

It is shown that SraL sRNA (RyjA), a sRNA present in many bacteria, down-regulates the expression of the chaperone Trigger Factor (TF), encoded by the tig gene and one of the three major ribosome-associated chaperones. SraL interacts directly with the 5′ UTR of the tig mRNA a few nucleotides upstream of the Shine-Dalgarno region. This constitutes the first report of a small RNA affecting protein folding.
Keywords: S. Typhimurium, RyjA, σS, PPIase, SraL
Abstract
Small noncoding RNAs (sRNAs) are usually expressed in the cell to face a variety of stresses. In this report we disclose the first target for SraL (also known as RyjA), a sRNA present in many bacteria, which is highly induced in stationary phase. We also demonstrate that this sRNA is directly transcribed by the major stress σ factor σS (RpoS) in Salmonella enterica serovar Typhimurium. We show that SraL sRNA down-regulates the expression of the chaperone Trigger Factor (TF), encoded by the tig gene. TF is one of the three major chaperones that cooperate in the folding of the newly synthesized cytosolic proteins and is the only ribosome-associated chaperone known in bacteria. By use of bioinformatic tools and mutagenesis experiments, SraL was shown to directly interact with the 5′ UTR of the tig mRNA a few nucleotides upstream of the Shine-Dalgarno region. Namely, point mutations in the sRNA (SraL*) abolished the repression of tig mRNA and could only down-regulate a tig transcript target with the respective compensatory mutations. We have also validated in vitro that SraL forms a stable duplex with the tig mRNA. This work constitutes the first report of a small RNA affecting protein folding. Taking into account that both SraL and TF are very well conserved in enterobacteria, this work will have important repercussions in the field.
INTRODUCTION
Small noncoding RNAs (sRNAs) perform a wide diversity of regulatory functions in both prokaryotic and eukaryotic cells. The majority of the sRNAs act by base-pairing with mRNA targets (antisense sRNAs) or by binding to proteins to modify their activity (for a review, see Storz et al. 2011). Most of the antisense sRNAs are trans-encoded since they are encoded in a separate locus in relation with the mRNA target. Consequently, these sRNAs exhibit only partial complementarity with the target and usually require the RNA chaperone Hfq for base-pairing. Typically, trans-encoded sRNAs are induced under environmental stress conditions and also upon entry into stationary phase of growth in order to up- or down-regulate their target(s) (Gottesman and Storz 2011).
A plethora of sRNAs have been identified in the last years; for instance, in Salmonella enterica serovar Typhimurium (S. Typhimurium), 140 sRNAs were reported in early stationary phase of growth by using a combination of RNA-seq and dRNA-seq analyses and Hfq-coIP-seq approach (Kröger et al. 2012).
sRNAs are generally highly controlled at the transcriptional level. Nearly one-third of the functional characterized sRNAs contribute to the control of the outer membrane protein (OMP) production. Some of these sRNAs are under the control of the σ factor RpoE (also known as σE or σ24) (Johansen et al. 2006; Papenfort et al. 2006; Udekwu and Wagner 2007; Johansen et al. 2008), which regulates gene expression upon the accumulation of misfolded OMPs in the periplasmic space (Mecsas et al. 1993; Missiakas et al. 1996; Raivio and Silhavy 1999). However, only a few sRNAs have been reported to be transcribed by the σ factor RpoS (also known as σS or σ38) (Opdyke et al. 2004; Padalon-Brauch et al. 2008; Fröhlich et al. 2012). This major stress σ factor regulates 10% of the Escherichia coli genes (Weber et al. 2005) and is induced under several stress conditions, namely, the entry into the stationary phase of growth (Battesti et al. 2011). RpoS is known to play important roles in the virulence of many bacterial pathogens, including S. Typhimurium (Dong and Schellhorn 2010).
SraL (also known as RyjA) is a 140-nucleotide (nt) antisense sRNA first described in 2001 in two exhaustive genetic studies (Argaman et al. 2001; Wassarman et al. 2001), in which a combination of different approaches was used in order to identify novel sRNAs in E. coli. Subsequently, this sRNA was also detected in S. Typhimurium (Viegas et al. 2007; Ortega et al. 2012). SraL sRNA is localized between soxR (Amábile-Cuevas and Demple 1991) and a gene encoding a putative glutathione S-transferase (STM4267), but it is transcribed in the opposite strand. SraL sRNA was only detected in cells during the entry into stationary phase (Argaman et al. 2001; Wassarman et al. 2001; Viegas et al. 2007), and its expression is particularly high in stationary phase (OD600 2 + 6 h). Moreover, its expression was also highly detected under Salmonella pathogenicity island (SPI)-2–inducing conditions (Viegas et al. 2007), which indicates a possible role for SraL in Salmonella virulence since SPI-2 genes are important for intramacrophage survival and systemic disease. SraL is also expressed in intracellular S. Typhimurium persisting inside eukaryotic cells (Ortega et al. 2012). The study of the post-transcriptional regulation of SraL through the use of several Salmonella ribonucleases mutants showed that this sRNA is controlled by RNases such as PNPase and the degradosome complex (Viegas et al. 2007). Moreover, it was shown that Poly(A) Polymerase I (PAP I) has a major impact in the control of the stability of this sRNA (Viegas et al. 2007). This fact was in agreement with previous 3′ RACE experiments that revealed the existence of 3′ A-tails of different lengths in the E. coli SraL transcript (Argaman et al. 2001).
In this work, we have determined that RpoS (the major stationary phase regulator) is a transcriptional regulator of the highly conserved sRNA SraL in S. Typhimurium. SraL transcription is dependent on the presence of RpoS in the cell, and we have proved that this regulation is direct since RpoS directly binds to the promoter of the sraL gene. We have also investigated the biological role of SraL since no targets were yet discovered for this sRNA. A proteomic analysis using a S. Typhimurium SraL null mutant and a SraL overexpressing strain detected Trigger Factor (TF) as a possible target. TF is one of the three major cytosolic chaperone proteins found in all eubacteria and assists in protein folding (Hesterkamp et al. 1996). This chaperone is the only ribosome-associated chaperone known in bacteria (Hoffmann et al. 2010). By using mutational analysis both in vivo and in vitro, we have determined that SraL represses tig mRNA through a short stretch of complementarity in the tig 5′ UTR near the Shine-Dalgarno region. The results obtained in this study constitute the first link between sRNAs and protein folding.
RESULTS
SraL sRNA is conserved among several enteric bacteria
SraL sRNA (also known as RyjA) was first discovered in two independent studies in E. coli, in which the use of comparative genomics and microarrays allowed the identification of novel sRNAs (Argaman et al. 2001; Wassarman et al. 2001). More recently, it was confirmed that this sRNA is also expressed in S. Typhimurium (Viegas et al. 2007; Ortega et al. 2012).
Since SraL expression was detected in both E. coli and S. Typhimurium, we performed an extensive search over the genomes of other enteric bacteria using BlastN. This sRNA was shown to be highly conserved, being identified in bacteria such as Shigella, Citrobacter, and Klebsiella (Fig. 1A).
FIGURE 1.

Analysis of SraL sequence, structure, and expression. (A) Alignment of the sraL gene including the upstream promoter region in several enterobacteria. All nucleotides are colored regarding their degree of conservation (red indicates high conservation; blue, partial conservation; black, little or no conservation). The asterisks (*) below the sequences indicate the nucleotides conserved between all the species analyzed. The putative −10 and −35 boxes of the sraL promoter are indicated (Argaman et al. 2001). The consensus sequences of −10 and −35 boxes of RpoS and the RpoS-specific promoter sequence features located around the −10 core promoter element (A/T-rich discriminator) are indicated (Typas et al. 2007). Parts of the −35 and −10 elements that are often degenerate in RpoS-dependent promoters are shown in italics (the least conserved nucleotides in lowercase letters) (Typas et al. 2007). +1 marks the transcriptional start site. The Rho (ρ)-independent terminator is indicated by arrows. The SraL sRNA interaction region with tig mRNA is indicated in yellow. Y represents a C or a T; STM, Salmonella enterica serovar Typhimurium; STY, Salmonella enterica serovar Typhi; SBO, Salmonella bongori; SBD, Shigella boydii; SFL, Shigella flexneri; SDR, Shigella dysenteriae; ECO, Escherichia coli; CRT, Citrobacter rodentium; CKO, Citrobacter koseri; ENT, Enterobacter; and KPN, Klebsiella pneumoniae. (B) S. Typhimurium SraL sRNA structure predicted by the Mfold program (Zuker 2003). (C) SraL sRNA expression under different growth conditions. Cells were grown in LB at 37°C until an OD600 of 0.5 (OD0.5), 1 (OD1), and 2 (OD2); 3 h after OD2 (OD2 + 3 h); and 6 h after OD2 (OD2 + 6 h). Cells were also grown under conditions of induction of SPI-1 and SPI-2 genes. For heat and cold shock, cells were grown in LB until an OD600 of 0.5 and then subjected to cold shock (10°C) for 30 min and 4 h (CS 30′ and CS 4 h, respectively) and heat shock (42°C) for 15 min (HS 15′). Fifteen micrograms of total RNA were run on a 6% PAA/8.3 M urea gel. SraL was detected using a riboprobe; probing for 5S rRNA was used as a loading control.
By use of the MFold program (http://mfold.rna.albany.edu/?q=mfold) (Zuker 2003), we predicted related secondary structures for SraL sRNA of several enteric bacteria. The most stable predicted structure of the sRNA in S. Typhimurium is shown in Figure 1B. Despite some small differences, the majority of SraL structures represented show a high resemblance, and all contain the same Rho-independent terminator (Fig. 1B; Supplemental Fig. S1).
We compared the expression of S. Typhimurium SraL under different growth conditions. When grown in LB at 37°C, SraL is mostly detected in the cells upon entry in stationary phase of growth (Fig. 1C). Moreover, this sRNA is also expressed in conditions that induce SPI-1 and SPI-2, indicating a possible role of SraL in Salmonella virulence (Fig. 1C). More specifically, SraL seems to be necessary after internalization of this bacterium into host cells since its expression is much higher under SPI-2–inducing conditions. In fact, it was recently shown that SraL is expressed in intracellular bacteria located in fibroblasts at 24 h post-infection (Ortega et al. 2012). SraL is also expressed in cells subjected to heat shock, as well as 4 h after growth in cold-shock conditions and under SPI-1–inducing conditions, albeit at much lower levels (Fig. 1C).
SraL sRNA is directly regulated by σS
Since SraL sRNA is conserved among Enterobacteriaceae and its expression is induced preferentially in stationary phase, we hypothesized that it could be part of the general stress response orchestrated by the σS factor of the RNA polymerase that operates in this growth phase. To this aim, we first examined the sraL promoter in search of conserved sequence elements that show specific features of promoters of bona fide RpoS-regulated genes. From the alignment of the immediately 75 nt upstream sequence of sraL in several enteric bacteria, we noticed some traits that are characteristic of an RpoS-regulated promoter (Fig. 1A; Typas et al. 2007). In this regard, we observed a conserved −10 box that fits well with the consensus sequence retrieved from experimentally determined RpoS-regulated genes, including the A/T-rich motif downstream from the −10 box (Fig. 1A; Weber et al. 2005; Typas et al. 2007). Moreover, the −35 box is also characteristic of an RpoS-regulated promoter. These observations suggested a plausible selectivity of RpoS for the sraL promoter.
To test experimentally the putative RpoS dependence on SraL expression, we first constructed an rpoS null mutant by P22 transduction from SV4210 strain (Tierrez and Garcia-del Portillo 2004) and also a complemented strain in which rpoS was cloned into a constitutive expression plasmid. Then, we compared SraL levels among the wild-type, the isogenic rpoS null mutant, and the complemented rpoS mutant strain throughout stationary phase, the growth condition where SraL is highly expressed (see Fig. 1C). Results presented in Figure 2A (upper panel) show that SraL sRNA is practically absent in the rpoS null mutant in this growth condition. In fact, reverse transcription and real-time quantitative PCR revealed that in stationary phase SraL is about 500 times less abundant in the rpoS mutant than in the wild-type strain (data not shown). Consistently, SraL expression is partially restored in the rpoS mutant upon ectopic expression of a wild-type rpoS allele from a constitutive promoter (Fig. 2A, upper panel). This might be due to the fact that ectopic RpoS is expressed at a much lower level in the complemented strain than in the wild-type strain (Fig. 2A, lower panel). Even so, these low levels of RpoS protein suffice to restore SraL expression (Fig. 2A). These results indicate that SraL expression in stationary phase is highly dependent on RpoS activity.
FIGURE 2.

Analysis of SraL sRNA regulation by RpoS. (A) SraL expression is dependent on RpoS. (Upper panel) SraL levels were detected by Northern blot using 15 μg of total RNA isolated at indicated time points during growth from S. Typhimurium wild-type (wt) and rpoS mutant strains and rpoS mutant strain carrying a constitutive plasmid expressing the wild-type rpoS allele (pISVA-002); 5S rRNA was used as loading control. (Lower panel) RpoS protein expression was monitored by Western blot analysis. Samples correspond to 5 × 107 bacteria at the indicated time points. GroEL was used as a loading control. (B) Transcriptional activity of sraL promoter in late stationary phase. Samples from overnight grown cultures of wild-type SL1344 and the isogenic rpoS null mutant transformed with a plasmid expressing the transcriptional fusion of sraL promoter to lacZ reporter gene (pISVA-003) were used to assess β-galactosidase activity. (C) Transcriptional activity of sraL promoter upon osmotic shock. Bacterial cultures were grown to reach exponential growth phase (OD 0.3), and then NaCl was added to a final concentration of 0.5 M and let grow for one more hour. β-Galactosidase activity was measured before (t0, white bar) and after the treatment (+NaCl, black bar). A control culture with no addition of NaCl was also carried in parallel (control, dashed-light gray bar). Bars correspond to the mean ± SD of three biological replicates. ***P < 0.001 by Student’s t-test; n.s. indicates nonsignificant.
To further examine the SraL regulation by RpoS, we analyzed sraL promoter response in a transcriptional fusion to lacZ reporter gene in both wild-type and rpoS mutant genetic backgrounds. RpoS is known to be induced during entry into stationary phase and/or many other stress conditions. Thus, we first analyzed the transcriptional activity of sraL promoter in stationary phase, and observed a significantly lower sraL promoter-driven β-galactosidase activity when RpoS is not available (Fig. 2B). To rule out any possible bias derived from the growth phase in which these analyses were performed, we investigated the RpoS dependence of sraL expression under high osmolarity, a stress condition that triggers an RpoS-mediated response (Hengge-Aronis et al. 1993). Bacteria were grown to early exponential phase and then 0.5 M NaCl was added, maintaining the bacteria in these stress conditions for 1 h. As a result of the increase in osmolarity, sraL transcriptional activity underwent an almost threefold induction in the wild-type strain, while in the rpoS mutant strain, the sraL promoter expression remained unchanged (Fig. 2C). Consistent with our previous observations on the SraL expression pattern during bacterial growth, the transcriptional activity of sraL promoter was much higher in stationary phase (Fig. 2B) than in exponential growth phase (Fig. 2C). These data suggest that the increase in SraL expression in stationary phase is the result of transcriptional regulation mediated by RpoS.
Up to now, our analysis supports that SraL expression is regulated by RpoS, but it does not differentiate between a direct or indirect regulation. To address this question, we analyzed in vivo the existence of binding of RpoS to the sraL promoter by chromatin immunoprecipitation (ChIP) assays (Raffaelle et al. 2005). The extent of sraL promoter enrichment in the immunoprecipitates (IPs), which is indicative of the binding in vivo of the σ factor to the promoter, was determined by real-time quantitative PCR. We first confirmed the suitability and the specificity of the monoclonal antibody for the immunoprecipitation of RpoS. Immunoprecipitation of uncrosslinked wild-type and rpoS mutant bacterial samples revealed that the antibody has a high affinity and specificity for RpoS, since no immunoreactive bands were visualized in rpoS mutant IPs, while a strong signal around the expected molecular weight for RpoS was obtained with the wild-type strain (Fig. 3A). Two additional bands with a lower mobility were also immunoprecipitated (Fig. 3A, asterisks). Nevertheless, as they are not detected in the rpoS mutant input or IP samples, we reasoned that these immunoreactive bands might correspond to RpoS aggregates rather than an unspecific contaminating protein. These results confirm that the antibody displays a high affinity for RpoS and that it can be used to precipitate specifically DNA–RpoS complexes in vivo in ChIP assays. To assess the specificity of the ChIP assay, we first used osmY promoter as a target DNA sequence (Fig. 3B). OsmY is a periplasmic protein of unknown function previously shown to be regulated by RpoS, and we have used it here as a positive control (Hengge-Aronis et al. 1993; Yim et al. 1994). Consistently, we found a 10-fold enrichment of osmY sequence in RpoS IPs, which indicates a relative high occupancy of osmY promoter by RpoS (Fig. 3B). Interestingly, sraL target sequence was more than 100 times enriched in RpoS IPs compared with the input, which strongly supports the binding of RpoS to sraL promoter in vivo (Fig. 3B). The higher enrichment of sraL promoter in RpoS IPs compared with that of osmY suggests that the transcriptional activity of sraL promoter is larger at the stationary phase, which points out the relevance of the induction of this sRNA at this specific growth phase. No enrichment in rnpB sequence, used here as a negative control, was observed in RpoS IPs. Collectively, these results strongly support that the increased expression levels of SraL sRNA observed during stationary phase result from a transcriptional induction directly mediated by the master regulator of the general stress response RpoS.
FIGURE 3.

In vivo binding of RpoS to the sraL promoter at stationary phase. (A) Anti-RpoS antibody immunoprecipitates the protein with high affinity and specificity. Western blot analysis using mouse monoclonal anti-RpoS antibody of protein samples coming from total extracts (input) and immunoprecipitates either with the anti-RpoS antibody (IP) or with no antibody (AB−). Protein extracts were obtained from wild-type SL1344 and the isogenic rpoS mutant strain. The arrowhead indicates the specific band corresponding to RpoS. Asterisks indicate other immunoreactive bands. IgG HC and LC indicate the heavy and light chains of the immunoglobulin used in the immunoprecipitation, respectively. (B) RpoS binds to sraL promoter in vivo. Chromatin immunoprecipitation (ChIP) assay with anti-RpoS antibody using the wild-type SL1344 (wt) and the isogenic rpoS mutant (rpoS) strains. DNA extracted from the samples was employed as a template for real-time quantitative PCR determination of target sequences (osmY, sraL, rnpB). The amount of target DNA was normalized to 16S (rrs genes) within each sample, and the relative enrichment is referred to the input sample of wild-type strain. Note that the relative enrichment is represented in log10 scale.
SraL sRNA down-regulates the expression of tig mRNA
Although there are some studies about SraL sRNA, the biological function of this sRNA was not yet revealed. To identify SraL targets, we analyzed the proteome in S. Typhimurium strains expressing different levels of SraL. We performed this analysis using cells in stationary phase of growth (OD2 + 6 h), the condition in which this sRNA is more expressed (see Fig. 1C; Viegas et al. 2007). We have constructed a sraL null mutant strain (in which we deleted the entire sequence of the gene) and an overexpressing strain in which the SraL region was cloned into a constitutive expression plasmid. Under our growth conditions, these strains have no significant differences in growth in comparison with the wild-type strain (data not shown). By Northern blot analysis, we could confirm the absence of SraL in the mutant strain and also its overexpression in the strain carrying the SraL overexpressing plasmid (Fig. 4, lower panel).
FIGURE 4.

Regulation of tig mRNA by SraL sRNA. Total cellular RNA was extracted from the S. Typhimurium strains indicated grown in LB at 37°C until 6 h after OD600 of 2. (Upper panel) Twenty micrograms of total RNA was separated on a 1.3% formaldehyde/Agarose gel. The gel was then blotted to a Hybond-N+ membrane and hybridized with the corresponding tig riboprobe. Full-length transcripts were quantified using a Molecular Dynamics PhosphorImager. The amount of RNA in the wild-type was set as one. The ratio between the amounts of RNA of each strain and the wild-type is represented (relative levels). A representative membrane is shown, and the values indicated correspond to the average of several Northern blot experiments with RNAs from at least two independent extractions. The membrane was stripped and then probed for 16S rRNA as loading control. (ND) Nondetectable. (Middle panel) Reverse transcription (RT) PCR experiments were carried out with specific primers for tig using 75 ng of total RNA extracted from the wild-type and derivatives, as indicated in each lane. RT-PCR primers specific for 16S rRNA show that there were not significant variations in the amount of RNA used in each sample. (Lower panel) Fifteen micrograms of RNA was separated on a 6% PAA/8.3 M urea gel. The gel was then blotted to a Hybond-N+ membrane and hybridized with the corresponding SraL riboprobe. Probing for 5S rRNA confirmed equal loading.
A total of 713 proteins were identified and quantified across the three strains analyzed (see Supplemental Table S2). Ten of these proteins were observed to change among the three bacterial strains following a logical regulatory trend: Compared with the wild-type, either they were less represented in the mutant and overrepresented in the complemented strain or vice-versa. These 10 putative targets were chosen to proceed with analyses at the RNA level (see Supplemental Fig. S2). Through the results obtained by reverse transcription (RT) PCR, three of the 10 putative targets matched the proteomic results (NuoG, RfbH, and Tig). Moreover, by using the IntaRNA algorithm (http://www.bioinf.uni-freiburg.de/Software/) (Busch et al. 2008), all of these three putative targets were predicted to base pair with SraL sRNA. Out of these three candidates, we proceeded with the analysis of TF, encoded by the STM0447 (tig) gene, since it appeared to be the most consistent putative target to pursue (Supplemental Fig. S2). At the protein level, there was difference of about twofold between the sraL deletion mutant and the SraL overexpressing strain, and the same tendency was obtained at the RNA level (see Supplemental Fig. S2).
TF is found in all eubacteria and is the first chaperone encountered cotranslationally by most of the nascent chains since it is localized at the exit of the ribosome tunnel (Stoller et al. 1995). This localization enables its binding to nascent polypeptides and prevents improper intra- and/or intermolecular interactions of the chains emerging on the surface of the ribosome (Valent et al. 1995). TF was also shown to be a peptidyl-prolyl cis/trans isomerase (PPIase) and therefore accelerates proline-limited steps in protein folding with a very high efficiency (Stoller et al. 1995; Hesterkamp et al. 1996).
We proceeded with a Northern blot analysis of the same three strains that were analyzed by proteomic and RT-PCR. Two specific tig transcripts were detected in the tig+ strains that were absent in the tig deletion mutant. The larger transcript was detected near the 16S rRNA (∼1.5 kb in size; used as loading control) (Mattatall and Sanderson 1996). The other tig transcript was smaller in size. In E. coli two different tig promoters were described that originate two transcripts with ∼1.5 kb and 1.37 kb (Aldea et al. 1989; Mendoza-Vargas et al. 2009), which seem to match with the sizes of tig transcripts detected in Salmonella. The quantification of the transcripts obtained by Northern blot analysis revealed that when SraL is absent, tig mRNA levels increase about twofold compared with the wild-type (Fig. 4, upper and middle panels). Moreover, there was a 50% reduction of the tig mRNA levels when SraL is transcribed from an overexpressing plasmid. Hereupon, SraL seems to negatively control either directly or indirectly the tig mRNA levels in the conditions tested.
SraL base pairs with tig
To further investigate the role of SraL sRNA in the regulation of tig mRNA, we performed a bioinformatic prediction to identify the interaction region between the sRNA and this target by using IntaRNA (Busch et al. 2008) and RNAHybrid (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/) (Rehmsmeier et al. 2004). Both algorithms were able to predict an imperfect SraL–tig interaction composed by two short segments (7 and 3 bp) (Fig. 5A). Additionally, the predicted interaction between the sRNA and its target corresponds to a well-conserved region in both RNAs (Figs 1A; Supplemental Fig. S2). To test whether pairing was direct and whether the predicted region was required for the interaction, three base changes were introduced in sraL chromosomal region (SraL*) in the predicted base-pairing site with the tig mRNA (Fig. 5A). We ensured by bioinformatic predictions that these mutations do not modify the structure of the sRNA (cf. Figs. 1B and 5B). These mutations in the interaction site of the sRNA should prevent the regulation of SraL over tig mRNA. The effect of these point mutations on the down-regulation of tig mRNA was tested by Northern blot, and in fact, the point mutations in the sRNA (SraL*) abolished the repression of tig mRNA (Fig. 5C, upper panel). To validate the previous result, we introduced three point mutations in the target (tig*) at the positions corresponding to the mutations in the sRNA, such that full complementarity would be restored when combining both mutations. The results obtained show that wild-type SraL is only able to repress wild-type tig mRNA and not tig* mRNA (Fig. 5C, upper panel, cf. lanes 1 and 4). Additionally, SraL* efficiently down-regulates tig*, but it does not down-regulate the wild-type tig mRNA (Fig. 5C, upper panel, cf. lanes 5 and 3). Thus, the down-regulation of tig* can only be restored when SraL carries the corresponding compensatory mutations enabling the base-pairing between both RNAs. These results provide important additional evidence that confirms that SraL negatively regulates TF directly, by interacting with its mRNA.
FIGURE 5.

Analysis of the interaction between SraL sRNA and tig mRNA. (A) Predicted interaction region between SraL sRNA and tig mRNA. The Shine-Dalgarno region and the start codon of tig are indicated. Chromosomal point mutations to generate SraL* and tig* alleles are indicated. (B) S. Typhimurium SraL* sRNA structure predicted by Mfold program (Zuker 2003). (C) Mutations in SraL and tig in the interaction region between both RNAs validate SraL–tig interaction. Total cellular RNA was extracted from the S. Typhimurium strains indicated grown in LB at 37°C until 6 h after OD600 of 2. (Upper panel) The expression level of tig mRNA was determined by using a 1.3% formaldehyde/Agarose gel. The amount of RNA in wild-type was set as one. The ratio between the RNA amount of each strain and the wild-type is represented (relative levels). A representative membrane is shown, and the values indicated correspond to the average of several Northern blot experiments with RNAs from at least two independent extractions. The membrane was stripped and then probed for 16S rRNA as loading control. The symbol ¥ in the picture indicated the position of the 16S rRNA. (Lower panel) Fifteen micrograms of RNA was separated on a 6% PAA/8.3 M urea to determine the expression level of both SraL and SraL*; probing of 5S rRNA was used as a loading control.
SraL sRNA forms a duplex with tig RNA in vitro
To examine whether SraL binds to tig mRNA, we investigated the duplex formation between the two RNAs in vitro by performing gel mobility shift assays. A fixed concentration of [32P]-labeled tig RNA was incubated with increasing concentrations of unlabeled SraL RNA for 1 h. The duplex formation was analyzed by gel electrophoresis on a native polyacrylamide gel. When wild-type tig RNA was incubated with wild-type SraL, the formation of a retarded SraL–tig RNA complex was obtained with the increasing concentration of SraL sRNA (Fig. 6, left panel). We have also tested the effect of the introduction of mutations in the base-pairing region on the duplex formation. When SraL* RNA was incubated with wild-type tig RNA duplex formation was no longer observed (Fig. 6, middle panel), while the tig* RNA (carrying the compensatory mutations) restored the duplex formation with SraL* RNA (Fig. 6, right panel). These data are consistent with the in vivo results, indicating that SraL sRNA directly interacts with the tig mRNA through the predicted interaction region.
FIGURE 6.
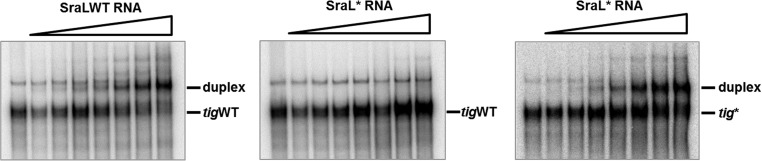
Analysis of the duplex formation between tig RNA and SraL RNA by gel mobility shift assays. [32P]-labeled tig RNA (0.015 pmol) was incubated at 37°C for 1 h with increasing concentrations of unlabeled SraL RNA. The duplex formation was monitored by gel mobility shift assay on native polyacrylamide gels. The concentration range of unlabeled SraL RNA was 0, 22, 44, 88, 175, 350, 700, and 1400 nM. (Left panel) [32P]-labeled tigWT RNA was incubated with SraLWT RNA. (Middle panel) [32P]-labeled tigWT RNA was incubated with SraL* RNA. (Right panel) [32P]-labeled tig* RNA was incubated with SraL* RNA.
DISCUSSION
Trans-encoded sRNAs are known to regulate several genes involved in stress responses. Computational and experimental methodologies have allowed the association of several of these sRNAs with important regulons of both E. coli and Salmonella. The RpoS regulon includes genes with functions in carbon metabolism, stress resistance, cell envelope integrity, morphology, stationary phase, and virulence (Dong and Schellhorn 2010; Battesti et al. 2011). In this report we have included the SraL sRNA in the RpoS regulon since SraL was shown to be directly regulated by this σ factor. An RpoS-recognized promoter is normally identified by a series of characteristic features (Typas et al. 2007). The predicted SraL promoter region (Argaman et al. 2001) presents several of these features, namely, the −35 and −10 box and the extended −10 motif TAA. Moreover, these features are also present in the several enteric bacteria analyzed. Accordingly, it is possible to admit that besides its expression in several other enterobacterial species, this sRNA is also directly transcribed by RpoS in these bacteria. There are only a few studies reporting the control of other sRNAs by RpoS (Opdyke et al. 2004; Padalon-Brauch et al. 2008). However, up to now there is only the case of SdsR sRNA (that controls the synthesis of the major Salmonella porin OmpD) that is controlled by RpoS and is conserved in a broad range of enteric bacteria (Fröhlich et al. 2012). Therefore, SraL constitutes the second example of a conserved sRNA that is controlled by RpoS. In previous work, we had shown that SraL is post-transcriptionally controlled by PNPase, the degradosome complex, and also by polyadenylation (Viegas et al. 2007). Therefore, after this report we can conclude that SraL is a tightly regulated sRNA both at transcriptional and post-transcriptional levels.
After the discovery of MicF sRNA and subsequent unraveling of its function (Mizuno et al. 1984), more than 100 sRNAs were identified. However, the biological function of many of these sRNAs is still unknown. In this study we present for the first time a target for SraL sRNA. We show that SraL contributes to the regulation of the expression of the chaperone TF in late stationary phase. SraL inhibits tig expression at the post-transcriptional level by an antisense mechanism that implicates the base-pairing between a region in the 5′-end of SraL and a few nucleotides before the ribosome binding site (RBS) of the tig mRNA. This interaction region between the sRNA and its target was confirmed through the introduction of point mutations in both RNAs. The mutated version of SraL was not able to down-regulate the expression of the wild-type tig mRNA. Nevertheless, the insertion of compensatory mutations in a mutated version of tig mRNA restored the regulation of SraL over tig mRNA. These results were also confirmed in vitro by gel mobility shift assays. The wild-type tig RNA was not able to interact with the mutated version of SraL. Conversely, we observed the formation of a duplex between the mutated tig RNA and the mutated version of SraL.
In a previous work, we studied the influence of the chaperone Hfq in the stability of SraL at stationary phase and concluded that in the absence of this chaperone SraL has a faster decay rate (Viegas et al. 2007). In the present work, we have confirmed that the steady-state levels of SraL are lower in the hfq deletion mutant strain both at early stationary phase (OD2) and at stationary phase (OD2 + 6 h) (Supplemental Fig. S4, lower left panel). On the other hand, a deep sequencing analysis study reported that at the early stationary phase (OD600 of 2), tig mRNA levels are also affected by the absence of Hfq (3.5-fold higher in the hfq deletion mutant strain) (Sittka et al. 2008). We have confirmed this result by Northern blot analysis (Supplemental Fig. S4, upper left panel). However, at the stationary phase (OD2 + 6 h), the absence of Hfq did not show a strong effect in the expression of tig mRNA, even though there is a slight increase in tig mRNA levels probably due to the lower levels of SraL sRNA. Moreover, we have seen that at stationary phase in the double sraL−/hfq− mutant, the levels of tig mRNA are very low compared with those of the SraL mutant (see Supplemental Fig. S4, right panel). Therefore, it seems to exist at stationary phase another level of tig mRNA regulation by Hfq, direct or through an additional pathway/sRNA.
Unlike what happens in many cases of riboregulation, the region of interaction between SraL sRNA and tig mRNA does not overlap the RBS and the tig mRNA start codon. However, interactions involving nucleotides in the mRNA leader in the vicinity of the RBS and/or the start codon have been also shown to inhibit translation (Liu et al. 1997; Babitzke and Gollnick 2001; Chen et al. 2004). Therefore, it is plausible to assume that it is also the case in this regulation. Based on the results obtained with the proteomic analysis, the level of TF protein was also shown to be higher in the absence of SraL in the cell.
TF is one of the three major chaperones (along with DnaK and GroEL) that cooperate in the folding of the newly synthesized cytosolic proteins (Lecker et al. 1989; Kandror et al. 1995; Stoller et al. 1995; Deuerling et al. 1999). Moreover, it was very recently reported that this chaperone can also unfold preformed structures and reverse premature misfolds, giving nascent chains a new opportunity for productive folding (Hoffmann et al. 2012). It possesses PPIase activity and accelerates proline-limited steps in protein folding with a very high efficiency (Stoller et al. 1995). This reaction is often a rate-limiting step in the folding of certain polypeptides. Even though it is dispensable for growth, TF is a very important protein since it is the first chaperone encountered by the majority of nascent peptide chains due to its location in contact with the large subunit of the ribosomes (Stoller et al. 1995). Therefore, this protein associates cotranslationally with most of the nascent polypeptides. TF competes with DnaK in the chaperoning of newly synthesized peptides (Deuerling et al. 1999; Teter et al. 1999), which explains why it is not an essential protein. The importance of TF in bacterial metabolism is indicated by the presence of a tig gene in Mycoplasma genitalium (Bang et al. 2000). This bacterium is believed to be free from genetic redundancy and thus contains only the minimal set of genes required for life. This chaperone appears to be the only PPIase of this organism (Bang et al. 2000).
This study presents for the first time a regulatory role of SraL sRNA in S. Typhimurium. Despite some significant differences over the sequences of both SraL and tig genes in Enterobacteriaceae, the interaction region between the two RNAs corresponds to a region very well conserved. Thus, it is possible that this regulation of SraL sRNA over tig mRNA also occurs in many other enteric bacteria. The biological significance of the regulatory pathway involving SraL and TF is not totally clear. During the stationary phase, the overall rate of protein synthesis is reduced compared with an exponentially growing culture (Albertson et al. 1990; Kuzj et al. 1998), concomitant with a decrease in the levels of ribosomes (Lambert et al. 1983). This happens because the cell avoids the production of unnecessary proteins when cells are not growing. Since TF is associated with the ribosomes and plays a key role in the folding of nascent peptides, it is possible that it is less required in stationary phase. In fact, results from our laboratory have shown that tig mRNA levels are higher at exponential phase (data not shown). Since TF is constitutively expressed in the cell, the RpoS induction of SraL sRNA under stationary phase seems to occur to avoid the superfluous production of this chaperone.
Since both SraL and TF are very well conserved in enterobacteria, this report will have a significant impact in the field. Moreover, this study constitutes the first report connecting small RNAs with protein folding.
MATERIALS AND METHODS
Oligonucleotides
All oligonucleotides used in this study are listed in Supplemental Table S1 in the Supplemental Material and were synthesized by STAB Vida and Sigma-Aldrich.
Bacterial strains and plasmids
All bacterial strains and plasmids used in this study are listed in the Tables 1 and 2, respectively. All Salmonella strains used are isogenic derivates of the wild-type S. Typhimurium strain SL1344. The sraL (CMA-651) and tig (CMA-652) null mutants were constructed using the primer pairs pIS-001/pIS-002 and pIS-005/pIS-006, respectively, and following the λ-red recombinase method described previously (Datsenko and Wanner 2000) with few modifications, as previously described (Viegas et al. 2007). All chromosomal mutations were subsequently transferred to a fresh genetic background (SL1344 strain) by P22 HT105/1 int-201 transduction (Schmieger 1971). The chloramphenicol-resistance (CmR) cassette of plasmid pKD3 replaces nucleotides −9 to +120 of the sraL gene and −16 to +1303 of tig. The gene deletions were verified by colony PCR using the primer pair pIS-003/pIS-004 for sraL and pIS-007/pIS-008 for tig. The S. Typhimurium rpoS null mutant (CMA-653) was obtained by P22 transduction from the SV4210 strain (Tierrez and Garcia-del Portillo 2004).
TABLE 1.
List of strains used in this work

TABLE 2.
List of plasmids used in this work
The chromosomal mutants with substitutions/point mutations in sraL (CMA-655) and tig (CMA-657) were constructed by a multiple-step PCR process. The strain CMA-654 was constructed by inserting a CmR cassette 59 nt upstream of the sraL transcription start site, in the intergenic region between the sraL and STM4267 genes. To construct the strain CMA-656, we inserted a CmR cassette 65 nt upstream of the tig transcription start site, in the intergenic region between the bolA and tig genes. The CmR cassettes were amplified from plasmid pKD3 using the primer pairs pIS-025/pIS026 for sraL and pIS-031/pIS-032 for tig. The resulting products were integrated into SL1344 wild-type strain chromosome. Then, DNA fragments containing the CmR cassette and the nucleotides substitutions in sraL were prepared by using two primer pairs pIS-027/pIS-028 and pIS-029/pIS-030 and using CMA-654 genomic DNA as a template. In the case of tig, the DNA fragments were prepared by using the two primer pairs pIS-033/pIS-034 and pIS-035/pIS-036 and CMA-656 genomic DNA as a template. The two resulting DNA fragments were mixed and used as PCR templates to amplify the DNA fragment containing CmR cassette and the nucleotides substitutions using the primer pair pIS-027/pIS-030 for sraL and pIS-033/pIS-036 for tig. The resulting DNA fragments were purified and integrated into SL1344 wild-type chromosome by the λ-red recombinase method (Datsenko and Wanner 2000). The mutants were subsequently transferred to a fresh genetic background (SL1344 strain) by P22 HT105/1 int-201 transduction (Schmieger 1971). The double mutant (CMA-658) was constructed using the same transduction method. The presence of the expected substitutions was verified by DNA sequencing.
For construction of pISVA-001 plasmid expressing SraL, a PCR fragment containing the entire sraL sequence was amplified from SL1344 chromosome using the primer pair pIS-009/pIS-010. The resultant PCR fragment carrying a 5′-phosphate at one end was cleaved with KpnI and ligated into the constitutive pZE12luc plasmid (blunt/KpnI site) (Lutz and Bujard 1997). In this plasmid, the initiation site of the encoded RNA lies at position +1 of the constitutive PLlacO promoter of pZE12luc plasmid.
For the rpoS complementation plasmid pISVA-002, a PCR fragment containing the entire rpoS sequence was amplified from SL1344 chromosome using the primer pair pIS-012/pIS-013 and was cloned into the XbaI and HindIII sites of the plasmid pWSK29 (Wang and Kushner 1991).
For the construction of plasmid pISVA-003 (PSraL::lacZ), a fragment of the 5′-UTR region of SraL gene, including promoter signals, was amplified by PCR with primers pIS-014 and pIS-015 (containing the restriction sites for XbaI and BamHI, respectively). Both the insert and pSP417 vector were digested with XbaI and BamHI enzymes and ligated. The cloned sequence includes a region of 195 bp before the start of the gene (including promoter signals) and 9 bp of sraL sequence.
Competent E. coli DH5α cells (New England Biolabs) were used for cloning procedures during plasmid construction.
Bacterial growth
All strains were grown in Luria-Bertani (LB) broth at 37°C and 220 rpm throughout this study. SOC medium was used to recover transformants after heat shock (in the case of E. coli) or electroporation (in the case of Salmonella), before plating. Conditions indicated as SPI-1– and SPI-2–inducing conditions corresponded to growth in high-salt (0.3 M NaCl) LB medium with low oxygen in sealed Falcon tubes, as described for SPI-1 induction (Sittka et al. 2007), and in PCN minimal medium (1 mM phosphate buffer at pH 5.8) as described for SPI-2 induction (Lober et al. 2006).
Growth medium was supplemented with the following antibiotics when appropriate: ampicillin (150 µg/mL), chloramphenicol (25 µg/mL), and streptomycin (90 µg/mL). For heat shock treatment, cells grown at 30°C to an OD600 of 0.5 were transferred for 15 min to 42°C. For cold shock treatment, cultures at an OD600 of 0.5 were transferred from 37°C to 10°C for 30 min and 4 h.
To apply osmotic shock, cells were grown at 37°C to an OD600 of 0.3. NaCl was added to the culture at a final concentration of 0.5 M.
RNA extraction, Northern blot, and RT-PCR analysis
Overnight cultures were diluted 1/100 in fresh medium and grown to the indicated cell densities at OD600 (growth medium and conditions are detailed in the respective figure legends). Culture samples were collected, mixed with 1 volume of stop solution (10 mM Tris at pH 7.2, 25 mM NaNO3, 5 mM MgCl2, 500 µg/mL chloramphenicol), and harvested by centrifugation (10 min, 6000g, 4°C). RNA was isolated using the phenol/chlorophorm extraction method, precipitated in ethanol, resuspended in water, and quantified on a Nanodrop 1000 machine (NanoDrop Technologies).
For Northern blot analysis, 15 µg of total RNA was separated under denaturing conditions either by 8.3 M urea/6% polyacrylamide gel in TBE buffer or by 1.3% Agarose MOPS/formaldehyde gel. For polyacrylamide gels, transfer of RNA onto Hybond-N+ membranes (GE Healthcare) was performed by electroblotting (1 h 50 min, 24 V, 4°C) in TAE buffer. For Agarose gels, RNA was transferred to Hybond-N+ membranes by capillarity using 20× SSC as transfer buffer. In both cases, RNA was UV crosslinked to the membrane immediately after transfer. Membranes were then hybridized in PerfectHyb Buffer (Sigma) at 68°C for riboprobes and 43°C in the case of oligoprobes. After hybridization, membranes were washed according to the method previously described (Viegas et al. 2007). Signals were visualized by PhosphorImaging (Storm Gel and Blot Imaging System, Amersham Bioscience) and analyzed using the ImageQuant software (Molecular Dynamics).
RT-PCR reactions were performed using total RNA with the OneStep RT-PCR kit (Quiagen). Reactions were mainly carried out according to the supplier’s instructions. Modifications were introduced regarding the amount of RNA and number of PCR cycles, depending on gene expression levels. The primer pair pIS-016/pIS017 was used to analyze tig expression. As a control, 16S rRNA was amplified with specific primers pIS-018/pIS-019. Prior to RT-PCR, all RNA samples were treated with Turbo DNA free Kit (Ambion). Control experiments, run in the absence of reverse transcriptase, yielded no product.
Hybridization probes
Primers for templates amplification are listed in Supplemental Table S1. Labeling of the riboprobes and oligoprobes were performed according to the method previously described (Viegas et al. 2007). The riboprobes were obtained using the primer pair pIS-021/pIS-022 for SraL riboprobe and pIS-017/pIS-020 for tig riboprobe. 5S rRNA and 16S rRNA were detected by the 5′-end-labeled oligonucleotides pIS-023 and pIS-024, respectively.
In vitro transcription and gel mobility shift assay
DNA templates for the in vitro transcription of the substrates were generated by PCR using chromosomal DNA from SL1344 wild-type strain for the wild-type transcripts, and the chromosomal DNA from CMA-655 and CMA-657, in the case of SraL and tig, respectively, for the transcripts with the point mutations. sraL was amplified with the primer pair pIS-037/pIS-038 and tig with pIS-039/pIS-040. For the synthesis of internally labeled tig and nonlabeled sraL transcripts, in vitro transcription was carried out using the purified PCR products using equimolar concentrations of all four ribonucleotides with the Riboprobe in vitro Transcription System (Promega) and T7 RNA polymerase. The internally labeled tig transcripts were purified by electrophoresis on an 8.3 M urea/5% polyacrylamide gel. The gel slices were crushed, and RNA was eluted with elution buffer (3 M ammonium acetate at pH 5.2, 1 mM EDTA, 2.5% [v/v] phenol at pH 4.3) overnight at room temperature. The RNA was ethanol precipitated and resuspended in RNase free water. The unlabeled sraL transcripts were run on an 8.3 M urea/6% polyacrylamide gel, identified by ethidium bromide staining, and cut out from the gel. The RNA was eluted from the gel according to the method described above.
Gel mobility shift assays were performed with 0.015 pmol of [32P]-labeled tigWT or tig* RNA in 1× binding buffer (20 mM Tris-acetate at pH 7.6, 100 mM sodium acetate, 5 mM magnesium acetate, 20 mM EDTA). The labeled RNA transcripts were incubated with increasing concentrations of unlabeled RNA (SraLWT or SraL*) in 10 μL for 1 h at 37°C. The binding reactions were mixed with 2 μL of loading dye (48% glycerol, 0.01% bromophenol blue) and loaded on native 4% polyacrylamide gels in 0.5× TBE buffer at 200V at 4°C. After electrophoresis, gels were dried and analyzed using a PhosphorImaging (Storm Gel and Blot Imaging System, Amersham Bioscience) and analyzed using the ImageQuant software (Molecular Dynamics).
Protein extraction and Western Blot analysis
Bacteria were resuspended in the appropriate volume of Laemmli sample buffer (1.3% SDS, 10% [v/v] glycerol, 50 mM Tris/HCl, 1.8% β-mercaptoethanol, 0.02% bromophenol blue at pH 6.8) to get ≈107 bacteria per microliter. RpoS protein was detected using the mouse monoclonal anti-σ S 1RS1 antibody (Santa Cruz Biotechnology) at 1:5000 dilution in antibody dilution buffer (50 mM Tris-HCl at pH 7.5, 0.1% Tween-20, 3% BSA, 1 mM sodium azide) and a goat anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibody (Bio-Rad Life). For recognition of the chaperonin GroEL, an anti-GroEL rabbit policlonal antibody was used (dilution 1:10,000, Sigma) and a goat anti-mouse HRP-conjugated secondary antibody (Bio-Rad Life). Membranes were developed with 1/10 diluted ECL prime reagent (GE Healthcare) and visualized using the ChemiDoc XRS+ imaging system and the Quantity One software (Bio-Rad Life).
β-Galactosidase assays
β-Galactosidase activity was determined essentially according to the method first described by Miller with minor modifications (Maloy 1990). In brief, 100 μL of culture was added to 655 μL of cold buffer Z (100 mM Na2HPO4/NaH2PO4 at pH 7, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol), and chloroform-SDS was used to permeabilize the cells. The reaction was started by the addition of the chromogenic substrate ortho-Nitrophenyl-β-galactoside to a final concentration of 0.8 mg/mL, conducted until it reaches a pale yellow color at 30°C, and stopped with Na2CO3. Prior to recording absorbance at 420 nm, samples were cleared by centrifugation. Optical density of the bacterial culture was also recorded at the time of the extraction of the sample. β-Galactosidase activity in Miller units was calculated as follows: (1,000 × A420)/(t × v × OD600), where t corresponds to the reaction time in minutes and v to the sample volume in milliliters.
ChIP assays
Ten milliliters of overnight grown wild-type SL1344 and isogenic rpoS mutant cultures was exposed to 150 μg/mL rifampicin for 30 min to trap RNA polymerase at gene promoters. Cells were subjected to chemical crosslinking in vivo by adding formaldehyde and phosphate buffer (pH 7.6) to a final concentration of 1% and 10 mM, respectively. Noncrosslinked control samples of both strains were processed in parallel. Crosslinking was left to proceed for 30 min at 37°C with shacking and then quenched with 100 mM glycine for 30 min at 4°C. Bacteria were recovered by centrifugation and washed twice with cold PBS. Bacterial pellets were resuspended in 1 mg/mL lysozyme in 0.2× IP buffer containing EDTA-free protease inhibitors cocktail (Roche) and maintained for 10 min at 37°C. One volume of 2× IP buffer (200 mM Tris-HCl at pH 8.0, 600 mM NaCl, 4% Triton X-100) was added, and the samples were sonicated in a B. Braun sonifier (Labsonic U model; duty cycle 0.7, output 0.49). DNA in cleared lysates was further digested with 0.1 unit of micrococcal nuclease (New England Biolabs) and 0.5 μg of RNase A in the presence of 5 mM CaCl2 and 0.1 mg/mL BSA for 10 min at 37°C. The digestion was stopped with 10 mM EDTA. DNA shearing was followed by Agarose electrophoresis after reversing the crosslinking of an aliquot for 6 h at 65°C.
Prior to immunoprecipitation, 1/10 volume of the total extract was taken to be used as input sample control. The extracts were then precleared with 20 μL of a 50% slurry containing 1:1 mix of protein-A and protein-G Sepharose (Sigma) in 1× IP buffer for 4 h at 4°C with rotation. Immunoprecipitation was carried out with 2 μL of monoclonal mouse anti-σ S 1RS1 antibody (Santa Cruz Biotechnology) overnight at 4°C. All samples (no-antibody or preclearing controls and IPs) were washed once with LiCl wash buffer (250 mM LiCl, 100 mM Tris-HCl at pH 8, 2% TritonX-100), twice with 0.6 M NaCl buffer (100 mM Tris-HCl at pH 8, 600 mM NaCl, 2% TritonX-100), twice with 1× IP buffer, and once with TE buffer (10 mM Tris-HCl at pH 8, 1 mM EDTA). To elute complexes from the protein-A and -G Sepharose, beads were resuspended in 30 μL of ChIP elution buffer (50 mM Tris-HCl at pH 8, 10 mM EDTA, 1% SDS) and incubated for 30 min at 65°C. The complexes were then incubated 6 h at 65°C to reverse crosslinking. Half of the sample was used to assess the efficiency of the immunoprecipitation by Western blot. DNA was obtained from the other half of the sample by phenol/chloroform extraction and precipitated with isopropanol using 20 μg of glycogen (Roche) as a carrier, and the pellet was dissolved in 12 μL of nuclease-free water.
For real-time quantitative PCR analysis of target DNA enrichment, a 1/50 dilution sample of IP and no-antibody control were used as template. In the case of input and flow-through samples, we used a 1/200 dilution. Reactions were performed with the Power Sybr Green PCR master mix (Applied Biosystems) in a 10 μL final volume and run in an ABI Prism 7,500 instrument (Applied Biosystems) using standard reaction conditions recommended by the manufacturer (10 min at 95°C; 45 cycles of 15 sec at 95°C, and 1 min at 60°C; dissociation curve of 15 sec at 95°C, 1 min at 60°C, and a progressive temperature increase until 95°C). Each sample was run in triplicate. Oligonucleotides osmY-F, osmY-R, sraL-F, sraL-R, rnpB-F, rnpB-R, 16S-F, and 16S-R were used to amplify the corresponding target DNA at 0.5 μM final concentration and are included in Supplemental Table S1. For data analysis, the mean Ct value of technical replicates showing a standard deviation below 0.1 for target DNA was normalized to the mean Ct for rrs (16S) in the same sample (Cttarget-Ctrrs). These values were referred to wild-type input sample and the anti-logarithm calculated.
Proteomic analysis
For the proteomic analysis, the cells were grown in the same conditions as for the RNA extraction (see above). Then, 2 OD units of each culture were transferred to a tube containing 0.2 volumes of stop solution (5% phenol/95% ethanol) and kept on ice for 30 min. Cells were spun down by centrifugation for 10 min at 3200g 4°C, washed with 1:5-diluted stop solution, centrifuged again, and stored at −80°C. Pellets were lysed in Laemmli sample buffer without bromophenol blue dye (see above), and the total protein estimated using the Bradford reagent (Bio-Rad). Approximately 30 μg of total protein (corresponding to ∼0.3 ODs) was run in a SDS-PAGE 12% gel. Loading equivalence among the samples was confirmed by checking GroEL levels by Western blot analysis. Five slices from mid-run gels covering molecular masses from ∼150 kDa to 10 kDa were submitted to in-gel tryptic digestion. The tryptic peptide mixtures were processed for protein identification by liquid chromatography in a C-18 reversed-phase nano-column (100-μm innerdiameter × 12 cm, Mediterranea Sea, Teknokroma) and real-time ionization and peptide fragmentation on an LTQ-Orbitrap XL ETD mass spectrometer (Thermo Fisher Scientific). For protein identification, tandem mass spectra were analyzed using SEQUEST (Thermo Fisher Scientific, versión 1.0.43.2) and X! Tandem (The GPM, version 2007.01.01.1) using SALTY proteome (UniProtKB, Taxon nr.99287) as reference. Scaffold (version Scaffold_3_00_03, Proteome Software) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95.0% probability. Protein identifications were accepted if they could be established at >95.0% probability and contained at least two identified peptides. For a further description of the experimental details of the procedure, see García-del Portillo et al. (2011).
Sequence retrieval and alignments
BlastN was used for sequence alignments (http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi) of the following genome sequences: S. Typhimurium LT2 (NC_003197), Salmonella enterica serovar Typhi Ty2 (NC_004631), Salmonella bongori NCTC 12419 (NC_015761), Shigella boydii CDC 3083-94 (NC_010658), Shigella flexneri 2a str. 301 (NC_004337), Shigella dysenteriae Sd197 (NC_007606), Escherichia coli K12 (NC_000913), Citrobacter rodentium ICC168 (NC_013716), Citrobacter koseri ATCC BAA-895 (NC_009792), Enterobacter sp. 638 (NC_009436), and Klebsiella pneumoniae 342 (NC_011283). Alignments were made using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Andreia Aires, Pablo García-Bravo, and Diana Barroso for technical assistance. We thank Enrique Calvo and J.A. López from the Proteomics Unit of the Centro Nacional de Investigaciones Cardiovasculares for proteomic identification. I.J. Silva (SFRH/BD/43211/2008) was a recipient of a FCT (Fundação para a Ciência e Tecnologia) Doctoral Fellowship; S.C. Viegas (SFRH/BPD/30766/2006) was a recipient of a FCT Post-Doctoral Fellowship. This work was supported by grants from Fundação para a Ciência e Tecnologia (FCT; including grant PEst-OE/EQB/LA0004/2011) and “Acção Integrada” E 80/12 from CRUP and FP7-KBBE-2011-1-289326 from European Commission (to C.M.A., Portugal) and grants BIO2010-18885, CSD2008-00013, and “Acción Integrada” PRI-AIBPT-2011-1136 from the Spanish Ministry of Economy and Competitiveness (to F.G.-P.).
REFERENCES
- Albertson NH, Nyström T, Kjelleberg S 1990. Macromolecular synthesis during recovery of the marine Vibrio sp. S14 from starvation. J Gen Microbiol 136: 2201–2207 [Google Scholar]
- Aldea M, Garrido T, Hernandez-Chico C, Vicente M, Kushner SR 1989. Induction of a growth-phase-dependent promoter triggers transcription of bolA, an Escherichia coli morphogene. EMBO J 8: 3923–3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amábile-Cuevas CF, Demple B 1991. Molecular characterization of the soxRS genes of Escherichia coli: Two genes control a superoxide stress regulon. Nucleic Acids Res 19: 4479–4484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S 2001. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol 11: 941–950 [DOI] [PubMed] [Google Scholar]
- Babitzke P, Gollnick P 2001. Posttranscription initiation control of tryptophan metabolism in Bacillus subtilis by the trp RNA-binding attenuation protein (TRAP), anti-TRAP, and RNA structure. J Bacteriol 183: 5795–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang H, Pecht A, Raddatz G, Scior T, Solbach W, Brune K, Pahl A 2000. Prolyl isomerases in a minimal cell. Catalysis of protein folding by trigger factor from Mycoplasma genitalium. Eur J Biochem 267: 3270–3280 [DOI] [PubMed] [Google Scholar]
- Battesti A, Majdalani N, Gottesman S 2011. The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol 65: 189–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, Richter AS, Backofen R 2008. IntaRNA: Efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 24: 2849–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhang A, Blyn LB, Storz G 2004. MicC, a second small-RNA regulator of Omp protein expression in Escherichia coli. J Bacteriol 186: 6689–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B 1999. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature 400: 693–696 [DOI] [PubMed] [Google Scholar]
- Dong T, Schellhorn HE 2010. Role of RpoS in virulence of pathogens. Infect Immun 78: 887–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich KS, Papenfort K, Berger AA, Vogel J 2012. A conserved RpoS-dependent small RNA controls the synthesis of major porin OmpD. Nucleic Acids Res 40: 3623–3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-del Portillo F, Calvo E, D’Orazio V, Pucciarelli MG 2011. Association of ActA to peptidoglycan revealed by cell wall proteomics of intracellular Listeria monocytogenes. J Biol Chem 286: 34675–34689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S, Storz G 2011. Bacterial small RNA regulators: Versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol 3: a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge-Aronis R, Lange R, Henneberg N, Fischer D 1993. Osmotic regulation of rpoS-dependent genes in Escherichia coli. J Bacteriol 175: 259–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesterkamp T, Hauser S, Lutcke H, Bukau B 1996. Escherichia coli trigger factor is a prolyl isomerase that associates with nascent polypeptide chains. Proc Natl Acad Sci 93: 4437–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Bukau B, Kramer G 2010. Structure and function of the molecular chaperone Trigger Factor. Biochim Biophys Acta 1803: 650–661 [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Becker AH, Zachmann-Brand B, Deuerling E, Bukau B, Kramer G 2012. Concerted action of the ribosome and the associated chaperone trigger factor confines nascent polypeptide folding. Mol Cell 48: 63–74 [DOI] [PubMed] [Google Scholar]
- Hoiseth SK, Stocker BA 1981. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291: 238–239 [DOI] [PubMed] [Google Scholar]
- Johansen J, Rasmussen AA, Overgaard M, Valentin-Hansen P 2006. Conserved small non-coding RNAs that belong to the σE regulon: Role in down-regulation of outer membrane proteins. J Mol Biol 364: 1–8 [DOI] [PubMed] [Google Scholar]
- Johansen J, Eriksen M, Kallipolitis B, Valentin-Hansen P 2008. Down-regulation of outer membrane proteins by noncoding RNAs: Unraveling the cAMP–CRP- and σE-dependent CyaR–ompX regulatory case. J Mol Biol 383: 1–9 [DOI] [PubMed] [Google Scholar]
- Kandror O, Sherman M, Rhode M, Goldberg AL 1995. Trigger factor is involved in GroEL-dependent protein degradation in Escherichia coli and promotes binding of GroEL to unfolded proteins. EMBO J 14: 6021–6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kröger C, Dillon SC, Cameron AD, Papenfort K, Sivasankaran SK, Hokamp K, Chao Y, Sittka A, Hébrard M, Händler K, et al. 2012. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc Natl Acad Sci 109: E1277–E1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzj AE, Medberry PS, Schottel JL 1998. Stationary phase, amino acid limitation and recovery from stationary phase modulate the stability and translation of chloramphenicol acetyltransferase mRNA and total mRNA in Escherichia coli. Microbiology 144 (Pt 3): 739–750 [DOI] [PubMed] [Google Scholar]
- Lambert JM, Boileau G, Howe JG, Traut RR 1983. Levels of ribosomal protein S1 and elongation factor G in the growth cycle of Escherichia coli. J Bacteriol 154: 1323–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker S, Lill R, Ziegelhoffer T, Georgopoulos C, Bassford PJ Jr, Kumamoto CA, Wickner W 1989. Three pure chaperone proteins of Escherichia coli—SecB, trigger factor and GroEL—form soluble complexes with precursor proteins in vitro. EMBO J 8: 2703–2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MY, Gui G, Wei B, Preston JF III, Oakford L, Yuksel U, Giedroc DP, Romeo T 1997. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J Biol Chem 272: 17502–17510 [DOI] [PubMed] [Google Scholar]
- Lober S, Jackel D, Kaiser N, Hensel M 2006. Regulation of Salmonella pathogenicity island 2 genes by independent environmental signals. Int J Med Microbiol 296: 435–447 [DOI] [PubMed] [Google Scholar]
- Lutz R, Bujard H 1997. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res 25: 1203–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy SR 1990. Experimental techniques in bacterial genetics. Jones and Bartlett Publishers, Boston [Google Scholar]
- Mattatall NR, Sanderson KE 1996. Salmonella typhimurium LT2 possesses three distinct 23S rRNA intervening sequences. J Bacteriol 178: 2272–2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecsas J, Rouviere PE, Erickson JW, Donohue TJ, Gross CA 1993. The activity of σE, an Escherichia coli heat-inducible σ-factor, is modulated by expression of outer membrane proteins. Genes Dev 7: 2618–2628 [DOI] [PubMed] [Google Scholar]
- Mendoza-Vargas A, Olvera L, Olvera M, Grande R, Vega-Alvarado L, Taboada B, Jimenez-Jacinto V, Salgado H, Juarez K, Contreras-Moreira B, et al. 2009. Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLoS One 4: e7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiakas D, Betton JM, Raina S 1996. New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol Microbiol 21: 871–884 [DOI] [PubMed] [Google Scholar]
- Mizuno T, Chou MY, Inouye M 1984. A unique mechanism regulating gene expression: Translational inhibition by a complementary RNA transcript (micRNA). Proc Natl Acad Sci 81: 1966–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opdyke JA, Kang JG, Storz G 2004. GadY, a small-RNA regulator of acid response genes in Escherichia coli. J Bacteriol 186: 6698–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega AD, Gonzalo-Asensio J, Garcia-Del Portillo F 2012. Dynamics of Salmonella small RNA expression in non-growing bacteria located inside eukaryotic cells. RNA Biol 9: 469–488 [DOI] [PubMed] [Google Scholar]
- Padalon-Brauch G, Hershberg R, Elgrably-Weiss M, Baruch K, Rosenshine I, Margalit H, Altuvia S 2008. Small RNAs encoded within genetic islands of Salmonella typhimurium show host-induced expression and role in virulence. Nucleic Acids Res 36: 1913–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K, Pfeiffer V, Mika F, Lucchini S, Hinton JC, Vogel J 2006. σE-Dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol Microbiol 62: 1674–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podkovyrov SM, Larson TJ 1995. A new vector-host system for construction of lacZ transcriptional fusions where only low-level gene expression is desirable. Gene 156: 151–152 [DOI] [PubMed] [Google Scholar]
- Raffaelle M, Kanin EI, Vogt J, Burgess RR, Ansari AZ 2005. Holoenzyme switching and stochastic release of σ factors from RNA polymerase in vivo. Mol Cell 20: 357–366 [DOI] [PubMed] [Google Scholar]
- Raivio TL, Silhavy TJ 1999. The σE and Cpx regulatory pathways: Overlapping but distinct envelope stress responses. Curr Opin Microbiol 2: 159–165 [DOI] [PubMed] [Google Scholar]
- Rehmsmeier M, Steffen P, Höchsmann M, Giegerich R 2004. Fast and effective prediction of microRNA/target duplexes. RNA 10: 1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieger H 1971. The fate of the bacterial chromosome in P22-infected cells of Salmonella typhimurium. Mol Gen Genet 110: 238–244 [DOI] [PubMed] [Google Scholar]
- Sittka A, Pfeiffer V, Tedin K, Vogel J 2007. The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol Microbiol 63: 193–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JC, Vogel J 2008. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet 4: e1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoller G, Rücknagel KP, Nierhaus KH, Schmid FX, Fischer G, Rahfeld JU 1995. A ribosome-associated peptidyl-prolyl cis/trans isomerase identified as the trigger factor. EMBO J 14: 4939–4948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Vogel J, Wassarman KM 2011. Regulation by small RNAs in bacteria: Expanding frontiers. Mol Cell 43: 880–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teter SA, Houry WA, Ang D, Tradler T, Rockabrand D, Fischer G, Blum P, Georgopoulos C, Hartl FU 1999. Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell 97: 755–765 [DOI] [PubMed] [Google Scholar]
- Tierrez A, Garcia-del Portillo F 2004. The Salmonella membrane protein IgaA modulates the activity of the RcsC-YojN-RcsB and PhoP-PhoQ regulons. J Bacteriol 186: 7481–7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Becker G, Hengge R 2007. The molecular basis of selective promoter activation by the σS subunit of RNA polymerase. Mol Microbiol 63: 1296–1306 [DOI] [PubMed] [Google Scholar]
- Udekwu KI, Wagner EG 2007. σE controls biogenesis of the antisense RNA MicA. Nucleic Acids Res 35: 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valent QA, Kendall DA, High S, Kusters R, Oudega B, Luirink J 1995. Early events in preprotein recognition in E. coli: Interaction of SRP and trigger factor with nascent polypeptides. EMBO J 14: 5494–5505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viegas SC, Pfeiffer V, Sittka A, Silva IJ, Vogel J, Arraiano CM 2007. Characterization of the role of ribonucleases in Salmonella small RNA decay. Nucleic Acids Res 35: 7651–7664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RF, Kushner SR 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100: 195–199 [PubMed] [Google Scholar]
- Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S 2001. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev 15: 1637–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber H, Polen T, Heuveling J, Wendisch VF, Hengge R 2005. Genome-wide analysis of the general stress response network in Escherichia coli: σS-dependent genes, promoters, and σ factor selectivity. J Bacteriol 187: 1591–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim HH, Brems RL, Villarejo M 1994. Molecular characterization of the promoter of osmY, an rpoS-dependent gene. J Bacteriol 176: 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]