

Abstract
We demonstrate experimentally that anthrax toxin complexes rupture artificial lipid bilayer membranes when isolated from the blood of infected animals. When the solution pH is temporally acidified to mimic that process in endosomes, recombinant anthrax toxin forms an irreversibly bound complex, which also destabilizes membranes. The results suggest an alternative mechanism for the translocation of anthrax toxin into the cytoplasm.
INTRODUCTION
Bacillus anthracis infects cells as an A-B toxin. The process begins when the B-component (83 kDa protective antigen PA83 protein) binds to receptors on cell membranes1, 2, 3 and is cleaved by a furin-type protease on the cell surface. The 63 kg/mol fragment, PA63, remains receptor bound4, 5 and forms a nanometer-scale heptameric pre-pore,6 (PA63)7, that binds up to three A-components, the 90 kDa lethal factor (LF), or the 89 kDa edema factor (EF) molecules.7 PA63 then forms nanometer-scale pores in membranes. This process and aggregation of PA receptors triggers endocytosis,8 which delivers the toxin complexes into endosomes. The reduced pH in these vesicles9 reportedly promotes the dissociation of toxin complexes from their cell receptors and insertion of PA63 oligomers into the membrane of intraluminal vesicles.10 A minor fraction of PA63 forms an octamer, which is less capable of forming channels, more robust in serum, and less cytotoxic.11, 12, 13
In the cytoplasm, LF and EF exert different deleterious effects on cells.14, 15, 16, 17 Isolation of cytosolic and membrane fractions of time-course samples from intoxicated macrophages revealed that EF remained bound to the membrane fraction, while LF was found in the cytosolic fraction.18 Understanding how they are transported into cells is of significant interest and the focus of this paper.
Several groups hypothesized that the transport of LF and EF into the cell requires their translocation through the PA63 pore,19, 20 which has a diameter of ≈1.2–2 nm.21, 22, 23 Previous electrophysiological studies demonstrated that full-length LF and EF (≈760 amino acids each)24, 25, 26, 27 and their N-terminal fragments (LFN and EFN, ≈254 amino acids each)28, 29, 30, 31, 32 bind strongly but reversibly to the channel and reduce the pore conductance. Reversal of the LFN-induced channel current blockade is kinetically dependent on the bulk pH and transmembrane pH gradients.28 It has been suggested that the blocking and subsequent unblocking of the channel by LFN and EFN is due to the complete translocation of these proteins through the channel. However, if these models are relevant to their full-length analogs, then LF and EF would have to unfold, detach from the channel, completely transport through the channel, and subsequently refold into fully functional enzymes in the cytosol, none of which has been demonstrated.
The transport of biopolymers (e.g., DNA and proteins) through single nanometer-scale pores, ion channels, and complex membrane protein machinery is of considerable experimental and theoretical interest.33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53 The anthrax toxin translocase mechanism, however, cannot account for all of the interactions between LF or EF and the PA63 channel. For example, anthrax toxin harvested from the blood of infected animals can be isolated in a complex of PA63 oligomer, polyglutamic acid-based capsule material, and LF or EF.24, 25, 54 This harvested complex has adverse effects when exposed to macrophages in vitro and when injected into animals.55 Interestingly, LF is enzymatically active in a complex with the PA63 channel25 and the complex reconstitutes into planar bilayer membranes.24 The latter result led to the hypothesis that the anthrax toxin complex (LF or EF bound to the channel) might enter the cytoplasm and cause cell intoxication.25
Here, we demonstrate that anthrax toxin can rupture artificial lipid bilayer membranes. In addition, we show that transmembrane pH gradient similar to that of an acidified endosome causes recombinant LF or EF to bind essentially irreversibly to the channel. The results suggest an alternative mechanism in which the anthrax toxin catalyzes the rupture of the endosomal membrane, thereby releasing the toxins into the cytoplasm.25 Figure 1 illustrates the putative translocase and rupture mechanisms for LF and EF transport from the endosomal interior to the cytoplasm.
Figure 1.

A schematic illustration of two putative mechanisms for PA63 channel-mediated LF and EF transport into the cytoplasm. One model suggests that LF and EF thread through the pore.19, 20 The results shown here suggest that anthrax toxin complexes (i.e., LF or EF bound to the PA63 channel) rupture membranes. A previous study demonstrated that LF in the complex is enzymatically active.25
MATERIALS AND METHODS
Reagents
Buffered electrolyte solutions were prepared with potassium chloride (KCl, J.T. Baker, Phillipsburg, NJ), morpholinoethanesulfonic acid monohydrate (MES, Aldrich, St. Louis, MO), and deionized water (Millipore Milli-Q Synthesis, Billerica, MA). The membrane-forming solution contained diphytanoyl phosphatidylcholine (DiPhyPC, Avanti Polar Lipids, Alabaster, AL) in pentane (Burdick and Jackson, Muskegon, MI). Recombinant forms of Bacillus anthracis PA63, LF, and EF were obtained from List Biological Laboratories (Campbell, CA) and reconstituted per the manufacturer's instructions. Hexadecane, poly(allylamine hydrochloride) (PAH, 56 kg/mol), poly(styrene sulfonate) (PSS, 70 kg/mol), D- and L-polyglutamic acid (PGlu, 25 kg/mol), and both chiral forms of poly(lysine) (PLys, 28 kg/mol) were obtained from Sigma-Aldrich (St. Louis, MO). Ionic current recordings were acquired with an Axon Instruments Axopatch 200B patch-clamp amplifier and Digidata 1322 analog to digital converter that were controlled with Axon pClamp 9 software (Molecular Devices, Sunnyvale, CA). The current recordings were analyzed with in-house software.
Preparation of animal-isolated anthrax toxin
Studies were conducted with B. anthracis Ames spores prepared as previously described.56, 57 Briefly, the spores were produced in flask cultures of Leighton and Doi medium, harvested by centrifugation, washed in sterile water for injection, and purified on a single-step gradient of 60% Hypaque-76 (Nycomed, Inc., Princeton, NJ), then stored until use at 4 °C in 1% phenol. The spores were used to challenge naive guinea pigs, rabbits, and monkeys by the aerosol route.58, 59
During the terminal stages of infection, blood from the moribund animals was collected in Vacutainer tubes containing EDTA (Beckton Dickinson, Franklin Lakes, NJ) to inhibit calcium-dependent protease activity on PA.60 Blood cells were removed by centrifugation and the plasma was filtered through 0.22 μm syringe filters (Millipore, Billerica, MA) and stored at 4 °C no longer than 2 days for analysis by Western blot, fractionation by column chromatography, and LF protease assays. Aliquots were maintained at −70 °C for long-term storage.
Filter-sterilized plasma from each infected animal was diluted in phosphate buffered saline solution (PBS), and applied to a Superose 6 size exclusion column (GE Healthcare, Piscataway NJ), at a flow rate of 0.5 ml/min. Capsule from B. anthracis Ames was prepared as described earlier.61 Purified capsule, capsule-toxin complex, PA, and LF from fractionated plasma were detected by Western blot55 or by methylene blue staining of capsule.62 Plasma fractions were also subjected to PA capture ELISA with a purified anti-PA antibody (IgG) produced in goats. Lethal toxin complexes in plasma were captured with the immobilized goat anti-PA and then assayed for other associated bacterial antigens. Briefly, wells of microdilution plates (Linbro/Titertek) were coated with purified goat anti-PA IgG in 0.05 M sodium borate buffer, pH 9.0, and blocked with PBS containing 0.5% gelatin, 0.3% Tween 20, and 1% bovine serum albumin (PBGT-BSA). Horseradish peroxidase (HRP)-conjugated monoclonal antibodies to PA63, PA20, LF, and capsule (FDF-1B9) were used as secondary antibodies. The chromogenic substrate 2, 2-azino-bis (3-ethylbenzthiazoline-6-sulfonic acid) (Sigma, St. Louis MO) was used, and absorbance was determined with a Bio-Tek 308 micro-plate reader (Bio-Rad) at 405 nm. LF protease activity in the Lethal Toxin complexes was determined as previously described.25 Plasma fractions containing capsule/anthrax toxin complexes were then used for electrophysiology experiments on artificial lipid bilayer membranes described herein.
Artificial planar membrane formation
Planar lipid bilayer membranes were prepared similarly to classical black lipid membrane techniques.63 A two-well Teflon chamber was separated by a 25 μm thick Teflon partition. The DiPhyPC bilayer membranes were formed across a hexadecane-primed 120-μm diameter hole in the partition. A transmembrane potential was applied via two Ag/AgCl electrodes bathed in 3 M KCl and isolated with Vycor glass frits (Koslow Scientific Company, Englewood, NJ). Each well held 2 ml of buffer (100 mM KCl, 5 mM MES). The stability of the membrane was verified over ±200 mV before performing the experiments described herein. The notation pHc|t indicates the pH of the cis and trans solutions, respectively (e.g., pHc|t 7.2|5.5 represents pHcis 7.2 and pHtrans 5.5).
Droplet membrane formation
A sample well was formed by drilling a 10 mm hole in a 75 mm × 25 mm × 1 mm microscope slide and attaching a #1.5 glass cover slip to one side sealing the hole and creating a ≈75 μl volume. The well was filled with 10 mg/ml DiPhyPC in hexadecane (99.8% purity, Sigma-Aldrich, St. Louis, MO). The ends of two 76 μm diameter Ag wires (A-M Systems, Carlsborg, WA) were etched using a modification of previously described techniques.64, 65 Briefly, a 50 ml Pt crucible was filled with a solution of 28% ammonium hydroxide. The Ag wire was mounted to a vertical translator and connected to one electrode of an adjustable step-down transformer. After connecting the crucible to the other transformer electrode, the wire was lowered into the solution so that 1–2 mm of the wire's end was submerged in the solution. The transformer was turned on and adjusted to ≈60 V (at 60 Hz) and the wire was manually extracted from the solution at ≈1 mm/s. This formed silver tips with a ≈10 μm radius of curvature. The tips were then chloridized by immersing them in a bleach solution for several minutes. The tips were set in separate micropipette holders (Warner Instruments, Hamden, CT), attached to manual translation stages (Newport, Irvine, CA), and positioned so that the ends of the tips were immersed in the lipid solution and held ≈500 μm apart. A femtopipette tip (Eppendorf, Hamburg, Germany) delivered 100 μm diameter aqueous droplets to the ends of each tip. The droplets spontaneously adhered to the end of each tip and a lipid monolayer assembled at the water-solvent interface. After waiting several minutes for the monolayer to form, the droplets were brought into contact with each other and a bilayer membrane spontaneously formed,66, 67 as judged by conductance and capacitance measurements. Each droplet contained the aqueous buffer (100 mM KCl, 5 mM MES) at pH 7.2.
Integration of recombinant PA63 artificial membranes
Ion channels were reconstituted into the planar bilayer membranes by adding 0.5–1.0 μl of 0.2 mg/ml PA63 (in 10 mM bis Tris phosphate at pH 8.0) to pH 7.2 buffer in one well (cis chamber). Channel formation was monitored at +50 mV, which drove cations from the cis to the trans chamber. When the conductance exceeded that of 25 channels, the cis chamber was flushed extensively with fresh buffer to remove excess PA63. The instantaneous current–voltage (I–V) relationship was determined at pH 7.2 by averaging over 400 ms of the current time series for each voltage (±150 mV in 10 mV increments).
The pH dependence of PA63 conductance and LF binding
The pH gradients across PA63-containing membranes were first adjusted by perfusing the chambers with buffer at the desired pH. The instantaneous I–V relationship was determined again to account for pH-induced deviations in the channel conductance. The LF concentration was increased by the stepwise addition of a concentrated (≈10 μM) LF solution. The chambers were thoroughly mixed by stirring. After the ionic current monitored at V = +50 mV had stabilized, an instantaneous I–V relationship was acquired for each LF addition.
Effect of EF/ LF present during acidification
Continuous monitoring of the channel conductance at V = +50 mV was performed by adjusting the pH and EF or LF content of the cis solution while maintaining the trans solution at pH 7.2. In these experiments, PA63 was in the cis chamber (pH 7.2) until a conductance equivalent of ≈60 channels was obtained. After extensive flushing with fresh pH 7.2 buffer, the I–V relationship was measured. After the cis addition of 1 nM LF, the channel conductance was monitored for ≈20 min prior to rinsing the cis chamber with either pH 7.2 buffer or pH 5.5 buffer containing 1 nM LF (created immediately before perfusion). Each subsequent cis rinsing step was followed by monitoring the current response for ≈20 min. To facilitate the recovery of channel activity, three 30 s pulses at V = −50 mV were applied during the monitoring period. After returning the cis solution to [LF] = 0 and pH 7.2, the current recordings were monitored for 60 min. The I–V relationship was measured at the beginning and end of each experiment (pH 7.2 and [LF] ≈ 0). This procedure was repeated for EF.
Planar membrane rupture by recombinant proteins
PA63-containing membranes were exposed to 1 nM of EF or LF at pH 7.2. For controls, the EF or LF was flushed from the cis chamber with fresh pH 7.2 buffer. For acidic conditioning, the EF or LF concentration was maintained at 1 nM while the pH was lowered to pH 5.5 by perfusing with pH 5.5 buffer containing 1 nM EF or LF. After acidic conditioning, the EF or LF was removed with fresh pH 5.5 buffer. The pH of the solution was adjusted accordingly per the experiment. The membrane rupturing capability was tested by applying a holding potential for ≤10 min.
Planar membrane rupture by cationic polymers
The effect of ionic polymers was tested by adding a cationic polymer (PGlu, PSS, PAH, or PLys) to the cis side of planar bilayer membranes that contain PA63. The instantaneous I–V relationship of the PA63 channels was measured in the presence of each polymer up to a concentration of 10 nM. The membrane rupturing capability was tested by applying a holding potential for ≤10 min.
Planar membrane rupture by animal isolates
Toxins were integrated into planar membranes by pipetting ∼5 μl of the purified complex directly over a planar membrane bathed in pH 7.2 buffer. At least 3 purified fractions of toxin-capsule complex harvested from guinea pigs, rabbits, and monkeys were tested. Channel integration was verified by a detectable increase in the ionic current at V = +50 mV, and the I–V relationships and membrane stability were tested.
Droplet membrane rupture by animal isolates
Toxins were integrated into droplet membranes via a femtojet pipetting system (Eppendorf, Hamburg, Germany). Only the last fractions of the toxin-capsule complex harvested from guinea pigs, rabbits, and monkeys were tested because they were the only samples of low enough density that could be delivered by the femtojet. Again, the I–V relationships and membrane stability were obtained.
Molecular modeling
Discovery Studio 2.0 (Accelrys, San Diego, CA) was used to calculate the pKa values for all the ionizable amino acids in the reported theoretical model of the PA63 channel pore (PDB code 1V36). The protonated state of the pore model was constructed and energy was refined using InsightII and Discover 3.0 (Accelrys, San Diego, CA). Electrostatic potential surfaces were calculated and visualized using Delphi and InsightII (Accelrys, San Diego, CA).
RESULTS
Artificial lipid bilayer membrane rupture induced by anthrax toxin harvested from infected animals
Anthrax toxin harvested from late-stage infected animals is associated with capsule material.54 Previous in vitro electrophysiology studies involving animal-harvested toxin demonstrated that the instantaneous I–V relationship of the reconstituted ion channels in those samples was highly rectifying and virtually identical to that of recombinant LF bound to the PA63 ion channel.24
In this study, 5–10 μl aliquots of toxin-capsule complex were injected directly over the planar membrane without stirring, instead of mixing into the bathing solution.24 Direct delivery of the toxin complex avoided dilution of the sample and caused more vigorous channel insertion. As expected, the instantaneous I–V relationship (Fig. 2a, (•)) shows that the harvested complex initially forms highly rectifying channels consistent with previously observed behavior for the LF:PA63 channel complex. Over the next 90 min, the ionic current rectification decreases (Fig. 2a, open symbols), which suggests the slow dissociation of components in the anthrax toxin complex under the conditions used here (100 mM KCl, 5 mM MES, pHc|t 7.2|7.2). Similar results were obtained with toxin-capsule complex isolated from anthrax-infected guinea pigs, rabbits, and monkeys.
Figure 2.

Interaction of animal-harvested anthrax toxin with artificial lipid bilayer membranes. (a) The I–V relationship of anthrax toxin was initially strongly rectifying (•). The degree of rectification decreased with increasing time after sample addition (10 min (□), and 90 min (▵). Inset: schematic illustration of LF bound to a PA63 channel in an artificial lipid bilayer membrane. (b) Ionic current time series recordings and video micrographs (inset) at V = 180 mV demonstrate that anthrax toxin harvested from infected rabbits causes the membrane to become unstable and eventually rupture (†). The grey rods in the micrographs are Ag/AgCl electrodes, and the solutions contained 100 mM KCl, 5 mM MES at pH 7.2. Capsule material was present in the isolated fractions of anthrax toxin.
When a constant potential of |V| > 70 mV was applied within 15 min after injecting the toxin-capsule complex, the ionic current progressed to the amplifier's current limit, suggesting that the planar bilayer membranes ruptured (data similar to Fig. 2b). At positive applied potentials, the strongly rectified channel had a low current value with little noise at the onset of the experiment. After several minutes, the current noise increased, akin to membrane electroporation.68, 69, 70, 71 Once destabilized, the membrane spontaneously ruptured (cf. Fig. 2b, inset).
The anthrax toxin-capsule complex was purified as a long band that was divided into fractions. The earliest fractions contained the greatest concentrations of PA63, highest LF activity, and the largest capsule material polymers. All fractions of the toxin-capsule complex purified from anthrax-infected rabbits, guinea pigs, and monkeys caused >90% of the planar membranes to rupture, if a relatively high transmembrane voltage (e.g., |V| ≥ 100 mV) was applied within 15 min of injection. The membranes did not rupture if the potential was applied more than 15 min after injection (data not shown). The loss of rupture capability is likely caused by the diffusion of a component into the bulk. In a previous study, when the toxin was mixed into the 2 ml bathing solution and allowed to diffuse to the membrane, it did not cause membrane rupture up to |V| = 130 mV.24
Effect of pH gradients on recombinant anthrax toxin
In vivo, a pH gradient across the endosomal membrane (the endosome interior is acidic with respect to the cytosol) coincides with the deleterious results of the anthrax toxin.72 Thus, we attempted to recreate this membrane rupture phenomenon with recombinant EF, LF, and PA63 by examining the effects of transmembrane pH gradients on anthrax toxin in artificial lipid bilayer membranes.
In the absence of LF, the pH values of the cis- and trans-side solutions both markedly affect the channel's instantaneous I–V relationship (Fig. 3, open symbols). Note that the I–V relationship obtained in the presence of an unphysiological pH gradient is superlinear (Fig. 3c), whereas those with physiologically relevant gradients are sublinear (Figs. 3a, 3b, 3d). A structural model of the PA63 channel22 provides a likely explanation for the pH-dependent channel conductance. The charges of ionizable amino acid side chains near the cap domain pore entrance (His211) and in the cap lumen (E398, E465, D472) will change as the bulk pHcis is reduced from 7.2 to 5.5 (Table 1). Similarly, His304 and His310, which are located near the trans pore entrance, will be affected by shifts in pH. A change in the charges on these side chains would markedly alter the barriers to ion transport through the pore.73 Therefore, all PA63 channels were reconstituted into the membrane at pHc|t 7.2|7.2 as a reference point for channel performance.
Figure 3.

The effect of pH gradients on the PA63 channel instantaneous I–V relationship in the absence and presence of LF (a)–(d). The cis and trans compartments were buffered at either pH 7.2 or 5.5, as indicated in each panel. The LF concentration in the cis chamber was either zero (□) or 1 nM (•). Normalized current for the LF-free (e) and 1 nM LF data (f) highlights relative changes due to pH. Colorization indicates the pH condition as used in (a)–(d), normalized at 10 mV and 120 mV, respectively. The error bars, which represent the standard deviation (n ≥ 7), are generally smaller than the symbols.
Table 1.
Calculated pKa values of residue side chains in the PA63 channel lumen.
| PA63 residue | pKa of side chain | ||
|---|---|---|---|
| Asp237 | 6.28 | ||
| Glu276 | 3.61 | ||
| Asp302 | 4.40 | ||
| His304 | 6.95 | ||
| Asp308 | 3.99 | ||
| His310 | 7.26 | ||
| Asp315 | 3.61 | ||
| Asp335 | 3.45 | ||
| Glu343 | 4.30 | ||
| Glu398 | 4.89 | ||
| Asp425 | 2.81 | ||
| Asp426 | 3.79 | ||
| Glu465 | 5.53 | ||
| Asp472 | 5.14 | ||
| Glu479 | 5.83 | ||
| Glu502 | 5.95 |
The addition of LF to the cis compartment caused a marked reduction in channel conductance at positive potentials for the pH gradients shown here (Fig. 3, solid symbols). The degree by which LF reduced the current for negative potentials depended on the direction and magnitude of the pH gradient. When only the cis compartment was acidified (Fig. 3b), LF also reduced the conductance for negative voltages. For non-physiological pH gradients (pHc|t 7.2|5.5 and 5.5|5.5, Figs. 3c, 3d, respectively), LF had little effect on the channel conductance at negative applied potentials, as was observed for symmetric solutions at neutral pH (Fig. 3a). The ability of LF to reduce the pore conductance at positive and negative potentials is greatest for the pH gradient that mimics that across an acidified endosome membrane.
Replacing the solution in the cis chamber with LF-free buffer caused the dissociation of LF from the channel with pH gradients of pHc|t 7.2|7.2, 5.5|5.5, and 7.2|5.5 (data not shown). In contrast, when the complex was formed at pHc|t 5.5|7.2, the channels remained blocked for >1 h after removal of LF from the bulk. Thus, despite the slight increase of the apparent dissociation constant at pHc|t 5.5|7.2, LF and the channel were essentially irreversibly bound when the proteins interacted after the pHcis was decreased. This strong binding directly coincides with a dramatic reduction in channel conductance at negative potentials that is observed at pHc|t 5.5|7.2 (Fig. 3b). Therefore, while the apparent dissociation constant determined at positive potentials may characterize the LF prepore or external binding site, the loss of conductance at negative potentials may indicate a second, stronger binding site.
To better understand the essentially irreversible binding of LF (or EF) to the channel, we monitored the evolution of this state with continuous ionic current recordings. The effect of cis-side acidification on the LF:channel complex formed at pHc|t 7.2|7.2 is illustrated in Fig. 4. The membrane contained the conductance equivalent of ≈60 channels. The addition of 1 nM LF to the cis compartment markedly reduced the channel conductance (Fig. 4a, top). After removing LF, the conductance was still low relative to that of the open channels. Reducing the pHcis to 5.5 caused the current to slowly increase (Fig. 4a, middle), which is consistent with dissociation of LF (Fig. 8). Brief potential reversals (Fig. 4a, time axis breaks) caused a further increase in current, because of additional dissociation of LF from the channel and/or the reversal of channel gating.24 The current increased further when pHcis was returned to 7.2 (Fig. 4a, bottom) likely due to the pH-dependence of the channel conductance (Fig. 3) and additional LF dissociation. Potential reversals (Fig. 4a, bottom, time axis break) again caused the current to increase slightly. About 30 min after returning the solutions to the initial condition, the conductance typically recovered to ∼90% of the initial value (cf. Fig. 4a, ◯ and ![]() ). Thus, the number of functional channels in the membrane was conserved, and nearly all of the complex had dissociated. Using the same experimental protocols, similar results were obtained with EF (data not shown). No rupture events were observed for membranes with either recombinant EF or LF bound to the channel processed in this manner.
). Thus, the number of functional channels in the membrane was conserved, and nearly all of the complex had dissociated. Using the same experimental protocols, similar results were obtained with EF (data not shown). No rupture events were observed for membranes with either recombinant EF or LF bound to the channel processed in this manner.
Figure 4.
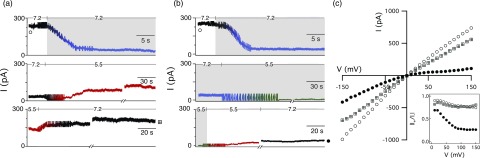

Time course of the PA63 channel conductance when (a) LF was removed before or (b) maintained [LF] = 1 nM during cis-side acidification. (Top) The conductance equivalent of ≈60 PA63 channels was reconstituted into a planar bilayer membrane at pHc|t 7.2|7.2 (black). Then, 1 nM LF was added to the cis chamber (blue). (Middle) The pHc|t 5.5|7.2 gradient was formed by perfusing the cis chamber with pH 5.5 buffer that contained either [LF] = 0 (red) or [LF] = 1 nM (green). (Bottom) The neutral pH condition (pHc|t 7.2|7.2) (black) was restored by perfusing the cis chamber with pH 7.2 buffer. If LF was present, the cis chamber was first purfused with pH 5.5 buffer (red) then pH 7.2 buffer (black). The ionic current was monitored for ≈20 min after each perfusion and ∼60 min after the final perfusion. The applied potential was V = +50 mV. Breaks in the current recordings correspond to ≈30 s pulses at V = −50 mV. The periodic noise corresponds to magnetic stirring during perfusion. (c) Instantaneous I–V measurements and the current rectification ratio (inset) taken at the beginning (◯) and end (,•) of the ionic current series confirm that the complex became essentially irreversibly bound only when maintained [LF] = 1 nM during cis-side acidification. Similar results were obtained with EF (not shown).
Figure 8.

The pH dependence of the LF:PA63 channel binding constant as estimated from the channel conductance at V = +70 mV. The symbols correspond to the pH conditions described in Fig. 3 (pHc|t 7.2|7.2 □; pHc|t 5.5|7.2 ○). The solid lines are the least-squares best fits of a simple binding equation for 1:1 stoichiometry (i.e., 1/(1 + [LF]/Kd), where Kd is the reaction dissociation constant. (Inset) The pH dependence of Kd. The pH in the trans chamber was 7.2. The dashed line is to guide the eye. The error bars represent the standard deviation from n = 3–5 membranes.
The experiment in Fig. 4a, however, does not completely mimic in vivo conditions because the concentration of LF within the endosome is implied to be zero. A single LF molecule confined in an endocytotic vesicle (≈0.2 fL74) corresponds to a bulk LF concentration of ∼8 nM. If the rate constant for association is assumed to be kon ≈ 109 M−1 s−1, by mass action, the complex would reform in ≈0.1 s after its dissociation. Given the approximately fourfold increase apparent dissociation constant at pHc|t 5.5|7.2, acidification of the endosome should promote the dissociation of LF from the channel, as we indeed observed in Fig. 4a. In the in vitro mimic where LF is removed prior to acidification, the ≈180 LF molecules initially bound to the ≈60 channels equate to ≈0.3 aM LF in the 1 ml chamber; ≈10 orders of magnitude below extrapolated [LF]bulk. Therefore, the [LF]bulk should be controlled to better mimic the finite volume of the endosome.
To test this hypothesis, we repeated the experiment to maintain the [LF]bulk at 1 nM during the acidification of the cis compartment. Initially, LF caused a significant decrease in the current flowing through ≈60 channels (Fig. 4b, top). The channels remained occluded after the cis chamber was perfused with a pH 5.5 buffer containing 1 nM LF and cycling the applied potential (Fig. 4b, middle), because the bulk still contained LF. Surprisingly, removing the LF with fresh pH 5.5 buffer caused only a minor increase in the current (Fig. 4b, bottom), which remained low even after a subsequent rinsing with pH 7.2 buffer (Fig. 4b, •). About 90 min after re-establishing the initial conditions (pHc|t 7.2|7.2, [LF]bulk = 0), the final channel conductance was only ≈17% of the initial value (cf. Fig. 4b, ◯ and •) and similar to the channel current for 1 nM LF at pHcis 7.2 (Fig. 4b, top right).
The sharp contrast between the results in Figs. 4a, 4b suggests that maintaining the LF concentration during acidification has a dramatic effect on the anthrax toxin interactions, which is presented more clearly in the channel's I–V relationships and rectification ratios. When virtually all of the LF is removed before acidification (Fig. 4a), the shape of the I–V relationship was not altered significantly by changing the pH gradient, with the exception of small decrease in the number of channels (cf. Fig. 4c, ◯ and ![]() ). Thus, LF dissociated from the channels and either diffused into the cis chamber or translocated through the pores. In contrast, the ion channels were strongly rectifying (Fig. 4c, •) when [LF]bulk = 1 nM during acidification (Fig. 4b). The LF was essentially irreversibly bound at +50 mV, a potential that presumably initiates LFN translocation.28, 75 Similar results were obtained with EF (data not shown).
). Thus, LF dissociated from the channels and either diffused into the cis chamber or translocated through the pores. In contrast, the ion channels were strongly rectifying (Fig. 4c, •) when [LF]bulk = 1 nM during acidification (Fig. 4b). The LF was essentially irreversibly bound at +50 mV, a potential that presumably initiates LFN translocation.28, 75 Similar results were obtained with EF (data not shown).
Membrane rupture with recombinant anthrax toxin
The irreversibly bound complex of LF:PA63 channel or EF: PA63 channel causes planar bilayer membranes to rupture for |V| ≥ 120 mV (Figs. 5a, 5b). The membranes ruptured even after excess LF (or EF) was removed from the bulk at pH 5.5 (data not shown) or when pHcis was subsequently returned to pH 7.2 (data not shown). Over 80% of the membranes ruptured when the recombinant toxin formed an irreversible complex (i.e., when LF or EF was in the bulk during acidification of the cis side.
Figure 5.

Rupture events of planar membranes caused by complexes of either essentially irreversibly bound recombinant (a) LF:PA63 channel and (b) EF:PA63 channel. Membrane rupture was also achieved by exposing the channel to the cis addition of the polycations (c) 28 kg/mol poly-L-lysine, and (d) 56 kg/mol poly(allylamine hydrocholoride). Chirality and cis/trans addition did not inhibit the membrane rupture capability (not shown). The membranes rupture events are denoted by (†). The solutions on either side of the membrane contained 100 mM KCl and 5 mM MES at pH 7.2. Membrane rupture with irreversibly bound recombinant proteins was also achievable at pHc|t 5.5|7.2 (not shown). The applied potential was |V| = 120 mV for recombinant proteins or |V| = 50 mV for the polycations.
We challenged recombinant PA63 channels with charged polymers to determine if they bind to the channel and subsequently rupture the membrane. The N-terminii of LF and EF are relatively rich in cationic residues76 and cationic polymers interact strongly with the pore.77, 78 The polycations PLys and PAH (up to 10 nM) ruptured membranes for |V| ≥ 50 mV (Figs. 5c, 5d), whereas anionic polymers PSS and PGlu did not (data not shown). The results suggest that positive charges on pore-permeant polymers play a key role in the rupture process, likely by binding to negative charges in the pore.22, 76 As a control, in the absence of PA63 channels, planar bilayer membranes were unaffected by the charged polymers and stable for |V| up to 200 mV.
Out of 70 attempts of inducing membrane rupture with anthrax toxin harvested from infected animal, recombinant anthrax toxin, and polycations bound to recombinant PA63 channels, membrane rupture was achievable at a >90% efficacy when |V| ≥ 50 mV (28/31 animal isolates, 6/6 recombinant LF, 4/5 recombinant EF, 14/15 PLys, and 13/13 PAH). At applied potentials ≈120 mV, the median time to rupture a planar bilayer membrane was ≈1 s.
DISCUSSION
Models for anthrax toxin translocation
It has been suggested that LFN and EFN, and by inference full length LF and EF, are driven completely through the pore. However, conclusively proving polymer translocation through nanometer-scale pores is non-trivial. Polymerase chain reaction amplification demonstrated that negatively charged polynucleotides are driven electrophoretically through the 2 nm diameter Staphylococcus aureus α-Hemolysin channel.36, 49 Support for these measurements was provided by theories for polymer entry79 and translocation through mathematically defined thin holes40, 41, 44, 53 and finite length nanopores.39, 43, 50, 53 Experimentally proving translocation of a trace amount of protein is more difficult, because there is no means to amplify protein. Fischer and colleagues claimed that they could detect as few as 100 LFN proteins (800 aM) at a rate of ≈2 s−1 with the channel,80 while the mean time to detect individual LFN molecules at that low concentration is ≈106 s.81
LFN translocation through the PA63 channel28, 30, 31, 32, 80, 82, 83, 84, 85, 86, 87 has been hypothesized to occur either by electrodiffusion as charged rods75 or via a “Brownian ratchet.”20 Both models make assumptions about how the energy barriers to LFN transport are overcome. In each, protein unfolding is assumed to be a limiting barrier. Previous work suggests, but does not conclusively demonstrate, that LFN partially unfolds in a highly acidic environment (pH 2) and in the presence of guanidinium chloride.29 The Brownian ratchet model postulates that the energy for translocation is the protonation/deprotonation-induced conformational changes in LFN.
Translocation in these models is inferred from the recovery of LFN-induced current blockade. The possibility that LFN dissociates from the channel under those conditions is dismissed as unlikely.28 In their in vitro experiments, LFN was typically removed from the bulk solution prior to adjusting the pH gradient.28 As we demonstrated here, the presence of LF in the bulk during acidification markedly changes the interaction between LF and the channel (Figs. 4a, 4b). The in vitro pH gradient formation used in the previous studies varies substantially, but follows two general patterns: (a) static low pH values on both sides of the membrane (e.g., pHc|t 5.5|5.575) or (b) starting with the symmetric acid condition and forming a pH gradient by raising the pH of trans side either by a base shock or perfusion (e.g., pHc|t 5.5|5.5 to pHc|t 5.5|7.228). Note that the latter is precisely opposite to what occurs in vivo. If the translocation models are to hold, they must measure up to experiments with full-length LF and EF using conditions that accurately mimic the acidification of the endosome. Moreover, full-length LF and EF are three times larger than LFN, and have catalytic domains88 that must remain functional after putative transport.
Irreversible binding of anthrax toxin
The results described herein demonstrate that the interactions of the anthrax toxin are more complicated than previously suggested.24, 25 Specifically, the presence of LF and EF during acidification determines the penultimate binding state. Prematurely removing LF from the bulk and increasing the potential to +50 mV slowly reverses the LF-induced current blockade (Fig. 4a), which was also observed with LFN.28 If the reservoir is large (≈2 ml), LF will dissociate from the complex and become infinitely diluted. Within the endosome (≈0.2 fL), however, the dissociation of a single LF molecule corresponds to ≈8 nM that would cause a rapid re-formation of the anthrax toxin. A conformational rearrangement in the structures of LF, EF, and/or the channel at lower pH is likely responsible for the formation of the irreversible complex (Fig. 4b).
Changes in the protonation state of ionizable amino acid residues of the anthrax toxin are likely responsible for the irreversibly bound complex. The pre-pore binding site,76, 89 however, is not a likely candidate. A decrease in pH would increase the positive charge on both the channel cap domain and LF (H211 of PA63 and H229/H197 of LF, Fig. 6) and thereby favor dissociation (Figs. 4a, 8). The coincidence of the irreversible binding and dramatic reduction in channel conductance at negatives potentials at pHc|t 5.5|7.2 (Fig. 2b) suggests the existence of a second, stronger binding site. We demonstrated here that recombinant anthrax proteins become irreversibly bound, which leads to membrane rupture (Figs. 5a, 5b). Because cationic polymers also produce these effects, the binding of cationic regions of the N-terminii of LF and EF to the channel lumen may be a major causative agent of membrane rupture. In addition, because LF bound to the PA63 channel is biologically and functionally active,25 it is conceivable that this anthrax toxin complex does not need to dissociate to be an enzymatically active agent in the cytosol.
Figure 6.

Hypothetical pH-induced changes to the putative binding pocket for the PA63 channel and LF. (Left) A top-down view for part of the theoretical model of the PA63 channel22 and the crystal structure of LF76 oriented to illustrate the proposed binding site. The colored regions correspond to the subunits shown in the right panel. (Right) A “folded-open” representation of the LF:PA63 channel binding pocket that includes a PA63 dimer and LF. The space-fill region emphasizes the residues at the binding site. The electrostatic potentials were computed at (a) pH 7.2, (b) pH 5.5, and (c) their difference. Negative and positive electrostatic surface potentials are denoted by red and blue, respectively.
Anthrax toxin and membrane destabilization
The question is whether the electrostatic potential across the endosomal membrane is sufficient to rupture membranes. Large transmembrane potentials (|V| ≥ 250 mV) can rupture solvent containing and “solvent-free” artificial planar lipid bilayer membranes.68, 69, 70, 71, 90, 91, 92, 93, 94, 95 The results shown here demonstrate that voltage-induced membrane rupture can occur at lower applied potentials with anthrax toxin-capsule complex (Fig. 2b), or LF, EF, or cationic polypeptides bound to the PA63 channel (Fig. 5).
Over the lifetime of an endosome, the pH inside decreases from pH 7.2 to ≈pH 4 due to ATP-driven proton pumps.96 If the proton was the only membrane-permeant species at steady state, the transmembrane potential, Δψ, could be as large as 195 mV at T = 37 °C, as described by the Nernst equation.97 However, other ions and their transporters lower the potential to (27 ± 16) mV (positive inside)98 which is consistent with the expected range estimated through the Goldman-Hodgkin-Katz equation96, 97 or from a kinetic analysis of the known channels and transporters in the membranes.99 If we assume the distribution of the Δψ values is Gaussian, then ≈2% of the endosomes will have transmembrane potentials that are sufficiently great (|V| ≥ 70 mV) that would support the putative LFN or EFN translocation model or the membrane rupture model suggested herein (Fig. 1).
The larger potential required to rupture artificial lipid membranes might be due to a need to match the thickness of the channel's membrane-spanning region and the bilayer.100 Artificial membranes are often comprised of one lipid type and contain residual solvent that is thinned by an applied potential.101 The need to thin an artificial membrane is supported by the minimum voltage required for anthrax toxin from animal isolates to rupture droplet (|V| = 180 mV) or planar membranes (|V| = 120 mV). Droplet membranes should require a greater force to thin than planar membranes, because solvent in the reservoir can more rapidly infiltrate the membrane. There is a precedent for voltage-dependent thinning of artificial lipid bilayers that allows peptides to disrupt membranes. For example, warnericin RK acts like a detergent that permeabilizes Legionella membranes.102 In DiPhyPC membranes, the polypeptide behaves like poorly defined ion channels at low potentials. At potentials >60 mV, an erratic conductance leads to membrane rupture, as we observed here with anthrax toxin (e.g., Figs. 2b, 5a, 5b). Because endosomal membranes are solvent-free, the threshold potential for anthrax toxin to disrupt them might be significantly lower or even zero.
Intoxication via membrane destabilization has precedents.103 A variety of agents (chitosan,104 cationic peptides,105, 106, 107, 108, 109, 110, 111, 112 cationic polymers,113, 114 and an HIV gp41 peptide115) are used to transfer genes into a cell. In those cases, osmoticants and lysosomotropic agents draw water into the endosome, causing it to swell and burst.116 In addition, various naturally occurring102, 117, 118 and designer peptides119, 120 use a detergent-like action to solubilize, destabilize, and rupture membranes.118, 121
Hypothetical mechanism
Our results demonstrate that the irreversible binding of LF or EF to the PA63 channel causes membrane rupture, likely due to a second binding site in the channel lumen. The first 60 residues of N terminus-truncated His6 LFN constructs apparently can access the pore lumen and in part extend past the trans mouth.122 Because the pore is narrow (diameter <2 nm),22, 23, 123 a polypeptide in it would likely be an extended chain. When the unstructured residues of the EF and LF proteins fill the pore, the cationic regions of EF or LF are in the proximity of the E335/D343 rings of the channel, the His residues of LFN (H4 and H10) and EFN (H4) align with the His rings (H304 and H310) in the pore, and hydrophobic residues of EFN and LFN are within the membrane-binding region of the channel, see Fig. 7.
Figure 7.

Protein topology of the disordered N-terminus of EF/LF and the lumen of the PA63 channel β-barrel. (Top) Extended chain representation of the first 30 disordered amino acids (right to left) from the N-terminus of LF. (Middle) The lumen of the β-barrel from a model of its structure. (Bottom) Extended chain representation of the first 30 disordered amino acids (right to left) from the N-terminus of EF. PyMol was used to generate the topological representation based on sequences of EF,124 LF,76 and whole model of (PA63)7.22 Residues are colored by the following classifications: Marine – His, Purple – Basic, Yellow – Acidic, Grey – Hydrophobic. Blue and Red denote N and O moieties, respectively. The bar represents the membrane spanning region of the β-barrel.
Histidines may play an important role in membrane disruption. The essentially irreversible binding state of the anthrax toxin complex, which is required for membrane rupture by recombinant proteins, occurs when pHcis is decreased below the pKa of histidine's imidazole (6.0). Interestingly, the addition of a His6-tag increases the binding strength of LFN by approximately threefold at pHc|t 5.5|5.575 and restores the binding activity to neutral and negatively charged mutants of LFN27.125 His residues are also responsible for pH-dependent rearrangement of proteins, such as the pH-dependent coil-helix transition of glycinamide ribonucleotide transformylase,126 acidic activation of His-rich synthetic peptides aimed at disrupting microbial membranes,127 and enhancing gene transfer though membrane permeabilization of human tumor cells.128, 129
There are several potential interactions that may contribute to the destabilization of the membrane spanning region of the PA63 pore and the membrane. The histidine residues, H4, and H10 of LF (H4 and H15 of EF) might shuttle protons to the H303 and H310 residues of PA (Table 1). This could create a strong interaction between the histidines and the neighboring glutamic acid residues (E302 and E308), with the potential to disrupt the β-barrel structure which in turn could conceivably give access to the hydrophobic residues on the PA pore (F313 and F314).
The results herein demonstrate that recombinant anthrax toxins require a pH gradient similar to that in acidified endosomes to rupture the membrane, but anthrax toxin from animal isolates did not. This mechanism for protein-mediated rupture does not directly account for the membrane rupture by animal-harvested anthrax toxin at neutral pH, until the effects of capsule material are considered. The polyglutamic acid capsule may act as a localized acidic buffer at the LF:PA63 channel, thereby establishing a localized pH gradient. This is supported by the time-dependent loss of membrane rupture capability by animal-harvested toxin (Fig. 2a), which we attribute to the diffusion of capsule material and that the anthrax toxin might not have been processed in a pH gradient in vivo.
CONCLUSIONS
We demonstrated that transmembrane pH gradients alter the ion conducting properties of the PA63 channel and the interactions of LF and the channel. For pH gradients that mimic those across the endosomal membrane (pHc|t 5.5|7.2), LF binds less strongly to the channel in the absence of bulk LF, but binds irreversibly to the PA63 channel when LF is present in the bulk during acidification. In addition, lipid bilayer membranes rupture in the presence of a transmembrane potential and either anthrax toxin harvested from late-stage infected animals (Fig. 2b), complexes of recombinant LF:PA63 channel, EF:PA63 channel, or polycations bound to the channel (Figs. 5a, 5b, 5c, 5d).
The membrane rupture mechanism is based on observations made with anthrax toxins in planar bilayer and droplet membranes under conditions that mimic the pH gradients in the process of endosomal acidification. The >80% reproducibility of anthrax toxin-induced membrane disruption with both full-length recombinant proteins and animal-harvested samples is compelling.
The results described here should help further elucidate the mechanism by which LF and EF enter the cytosol. However, the question remains whether these pathogens do so via membrane rupture; by unfolding, threading through the PA63 channel,20, 75 and refolding in the cytosol; or perhaps by a yet undetermined process. Nevertheless, the irreversible binding of LF and EF to the channel puts an additional and significant constraint on the LF/EF translocation model inferred from N-terminal fragments.
ACKNOWLEDGMENTS
This work is supported, in part, by a NIST-NRC Research Associateship (B.J.N.), a NIH-NIST-NRC Research Associateship (A.B.), and the NIST Office of Law Enforcement Standards (J.J.K.). Certain commercial equipment, instruments, or materials are identified in this paper to adequately specify the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose. In conducting the research described in this paper, the investigators adhered to the “Guide for Care and Use of Laboratory Animals” as promulgated by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council. The facilities are fully accredited by the American Association for Accreditation of Laboratory Animal Care.
APPENDIX: pH DEPENDENCE OF ANTHRAX TOXIN BINDING CONSTANT
The apparent dissociation constant of LF:PA63 channel (Kd ≈ 20 pM, pHc|t 7.2|7.2) was estimated from the ratio of the instantaneous ionic current at V = +70 mV for different LF concentrations (Fig. 8, □). Note that positive potentials were used to estimate the apparent Kd for anthrax toxin because the effect on the channel conductance is more pronounced and less prone to gating. For the pH gradient pHc|t 5.5|7.2, the Kd increased approximately fourfold (Fig. 8, ○, also see inset), which suggests the binding of the two toxins became slightly weaker as the pH decreased. These results are consistent with our earlier estimate of the dissociation constant, Kd ≈ 40 pM, for pHc|t 6.6|6.6.24, 25 The pH dependence of the interaction between LF and the channel shown here and work by others,24, 86, 125, 130 suggest that the attraction between the anthrax toxin is, in part, electrostatic.
References
- Bradley K., Mogridge J., Mourez M., Collier R., and Young J., Nature (London) 414, 225 (2001). 10.1038/n35101999 [DOI] [PubMed] [Google Scholar]
- Scobie H., Rainey G., Bradley K., and Young J., Proc. Natl. Acad. Sci. U.S.A. 100, 5170 (2003). 10.1073/pnas.0431098100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martchenko M., Jeong S.-Y., and Cohen S. N., Proc. Natl. Acad. Sci. U.S.A. 107, 15583 (2010). 10.1073/pnas.1010145107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy S., Bresnahan P., Leppla S., Klimpel K., and Thomas G., J. Biol. Chem. 267, 16396 (1992). [PubMed] [Google Scholar]
- Klimpel K. R., Molloy S. S., Thomas G., and Leppla S. H., Proc. Natl. Acad. Sci. U.S.A. 89, 10277 (1992). 10.1073/pnas.89.21.10277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petosa C., Collier R. J., Klimpel K. R., Leppla S. H., and Liddington R. C., Nature (London) 385, 833 (1997). 10.1038/385833a0 [DOI] [PubMed] [Google Scholar]
- Elliott J. L., Mogridge J., and Collier R. J., Biochemistry 39, 6706 (2000). 10.1021/bi000310u [DOI] [PubMed] [Google Scholar]
- Abrami L., Liu S. H., Cosson P., Leppla S., and van der Goot F., J. Cell Biol. 160, 321 (2003). 10.1083/jcb.200211018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J., Nat. Rev. Mol. Cell Biol. 2, 721 (2001). 10.1038/35096054 [DOI] [PubMed] [Google Scholar]
- Abrami L., Lindsay M., Parton R. G., Leppla S. H., and van der Goot F. G., J. Cell Biol. 166, 645 (2004). 10.1083/jcb.200312072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintzer A. F., Sterling H. J., Tang I. I., Williams E. R., and Krantz B. A., PLoS ONE 5, e13888 (2010). 10.1371/journal.pone.0013888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintzer A. F., Sterling H. J., Tang I. I., Abdul-Gader A., Miles A. J., Wallace B. A., Williams E. R., and Krantz B. A., J. Mol. Biol. 399, 741 (2010). 10.1016/j.jmb.2010.04.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld G. K., Thoren K. L., Kintzer A. F., Sterling H. J., Tang I. I., Greenberg S. G., Williams E. R., and Krantz B. A., Nat. Struct. Mol. Biol. 17, 1383 (2010). 10.1038/nsmb.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duesbery N. S., Science 280, 734 (1998). 10.1126/science.280.5364.734 [DOI] [PubMed] [Google Scholar]
- Friedlander A. M., J. Biol. Chem. 261, 7123 (1986). [PubMed] [Google Scholar]
- Leppla S. H., Proc. Natl. Acad. Sci. U.S.A. 79, 3162 (1982). 10.1073/pnas.79.10.3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Arocho F. J., Fulcher J. A., Lee B., and Bradley K. A., Mol. Microbiol. 61, 324 (2006). 10.1111/j.1365-2958.2006.05232.x [DOI] [PubMed] [Google Scholar]
- Guidi-Rontani C., Weber-Levy M., Mock M., and Cabiaux V., Cell. Microbiol. 2, 259 (2000). 10.1046/j.1462-5822.2000.00057.x [DOI] [PubMed] [Google Scholar]
- Young J. A. T. and Collier R. J., Annu. Rev. Biochem. 76, 243 (2007). 10.1146/annurev.biochem.75.103004.142728 [DOI] [PubMed] [Google Scholar]
- Feld G. K., Brown M. J., and Krantz B. A., Protein Sci. 21, 606 (2012). 10.1002/pro.2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein R. and Finkelstein A., J. Gen. Physiol. 96, 905 (1990). 10.1085/jgp.96.5.905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. L., J. Biomol. Struct. Dyn. 22, 253 (2004). 10.1080/07391102.2004.10531226 [DOI] [PubMed] [Google Scholar]
- Nablo B. J., Halverson K. M., Robertson J. W. F., Nguyen T. L., Panchal R. G., Gussio R., Bavari S., Krasilnikov O. V., and Kasianowicz J. J., Biophys. J. 95, 1157 (2008). 10.1529/biophysj.107.121715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halverson K. M., Panchal R. G., Nguyen T. L., Gussio R., Little S. F., Misakian M., Bavari S., and Kasianowicz J. J., J. Biol. Chem. 280, 34056 (2005). 10.1074/jbc.M507928200 [DOI] [PubMed] [Google Scholar]
- Panchal R. G., Halverson K. M., Ribot W., Lane D., Kenny T., Abshire T. G., Ezzell J. W., Hoover T. A., Powell B., Little S., Kasianowicz J. J., and Bavari S., J. Biol. Chem. 280, 10834 (2005). 10.1074/jbc.M412210200 [DOI] [PubMed] [Google Scholar]
- Neumeyer T., Tonello F., Dal Molin F., Schiffler B., and Benz R., J. Biol. Chem. 281, 32335 (2006). 10.1074/jbc.M606552200 [DOI] [PubMed] [Google Scholar]
- Neumeyer T., Tonello F., Dal Molin F., Schiffler B., Orlik F., and Benz R., Biochemistry 45, 3060 (2006). 10.1021/bi0524316 [DOI] [PubMed] [Google Scholar]
- Krantz B., Finkelstein A., and Collier R. J., J. Mol. Biol. 355, 968 (2006). 10.1016/j.jmb.2005.11.030 [DOI] [PubMed] [Google Scholar]
- Krantz B., Trivedi A., Cunningham K., Christensen K., and Collier R. J., J. Mol. Biol. 344, 739 (2004). 10.1016/j.jmb.2004.09.067 [DOI] [PubMed] [Google Scholar]
- Krantz B. A., Melnyk R. A., Zhang S., Juris S. J., Lacy D. B., Wu Z., Finkelstein A., and Collier R. J., Science 309, 777 (2005). 10.1126/science.1113380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Finkelstein A., and Collier R. J., Proc. Natl. Acad. Sci. U.S.A. 101, 16756 (2004). 10.1073/pnas.0405754101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Udho E., Wu Z., Collier R., and Finkelstein A., Biophys. J. 87, 3842 (2004). 10.1529/biophysj.104.050864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl F.-U., Pfanner N., Nicholson D. W., and Neupert W., Biochim. Biophys. Acta 988, 1 (1989). 10.1016/0304-4157(89)90002-6 [DOI] [PubMed] [Google Scholar]
- Wagner E., Plank C., Zatloukal K., Cotten M., and Birnstiel M. L., Proc. Natl. Acad. Sci. U.S.A. 89, 7934 (1992). 10.1073/pnas.89.17.7934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider H.-C., Berthold J., Bauer M. F., Dietmeier K., Guiard B., Brunner M., and Neupert W., Nature (London) 371, 768 (1994). 10.1038/371768a0 [DOI] [PubMed] [Google Scholar]
- Kasianowicz J. J., Brandin E., Branton D., and Deamer D., Proc. Natl. Acad. Sci. U.S.A. 93, 13770 (1996). 10.1073/pnas.93.24.13770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert W., Annu. Rev. Biochem. 66, 863 (1997). 10.1146/annurev.biochem.66.1.863 [DOI] [PubMed] [Google Scholar]
- Hill K., Model K., Ryan M., Dietmeier K., Martin F., Wagner R., and Pfanner N., Nature (London) 395, 516 (1998). 10.1038/26780 [DOI] [PubMed] [Google Scholar]
- Lubensky D. and Nelson D., Biophys. J. 77, 1824 (1999). 10.1016/S0006-3495(99)77027-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthukumar M., J. Chem. Phys. 111, 10371 (1999). 10.1063/1.480386 [DOI] [Google Scholar]
- de Gennes P. G. P., Proc. Natl. Acad. Sci. U.S.A. 96, 7262 (1999). 10.1073/pnas.96.13.7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnah S. C., Wagner R., Sveshnikova N., Harrer R., and Soll J., Biophys. J. 83, 899 (2002). 10.1016/S0006-3495(02)75216-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthukumar M., J. Chem. Phys. 118, 5174 (2003). 10.1063/1.1553753 [DOI] [Google Scholar]
- DiMarzio E. and Kasianowicz J., J. Chem. Phys. 119, 6378 (2003). 10.1063/1.1603725 [DOI] [Google Scholar]
- Grigoriev S. M., Muro C., Dejean L. M., Campo M. L., Martinez-Caballero S., and Kinnally K. W., Int. Rev. Cytol. 238, 227 (2004). 10.1016/S0074-7696(04)38005-8 [DOI] [PubMed] [Google Scholar]
- Neupert W. and Herrmann J. M., Annu. Rev. Biochem. 76, 723 (2007). 10.1146/annurev.biochem.76.052705.163409 [DOI] [PubMed] [Google Scholar]
- Martinez-Caballero S., Peixoto P. M. V., Kinnally K. W., and Campo M. L., Anal. Biochem. 362, 76 (2007). 10.1016/j.ab.2006.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokranjac D. and Neupert W., Biochim. Biophys. Acta 1777, 758 (2008). 10.1016/j.bbabio.2008.04.009 [DOI] [PubMed] [Google Scholar]
- Kasianowicz J. J., Robertson J. W. F., Chan E. R., Reiner J. E., and Stanford V. M., Annu. Rev. Anal. Chem. 1, 737 (2008). 10.1146/annurev.anchem.1.031207.112818 [DOI] [PubMed] [Google Scholar]
- Wong C. T. A. and Muthukumar M., J. Chem. Phys. 128, 154903 (2008). 10.1063/1.2897932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokranjac D. and Neupert W., Biochim. Biophys. Acta 1793, 33 (2009). 10.1016/j.bbamcr.2008.06.021 [DOI] [PubMed] [Google Scholar]
- Wagener N., Ackermann M., Funes S., and Neupert W., Mol. Cell 44, 191 (2011). 10.1016/j.molcel.2011.07.036 [DOI] [PubMed] [Google Scholar]
- Muthukumar M., Polymer Translocation (CRC, Boca Raton, 2011). [Google Scholar]
- Ezzell J. W., Abshire T. G., Panchal R., Chabot D., Bavari S., Leffel E. K., Purcell B., Friedlander A. M., and Ribot W. J., Infect. Immun. 77, 749 (2009). 10.1128/IAI.00764-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribot W. J., Panchal R. G., Brittingham K. C., Ruthel G., Kenny T. A., Lane D., Curry B., Hoover T. A., Friedlander A. M., and Bavari S., Infect. Immun. 74, 5029 (2006). 10.1128/IAI.00275-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz D., Jaax N., Lawrence W., Davis K., Pitt M., Ezzell J., and Friedlander A., Lab. Invest. 73, 691 (1995). [PubMed] [Google Scholar]
- Ivins B., Welkos S., Knudson G., and Little S., Infect. Immun. 58, 303 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt M., Little S., Ivins B., Fellows P., Barth J., Hewetson J., Gibbs P., Dertzbaugh M., and Friedlander A., Vaccine 19, 4768 (2001). 10.1016/S0264-410X(01)00234-1 [DOI] [PubMed] [Google Scholar]
- Zaucha G., Pitt M., Estep J., Ivins B., and Friedlander A., Arch. Pathol. Lab. Med. 122, 982 (1998). [PubMed] [Google Scholar]
- Ezzell J. W. and Abshire T. G., J. Gen. Microbiol. 138, 543 (1992). 10.1099/00221287-138-3-543 [DOI] [PubMed] [Google Scholar]
- Chabot D., Scorpio A., Tobery S., Little S., Norris S., and Friedlander A., Vaccine 23, 43 (2004). 10.1016/j.vaccine.2004.05.029 [DOI] [PubMed] [Google Scholar]
- Kimura K. and Itoh Y., Appl. Environ. Microbiol. 69, 2491 (2003). 10.1128/AEM.69.5.2491-2497.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montal M. and Mueller P., Proc. Natl. Acad. Sci. U.S.A. 69, 3561 (1972). 10.1073/pnas.69.12.3561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickmann K., Demming F., and Jersch J., Rev. Sci. Instrum. 67, 845 (1996). 10.1063/1.1146655 [DOI] [Google Scholar]
- Iwami M., Uehara Y., and Ushioda S., Rev. Sci. Instrum. 69, 4010 (1998). 10.1063/1.1149215 [DOI] [Google Scholar]
- Funakoshi K., Suzuki H., and Takeuchi S., Anal. Chem. 78, 8169 (2006). 10.1021/ac0613479 [DOI] [PubMed] [Google Scholar]
- Malmstadt N., Nash M. A., Purnell R. F., and Schmidt J. J., Nano Lett. 6, 1961 (2006). 10.1021/nl0611034 [DOI] [PubMed] [Google Scholar]
- Winterhalter M. and Helfrich W., Phys. Rev. A 36, 5874 (1987). 10.1103/PhysRevA.36.5874 [DOI] [PubMed] [Google Scholar]
- Klotz K., Winterhalter M., and Benz R., Biochim. Biophys. Acta 1147, 161 (1993). 10.1016/0005-2736(93)90327-V [DOI] [PubMed] [Google Scholar]
- Wilhelm C., Winterhalter M., and Zimmermann U., Biophys. J. 64, 121 (1993). 10.1016/S0006-3495(93)81346-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterhalter M., Klotz K. H., Benz R., and Arnold W. M., IEEE Trans. Ind. Appl. 32, 125 (1996). 10.1109/28.485823 [DOI] [Google Scholar]
- Milne J. C. and Collier R. J., Mol. Microbiol. 10, 647 (1993). 10.1111/j.1365-2958.1993.tb00936.x [DOI] [PubMed] [Google Scholar]
- Merzlyak P., Capistrano M., Valeva A., Kasianowicz J., and Krasilnikov O., Biophys. J. 89, 3059 (2005). 10.1529/biophysj.105.066472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley I. G., Carroll K., Bittova L., and Pfeffer S., Mol. Biol. Cell 15, 5420 (2004). 10.1091/mbc.E04-08-0747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basilio D., Kienker P. K., Briggs S. W., and Finkelstein A., J. Gen. Physiol. 137, 521 (2011). 10.1085/jgp.201110627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannifer A., Wong T., Schwarzenbacher R., Renatus M., Petosa C., Bienkowska J., Lacy D., Collier R. J., Park S., Leppla S., Hanna P., and Liddington R., Nature (London) 414, 229 (2001). 10.1038/n35101998 [DOI] [PubMed] [Google Scholar]
- Mourez M., Kane R. S., Mogridge J., Metallo S., Deschatelets P., Sellman B. R., Whitesides G. M., and Collier R. J., Nat. Biotechnol. 19, 958 (2001). 10.1038/nbt1001-958 [DOI] [PubMed] [Google Scholar]
- Rai P., Padala C., Poon V., Saraph A., Basha S., Kate S., Tao K., Mogridge J., and Kane R. S., Nat. Biotechnol. 24, 582 (2006). 10.1038/nbt1204 [DOI] [PubMed] [Google Scholar]
- Ambjornsson T., Apell S., Konkoli Z., DiMarzio E., and Kasianowicz J., J. Chem. Phys. 117, 4063 (2002). 10.1063/1.1486208 [DOI] [Google Scholar]
- Fischer A., Holden M. A., Pentelute B. L., and Collier R. J., Proc. Natl. Acad. Sci. U.S.A. 108, 16577 (2011). 10.1073/pnas.1113074108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg H. and Purcell E., Biophys. J. 20, 193 (1977). 10.1016/S0006-3495(77)85544-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier R. J., Mol. Aspects Med. 30, 413 (2009). 10.1016/j.mam.2009.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basilio D., Juris S. J., Collier R. J., and Finkelstein A., J. Gen. Physiol. 133, 307 (2009). 10.1085/jgp.200810170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowiak B. E., Fischer A., and Collier R. J., J. Biol. Chem. 285, 8130 (2010). 10.1074/jbc.M109.093195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zornetta I., Brandi L., Janowiak B., Dal Molin F., Tonello F., Collier R. J., and Montecucco C., Cell. Microbiol. 12, 1435 (2010). 10.1111/j.1462-5822.2010.01480.x [DOI] [PubMed] [Google Scholar]
- Pentelute B. L., Sharma O., and Collier R. J., Angew. Chem., Int. Ed. 50, 2294 (2011). 10.1002/anie.201006460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentelute B. L., Barker A. P., Janowiak B. E., Kent S. B. H., and Collier R. J., ACS Chem. Biol. 5, 359 (2010). 10.1021/cb100003r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoop W., Xiong Y., Wiltsie J., Woods A., Guo J., Pivnichny J., Felcetto T., Michael B., Bansal A., Cummings R., Cunningham B., Friedlander A., Douglas C., Patel S., Wisniewski D., Scapin G., Salowe S., Zaller D., Chapman K., Scolnick E., Schmatz D., Bartizal K., MacCoss M., and Hermes J., Proc. Natl. Acad. Sci. U.S.A. 102, 7958 (2005). 10.1073/pnas.0502159102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings-Antipov L. D., Song L., and Collier R. J., Proc. Natl. Acad. Sci. U.S.A. 108, 1868 (2011). 10.1073/pnas.1018965108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz R., Beckers F., and Zimmermann U., J. Membr. Biol. 48, 181 (1979). 10.1007/BF01872858 [DOI] [PubMed] [Google Scholar]
- Abidor I. G., Arakelyan V. B., Chernomordik L. V., Chizmadzhev Y. A., Pastushenko V. F., and Tarasevich M. P., J. Electroanal. Chem. Interfacial Electrochem. 104, 37 (1979). 10.1016/S0022-0728(79)81006-2 [DOI] [Google Scholar]
- Chernomordik L. V. and Abidor I. G., Bioelectrochem. Bioenerg. 7, 617 (1980). 10.1016/0302-4598(80)80028-6 [DOI] [Google Scholar]
- Abidor I. G., Chernomordik L. V., and Sukharev S. I., Bioelectrochem. Bioenerg. 9, 141 (1982). 10.1016/0302-4598(82)80170-0 [DOI] [Google Scholar]
- Chernomordik L. V., Sukharev S. I., Abidor I. G., and Chizmadzhev Y. A., Biochim. Biophys. Acta 736, 203 (1983). 10.1016/0005-2736(83)90285-7 [DOI] [Google Scholar]
- Chernomordik L., Kozlov M., Melikyan G., Abidor I., Markin V., and Chizmadzhev Y., Biochim. Biophys. Acta 812, 643 (1985). 10.1016/0005-2736(85)90257-3 [DOI] [Google Scholar]
- Scott C. C. and Gruenberg J., Bioessays 33, 103 (2011). 10.1002/bies.201000108 [DOI] [PubMed] [Google Scholar]
- Hille B., Ion Channels of Excitable Membranes, 3rd ed. (Sunderland, Massachusetts, 2001). [Google Scholar]
- Sonawane N., Thiagarajah J., and Verkman A., J. Biol. Chem. 277, 5506 (2002). 10.1074/jbc.M110818200 [DOI] [PubMed] [Google Scholar]
- Rybak S., Lanni F., and Murphy R., Biophys. J. 73, 674 (1997). 10.1016/S0006-3495(97)78102-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duclohier H. and Wróblewski H., J. Membr. Biol. 184, 1 (2001). 10.1007/s00232-001-0077-2 [DOI] [PubMed] [Google Scholar]
- Niles W. D., Levis R. A., and Cohen F. S., Biophys. J. 53, 327 (1988). 10.1016/S0006-3495(88)83110-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdon J., Falge M., Maier E., Bruhn H., Steinert M., Faber C., Benz R., and HEchard Y., Biophys. J. 97, 1933 (2009). 10.1016/j.bpj.2009.06.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkouhi A. K., Scholte M., Storm G., and Haisma H. J., J. Controlled Release 151, 220 (2011). 10.1016/j.jconrel.2010.11.004 [DOI] [PubMed] [Google Scholar]
- Moreira C., Oliveira H., Pires L. R., Simões S., Barbosa M. A., and Pêgo A. P., Acta Biomater. 5, 2995 (2009). 10.1016/j.actbio.2009.04.021 [DOI] [PubMed] [Google Scholar]
- Wyman T., Nicol F., Zelphati O., Scaria P., Plank C., and Szoka F., Biochemistry 36, 3008 (1997). 10.1021/bi9618474 [DOI] [PubMed] [Google Scholar]
- Kichler A., Leborgne C., Marz J., Danos O., and Bechinger B., Proc. Natl. Acad. Sci. U.S.A. 100, 1564 (2003). 10.1073/pnas.0337677100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikov K. and Chernomordik L. V., Cell. Mol. Life Sci. 62, 2739 (2005). 10.1007/s00018-005-5293-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatefi A., Megeed Z., and Ghandehari H., J. Gene Med. 8, 468 (2006). 10.1002/jgm.872 [DOI] [PubMed] [Google Scholar]
- Kichler A., Leborgne C., Danos O., and Bechinger B., J. Mol. Med. 85, 191 (2006). 10.1007/s00109-006-0119-4 [DOI] [PubMed] [Google Scholar]
- Min S.-H., Lee D. C., Lim M. J., Park H. S., Kim D. M., Cho C. W., Yoon D. Y., and Yeom Y. I., J. Gene Med. 8, 1425 (2006). 10.1002/jgm.973 [DOI] [PubMed] [Google Scholar]
- Lebleu B., Moulton H. M., Abes R., Ivanova G. D., Abes S., Stein D. A., Iversen P. L., Arzumanov A. A., and Gait M. J., Adv. Drug Delivery Rev. 60, 517 (2008). 10.1016/j.addr.2007.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abes R., Arzumanov A., Moulton H., Abes S., Ivanova G., Gait M. J., Iversen P., and Lebleu B., J. Pept. Sci. 14, 455 (2008). 10.1002/psc.979 [DOI] [PubMed] [Google Scholar]
- Lin C. and Engbersen J. F. J., J. Controlled Release 132, 267 (2008). 10.1016/j.jconrel.2008.06.022 [DOI] [PubMed] [Google Scholar]
- Boussif O., Lezoualc'h F., Zanta M., Mergny M. D., Scherman D., Demeneix B., and Behr J., Proc. Natl. Acad. Sci. U.S.A. 92, 7297 (1995). 10.1073/pnas.92.16.7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon E. J., Bergen J. M., and Pun S. H., Bioconjugate Chem. 19, 920 (2008). 10.1021/bc700448h [DOI] [PubMed] [Google Scholar]
- Miller D. K., Griffiths E., Lenard J., and Firestone R. A., J. Cell Biol. 97, 1841 (1983). 10.1083/jcb.97.6.1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerklotz H. and Seelig J., Biophys. J. 81, 1547 (2001). 10.1016/S0006-3495(01)75808-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff M., Nature (London) 415, 389 (2002). 10.1038/415389a [DOI] [PubMed] [Google Scholar]
- Corin K., Baaske P., Ravel D. B., Song J., Brown E., Wang X., Wienken C. J., Jerabek-Willemsen M., Duhr S., Luo Y., Braun D., and Zhang S., PLoS ONE 6, e25067 (2011). 10.1371/journal.pone.0025067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand B., Laurencin M., Sarkis J., Duval E., Mouret L., Hubert J.-F., Collen M., Vié V., Zatylny-Gaudin C., Henry J., Baudy-Floc'h M., and Bondon A., Biochim. Biophys. Acta 1808, 106 (2011). 10.1016/j.bbamem.2010.08.020 [DOI] [PubMed] [Google Scholar]
- Bechinger B. and Lohner K., Biochim. Biophys. Acta 1758, 1529 (2006). 10.1016/j.bbamem.2006.07.001 [DOI] [PubMed] [Google Scholar]
- Basilio D., Jennings-Antipov L. D., Jakes K. S., and Finkelstein A., J. Gen. Physiol. 137, 343 (2011). 10.1085/jgp.201010578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein R. and Finkelstein A., J. Gen. Physiol. 96, 943 (1990). 10.1085/jgp.96.5.943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson D. L., Tippetts M. T., and Leppla S. H., Gene 73, 363 (1988). 10.1016/0378-1119(88)90501-X [DOI] [PubMed] [Google Scholar]
- Brown M. J., Thoren K. L., and Krantz B. A., J. Biol. Chem. 286, 23189 (2011). 10.1074/jbc.M111.231167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikis D., Elcock A. H., Jennings P. A., and McCammon J. A., Protein Sci. 10, 2363 (2001). 10.1110/ps.17201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason A. J., Gasnier C., Kichler A., Prevost G., Aunis D., Metz-Boutigue M. H., and Bechinger B., Antimicrob. Agents Chemother. 50, 3305 (2006). 10.1128/AAC.00490-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midoux P., Kichler A., Boutin V., Maurizot J. C., and Monsigny M., Bioconjugate Chem. 9, 260 (1998). 10.1021/bc9701611 [DOI] [PubMed] [Google Scholar]
- Midoux P. and Monsigny M., Bioconjugate Chem. 10, 406 (1999). 10.1021/bc9801070 [DOI] [PubMed] [Google Scholar]
- Orlik F., Schiffler B., and Benz R., Biophys. J. 88, 1715 (2005). 10.1529/biophysj.104.050336 [DOI] [PMC free article] [PubMed] [Google Scholar]