

Abstract
Growth of Listeria monocytogenes on refrigerated, ready-to-eat food is a significant food safety concern. Natural antimicrobials, such as nisin, can be used to control this pathogen on food, but little is known about how other food-related stresses may impact how the pathogen responds to these compounds. Prior work demonstrated that exposure of L. monocytogenes to salt stress at 7°C led to increased expression of genes involved in nisin resistance, including the response regulator liaR. We hypothesized that exposure to salt stress would increase subsequent resistance to nisin and that LiaR would contribute to increased nisin resistance. Isogenic deletion mutations in liaR were constructed in 7 strains of L. monocytogenes, and strains were exposed to 6% NaCl in brain heart infusion broth and then tested for resistance to nisin (2 mg/ml Nisaplin) at 7°C. For the wild-type strains, exposure to salt significantly increased subsequent nisin resistance (P < 0.0001) over innate levels of resistance. Compared to the salt-induced nisin resistance of wild-type strains, ΔliaR strains were significantly more sensitive to nisin (P < 0.001), indicating that induction of LiaFSR led to cross-protection of L. monocytogenes against subsequent inactivation by nisin. Transcript levels of LiaR-regulated genes were induced by salt stress, and lmo1746 and telA were found to contribute to LiaR-mediated salt-induced nisin resistance. These data suggest that environmental stresses similar to those on foods can influence the resistance of L. monocytogenes to antimicrobials such as nisin, and potential cross-protective effects should be considered when selecting and applying control measures for this pathogen on ready-to-eat foods.
INTRODUCTION
The food-borne pathogen Listeria monocytogenes must survive a number of environmental stresses in order to be successfully transmitted to a human host. On foods, L. monocytogenes is exposed to many stresses, and it is capable of surviving and even growing under stresses such as high salt and low temperature (1). L. monocytogenes contamination of ready-to-eat (RTE) foods is of particular concern, since this pathogen is capable of multiplying to high levels during refrigerated storage (2). One of the critical points in controlling L. monocytogenes in the food supply focuses on reducing or eliminating its ability to multiply on RTE refrigerated foods (3).
Natural antimicrobials, such as nisin, can be used to control L. monocytogenes on RTE foods (4–6). In many cases, RTE foods pose additional environmental stresses to this pathogen, such as osmotic stress, organic acid stress, and cold stress. Some of these stresses are known to affect nisin resistance; for example, prior acid stress increases subsequent resistance of L. monocytogenes to nisin (7, 8). Other food-related stresses may impact how L. monocytogenes survives nisin stress, and other than cell wall (9) and cell membrane (8) modifications, the molecular mechanisms that lead to cross-protection against nisin are not well described.
A number of mechanisms are known to contribute to the innate nisin resistance of L. monocytogenes, including expression of the glutamate decarboxylase GadD1 (10), MprF (11), VirRS (12), and LiaR (13). LiaR is the response regulator of the LiaFSR three-component system, which regulates expression of 27 genes in L. monocytogenes (14). LiaR-regulated genes include some with previously described roles in nisin resistance, including lmo2229, which encodes a penicillin binding protein (13), and telA, which encodes a protein involved in toxic ion resistance (15). Previous work by our group indicated that the cell envelope stress response system LiaFSR is induced by L. monocytogenes during adaptation to salt stress at low temperature (16).
Taken together, these data suggest that salt stress at low temperature could provide cross-protection against nisin and that a potential mechanism of cross-protection is activation of the cell envelope stress response controlled by LiaR. To test these hypotheses, we generated isogenic liaR deletion mutants in 7 different L. monocytogenes strains and assessed nisin resistance for strains with and without prior exposure to salt stress. Our data suggest that environmental stresses similar to those present on foods can influence the resistance of L. monocytogenes to control measures such as the antimicrobial nisin, and potential cross-protective effects should be considered when selecting and applying control measures for this pathogen on ready-to-eat foods.
MATERIALS AND METHODS
Strains and mutant construction.
L. monocytogenes strains from genetic lineages I and II were used in this study (Table 1). In-frame deletion mutations were constructed in the liaR coding region (Table 1) in all strains using the splicing-by-overlap-extension (SOE) method as previously described (16). Additionally, in-frame deletion mutations were constructed in the coding regions of LMOh7858_0955 (sigB), LMOh7858_2364 (lmo2229), LMOh7858_2079 (telA), and LMOh7858_1871 (lmo1746) in strain H7858 (Table 1). Strain H7858, a serotype 4b strain from the 1998 hot dog outbreak (17, 18), was selected as a representative lineage I strain for further genetic manipulations and gene expression assays.
Table 1.
Strains and plasmids used in this study
| Strain/plasmid | Strain alias/relevant genotype | Reference(s) |
|---|---|---|
| FSL F6-0366 | H7858 | 17, 18 |
| FSL J1-0194 | 33 | |
| FSL R2-0503 | G6054 | 34 |
| FSL F2-0539 | EGDe | 18 |
| FSL R2-0499 | J0161 | 34 |
| FSL R2-0559 | F6854 | 18, 35 |
| FSL X1-0001 | 10403S | 36 |
| FSL B2-0086 | 10403S Δlmo1022 (ΔliaR) | This work |
| FSL B2-0315 | H7858 ΔLMOh7858_1089 (ΔliaR) | This work |
| FSL B2-0343 | FSL J1-194 ΔLMOh7858_1089 (ΔliaR) | This work |
| FSL B2-0347 | EGDe Δlmo1022 (ΔliaR) | This work |
| FSL B2-0348 | J0161 Δlmo1022 (ΔliaR) | This work |
| FSL B2-0350 | F6854 Δlmo1022 (ΔliaR) | This work |
| FSL B2-0351 | G6054 ΔLMJG_00808 (ΔliaR) | This work |
| FSL K5-0018 | H7858 ΔLMOh7858_0955 (ΔsigB) | This work |
| FSL K5-0019 | H7858 ΔLMOh7858_1089 (ΔliaR) | This work |
| ΔLMOh7858_0955 (ΔsigB) | ||
| FSL W9-0009 | H7858 ΔLMOh7858_2364 (Δlmo2229) | This work |
| FSL B2-0390 | H7858 ΔLMOh7858_2079 (ΔtelA) | This work |
| FSL B2-0388 | H7858 ΔLMOh7858_1871 (Δlmo1746) | This work |
| pBMB78 | ΔLMOh7858_1089 (ΔliaR) | 16 |
| pBMB25 | Δlmo1022 (ΔliaR) | This work |
| pBMB83 | ΔLMJG_00808 (ΔliaR) | This work |
| pEMW7 | ΔLMOh7858_2364 | This work |
| pBMB88 | ΔLMOh7858_2079 | This work |
| pBMB87 | ΔLMOh7858_1871 | This work |
| pHFO01 | ΔLMOh7858_0955 (ΔsigB) | 37 |
Growth conditions.
L. monocytogenes strains were stored at −80°C in brain heart infusion (BHI) broth with 15% glycerol, and these stocks were used for all experiments. Strains were inoculated onto BHI agar from frozen stocks, and plates were incubated at 37°C for 22 to 24 h. A single colony was inoculated into 5 ml BHI broth, which was then incubated at 37°C, 230 rpm, for 20 h. Cultures were transferred 1:100 to BHI at 7°C and incubated for 50 h (exponential-phase cultures; optical density at 600 nm [OD600] = 0.23 ± 0.01) or for 76 h (stationary-phase cultures; OD600 = 0.80 ± 0.02, where the maximum OD600 at 7°C was 0.88) without aeration at 7°C. Exponential-phase cultures were transferred 1:10 into both BHI and BHI plus 6% NaCl and incubated at 7°C for 200 min. This incubation period corresponds with the maximum expression of liaR induced by salt stress (16). Stationary-phase cultures were transferred 1:50 into both BHI and BHI plus 6% NaCl and incubated at 7°C for 200 min. Stationary-phase cultures were transferred at a higher dilution ratio than exponential-phase cultures to achieve similar numbers of cells in BHI and BHI plus 6% NaCl to minimize any cell density-dependent effects.
Nisin resistance assays.
After 200 min of incubation, cultures in BHI were transferred 1:5 into BHI plus 2 mg/ml Nisaplin (Danisco) (equivalent to 50 ppm nisin, since Nisaplin contains ∼2.5% nisin), and cultures in BHI plus 6% NaCl were transferred 1:5 into BHI plus 6% NaCl plus 2 mg/ml Nisaplin. All cultures were sampled prior to nisin exposure and after 24 h of incubation at 7°C in medium containing nisin. Cultures were plated on BHI agar using an Autoplate 4000 instrument (Spiral Biotech, Norwood MA). Plates were incubated overnight at 37°C, and colonies were enumerated using a Q-Count instrument (Spiral Biotech). The limit of detection for the survival assays was 85 CFU/ml. Survival experiments were conducted for three independent cultures of each strain tested, and data were used to calculate the log decrease in cell density after 24 h of exposure to nisin under each test condition.
RNA extraction.
RNA was extracted from cultures of strain H7858 and its isogenic liaR mutant before and after 200 min of exposure to BHI plus 6% NaCl at 7°C. Cells were collected, and RNA synthesis and degradation were stopped by adding 10 ml acid phenol-ethanol (1:10 [vol/vol]) to 100 ml of culture, mixing, and centrifuging at 10,000 rpm for 20 min at 4°C. Cell pellets were suspended in 5 ml TriReagent (Life Technologies, Foster City, CA) and homogenized for 4 min in a Beadbeater-8 instrument (Biospec Products, Bartlesville, OK) with 0.1-mm acid-washed zirconium beads. After homogenization, the manufacturer's protocol was used to extract RNA. After extraction, RNA was treated with RQ1 DNase (Promega, Madison WI), followed by purification with RNeasy mini columns (Qiagen, Valencia, CA). RNA quality was assessed on a Bioanalyzer system (Agilent, Santa Clara, CA), and only samples with an RNA integrity number (RIN) of >8.0 were used for subsequent analyses.
Q-PCR.
cDNA was synthesized from 500 ng total RNA using the TaqMan reverse transcription kit (Life Technologies). Reverse transcription reaction mixtures contained 1× TaqMan buffer, 5.5 mM magnesium chloride, 500 μM (each) deoxynucleoside triphosphate (dNTP), 2.5 μM random hexamers, 4 U RNase inhibitor, and 12.5 U MultiScribe reverse transcriptase and were carried out under the following conditions: 10 min at 25°C, 30 min at 48°C, and 5 min at 95°C. Reaction mixtures containing all components except reverse transcriptase were prepared for all RNA samples to determine background levels of DNA. Ten-fold serial dilutions of cDNA were used as the input for quantitative PCR (Q-PCR) assays. TaqMan primers and probes were designed using the software program Primer Express v1.0 (Life Technologies) (see Table S1 in the supplemental material). Q-PCR mixtures contained 1× Universal TaqMan master mix (Applied Biosystems), 900 nmol (each) primer, and 250 nmol TaqMan probe and were run on the ABI Prism 7000 system (Applied Biosystems) under the following conditions: 40 cycles at 95°C for 15 s and 60°C for 1 min. Threshold cycle (CT) and reaction efficiencies were determined using the ABI SDS v1.0 software program. Q-PCRs were carried out in duplicate for each cDNA sample tested. Target gene copy numbers were quantified using genomic DNA standard curves and normalized to copy numbers of rpoB. mRNA copy numbers were log10 transformed, and the ratio of transcript levels in BHI plus 6% NaCl (test condition) to transcript levels in BHI (reference condition) were calculated for each gene.
Statistical analyses.
The log decrease in cell density after 24 h of exposure to 2 mg/ml Nisaplin was calculated as the difference in log10 CFU/ml at time zero and after 24 h in medium with nisin. The Shapiro-Wilk test for normality was used to determine if the log decrease after 24 h for each growth phase (exponential or stationary phase) fit a normal distribution. All data sets were found to be normally distributed, and analysis of variance (ANOVA) was implemented using the mixed procedure in the software program SAS v.9.3 (SAS Institute, Cary, NC) with a linear model that included strain, replicate, and treatment (with or without prior salt exposure), all as fixed effects. The Tukey multiple-correction procedure was applied to all analysis-of-variance results. Adjusted P values of <0.05 were considered significant.
For Q-PCR data, significant differences in transcript levels induced by salt between the wild-type H7858 and ΔliaR strains were determined for each gene using ANOVA with a linear model that included strain and replicate as fixed effects. P values of <0.05 were considered significant.
RESULTS
Exposure to salt stress increases subsequent resistance to nisin.
To determine the effect of salt exposure on subsequent nisin resistance, we compared the survival of L. monocytogenes in BHI with nisin for cultures with and without prior exposure to 6% NaCl. Without exposure to salt, exponential-phase cells were sensitive to nisin, with an average decrease in cell density of 3.7 ± 0.6 log10 CFU/ml after 24 h for the seven wild-type strains of L. monocytogenes (Fig. 1). When exponential-phase cells were exposed to 6% NaCl in BHI for 200 min and then exposed to nisin, nisin resistance increased significantly (P < 0.0001), with an average decrease in cell density of 1.6 ± 0.8 log10 CFU/ml after 24 h. To determine if salt-induced nisin resistance was growth phase dependent, we also measured nisin resistance of stationary-phase cells. Wild-type stationary-phase cells were also sensitive to nisin, with an average decrease in cell density of 3.9 ± 0.6 log10 CFU/ml after 24 h (Fig. 2). Exposure of stationary-phase cells to 6% NaCl in BHI for 200 min prior to nisin exposure also significantly increased nisin resistance (P < 0.0001), with an average decrease in cell density of 2.6 ± 0.6 log10 CFU/ml after 24 h. While salt exposure did lead to increased nisin resistance, significant variation in survival among strains was observed (P < 0.001). For exponential-phase cells, the decrease in log10 CFU/ml after 24 h ranged from 0.3 to 3.2, and for stationary-phase cells, it ranged from 1.1 to 3.9.
Fig 1.
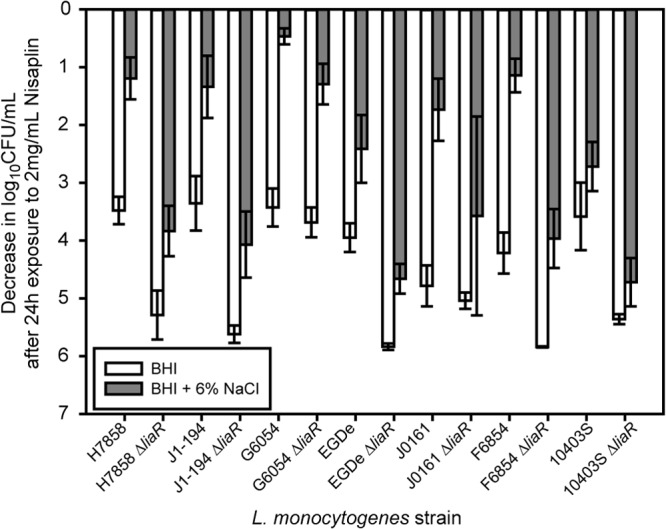
Log decrease in cell density for exponential-phase cells exposed to 2 mg/ml Nisaplin in BHI for 24 h at 7°C. The average and standard deviation for three independent replicates are plotted for each strain. White bars represent cultures that were exposed only to BHI prior to exposure to Nisaplin, and gray bars represent cultures that were exposed to BHI plus 6% NaCl before exposure to Nisaplin.
Fig 2.
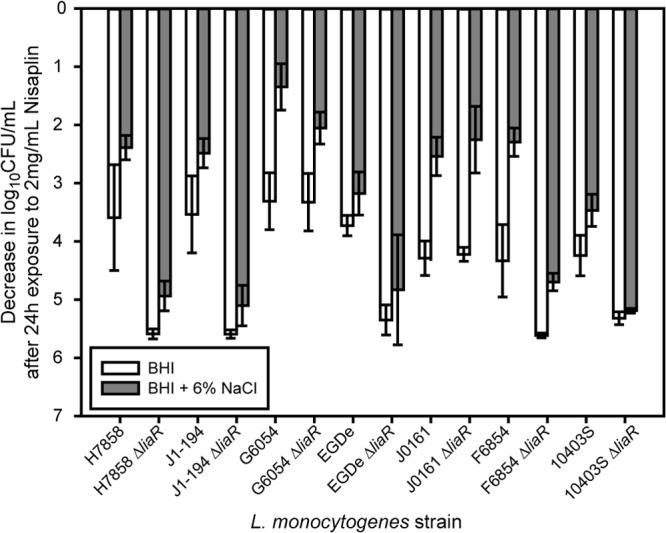
Log decrease in cell density for stationary-phase cells exposed to 2 mg/ml Nisaplin in BHI for 24 h at 7°C. The average and standard deviation for three independent replicates are plotted for each strain. White bars represent cultures that were exposed only to BHI prior to exposure to Nisaplin, and gray bars represent cultures that were first exposed to BHI plus 6% NaCl before exposure to Nisaplin.
ΔliaR strains are sensitive to nisin even when exposed to salt stress.
To determine if LiaR plays a role in salt-induced nisin resistance, we compared the survival of ΔliaR mutants to that of wild-type strains. Without salt exposure, exponential-phase ΔliaR strains were significantly more sensitive to nisin than wild-type strains (P < 0.0001), with an average decrease in cell density of 5.2 ± 0.7 log10 CFU/ml after 24 h for the 7ΔliaR strains (Fig. 1). Exposure of exponential-phase ΔliaR strains to BHI plus 6% NaCl increased nisin resistance, with an average decrease in cell density of 4.0 ± 1.1 log10 CFU/ml after 24 h. While salt exposure increased nisin resistance of ΔliaR strains, the increase in survival was significantly lower (P < 0.0001) than was seen for salt-induced nisin resistance of exponential-phase wild-type strains (Fig. 1).
Without salt exposure, stationary-phase ΔliaR strains were also significantly more sensitive to nisin than wild-type strains (P < 0.0001), with an average decrease in cell density of 5.1 ± 0.9 log10 CFU/ml after 24 h (Fig. 2). Exposure of stationary-phase ΔliaR strains to BHI plus 6% NaCl increased nisin resistance, with an average decrease in cell density of 4.1 ± 1.4 log10 CFU/ml after 24 h. While salt exposure increased nisin resistance of stationary-phase ΔliaR strains, the increase in survival was significantly less (P < 0.0001) than was seen for salt-induced nisin resistance of stationary-phase wild-type strains (Fig. 2). Loss of LiaR leads to significantly lower salt-induced nisin resistance in both exponential- and stationary-phase cells.
SigB and LiaR contribute to salt-induced nisin resistance.
In L. monocytogenes, the stress response sigma factor SigB has known roles in salt tolerance (19) and in nisin resistance (20). Since loss of LiaR did not completely eliminate salt-induced nisin resistance, we hypothesized that SigB plays a role, along with LiaR, in salt-induced nisin resistance. To test this hypothesis, ΔsigB and ΔsigB ΔliaR mutants of strain H7858 were created, and nisin resistance was measured. Without salt exposure, exponential-phase cells of both the ΔliaR and ΔliaR ΔsigB mutants had significantly lower (adjusted P = 0.0001) nisin resistance (i.e., a larger decrease in cell numbers after nisin treatment) than wild-type H7858 (Fig. 3A). The ΔsigB strain had nisin resistance similar to that of H7858 (Fig. 3A), indicating that SigB does not play a role in the innate nisin resistance of L. monocytogenes. For exponential-phase cells exposed to salt, nisin resistance of ΔsigB, ΔliaR, and ΔliaR ΔsigB mutants was significantly lower (adjusted P = 0.026) than that of H7858. Nisin resistance after salt exposure was not significantly different (adjusted P = 0.223) among the ΔsigB, ΔliaR, and ΔliaR ΔsigB mutants, indicating that both SigB and LiaR contribute to salt-induced nisin resistance in exponential-phase cells.
Fig 3.
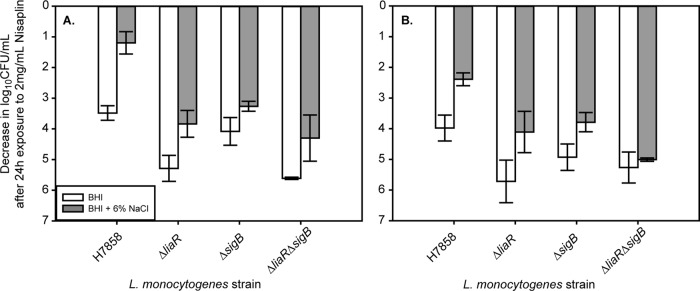
Log decrease in cell density for exponential-phase (A) or stationary-phase (B) cells of H7858 and isogenic ΔliaR, ΔsigB, and ΔliaR ΔsigB mutants exposed to 2 mg/ml Nisaplin in BHI for 24 h at 7°C. The average and standard deviation for three independent replicates are plotted for each strain. White bars represent cultures that were exposed only to BHI prior to exposure to Nisaplin, and gray bars represent cultures that were first exposed to BHI plus 6% NaCl before exposure to Nisaplin.
Without salt exposure, stationary-phase cells of the ΔliaR and ΔliaR ΔsigB mutants both had significantly lower (adjusted P = 0.03) nisin resistance than wild-type H7858, while the ΔsigB strain had nisin resistance similar to that of H7858 (Fig. 3B). Nisin resistance of the ΔsigB, ΔliaR, and ΔliaR ΔsigB mutants with salt exposure was significantly lower than that of H7858 (adjusted P = 0.026), and while the ΔsigB mutant had higher nisin resistance than the ΔliaR ΔsigB mutant, the resistance of the ΔliaR mutant and that of the ΔliaR ΔsigB mutant were similar. SigB thus seems to play a role in salt-induced nisin resistance in stationary-phase cells, but this seems at least partially independent of LiaR.
Transcript levels of LiaR-regulated genes are induced by salt stress.
Q-PCR was used to quantify salt-induced changes in transcript levels of selected genes known to be regulated by LiaR. liaI and liaH have been shown to be strongly induced by LiaR in both L. monocytogenes and Bacillus subtilis (14, 21) and were selected as positive controls. Genes known to be regulated by LiaR and with potential roles in nisin resistance were selected to determine if they are induced by LiaR under salt stress conditions. These genes included lmo2229, which encodes a penicillin binding protein and has been shown to have increased expression linked to nisin resistance (13), lmo1967, which encodes a toxic ion resistance protein, TelA, for which a deletion mutant has increased nisin sensitivity (15), and lmo1746, which encodes an ABC transporter permease and is predicted to be part of an antimicrobial peptide detoxification module (22). In strain H7858, liaR transcript levels increased 1.8-fold ± 0.4-fold, liaI transcript levels increased 29-fold ± 16-fold, and liaH transcript levels increased 49-fold ± 37-fold after exposure to salt, confirming that the LiaR regulon is induced during salt stress (Fig. 4).
Fig 4.
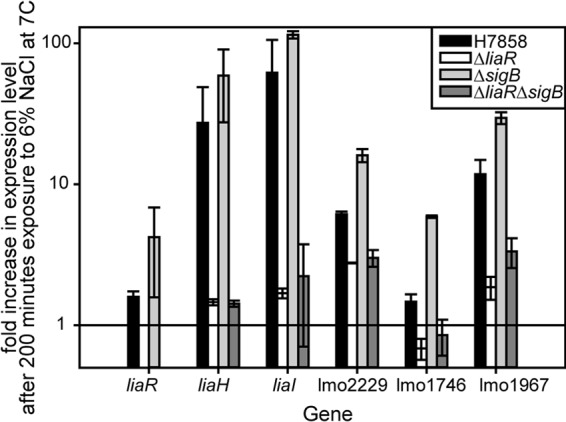
Salt-induced changes in transcript levels of genes known to be regulated by LiaR in strain H7858 and isogenic ΔliaR, ΔsigB, and ΔliaR ΔsigB mutants. The average and standard deviation for three independent replicates are plotted for each strain for each gene.
Transcript levels of liaI (P = 0.04) and liaH (P = 0.03) were significantly lower for the ΔliaR mutant, confirming the loss of function of the LiaR regulator. An absence of SigB did not significantly influence transcript levels of liaR, liaI, or liaH, since the ΔsigB strain had similar transcript levels of these genes compared to H7858. The liaI and liaH transcript levels for the ΔliaR ΔsigB strain were significantly lower than those for H7858 but were not significantly different from those for the ΔliaR strain (Fig. 4).
Salt-induced gene expression of lmo2229, lmo1746, and telA was influenced by LiaR. Transcript levels of lmo2229 (P = 0.004) and lmo1746 (P = 0.0019) were significantly lower for the ΔliaR mutant than for H7858; telA transcript levels were numerically lower for the ΔliaR mutant than for H7858, but this difference was not significant (P = 0.16) (Fig. 4). Salt-induced gene expression of lmo2229, lmo1746, and telA was also influenced by the absence of SigB. Transcript levels of these three genes were significantly higher (P = 0.007) for the ΔsigB strain than for H7858. These results indicate that while SigB has a role in salt-induced nisin resistance, it is not due to positive regulation of the genes tested here.
Lmo1746 and TelA contribute to LiaR-mediated salt-induced nisin resistance.
Since transcript levels of lmo2229, lmo1746, and telA were all LiaR dependent under salt stress, we hypothesized that these genes play a role in salt-induced nisin resistance mediated by LiaR. Δlmo1746, Δlmo2229, and ΔtelA mutants of H7858 were generated, and nisin resistance was measured. For exponential-phase cells (without salt exposure), only loss of lmo1746 led to nisin resistance that was significantly lower than that of wild-type H7858 (adjusted P < 0.0001) (Fig. 5A). After salt exposure, nisin resistance of the Δlmo1746 and ΔtelA mutants was significantly lower than that of H7858 (adjusted P = 0.0047), while the Δlmo2229 mutant exhibited similar nisin resistance to H7858 (Fig. 5A). For stationary-phase cells (without salt exposure) treated with nisin, results were similar to those of exponential-phase cells, where only loss of lmo1746 led to nisin resistance that was significantly lower than that of wild-type H7858 (adjusted P < 0.0057) (Fig. 5B). With salt exposure, nisin resistance of H7858 and that of the Δlmo1746 mutant were similar, while resistance of the Δlmo2229 and ΔtelA mutants was significantly greater than that of H7858 (adjusted P = 0.04), suggesting growth phase-dependent effects of lmo2229 and telA on nisin resistance (Fig. 5B).
Fig 5.
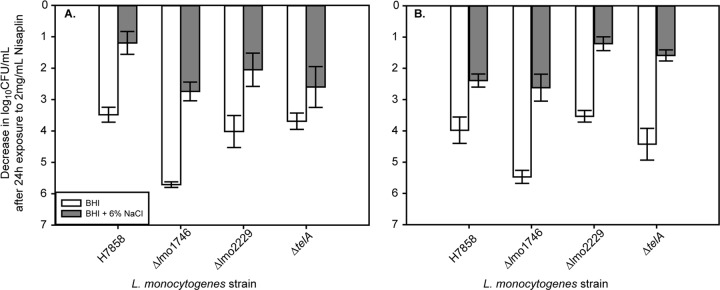
Log decrease in cell density for exponential-phase (A) or stationary-phase (B) cells of H7858 and isogenic Δlmo1746, Δlmo2229, and ΔtelA mutants exposed to 2 mg/ml Nisaplin in BHI for 24 h at 7°C. The average and standard deviation for three independent replicates are plotted for each strain. White bars represent cultures that were exposed only to BHI prior to exposure to Nisaplin, and gray bars represent cultures that were first exposed to BHI plus 6% NaCl before exposure to Nisaplin.
DISCUSSION
While it is well established that the antimicrobial peptide nisin can be used as a control measure for L. monocytogenes on RTE foods, the effects of environmental conditions on L. monocytogenes nisin resistance in RTE foods are less well established, particularly at a mechanistic level. Our data reported here indicate that exposure of L. monocytogenes to salt stress at low temperature significantly impacts subsequent resistance to nisin. In response to salt stress, L. monocytogenes undergoes many alterations in gene expression, including activation of the cell envelope stress response controlled by LiaR (16), which, as shown here, plays a significant role in salt-induced nisin resistance.
Salt-induced cross-protection against nisin is independent of growth phase.
The antimicrobial effects of nisin on L. monocytogenes have been evaluated in laboratory media (4, 23), on foods (5, 6), and in animal models of infection (24). L. monocytogenes does exhibit a level of innate resistance to nisin, which can vary from strain to strain (4, 25). Nisin resistance can also be induced above the level of innate resistance. Factors that are known to influence nisin resistance include the growth phase and other environmental stresses. Stationary-phase cells exhibit higher resistance to nisin than exponential-phase cells (26). Exposure to mild acid (7) and repeated exposure to sublethal levels of nisin (13) can also increase resistance to lethal levels of nisin. Temperature also influences nisin resistance; the nisin resistance of L. monocytogenes strain ScottA was found to be very low when grown at 10°C compared to that when grown at 37°C, but the addition of 2 to 3.5% NaCl provided a protective effect and increased the frequency of nisin resistance at low temperature (23). Here we demonstrated that both exponential- and stationary-phase cells exposed to salt stress at 7°C exhibited cross-protective effects in the form of increased survival under subsequent nisin stress at low temperature.
While induced nisin resistance has typically been demonstrated for a single strain of L. monocytogenes, here we investigated multiple strains and showed that salt-induced nisin resistance occurs across diverse genetic backgrounds. Assessing the extent of induced nisin resistance among multiple strains is important in light of previous studies demonstrating variation in innate nisin resistance linked to a genomic island that is variably present in ∼50% of L. monocytogenes genomes (10). Our data indicate that while salt-induced nisin resistance does occur in all the strains tested, variation in the extent of the protective effect of salt does exist.
Induction of LiaR by salt stress significantly contributes to cross-protection against nisin.
The LiaFSR three-component system was first described for Bacillus subtilis as a system responding to cell wall-acting antibiotics that interfere with the lipid II cycle in the cytoplasmic membrane (21, 27). In L. monocytogenes, LiaFSR is known to be induced by cell wall-acting antibiotics (14) and by salt stress at low temperature (16). LiaS, the histidine kinase, was found to be expressed at significantly higher levels in a spontaneous nisin-resistant strain of L. monocytogenes (28), indicating a role for this cell envelope stress response system in L. monocytogenes nisin resistance. We demonstrated here that salt-induced cross-protection against nisin is caused in part by induction of LiaR during salt stress. Loss of LiaR led to significantly lower nisin resistance even after salt stress, and this effect was independent of the growth phase.
In addition to salt-induced resistance, LiaFSR also appears to contribute to innate nisin resistance. Without prior exposure to salt, the ΔliaR strains were more sensitive to nisin than the wild-type strains. This is in contrast to results in a recent study by Collins et al., where they found that deletion of liaS led to increased nisin resistance in strain LO28 at 37°C (29). These differences could be due to the inactivation of different members of the three-component system between the studies. Since the Collins et al. study was assessing innate, rather than induced, resistance and was conducted at 37°C, it may be that LiaFSR plays a different role in nisin resistance dependent on the inducing conditions as well as the growth temperature. Data presented here support this conclusion, since Δlmo2229 and ΔtelA mutants exhibited different levels of salt-induced nisin resistance dependent on the growth phase.
Various contributions of selected LiaR-regulated genes to salt-induced nisin resistance.
LiaR has been reported to regulate 27 genes as part of the cell envelope stress response in L. monocytogenes (14). Some of these genes, such as telA (15) and lmo2229 (29), have identified roles in innate resistance to nisin. telA forms an operon with lmo1966 and encodes a toxic ion resistance transporter that also plays a role in resistance to cell wall-acting antibiotics (15). Lmo2229 is a penicillin binding protein and was first described as contributing to nisin resistance due to overexpression in a spontaneous nisin-resistant strain of L. monocytogenes (13). Here we demonstrated that increased transcription of these genes under salt stress was in part due to activation by LiaR. In the absence of LiaR, salt-induced transcript levels of telA and lmo2229 were significantly reduced from those of the wild type, but loss of function of these genes had different effects on nisin resistance. Loss of lmo2229 did not impact nisin resistance, either innate or salt induced, and while loss of telA did not impact innate resistance, it did decrease salt-induced nisin resistance, though only in exponential-phase cells. These data suggest that telA may play a different role in the LiaR regulon dependent on the growth phase, and they also indicate that there may be other proteins that can have a compensatory effect, alleviating the loss of function of lmo2229.
Another member of the LiaFSR regulon that we assessed for its impact on nisin resistance was lmo1746, annotated as a permease of an ABC transporter. lmo1746 and lmo1747 (encoding the ATP binding subunit of the ABC transporter) are similar to bceAB in B. subtilis, which encodes an antimicrobial peptide detoxification module (30). BceAB is regulated by the two-component system BceRS, and these antimicrobial detoxification module–two-component system pairs are common among members of the Firmicutes (22). Loss of lmo1746 had significant negative effects on both innate and salt-induced nisin resistance in both exponential- and stationary-phase cells. It was recently speculated that Lmo1746-47, along with AnrAB and VirRS, may form an antimicrobial sensing and detoxification module similar to the VraDE-BraSR-BraDE circuit in Staphylococcus aureus (31). In this circuit, the BraDE ABC transporter is responsible for sensing the antimicrobial peptide and relaying the signal to activate the BraSR two-component system and therefore the BraDE transporter responsible for detoxification (31). Our results provide the first description of the contribution of lmo1746 to nisin resistance in L. monocytogenes, and the potential involvement of this locus in a detoxification module circuit remains to be investigated.
Conclusions.
Environmental stresses similar to those present on foods can lead to cross-protection of L. monocytogenes against nisin. While previous reports identified cell wall modifications (9), alterations in membrane fluidity (8), and the F1F0 ATPase (32) as cellular mechanisms contributing to nisin resistance in L. monocytogenes, we have demonstrated a role for the cell envelope stress response system LiaFSR in nisin resistance. Our data suggest that stresses that induce the cell envelope stress response could lead to increased resistance to antimicrobial peptides, decreasing the efficacy of these L. monocytogenes control measures. The potential for cross-protective effects should be taken into consideration when developing and implementing control measures with foods.
Supplementary Material
ACKNOWLEDGMENTS
We thank Barbara Bowen, Jihun Kang, and Emily Wright for constructing the deletion mutants used in this study. We also thank Julie Sherwood for assistance with the nisin survival experiments.
Support for this work was provided by award number R01A1052151 from the National Institute of Allergy and Infectious Diseases (to K.J.B.), by Agriculture and Food Research Initiative grant no. 2010-65201-20575 from the U.S. Department of Agriculture, National Institute of Food and Agriculture, Food Safety Program (to K.J.B.), and by funds from the North Dakota Agricultural Experiment Station (to T.M.B.).
Footnotes
Published ahead of print 12 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01797-13.
REFERENCES
- 1.Swaminathan B, Cabanes D, Zhang W, Cossart P. 2007. Listeria monocytogenes. In Doyle MP, Beuchat LR. (ed), Food microbiology: fundamentals and frontiers, 3rd ed. ASM Press, Washington, DC [Google Scholar]
- 2.FDA 2003. Quantitative assessment of relative risk to public health from foodborne Listeria monocytogenes among selected categories of ready-to-eat foods. FDA, Silver Spring, MD [Google Scholar]
- 3.Pradhan AK, Ivanek R, Grohn YT, Geornaras I, Sofos JN, Wiedmann M. 2009. Quantitative risk assessment for Listeria monocytogenes in selected categories of deli meats: impact of lactate and diacetate on listeriosis cases and deaths. J. Food Prot. 72:978–989 [DOI] [PubMed] [Google Scholar]
- 4.Ferreira MA, Lund BM. 1996. The effect of nisin on Listeria monocytogenes in culture medium and long-life cottage cheese. Lett. Appl. Microbiol. 22:433–438 [DOI] [PubMed] [Google Scholar]
- 5.Franklin NB, Cooksey KD, Getty KJ. 2004. Inhibition of Listeria monocytogenes on the surface of individually packaged hot dogs with a packaging film coating containing nisin. J. Food Prot. 67:480–485 [DOI] [PubMed] [Google Scholar]
- 6.Nilsson L, Huss HH, Gram L. 1997. Inhibition of Listeria monocytogenes on cold-smoked salmon by nisin and carbon dioxide atmosphere. Int. J. Food Microbiol. 38:217–227 [DOI] [PubMed] [Google Scholar]
- 7.Bonnet M, Montville TJ. 2005. Acid-tolerant Listeria monocytogenes persist in a model food system fermented with nisin-producing bacteria. Lett. Appl. Microbiol. 40:237–242 [DOI] [PubMed] [Google Scholar]
- 8.van Schaik W, Gahan CG, Hill C. 1999. Acid-adapted Listeria monocytogenes displays enhanced tolerance against the lantibiotics nisin and lacticin 3147. J. Food Prot. 62:536–539 [DOI] [PubMed] [Google Scholar]
- 9.Crandall AD, Montville TJ. 1998. Nisin resistance in Listeria monocytogenes ATCC 700302 is a complex phenotype. Appl. Environ. Microbiol. 64:231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Begley M, Cotter PD, Hill C, Ross RP. 2010. Glutamate decarboxylase-mediated nisin resistance in Listeria monocytogenes. Appl. Environ. Microbiol. 76:6541–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thedieck K, Hain T, Mohamed W, Tindall BJ, Nimtz M, Chakraborty T, Wehland J, Jansch L. 2006. The MprF protein is required for lysinylation of phospholipids in listerial membranes and confers resistance to cationic antimicrobial peptides (CAMPs) on Listeria monocytogenes. Mol. Microbiol. 62:1325–1339 [DOI] [PubMed] [Google Scholar]
- 12.Mandin P, Fsihi H, Dussurget O, Vergassola M, Milohanic E, Toledo-Arana A, Lasa I, Johansson J, Cossart P. 2005. VirR, a response regulator critical for Listeria monocytogenes virulence. Mol. Microbiol. 57:1367–1380 [DOI] [PubMed] [Google Scholar]
- 13.Gravesen A, Sorensen K, Aarestrup FM, Knochel S. 2001. Spontaneous nisin-resistant Listeria monocytogenes mutants with increased expression of a putative penicillin-binding protein and their sensitivity to various antibiotics. Microb. Drug Resist. (Larchmont, NY) 7:127–135 [DOI] [PubMed] [Google Scholar]
- 14.Fritsch F, Mauder N, Williams T, Weiser J, Oberle M, Beier D. 2011. The cell envelope stress response mediated by the LiaFSR three-component system of Listeria monocytogenes is controlled via the phosphatase activity of the bifunctional histidine kinase LiaS. Microbiology 157:373–386 [DOI] [PubMed] [Google Scholar]
- 15.Collins B, Joyce S, Hill C, Cotter PD, Ross RP. 2010. TelA contributes to the innate resistance of Listeria monocytogenes to nisin and other cell wall-acting antibiotics. Antimicrob. Agents Chemother. 54:4658–4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergholz TM, Bowen B, Wiedmann M, Boor KJ. 2012. Listeria monocytogenes shows temperature dependent and independent responses to salt stress, including responses that induce cross-protection to other stresses. Appl. Environ. Microbiol. 78:2602–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.CDC 1999. Update: multistate outbreak of listeriosis—United States, 1998–1999. JAMA 281:317–318 [PubMed] [Google Scholar]
- 18.Nelson KE, Fouts DE, Mongodin EF, Ravel J, DeBoy RT, Kolonay JF, Rasko DA, Angiuoli SV, Gill SR, Paulsen IT, Peterson J, White O, Nelson WC, Nierman W, Beanan MJ, Brinkac LM, Daugherty SC, Dodson RJ, Durkin AS, Madupu R, Haft DH, Selengut J, Van Aken S, Khouri H, Fedorova N, Forberger H, Tran B, Kathariou S, Wonderling LD, Uhlich GA, Bayles DO, Luchansky JB, Fraser CM. 2004. Whole genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen Listeria monocytogenes reveal new insights into the core genome components of this species. Nucleic Acids Res. 32:2386–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker LA, Cetin MS, Hutkins RW, Benson AK. 1998. Identification of the gene encoding the alternative sigma factor sigmaB from Listeria monocytogenes and its role in osmotolerance. J. Bacteriol. 180:4547–4554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Begley M, Hill C, Ross RP. 2006. Tolerance of Listeria monocytogenes to cell envelope-acting antimicrobial agents is dependent on SigB. Appl. Environ. Microbiol. 72:2231–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jordan S, Junker A, Helmann JD, Mascher T. 2006. Regulation of LiaRS-dependent gene expression in Bacillus subtilis: identification of inhibitor proteins, regulator binding sites, and target genes of a conserved cell envelope stress-sensing two-component system. J. Bacteriol. 188:5153–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dintner S, Staron A, Berchtold E, Petri T, Mascher T, Gebhard S. 2011. Coevolution of ABC transporters and two-component regulatory systems as resistance modules against antimicrobial peptides in Firmicutes bacteria. J. Bacteriol. 193:3851–3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeMartinis ECP, Crandall AD, Mazzotta AS, Montville TJ. 1997. Influence of pH, salt, and temperature on nisin resistance in Listeria monocytogenes. J. Food Prot. 60:420–423 [DOI] [PubMed] [Google Scholar]
- 24.Campion A, Casey PG, Field D, Cotter PD, Hill C, Ross RP. 2013. In vivo activity of nisin A and nisin V against Listeria monocytogenes in mice. BMC Microbiol. 13:23. 10.1186/1471-2180-13-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rasch M, Knochel S. 1998. Variations in tolerance of Listeria monocytogenes to nisin, pediocin PA-1 and bavaricin A. Lett. Appl. Microbiol. 27:275–278 [DOI] [PubMed] [Google Scholar]
- 26.Jydegaard AM, Gravesen A, Knochel S. 2000. Growth condition-related response of Listeria monocytogenes 412 to bacteriocin inactivation. Lett. Appl. Microbiol. 31:68–72 [DOI] [PubMed] [Google Scholar]
- 27.Mascher T, Zimmer SL, Smith TA, Helmann JD. 2004. Antibiotic-inducible promoter regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob. Agents Chemother. 48:2888–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gravesen A, Kallipolitis B, Holmstrom K, Hoiby PE, Ramnath M, Knochel S. 2004. pbp2229-mediated nisin resistance mechanism in Listeria monocytogenes confers cross-protection to class IIa bacteriocins and affects virulence gene expression. Appl. Environ. Microbiol. 70:1669–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collins B, Guinane CM, Cotter PD, Hill C, Ross RP. 2012. Assessing the contributions of the LiaS histidine kinase to the innate resistance of Listeria monocytogenes to nisin, cephalosporins, and disinfectants. Appl. Environ. Microbiol. 78:2923–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohki R, Giyanto Tateno K, Masuyama W, Moriya S, Kobayashi K, Ogasawara N. 2003. The BceRS two-component regulatory system induces expression of the bacitracin transporter, BceAB, in Bacillus subtilis. Mol. Microbiol. 49:1135–1144 [DOI] [PubMed] [Google Scholar]
- 31.Gebhard S, Mascher T. 2011. Antimicrobial peptide sensing and detoxification modules: unravelling the regulatory circuitry of Staphylococcus aureus. Mol. Microbiol. 81:581–587 [DOI] [PubMed] [Google Scholar]
- 32.Bonnet M, Rafi MM, Chikindas ML, Montville TJ. 2006. Bioenergetic mechanism for nisin resistance, induced by the acid tolerance response of Listeria monocytogenes. Appl. Environ. Microbiol. 72:2556–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sauders BD, Fortes ED, Morse DL, Dumas N, Kiehlbauch JA, Schukken Y, Hibbs JR, Wiedmann M. 2003. Molecular subtyping to detect human listeriosis clusters. Emerg. Infect. Dis. 9:672–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fugett E, Fortes E, Nnoka C, Wiedmann M. 2006. International Life Sciences Institute North America Listeria monocytogenes strain collection: development of standard Listeria monocytogenes strain sets for research and validation studies. J. Food Prot. 69:2929–2938 [DOI] [PubMed] [Google Scholar]
- 35.CDC 1989. Listeriosis associated with consumption of turkey franks. MMWR Morb. Mortal. Wkly. Rep. 38:267–268 [PubMed] [Google Scholar]
- 36.Bishop DK, Hinrichs DJ. 1987. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J. Immunol. 139:2005–2009 [PubMed] [Google Scholar]
- 37.Oliver HF, Orsi RH, Wiedmann M, Boor KJ. 2010. Listeria monocytogenes σB has a small core regulon and a conserved role in virulence but makes differential contributions to stress tolerance across a diverse collection of strains. Appl. Environ. Microbiol. 76:4216–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.