

ABSTRACT
Vibrio cholerae is an aquatic organism that causes the severe acute diarrheal disease cholera. The ability of V. cholerae to cause disease is dependent upon the production of two critical virulence determinants, cholera toxin (CT) and the toxin-coregulated pilus (TCP). The expression of the genes that encode for CT and TCP production is under the control of a hierarchical regulatory system called the ToxR regulon, which functions to activate virulence gene expression in response to in vivo stimuli. Cyclic dipeptides have been found to be produced by numerous bacteria, yet their biological function remains unknown. V. cholerae has been shown to produce cyclo(Phe-Pro). Previous studies in our laboratory demonstrated that cyclo(Phe-Pro) inhibited V. cholerae virulence factor production. For this study, we report on the mechanism by which cyclo(Phe-Pro) inhibited virulence factor production. We have demonstrated that exogenous cyclo(Phe-Pro) activated the expression of leuO, a LysR-family regulator that had not been previously associated with V. cholerae virulence. Increased leuO expression repressed aphA transcription, which resulted in downregulation of the ToxR regulon and attenuated CT and TCP production. The cyclo(Phe-Pro)-dependent induction of leuO expression was found to be dependent upon the virulence regulator ToxR. Cyclo(Phe-Pro) did not affect toxR transcription or ToxR protein levels but appeared to enhance the ToxR-dependent transcription of leuO. These results have identified leuO as a new component of the ToxR regulon and demonstrate for the first time that ToxR is capable of downregulating virulence gene expression in response to an environmental cue.
IMPORTANCE
The ToxR regulon has been a focus of cholera research for more than three decades. During this time, a model has emerged wherein ToxR functions to activate the expression of Vibrio cholerae virulence factors upon host entry. V. cholerae and other enteric bacteria produce cyclo(Phe-Pro), a cyclic dipeptide that we identified as an inhibitor of V. cholerae virulence factor production. This finding suggested that cyclo(Phe-Pro) was a negative effector of virulence factor production and represented a molecule that could potentially be exploited for therapeutic development. In this work, we investigated the mechanism by which cyclo(Phe-Pro) inhibited virulence factor production. We found that cyclo(Phe-Pro) signaled through ToxR to activate the expression of leuO, a new virulence regulator that functioned to repress virulence factor production. Our results have identified a new arm of the ToxR regulon and suggest that ToxR may play a broader role in pathogenesis than previously known.
Introduction
Vibrio cholerae is a Gram-negative bacterium and facultative human pathogen that causes an estimated 3 million to 5 million cases of the acute diarrheal disease cholera each year (1). Humans acquire V. cholerae from environmental reservoirs by ingestion of V. cholerae-contaminated food or water. Upon ingestion, V. cholerae enters the small intestine, where a complicated regulatory cascade, called the ToxR regulon, activates virulence gene expression (2). The most important virulence genes activated by the ToxR regulon encode the production of cholera toxin (CT) and the toxin-coregulated pilus (TCP). CT is an enterotoxin responsible for the acute diarrhea that is associated with cholera, while the TCP is a type IV pilus that is required for colonization (3).
The ToxR regulon is a hierarchical regulatory system that activates virulence gene expression in response to environmental stimuli (2). Induction of the ToxR regulon starts with activation of tcpPH expression by AphA and AphB (4, 5). AphA and AphB are cytoplasmic regulatory proteins that cooperatively bind to the tcpPH promoter and activate its expression. Once TcpP is produced, it functions in conjunction with ToxR to activate toxT expression. TcpP and ToxR are structurally similar membrane-bound DNA binding proteins that are thought to modulate gene expression in response to external stimuli (6, 7). When stimulated, TcpP and ToxR bind to the toxT promoter and activate its expression. ToxT then directly activates the expression of the genes that encode for CT and TCP production (8). Late in infection, the ToxR regulon appears to be repressed prior to exiting the host, while other genes that are important for infectivity and environmental survival are upregulated (9–12). The mechanisms that effect the expression of genes late in infection are unknown.
Cyclic dipeptides (CDPs) are widespread in nature and are produced by a variety of bacteria (13–16). However, in most bacteria neither the biological function nor the mechanism of CDP biosynthesis is known. CDPs have been reported to modulate the expression of genes controlled by LuxR-type quorum sensing (QS) systems (14–16), a finding that suggested that CDPs may function as bacterial signaling molecules (16). Consistent with this idea, Vibrio spp. produce cyclo(Phe-Pro) (cFP), a CDP that functions as an effector of V. cholerae virulence gene expression (17, 18). cFP accumulates in V. cholerae culture supernatants in a growth-dependent manner and reaches a maximum concentration of ~0.8 mM as the cultures transition to stationary-phase growth (17). Work in our laboratory previously showed that the addition of cFP to cultures grown under virulence gene-inducing conditions inhibited tcpP expression and attenuated CT and TCP production (18). These results, coupled with the observation that cFP activated the expression of the ToxR-regulated ompU porin (17, 18), suggested that cFP may be affecting virulence gene expression via the ToxR regulon.
In this study, we sought to elucidate the cFP-dependent signal transduction cascade that led to attenuated virulence gene expression. The results of this work revealed that cFP activated a novel ToxR-dependent signaling cascade. Our results showed that cFP signaled through ToxR to activate leuO expression. LeuO is a LysR-family DNA binding protein that functions as a global regulator in other enteric pathogens (19, 20) but had not been previously associated with virulence in V. cholerae. Upregulation of leuO resulted in repressed aphA transcription which led to downregulation of the ToxR regulon and attenuated CT and TCP production. Our results have identified a new branch of the ToxR regulon and show that ToxR is capable of downregulating virulence factor production in response to an environmental cue.
RESULTS
cFP inhibits CT and TCP production in multiple epidemic V. cholerae strains.
Previous analysis showed that cFP inhibited CT and TCP production in O1 El Tor strain N16961. We therefore tested cFP for inhibitory activity against two additional epidemic El Tor strains (21, 22). The respective strains were grown overnight under AKI conditions in the presence or absence of 1 mM cFP before being processed for CT and TcpA quantification. The results showed that cFP inhibited CT and TCP production in strains HK1 and MO10 to a similar (HK1) or greater (MO10) extent than was observed in N16961 (see Fig. S1 in the supplemental material). These data suggest that cFP inhibition of virulence factor production is not a strain-specific phenomenon and is conserved among a broad range V. cholerae strains.
cFP induces leuO transcription.
Previous work in our laboratory showed that cFP inhibited tcpP expression (18). This suggested that cFP might be affecting genes that were upstream of tcpP in the ToxR regulon. To identify upstream genes, we performed microarray analysis of V. cholerae grown under AKI conditions in the presence and absence of cFP. Gene expression was assayed at 3 h in order to determine the effect of cFP on genes that were induced prior to tcpP. The microarray analysis resulted in the identification of 18 genes that were differentially expressed in the presence of cFP (see Table S1 in the supplemental material). This is likely a conservative estimate of cFP-responsive genes, since known cFP-responsive genes (e.g., tcpPH and the genes involved in CT and TCP biosynthesis) were not identified as differentially expressed. The omission of these genes was likely due to a combination of experimental variability and the early time point used for analysis. Two of the differentially expressed genes, vexR and leuO, encoded regulatory proteins that could be involved in cFP signaling. VexR appears to be a regulator of the VexAB RND efflux system and was not considered for further study here (23, 24). In contrast, leuO was deemed a high-priority gene for follow-up due to its role as a global regulator in other bacteria (19, 20).
The effect of cFP on leuO was confirmed by quantitative reverse transcription-PCR (qRT-PCR). Wild-type (WT) V. cholerae was grown under AKI conditions in the presence and absence of cFP, and RNA was isolated at 3 h and 6 h. These two time points were selected to confirm that changes in gene expression were not growth phase dependent. The results confirmed the microarray data and showed that cFP induced leuO expression by 2.0-fold at 3 h and 4.1-fold at 6 h (Fig. 1). cFP also repressed tcpP expression at 3 h and 6 h by 3.4- and 4.6-fold, respectively, confirming our published results (18). Since cFP inhibited tcpP expression, we tested if cFP affected aphA and aphB expression. ToxR was included as a control since it appears to be constitutively expressed under most growth conditions. The results showed that cFP did not appreciably affect aphB or toxR expression (Fig. 1). In contrast, cFP inhibited aphA transcription by 2.1-fold at 3 h and 2.8-fold at 6 h. This suggested that cFP likely effected tcpP expression by downregulating aphA. Previous analysis using a plasmid-based reporter did not reveal cFP repression of aphA (18). This discrepancy may have resulted from plasmid copy number effects or other regulatory mechanisms that are known to affect aphA (25).
FIG 1 .
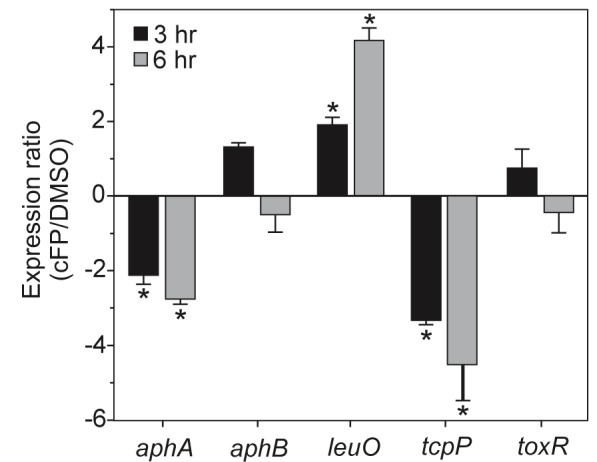
Effect of cFP on gene expression in V. cholerae. The WT was grown under AKI conditions in the presence of 1 mM cFP or an equivalent volume of DMSO. Total RNA was isolated at 3 h and 6 h and used for qRT-PCR. The expression ratios of the indicated genes in the cFP-treated versus DMSO control cultures were normalized to 16s RNA and are presented as the means and SEM for three biological replicates. *, P < 0.01 from a hypothetical value of 1.0.
LeuO represses CT and TCP production.
Since leuO was induced by cFP, we sought to determine if leuO functioned in virulence regulation. We therefore tested the effect of leuO expression on CT and TCP production. The WT containing pBAD18-leuO or the pBAD18 control was grown under AKI conditions in the presence of 0.02% or 0.08% arabinose before being assayed for CT and TcpA production. The results showed that leuO overexpression attenuated CT and TcpA production (Fig. 2A). These findings suggested that LeuO was a CT and TCP repressor.
FIG 2 .
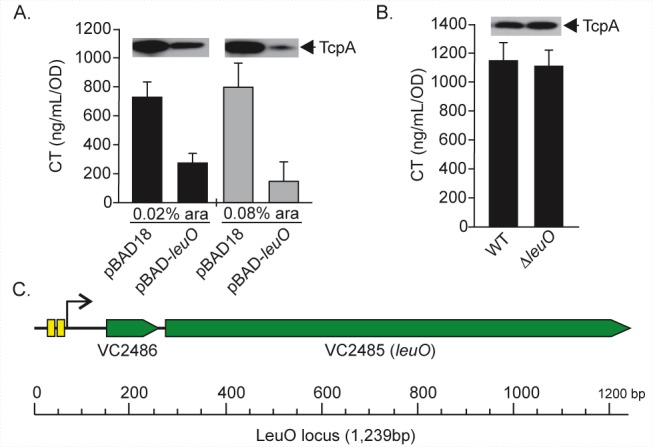
Effect of leuO on CT and TcpA production. The indicated strains were grown overnight under AKI conditions, normalized by OD, and used for CT ELISA and TcpA Western blotting. (A) CT and TcpA production in the WT (pBAD18) or WT (pBAD-leuO) strain grown with 0.02% or 0.08% arabinose. (B) CT and TcpA production in the WT and the ΔleuO mutant. The results are representative of three experiments ± SD. (C) Schematic diagram of the V. cholerae leuO locus. The gene encoding LeuO (VC2485) is downstream from VC2486 in an apparent two-gene operon. VC2486 and leuO are separated by 15 nucleotides (nt) and appear to be expressed from a common promoter that is located upstream of VC2486. The two ToxR-binding site consensus sequences in the leuO promoter are indicated by the yellow boxes.
LeuO is encoded in an apparent operon consisting of VC2486 and VC2485 (leuO) (Fig. 2C). VC2486 is a 102-bp open reading frame (ORF) located upstream of leuO and is annotated as encoding a hypothetical protein. Analysis of the leuO locus using BPROM promoter prediction software (http://www.softberry.com) indicated the presence of a single promoter upstream of VC2486. Consistent with this, VC2486 was coregulated with leuO in the microarray experiments (see Table S1 in the supplemental material). This suggested that VC2486 could affect CT and TCP production independent of leuO. To address this possibility, we repeated the above-described experiments with the WT containing pXB297 (pBAD18Km::VC2486) or pXB298 (pBAD18Km::leuO). The results showed that overexpression of VC2486 did not affect CT or TCP production, whereas overexpression of leuO inhibited CT and TCP production in an arabinose dose-dependent manner that was similar to results when both genes were expressed together in pXB269 (Fig. 2A and data not shown). These results confirmed that LeuO repressed CT and TCP production. Consistent with this conclusion, leuO deletion did not affect CT or TcpA production (Fig. 2B).
LeuO represses aphA expression.
We tested if LeuO affected aphA expression by using qRT-PCR to compare aphA expression in the WT and a ∆leuO mutant. The results showed a 2.4-fold increase in aphA expression and a 2.7-fold increase in tcpP expression in the ∆leuO strain relative to that in the WT (Fig. 3A). The fact that leuO deletion resulted in increased tcpP expression indicated that LeuO was an aphA repressor. We then overexpressed leuO in the WT under AKI conditions and quantified aphA, aphB, tcpP, and toxR expression by qRT-PCR. This showed that leuO overexpression resulted in a 4.4-fold decrease in aphA expression and a 6.6-fold decrease in tcpP transcription (Fig. 3B), further confirming that LeuO was an aphA repressor. In contrast, leuO overexpression did not have a significant effect on aphB or toxR. Overexpression of leuO also inhibited TcpP production relative to that of the control during growth under AKI conditions (Fig. 3C). Based on these results, we concluded that LeuO was an aphA repressor.
FIG 3 .
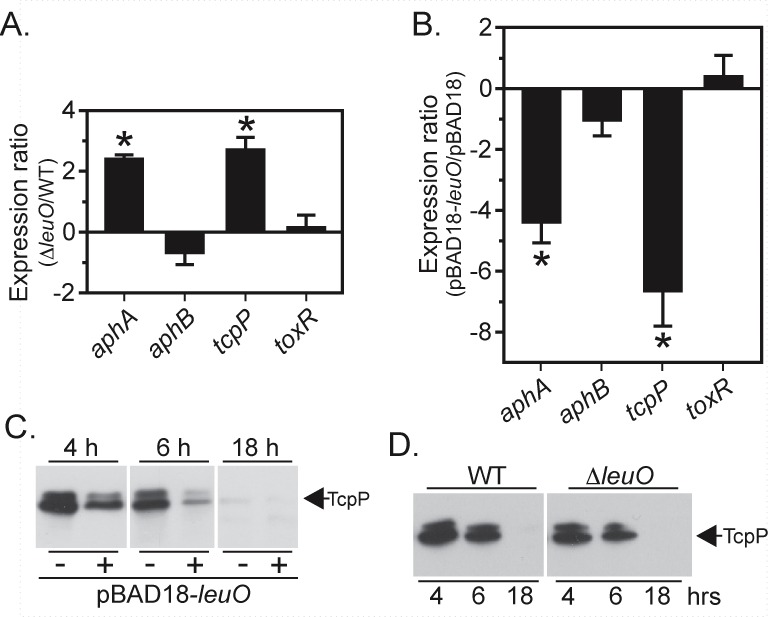
Effect of leuO on aphA and tcpP expression. (A) Quantification of gene expression by qRT-PCR. The WT or ΔleuO strain grown under AKI conditions. (B) Effect of leuO on the expression of the indicated genes. The WT (pBAD18) and WT (pBAD18-leuO) strains were grown under AKI conditions for 3 h, when RNA was isolated and used for qRT-PCR. The means and SEM for three biological replicates are presented. *, P < 0.01 from a hypothetical value of 1.0. (C) TcpP Western blot of WT(pBAD18) and WT(pBAD18-leuO) grown under AKI conditions with 0.02% arabinose. Aliquots were collected at the indicated times and normalized by OD before analysis. (D) TcpP Western blot of the WT and ΔleuO strains grown under AKI conditions for the indicated times. The samples were processed as described above. Samples presented within panels C and D were run on the same gel and exposed to X-ray film for identical times. The resulting X-ray film images were then converted to gray-scale TIFF files, and the contrast and brightness were adjusted before being cropped to generate panels C and D.
The expression of tcpP is temporally regulated, with maximal expression occurring at low cell density and then declining as the culture enters stationary phase (26). Since LeuO appeared to function as a tcpP repressor, we tested if leuO affected temporal tcpP expression (26, 27). TcpP production was assayed over time in the WT and the ΔleuO mutant during growth under AKI conditions. The results showed that TcpP production was inversely correlated with cell density (Fig. 3D). TcpP production was highest at early exponential growth (4 h) and then declined at mid-logarithmic phase (6 h) and was not detected in stationary-phase cultures (18 h). TcpP production in the leuO mutant was similar to that in the WT, which indicated that leuO did not affect TcpP production during growth under AKI conditions. We did not observe increased TcpP production in the leuO mutant, as would be predicted based on the qRT-PCR results in Fig. 3A. This likely resulted from the inherent variability and differences in detection sensitivity associated with each method. Temporal TcpP production in the leuO mutant suggests that other factors also affect TcpP production during growth under AKI conditions (28, 29).
cFP activity depends on LeuO.
If cFP inhibited CT and TCP production via leuO, we hypothesized that leuO deletion would abrogate cFP activity. We therefore compared the effects of cFP on CT and TCP production in the WT and the ΔleuO mutant. The results showed that cFP inhibited CT and TCP production in the WT in a dose-dependent manner as previously reported (18). However, cFP inhibition of CT/TCP production was largely but not completely lost in the leuO mutant (Fig. 4A and B). Similarly, cFP inhibited TcpP production in a dose-dependent manner in the WT but much less so in the leuO mutant (Fig. 4C). These results show that leuO is a major factor in the cFP-dependent inhibition of virulence factor expression. However, the fact that leuO deletion did not completely abrogate cFP activity suggests that cFP also affects virulence factor production by a leuO-independent mechanism.
FIG 4 .
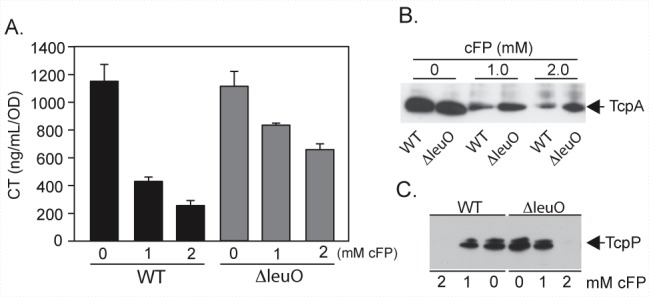
LeuO contributes to the antivirulence activity of cFP. WT and ΔleuO were grown under AKI conditions in the presence of 0, 1, or 2 mM cFP for 18 h and normalized by OD. (A) CT production. The results are the mean ± SD of three experiments. (B) TcpA Western blot. (C) TcpP Western blot of the WT or ΔleuO strain following 6 h of growth in 0, 1, or 2 mM cFP. The Western blot results are representative of three independent experiments.
ToxR is required for leuO expression.
cFP was previously shown to induce expression of the ToxR-regulated ompU porin (17, 18). This suggested that ToxR may function in cFP signaling and possibly leuO regulation. We tested this hypothesis by quantifying leuO-lacZ expression in WT, ∆toxRS, ∆toxT, and ∆tcpPH strains during growth under AKI conditions. The results showed that leuO was expressed at a low level in the WT at 3 h and that its expression increased with cell growth and reached a maximum expression level at stationary phase (Fig. 5A). Expression of leuO in the toxT and tcpPH mutants was similar to that in the WT, suggesting that neither of these genes affects leuO. In contrast, leuO expression in the toxRS mutant was dramatically reduced relative to that in the WT, indicating that ToxR was a positive regulator of leuO and that ToxR was responsible for the growth-dependent upregulation of leuO. Although leuO expression was dramatically reduced in the toxR mutant, leuO was still expressed at a low basal level and was induced at stationary phase. This suggests that other factors in addition to ToxR likely contribute to leuO expression.
FIG 5 .
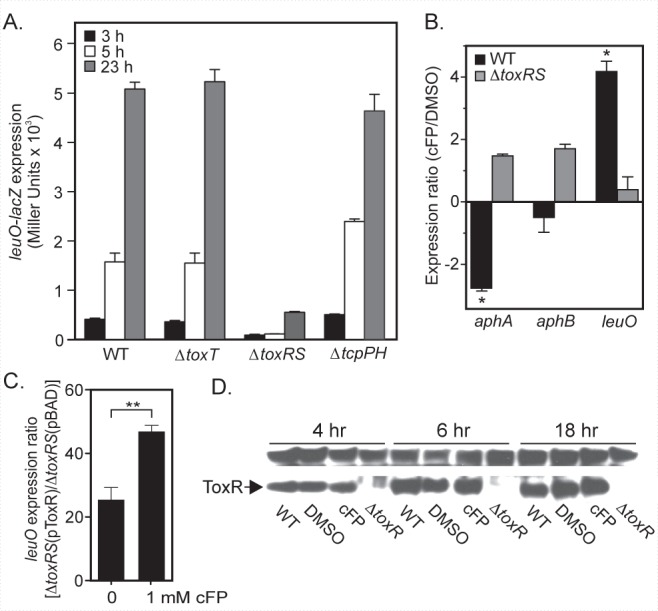
ToxR regulation of leuO expression. (A) Expression of leuO-lacZ in the WT, ΔtoxT, ΔtoxRS, and ΔtcpPH strains. The strains were grown under AKI conditions and assayed for β-galactosidase activity at the indicated times. The results are representative of three experiments ± SD. (B) Effect of 1 mM cFP on aphA, aphB, and leuO expression of the WT and ΔtoxRS strains following growth under AKI conditions. RNA was isolated at 3 h and used for qRT-PCR. The expression ratios are the cFP-treated versus DMSO control culture and represent the means ± SEM for three biological replicates. *, P < 0.01 from a hypothetical value of 1.0. (C) Expression of leuO in the ΔtoxRS strain following growth in the presence and absence of cFP. The ΔtoxRS strain containing pBAD18 or pBAD18-toxRS was grown under AKI conditions in broth containing 0.02% arabinose and 1 mM cFP or an equivalent volume of DMSO. RNA was isolated at 3 h and used for qRT-PCR. The leuO expression ratios in the ΔtoxRS(pBAD18-toxRS) versus ΔtoxRS(pBAD18) strains are presented. The results are the mean for three biological replicates ± SEM. “**” indicates significance at P < 0.001. (D) ToxR Western blot of the toxR mutant or the WT grown under AKI conditions in the absence or presence of 1 mM cFP. Culture aliquots were collected at the indicated time points, normalized by OD, and processed for ToxR Western blotting. The band above ToxR is a nonspecific immunoreactive protein that serves as a loading control.
ToxR is required for cFP induction of leuO expression.
Our data indicated both cFP and ToxR activated leuO expression. We therefore tested if cFP activity was dependent on ToxR by quantifying leuO and aphA expression in the WT and the ΔtoxRS mutant during growth in the presence and absence of cFP. The results showed that the addition of cFP caused a 4.2-fold increase in leuO expression in the WT (Fig. 5B). In contrast, cFP did not have a significant effect on leuO expression in the toxRS mutant. Likewise, the addition of cFP caused a 2.8-fold decrease in aphA expression in the WT but did not significantly affect aphA expression in the toxRS mutant. Taken together, these results strongly suggest that cFP signals through ToxR to activate leuO expression and to repress aphA expression.
Since our data suggested that cFP did not affect toxR expression (Fig. 1), we hypothesized that cFP may affect the ability of ToxR to activate leuO transcription. If this was true, we posited that cFP would enhance leuO expression in a complemented ΔtoxRS strain. We therefore tested the effect of cFP on leuO expression in the ΔtoxRS strain DT730 containing pBAD18::toxRS or the control vector (pBAD18) during growth under AKI conditions in the presence of arabinose. The results showed that toxRS expression complemented DT730 for leuO expression in the absence of cFP as exhibited by the 25.6-fold increase in leuO expression relative to that for the vector control (Fig. 5C); this further confirmed that ToxR was a positive regulator of leuO. The addition of cFP to the AKI broth caused an ~2-fold increase in leuO expression relative to that of the same strains grown without cFP (Fig. 5C). This suggested that cFP enhanced the ability of ectopically expressed ToxR to activate leuO expression. Collectively these data suggest that the cFP-dependent induction of leuO expression was likely a result of cFP stimulation of ToxR transcriptional activity.
The above data did not exclude the possibility that cFP also increased the cellular ToxR pool. We therefore determined the effect of cFP on ToxR production during growth under AKI conditions. The results showed that cFP did not affect ToxR production relative to that of the WT or the dimethyl sulfoxide (DMSO) control at any time point (Fig. 5D). The amount of ToxR in the cells did appear to increase during growth. An increase in ToxR was apparent between 4 and 6 h, which correlated with leuO induction (Fig. 5A). This suggests that the growth-dependent increase in ToxR could contribute to the growth-dependent increase in leuO expression.
The periplasmic domain of ToxR is required for cFP induction of leuO expression.
We tested the hypothesis that the ToxR periplasmic domain (PPD) was involved in cFP signaling. This was accomplished by determining whether loss of the ToxR PPD affected cFP activity. These experiments used a mutant version of ToxR, called ToxRmem, that lacked the PPD. Previous studies have shown that the PPD deletion in ToxRmem did not affect ToxR regulon expression during growth under AKI conditions (30).
We first confirmed that ToxRmem was functional in V. cholerae by testing whether ectopic expression of toxRmemS could complement a ΔtoxRS mutant for OmpU production. We cultured WT and ΔtoxRS strains containing pBAD18, pBAD18-toxRS, or pBAD18-toxRmemS under AKI conditions in the presence and absence of arabinose. Following overnight growth, normalized aliquots from each culture were resolved by SDS-PAGE. Porin production was then visualized by Coomassie brilliant blue dye staining and by OmpU Western blotting. The results showed that WT V. cholerae produced only OmpU, whereas the complemented ΔtoxRS strains grown in the absence of arabinose produced only OmpT (see Fig. S2 in the supplemental material). This was expected, since ToxR is an ompU activator and ompT repressor (31, 32). The addition of arabinose to the AKI broth resulted in a shift from OmpT to OmpU production in the ΔtoxRS strains containing pBAD18-toxRS and pBAD18-toxRmemS, while the empty vector control (i.e., pBAD18) produced only OmpT (see Fig. S2). These data confirmed the previously published data (30) and showed that ToxRmem is functional and can complement for loss of ΔtoxRS during growth under AKI conditions.
We next tested whether ToxRmem affected cFP signaling. We cultured the ΔtoxRS strain containing pBAD18, pBAD18-toxRS, or pBAD18-toxRmemS under AKI conditions (plus arabinose) in the presence and absence of cFP for 4 h and assayed for leuO and tcpP expression by qRT-PCR. The results showed that cFP induced leuO expression and repressed tcpP expression in the WT (Fig. 6) and that cFP activity was lost in the ΔtoxRS (pBAD18) control strain. The presence of pBAD18-toxRS restored cFP-dependent effects on leuO and tcpP expression in the ΔtoxRS mutant, confirming that toxRS was required for cFP signaling. In contrast, the presence of pBAD18-toxRmemS in the ΔtoxRS strain failed to restore cFP activity. This finding suggests that the periplasmic domain of ToxR is required for cFP activity.
FIG 6 .
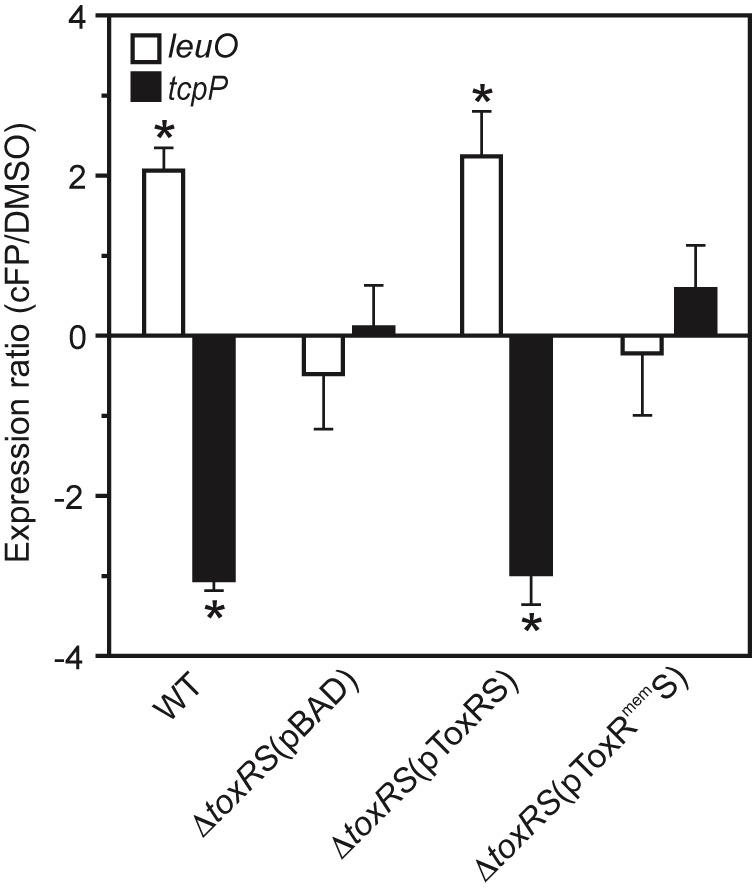
The periplasmic domain of ToxR is required for cFP activity. WT or ΔtoxRS V. cholerae containing the indicated plasmids was grown under AKI conditions in the presence of arabinose and 1 mM cFP or an equivalent volume of DMSO. Total RNA was isolated at 4 h and used for qRT-PCR. The expression ratios for leuO (white bars) and tcpP (black bars) in the cFP-treated versus DMSO control cultures were normalized to 16s RNA and are presented as the means and SEM for three biological replicates. *, P < 0.01 from a hypothetical value of 1.0.
ToxR consensus binding sites are present in the leuO promoter.
ToxR regulates the expression of many genes by binding to a consensus sequence located in the promoter of the target genes (33). Analysis of the leuO promoter showed the presence of two ToxR consensus sequence sites, located at −126 to −112 and −104 to −90, that were 100% conserved with the published consensus sequence (Fig. 7A). The presence of the ToxR consensus sequences in the leuO promoter was consistent with ToxR being a leuO activator and suggested that ToxR may function directly at the leuO promoter. To test this possibility, we expressed toxRS in the presence of a leuO-lux reporter (pJB906) in Escherichia coli. The results showed that in the absence of arabinose, leuO-lux was expressed at a low basal level (Fig. 7B). However, when toxRS expression was induced, we saw a dramatic arabinose dose-dependent increase in leuO-lux expression. Taken together, these results are consistent with ToxR acting directly on the leuO promoter. We were unable to test if cFP affected ToxRS activation of leuO-lux expression since cFP inhibits luminescence production in vitro.
FIG 7 .
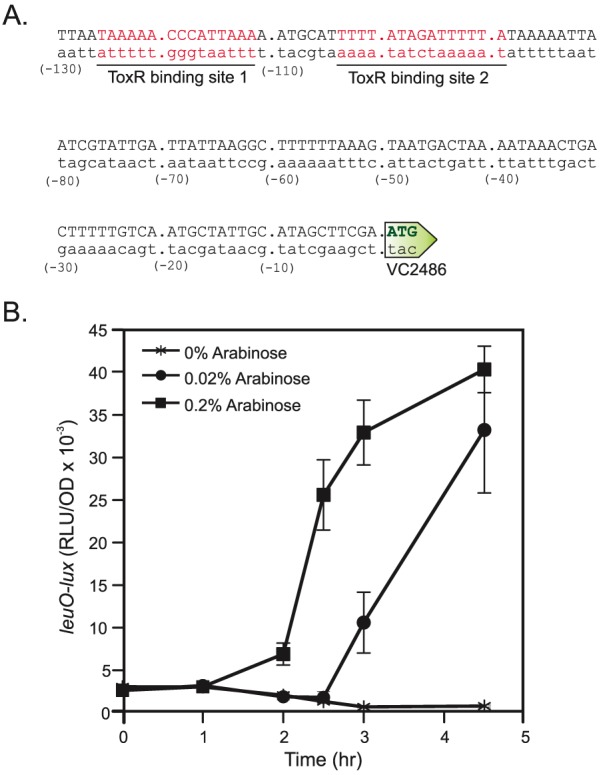
ToxR activates leuO expression. (A) DNA sequence of the leuO locus promoter region showing the location of the two ToxR-binding site consensus sequences in red. The ATG start codon for VC2486 is indicated by the green arrow. (B) Effect of toxRS on leuO expression in E. coli. E. coli containing a leuO-lux reporter (pJB906) and pBAD18-toxRS was grown in LB broth containing the indicated amounts of arabinose to induce toxRS expression. At the indicated time points, the relative luminescence (RLU) was determined and normalized by OD600. The results are representative of three experiments ± SD.
Expression of leuO in vivo.
We tested the ΔleuO mutant in the infant mouse competition model to determine if leuO affected colonization. The results showed that the ΔleuO mutant competed equally with the WT for colonization, which indicated that leuO was dispensable for colonization (see Fig. S3A in the supplemental material). This finding is consistent with the apparent function of leuO as a virulence repressor that functions late in infection. To confirm that leuO was expressed in vivo, we challenged infant mice with V. cholerae containing pJB906 (leuO-lux) and imaged bioluminescence production in the mice following overnight incubation. The results showed luminescence production in the gut of mice challenged with V. cholerae bearing the leuO-lux reporter, whereas there was no luminescence production in mice challenged with the empty vector control (pCM10) (see Fig. S3B). From this, we concluded that leuO was expressed in the infant mouse small intestine during colonization.
DISCUSSION
The ToxR regulon has been a focus of cholera research for more than three decades. From this work, a model has emerged whereby the ToxR regulon functions to activate virulence gene expression in response to in vivo stimuli. In this work, we showed for the first time that the ToxR regulon can also repress virulence factor production. Our data, in conjunction with published results (17, 18), support a model where ToxR functions in a novel regulatory circuit that links cFP to virulence gene expression. We showed that in response to extracellular cFP, ToxR significantly upregulated leuO expression. LeuO then downregulated the ToxR regulon by repression of aphA expression.
The role of ToxR in cFP signaling was evidenced by the findings that toxR deletion decreased leuO expression (Fig. 5A) and abolished cFP-dependent induction of leuO expression (Fig. 5B) and that cFP activated leuO transcription by ectopically expressed toxR in a complemented toxR mutant (Fig. 5C and 6). Taken together, these results strongly suggested that cFP signaled through ToxR to upregulate leuO expression. The linkage of ToxR to cFP signaling is significant, since cFP is unlikely to enter V. cholerae by hydrophobic uptake. Therefore, V. cholerae requires a means to sense cFP in the external environment. ToxR is a protein that could fulfill this role. ToxR is a membrane protein that contains a periplasmic sensing domain that is linked to a cytoplasmic DNA binding domain by a transmembrane spanning domain (7). The orientation of ToxR in the membrane is thought to facilitate the transduction of environmental stimuli across the cytoplasmic membrane to modulate the expression of ToxR-regulated genes (34). Consistent with this, a number of extracellular stimuli have been shown to modulate ToxR-regulated genes (35). This fact adds significance to our finding that cFP activity was dependent upon the ToxR PPD.
The observation that cFP activity was dependent upon the ToxR PPD was consistent with the presumed role of the PPD in signal transduction. However, the mechanism by which cFP affects ToxR is unclear. The function of the ToxR PPD in gene regulation is not entirely clear. The PPD was found to be dispensable for ompT and ompU regulation and virulence gene expression during growth under virulence gene-inducing conditions (30). In contrast, the PPD was required for proper porin expression during growth in minimal medium via a process that was linked to disulfide bond formation (36). The latter finding is consistent with the hypothesis that the PPD functions in environmental sensing. It is tempting to speculate that cFP may interact directly with ToxR via the PPD; however, we cannot rule out the possibility that cFP acts indirectly on ToxR through other proteins or by effects on the cytoplasmic membrane. Work is ongoing to discriminate between these possibilities.
The requirement for ToxR in cFP signaling suggests that ToxR may have broader functions in pathogenesis than was previously known. The current model for the ToxR regulon shows that ToxR plays an essential role in activating virulence factor production early in infection (2). The work presented here shows that ToxR could also play a role in downregulating virulence factor production. Studies in humans and animals have shown that the genes responsible for CT and TCP production were poorly expressed late in infection (9–11, 37), which indicated the existence of a mechanism to downregulate CT and TCP production late in infection. While the quorum sensing systems and ToxT and TcpP proteolysis have been implicated in this process (27, 29, 38), we hypothesize that the ToxR-dependent mechanism described in this work could also contribute to this phenotype. This hypothesis is supported by the observation that leuO was expressed in the infant mouse intestine (see Fig. S3 in the supplemental material), a finding that was consistent with published data indicating that the leuO operon was also expressed in human-shed V. cholerae (9) and in rabbit ileal loops (11, 39). Late in infection, V. cholerae also appears to be exposed to conditions under which cFP could be relevant (17, 18). For example, V. cholerae titers in the intestinal lumen can exceed 108 CFU/ml late in infection, and human- and animal-derived V. cholerae exhibits genetic signatures that are consistent with stationary-phase cells (9–11, 37). While these data are suggestive, additional work will be required to determine if ToxR is affecting the expression of genes late in infection.
LeuO had not been previously implicated in V. cholerae virulence. LeuO was first identified as a leucine operon activator in Salmonella (40). Subsequently, leuO was shown to function as a stationary-phase global regulator that affected the expression of multiple phenotypes, including virulence (19). We speculate that LeuO functions similarly in V. cholerae. This conclusion is supported for the following observations: leuO was induced at stationary phase (Fig. 5A), leuO overexpression inhibited virulence factor production (Fig. 2A), and leuO was associated with V. cholerae biofilm production (41). It is interesting to note that in E. coli LeuO functioned to regulate the expression of many genes by H-NS antagonism (20). This does not appear to be the case with V. cholerae virulence, since H-NS is also a virulence repressor (42–44). It appears that the role of LeuO in virulence may extend to other Vibrio spp., since recent reports have linked toxR and leuO to production of the type III secretion system in Vibrio parahaemolyticus (45, 46). Given that V. parahaemolyticus also produces cFP (17), it is possible that the ToxR-dependent cFP signaling pathway described in this work also functions in V. parahaemolyticus.
The cFP-dependent system appears to be redundant with the V. cholerae QS systems for virulence regulation, since both systems function to repress aphA. It is interesting to speculate that the cFP-dependent system could function late in infection as an alternative method to downregulate the ToxR regulon in QS-negative strains that are widespread among toxigenic V. cholerae isolates (22, 47). The similarity between the cFP-dependent system and the QS systems leads to the question of whether the cFP-dependent system is a QS system. This is unclear at present because we have not yet been able to identify the gene(s) responsible for cFP biosynthesis. In the absence of a cFP-negative mutant, we cannot determine whether cFP functions as a diffusible signal. We also cannot rule out the alternative hypothesis that cFP functions as an intracellular cue. For example, it is possible that cFP is produced in response to the metabolic status of the cell (e.g., amino acid limitation). This would be consistent with the induction of leuO at stationary phase and the reported roles of ToxR and LeuO in responding to environmental stimuli, including specific amino acids (19, 48, 49). This scenario could also explain leuO expression in vivo, since both human- and animal-derived V. cholerae bacteria exhibit genetic signatures indicative of growth in a nutrient-limiting environment (9, 11, 37).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this study are listed in Table S2 in the supplemental material. Strain JB58 (V. cholerae O1 El Tor strain N16961 ΔlacZ Smr) was used as the wild type (WT) for all experiments unless noted otherwise. E. coli EC100λpir and SM10λpir were the hosts for plasmid construction and conjugation, respectively. All bacterial strains were grown in Luria-Bertani (LB) broth or on LB agar at 37°C. V. cholerae was grown under AKI conditions for induction of the ToxR regulon as previously described (50). Antibiotics were used at the following concentrations: carbenicillin (Cb), 100 µg/ml; kanamycin (Km), 50 µg/ml, streptomycin (Sm), 100 µg/ml. Synthetic cFP (Bachem, Bubendorf, Switzerland) was used for all experiments and was dissolved in DMSO. Arabinose was added to media at the indicated concentrations to induce expression from the arabinose-regulated promoter in pBAD18 and pBAD18Km.
Plasmid and mutant construction.
V. cholerae O1 El Tor strain N16961 genomic DNA was used as a template for PCR amplification and cloning of all V. cholerae genes and gene promoters. Expression plasmid pXB269 was constructed by cloning the V. cholerae leuO locus into the multiple cloning site of pBAD18Km by PCR using the pBAD-leuO-F and pBAD-leuO-R primers (see Table S2 in the supplemental material). pXB297 and pXB298 were generated, respectively, by self religation of pXB269 that had been digested with SphI/EcoRI or XcmI/EcoRI. The toxRS expression plasmid pXB289 was created by cloning the toxRS gene into pBAD18 using the pBAD-toxR-F and pBAD-toxR-R primers. The lacZ fusion plasmid pXB266 was created by cloning the leuO promoter into pTL61T using the PleuO-F/PleuO-R primers (see Table S2). The lux reporter plasmid pJB906 was also created using the PleuO-F/PleuO-R PCR amplicon, which was digested with EcoRI-BamHI and cloned into the same restriction site in pCM10 to generate pJB906. The leuO deletion plasmid pWM91ΔleuO was used as previously described (41) to create strain XBV222. Plasmid pXB286 was constructed by crossover PCR as previously described using the respective pBAD-toxRmem PCR primers listed in Table S2 (51, 52).
Method for microarray analysis.
V. cholerae strain JB58 was grown under AKI conditions in the presence and absence of 1 mM cFP for 3 h when total RNA was purified from the cultures using Trizol (Invitrogen) according to the manufacturer’s instructions. The resulting RNA was treated with DNase and further purified on an RNeasy column according to the manufacturer’s directions (Qiagen) before being used for the microarray profiling experiments. The microarrays were provided by the Pathogen Functional Genomics Resource Center (Rockville, MD) and consisted of glass slides with 3,754 V. cholerae genes represented by 70-bp oligonucleotides that were spotted in triplicate. Processing of the mRNA and hybridization to the microarrays were performed as described at http://pfgrc.jcvi.org/index.php/microarray/protocols.html. A total of four microarrays were performed, including two dye swap experiments. The resulting microarrays were scanned using a GenePix 4000B scanner (Axon Instruments, Union City, CA), and the data were analyzed using the TM4 microarray suite (J. Craig Venter Institute). The scanned images were first processed using TM4 Spotfinder software using local background subtraction to generate the intensity data for each probe. The resulting data were normalized in the TM4 MIDAS program using the default normalization settings with a low-intensity filter of 75. Significant genes were then identified from the MIDAS output by applying a one-class t test analysis using the TM4 MeV software tool, and significant genes exhibiting a change in expression of ≥1.5-fold were selected for further analysis.
Quantitative real-time PCR, gene reporter assays.
V. cholerae cultures were grown under AKI conditions, and samples were collected at various time points for the qRT-PCR studies. Cells were pelleted at 5,000 × g for 10 min at 4°C, followed by RNA extraction as described above. First-strand synthesis was carried out using SuperScript III reverse transcriptase (Invitrogen) and random primers at 50°C for 1 h in the presence of 5 mM dithiothreitol (DTT). The reaction mixtures were then purified using a Wizard SV gel and PCR cleanup system (Promega, Madison, WI). cDNA was quantified photometrically and adjusted to 25 ng/µl. Gene-specific primers (see Table S2 in the supplemental material) were used to amplify selected genes. The expression levels of specific genes were quantified by amplifying 25 ng cDNA with 0.5-µM primers using the SYBR green PCR mix on a StepOnePlus real-time PCR System (Applied Biosystems). The relative expression levels for the cFP-treated and DMSO control cultures or the mutant and wild-type cultures were calculated by using the 2−∆∆CT method (53). The presented results are the means ± standard errors of the means (SEM) for three biological replicates, with each biological replicate generated from three technical replicates. 16s RNA was used as the internal control. Promoter reporter assays using transcriptional fusions of the indicated promoters to β-galactosidase in pTL61T were performed on cells grown under AKI growth conditions. Culture aliquots were collected at the indicated time points and processed in triplicate before β-galactosidase activity was quantified.
Luminescence reporter assay.
E. coli EC100λpir carrying the reporter plasmid pJB906 (PleuO-lux) and either pBAD18 or pXB289 (pBAD18::toxRS) was cultured overnight. The cultures were then diluted 1:100 into LB broth containing antibiotics and 0%, 0.02%, or 0.2% arabinose and incubated in 96-well plates with shaking at 37°C. A Bio-Tek Synergy HT plate reader was then used to quantify expression of the leuO-lux reporter at the times indicated. Luminescence was expressed as relative light units (RLU) divided by the optical density at 600 nm (OD600). The results are the averages for three replicates ± standard deviations (SD) and representative of three independent experiments.
Quantification of cholera toxin, TcpA, TcpP, ToxR, and OmpU.
CT production was determined by GM1 enzyme-linked immunosorbent assay (ELISA) as previously described using purified CT (Sigma) as a standard (50). The production of TcpA, TcpP, ToxR, and OmpU was determined by Western immunoblotting as previously described (50) using polyclonal rabbit antisera against TcpA, TcpP, ToxR, and OmpU; antisera against these proteins were graciously provided by Jun Zhu (anti-TcpA and anti-ToxR), Victor DiRita (anti-TcpP), and James Kaper (anti-OmpU). Immunoreactive proteins on the Western blots were visualized using the SuperSignal West Pico chemiluminescent detection kit (Pierce Biotechnology).
Infant mouse competition assay.
Overnight cultures of XBV222 (ΔlacZ ΔleuO) and WT strain JB3 (lacZ+ leuO+) were mixed at a 1:1 ratio before being delivered perorally to infant mice as previously described (50). An aliquot of the inoculum mixture was also serially diluted and plated on LB agar plates containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) to verify the input ratio of the two strains. The mice were incubated overnight at 30°C in a humidified chamber before being sacrificed. The small intestines were collected, homogenized, and serially diluted before being spread on LB agar plates containing Sm and X-Gal. Following overnight incubation at 37° C, the colonies were counted and scored as XBV222 (white colonies) or WT (blue colonies). A competitive index (CI) was then calculated as the ratio of the WT to XBV222 in the input divided by the ratio of the WT to XBV222 in the output. The CI for the in vitro growth competition assays were generated similarly.
In vivo imaging.
A previously described lux reporter system (54) was used to determine if leuO was expressed in vivo during colonization. To accomplish this, the leuO-lux reporter plasmid pJB906 and the empty vector control pCM10 were introduced into V. cholerae. Overnight broth cultures of V. cholerae containing the respective plasmids were diluted 200-fold into phosphate-buffered saline (PBS), and a 50-µl inoculum was then delivered by gavage to infant mice. Bioluminescence production from the leuO-lux reporter was then assayed 18 h later with an Ivis Spectrum imaging system (Caliper Life Sciences, Hopkinton, MA) according to the manufacturer’s directions.
Microarray data accession number.
The microarray data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE38272.
SUPPLEMENTAL MATERIAL
cFP activity in El Tor Vibrio cholerae isolates. V. cholerae O1 El Tor strains HK1 and N16961 or O139 El Tor strain MO10 was cultured overnight under virulence gene-inducing condition (i.e., AKI conditions) in the presence or absence of 1 mM cFP before being analyzed for CT and TCP production. (A) TcpA Western blot. (B) CT production. Download
OmpU production in V. cholerae ΔtoxRS expressing ectopic toxRS or toxRmemS. WT and ΔtoxRS V. cholerae strains containing the indicated plasmids were cultured under AKI growth conditions in the presence or absence of 0.1% arabinose (as indicated in parentheses). Following overnight growth, the cultures were normalized by optical density before equal quantities of each strain were collected by centrifugation. The cell pellets were resuspended in 3× SDS buffer and boiled for 10 min. Cell debris was removed by centrifugation before equivalent volumes of the solubilized protein samples were resolved on an SDS-7.5% PAGE gel. The gel was then either stained with Coomassie brilliant blue (upper panel) or transferred to a polyvinylidene difluoride (PVDF) membrane for Western blot analysis using polyclonal antisera to OmpU (lower panel). The positions of OmpU and OmpT are indicated. Download
Analysis of V. cholerae leuO in the infant mouse model. (A) Infant mouse competition assay. The competition assay was performed as described in Materials and Methods to determine if leuO was required for V. cholerae colonization of the infant mouse small intestine. The competitive index represents the ratio of the ΔleuO strain to the WT recovered from the small intestine divided by the ratio of the ΔleuO strain to the WT strain in the input. Each symbol represents one mouse. The horizontal line and error bars represent the means ± SD, respectively. (B) V. cholerae expresses leuO in vivo. Overnight culture of the WT containing pCM10 or pJB906 (pCM10::leuO-lux) was administered by gavage to 5-day-old infant mice. Bioluminescence imaging of the mice was performed at 18 h with an Ivis Spectrum imaging system. The heat bar indicates the bioluminescent intensity scale. Download
Vibrio cholerae genes that were differentially expressed in the presence of cFP; V. cholerae was grown under AKI conditions in the presence and absence of cFP for 3 h before being processed for microarray analysis as described in Materials and Methods
Strains, plasmids, and oligonucleotides used in this study
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) award R01AI091845. D.L.T. was supported by NIH training grant T32AI049820.
Footnotes
Citation Bina XR, Taylor DL, Vikram A, Ante VM, Bina JE. 2013. Vibrio cholerae ToxR downregulates virulence factor production in response to cyclo(Phe-Pro). mBio 4(5):e00366-13. doi:10.1128/mBio.00366-13.
REFERENCES
- 1. Kaper JB, Morris JG, Jr, Levine MM. 1995. Cholera. Clin. Microbiol. Rev. 8:48–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Childers BM, Klose KE. 2007. Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol. 2:335–344 [DOI] [PubMed] [Google Scholar]
- 3. Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc. Natl. Acad. Sci. U. S. A. 84:2833–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kovacikova G, Skorupski K. 1999. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J. Bacteriol. 181:4250–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Skorupski K, Taylor RK. 1999. A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol. Microbiol. 31:763–771 [DOI] [PubMed] [Google Scholar]
- 6. Häse CC, Mekalanos JJ. 1998. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 95:730–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miller VL, Taylor RK, Mekalanos JJ. 1987. Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell 48:271–279 [DOI] [PubMed] [Google Scholar]
- 8. DiRita VJ, Parsot C, Jander G, Mekalanos JJ. 1991. Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 88:5403–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bina J, Zhu J, Dziejman M, Faruque S, Calderwood S, Mekalanos J. 2003. ToxR regulon of Vibrio cholerae and its expression in vibrios shed by cholera patients. Proc. Natl. Acad. Sci. U. S. A. 100:2801–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Larocque RC, Harris JB, Dziejman M, Li X, Khan AI, Faruque AS, Faruque SM, Nair GB, Ryan ET, Qadri F, Mekalanos JJ, Calderwood SB. 2005. Transcriptional profiling of Vibrio cholerae recovered directly from patient specimens during early and late stages of human infection. Infect. Immun. 73:4488–4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Merrell DS, Butler SM, Qadri F, Dolganov NA, Alam A, Cohen MB, Calderwood SB, Schoolnik GK, Camilli A. 2002. Host-induced epidemic spread of the cholera bacterium. Nature 417:642–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schild S, Tamayo R, Nelson EJ, Qadri F, Calderwood SB, Camilli A. 2007. Genes induced late in infection increase fitness of Vibrio cholerae after release into the environment. Cell Host Microbe 2:264–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prasad C. 1995. Bioactive cyclic dipeptides. Peptides 16:151–164 [DOI] [PubMed] [Google Scholar]
- 14. Degrassi G, Aguilar C, Bosco M, Zahariev S, Pongor S, Venturi V. 2002. Plant growth-promoting Pseudomonas putida WCS358 produces and secretes four cyclic dipeptides: cross-talk with quorum sensing bacterial sensors. Curr. Microbiol. 45:250–254 [DOI] [PubMed] [Google Scholar]
- 15. Holden MT, Ram Chhabra S, de Nys R, Stead P, Bainton NJ, Hill PJ, Manefield M, Kumar N, Labatte M, England D, Rice S, Givskov M, Salmond GP, Stewart GS, Bycroft BW, Kjelleberg S, Williams P. 1999. Quorum-sensing cross talk: isolation and chemical characterization of cyclic dipeptides from Pseudomonas aeruginosa and other gram-negative bacteria. Mol. Microbiol. 33:1254–1266 [DOI] [PubMed] [Google Scholar]
- 16. Shiner EK, Rumbaugh KP, Williams SC. 2005. Inter-kingdom signaling: deciphering the language of acyl homoserine lactones. FEMS Microbiol. Rev. 29:935–947 [DOI] [PubMed] [Google Scholar]
- 17. Park DK, Lee KE, Baek CH, Kim IH, Kwon JH, Lee WK, Lee KH, Kim BS, Choi SH, Kim KS. 2006. Cyclo(Phe-Pro) modulates the expression of ompU in Vibrio spp. J. Bacteriol. 188:2214–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bina XR, Bina JE. 2010. The cyclic dipeptide cyclo(Phe-Pro) inhibits cholera toxin and toxin-coregulated pilus production in O1 El Tor Vibrio cholerae. J. Bacteriol. 192:3829–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dillon SC, Espinosa E, Hokamp K, Ussery DW, Casadesús J, Dorman CJ. 2012. LeuO is a global regulator of gene expression in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 85:1072–1089 [DOI] [PubMed] [Google Scholar]
- 20. Shimada T, Bridier A, Briandet R, Ishihama A. 2011. Novel roles of LeuO in transcription regulation of E. coli genome: antagonistic interplay with the universal silencer H-NS. Mol. Microbiol. 82:378–397 [DOI] [PubMed] [Google Scholar]
- 21. Dziejman M, Serruto D, Tam VC, Sturtevant D, Diraphat P, Faruque SM, Rahman MH, Heidelberg JF, Decker J, Li L, Montgomery KT, Grills G, Kucherlapati R, Mekalanos JJ. 2005. Genomic characterization of non-O1, non-O139 Vibrio cholerae reveals genes for a type III secretion system. Proc. Natl. Acad. Sci. U. S. A. 102:3465–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Joelsson A, Liu Z, Zhu J. 2006. Genetic and phenotypic diversity of quorum-sensing systems in clinical and environmental isolates of Vibrio cholerae. Infect. Immun. 74:1141–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bina JE, Provenzano D, Wang C, Bina XR, Mekalanos JJ. 2006. Characterization of the Vibrio cholerae vexAB and vexCD efflux systems. Arch. Microbiol. 186:171–181 [DOI] [PubMed] [Google Scholar]
- 24. Bina XR, Provenzano D, Nguyen N, Bina JE. 2008. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect. Immun. 76:3595–3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shao Y, Bassler BL. 2012. Quorum-sensing non-coding small RNAs use unique pairing regions to differentially control mRNA targets. Mol. Microbiol. 83:599–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kanjilal S, Citorik R, LaRocque RC, Ramoni MF, Calderwood SB. 2010. A systems biology approach to modeling Vibrio cholerae gene expression under virulence-inducing conditions. J. Bacteriol. 192:4300–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. 2002. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 99:3129–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beck NA, Krukonis ES, DiRita VJ. 2004. TcpH influences virulence gene expression in Vibrio cholerae by inhibiting degradation of the transcription activator TcpP. J. Bacteriol. 186:8309–8316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matson JS, DiRita VJ. 2005. Degradation of the membrane-localized virulence activator TcpP by the YaeL protease in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 102:16403–16408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crawford JA, Krukonis ES, DiRita VJ. 2003. Membrane localization of the ToxR winged-helix domain is required for TcpP-mediated virulence gene activation in Vibrio cholerae. Mol. Microbiol. 47:1459–1473 [DOI] [PubMed] [Google Scholar]
- 31. Li CC, Crawford JA, DiRita VJ, Kaper JB. 2000. Molecular cloning and transcriptional regulation of ompT, a ToxR-repressed gene in Vibrio cholerae. Mol. Microbiol. 35:189–203 [DOI] [PubMed] [Google Scholar]
- 32. Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goss TJ, Morgan SJ, French EL, Krukonis ES. 2013. ToxR recognizes a direct repeat element in the toxT, ompU, ompT, and ctxA promoters of Vibrio cholerae to regulate transcription. Infect. Immun. 81:884–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peterson KM. 2002. Expression of Vibrio cholerae virulence genes in response to environmental signals. Curr. Issues Intest. Microbiol. 3:29–38 [PubMed] [Google Scholar]
- 35. Gardel CL, Mekalanos JJ. 1994. Regulation of cholera toxin by temperature, pH, and osmolarity. Methods Enzymol. 235:517–526 [DOI] [PubMed] [Google Scholar]
- 36. Fengler VH, Boritsch EC, Tutz S, Seper A, Ebner H, Roier S, Schild S, Reidl J. 2012. Disulfide bond formation and ToxR activity in Vibrio cholerae. PLOS One 7:e47756. 10.1371/journal.pone.0047756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu Q, Dziejman M, Mekalanos JJ. 2003. Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc. Natl. Acad. Sci. U. S. A. 100:1286–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abuaita BH, Withey JH. 2011. Termination of Vibrio cholerae virulence gene expression is mediated by proteolysis of the major virulence activator, ToxT. Mol. Microbiol. 81:1640–1653 [DOI] [PubMed] [Google Scholar]
- 39. Nielsen AT, Dolganov NA, Rasmussen T, Otto G, Miller MC, Felt SA, Torreilles S, Schoolnik GK. 2010. A bistable switch and anatomical site control Vibrio cholerae virulence gene expression in the intestine. PLOS Pathog. 6:e1001102 http://dx.doi.org/doi:10.1371/journal.ppat.1001102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hertzberg KM, Gemmill R, Jones J, Calvo JM. 1980. Cloning of an EcoRI-generated fragment of the leucine operon of Salmonella typhimurium. Gene 8:135–152 [DOI] [PubMed] [Google Scholar]
- 41. Moorthy S, Watnick PI. 2005. Identification of novel stage-specific genetic requirements through whole genome transcription profiling of Vibrio cholerae biofilm development. Mol. Microbiol. 57:1623–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nye MB, Pfau JD, Skorupski K, Taylor RK. 2000. Vibrio cholerae H-NS silences virulence gene expression at multiple steps in the ToxR regulatory cascade. J. Bacteriol. 182:4295–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nye MB, Taylor RK. 2003. Vibrio cholerae H-NS domain structure and function with respect to transcriptional repression of ToxR regulon genes reveals differences among H-NS family members. Mol. Microbiol. 50:427–444 [DOI] [PubMed] [Google Scholar]
- 44. Stonehouse EA, Hulbert RR, Nye MB, Skorupski K, Taylor RK. 2011. H-NS binding and repression of the ctx promoter in Vibrio cholerae. J. Bacteriol. 193:979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gode-Potratz CJ, Chodur DM, McCarter LL. 2010. Calcium and iron regulate swarming and type III secretion in Vibrio parahaemolyticus. J. Bacteriol. 192:6025–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Whitaker WB, Parent MA, Boyd A, Richards GP, Boyd EF. 2012. The Vibrio parahaemolyticus ToxRS regulator is required for stress tolerance and colonization in a novel orogastric streptomycin-induced adult murine model. Infect. Immun. 80:1834–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Y, Wang H, Cui Z, Chen H, Zhong Z, Kan B, Zhu J. 2011. The prevalence of functional quorum-sensing systems in recently emerged Vibrio cholerae toxigenic strains. Environ. Microbiol. Rep. 3:218–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Skorupski K, Taylor RK. 1997. Control of the ToxR virulence regulon in Vibrio cholerae by environmental stimuli. Mol. Microbiol. 25:1003–1009 [DOI] [PubMed] [Google Scholar]
- 49. Majumder A, Fang M, Tsai KJ, Ueguchi C, Mizuno T, Wu HY. 2001. LeuO expression in response to starvation for branched-chain amino acids. J. Biol. Chem. 276:19046–19051 [DOI] [PubMed] [Google Scholar]
- 50. Taylor DL, Bina XR, Bina JE. 2012. Vibrio cholerae VexH encodes a multiple drug efflux pump that contributes to the production of cholera toxin and the toxin co-regulated pilus. PLoS One 7:e38208. 10.1371/journal.pone.0038208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Imai Y, Matsushima Y, Sugimura T, Terada M. 1991. A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res. 19:2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bina JE, Mekalanos JJ. 2001. Vibrio cholerae tolC is required for bile resistance and colonization. Infect. Immun. 69:4681–4685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 54. Morin CE, Kaper JB. 2009. Use of stabilized luciferase-expressing plasmids to examine in vivo-induced promoters in the Vibrio cholerae vaccine strain CVD 103-HgR. FEMS Immunol. Med. Microbiol. 57:69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
cFP activity in El Tor Vibrio cholerae isolates. V. cholerae O1 El Tor strains HK1 and N16961 or O139 El Tor strain MO10 was cultured overnight under virulence gene-inducing condition (i.e., AKI conditions) in the presence or absence of 1 mM cFP before being analyzed for CT and TCP production. (A) TcpA Western blot. (B) CT production. Download
OmpU production in V. cholerae ΔtoxRS expressing ectopic toxRS or toxRmemS. WT and ΔtoxRS V. cholerae strains containing the indicated plasmids were cultured under AKI growth conditions in the presence or absence of 0.1% arabinose (as indicated in parentheses). Following overnight growth, the cultures were normalized by optical density before equal quantities of each strain were collected by centrifugation. The cell pellets were resuspended in 3× SDS buffer and boiled for 10 min. Cell debris was removed by centrifugation before equivalent volumes of the solubilized protein samples were resolved on an SDS-7.5% PAGE gel. The gel was then either stained with Coomassie brilliant blue (upper panel) or transferred to a polyvinylidene difluoride (PVDF) membrane for Western blot analysis using polyclonal antisera to OmpU (lower panel). The positions of OmpU and OmpT are indicated. Download
Analysis of V. cholerae leuO in the infant mouse model. (A) Infant mouse competition assay. The competition assay was performed as described in Materials and Methods to determine if leuO was required for V. cholerae colonization of the infant mouse small intestine. The competitive index represents the ratio of the ΔleuO strain to the WT recovered from the small intestine divided by the ratio of the ΔleuO strain to the WT strain in the input. Each symbol represents one mouse. The horizontal line and error bars represent the means ± SD, respectively. (B) V. cholerae expresses leuO in vivo. Overnight culture of the WT containing pCM10 or pJB906 (pCM10::leuO-lux) was administered by gavage to 5-day-old infant mice. Bioluminescence imaging of the mice was performed at 18 h with an Ivis Spectrum imaging system. The heat bar indicates the bioluminescent intensity scale. Download
Vibrio cholerae genes that were differentially expressed in the presence of cFP; V. cholerae was grown under AKI conditions in the presence and absence of cFP for 3 h before being processed for microarray analysis as described in Materials and Methods
Strains, plasmids, and oligonucleotides used in this study