve was also performed and subtracted from each curve.
RESULTS
MD Simulation
For the substrate-free WT ProRS, the root-mean-square-deviation (RMSD) of each frame of the 25-ns MD simulation trajectory was computed relative to the initial coordinates (Fig. 2). RMSD values of these frames fluctuated with a mean value of 1.5–2.0 Å during the last 20 ns of the simulations and similar fluctuations in RMSD values were obtained for all three replicate simulations. These variations are consistent with previous studies on substrate-unbound AARSs (42–44).
Figure 2.
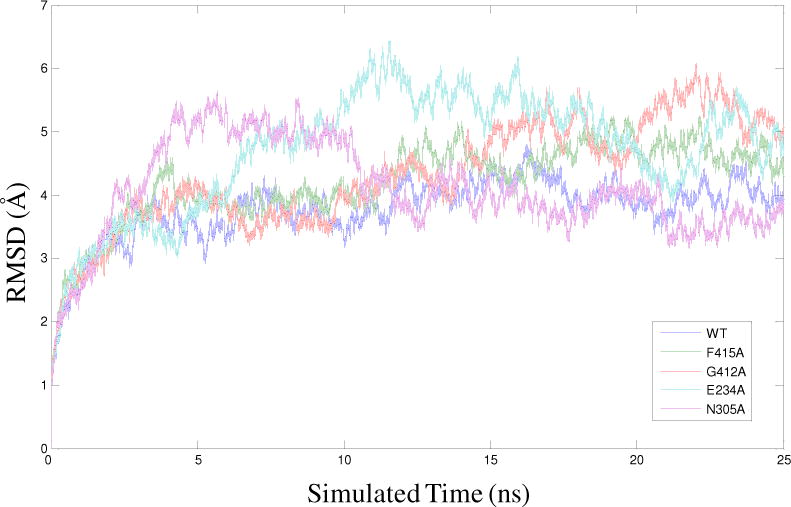
RMSD of the Cα atoms from their initial coordinate as a function of time for WT Ec ProRS and four variants. Calculations of RMSDs were performed using 25 ns MD simulation data.
Dynamic Cross-Correlation Analysis
The dynamic cross-correlation matrix (C) for the WT enzyme (Fig. S1) was generated using the first three principal components. Analysis of the cross-correlation of fluctuations of residues revealed both inter- and intra-domain dynamic correlation. The aminoacylation domain and the INS are mainly engaged in anticorrelated motions; i.e., their displacements are in opposite directions (Cij < 0). However, various structural elements within the aminoacylation domain and the INS are engaged mainly in correlated motion (Cij > 0). The C matrix (Fig. S1) was used to extract residues that are engaged in strong correlated motion (Cij ≥ 0.8).
Conserved and Co-evolved Residues
Earlier, we showed that evolutionarily constrained residues are important for maintaining coupled-domain dynamics in Tt LeuRS (20). To identify the conserved residues responsible for coupled domain-dynamics between aminoacylation and editing active sites of Ec ProRS, the value of each residue, a quantitative measure of the conservation of a residue at the ith position of the sequence, was calculated using SCA (21, 22, 33). The values were obtained as a normalized one-dimensional vector (Fig. S2). The number of conserved residues in Ec ProRS obtained at different cut-offs is reported in Table 1. In addition, a 567×152 coupling matrix containing the evolutionary coupling indices ( of the 567 total residues for 152 perturbation sites (21, 22) was obtained from SCA (Fig. 3). It is evident that only a small fraction of Ec ProRS residues have high values. The multiplication of this 567×152 SCA matrix with the truncated 567×152 MD matrix yields the CDC matrix (eq. 5). The numbers of evolutionarily and dynamically coupled residues determined for various CDCij cut-offs are listed in Table 1. These pools of conserved and coevolved residues were further used to identify long-range interaction networks described below.
Table 1.
Number of conserved and co-evolved residues with in Ec ProRS that are engaged in correlated motion. These residue numbers were determined by varying cutoff values for (evolutionary conservation constant) and CDCij, ( ; coevolutionary dynamic coupling constant), respectively, while maintaining the correlation coefficient of residue-residue fluctuations (Cij) greater than or equal to 0.8.
|
|
≥ 0.50 | ≥ 0.55 | ≥ 0.60 | ≥ 0.65 | ≥ 0.70 | |
|---|---|---|---|---|---|---|
| Number of Conserved Residues | 207 | 182 | 151 | 104 | 61 | |
| CDCij | ≥ 0.40 | ≥ 0.45 | ≥ 0.50 | ≥ 0.55 | ≥ 0.60 | |
| Number of Co-evolved Residues | 96 | 69 | 48 | 28 | 18 |
Figure 3.
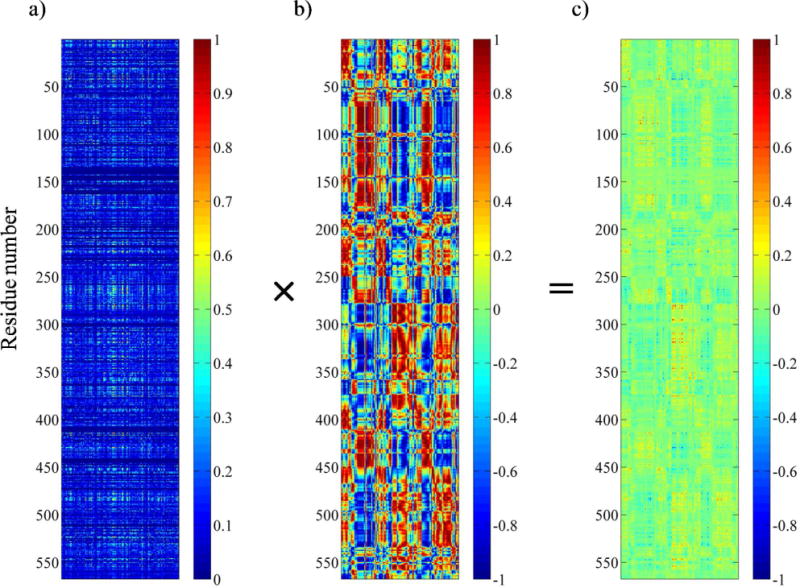
Generation of coevolutionary dynamic coupling (CDC) matrix from SCA coupling matrix and truncated dynamic cross-correlation matrix. a) The unclustered SCA coupling matrix from the SCA of ProRS family. The X-axis represents the 152 perturbation sites, while the Y-axis corresponds to the 1–567 residues of the Ec ProRS. The color gradient, as indicated in the color bar, is as follows: blue squares represent the lowest and red squares represent the highest statistical coupling energies, . b) The truncated MD cross-correlation matrix generated by taking only those columns of perturbation sites (residues) that are present in the SCA matrix. The cross-correlation values range between +1 (correlated) to −1 (anticorrelated). c) The SCA•MD plot obtained as the CDC matrix by multiplying individual elements of the SCA matrix with the corresponding elements of the truncated MD matrix. Values range from +1.0 (co-evolved and thermally correlated) to −1.0 (co-evolved and thermally anticorrelated).
Interaction Networks Across Domains
To map the interaction networks between the aminoacylation domain (C443/R450) and the INS (K279), Dijkstra’s algorithm (23) was applied to the pool of dynamically and evolutionarily (conserved/co-evolved) coupled residues (Scheme 1), as described in the Methods section. Since the average van der Waal’s radius of an amino acid is ~ 3 Å and the acceptable distance for the non-covalent interactions is 2.0–3.0 Å, a strong non-covalent interaction will be prevalent when two Cα atoms are separated by a distance of 8.0–9.0 Å. Therefore, in this study the distance cutoff for the potential interactions between the neighboring Cα atoms was varied between 8.0–9.0 Å.
Several residue-residue interaction networks were identified between the aminoacylation and INS domains. These contiguous interaction networks were obtained by using various cutoff values for , Cij, and as listed in Table 2 while keeping CDCij ≥ 0.4 ( ; , ). The four probable contiguous pathways (path I – IV), identified based on the distance between the starting and ending residues (cost), as well as the degree of dynamic correlation between networking residues (average correlation coefficient value), are listed in Table 2 and shown in Fig. 4. These predicted paths (interaction networks) are dominated by polar residues. Moreover, close scrutiny of residues in the predicted pathways revealed that conserved residues are dominant over co-evolved residues.
Table 2.
Probable pathways of communication between the aminoacylation and the INS domains in Ec ProRS as identified using STCAa
| Pathways | Parameters | Residue Networks | Cost (Å) |
|---|---|---|---|
|
C443→K279 I (8 residues) |
Cij ≥ 0.8 CDCij = 0.4 |
C443→F415→T199→D198→E234 →N389→H302→K279 |
43.9 (0.62,0.51,60) |
|
|
|||
|
II (9 residues) |
Cij ≥ 0.8 CDCij = 0.4 |
C443→F415→T199→D 198→E234→ N305→L304→L281→K279 |
46.6 (0.66,0.59, 0.62) |
|
| |||
|
R450→K279 III (10 residues) |
Cij ≥ 0.8 CDCij = 0.4 |
R450→V411→G412→E209→D198 →E234→N305→L304→L281→K279 |
51.5 (0.47,0.60, 0.63) |
|
|
|||
|
IV (10 residues) |
Cij ≥ 0.8 CDCij = 0.4 |
R450→I409→E218→D219→N232→ E234→N305→L304→L281→K279 |
53.3 (0.74,0.65, 0.64) |
Cij, correlation coefficient of residue-residue fluctuations; , evolutionary conservation constant; , evolutionary coupling constant; , coevolutionary dynamic coupling constant; , distance cutoff. Numbers in parentheses in column 4 represent average Cij between adjacent residues in a given path obtained from the dynamic cross-correlation matrix C for the three replica simulations. The residues in bold represent the terminal residues, all but residues shown in italics are conserved residues; residues in italics represent the co-evolved residues. The underlined residues were chosen for mutational studies.
Figure 4.
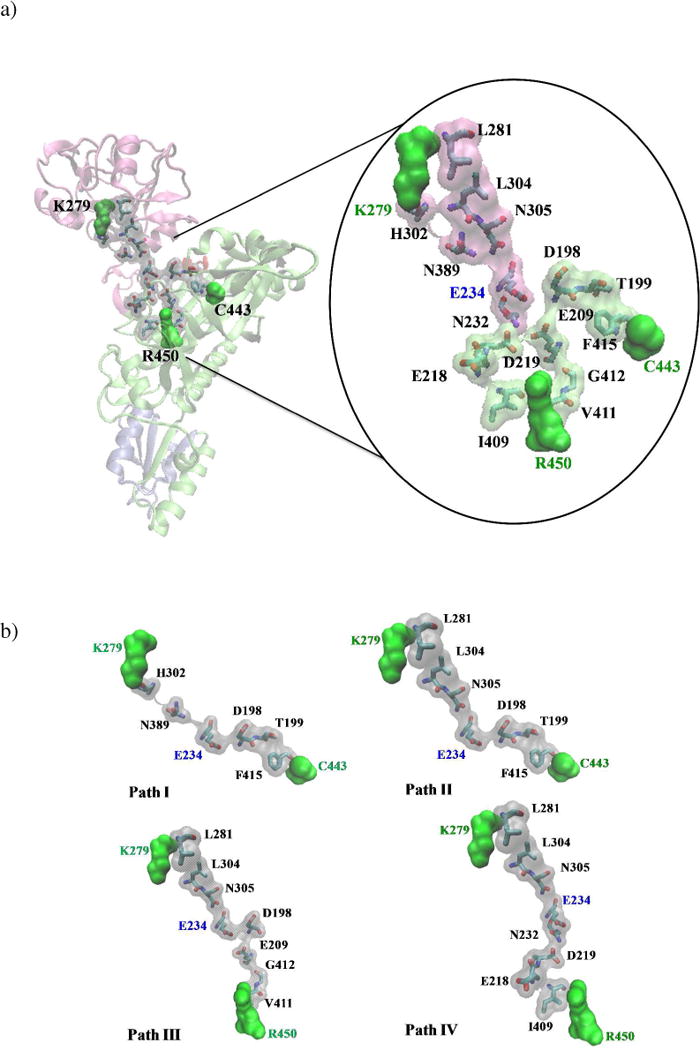
a) Representation of residue-residue interaction networks between the substrate binding domain (R450/C443) and the INS (K279) identified in this study. Terminal residues in the pathways are shown in green space-filling surface representation. b) Paths I – IV are shown in space-filling surface representations.
To evaluate the proposed communication pathways, mutational studies were carried out on some of the conserved residues (Table 2, underlined) that are present in more than one of the predicted pathways. The impact of altering pathway residues on amino acid activation, tRNA aminoacylation, and editing reactions was investigated.
Amino Acid Activation
First, the WT enzyme and six single mutants [F415A, G412A, and D198A (aminoacylation domain); N305A, H302A, and E234A (INS)] were examined for their role in amino acid activation. Using the ATP-PPi exchange reaction, we found that mutation of some of these residues to alanine had considerable impact on proline activation (Table 3). Alanine substitution of F415 of the catalytic domain, which undergoes a conformational change upon prolyl-adenylate binding (15), resulted in an ~ 400-fold decrease in kcat/KM. Mutation of residues in the INS domain also had significant impact on amino acid activation. For example, significantly lower levels of amino acid activation efficiency were observed for N305A (~ 50-fold reduced) and E234A (~ 10-fold reduced) variants. To examine if residues in the predicted pathways are indeed coupled, amino acid activation efficiencies of double mutants were also evaluated. The H302A/G412A variant, obtained by alanine substitution of residues that belong to the two separate pathways (H302, path I and G412, path III), exhibited over 4-fold decreased proline activation efficiency (Table 3); the H302A and G412A single mutants exhibited a 2.1-fold and 1.6-fold reduction in amino acid activation efficiency, respectively. The free energy analysis [(ΔΔG = −RT ln(fold-decreased in kcat/KM), where R is the gas constant, 1.986 cal K− 1mol−1 and T is 310 K] (42) showed that the free energy change of the double mutant (0.88 kcal/mol) is somewhat greater than the sum of the free energy change of the H302A (0.46 kcal/mol) and G412A (0.29 kcal/mol) mutants (Table 3). This difference in free energy change implies weak coupling between H302 and G412 (45). Proline activation was not detected for the other two double mutants tested, N305A/G412A (path III) and E218A/N305A (path IV). The lack of activity of the path III variant suggests strong coupling between N305A and G412A since the effect of the double mutant (ΔΔG > 3.7 based on level of detection of this assay) is greater than expected based on the single mutant effects (2.4 kcal/mol and 0.29 kcal/mol, for N305A and G412A, respectively). In a recent study, we showed that proline activation efficiency was reduced by ~ 50-fold in the E218A variant (16). Thus, the lack of activity of the path IV double mutants is not surprising. No conclusions can be drawn regarding energetic coupling between E218A and N305A since the combined ΔΔG (5 kcal/mol) for the two single mutants is beyond the detection limit of this assay and no activity was detected for the double mutant.
Table 3.
Kinetic parameters for amino acid activation by WT and mutant variants of Ec ProRSa
| Ec ProRS | kcat (s−1) | KM (mM) | kcat/KM (mM−1 sec−1) | kcat/KM (relative) | Fold-Decrease | ΔΔG (kcal/mol) |
|---|---|---|---|---|---|---|
| WT | 12.6 ± 4.9 | 0.18 ± 0.03 | 71 | 1.0 | - | - |
| D198A | 6.98 ± 0.37 | 0.33 ± 0.01 | 21 | 0.30 | 3.4 | 0.75 |
| E218Ab | 4.4 ± 2.3 | 3.40 ± 0.68 | 1.3 | 0.02 | 55 | 2.5 |
| E234A | 6.7 ± 1.9 | 1.03 ± 0.25 | 6.5 | 0.09 | 11 | 1.5 |
| H302A | 7.3 ± 1.9 | 0.22 ± 0.04 | 33 | 0.46 | 2.1 | 0.46 |
| N305A | 0.61 ± 0.18 | 0.45 ± 0.18 | 1.4 | 2.0 × 10−2 | 51 | 2.4 |
| G412A | 12.8 ± 0.57 | 0.300 ± 0.003 | 43.0 | 0.61 | 1.6 | 0.29 |
| F415A | 0.131 ± 0.010 | 0.76 ± 0.29 | 0.17 | 2.4 × 10−3 | 400 | 3.7 |
| H302A/G412A | 10.7 ± 0.80 | 0.62 ± 0.26 | 17 | 0.24 | 4. 2 | 0.88 |
| N305A/G412A | ND | ND | ND | - | - | - |
| E218A/N305A | ND | ND | ND | - | - | - |
Results are the average of 3 trials with the standard deviation indicated. ΔΔG was calculated according to the equation ΔΔG = −RT ln (fold-decrease of kcat/KM), where R is the gas constant, 1.986 cal K−1 mol−1 and T is 310 K. ND indicates not detectable under the experimental conditions used.
Data is from reference (16).
Aminoacylation
The aminoacylation activity of single and double mutants was examined by steady-state kinetic assays under conditions where synthesis of Pro-tRNAPro was linear with time and the initial rate of reaction was proportional to kcat/KM. The aminoacylation activity of F415A and N305A variants was nearly abolished with rates 70-fold slower than WT (Fig. 5a). The overall aminoacylation activity was also slightly reduced for the H302A and E234A mutant variants (2-fold) and the D198A (5.5-fold) mutant variant (Fig. 5a and b). Interestingly, mutation of G412 to alanine resulted in significantly reduced aminoacylation capability for both the single (G412A – 7-fold) and double (H302A/G412A – 5.5-fold) mutants (Fig. 5b); although only ~ 2- to 4-fold reduction in proline activation efficiency was observed for these variants, which were weakly coupled (Table 3). When ΔΔG is calculated for the aminoacylation reaction (Table 4), strong coupling is observed between H302 and G412 since the ΔΔG for the double mutant H302A/G412A (1.0 kcal/mol) is significantly less (sub-additive) than the sum of the two single mutants H302A and G412A (ΔΔG = 0.4 kcal/mol + 1.2 kcal/mol = 1.6 kcal/mol). Aminoacylation activity was not detected for the double mutants N305A/G412A and E218A/N305A (Fig. 5). Based on the detection limit of the aminoacylation assays, no conclusions can be drawn regarding the coupling of these residues. Taken together, these observations suggest that mutation of putative pathway residues has distinct impacts on the two steps of the tRNA aminoacylation reaction.
Figure 5.
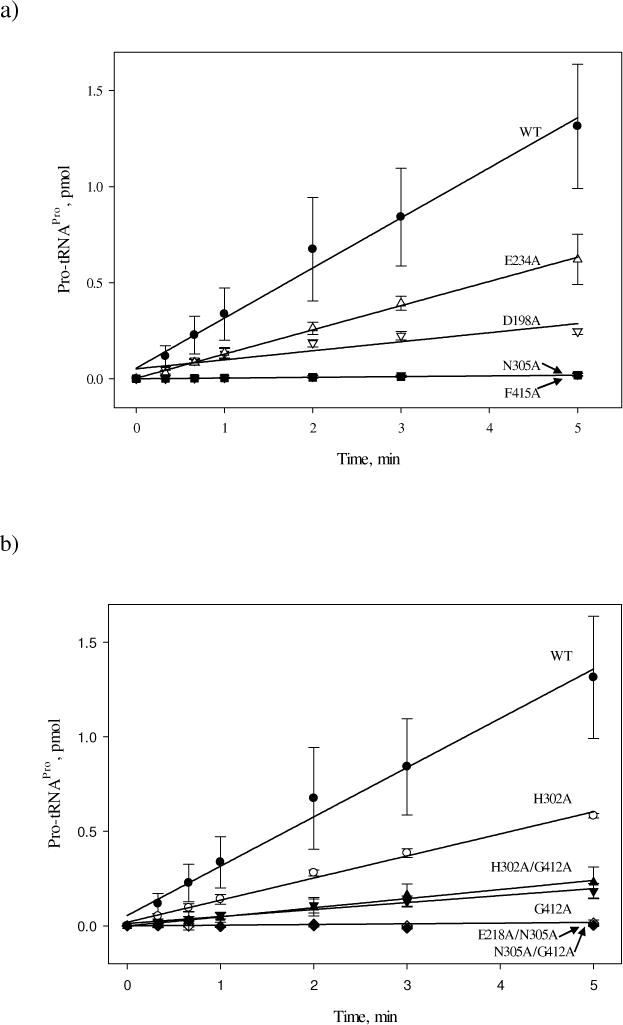
Initial rates of aminoacylation of tRNAPro with proline by WT and single and double point variants of Ec ProRS. For clarity, the results are presented in two panels, (a) and (b). The assays were performed at room temperature with 0.5 μM tRNAPro and 100 nM Ec ProRS. Linear fits of the data are shown.
Table 4.
Kinetic parameters for aminoacylation by WT and mutant variants of Ec ProRSa
| Ec ProRS | kcat/KM (relative) | Fold-Decrease | ΔΔG (kcal/mol) |
|---|---|---|---|
| WT | 1 | 1 | - |
| D198A | 0.18 | 5.5 | 1.0 |
| E218Ac | 0.71 | 1.4 | 0.20 |
| E234A | 0.48 | 2.1 | 0.43 |
| H302A | 0.45 | 2.2 | 0.46 |
| N305A | 0.014 | 70 | 2.5 |
| G412A | 0.14 | 7.1 | 1.2 |
| F415A | 0.014 | 70 | 2.5 |
| H302A/G412A | 0.18 | 5.5 | 1.0 |
| N305A/G412A | ND | - | - |
| E218A/N305A | ND | - | - |
Aminoacylation assays were performed under conditions where the initial rates were proportional to RNA concentrations. This indicated that V0/[S] was an accurate reflection of kcat/KM. The relative kcat/KM values were normalized and set to 1 for WT. ΔΔG was calculated according to the equation ΔΔG = −RT ln (fold-decrease of kcat/KM), where R is the gas constant, 1.986 cal K−1 mol−1 and T is 298 K. ND indicates not detectable under the experimental conditions used.
Data is from reference (16).
Pre-transfer Editing
The pre-transfer editing reaction was studied by monitoring enhanced hydrolysis of ATP in the presence of alanine (13) relative to that observed in the absence of amino acid. Reduced pre-transfer editing activity was observed for all mutants except G412A, which demonstrated similar levels of pre-transfer editing as WT ProRS (Fig. 6). This result may be due to the variant’s capability to activate alanine slightly more efficiently than the WT enzyme (data not shown).
Figure 6.
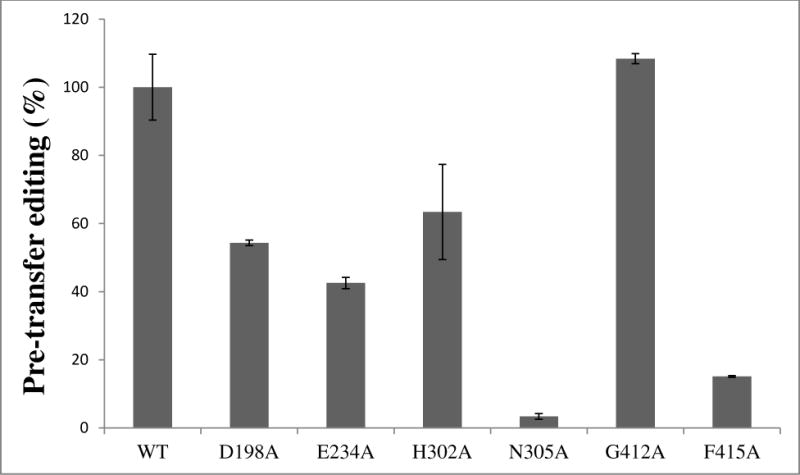
Relative alanine pre-transfer editing activity of WT and mutant variants of Ec ProRS. The assay was performed at 37 °C using 4 μM ProRS and 500 mM alanine. Results are reported as percent activity relative to WT, which was set to 100%.
Post-transfer Editing
The impact of mutations on Ala-tRNAPro hydrolysis was also studied (Fig. 7). A significant reduction in post-transfer activity was observed for the N305A mutant (~4-fold reduced relative to WT). This residue resides at the interface between the INS and activation domains, far from the editing active-site. Most of the other mutant ProRS variants tested (D198A, E234A, H302A, G412A, and E218A/N305A) displayed editing activity that was similar to that of WT ProRS (data not shown). The H302A/G412A and N305A/G412A double mutants exhibited a small (~3-fold and ~2-fold, respectively) reduction in activity. Interestingly, the F415A mutant, which was severely defective in proline activation and aminoacylation, hydrolyzed mischarged Ala-tRNAPro at a rate nearly 9-fold greater than that of WT enzyme (Fig. 7).
Figure 7.
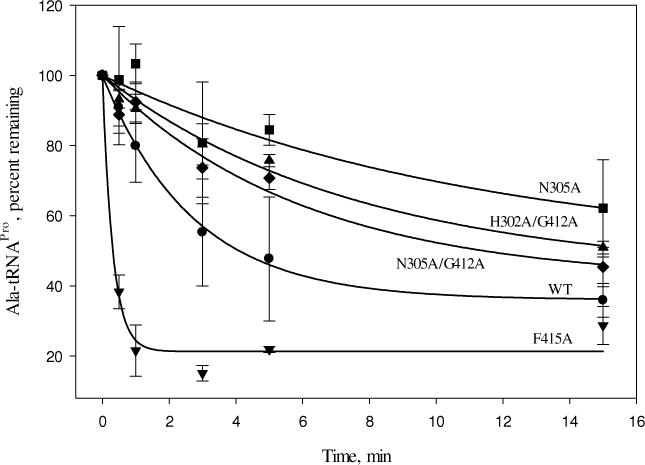
Deacylation of Ala-tRNAPro by WT and mutant variants of Ec ProRS. The assays were performed at room temperature with 1 μM G1:C72/U70 [14C]Ala-tRNAPro and 0.5 μM Ec ProRS.
Protein Flexibility and PBL Dynamics
To examine if the alteration in enzyme activity observed for the ProRS variants correlates with a change in protein dynamics caused by mutation of on-pathway residues, we analyzed the backbone flexibility and collective dynamics of these systems. This was accomplished by using MD simulations of the four selective substrate-free mutants, namely, E234A, N305A, G412A, and F415A, for which significant change in catalysis was observed. In each case, three replicates of 25 ns MD simulations were carried out. To assess if each system has reached an equilibrated state, the RMSD of each frame of the MD simulation trajectory was computed from their respective initial coordinates for all the four mutants (Fig. 2). The RMSD values were observed to fluctuate within 2.0 Å during the production period (final 20 ns) of the simulations indicating stability for each system. Also, the quality of simulations were assessed by computing the RMS fluctuation of each amino acid from the time-averaged structure using the last 20 ns MD simulation data. The RMS fluctuations for each replica, as well as the replica-averaged fluctuations of the WT and the four ProRS variants, are reported in Fig. 8. Analysis of these RMS fluctuation data indicates that the backbone flexibilities are quite reproducible for each of these protein systems, with only a propagated uncertainty of 0.2–0.3 Å for the three replica simulations (Fig. 8). These results indicate that all simulations have reached equilibrated states.
Figure 8.
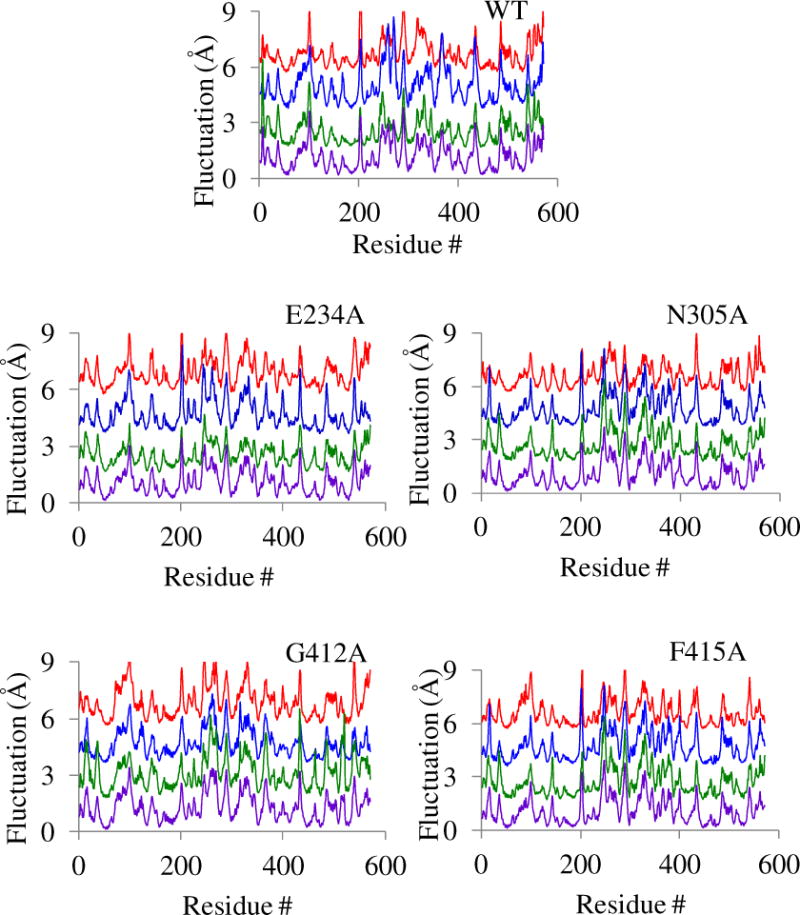
Root-mean-square fluctuations of individual amino acids for WT ProRS and four variants. In each stacked plot, the RMS fluctuations of Cα atoms calculated from the time-averaged structures over the last 20 ns MD trajectories are shown. Fluctuations for the three replicas are separated by 2 Å and color coded for clarity; replica 1 (red), replica 2 (blue), and replica 3 (green). In each case the bottom plot (purple) represents the replica-averaged RMS fluctuations. The calculated propagated uncertainties are 0.3 Å for WT, 0.2 Å for E234A, 0.2 Å for N305A, 0.3 Å for G412A, and 0.3 Å for F415A.
Next, we probed the impact of these mutations on the collective dynamics. For each system, PCA was carried out using the combined 60 ns trajectory (last 20 ns of three replicates) and the first three clusters representing the predominant collective dynamics were extracted. RMS fluctuations of Cα atoms were computed from their respective average structures, normalized, and averaged over the three clusters. The impact of mutation of on-pathway residues on the protein flexibility was examined by computing the difference of these cluster-averaged RMS fluctuations between the WT and a specific mutant (Fig. 9). Analysis of the results indicates that the flexibility of the protein backbone was altered by varied extents due to alanine substitution at these sites. The overall flexibility of the catalytic domain residues including the PBL was found to be considerably impacted by the alanine substitution of N305 (Fig. 9), which is located in the editing domain and has been observed to have significantly reduced proline activation and aminoacylation activities. The fluctuations of the editing domain residues were also impacted in all four mutants, with the most significant changes observed for N305A, G412A, and F415A. A noticeable alteration in editing activity was also observed for these mutants indicating that coupled thermal motions are important for Ec ProRS function.
Figure 9.
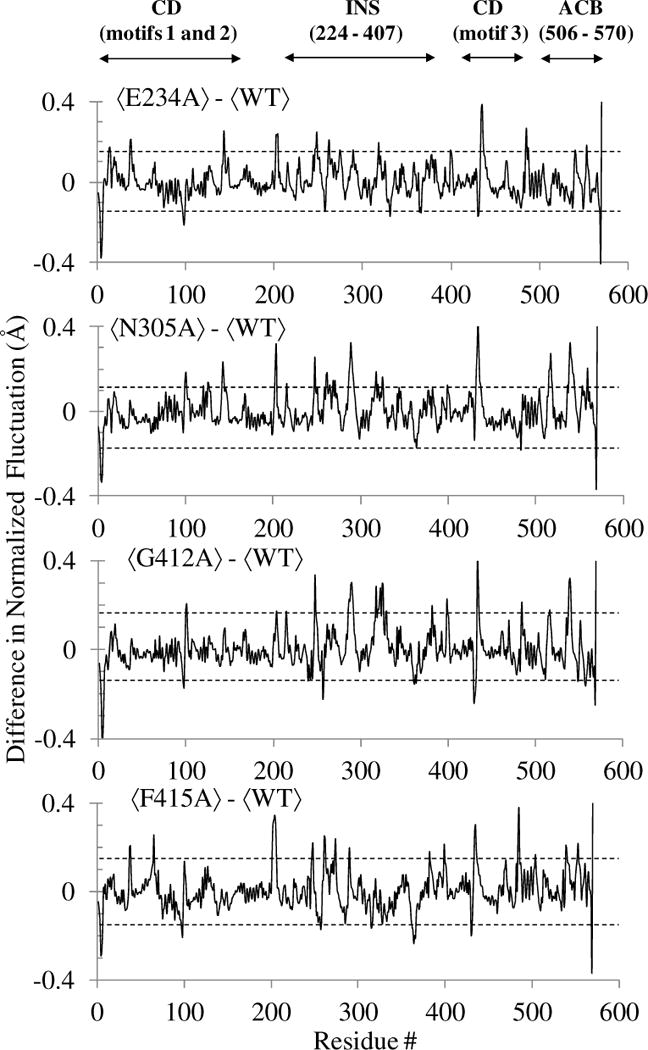
Change in the normalized RMS fluctuations of Cα atoms observed in the collective dynamics of the four mutants with respect to WT Ec ProRS. The angular bracket indicates that the RMS fluctuations are averaged over the first three clusters representing the predominant collective protein motions. The propagated uncertainties for each of these plots are within 0.15 Å and are shown with two parallel dotted lines. The abbreviations used are: CD, catalytic (aminoacylation) domain; INS, insertion domain; and ACB, anticodon binding domain.
The PBL is critical for substrate binding and catalysis. Therefore, in order to examine the impact of these point mutations on the collective dynamics of the PBL, PCA of WT Ec ProRS and the same four Ec ProRS variants (F415A, G412A, E234A, and N305A) was carried out using the last 20 ns MD simulation data for the Cα atoms of the PBL. The root-mean-square-projections (RMSP) (16) of the PBL for the WT and mutant proteins are shown in Fig. 10. In the present study, the RMSP (averaged over three replicate simulations) represents the collective displacement of the loop along the principal components 1 to 10 (i.e. PC1 to PC10), which are Cα eigenvectors. The PCA shows that each mutation had an impact on the collective dynamics of the PBL. The displacement of the PBL along PC1 is significantly altered for all the mutant proteins compared to the WT enzyme. However, the error in PBL displacement along PC1 for E234A is large, making it difficult to ascertain the precise effect of this mutation on the PBL dynamics and further studies are required. Noticeable changes in PBL displacement were also observed for PC2 and PC3. Therefore, this analysis indicates that mutation of the pathway residues could impact PBL dynamics and potentially alter substrate binding.
Figure 10.
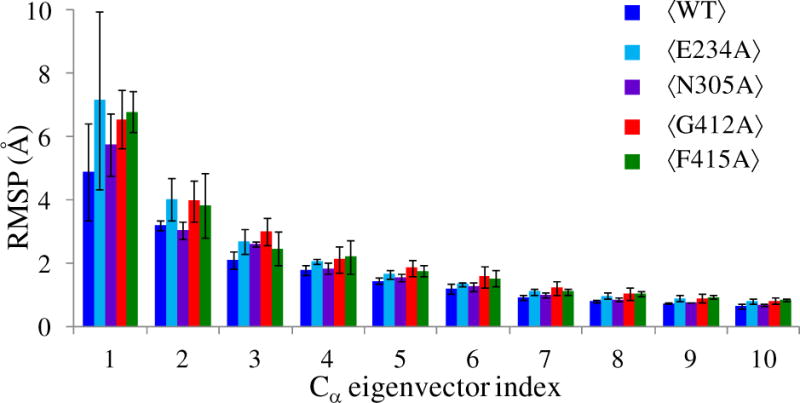
Analysis of root-mean-square projections (RMSP) of the PBL Cα atoms for WT ProRS and the four variant proteins. The angular bracket indicates that the RMSP values are averaged over the three replica simulations. These average RMSPs and standard deviations for eigenvectors 1 – 10 are shown.
DISCUSSION
Protein Dynamics and Site-to-Site Communication
A protein’s internal dynamics is critical for many important biochemical processes ranging from catalysis to allostery. Various studies have demonstrated that coupling of dynamics between domains is a prerequisite to the coordination of biological events occurring in distant sites (17–20, 46–48). It has been reported that correlated backbone motion provides pathways for transfer of structural/dynamics information (36). Alternatively, long-range intra-protein communication can be transmitted by correlated side-chain fluctuations (35). These studies suggest that both local interactions, as well as global dynamics are critical for long-range communication. Moreover, evolutionary studies on myosin motor protein and other proteins systems including G protein-coupled receptors and hemoglobin have demonstrated that conserved residues are key contributors to allosteric communication (21, 22, 49). Recently, nuclear magnetic resonance experiments have provided information on the dynamic processes through which the KIX domain of CREB binding protein communicates allosteric information (50). This study also revealed that the information is transmitted through an evolutionarily conserved network of residues. Similarly, we showed that coupled domain motions are mediated by conserved and evolutionarily coupled residues in Tt LeuRS (20). Recent experimental (42, 51) and computational (20, 44, 52) studies on other AARS systems have also revealed the existence of residue-residue interaction networks that promote inter-domain communication.
An earlier study using principal component analysis on the trajectory of MD simulations of prokaryotic-like ProRSs demonstrated that the INS is engaged in coupled motion with various structural elements of the aminoacylation domain including the PBL (16). The collective dynamics of the PBL was found to be altered by the deletion of the INS or point mutation at the INS-aminoacylation domain junction (vide infra)(16). Because the PBL undergoes a large-scale conformational transition upon substrate binding (15), we sought to understand the molecular mechanism by which the PBL dynamics is modulated by distant protein segments. The STCA method was specifically applied to identify pre-existing residue-residue interactions between the INS and aminoacylation domains of Ec ProRS that could modulate PBL dynamics. Four probable pathways of communication between the aminoacylation domain (C443/R450) and INS (K279) (Table 2, Fig. 4) were identified. Residues involved in these pathways were found to be predominantly conserved and polar (Table 2). In addition, the majority of pathway residues are non-catalytic, suggesting that these residues are conserved in the ProRS family to maintain functional dynamics.
Enzymatic Function and Mutation
The impact of alanine substitution of pathway residues on amino acid activation is significant (Table 3). Earlier studies showed that mutation of two separate pathway residues, E218 (path IV) and C443 (paths I and II), resulted in significantly lower activation efficiencies compared to WT ProRS (16, 38). Interestingly, mutation of residues E234 and N305, which are located farther away from the aminoacylation active site, also impacts amino acid activation. For example, a 50-fold decrease in amino acid activation efficiency was observed for the N305A variant compared to the WT enzyme. In general, the mutations that resulted in decreased amino acid activation also resulted in similar reductions in aminoacylation, as well as in pre- and post-transfer editing activities, which strongly suggests that the predicted pathways of inter-domain communication are important for substrate binding and catalysis. In one case, a catalytic domain mutant (F415A) severely defective in amino acid activation, aminoacylation and pre-transfer editing, which are all believed to occur in the aminoacylation active site, was more active than WT ProRS in post-transfer editing, an activity that resides in the INS domain. This suggests that F415 is coupled to the INS. Moreover, mutations of residues at the INS-aminoacylation domain interface (N305A and E234A) have greater impact on overall enzyme function, consistent with inter-domain communication occurring between the INS and aminoacylation domain through their interface region.
Amino acid activation results for the double mutant N305A/G412A (ΔΔG > 3.7, based on level of detection) also support coupling between pathway residues even though they are located in distant domains. This mutant is totally inactive in amino acid activation, an effect that is more severe than predicted based on the single mutations (ΔΔG = 2.5 + 0.31 = 2.81 kcal/mol). Moreover, aminoacylation activity of this double mutant was completely abolished. Note that this double mutant is active in hydrolyzing Ala-tRNAPro; only a 2-fold reduction in post-transfer editing activity was observed. The present study also revealed cross-talk between the predicted pathways. Free energy analysis for the double mutant H302A (path I)/G412A (path III) for the aminoacylation reaction indicates sub-additivity suggesting coupling between H302 and G412, which implies communication between pathways. Close scrutiny of the predicted pathways showed that they are intertwined through residue E234, a component of all four of the predicted pathways. This observation suggests that E234, which is located in a bottleneck position of the beta-strand connecting the catalytic and the INS domains, may serve as a key hinge in site-to-site communication.
Coupled-Dynamics between the Two Functional Sites
If the identified residue networks facilitate coupled-domain dynamics, then a mutation along these predicted paths is expected to have an observable impact on the protein dynamics. This hypothesis was supported by the RMS fluctuation analysis of the WT and mutant ProRS variants, which demonstrates that alanine substitution of on-pathway residues have considerable impact on the flexibility of various domains of Ec ProRS (Fig. 9). In addition, the collective dynamics of the PBL residues were also impacted by these mutations (Fig. 10). Thus, noncatalytic residues present in the communication pathways are indeed important for propagating substrate-induced conformational transitions from the active site pocket to the catalytically important PBL. Taken together, the in silico mutational study supports the role of the predicted interaction networks in maintaining distant coupled domain dynamics.
In addition, analysis of cross-correlation coefficients between adjacent pairs of residues in each pathway revealed an interesting pattern of dynamic correlations (Fig. 11). Adjacent residues in a pathway are primarily engaged in correlated motion, however, two distinct clusters of residues are observed. Residues within each cluster are engaged in strong correlated motion about the hinge residue (E234) between the INS and the aminoacylation domain. This observation is significant as it portrays a unique feature of coupled-domain dynamics in Ec ProRS; although correlated motions exist within each cluster, the coupling of these motions ultimately results in anti-correlated dynamics between the two domains (Fig. S1). The existence of anticorrelated motion between the INS and the aminoacylation domain apparently modulates the PBL conformational transition from “open” to “closed” state that is required for substrate binding and for protecting the Pro-AMP from hydrolysis.
Figure 11.
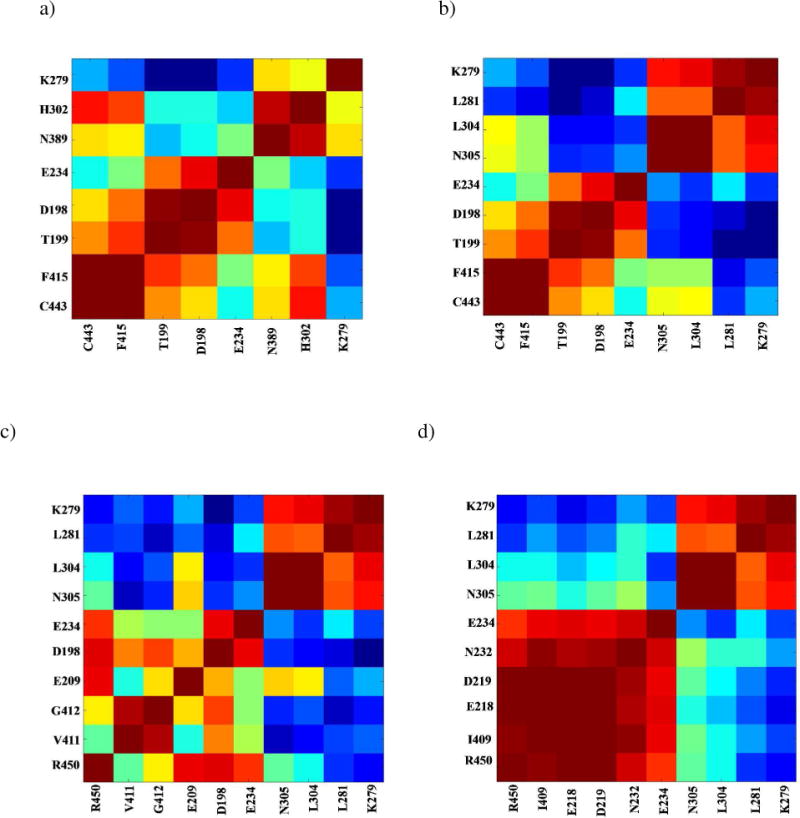
Coupling of thermal motions between residues in various pathways. Cross-correlations in fluctuation between residue pairs in a) path I, b) path II, c) path III, and d) path IV, extracted from the C matrix of the MD simulation. A strongly correlated motion between residues is colored red, whereas strongly anti-correlated motions are colored blue.
Proteins are dynamical in nature; mutation of any residue will have some impact on protein dynamics and may affect its function. Although mutation of catalytic residues is expected to impact an enzyme’s function, in the present study, we found that site-directed changes of noncatalytic residues that are part of key communication pathways also affect enzyme function. Most of these residues in the predicted pathways not only impact the function of the domain in which they are located, but also affect the distant domain, with which they are dynamically coupled. For example, alanine substitution of F415 of the aminoacylation domain has a significant impact on the editing reaction. Similarly, N305A (INS domain residue) exhibited significantly reduced efficiencies in proline activation and aminoacylation of tRNAPro. On the other hand, residues not belonging to the predicted communication pathways, are expected to have little impact on the function of a distant domain. Previous experimental results showed that alanine substitution of several residues of the INS domain (viz. T257, H369, and D386), resulted in only a minor change in the aminoacylation activity (≤ 2.5-fold decrease) and proline activation remained unaffected by these mutations (7). These three residues are not members of the predicted pathways. Analysis of the dynamic cross-correlation matrix indeed revealed low average correlation coefficients (−0.15 to −0.29) of these residues with respect to residues in the predicted pathways. In summary, coupled motions between INS and the aminoacylation domain are maintained by some defined residue-residue interaction networks and play important roles in both aminoacylation and editing functions.
CONCLUSIONS
There are two proposed models (53) for long-range site-to-site communication - the “induced-fit” model where a substrate-induced conformational change is propagated through a single pathway of residue-residue interactions, and the “population-shift” model according to which a perturbation at a distant site alters the conformational equilibrium through multiple “pre-existing” pathways of residue-residue interactions. Separate experimental and computational studies have suggested that allosteric signal propagation is mediated by a network of coupled residues and could be regulated by enthalpic (conformational) and/or entropic (dynamic) changes (53–57). In the present study, an effort was made to identify the pre-existing network of residue-residue interactions that could facilitate substrate induced conformational changes from one site to another in Ec ProRS through correlated motion.
It is well established that the structural and dynamic information of a protein is encoded in its primary sequence. Therefore, the substrate-free structure can be used to extract information regarding dynamically coupled residues. Moreover, amino acid residues that are important for structure/function/dynamics should be either conserved or have co-evolved resulting in a functionally more efficient enzyme. Therefore, integration of evolutionary and dynamic information is essential for identifying residues important for maintaining coupled dynamics and thereby, function of proteins. The STCA method takes into account both dynamic and evolutionary information in predicting pre-existing pathways of inter-domain communication in multi-domain proteins like Ec ProRS.
Results from the STCA analysis suggest that Ec ProRS employs multiple pathways of communication between the INS and aminoacylation domains to modulate the catalytically important PBL dynamics. A significant number of residues participating in inter-domain communication are evolutionarily conserved. Altering these conserved residues has a considerable impact on enzyme function. Moreover, in silico mutation followed by principal component analyses suggested that residues on the predicted pathways of communication alter protein dynamics. Taken together, the present study has identified residues that are likely to be involved in maintaining the coupled dynamics between functional domains of Ec ProRS. Moreover, this study supports the notion that to facilitate long-range site-to-site communication, multi-domain proteins like Ec ProRS use parallel paths of residue-residue interactions. Additional mutational studies are underway to explore the relative importance of these pathways.
Supplementary Material
Acknowledgments
Funding
This work was supported in part by XSEDE [grant numbers CHE110018 (S.B.) and MCB110173 (S.H.)] and National Institute of Health [grant numbers GM049928 (K.M.-F.) and GM085779 (S.H.)] and by the Office of Research and Sponsored Programs of the University of Wisconsin-Eau Claire, Eau Claire, WI.
Abbreviations used
- AARS
aminoacyl-tRNA synthetase
- Ec
Escherichia coli
- INS
insertion domain
- MD
molecular dynamics
- PBL
proline-binding loop
- PCA
principal component analysis
- Pro-AMP
prolyl-adenylate
- ProRS
prolyl-tRNA synthetase
- RMS
root-mean-square
- RMSD
root-mean-square deviation
- RMSP
root-mean-square projection
- SCA
statistical coupling analysis
- STCA
statistical-thermal coupling analysis
- Tt LeuRS
Thermus thermophilus leucyl-tRNA synthetase
- WT
wild-type
Footnotes
SUPPORTING MATERIALS
Dynamic cross-correlation matrix of WT Ec ProRS obtained from the PCA-based cluster analysis, a normalized plot of vs. residue number obtained from the statistical coupling analysis, and a table containing the list of primers used in this study are presented as supporting materials. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cusack S, Yaremchuk A, Krikliviy I, Tukalo M. tRNA(Pro) anticodon recognition by Thermus thermophilus prolyl-tRNA synthetase. Structure. 1998;6:101–108. doi: 10.1016/s0969-2126(98)00011-2. [DOI] [PubMed] [Google Scholar]
- 2.Stehlin C, Burke B, Yang F, Liu H, Shiba K, Musier-Forsyth K. Species-specific differences in the operational RNA code for aminoacylation of tRNAPro. Biochemistry. 1998;37:8605–8613. doi: 10.1021/bi980364s. [DOI] [PubMed] [Google Scholar]
- 3.Musier-Forsyth K, Burke B, Cusack S. Prolyl-tRNA synthetases. Georgetown, Texas, U.S.A.: 2005. Landes Biosciences/ Eurekah.com. [Google Scholar]
- 4.Ahel I, Stathopoulos C, Ambrogelly A, Sauerwald A, Toogood H, Hartsch T, Soll D. Cysteine activation is an inherent in vitro property of prolyl-tRNA synthetases. J Biol Chem. 2002;277:34743–34748. doi: 10.1074/jbc.M206928200. [DOI] [PubMed] [Google Scholar]
- 5.Beuning PJ, Musier-Forsyth K. Hydrolytic editing by a class II aminoacyl-tRNA synthetase. Proc Natl Acad Sci USA. 2000;97:8916–8920. doi: 10.1073/pnas.97.16.8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mascarenhas A, Martinis SA, An S, Rosen AE, Musier-Forsyth K. Fidelity Mechanisms of the Aminoacyl-tRNA Synthetases. In: L RU, C K, editors. Protein Engineering. Springer Verlag; New York: 2009. pp. 153–200. [Google Scholar]
- 7.Wong FC, Beuning PJ, Nagan M, Shiba K, Musier-Forsyth K. Functional role of the prokaryotic proline-tRNA synthetase insertion domain in amino acid editing. Biochemistry. 2002;41:7108–7115. doi: 10.1021/bi012178j. [DOI] [PubMed] [Google Scholar]
- 8.Wong FC, Beuning PJ, Silvers C, Musier-Forsyth K. An isolated class II aminoacyl-tRNA synthetase insertion domain is functional in amino acid editing. J Biol Chem. 2003;278:52857–52864. doi: 10.1074/jbc.M309627200. [DOI] [PubMed] [Google Scholar]
- 9.An S, Musier-Forsyth K. Trans-editing of Cys-tRNAPro by Haemophilus influenzae YbaK protein. J Biol Chem. 2004;279:42359–42362. doi: 10.1074/jbc.C400304200. [DOI] [PubMed] [Google Scholar]
- 10.An S, Musier-Forsyth K. Cys-tRNA(Pro) editing by Haemophilus influenzae YbaK via a novel synthetase. YbaK.tRNA ternary complex. J Biol Chem. 2005;280:34465–34472. doi: 10.1074/jbc.M507550200. [DOI] [PubMed] [Google Scholar]
- 11.Ahel I, Korencic D, Ibba M, Soll D. Trans-editing of mischarged tRNAs. Proc Natl Acad Sci USA. 2003;100:15422–15427. doi: 10.1073/pnas.2136934100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.SternJohn J, Hati S, Siliciano PG, Musier-Forsyth K. Restoring species-specific posttransfer editing activity to a synthetase with a defunct editing domain. Proc Natl Acad Sci USA. 2007;104:2127–2132. doi: 10.1073/pnas.0611110104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hati S, Ziervogel B, Sternjohn J, Wong FC, Nagan MC, Rosen AE, Siliciano PG, Chihade JW, Musier-Forsyth K. Pre-transfer editing by class II prolyl-tRNA synthetase: role of aminoacylation active site in “selective release” of noncognate amino acids. J Biol Chem. 2006;281:27862–27872. doi: 10.1074/jbc.M605856200. [DOI] [PubMed] [Google Scholar]
- 14.Zhang CM, Hou YM. Domain-domain communication for tRNA aminoacylation: the importance of covalent connectivity. Biochemistry. 2005;44:7240–7249. doi: 10.1021/bi050285y. [DOI] [PubMed] [Google Scholar]
- 15.Crepin T, Yaremchuk A, Tukalo M, Cusack S. Structures of two bacterial prolyl-tRNA synthetases with and without a cis-editing domain. Structure. 2006;14:1511–1525. doi: 10.1016/j.str.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Sanford B, Cao BV, Johnson JM, Zimmerman K, Strom AM, Mueller RM, Bhattacharyya S, Musier-Forsyth K, Hati S. Role of coupled-dynamics in the catalytic activity of prokaryotic-like prolyl-tRNA synthetases. Biochemistry. 2012;51:2146–2156. doi: 10.1021/bi300097g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bu Z, Biehl R, Monkenbusch M, Richter D, Callaway DJ. Coupled protein domain motion in Taq polymerase revealed by neutron spin-echo spectroscopy. Proc Natl Acad Sci USA. 2005;102:17646–17651. doi: 10.1073/pnas.0503388102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu H, Ma L, Yang Y, Cui Q. Mechanochemical coupling in the myosin motor domain. II Analysis of critical residues. PLoS Comput Biol. 2007;3:e23. doi: 10.1371/journal.pcbi.0030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chennubhotla C, Yang Z, Bahar I. Coupling between global dynamics and signal transduction pathways: a mechanism of allostery for chaperonin GroEL. Mol Biosyst. 2008;4:287–292. doi: 10.1039/b717819k. [DOI] [PubMed] [Google Scholar]
- 20.Weimer KM, Shane BL, Brunetto M, Bhattacharyya S, Hati S. Evolutionary basis for the coupled-domain motions in Thermus thermophilus leucyl-tRNA synthetase. J Biol Chem. 2009;284:10088–10099. doi: 10.1074/jbc.M807361200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 22.Suel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 23.Dijkstra EW. A note on two problems in connexion with graphs. Numerische Mathematik. 1959;1:269–271. [Google Scholar]
- 24.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 25.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926. [Google Scholar]
- 26.MacKerell ADJ, Bashford D, Bellott M, Dunbrack RLJ, Evanseck JD, Field MJ, Fischer S, Gao J, Gou J, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WEI, Roux B, Schelenkrich M, Smith JC, Stote R, Straub J, Watanbe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. Allatom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 27.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy J, Laughton CA. Long-timescale molecular-dynamics simulations of the major urinary protein provide atomistic interpretations of the unusual thermodynamics of ligand binding. Biophys J. 2010;99:218–226. doi: 10.1016/j.bpj.2010.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Aalten DM, Amadei A, Linssen AB, Eijsink VG, Vriend G, Berendsen HJ. The essential dynamics of thermolysin: confirmation of the hinge-bending motion and comparison of simulations in vacuum and water. Proteins. 1995;22:45–54. doi: 10.1002/prot.340220107. [DOI] [PubMed] [Google Scholar]
- 30.Mueller RM, North MA, Yang C, Hati S, Bhattacharyya S. Interplay of flavin’s redox states and protein dynamics: an insight from QM/MM simulations of dihydronicotinamide riboside quinone oxidoreductase 2. J Phys Chem B. 2011;115:3632–3641. doi: 10.1021/jp1107922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amadei A, Linssen AB, Berendsen HJ. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 32.Glykos NM. Software news and updates. Carma: a molecular dynamics analysis program. J Comput Chem. 2006;27:1765–1768. doi: 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- 33.Hatley ME, Lockless SW, Gibson SK, Gilman AG, Ranganathan R. Allosteric determinants in guanine nucleotide-binding proteins. Proc Natl Acad Sci USA. 2003;100:14445–14450. doi: 10.1073/pnas.1835919100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dubay KH, Bothma JP, Geissler PL. Long-range intra-protein communication can be transmitted by correlated side-chain fluctuations alone. PLoS Comput Biol. 2011;7:e1002168. doi: 10.1371/journal.pcbi.1002168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fenwick RB, Esteban-Martin S, Salvatella X. Understanding biomolecular motion, recognition, and allostery by use of conformational ensembles. Eur Biophys J. 2011;40:1339–1355. doi: 10.1007/s00249-011-0754-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burke B, Lipman RS, Shiba K, Musier-Forsyth K, Hou YM. Divergent adaptation of tRNA recognition by Methanococcus jannaschii prolyl-tRNA synthetase. J Biol Chem. 2001;276:20286–20291. doi: 10.1074/jbc.m100456200. [DOI] [PubMed] [Google Scholar]
- 38.Stehlin C, Heacock DH, 2nd, Liu H, Musier-Forsyth K. Chemical modification and site-directed mutagenesis of the single cysteine in motif 3 of class II Escherichia coli prolyl-tRNA synthetase. Biochemistry. 1997;36:2932–2938. doi: 10.1021/bi962295s. [DOI] [PubMed] [Google Scholar]
- 39.Fersht AR, Ashford JS, Bruton CJ, Jakes R, Koch GL, Hartley BS. Active site titration and aminoacyl adenylate binding stoichiometry of aminoacyl-tRNA synthetases. Biochemistry. 1975;14:1–4. doi: 10.1021/bi00672a001. [DOI] [PubMed] [Google Scholar]
- 40.Liu H, Musier-Forsyth K. Escherichia coli proline tRNA synthetase is sensitive to changes in the core region of tRNA(Pro) Biochemistry. 1994;33:12708–12714. doi: 10.1021/bi00208a023. [DOI] [PubMed] [Google Scholar]
- 41.Heacock D, Forsyth CJ, Shiba K, Musier-Forsyth K. Bioorganic Chemistry. 1996;24:273–289. [Google Scholar]
- 42.Ghosh A, Sakaguchi R, Liu C, Vishveshwara S, Hou YM. Allosteric communication in cysteinyl tRNA synthetase: a network of direct and indirect readout. J Biol Chem. 2011;286:37721–37731. doi: 10.1074/jbc.M111.246702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Budiman ME, Knaggs MH, Fetrow JS, Alexander RW. Using molecular dynamics to map interaction networks in an aminoacyl-tRNA synthetase. Proteins. 2007;68:670–689. doi: 10.1002/prot.21426. [DOI] [PubMed] [Google Scholar]
- 44.Ghosh A, Vishveshwara S. A study of communication pathways in methionyl- tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc Natl Acad Sci USA. 2007;104:15711–15716. doi: 10.1073/pnas.0704459104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horovitz A. Double-mutant cycles: a powerful tool for analyzing protein structure and function. Fold Des. 1996;1:R121–126. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- 46.Daily MD, Gray JJ. Allosteric communication occurs via networks of tertiary and quaternary motions in proteins. PLoS Comput Biol. 2009;5:e1000293. doi: 10.1371/journal.pcbi.1000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fidelak J, Ferrer S, Oberlin M, Moras D, Dejaegere A, Stote RH. Dynamic correlation networks in human peroxisome proliferator-activated receptor-gamma nuclear receptor protein. Eur Biophys J. 2010;39:1503–1512. doi: 10.1007/s00249-010-0608-9. [DOI] [PubMed] [Google Scholar]
- 48.Zheng W, Liao JC, Brooks BR, Doniach S. Toward the mechanism of dynamical couplings and translocation in hepatitis C virus NS3 helicase using elastic network model. Proteins. 2007;67:886–896. doi: 10.1002/prot.21326. [DOI] [PubMed] [Google Scholar]
- 49.Tang S, Liao JC, Dunn AR, Altman RB, Spudich JA, Schmidt JP. Predicting allosteric communication in myosin via a pathway of conserved residues. J Mol Biol. 2007;373:1361–1373. doi: 10.1016/j.jmb.2007.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bruschweiler S, Schanda P, Kloiber K, Brutscher B, Kontaxis G, Konrat R, Tollinger M. Direct observation of the dynamic process underlying allosteric signal transmission. J Am Chem Soc. 2009;131:3063–3068. doi: 10.1021/ja809947w. [DOI] [PubMed] [Google Scholar]
- 51.Rodriguez-Hernandez A, Perona JJ. Heat maps for intramolecular communication in an RNP enzyme encoding glutamine. Structure. 19:386–396. doi: 10.1016/j.str.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 52.Bhattacharyya M, Vishveshwara S. Probing the allosteric mechanism in pyrrolysyl-tRNA synthetase using energy-weighted network formalism. Biochemistry. 2011;50:6225–6236. doi: 10.1021/bi200306u. [DOI] [PubMed] [Google Scholar]
- 53.del Sol A, Tsai CJ, Ma B, Nussinov R. The origin of allosteric functional modulation: multiple pre-existing pathways. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 55.Tsai CJ, del Sol A, Nussinov R. Allostery: absence of a change in shape does not imply that allostery is not at play. J Mol Biol. 2008;378:1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Daily MD, Gray JJ. Local motions in a benchmark of allosteric proteins. Proteins. 2007;67:385–399. doi: 10.1002/prot.21300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.