

Abstract
We have cloned a fusion partner of the MLL gene at 11q23 and identified it as the gene encoding the human formin-binding protein 17, FBP17. It maps to chromosome 9q34 centromeric to ABL. The gene fusion results from a complex chromosome rearrangement that was resolved by fluorescence in situ hybridization with various probes on chromosomes 9 and 11 as an ins(11;9)(q23;q34)inv(11)(q13q23). The rearrangement resulted in a 5′-MLL/FBP17-3′ fusion mRNA. We retrovirally transduced murine-myeloid progenitor cells with MLL/FBP17 to test its transforming ability. In contrast to MLL/ENL, MLL/ELL and other MLL-fusion genes, MLL/FBP17 did not give a positive readout in a serial replating assay. Therefore, we assume that additional cooperating genetic abnormalities might be needed to establish a full malignant phenotype. FBP17 consists of a C-terminal Src homology 3 domain and an N-terminal region that is homologous to the cell division cycle protein, cdc15, a regulator of the actin cytoskeleton in Schizosaccharomyces pombe. Both domains are separated by a consensus Rho-binding motif that has been identified in different Rho-interaction partners such as Rhotekin and Rhophilin. We evaluated whether FBP17 and members of the Rho family interact in vivo with a yeast two-hybrid assay. None of the various Rho proteins tested, however, interacted with FBP17. We screened a human kidney library and identified a sorting nexin, SNX2, as a protein interaction partner of FBP17. These data provide a link between the epidermal growth factor receptor pathway and an MLL fusion protein.
The chromosomal region 11q23 is one of the most promiscuous chromosomal sites. The gene MLL (also known as ALL-1, HRX, or HRTX) that is involved in these translocations was identified in 1991 (1). More than 40 different fusion partners have been identified on the cytogenetic level. Among those identified, the translocations t(4;11)(q21;q23), t(9;11)(p22;q23), and t(11;19)(q23;p13) are the most common, and their identification by cytogenetics, fluorescence in situ hybridization (FISH), or reverse transcriptase–PCR (RT-PCR) is an essential routine procedure in many therapy trials. To date, 26 such MLL fusion partners have been cloned, but the physiological role and function is known of only few of them (2, 3).
In acute myeloid leukemias (AML), the chromosomal region 9q may be affected by various types of aberration. The most common types are deletions and occur either alone or as part of complex karyotype changes (http://www.infobiogen.fr/services/chromcancer/Anomalies/del9q.html). In addition, three specific reciprocal translocations, t(9;22)(q34;q11), t(6;9)(p23;q34), and t(7;9)(q34;q34) affect the genes ABL, CAN, and TAN-1, respectively, that are located in the telomeric region 9q34 (4–6).
The recent case analysis of 550 persons with acute leukemias or or myelodysplastic syndromes and 11q23 abnormalities by the European Concerted Action Workshop has greatly extended our knowledge about their cytogenetic, biological, and clinical features. On behalf of the workshop participants, Harrison et al. (7) reported the identification of 10 previously uncharacterized chromosomal partner sites. Despite the large collection of cases and their intensive review by experienced cytogeneticists, no translocation with or suspicious rearrangement of 9q34 was detected.
Herein, we report the fusion of a gene that encodes the human formin-binding protein 17 (FBP17) to MLL at 11q23 in a 14-month-old child with AML. After the identification of a chimeric MLL fusion gene, we tested its transforming ability with the help of retroviral gene transfer. Finally, we determined the protein expression of the normal wild-type FBP17 in a variety of tissues and searched for its intracellular interaction partners by screening a human kidney library.
Materials and Methods
Case History.
AML-M4 was diagnosed in a 14-month-old boy with an infiltration of the gingiva. His WBC count was 3.5 × 1010 cells per liter with 7% blasts in the differential count. A bone marrow (BM) smear revealed 38% myeloblasts. His hemoglobin level was 123 g/liter, and his platelet count was 2.49 × 1011 cells per liter. He was treated according to a standard chemotherapy protocol and remains in remission 28 months after diagnosis.
Cytogenetic Analysis.
Unstimulated isolated BM and peripheral blood cells were cultured for 24–48 h. Chromosomes were prepared and G-banded according to standard procedures. Of 20 analyzed metaphases, 13 showed a normal karyotype and 7 were interpreted to harbor a paracentric inversion of the long arm of chromosome 11: 46, XY, inv(11)(q13q23).
Southern Blotting, FISH, Cell Culture, cDNA Transfection, Western Blotting, and Coimmunoprecipitation.
These techniques were performed according to standard protocols. The detailed description as well as the source of the FISH probes is published as supplemental data on the PNAS web site, www.pnas.org.
Rapid Amplification of cDNA Ends (RACE)-PCR, Bispecific PCR, and Nucleotide Sequencing.
We performed RACE-PCR with the Marathon cDNA amplification kit and nested PCR (CLONTECH). For the 3′ RACE-PCR, we used the two MLL-specific sense primers (MLL external, 5′-TCATCCCGCCTCAGCCACCTACTACAGGACCGC-3′; MLL internal 5′-KspI-CAAGAAAAGAAGTTCCCAAAACCACTCCTAGTGAGCC-3′) that were located in exon 5 of MLL (8). Cloning of the KspI- and NotI-digested PCR-products into a pBluescript II SK(+) vector (Stratagene) generated three clones that contained the 5′-MLL/FBP17–3′ fusion site. To confirm the presence of an MLL/FBP17 fusion transcript, we performed a bispecific RT-PCR with MLL- and FBP17-specific primers. The MLL primers described above were combined with two antisense FBP17-PCR primers (external 5′-ACTTCTTTGAAAGATTCCTGAGTTGC-3′, internal 5′-GCATAGCTGAGTTCAATCTCTGTCC-3′). Similarly, we tried to amplify the reciprocal FBP17/MLL fusion transcript with FBP17 sense primers (external 5′-GAGGAGCTGCGAGCCGGAGGAGG-3′, internal 5′-GCTTTCCGCCGTCGCGGGGATCTC-3′) that were combined with antisense MLL primers.
Isolation of the P1 Clone Containing FBP17.
The human P1 library was obtained from the Resource Center/Primary Database (RZPD at http://www.rzpd.de/). The 22 × 22 cm filters were hybridized with 330 ng of a labeled FBP17 cDNA that was generated by PCR. The FBP17 sense primer 5′-CCAGAGGAGAAGGCAAACCAGACC-3′ and the FBP17 antisense primer 5′-GCTGACTTCTGCTAATTTGTGATCC-3′ generated a 479-bp fragment. For probe labeling, the digoxygenin (DIG)-11dUTP was mixed with dTTP in the ratio of 1:1 during PCR cycling (94°C, 30 sec; 60°C, 45 sec; 72°C, 45 sec). Incorporation of DIG-11UTP was proved by dot-blot and agarose gel electrophoresis. The probe was dissolved in 13.2 ml of Easy Hyb (Roche, Mannheim, Germany) solution, achieving a final concentration of 25 ng/ml. The filters were hybridized for 16 h at 42°C. The detection was done at room temperature for 30 min with 13.2 μl of anti-DIG antibody that was dissolved in 132 ml of blocking buffer (Roche Diagnostics).
Immunohistochemistry.
Normal human tissues were obtained as biopsy specimens or less than 24 h after postmortem from the Department of Pathology, Justus Liebig University (Giessen, Germany). All individuals (n = 43) were between 4 weeks and 89 years old. The different tissues were either snap-frozen in liquid nitrogen, embedded in OCT Compound (Miles) and stored at −80°C until use or fixed in 4.5% (vol/vol) formalin, pH 7.0 for at least 24 h and then embedded in paraffin. The peptide used for the production of polyclonal antiserum (NH2-KQLESSKRRFERDC-COOH) corresponds to amino acids 194–207 of human FBP17. Synthesis of the peptide, immunization of rabbits, purification, and ELISA testing of the antiserum was commercially done by Eurogentec, Brussels. Preimmune serum of each rabbit was used as negative control.
Retroviral Gene Transfer and Methylcellulose Colony-Forming (CFC) Assay.
The MLL/FBP17 cDNA found in the patient was cloned in the pMSCV (CLONTECH) vector (9). For this purpose, the ENL portion of an MSCVneo-MLL/ENL plasmid that was originally used in the CFC assay (10) was replaced by FBP17 by using the SacI and XhoI recognition sites. The fusion product was sequenced to confirm the correct MLL/FBP17 reading frame. For comparison, we included the MLL/GRAF fusion gene that has recently been described (11) and two positive as well as two negative controls in the CFC assays. MLL/ENL and MLL, fused to the minimal transactivation domain of ENL (MLL/16), served as positive controls. MLL without a fusion partner and MLL fused to the KRAB repressor domain were used as negative controls (ref. 10 and R.S., unpublished data). The infection of lineage-depleted bone marrow from 5-fluorouracil-treated BS/BA mice and culture of the transduced progenitors in methylcellulose were exactly conducted as described by Lavau et al. (10) and Slany et al. (12).
Plasmid Constructs and Interaction Trap.
We used the commercially available Matchmaker LexA two-hybrid system (CLONTECH). The yeast strain EGY48[p8op-lacZ] (13), was cotransformed with the various Rho family members pB42-AD-cDNAs and pLEXA-FBP17 (GenBank accession no. AF265550). The following genes from the Rho family of small GTPases were amplified by PCR with cDNA from a healthy volunteer: RhoA (GenBank accession no. L25080), RhoB (NM_004040), RhoC (NM_005167), RhoG (NM_001665), cdc42 (NM_001791), TTF (NM_004310), Rac1 (NM_006908), and Rac2 (NM_002872). For primer sequences see Table 1, which is published as supplemental data. Cloning into pB42AD vector was done with the help of the recognition sites for EcoRI and XhoI (Roche Diagnostics) which were incorporated by the PCR primers used.
Results
Isolation of the MLL/FBP17 Fusion cDNA.
Southern blot and FISH analyses provided strong evidence that the cytogenetically detected inversion 11 disrupts the MLL gene (Figs. 1B and 2B). Therefore, we used rapid amplification of cDNA ends-PCR to search for the potential new MLL fusion partner. Sequence analysis revealed an exon substituting for MLL exon 8. These previously uncharacterized sequences, fused in-frame to MLL, were subsequently used to design specific antisense primers. These primers were combined with MLL Ex5 sense primers in a bispecific RT-PCR. The presence of the chimeric MLL-RNA in the patient's material was confirmed (Fig. 1C). Searches with blast indicated that we had cloned the human counterpart of the mouse fbp17. The mouse fbp17 is partially (231 aa) cloned and shows 90% homology to our predicted amino acid sequence. In addition, we found an identical human DNA sequence that was previously cloned from a human brain library and named KIAA0554 (GenBank accession no. AB011126). We complemented the KIAA0554 sequence on its 5′ end but failed in cloning the first ATG codon. In total, we recovered a 2,042-nt consensus sequence with an ORF of 679 aa (GenBank accession no. AF265550). The search for protein domains revealed a long region of homology to cdc15 (amino acids 96–290), a protein that serves as a key element for the organization of the cytoskeleton in S. pombe. At the extreme amino terminus of FBP17, we found a so-called FER-CIP4 homology domain that is present in the cdc42 interacting protein, a nonreceptor fps/fes related tyrosine-kinase, and other proteins that potentially regulate the small GTPase Rho (14). Moreover, amino acids 475–537 of the human FBP17 encode for a protein domain that was found in proteins like the serine/threonine kinase PKN, Rhophilin, and Rhotekin (15). At the carboxyl terminus (amino acids 612–669), FBP17 contains an Src homology 3 domain that is retained in the predicted MLL/FBP17 oncoprotein. The protein motifs of the predicted MLL/FBP17 fusion protein are depicted in Fig. 1A.
Figure 1.

(A) Schematic representation of the in-frame MLL/FBP17 gene fusion. In the predicted chimeric MLL/FBP17 fusion protein, the MLL zinc finger domain is disrupted and the MLL SET [Su(var) 3-9, Enhancer-of-zeste, Thritorax] domain is replaced by the FBP17 Src homology 3 domain. (B) Southern blot analysis revealed a rearrangement of the MLL gene in the index patient. Lane M, size standards; lane 1, the normal 8.3-kb MLL germ-line fragment (cell line HL60); lane 2, the rearranged MLL fragments of the leukemia sample. (C) Lane M, size marker VI (Roche Diagnostics). Lane 1, RT-PCR analysis with an MLL exon 5 sense primer and an FBP17 antisense primer detects a chimeric MLL/FBP17 transcript. The smaller faint PCR product is caused by alternative splicing of MLL exon 6. Lanes 2 and 3, negative controls. Lanes 4–6, sequence of the ABL gene amplified to ensure the integrity of the RNA from the patient and the two cell lines that were used for control (HL60 and THP1). (D) Western blot after transfection of 293T cells with chimeric MLL/FBP17 cDNA. Almost all MLL/FBP17 protein is localized into the nucleus (lane 3), whereas only a small amount is retained in the cytoplasm (lane 1). Lanes 2 and 4 are negative controls.
Figure 2.

Summary of cytogenetic findings and FISH analyses in the index patient. (A) Partial karyotype showing the chromosomes 9 and 11. In B–F, n = normal homologues, a = abnormal homologues. (B) FISH with 3′-MLL and 5′-MLL probes reveals that this gene is split. The 3′ portion remains at 11q23, whereas the 5′ portion is transferred to 11q13. (C) FISH with an MLL yeast artificial chromosome (YAC)and the FBP17 P1 artificial chromosome (PAC) shows that one part of the split MLL probe colocalizes with FBP17 at 11q13. The orientation of the MLL/FBP17 fusion could not be resolved. The fact that the splitting of the FBP17 probe is not seen indicates that either the remaining 5′ portion is too small for detection or that it is deleted. In any case, whether 5′-FBP17 is deleted or remains on chromosome 9q, there is no possibility of generating a reciprocal 5′-FB17/MLL-3′ fusion. Therefore, we were unable to amplify such a chimeric 5′-FBP17/MLL-3′ mRNA by bispecific RT-PCR. (D) FISH with the MLL YAC clone and C1943, a PAC clone that is located at 11q13, confirms the presence of a paracentric inv(11)(q13q23) and reveals that the second chromosome 11 breakpoint lies centromeric to C1943. (E) FISH with the FBP17 PAC and an ABL cosmid confirms that a break occurred between FBP17 and ABL and that ABL maps telomeric to FBP17. (F) FISH with the C1943 PAC and an 11q subtelomere probe confirms that the MLL/FBP17 colocalization on 11q13 results from an insertion and an inversion rather than from a translocation and an inversion, because the integrity of the telomeric region of chromosome 11 is maintained. In the schematic representation of the complex rearrangement that summarizes these findings (Lower Right), the short black arrows indicate breaks.
MLL/FBP17 Is Targeted to the Nucleus.
A prerequisite for a chimeric MLL oncoprotein to function as a transcription factor is its localization in the nucleus. We examined the distribution of MLL/FBP17 in the nucleus and cytoplasm, respectively. As shown in Fig. 1D, MLL/FBP17 is mainly found in the nucleus, despite the fact that FBP17 itself localizes in the cytoplasm exclusively (see also Fig. 3.).
Figure 3.

Distribution of the human FBP17 protein in paraffin-embedded tissue specimens. Cytoplasmatic immunoreactivity in squamous epithelia of the skin (A), ductal cells of the mammary gland (B), cardiomyocytes (D), Leydig cells of the testis (E), hepatocytes (F), gangliocytes of the myenteric plexus (G) and in glucagon-producing A cells within the islets of Langhans as detected by immunohistochemical single and doublestaining (FBP17 stains APAAP red) (I). Glucagon stains LSAB brown (K), red-brown stained product indicates strong FBP17 (red) expression in glucagon (brown) containing A cells (M). Weak FBP17 staining in vascular smooth muscle cells (C) and insulin-containing B cells (I, L, and M). Negative control (H). No staining of connective tissue (A, B, and G), endothelial cells (C, D, and F central vein), and testicular germ cells (E) (A—-M, ×40.)
Chromosomal Assignment of the FBP17 to 9(q34) and FISH Analysis of Patients with 9q34 Abnormality.
Screening of a human P1 library revealed a positive clone, ICRFP700L11103Q5. Rescreening of this particular clone for the presence of FBP17 was done by PCR and Southern blot analysis (data not shown). On normal metaphase chromosomes, this clone, as well as a single FBP17 cDNA, mapped to 9q34 (data not shown). Double-color FISH on samples from patients with chronic myeloid leukemia and a t(9;22)(q34;q11) revealed that FBP17 remains on 9q34. Thus, FBP17 maps centromeric to ABL (Fig. 2E and data not shown), as well as CAN and TAN-1, two other genes that are located distal to ABL and also participate in leukemia-specific translocations. Fig. 2 shows representative examples of the hybridization patterns that were obtained with various FISH probes in our index patient together with a scheme that summarizes the respective results. To look for additional patients with potential rearrangements of the FBP17 gene, we searched in our karyotype database for children with newly diagnosed AML (n = 708) and 9q34 aberrations (n = 10). One patient with a t(6;9)(p23;q34) and four rare cases of AML with t(9;22) and BCR/ABL rearrangement were excluded from further analysis. Material was available from four of the five remaining cases. In three of those four cases, the FBP17 probe mapped to 9q34, whereas in the fourth patient, a 5-year-old girl with an AML-M5 and a complex karyotype [46,XX,der(9)t(9;11)(p22;q23)?inv(9)(p22q34), der(11)t(9;11)-(p22;q23)], the 3′-MLL probe colocalized with FPB17 on 9q34. Because this patient was MLL/AF9 positive, this colocalization was rather a fortuitous by-product than a specific MLL/FBP17 fusion. We considered it to result from the pericentric inv(9)(p22q34) of the der(9)t(9;11) that transferred the 3′-MLL probe into the close vicinity of FBP17.
FBP17 Protein Expression in Human Tissues.
We tested the specificity of the polyclonal anti-FBP17 antiserum in frozen and in paraffin-embedded specimens of expressing and nonexpressing cell lines and tissues. Comparison of the immunostaining patterns of freshly fixed cryostat tissue sections with the respective formalin-fixed, paraffin-embedded material confirmed that the distribution and intensity of labeling was comparable in all cases analyzed. In total, more than 250 histological slides were analyzed by immunostaining. FBP17 was expressed exclusively within the cytoplasm of the cells. Epithelial cells from the respiratory system, gastrointestinal tract, urinary, and reproductive system especially showed a remarkably strong expression of FBP17. In addition, enteric ganglia, brown adipose tissue, cardiomyocytes, and glucagon-producing cells of the pancreas revealed a strong labeling with the anti-FBP17 antiserum (Fig. 3). With respect to hematopoietic tissues, FBP17 was seen mainly during erythropoiesis, whereas myelopoietic cells remained negative for FBP17 expression. The entire distribution patterns in normal human tissues are summarized in Table 2, which is published as supplemental data.
MLL/FBP17 Alone Has Only Low Transforming Capacity.
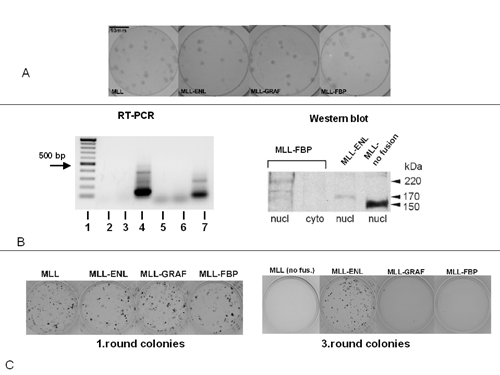
To study the potential oncogenic effect of MLL/FBP17, we retrovirally transduced BM cells whose growth properties were then evaluated in a serial replating assay. The transduced cells were plated in methylcellulose cultures supplemented with stem cell factor, IL-3, IL-6, and granulocyte/macrophage colony stimulating factor. A set of control experiments demonstrated the adequate expression of the various MLL fusion mRNAs and corresponding fusion proteins in the retroviral packaging cell line. These data and the titers obtained with the various retroviral constructs are summarized in Fig. 5 A–C, which is published as supplemental data. Every 7 days, the cells from the culture dish were rinsed from the methylcellulose and replated. In the first round of plating, numerous colonies were observed for cells transduced with truncated MLL, MLL/ENL, MLL/GRAF, MLL/FBP17 and even the empty pMSCV vector (see Fig. 5C). This fact reflects the normal clonogenic potential of the primitive myeloid progenitors. First-round colonies were then harvested and cells were replated in secondary and tertiary methylcellulose cultures. After three rounds of replating, we compared the growth of cells transduced with MLL/FBP17 to that of cells transduced with the control MSCVneo vector, truncated MLL alone, and MLL/KRAB (negative controls) or with MLL/ENL and MLL/16 that contain a fusion of MLL with the ENL transactivator domain (positive controls), respectively. As shown in Fig. 4A, the MLL/ENL- and MLL/16-transduced BM cells produced more than 100 colonies per 10,000 cells seeded, whereas only very few colonies were seen in the MLL/FBP17- and MLL/GRAF-transduced cells.
Figure 4.

(A) Results of the in vitro transformation/CFC test after three rounds of replating. Colonies generated per 10,000 input cells. Strong in vitro transformation activity of MLL/ENL and the fusion of MLL with the ENL transactivation domain (MLL/16). Only very few colonies are seen after retroviral transduction with MLL/FBP17. The MLL/GRAF fusion, generated by a recently described (11) translocation t(5;11)(q31;q23), was tested as well. A truncated form of MLL (no fusion) and a fusion of MLL to the KRAB repressor domain revealed no transforming activity. (B) Western blotting of yeast EGY48 cells with anti-RhoA-moAB and polyclonal anti-FBP17 antiserum. Lane 1, RhoA-fusion 44 kDa; lanes 2 and 3, negative controls with preimmunserum; lane M, molecular mass standard (Sigma); lane 4, FBP17-LexA 109 kDa. (C) Results of the two-hybrid screening. None of the Rho family members tested showed an interaction with FBP17. In addition, library screening revealed a specific interaction between SNX2 and FBP17 (blue-colored colonies). Neither FBP17 nor MLL/FBP17 interacted with mouse formin. Positive control shows the interaction between pLexA-p53 and pB42AD large T antigen. All colonies were plated on drop-out medium, without tryptophane, uracil, histidine, and leucine (D) Coimmunoprecipitation of FBP17 and SNX2. (Left) Lane 1, GFP-SNX2 after coprecipitation with myc-FBP17. Note the strong signal at 42 kDa. The detection was performed with an anti-myc antibody. The 42-kDa GFP-SNX2 protein was detected with an anti-GFP antibody by Western blotting (lane 4). In the control reaction, no coprecipitated SNX2 could be detected (lanes 2, 3, and 5). (Right) The myc-tagged FBP17 protein is expressed in similar amounts in the 293T cells cotransfected with GFP-SNX2 (lane 1) as well as in the negative control transfected only with myc-FBP17 (lane 2).
FBP17 Interacts with Sorting Nexin 2 (SNX2).
In light of the putative Rho-binding site located within FBP17, we applied the yeast two-hybrid methodology to look for an in vivo interaction between FBP17 and various members of the Rho family. To ensure that the proteins are properly expressed, we performed Western analysis of extracts from transformed yeast cells as shown for RhoA and FBP17 in Fig. 4B.
Neither the lacZ nor the Leu2 reporter genes were activated by any of the Rho members/pB42-AD cDNA, and FBP17/pLexA-BD cDNA. The tumor suppressor p53, a protein previously shown to interact with the large T antigen, was used as a positive control (Fig. 4C) and Lamin C was used as a negative control. We examined whether wild-type FBP17 or MLL/FBP17 bind to the mouse formin protein. By our two-hybrid assay, neither FBP17 nor MLL/FBP17 showed a specific interaction. Next, we tried to isolate the in vivo interaction partner of FBP17. These efforts were encouraged by the information that the interaction between formin and its binding proteins may not play a major physiological role. Rather, the proline-rich domains of the formins, molecular isoforms involved in murine limb and kidney development (16), can be considered as suitable tools to isolate the completely new class of proteins, the FBPs (M. Bedford, personal communication). Based on our own results of the strong FBP17 expression in the proximal tubulus of the kidney and the data provided by Nagase et al. (expression pattern of KIAA0554 in 13 different human tissues, http://www.kazusa.or.jp/huge/gfpage/KIAA0554), we decided to screen a human kidney library by the two-hybrid methodology. From a total of 3.5 × 106 transformants, we isolated four clones that were sequenced. Analysis by blast indicated that these clones contained the human SNX2 (GenBank accession no. NM_003100) in all four cases (Fig. 4C). To verify the two-hybrid interaction, the isolated library plasmid was retransformed with control baits, and cotransformants were streaked on selective drop-out medium.
Finally, we wanted to investigate whether the FBP17-SNX2 interaction would be persistent also in a natural mammalian cell environment. A green fluorescent protein (GFP) tag was attached to the amino terminus of SNX2 and similarly a myc tag was added to 3′-FBP17. 293T cells were transiently transfected with both GFP-SNX2 and myc-FBP17. Expression of full-length protein products was verified by Western blot analysis with anti-GFP and anti-myc antibodies. A Western analysis with a GFP epitope tag-specific antibody revealed a specific coprecipitation of GFP-SNX2 protein with immobilized myc-FBP17 (Fig. 4D).
Discussion
Formins are the protein products of the limb deformity (LD) locus and regulate the embryonic polarity in mouse (17). Mutations within the LD locus affect the patterning of distal limb structures and disrupt induction of metanephric kidneys (18). FBPs were originally identified as a group of proteins that have the capability to bind to the mouse LD proteins (19). Moreover, during an in vitro screening for possible other ligands of the FBPs, Bedford et al. (20) identified eight clones, one of which (clone 7) displayed structural features of the trithorax-like sequences, the Drosophila homologue of MLL. Herein, we show that the gene encoding FBP17 fuses in vivo to the truncated MLL gene. This gene fusion encodes a chimeric MLL/FBP17 protein that is, like other MLL-fusion proteins, targeted to the nucleus (21).
The serial replating assay in semisolid medium has increasingly become an important experimental system to examine the oncogenic potential of MLL fusion genes. Perhaps surprisingly, in this test the MLL/FBP17 fusion showed a barely detectable transforming capability. The MLL/FBP17- and MLL/GRAF-transduced BM cells did form few colonies in contrast to the negative controls. However, when compared with the colony number obtained with other MLL fusion genes, e.g., MLL/ENL, the significance of this marginal result is unclear (10, 22, 23). The observation that the various MLL partner genes confer a different transforming capacity to the respective MLL fusion protein that translates into varying colony numbers in the CFC test also has been made by Lavau et al (24). In these studies, a much lower number of colonies was found with MLL/ELL as compared with MLL/ENL. It is tempting to speculate that the incidence of the various MLL fusions in leukemias might indirectly reflect their individual transformation capacities. In those MLL fusions with a low propensity for transformation, additional genetic alterations might promote this process. Such potential factors may include mutations in the ras and p53 genes as well as dominant-negative Ikaros forms (25–28). However, the short latency periods of infant leukemias, in particular, suggest that the common MLL fusions encountered in these instances have a high intrinsic transformation capacity already (29).
FBP17 shares a large region of homology with the cell division cycle genes. Similar regions are present also in two other MLL fusion partners, namely MSF and hCDCrel (30, 31). Computer analysis suggests that FBP17 bears a consensus Rho-binding sequence and might, therefore, interact with this small GTPase domain. The fact that we failed to demonstrate a specific protein–protein interaction between FBP17 and members of the Rho family further underlines that such computerized sequence analysis should always be complemented by functional assays. The in vivo interaction between FBP17 and SNX2 described herein provides a link between an MLL fusion protein and the epidermal growth factor (EGF) receptor pathway. So far, six different SNXs in humans have been described and all of them share a 100-aa region of high homology (termed phox region; refs. 32 and 33). Generally, SNXs are involved in various aspects of protein trafficking, and the first member of the family, SNX1, was discovered with the two-hybrid method and a portion of the cytoplasmatic domain of the EGF receptor (EGFR) as bait (34). Interestingly, the interaction between EGFR and SNX1 leads to an enhanced degradation of EGFR and enables its transport to lysosomal organelles. The more generally expressed SNX2 coimmunoprecipitated with the receptor tyrosine kinases for EGF, platelet-derived growth factor, and insulin (32). Indirect evidence also links another MLL fusion partner, AF1p in the t(1;11)(p32;q23), to the EGFR pathway. The product of this gene is nearly 90% homologous to the murine EGFR-pathway substrate 15 (eps15). The eps15 is a cytoplasmatic target for phosphorylation by EGFR and has transforming activity by itself (35, 36).
To summarize, the FBP17 gene is fused to MLL, although the resulting chimeric MLL/FBP17 protein that is present in the nuclear compartment has only a low-transforming capacity. The normal FBP17 protein is widely expressed in a variety of tissues and interacts in yeast and mammalian cells with SNX2, a protein involved in the EGFR signal pathway. In the near future, both the evaluation of the functional consequences of the FBP17-SNX2 interaction in normal mammalian cells and the identification of the supposed additional cooperating genetic aberrations in leukemias with MLL/FBP17 fusions will be challenging issues.
Supplementary Material
Acknowledgments
This work was supported by Grant 10-1658-Bo 2 from the Mildred Scheel Stiftung for Cancer Research, Grant Bo1549/2-1 from the Deutsche Forschungsgemeinschaft, and by the Österreichische Kinderkrebshilfe.
Abbreviations
- FISH
fluorescence in situ hybridization
- RT-PCR
reverse transcriptase–PCR
- AML
acute myeloid leukemias
- FBP
formin binding protein
- BM
bone marrow
- CFC
colony-forming assay
- SNX
sorting nexin
- GFP
green fluorescent protein
- EGFR
epidermal growth factor receptor
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF265550).
References
- 1.Ziemin-van der Poel S, McCabe N R, Gill H J, Espinosa R, III, Patel Y, Harden A, Rubinelli P, Smith S D, LeBeau M M, Rowley J D, Diaz M O. Proc Natl Acad Sci USA. 1991;88:10735–10739. doi: 10.1073/pnas.88.23.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DiMartino J F, Cleary M L. Br J Haematol. 1999;106:614–626. doi: 10.1046/j.1365-2141.1999.01439.x. [DOI] [PubMed] [Google Scholar]
- 3.Rowley J D. Semin Hematol. 1999;36:59–72. [PubMed] [Google Scholar]
- 4.Rabbitts T H. Nature (London) 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 5.von Lindern M, Fornerod M, van Baal S, Jaegle M, de Wit T, Buijs A, Grosveld G. Mol Cell Biol. 1992;12:1687–1697. doi: 10.1128/mcb.12.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Klein A, Geurts van Kessel A H M, Grosveld G, Bartram C R, Hagemeijer A, Bootsma D, Spurr N K, Heisterkamp N, Groffen J, Stephenson J R. Nature (London) 1982;300:765–767. doi: 10.1038/300765a0. [DOI] [PubMed] [Google Scholar]
- 7.Harrison C J, Cuneo A, Clark R, Johansson B, Lafage-Pochitaloff M, Mugneret F, Moorman A V, Secker-Walker L M European 11q23 Workshop Participants. Leukemia. 1998;12:811–822. doi: 10.1038/sj.leu.2401017. [DOI] [PubMed] [Google Scholar]
- 8.Gu Y, Alder H, Nakamura T, Schichman S A, Prasad R, Canaani O, Saito H, Croce C M, Canaani E. Cancer Res. 1994;54:2327–2330. [PubMed] [Google Scholar]
- 9.Hawley R G, Lieu F H, Fong A Z, Hawley T S. Gene Ther. 1994;1:136–138. [PubMed] [Google Scholar]
- 10.Lavau C, Szilvassy S J, Slany R, Cleary M L. EMBO J. 1997;16:4226–4237. doi: 10.1093/emboj/16.14.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borkhardt A, Bojesen S, Haas O A, Fuchs U, Bartelheimer D, Loncarevic I, Bohle R M, Harbott J, Repp R, Jaeger U, et al. Proc Natl Acad Sci USA. 2000;97:9168–9173. doi: 10.1073/pnas.150079597. . (First Published July 25, 2000; 10.1073/pnas.150079597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slany R K, Lavau C, Cleary M L. Mol Cell Biol. 1998;18:122–129. doi: 10.1128/mcb.18.1.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Estojak J, Brent R, Golemis E A. Mol Cell Biol. 1995;15:5820–5829. doi: 10.1128/mcb.15.10.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aspenström P. Curr Biol. 1997;7:479–487. doi: 10.1016/s0960-9822(06)00219-3. [DOI] [PubMed] [Google Scholar]
- 15.Reid T, Furayashiki T, Ishizaki T, Watanabe G, Watanabe N, Fujisawa K, Morii N, Madaule P, Narumiya S. J Biol Chem. 1996;271:13556–13560. doi: 10.1074/jbc.271.23.13556. [DOI] [PubMed] [Google Scholar]
- 16.Woychik R P, Mass R L, Zeller R, Vogt T F, Leder P. Nature (London) 1990;346:850–853. doi: 10.1038/346850a0. [DOI] [PubMed] [Google Scholar]
- 17.Vogt T F, Jackson-Grusby L, Rush J, Leder P. Proc Natl Acad Sci USA. 1993;90:5554–5558. doi: 10.1073/pnas.90.12.5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mass R L, Zeller R, Woychik R P, Vogt T F, Leder P. Nature (London) 1990;346:853–855. doi: 10.1038/346853a0. [DOI] [PubMed] [Google Scholar]
- 19.Chan D C, Bedford M T, Leder P. EMBO J. 1996;15:1045–1054. [PMC free article] [PubMed] [Google Scholar]
- 20.Bedford M T, Chan D C, Leder P. EMBO J. 1997;16:2376–2383. doi: 10.1093/emboj/16.9.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joh T, Yamamoto K, Kagami Y, Kakuda H, Sato T, Yamamoto T, Takahashi T, Ueda R, Kaibuchi K, Seto M. Oncogene. 1997;15:1681–1687. doi: 10.1038/sj.onc.1201332. [DOI] [PubMed] [Google Scholar]
- 22.Lavau C, Du C C, Thirman M, Zeleznik-Le N. EMBO J. 2000;19:4655–4664. doi: 10.1093/emboj/19.17.4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DiMartino J F, Miller T, Ayton P, Landewe T, Hess J L, Cleary M L, Shilatifard A. Blood. 2000;96:3887–3893. [PubMed] [Google Scholar]
- 24.Lavau C, Luo R T, Du C C, Thirman M J. Proc Natl Acad Sci USA. 2000;97:10984–10989. doi: 10.1073/pnas.190167297. . (First Published September 19, 2000; 10.1073/pnas.190167297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahgoub N, Parker R I, Hosler M R, Close P, Winick N J, Masterson M, Shannon K M, Felix C. Genes Chromosomes Cancer. 1998;21:270–275. [PubMed] [Google Scholar]
- 26.Naoe T, Kubo K, Kiyoi H, Ohno R, Akao Y, Yoshida J, Kato K, Kojima S, Matsuyama T. Blood. 1993;82:2260–2261. [PubMed] [Google Scholar]
- 27.Sun L, Heerema N, Crotty L, Wu X, Navara C, Vassilev A, Sensel M, Reaman G H, Uckun F M. Proc Natl Acad Sci USA. 1999;96:680–685. doi: 10.1073/pnas.96.2.680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanza C, Gaidano G, Cimino G, Pastore C, Nomdedeu J, Volpe G, Vivenza C, Parvis G, Mazza U, Basso G, et al. Genes Chromosomes Cancer. 1996;15:48–53. doi: 10.1002/(SICI)1098-2264(199601)15:1<48::AID-GCC7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 29.Greaves M F. Lancet. 1997;349:344–349. doi: 10.1016/s0140-6736(96)09412-3. [DOI] [PubMed] [Google Scholar]
- 30.Osaka M, Rowley J D, Zeleznik-Le N J. Proc Natl Acad Sci USA. 1999;96:6428–6433. doi: 10.1073/pnas.96.11.6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Megonigal M D, Rappaport E F, Jones D H, Williams T M, Lovett B D, Kelly K M, Lerou P H, Moulton T, Budarf M L, Felix C A. Proc Natl Acad Sci USA. 1998;95:6413–6418. doi: 10.1073/pnas.95.11.6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haft C R, Sierra M D, Barr V A, Haft D H, Taylor S I. Mol Cell Biol. 1998;18:7278–7287. doi: 10.1128/mcb.18.12.7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haft C R, Sierra M L, Bafford R, Lesniak M A, Barr V R, Taylor S I. Mol Biol Cell. 2000;11:4105–4116. doi: 10.1091/mbc.11.12.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurten R C, Cadena D L, Gill G N. Science. 1996;272:1008–1010. doi: 10.1126/science.272.5264.1008. [DOI] [PubMed] [Google Scholar]
- 35.Bernard O A, Mauchauffe M, Mecucci C, Van den Berghe H, Berger R. Oncogene. 1994;9:1039–1045. [PubMed] [Google Scholar]
- 36.Fazioli F, Minichiello L, Matoskova B, Wong W T, Di-Fiore P P. Mol Cell Biol. 1993;13:5814–5828. doi: 10.1128/mcb.13.9.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}