

Abstract
Caffeine and other N-methylated xanthines are natural products found in many foods, beverages, and pharmaceuticals. Therefore, it is not surprising that bacteria have evolved to live on caffeine as a sole carbon and nitrogen source. The caffeine degradation pathway of Pseudomonas putida CBB5 utilizes an unprecedented glutathione-S-transferase-dependent Rieske oxygenase for demethylation of 7-methylxanthine to xanthine, the final step in caffeine N-demethylation. The gene coding this function is unusual, in that the iron-sulfur and non-heme iron domains that compose the normally functional Rieske oxygenase (RO) are encoded by separate proteins. The non-heme iron domain is located in the monooxygenase, ndmC, while the Rieske [2Fe-2S] domain is fused to the RO reductase gene, ndmD. This fusion, however, does not interfere with the interaction of the reductase with N1- and N3-demethylase RO oxygenases, which are involved in the initial reactions of caffeine degradation. We demonstrate that the N7-demethylation reaction absolutely requires a unique, tightly bound protein complex composed of NdmC, NdmD, and NdmE, a novel glutathione-S-transferase (GST). NdmE is proposed to function as a noncatalytic subunit that serves a structural role in the complexation of the oxygenase (NdmC) and Rieske domains (NdmD). Genome analyses found this gene organization of a split RO and GST gene cluster to occur more broadly, implying a larger function for RO-GST protein partners.
INTRODUCTION
Caffeine (1,3,7-trimethylxanthine) and other N-methylated xanthines are well known for applications in food and as pharmaceuticals that improve lung function for asthmatics and chronic obstructive pulmonary disease (COPD) sufferers. More recently, these compounds have been investigated for use as natural insecticides and in treatments for cancer, septic shock, and functional neutrophil disorders (1–3). Enzymatic methods for producing and degrading these N-methylated xanthines could have broader applications for health through both biosynthesis and environmental remediation of waste and by-products. Therefore, bacteria that have evolved to live on caffeine as the sole carbon and nitrogen source are of interest, as are their metabolic pathways toward N-methylated xanthines.
Pseudomonas putida CBB5 degrades caffeine, theophylline (1,3-dimethylxanthine), and related methylxanthines via sequential N-demethylation to xanthine (4–6). The ordered N-demethylation of caffeine to xanthine occurs in three steps catalyzed by enzymes belonging to the Rieske oxygenase (RO) family (5, 6), which are encoded by the Alx operon. Initially, two Rieske, non-heme Fe(II) monooxygenases, NdmA and NdmB, remove the N1- and N3-methyl groups, respectively, from caffeine to form 7-methylxanthine. Both enzymes require an unusually large 65-kDa redox-dense RO reductase, NdmD, which transfers electrons from NADH to NdmA and NdmB for oxygen activation.
The final step in the caffeine degradation pathway is N7-demethylation of 7-methylxanthine to xanthine. This N7-demethylation activity was inseparable from NdmD after four chromatographic steps (6). A highly enriched protein fraction containing this activity was comprised of NdmD and two additional major protein bands, as visualized by SDS-PAGE. These two additional peptides are encoded by two genes in the Alx operon, labeled orf7 and orf8, which flank ndmD on the CBB5 genome (Fig. 1A). The orf7 gene, here referred to as ndmC, was predicted to catalyze the N7-demethylation activity; however, this could not be confirmed, as all attempts to express ndmC in Escherichia coli resulted in inclusion body formation. The incomplete orf8 gene, now named ndmE, had no discernible function but displayed high similarity to various glutathione-S-transferases (GSTs).
Fig 1.
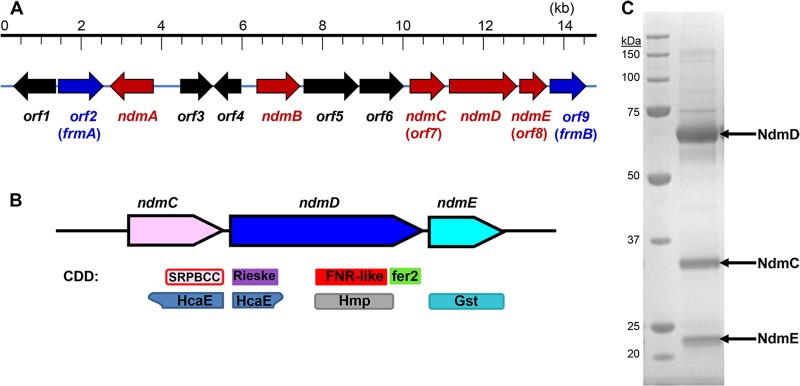
(A) Genetic organization of the 14.8-kb genomic DNA fragment containing the Alx operon for N-demethylation of caffeine to xanthine in P. putida CBB5. Arrows indicate the position and orientation of each orf. Red arrows represent genes required for N-demethylation, while blue arrows represent genes predicted to metabolize formaldehyde. (B) Gene organization for homologous ndmCDE gene clusters. One or two different ndmC homologs may appear in the gene cluster. Conserved domains from the Conserved Domains Database (CDD) are depicted below the genes. The SRPBCC superfamily contains the C-terminal catalytic domain of alpha oxygenases (pink with red border), HcaE members are ring-hydroxylating dioxygenases (blue-gray), the Rieske superfamily contains the N-terminal Rieske domain of alpha oxygenases (purple), the FNR-like superfamily is the ferredoxin reductase (FNR) domain that binds the NADH/FADH cofactor (red), HMP is the ferredoxin reductase family 1 (gray), fer2 is the 2Fe-2S cluster/ferredoxin domain (green), and Gst is the glutathione S-transferase (light blue). (C) SDS-PAGE of Sephacryl S200-purified His6-NdmCDE (right lane). Sizes of markers in the molecular mass ladder (left lane) are given on the left.
Here, we report that ndmE encodes a new type of GST that is absolutely required for N7-demethylation of 7-methylxanthine, the final step of caffeine degradation in P. putida CBB5. The N7-demethylase RO is unusual in itself because the iron-sulfur and non-heme iron domains that compose the normally functional oxygenase are encoded by two separate genes. The non-heme iron is contained in NdmC, while the iron-sulfur domain is fused to NdmD. NdmE is proposed to facilitate the formation of the NdmCDE complex, which catalyzes the N7-demethylation. This is the first report of a new class of GST-dependent ROs. Additional identification of similar uncharacterized gene clusters within genome databases suggests that there is a more generalized role for GSTs in oxygenation and/or biodegradation.
MATERIALS AND METHODS
Chemicals.
Caffeine, 7-methylxanthine, xanthine, ammonium acetate, acetic acid, and 2,4-pentanedione were purchased from Sigma-Aldrich (St. Louis, MO). Tryptone, yeast extract, Soytone, and agar were purchased from Becton, Dickinson and Company (Sparks, MD). NADH and isopropyl-β-d-thiogalactopyranoside (IPTG) were obtained from RPI Corp. (Mt. Prospect, IL). Restriction enzymes, Taq DNA polymerase, and Phusion high-fidelity DNA polymerase (the default polymerase used in all PCRs unless otherwise noted) were purchased from New England BioLabs (Ipswich, MA). High-pressure liquid chromatography (HPLC)-grade methanol (J. T. Baker, Phillipsburg, NJ) was used in all chromatographic studies.
Molecular biology procedures.
The full ndmE and orf9 sequences were determined by sequencing two PCR-amplified DNA fragments obtained from an EcoRI genomic DNA library (6) as described in the supplemental material. CBB5 strains harboring a single gene knockout of ndmC, ndmD, or ndmE were created by the method of Link et al. (7).
All cloning steps are described in full in the supplemental material. ndmC was cloned into the pET32a(+) plasmid at the NdeI and XhoI sites with a fused C-terminal hexahistidine (His6) tag to form plasmid p32CHis. ndmE was cloned into the pET28a(+) plasmid at the NdeI and EagI sites with a fused N-terminal His6 tag, resulting in plasmid p28HisE. Similarly, the ndmCDE genes, including native CBB5 intergenic DNA sequence surrounding ndmD, were cloned into the pET28a(+) plasmid at the NdeI and EagI sites with a His6 tag fused only to the N terminus of ndmC, creating plasmid p28HisCDE. ndmA and ndmB were cloned into the pACYCDuet-1 vector at the NcoI and BamHI sites (for ndmA) and the NdeI and KpnI sites (for ndmB), resulting in plasmid dAB.
Heterologous expression of ndm genes.
Plasmids p32CHis, p28HisE, and p28HisCDE were transformed into E. coli BL21(DE3) for overproduction of recombinant proteins. Plasmid dAB was transformed into E. coli BL21(DE3) already containing p28HisCDE. Expression of all genes was carried out in the same manner unless otherwise noted. The cells were grown in LB broth with appropriate antibiotic at 37°C and 225 rpm. When cell density reached an optical density at 600 nm (OD600) of 0.5, sterile iron(III) chloride was added to the culture at a final concentration of 10 μM, and the culture was shifted to 18°C and 250 rpm for incubation to increase the solubility of recombinant protein produced. Riboflavin was also added to cultures expressing ndmD to a final concentration of 2.5 μM. IPTG at a final concentration of 0.1 mM was added to induce gene expression when the OD600 reached 0.8 to 1.0. Induced cells were incubated at 18°C for 18 to 20 h and harvested by centrifugation at 4,000 × g for 10 min at 4°C. Cells were washed twice in 50 mM potassium phosphate (KPi) buffer (pH 7.5).
Purification of His6-NdmCDE.
Enzyme purification was performed at 4°C using an automated fast protein liquid chromatography system (ÄKTA FPLC system; Amersham Pharmacia Biotech). About 8 g cells containing His6-NdmCDE was suspended to 32 ml with 50 mM KPi buffer containing 300 mM NaCl and 10 mM imidazole (buffer A). Cells were lysed by passing twice through a chilled French press at 138 MPa. The lysates were centrifuged at 30,000 × g for 20 min, and supernatants were saved as cell extracts. Following cell lysis and centrifugation, the cell extract was loaded onto a 20-ml (bed volume) nickel-nitrilotriacetic acid (Ni-NTA) column (GE Healthcare) preequilibrated in Buffer A at a flow rate of 2 ml · min−1. The column was washed with 80 ml Buffer A to remove unbound protein. Bound protein was then eluted with 40 ml Buffer B (50 mM KPi with 300 mM NaCl and 125 mM imidazole) and concentrated using Amicon ultrafiltration units (molecular weight cutoff [MWCO], 10,000). The concentrated enzyme solution was dialyzed at 4°C in 50 mM KPi buffer containing 5% glycerol and 1 mM dithiothreitol (DTT) (KPGD buffer). All purified enzymes were stored short term on ice and at −80°C for long-term storage.
Resting cell assays.
CBB5 and its derivative strains containing single gene deletions were inoculated from frozen glycerol stocks and grown overnight in a modified M9 mineral salts medium containing 4 g · liter−1 Soytone and 2.5 g · liter−1 caffeine (M9CS medium) (4) at 30°C with 225 rpm rotary shaking. The cells were then inoculated into fresh M9CS medium (inoculum volume was 1% of the final volume) and again grown overnight. Upon reaching an OD600 of 1.5 to 2, cells were harvested by centrifugation at 4,000 × g for 10 min at 4°C. Cells were washed twice in 50 mM potassium phosphate (KPi) buffer (pH 7.5). Resting cell assays were conducted by resuspending cells in 2 ml 50 mM KPi buffer containing 500 μM caffeine or 7-methylxanthine and incubated at 30°C with shaking at 400 rpm in an incubated microplate shaker (VWR, Radnor, PA). A 100-μl sample was periodically removed and mixed with 100 μl acetonitrile to stop the reaction, and concentrations of metabolites and products were determined by HPLC.
Protein characterization.
Oxygen consumption, formaldehyde production, and methylxanthine N-demethylase activity were assayed as reported previously (5, 6). NdmC activity was also determined using a UV spectrophotometer. A typical 1-ml reaction mixture contained 0.5 mM 7-methylxanthine, 0.5 mM NADH, 50 μM Fe(NH4)2(SO4)2, and 160 μg of His6-NdmCDE. Apparent kinetic parameters of His6-NdmCDE were determined by measuring the initial rate of NADH oxidation (vo) in 50 mM KPi buffer (pH 7.5) at 30°C. The initial 7-methylxanthine concentrations ([S]) used were from 1 to 500 μM. The activity was determined by measuring the decrease in absorbance at 340 nm due to oxidation of NADH. An extinction coefficient of 6,200 M−1 · cm−1 for oxidized minus reduced NADH was used to quantify the activity. One unit of activity was defined as the amount of protein required to oxidize 1 μmol of NADH per min under defined reaction conditions.
Purity of proteins and molecular weight estimation were determined by running samples of approximately 10 μg protein for 55 min on 10% Bis-Tris gels with MOPS (morpholinepropanesulfonic acid) running buffer (Invitrogen Corporation, Carlsbad, CA). Gels were stained for viewing with GelCode blue safe protein stain (Thermo Fisher-Scientific, Waltham, MA). Protein concentration was determined by the Bradford method (8) using bovine serum albumin as the standard.
Analytical method.
Quantification of caffeine, 7-methylxanthine, and their metabolites was performed with a Shimadzu LC-20AT high-performance liquid chromatography (HPLC) system equipped with an SPD-M20A photodiode array detector and a Hypersil BDS C18 column (4.6 by 125 mm) as described previously (4–6).
Phylogenetic tree construction.
The close homologs to the ndmCDE genes were identified by searching the nonredundant protein database using BLASTP. Gene context was confirmed using GenBank nucleotide files for each of the homologs. All sequence alignments were produced using ClustalW (version 2.1) (9). Neighbor-joining trees, with 10,000 bootstrap samples, were constructed using Phylip software (version 3.69) (10).
Nucleotide sequence accession numbers.
The sequences for ndmC and ndmE have been deposited in GenBank under accession numbers JQ061129 and KC778191, respectively. Accession numbers of ndmA, ndmB, and ndmD have been previously reported (6).
RESULTS
Determination of domain architecture within NdmCDE.
The Alx operon encodes caffeine degradation in CBB5, including the ndmCDE genes. NdmC (orf7) is a 32.5-kDa protein homologous to the C terminus of many ROs. The Conserved Domains Database (CDD) (11) identified an SRPBCC (START/RHO_alpha_C/PITP/Bet_v1/CoxG/CalC) ligand-binding superfamily domain within this region (Fig. 1B). This superfamily domain usually has a deep, hydrophobic ligand binding pocket containing non-heme Fe(II) and is also present in NdmA and NdmB (6). The N-terminal Rieske center domain that generally accompanies such proteins is lacking in NdmC. Curiously, this domain is attached to the N terminus of NdmD. With the exception of this extra domain, NdmD has the typical RO reductase arrangement, consisting of ferredoxin reductase (FNR) and C-terminal ferredoxin domains (6).
Using a nested PCR approach with a CBB5 genomic DNA library, we determined the full ndmE (orf8) sequence and an additional complete open reading frame, orf9, located 3′ to ndmE (Fig. 1A). ndmE encoded a 25.5-kDa protein comprised of 222 amino acids, which is slightly larger than the 22-kDa protein band observed in a highly enriched protein fraction containing NdmC, NdmD, and NdmE from CBB5 (6). As suggested by the partial gene sequence, the full-length NdmE protein was homologous to many GSTs of various classes (Fig. 1B).
The theoretical 284-amino-acid sequence of orf9 displayed significant homology to S-formylglutathione hydrolases (frmB). FrmB and FrmA, an orf2 homolog, catalyze the glutathione (GSH)-dependent conversion of formaldehyde to formate (12) and provide a genetic correlation for utilization of formaldehyde produced by N-demethylation, which has been confirmed experimentally (6). It should be noted that although FrmAB use GSH, no GST is known to be associated with these reactions.
Functional expression and characterization of ndmCDE.
Although NdmD can be expressed alone as a partly (∼5%) soluble protein in E. coli, all attempts to express either NdmC or NdmE individually in E. coli resulted only in production of inclusion bodies (see Table S1 in the supplemental material). In order to detect soluble NdmC and NdmE, both proteins must be coexpressed with NdmD. The ndmCDE genes were cloned for overexpression in E. coli with an N-terminal His6 tag fused only to NdmC. When cell lysate containing His6-NdmCDE was passed through a Ni-NTA column, all three peptides bound to the column and eluted only with 125 mM imidazole. The His6-NdmCDE peptides also coeluted from a gel filtration column as a tight complex (Fig. 1C) with an estimated molecular mass of 360 kDa, suggesting that the complex is composed of trimers of each peptide (α3β3γ3). Interestingly, NdmA and NdmB, which are also dependent on NdmD for activity, do not complex with NdmCDE (see Fig. S1).
His6-NdmCDE was highly specific for N7-demethylation of 7-methylxanthine; no activity was observed toward caffeine or theobromine (3,7-dimethylxanthine), both of which also contain an N7-methyl group (data not shown). The apparent Km and kcat values of His6-NdmCDE for 7-methylxanthine were 15.3 ± 2.3 μM and 9.4 ± 0.3 min−1, respectively, with a catalytic efficiency (kcat/Km) of 0.61 ± 0.09 min−1 μM−1, similar to the efficiency of the purified enzyme from wild-type bacterium (5, 6). One O2 molecule is consumed per N7-demethylation step (see Fig. S2 in the supplemental material), resulting in stoichiometric formation of xanthine and formaldehyde (Fig. 2). Addition of 50 μM exogenous reduced GSH did not alter N7-demethylation activity. Also, His6-NdmCDE displayed no 1-chloro-2,4-dinitrobenzene-dependent conjugation of GSH, an activity displayed by many, but not all, GSTs. These data suggest that the N7-demethylation reaction occurs at the non-heme Fe(II) found in the binding pocket of NdmC, and that NdmE provides a structural, rather than catalytic, role.
Fig 2.
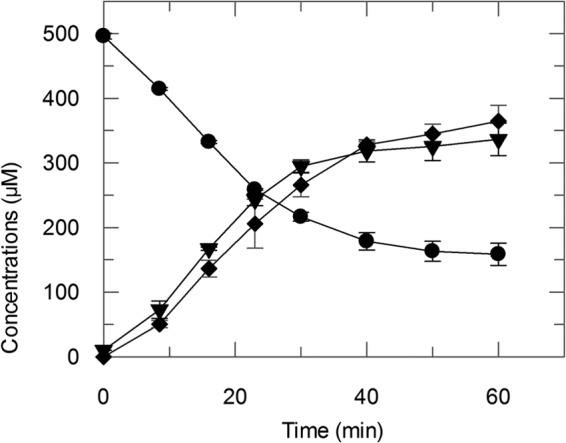
His6-NdmCDE consumed 337.4 ± 13.0 μM 7-methylxanthine (●) with concomitant formation of 336.5 ± 25.1 μM xanthine (▼) and 348.3 ± 8.3 μM formaldehyde (⧫). Concentrations are reported as means with standard deviations from triplicate results.
Individual gene knockout studies.
NdmCDE copurified from CBB5 under fairly stringent conditions, indicating that it exists as a complex. To provide additional proof that the NdmCDE complex is required for N-demethylation of 7-methylxanthine to xanthine, we individually knocked out each gene and assayed for N-demethylation activity. CBB5 resting cells completely N-demethylated 500 μM caffeine to xanthine in less than 2 h (Fig. 3A). Resting cells harboring the ndmC or ndmE knockout converted caffeine to 7-methylxanthine (Fig. 3B and C) but were unable to convert 7-methylxanthine to xanthine. CBB5ΔndmD resting cells showed very low activity toward caffeine (Fig. 3D), implying that there is a weak secondary reducing source for NdmA and NdmB in CBB5. All knockout strains exhibited a caffeine-negative growth phenotype, indicating that NdmCDE are required to live on caffeine.
Fig 3.

HPLC analyses of metabolites produced by resting cells of P. putida CBB5 and single-knockout strains. N-demethylation of caffeine (■) to theobromine (▲) and 7-methylxanthine (●) by resting cells of CBB5 (A), CBB5ΔndmC (B), CBB5ΔndmE (C), and CBB5ΔndmD (D). (E) P. putida CBB5 completely N7-demethylated 500 μM 7-methylxanthine (●) to xanthine within 75 min. (F) CBB5 strains with a single knockout of ndmC (○), ndmD (□), or ndmE (△) oxidized 60 to 125 μM 7-methylxanthine to 7-methyluric acid over 24 h. Each assay contained 0.5 mM caffeine or 7-methylxanthine, as well as freshly harvested and washed cells (OD600 of 2.5) in 50 mM potassium phosphate buffer (pH 7.5). Concentrations are reported as means with standard deviations from triplicate results.
To further confirm that the knockout strains cannot N7-demethylate 7-methylxanthine, we added 7-methylxanthine to resting cells. CBB5 consumed 500 μM 7-methylxanthine within 75 min (Fig. 3E). However, cells harboring a single deletion of ndmC, ndmD, or ndmE converted only 60 to 125 μM 7-methylxanthine to a previously unobserved product (Fig. 3F; also see Fig. S3 in the supplemental material) over 24 h. The retention time of the product (4.89 min) was slightly lower than that of 7-methylxanthine (5.47 min; see Fig. S3A) but was higher than that of xanthine (3.92 min). Also, the maximum UV absorbance of the product occurred at 232 and 286 nm, compared to 269 nm for 7-methylxanthine (see Fig. S3B) and 267 nm for xanthine. The slight reduction in retention time and maximum UV absorbance suggest that the product is the C-8 oxidized form of 7-methylxanthine, 7-methyluric acid. Unfortunately, there is no commercially available authentic standard to confirm that the product is 7-methyluric acid.
Phylogenetic analysis of ndmCDE.
The ndmCDE genes were used as Blast queries to identify potential homologs, resulting in 18 organisms (see Table S2 in the supplemental material) containing either one or two ndmC homologs of unknown function (C-terminal oxygenase domain) adjacent to homologs of ndmD (Rieske/reductase fusion) and ndmE (GST) (Fig. 1B). Organisms with two ndmC homolog genes belong to the Sinorhizobium and Mesorhizobium genera. Interestingly, Sinorhizobium medicae WSM419 is the only member of these genera with this gene cluster containing one ndmC homolog.
Neighbor-joining trees were prepared for the NdmCDE proteins to further investigate the evolutionary links between the various proteins (Fig. 4). The general morphology of the trees is very similar; the major difference is that some organisms have two NdmC homolog proteins. The proteins encoded by the ndmC gene closest to the ndmDE genes, labeled 2 and shown in blue in Fig. 4A, are positioned similarly to the NdmDE trees. The proteins encoded by the gene at the front of the cluster in the ndmCCDE arrangement (red in Fig. 4A) appear remote from their cluster counterparts. This suggests that these genes diverged after gene duplication with a potentially new function, and that the gene duplication and divergence occurred prior to gene dissemination.
Fig 4.
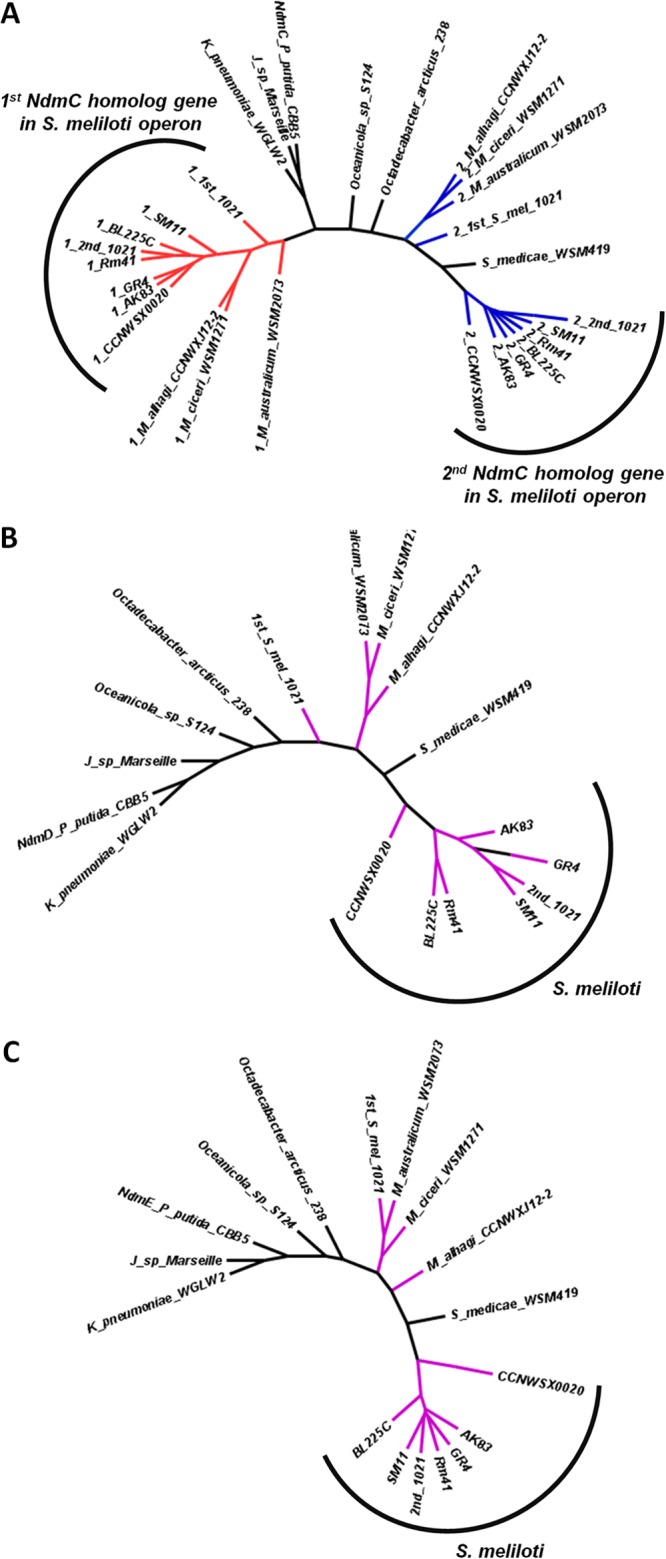
Neighbor-joining trees for homologous protein present in conserved gene clusters: NdmC (A), NdmD (B), and NdmE (C). Full strain names are listed in Table S2 in the supplemental material. Operons containing two NdmC proteins are colored: the first NdmC homolog is red, and the second is blue. NdmD and NdmE proteins in those strains are in purple.
DISCUSSION
In this report, we have identified and characterized the genes of P. putida CBB5 responsible for N7-demethylation of 7-methylxanthine to xanthine, which is the final step in the caffeine N-demethylation pathway (Fig. 5). While ndmC was previously hypothesized to encode an N7-demethylase, this could not be demonstrated, because previous attempts to express NdmC resulted only in formation of inclusion bodies (6). Soluble NdmC is obtained only when coexpressed with an RO reductase, NdmD, and a GST, NdmE, to form a large multisubunit protein complex. The reaction stoichiometry for N7-demethylation by NdmCDE is consistent with reactions catalyzed by monooxygenases and is similar to reactions catalyzed by NdmA and NdmB. As with NdmA and NdmB, the N7-demethylation is likely catalyzed at the non-heme iron in NdmC. NdmC itself is composed only of the SRPBCC catalytic domain of a traditional RO. Although NdmC lacks a Rieske [2Fe-2S] cluster, NdmD carries the extra Rieske [2Fe-2S] cluster at its N terminus. In spite of the unusual arrangement of domains in the ndmCDE gene cluster, the flow of electrons (see Fig. S4 in the supplemental material) appears to be typical of an RO (13).
Fig 5.
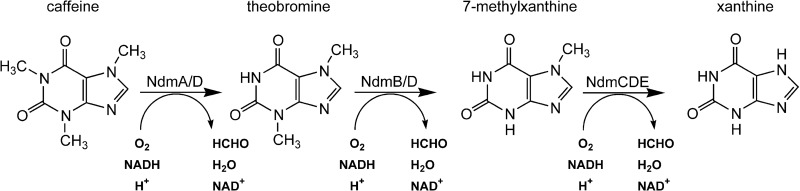
N-demethylation of caffeine to xanthine by P. putida CBB5. NdmA catalyzes the N1-demethylation of caffeine to theobromine, which is then N3-demethylated to 7-methylxanthine by NdmB. Both NdmA and NdmB couple with NdmD for electron transfer from NADH. 7-Methylxanthine is N7-demethylated to xanthine by the NdmCDE protein complex. Each reaction requires NADH and oxygen, with the methyl groups removed as formaldehyde.
NdmE was identified as a member of the GST superfamily (Fig. 1B), which contains proteins with great mechanistic and functional diversity housed within a conserved three-dimensional thioredoxin structure (14, 15). Catalytic functions within this superfamily include nucleophilic substitution, epoxide ring opening, ester thiolysis, isomerization, disulfide bond reduction, and hydroperoxidase reactions. GST superfamily members have also been found with noncatalytic functions, including intracellular transport, regulation of signaling cascades, transcriptional regulation, protein degradation and folding, and binding of a wide range of ligands (16–20). Newly identified functions of GSTs, including S-glutathionylation of proteins, indicate that other GST functionalities remain to be discovered (21).
Of the thousands of oxygenases known, this is the first report of the absolute requirement for a GST-like protein in any oxygenation reaction. Although GSH is not required for N7-demethylation of 7-methylxanthine by NdmCDE, it is possible that GSH is covalently bound to NdmE. GST proteins are almost exclusively found as dimers; however, the GST monomer has been hypothesized to be involved in protein-protein interactions (22). Recombinant protein biosynthesis, as a laboratory technique, often takes advantage of increased solubility when fused with GST (23). NdmE itself has no known redox groups. Although the exact function of the GST homolog NdmE has not been ascertained, tight complex formation found between NdmCDE and the inability to synthesize or purify these proteins individually in a soluble form suggest a key role for NdmE in complex formation and solubilization.
GST homologs of undetermined function have also been found in other operons, some containing RO enzymes. These operons are involved in the degradation of many xenobiotics and recalcitrant aromatic compounds, including gentisate (24), m-toluate and m-xylene (25), and the polycyclic aromatic hydrocarbons biphenyl, polychlorinated biphenyl, dibenzofuran, and dibenzo-p-dioxin (24–27). Unlike NdmE, these GST homologs do not appear to be essential for degradation of the target compounds of their operons. However, their presence is thought to confer advantages and allow function under specific conditions or with different substrates (20, 25, 27, 28).
Although other characterized bacterial GSTs are involved in degradation of xenobiotics and recalcitrant compounds, they either catalyze a specific reaction or are not necessary for the degradative pathway to function (20). To our knowledge, NdmE is the first characterized noncatalytic GST protein that is absolutely required for oxygenase activity and enables soluble NdmCDE expression in E. coli. NdmCDE also exists as a tight complex in the native organism, CBB5, and is required for bacteria to live on caffeine by enabling the final N7-demethylation of 7-methylxanthine to xanthine. The exact structural role that NdmE plays is still under investigation. However, there might be a larger role for NdmE-like proteins in the degradation of other natural products, xenobiotics, and complex hydrocarbons by bacteria, such as those recently released in the Gulf of Mexico (29).
Resting cells of CBB5 strains containing a single deletion of ndmC, ndmD, or ndmE were unable to metabolize 7-methylxanthine via N7-demethylation. Instead, these strains produced small amounts of 7-methyluric acid. Although oxidation of 7-methylxanthine at the C-8 position has not been previously observed in CBB5, CBB5 does oxidize theophylline and 1- and 3-methylxanthines to their respective methyluric acids as dead-end metabolites (4). Similarly, other bacterial strains have been shown to produce 7-methyluric acid from 7-methylxanthine (30, 31). This C-8 oxidation activity has previously been attributed to a broad-specificity xanthine dehydrogenase (4, 30, 31). That formation of 7-methyluric acid has not been previously observed in CBB5 is most likely due to the speed at which 7-methylxanthine is converted to xanthine by NdmCDE (Fig. 3E).
Further evidence that NdmCDE are required for N7-demethylation comes from a metabolically engineered, caffeine-addicted E. coli strain. The addition of a plasmid containing ndmABCD from CBB5 to the guanine auxotroph E. coli ΔguaB mutant failed to provide growth on caffeine. However, addition of an NdmE homolog gene from Janthinobacterium sp. strain Marseille enabled a caffeine-plus phenotype. This metabolically engineered strain N-demethylated caffeine to xanthine, which was further converted to guanine (32). These experiments agree with the knockout and purified protein experiments described here and indicate that NdmE is essential for the final N7-demethylation reaction and caffeine-plus growth phenotype.
We have also identified 18 additional organisms with a homologous ndmCDE gene cluster. Eleven of these organisms, which belong to either the Sinorhizobium or Mesorizobium genus, actually contain an additional ndmC homolog lacking a Rieske center. This is intriguing, since the organisms with two ndmC genes have taken a different evolutionary route than CBB5. In both cases, multiple oxygenases utilize the same RO reductase (NdmD homologs), but in the case of caffeine degradation, only one of the oxygenase proteins, NdmC, utilizes the Rieske center attached to the reductase (NdmD); NdmAB have their own Rieske centers. In the ndmCCDE cases, both of the oxygenases likely utilize the same Rieske domain attached to the NdmD homolog. Currently, none of these genes have been characterized; however, their divergence from the caffeine operon genes and difference in gene organization suggest a different function.
Dissemination of these genes could have been due to linear or horizontal gene transfer or a combination of the two. A 16S rRNA phylogenetic tree placed Sinorhizobium and Mesorhizobium genera on adjacent branches (33), indicating that an ndmC gene duplication event occurred prior to divergence with gene loss in other Sinorhizobium and Mesorhizobium genomes. Alternatively, Rhizobium genomes are known for repeated regions, frequent genome rearrangements, and megaplasmids (34–37). The presence of 2 sets of ndmCCDE gene clusters in Sinorhizobium meliloti 1021 and their position in the phylogenetic tree suggests that these two sets of genes were acquired independently by genomic rearrangement and explain the limited number of organisms containing ndmCDE genes. Octadecabacter arcticus 238 has transposase sequences flanking the gene region containing the ndmCDE homologs, thereby providing additional means of genetic mobilization. The caffeine degradation phenotype in CBB5 has not been observed to be transferable, mobile, or lost after culturing in a variety of culture conditions. Therefore, how these genes were acquired by a pseudomonad is not evident at this time.
In the case presented here, the Rieske domain has fused to the NdmD RO reductase protein. There are six additional cases lacking this fusion in which the oxygenase, composed of the SRPBCC domain and the Rieske domain, appear in separate proteins, with an adjacent GST and RO reductase. There are an additional 52 bacterial genomes in which the split oxygenase and GST cluster in close proximity without a close RO reductase. In fact, in the 76 present instances involving a split oxygenase, a GST protein is adjacent. Homologous regions in which the oxygenase appears as a single protein lack the GST. This implicates GST in functioning in some way to make the oxygenase, whose domains have been split into two separate proteins, into a functional oxygenase, which represents a completely new function for the GST superfamily.
In summary, P. putida CBB5 utilizes a new combination of RO and GST proteins for N7-demethylation of 7-methylxanthine to xanthine, which enables it to live on caffeine as the sole carbon and nitrogen source (Fig. 5). The traditional RO has been split in two: the catalytic (SRPBCC) domain is encoded by NdmC, while the Rieske domain has been fused to the N-terminal end of the RO reductase, NdmD. The NdmE GST protein is proposed to have a structural role, making it possible for an oxygenase that is split across two proteins to function. The tight NdmCDE complex acts as a highly specific 7-methylxanthine N7-demethylase, with one O2 molecule consumed per N7-methyl group removed as formaldehyde. This first report of a GST protein that is essential for an RO system adds more knowledge and a new function to the GST enzyme superfamily, and it suggests that biochemical studies of other RO systems need to include adjacent GST proteins for functionality.
Supplementary Material
Footnotes
Published ahead of print 28 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00585-13.
REFERENCES
- 1.Nathanson JA. 1984. Caffeine and related methylxanthines: possible naturally occurring pesticides. Science 226:184–187 [DOI] [PubMed] [Google Scholar]
- 2.Tuckey JA, Parry BR, McCall JL. 1992. Methylxanthines in surgery: a bright future? Aust. N. Z. J. Surg. 62:250–255 [DOI] [PubMed] [Google Scholar]
- 3.Malki AM, Gentry J, Evans SC. 2006. Differential effect of selected methylxanthine derivatives on radiosensitization of lung carcinoma cells. Exp. Oncol. 28:16–24 [PubMed] [Google Scholar]
- 4.Yu CL, Louie TM, Summers R, Kale Y, Gopishetty S, Subramanian M. 2009. Two distinct pathways for metabolism of theophylline and caffeine are coexpressed in Pseudomonas putida CBB5. J. Bacteriol. 191:4624–4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Summers RM, Louie TM, Yu CL, Subramanian M. 2011. Characterization of a broad-specificity non-haem iron N-demethylase from Pseudomonas putida CBB5 capable of utilizing several purine alkaloids as sole carbon and nitrogen source. Microbiology 157:583–592 [DOI] [PubMed] [Google Scholar]
- 6.Summers RM, Louie TM, Yu C-L, Gakhar L, Louie KC, Subramanian M. 2012. Novel, highly specific N-demethylases enable bacteria to live on caffeine and related purine alkaloids. J. Bacteriol. 194:2041–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Link AJ, Phillips D, Church GM. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 179:6228–6237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 9.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 10.Felsenstein J. 1989. PHYLIP–phylogeny inference package (version 3.2). Cladistics 5:164–166 [Google Scholar]
- 11.Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. 2013. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 41:D348–D352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herring CD, Blattner FR. 2004. Global transcriptional effects of a suppressor tRNA and the inactivation of the regulator frmR. J. Bacteriol. 186:6714–6720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mason JR, Cammack R. 1992. The electron-transport proteins of hydroxylating bacterial dioxygenases. Annu. Rev. Microbiol. 46:277–305 [DOI] [PubMed] [Google Scholar]
- 14.Atkinson HJ, Babbitt PC. 2009. Glutathione transferases are structural and functional outliers in the thioredoxin fold. Biochemistry 48:11108–11116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheehan D, Meade G, Foley VM, Dowd CA. 2001. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 360:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vuilleumier S. 1997. Bacterial glutathione S-transferases: what are they good for? J. Bacteriol. 179:1431–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Penninckx MJ, Elskens MT. 1993. Metabolism and functions of glutathione in microorganisms. Adv. Microb. Physiol. 34:239–301 [DOI] [PubMed] [Google Scholar]
- 18.Koonin EV, Tatusov RL, Altschul SF, Bryant SH, Mushegian AR, Bork P, Valencia A. 1994. Eukaryotic translation elongation factor 1γ contains a glutathione transferase domain–study of a diverse, ancient protein super family using motif search and structural modeling. Protein Sci. 3:2045–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dulhunty A, Gage P, Curtis S, Chelvanayagam G, Board P. 2001. The glutathione transferase structural family includes a nuclear chloride channel and a ryanodine receptor calcium release channel modulator. J. Biol. Chem. 276:3319–3323 [DOI] [PubMed] [Google Scholar]
- 20.Allocati N, Federici L, Masulli M, Di Ilio C. 2009. Glutathione transferases in bacteria. FEBS J. 276:58–75 [DOI] [PubMed] [Google Scholar]
- 21.Pajaud J, Kumar S, Rauch C, Morel F, Aninat C. 2012. Regulation of signal transduction by glutathione transferases. Int. J. Hepatol 2012:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vuilleumier S, Pagni M. 2002. The elusive roles of bacterial glutathione S-transferases: new lessons from genomes. Appl. Microbiol. Biotechnol. 58:138–146 [DOI] [PubMed] [Google Scholar]
- 23.Ghosh S, Rasheedi S, Rahim SS, Banerjee S, Choudhary RK, Chakhaiyar P, Ehtesham NZ, Mukhopadhyay S, Hasnain SE. 2004. Method for enhancing solubility of the expressed recombinant proteins in Escherichia coli. Biotechniques 37:418–423 [DOI] [PubMed] [Google Scholar]
- 24.Zhou N-Y, Fuenmayor SL, Williams PA. 2001. nag genes of Ralstonia (formerly Pseudomonas) sp. strain U2 encoding enzymes for gentisate catabolism. J. Bacteriol. 183:700–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bae M, Sul W, Koh S-C, Lee J, Zylstra G, Kim Y, Kim E. 2003. Implication of two glutathione S-transferases in the optimal metabolism of m-toluate by Sphingomonas yanoikuyae B1. Antonie Van Leeuwenhoek 84:25–30 [DOI] [PubMed] [Google Scholar]
- 26.Armengaud J, Timmis KN. 1997. Molecular characterization of Fdx1, a putidaredoxin-type [2Fe-2S] ferredoxin able to transfer electrons to the dioxin dioxygenase of Sphingomonas sp. RW1. Eur. J. Biochem. 247:833–842 [DOI] [PubMed] [Google Scholar]
- 27.Demanèche S, Meyer C, Micoud J, Louwagie M, Willison JC, Jouanneau Y. 2004. Identification and functional analysis of two aromatic-ring-hydroxylating dioxygenases from a sphingomonas strain that degrades various polycyclic aromatic hydrocarbons. Appl. Environ. Microbiol. 70:6714–6725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lloyd-Jones G, Lau PC. 1997. Glutathione S-transferase-encoding gene as a potential probe for environmental bacterial isolates capable of degrading polycyclic aromatic hydrocarbons. Appl. Environ. Microbiol. 63:3286–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kostka JE, Prakash O, Overholt WA, Green SJ, Freyer G, Canion A, Delgardio J, Norton N, Hazen TC, Huettel M. 2011. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the Deepwater Horizon oil spill. Appl. Environ. Microbiol. 77:7962–7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazzafera P, Olsson O, Sandberg G. 1996. Degradation of caffeine and related methylxanthines by Serratia marcescens isolated from soil under coffee cultivation. Microb. Ecol. 31:199–207 [DOI] [PubMed] [Google Scholar]
- 31.Yamaoka-Yano DM, Mazzafera P. 1999. Catabolism of caffeine and purification of a xanthine oxidade responsible for methyluric acids production in Pseudomonas putida L. Braz. J. Microbiol. 30:62–70 [Google Scholar]
- 32.Quandt EM, Hammerling MJ, Summers RM, Otoupal PB, Slater B, Alnahhas RN, Dasgupta A, Bachman JL, Subramanian MV, Barrick JE. 2013. Decaffeination and measurement of caffeine content by addicted Escherichia coli with a refactored N-demethylation operon from Pseudomonas putida CBB5. ACS Synth. Biol. 2:301–307 [DOI] [PubMed] [Google Scholar]
- 33.Reeve WG, Chain P, O'Hara G, Ardley J, Nandesena K, Bräu L, Tiwari R, Malfatti S, Kiss H, Lapidus A, Copeland A, Nolan M, Land M, Hauser L, Chang Y-J, Ivanova N, Mavromatis K, Markowitz V, Kyrpides NC, Gollagher M, Yates R, Dilworth M, Howieson J. 2010. Complete genome sequence of the Medicago microsymbiont Ensifer (Sinorhizobium) medicae strain WSM419. Stand. Genomic Sci. 2:77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo X, Flores M, Mavingui P, Fuentes SI, Hernández G, Dávila G, Palacios R. 2003. Natural genomic design in Sinorhizobium meliloti: novel genomic architectures. Genome Res. 13:1810–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flores M, González V, Pardo MA, Leija A, Martínez E, Romero D, Piñero D, Dávila G, Palacios R. 1988. Genomic instability in Rhizobium phaseoli. J. Bacteriol. 170:1191–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flores M, Mavingui P, Perret X, Broughton WJ, Romero D, Hernández G, Dávila G, Palacios R. 2000. Prediction, identification, and artificial selection of DNA rearrangements in Rhizobium: toward a natural genomic design. Proc. Natl. Acad. Sci. U. S. A. 97:9138–9143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaneko T, Nakamura Y, Sato S, Asamizu E, Kato T, Sasamoto S, Watanabe A, Idesawa K, Ishikawa A, Kawashima K, Kimura T, Kishida Y, Kiyokawa C, Kohara M, Matsumoto M, Matsuno A, Mochizuki Y, Nakayama S, Nakazaki N, Shimpo S, Sugimoto M, Takeuchi C, Yamada M, Tabata S. 2000. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 7:331–338 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.