

Abstract
During mitosis, kinetochores physically link chromosomes to the dynamic ends of spindle microtubules. This linkage depends on the Ndc80 complex, a conserved and essential microtubule-binding component of the kinetochore. As a member of the complex, the Ndc80 protein forms microtubule attachments through a calponin homology domain. Ndc80 is also required for recruiting other components to the kinetochore and responding to mitotic regulatory signals. While the calponin homology domain has been the focus of biochemical and structural characterization, the function of the remainder of Ndc80 is poorly understood. Here, we utilized a new approach that couples high-throughput sequencing to a saturating linker-scanning mutagenesis screen in Saccharomyces cerevisiae. We identified domains in previously uncharacterized regions of Ndc80 that are essential for its function in vivo. We show that a helical hairpin adjacent to the calponin homology domain influences microtubule binding by the complex. Furthermore, a mutation in this hairpin abolishes the ability of the Dam1 complex to strengthen microtubule attachments made by the Ndc80 complex. Finally, we defined a C-terminal segment of Ndc80 required for tetramerization of the Ndc80 complex in vivo. This unbiased mutagenesis approach can be generally applied to genes in S. cerevisiae to identify functional properties and domains.
Keywords: Hec1, Illumina, coiled coil, total internal reflection fluorescence (TIRF)
ACCURATE chromosome segregation depends on the attachment between kinetochores and the dynamic ends of spindle microtubules. This attachment requires the Ndc80 complex, an essential and conserved microtubule-binding component of the kinetochore. In vivo, inactivation or depletion of Ndc80 complex components leads to detached kinetochores and severe chromosome segregation defects (reviewed in Kline-Smith et al. 2005). Mirroring kinetochore-microtubule linkages in vivo, recent experiments show that attachments between purified Ndc80 complexes and microtubule tips can persist throughout rounds of microtubule growth and shortening, can withstand tensile forces, can modulate microtubule dynamics, and can respond to regulation by mitotic kinases (Guimaraes et al. 2008; Miller et al. 2008; Powers et al. 2009; Tien et al. 2010; DeLuca et al. 2011; Schmidt et al. 2012; Umbreit et al. 2012).
The Ndc80 complex (Figure 1) is a rod-shaped heterotetramer of Ndc80, Nuf2, Spc24, and Spc25 (Osborne et al. 1994; Janke et al. 2001; Wigge and Kilmartin 2001; Wei et al. 2005). The two ends of the complex have globular domains, formed by Ndc80/Nuf2 and Spc24/Spc25, respectively. All four components of the complex are predicted to participate in coiled-coil interactions that link these two globular ends. Structural studies of the complex at atomic resolution have been limited to the two globular domains, revealing a microtubule-binding calponin homology (CH) domain in Ndc80 (Wei et al. 2006, 2007; Ciferri et al. 2008).
Figure 1.
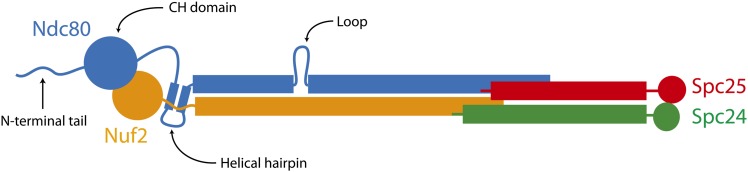
The Ndc80 complex contains Ndc80, Nuf2, Spc24, and Spc25.
These structural studies on the globular ends of the Ndc80 complex have provided high-resolution views of key protein–protein interfaces (Alushin et al. 2010; Malvezzi et al. 2013; Nishino et al. 2013). Together, they help explain how the Ndc80 complex plays a central role in kinetochore function by physically linking spindle microtubules to centromere-proximal kinetochore components. However, genetic and biochemical studies have suggested additional roles for the Ndc80 complex during mitosis. For example, mutations in NDC80 result in mislocalization of kinetochore- and microtubule-associated proteins, such as the Dam1 complex and Stu2 in yeast (He et al. 2001; Janke et al. 2002). It is still unknown how the Ndc80 complex interacts with many of its proposed binding partners, and much of the complex is composed of long coiled-coil elements that have not been amenable to structural characterization. To identify the functionally important regions within Ndc80, we employed an unbiased linker-scanning mutagenesis screen. This screen utilizes short insertions at random positions within a protein to determine putative functional domains. Previous applications of linker-scanning mutagenesis have been successful in generating mutants to examine cohesin complex formation and Cre recombinase activity, and to map the Rab8A interaction interface on JFC1 (Petyuk et al. 2004; Milutinovich et al. 2007; Pajunen et al. 2007). Here, we comprehensively covered NDC80 with 15-bp insertions at random positions, and the resulting mutant library was screened for viability in a red/white plasmid shuffle assay. High-throughput Illumina sequencing showed that lethal mutations fell into clusters, revealing several regions of Ndc80 that are essential for its function. These include the microtubule-binding domain, the helical hairpin, the tetramerization domain, and previously uncharacterized segments. Our approach, which couples unbiased mutagenesis to high-throughput sequencing, can be generally applied to reveal new functional domains of proteins in Saccharomyces cerevisiae.
Materials and Methods
Media
The compositions of YPD (yeast peptone dextrose rich) and SD (synthetic dextrose minimal) media were previously described (Burke et al. 2000). YPD-NAT medium is YPD with 25 µg/ml nourseothricin (clonNAT, Werner BioAgents). SD–lys medium was described previously (Nguyen et al. 1998). SD–ura low ade is SD medium containing 100 µg/ml tryptophan, 0.1% casamino acids, and 5 µg/ml adenine.
Plasmids
All plasmids used in this study are listed in Supporting Information, Table S1. QuikChange Lightning site-directed mutagenesis (Stratagene) was used to construct plasmids containing lethal mutations in NDC80.
Strains
All yeast strains used in this study were derived from W303 and are listed in Table S2. To make the strain used in the linker-scanning mutagenesis screen, JTY5-5C, the endogenous copy of NDC80 was deleted by PCR amplifying a NatMX cassette from pKG9 using primers with homology to the flanking DNA of NDC80. The deletion cassette was then transformed into the diploid strain JTY1 and selected for on YPD-NAT to generate JTY4. The deletion was checked by PCR to ensure replacement of NDC80 with the cassette. JTY4 was transformed with the “ADE3 plasmid” (pJT12) containing ADE3LYS2NDC80 and selected for on SD–lys. The transformants were sporulated and dissected to obtain JTY5-5C.
Linker-scanning mutagenesis
A “target plasmid” (pJT36) was constructed containing the wild-type NDC80 gene (including 245 upstream and 299 downstream base pairs), the ampicillin resistance gene, and the URA3 gene for selection. Importantly, this plasmid did not contain a NotI recognition sequence. Using the MuA transposase, an artificial transposon was inserted at a random location within the target plasmid (Finnzymes). Transposition efficiency was kept low so that, on average, each plasmid would contain only one transposon insertion. The transposon contains a kanamycin resistance gene flanked by the 8-bp NotI recognition sequence. Plasmids that contained a transposon were isolated by selection on kanamycin. These plasmids were then cut with NotI to remove the kanamycin cassette and then religated. The resulting “transposition library” contained plasmids that each have 15 bp inserted at a random location. Each 15-bp insertion contained 5 bp of duplicated target sequence and the NotI recognition sequence.
Red/white plasmid shuffle screen
To identify lethal insertions in NDC80, the transposition library was screened in S. cerevisiae using a red/white plasmid shuffle system (Davis 1992; Muller 1996). This screen was carried out in the strain JTY5-5C (ade2–1oc ade3Δ–100 ura3-1 ndc80Δ::NatMX), transformed with pJT12 (NDC80ADE3 in a 2-µm vector). In an ade2–1oc ade3Δ–100 background, yeast containing this ADE3 plasmid produce a red pigment when grown on low adenine plates (Bender and Pringle 1991). The transposition library was transformed into this strain and selected for growth on SD–ura low ade plates at 37°. Insertions that abolish the function of NDC80 are lethal and render cells dependent on the ADE3 plasmid with the wild-type copy of NDC80 for survival. Therefore, lethal mutations can be identified as those that yield solid red colonies. Insertions that do not disrupt the function of NDC80 have no effect on cell viability, making the ADE3 plasmid extraneous. These colonies have a sectored appearance—red in the center and white around the edges where the ADE3 plasmid was lost. Nonsectored red colonies, containing lethal mutations, were isolated and their plasmid DNA was extracted for sequencing.
We determined that the false-positive rate of the red/white plasmid shuffle screen is <0.3%. This was accomplished by transforming JTY5-5C [pJT12] with a plasmid containing wild-type NDC80 (pJT36) and counting the number of nonsectoring colonies. These false positives therefore represent colonies that were solid red despite the presence of a second wild-type copy of the gene. The representative lethal insertions selected for characterization (see below) were also retested in the red/white plasmid shuffle assay. JTY5-5C [pJT12] cells transformed with plasmids containing these insertions were confirmed to not sector.
Illumina sequencing
Illumina sequencing was used to determine the coverage of insertions in the transposition library and to identify the location of each lethal insertion obtained from the solid red colonies. Using the Expand Long Template PCR System (Roche), NDC80 was amplified both from the transposition library and the pooled lethal mutants. In this PCR, primers were designed to specifically amplify the mutant alleles from the transposition library plasmids and not NDC80 from the ADE3 plasmid (Table S3). Single-end Illumina libraries were prepared from PCR products that were sheared to an average size of 500 bp by sonication. The sheared DNA ends were repaired using the End-It DNA repair kit (Epicentre), and A-tailed for ligation of adaptors (Table S3). The products were size selected (250–350 bp) and enriched by PCR using the primers presented in Table S3. Unique 6-bp indices on the PCR primers permitted multiplexed Illumina sequencing to distinguish between products from the transposition library and the pooled lethal mutants. One sequencing run of the transposition library and both sequencing runs of the lethal insertion subsets were alternatively prepared using the TruSeq DNA Sample Preparation v2 Kit. Briefly, PCR products were sheared to an average size of 500 bp using a Covaris E210. The sheared DNA ends were repaired and adenylated using the TruSeq protocol. Adapters AD002, AD018, and AD019 (Table S3) were ligated onto the transposition library sample and the two lethal insertion subset samples, respectively. The products were size selected (400–500 bp) and enriched by PCR using the TruSeq PCR Master Mix. All sequencing was performed on an Illumina Genome Analyzer II, to obtain 36-bp single reads. Sequencing results are provided in Table S4.
Custom programs (available upon request) were used to analyze the FASTQ files for each Illumina sequencing run. The 36-bp reads were queried for the NotI recognition sequence; for each hit, the position of the NotI sequence within the read was recorded. The sequences flanking each NotI insertion were then aligned to the NDC80 gene using mrsFAST (Hach et al. 2010) to determine the position of the insertion in NDC80. Read coverage was obtained by aligning all reads (with and without NotI) to NDC80 using mrsFAST. Each 15-bp insertion was also translated to determine the protein sequence of the five inserted amino acids. See Table 1 for examples of insertion positions, resulting sequences, and the translated insertion residues.
Table 1. Representative lethal insertions in NDC80.
| Insertion position | Sequencea | Inserted residues | First mutation | In vivo expressionb | Kinetochore localizationc |
|---|---|---|---|---|---|
| 506 | TTCAAGTGGTTGCGGCCGCAGTGGTTATAT | LRPQW | Y170R | + | − |
| 656 | CACAAATTTCTGCGGCCGCAATTTCTTGGC | LRPQF | G220R | + | + |
| 839 | AAACTGTTAATGCGGCCGCAGTTAATTGAT | MRPQL | I280M | + | + |
| 940 | ATTCGTTCACTGCGGCCGCATTCACATAAT | CGRIH | I314C | + | + |
| 1148 | AAGATGAAATTGCGGCCGCAGAAATCCGAG | LRPQK | S383L | + | + |
| 1687 | GAAAACTCAATGCGGCCGCACTCAAATTAA | CGRTQ | I563C | + | + |
| 1957 | TATTGATATATGCGGCCGCAATATAACAAG | CGRNI | T653C | + | − |
We selected representative mutations from each lethal insertion cluster for further analysis. Each mutation represents the insertion with one of the highest number of NotI reads in the cluster. For further characterization, we required that each insertion was lethal at both the screening temperature of 37° and additionally 24°. Based on these criteria, we selected ins506, ins656, ins839, ins940, ins1148, ins1687, and ins1957 (Table 1) for further analysis.
Coiled-coil prediction and sequence alignment of Ndc80
The probabilities of coiled-coil formation for wild-type and mutant Ndc80 were predicted using Paircoil2 (McDonnell et al. 2006). To perform sequence alignments, NDC80 from S. cerevisiae and its orthologs in Saccharomyces bayanus, S. kudriavzevii, S. mikatae (Scannell et al. 2011), Lachancea (Kluyveromyces) thermotolerans, Kluyveromyces lactis, and Debaryomyces hansenii (http://genolevures.org) were translated using Transeq (Rice et al. 2000) and then aligned using Clustal-O (Blackshields et al. 2010). The similarity score was plotted for each position using Plotcon (Rice et al. 2000) with a window size of 21 bp.
Protein expression and purification
Recombinant S. cerevisiae Ndc80 and Dam1 complexes were expressed and purified as previously described (Miranda et al. 2005; Wei et al. 2005; Asbury et al. 2006; Powers et al. 2009).
Immunoprecipitation
JTY29-1C (NUF2-TAP) and JTY47-2B (SPC24-TAP) were transformed with plasmids encoding wild-type or mutant GFP–NDC80 (Table S1) and selected for growth on SD–ura at 24°. Controls for the immunoprecipitation include JTY29-1C and JTY47-2B transformed with pRS316, as well as JTY29-1B (NUF2) and JTY47-2A (SPC24) transformed with a wild-type GFP–NDC80 plasmid. Cells were grown to ∼100 Klett units in 50 ml SD–ura at 24° and washed with dH2O, and the pellets were frozen in liquid nitrogen. Pellets were resuspended in 500 µl lysis buffer (20 mM HEPES, pH 7.4, 300 mM NaCl, 100 µM GTP, 1 mM MgCl2, 1 mM dithiothreitol, 4 µg/ml pepstatin, 4 µg/ml leupeptin, 4 µg/ml aprotinin, 4 µg/ml chymostatin, 1 mM phenylmethanesulfonyl fluoride, 1 mM sodium pyrophosphate, 1 mM sodium fluoride, 1 mM β-glycerophosphate, 5% glycerol) and vortexed with ice-cold glass beads in 1-min intervals until >60% of cells were lysed. Triton X-100 was added to 0.5% and lysates were cleared by centrifugation at 18,000 × g for 20 min at 4°. An aliquot (50 µl) of 60 mg/ml Dynabeads (Invitrogen) conjugated with rabbit IgG (MP Biomedicals) was added to the clarified lysate and incubated for 30 min at 4°. Beads were then washed with 150 µl of wash buffer (20 mM HEPES, pH 7.4, 200 mM NaCl, 100 µM GTP, 1 mM MgCl2, 5% glycerol) three times and resuspended in 50 µl of SDS–PAGE sample buffer.
Fluorescence microscopy
For live-cell imaging, JTY12-25A (NUF2-mCherry NDC80) was transformed with plasmids encoding wild-type or mutant GFP–NDC80 (Table S1) and selected for growth on SD–ura at 24°. Cells were mounted for microscopy as previously described (Muller et al. 2005). Images of cells were taken at a single focal plane, binned 1 × 1, using a DeltaVision system (Applied Precision) equipped with an IX70 inverted microscope (Olympus), an U Plan Apo 100× objective (1.35 NA), and a CoolSnap HQ digital camera (Photometrics). Exposures were 0.4 sec for both GFP and mCherry. Images were processed as previously described (Shimogawa et al. 2010) using custom Matlab programs (available upon request) to isolate and quantify the fluorescence intensities of GFP and mCherry spots.
Total internal reflection fluorescence (TIRF) microscopy was performed as previously described (Tien et al. 2010), with the following modifications. A custom TIRF illumination system was constructed by modification of a commercial inverted microscope (Nikon Ti-U). Total internal reflection of 488 nm (Sapphire 488-100 CW, Coherent Inc.) and 561 nm (Sapphire 561-100 CW, Coherent Inc.) wavelength lasers was achieved using a through-the-objective arrangement with a 100× oil immersion 1.49 NA lens (Nikon CFI APO 100× Oil TIRF NA 1.49 WD 0.12 mm). Simultaneous imaging of GFP and Alexa Fluor 568 was captured by two cooled EM CCD cameras (iXon+ DU897, Andor Technology). Flow cell channels were constructed using double-sided sticky tape (Scotch), sandwiched between a glass slide (Gold Seal) and silanized coverslip (Corning). To bind taxol-stabilized microtubules, a modified “rigor” kinesin was flowed into the channel and bound nonspecifically to the coverslip. The flow cell channel was washed with 50 µl BB80 (80 mM Pipes, 120 mM K+, 1 mM MgCl2, 1 mM EGTA, 8 mg/ml BSA, pH 6.9) and 50 µl BB80T (BB80 with 10 µM taxol). Alexa Fluor 568-labeled microtubules diluted in BB80T were flowed in and allowed to bind for 5 min. The channel was washed once more with 50 µl BB80T. Proteins were then introduced, diluted in BB80T containing 0.1 mg/ml κ-casein, 200 µg/ml glucose oxidase, 35 µg/ml catalase, 25 mM glucose, and 5 mM dithiothreitol. GFP-tagged Ndc80 complex was assayed at 50 pM to resolve single molecules, and untagged Dam1 complex was added at 2.5 nM. Flow cell channels were sealed with nail polish and immediately imaged for 1500 frames at 10 frames per second. Software analysis of TIRF microscopy data was performed using custom software (available upon request) in Labview (National Instruments) and Igor Pro (WaveMetrics), as previously described (Tien et al. 2010).
Results
Construction of the linker-scanning transposition library
To discover new functional domains in the Ndc80 kinetochore protein (Figure 1), we performed a saturating screen that combined linker-scanning mutagenesis with a plasmid shuffle colorimetric assay and high-throughput sequencing. We first constructed a transposition library using an artificial transposition system (Figure 2A; Materials and Methods). This library contains a collection of plasmids, each with a single 15-bp insertion at a random location, corresponding to a five-amino-acid insertion in the translated protein product. Our NDC80 transposition library was generated from ∼11,000 transposition events on a 7.4-kb plasmid, which includes the 2.1-kb NDC80 gene. Each 15-bp insertion contained the 8-bp NotI recognition sequence, which was used to identify the position of the insertion within the gene.
Figure 2.

Workflow of the linker-scanning mutagenesis screen. (A) An Ndc80 transposition library, containing 15-bp insertions at random locations, was created using MuA transposition. (B) The transposition library was screened in S. cerevisiae using a red/white plasmid shuffle system. Lethal insertions that disrupt the function of Ndc80 appear as nonsectoring red colonies in the screen. From these red colonies, plasmids were isolated and the positions of the lethal insertions were determined by Illumina sequencing.
Insertion coverage of the NDC80 gene was determined by Illumina sequencing (Figure 3, A–D, and Figure S1). From three independent sequencing runs, we determined that transpositions targeted 1074 unique positions on NDC80, corresponding to 52% coverage of the 2076-bp gene (see Table S4 for a summary of the sequencing study). Furthermore, insertions were spread evenly across NDC80, with an average of 10.8 insertions per 21-bp window across the entire gene (Figure 3D, right plot). From the 1074 insertion positions identified in the library, the translated protein sequences were determined. The frame of each insertion and its surrounding wild-type NDC80 sequence ultimately dictates the identity of the amino acids inserted into the protein (Figure S2A). By experimental design, the insertions did not introduce stop codons and were always 15 bp long to prevent frame shifts in the resulting coding sequence (Figure S2A). The transposition library contained insertions at 444 unique codons, resulting in 64% coverage of the 691-amino-acid Ndc80 protein. Of these insertions, 30% were in frame 1, 26% were in frame 2, and 44% were in frame 3 (Figure S2B). The transposition library was then screened in S. cerevisiae to identify mutations that disrupt the function of Ndc80.
Figure 3.

Lethal insertions were found in distinct clusters in NDC80. (A) Bar diagram showing the positions of notable structural features in Ndc80. (B) Probability of coiled-coil formation as predicted by Paircoil2 (McDonnell et al. 2006). (C) Similarity of Ndc80 protein sequences between select fungal species (see Materials and Methods). (D) Left: the insertion density for the transposition library (blue line) and from the red colonies (red bars) is shown along NDC80. Insertion density is plotted as the number of unique insertion sites within a 21-bp window. Arrows indicate the positions of the representative insertions characterized. Right: from the insertion density plot, the distributions of insertion densities for the transposition library (blue line) and from the red colonies (red bars) are shown. (E) Immunoblot of cell lysate showing that lethal insertion mutants (GFP tagged) are expressed in vivo over the endogenous Ndc80 (untagged). (*) Nonspecific band from the α-Ndc80 antibody. Act1 serves as a loading control. (F) Coomassie-stained gel of recombinant Ndc80 complex containing lethal insertions. Recombinant Ndc80 complexes were purified by affinity chromatography and gel filtration. Mutant Ndc80 complexes migrated similarly to the wild-type complex in the gel filtration column and were collected at the same elution volume. The band for Ndc80 containing the ins1148 mutation reproducibly migrates higher than the wild-type protein.
Linker-scanning mutagenesis screen of Ndc80
A red/white plasmid shuffle system (Davis 1992; Muller 1996) was used to screen the transposition library for insertions detrimental to the function of NDC80 in S. cerevisiae (Figure 2B). In this screen, colonies containing library plasmids with lethal mutations in NDC80 do not sector and are solid red (see Materials and Methods). We screened 25,439 total colonies and isolated 959 red colonies (4%). These red colonies were pooled and the positions of the insertions were determined by Illumina sequencing (Table S4). Lethal insertions were found at 336 unique positions in 162 unique codons of NDC80, corresponding to 16% coverage of the gene and 23% of the protein. Unlike the transposition library, in which insertions were found evenly spread throughout the NDC80 gene, plasmids isolated from the red colonies contained insertions that mapped to distinct clusters (Figure S1). This was most evident on a coverage density map, showing the number of lethal insertions in 21-bp windows across NDC80 (Figure 3D). We defined a stretch of lethal insertions as a cluster if four or more insertions per 21-bp window were lethal (Figure 3D). In several windows within these clusters, every single insertion present in the transposition library was lethal. Surprisingly, lethal insertions were not enriched in any particular frame relative to the transposition library (Figure S2B). Furthermore, each of the three frames was well represented in all of the clusters identified (Figure S2C). These results suggest that the position of the insertion, rather than the identity of the residues, was most important in disrupting the function of Ndc80.
To verify that the clusters identified in our screen are not a result of random sampling, we divided the nonsectoring red colonies into two random pools. When sequenced independently (Table S4), both subsets had the same lethal insertion clusters as the original pool (Figure S3), indicating we have comprehensively screened the starting transposition library. Overall, our screen demonstrates that five-amino-acid insertions are tolerated throughout most of Ndc80 without disrupting its function. We did not identify lethal insertions in the putative “loop” (Figure 3A, blue bar), nor in a segment of Ndc80 that is disordered based on the human Ndc80 globular domain crystal structure (Figure 3A, black bar) (Ciferri et al. 2008). No lethal insertion clusters were found in the 113-amino acid N-terminal tail of Ndc80, which is dispensable in S. cerevisiae (Akiyoshi et al. 2009; Kemmler et al. 2009). Likewise, there were no lethal insertion clusters in the last ∼30 amino acid residues of Ndc80, consistent with the viability of the ndc80-1 temperature-sensitive allele, which contains a frameshift mutation that alters the last 18 amino acids of the protein (Wigge et al. 1998).
From the lethal insertion clusters identified in the linker-scanning mutagenesis screen, we selected representative mutations for further analysis. These lethal insertions are referred to as, for example, “ins506” to indicate that the first position of the insertion is nucleotide 506 of the mutant allele. A representative insertion from each cluster was independently verified to be lethal (see Materials and Methods). Insertions from the cluster around nucleotide 1400 of NDC80 were found to be temperature sensitive; six insertions from this cluster, ranging from nucleotide 1380 to 1406, were lethal at 37° but not 24°. Therefore, this cluster was omitted from further analysis. Here, we selected a total of seven representative mutations from the remaining lethal insertion clusters for analysis (Table 1). We first tested if these mutations perturb protein expression or assembly of Ndc80 into a heterotetrameric complex. In vivo, all seven lethal insertion mutant alleles were expressed (Figure 3E). In these cells, the endogenous wild-type copy of NDC80 was maintained for viability. A GFP tag on the mutant Ndc80 was used to distinguish it from the wild-type protein. In a recombinant expression system, six of the seven mutant Ndc80 proteins copurified as heterotetrameric complexes with Nuf2, Spc24, and Spc25 (Figure 3F). These results suggest that most of the insertions identified in this screen specifically disrupt the function of Ndc80 independent from protein production and folding.
Lethal insertions in the Ndc80 microtubule-binding domain
Two of the seven clusters of lethal insertions (represented by ins506 and ins656) are in the conserved microtubule-binding CH domain (Figure 3, A–D and Figure 4A). Based on homology to the human Ndc80 CH domain crystal structure (Ciferri et al. 2008), these lethal insertion clusters map to helices αC and αG, respectively. Even though these helices are adjacent in the crystal structure, the two mutations had drastically different effects. Ndc80 containing the ins506 mutation was expressed in vivo (Figure 3E), but failed to assemble into a recombinant complex. Consistent with our conclusion that the position of the insertion is likely most important in disrupting the function of Ndc80, we found that the ins511 mutation, which overlaps ins506 by 10 bp but is in a different frame, also impairs assembly of Ndc80 into a recombinant complex. By contrast, we successfully purified ins656 Ndc80 complex and further demonstrated that this complex binds microtubules in vitro (Figure 3F and Figure S4). Two additional clusters of lethal insertions (represented by ins839 and ins940) were found in a region of Ndc80 that folds into a helical hairpin (αH and αI) in the human crystal structure. The hairpin forms C-terminal to the Ndc80 CH domain and, together, they “sandwich” the globular domain of Nuf2 (Ciferri et al. 2008). Both of these mutant complexes bound to microtubules in vitro (Figure S4). Furthermore, the ins839 mutation altered the behavior of single molecules of the Ndc80 complex on microtubules, increasing their residence time and slowing their diffusion as compared to wild type (Figure 4, B and C). This observation suggests that although the hairpin is not part of the microtubule-binding interface, it indirectly affects the attachment between the Ndc80 complex and microtubules.
Figure 4.

The ability of the Dam1 complex to enhance binding of Ndc80 complexes to microtubules is abrogated by a lethal insertion in the helical hairpin. (A) Four lethal insertion clusters (red) are mapped onto homologous regions of the human Ndc80 (blue) crystal structure (PDB 2VE7; Ciferri et al. 2008). Lethal insertion clusters are labeled based on their representative insertion. The structure is illustrated with the UCSF Chimera package (Pettersen et al. 2004). (B) Residence time distributions of GFP-tagged Ndc80 complex on microtubules fit with single exponentials (dashed lines) to determine the dissociation rate constants, koff. Top: wild-type Ndc80 complex in the absence (black, n = 622) or presence (red, n = 252) of Dam1 complex. Bottom: ins839 Ndc80 complex in the absence (black, n = 374) or presence (red, n = 357) of Dam1 complex. (C) Plots of mean-squared displacement (MSD) ± SEM vs. time lag for the binding events in B. Linear fits to the data (dashed lines) were used to determine the diffusion constant, D.
The ins839 Ndc80 complex is insensitive to the presence of the Dam1 complex on microtubules
Ndc80 is required for recruitment of the essential Dam1 complex to kinetochores in vivo (Janke et al. 2002). In vitro, the Ndc80 and Dam1 complexes interact directly, in a microtubule-dependent manner (Lampert et al. 2010; Tien et al. 2010). The Dam1 complex causes the Ndc80 complex to dissociate more slowly from microtubules and to diffuse more slowly on the lattice (Tien et al. 2010). The ins839 mutation in the hairpin of Ndc80 has a similar but less dramatic effect, causing a twofold decrease in the diffusion and dissociation rate constants (Figure 4, B and C). Therefore, we asked whether the Dam1 complex can further alter the microtubule-binding properties of the Ndc80 complex with the ins839 mutation. Surprisingly, unlike wild-type Ndc80 complex and the other five lethal mutant complexes we purified, the ins839 Ndc80 complex was not influenced by the presence of the Dam1 complex (Figure 4, B and C, and Figure S4). Instead, the ins839 mutation makes the Ndc80 complex insensitive to the presence of the Dam1 complex. Whether this is a result of the mutation perturbing the microtubule-binding ability of the Ndc80 complex or disrupting an interaction with the Dam1 complex remains to be determined (see Discussion).
The ins1957 mutation in Ndc80 disrupts tetramerization of the Ndc80 complex
Localization of Ndc80 to the kinetochore depends on its assembly into an intact complex with Nuf2, Spc24, and Spc25. This complex is anchored to the kinetochore via interactions between Spc24/Spc25 and other components, such as the Mtw1 complex (De Wulf et al. 2003; Maskell et al. 2010; Petrovic et al. 2010; Hornung et al. 2011). From cross-linking and limited proteolysis experiments, it was proposed that the C termini of Ndc80/Nuf2 contact Spc24/Spc25 to assemble the Ndc80 complex (Ciferri et al. 2005; Wei et al. 2005; Maiolica et al. 2007). However, the proteins and residues involved in this putative tetramerization domain and the structural basis for assembly of the complex remain poorly understood.
Our linker-scanning mutagenesis screen identified three previously uncharacterized regions in Ndc80 that are essential for its function (represented by ins1148, ins1687, and ins1957). These regions are C-terminal to the CH domain, and all three representative mutant Ndc80 complexes bound microtubules similar to the wild-type complex (Figure S4). The ins1687 and ins1957 mutations represent broad, neighboring clusters of lethal insertions that cover an area including the putative tetramerization domain. We screened these and our other representative mutations for defective kinetochore localization of Ndc80. Using fluorescence microscopy, we measured the amount of GFP-tagged mutant Ndc80 that colocalized with Nuf2–mCherry at kinetochores (Figure 5, A and B). The ins656, ins839, ins940, ins1148, and ins1687 mutant GFP–Ndc80 proteins colocalized with Nuf2–mCherry, indicating that these insertions do not disrupt formation of the Ndc80 complex or its association with the kinetochore in vivo. By contrast, ins1957 GFP–Ndc80 was absent from kinetochores. Given its position at the C terminus of Ndc80, we hypothesized that the ins1957 mutation disrupts tetramerization of the complex. However, the ins1957 mutant copurifies with Nuf2, Spc24, and Spc25 in a recombinant Ndc80 complex (Figure 3F), suggesting that the mutation does not completely abolish complex formation. To address this question in vivo, we performed immunoprecipitation experiments with TAP tags fused to the endogenous Nuf2 or Spc24 (Figure 5C). We found that ins1957 GFP–Ndc80 co-immunoprecipitated with Nuf2-TAP, but not Spc24–TAP. As a control, wild-type and ins1687 GFP–Ndc80 co-immunoprecipitated with both Nuf2–TAP and Spc24–TAP. Consistent with its inability to form a recombinant complex, ins506 GFP–Ndc80 did not interact with Nuf2–TAP or Spc24–TAP in the co-immunoprecipitation assay and was absent from kinetochores (Figure 5B and C). These results show that ins506 GFP–Ndc80 cannot pair with Nuf2 and consequently fails to assemble into an intact complex. By contrast, ins1957 GFP–Ndc80 can pair with Nuf2 in vivo, but is unable to compete with endogenous Ndc80 for association with Spc24/Spc25 at kinetochores. Therefore, the lethal insertion cluster represented by ins1957 is likely the tetramerization domain, which mediates the association of Ndc80/Nuf2 with Spc24/Spc25 into intact complexes at kinetochores.
Figure 5.

A C-terminal segment of Ndc80 controls tetramerization of the Ndc80 complex. (A) Sample images of GFP–Ndc80 lethal insertion mutants colocalized with Nuf2–mCherry at kinetochores in vivo. Endogenous Ndc80 is untagged. Scale bar, 5 µm. (B) The fluorescence intensity of GFP and mCherry are quantified and shown. Intensity is plotted as the mean of fluorescence signal—mean of background signal. Error bars represent SEM. The total numbers of mCherry spots quantified are: wild type (n = 686), ins506 (n = 788), ins656 (n = 760), ins839 (n = 913), ins940 (n = 643), ins1148 (n = 372), ins1687 (n = 576), and ins1957 (n = 389). (C) Immunoprecipitation (IP) of GFP–Ndc80 lethal insertion mutants, and untagged endogenous Ndc80, with Nuf2-TAP or Spc24–TAP. (*) Nonspecific band from the α-Ndc80 antibody. On the immunoblots, input is 0.5% of the clarified lysate and TAP IP is 10% of the total volume.
Discussion
Linker-scanning mutagenesis specifically identifies regions essential for function
Here, we describe an approach to uncover new functional domains of essential proteins in S. cerevisiae. We accomplished this by combining linker-scanning mutagenesis, a plasmid shuffle assay, and high-throughput sequencing together in a comprehensive screen. We applied this screen to the conserved Ndc80 kinetochore protein, which contains a microtubule-binding globular domain and is predicted to contain long coiled-coil domains. While the short insertions resulting from transposon mutagenesis were generally well tolerated, several clusters of insertions were detrimental to Ndc80 function in vivo. These clusters of lethal insertions highlight important regions of the protein, many of which are not readily apparent by inspecting sequence conservation (Figure 3C and D).
New functions for the N terminus of Ndc80
Binding of Ndc80 to microtubules is accomplished by its N terminus, which contains an unstructured tail domain and a conserved CH domain that is commonly found on actin- and microtubule-binding proteins (Gimona et al. 2002; Korenbaum and Rivero 2002; Hayashi and Ikura 2003; Wei et al. 2007; Ciferri et al. 2008). In our linker-scanning mutagenesis screen, insertions were generally tolerated throughout the entire N terminus of Ndc80, with just two clusters of lethal insertions in the CH domain. A representative mutation from the first cluster (ins506) is expressed in vivo, but is absent from kinetochores and does not co-immunoprecipitate with Nuf2–TAP. This mutant impairs the formation of the Ndc80 complex, likely by disrupting the interaction between the Ndc80 and Nuf2 CH domains. This suggests that the CH domain plays a critical role in the dimerization of Ndc80 and Nuf2. The second cluster lies in an interior helix of the CH domain adjacent to the first cluster. A representative mutation from this cluster (ins656) surprisingly did not abolish microtubule binding by the recombinant complex or disrupt kinetochore localization in vivo. It is unlikely that this mutation is lethal because of subtle changes in the microtubule-binding ability of Ndc80, as several mutations that are known to impair microtubule binding by the human complex in vitro are tolerated in S. cerevisiae (Ciferri et al. 2008; Akiyoshi et al. 2009; Kemmler et al. 2009; Lampert et al. 2013; Umbreit et al. 2012). We propose that this mutation instead disrupts another essential function of the Ndc80 CH domain that is independent of microtubule binding.
Downstream of the CH domain, we mapped two clusters of lethal insertions to a putative helical hairpin motif. Together with the Ndc80 CH domain, this hairpin packs against the paired CH domain of Nuf2 in a crystal structure of the truncated human Ndc80 complex (Ciferri et al. 2008). One of these lethal insertions, ins839, slows the dissociation and diffusion rates of Ndc80 complexes on microtubules in vitro. These effects cannot be due to cooperative interactions between Ndc80 complexes on microtubules, as our experiments were performed at the single-molecule level. Moreover, given its position in the crystal structure, the hairpin is unlikely to contact microtubules directly. Therefore, we propose that the hairpin contributes indirectly to microtubule binding, perhaps through structural stabilization or organization of the microtubule-binding interface.
One key role of Ndc80 in vivo is to recruit the Dam1 complex to kinetochores (Janke et al. 2002), but the nature of this interaction remains unclear. In vitro, the wild-type Ndc80 complex interacts directly with the Dam1 complex in a microtubule-dependent manner (Lampert et al. 2010; Tien et al. 2010). We found previously that the addition of Dam1 complex decreases the dissociation and diffusion rates of single Ndc80 complexes on microtubules. Here we show that the ins839 mutation parallels these effects on the Ndc80 complex and that the addition of Dam1 complex does not further change the behavior of the ins839 Ndc80 complex on microtubules. Notably, while the dissociation and diffusion rates of single ins839 Ndc80 complexes on microtubules are lower than that of the wild-type complex, they are higher than that of the wild-type complex when assayed in the presence of Dam1 complex. This observation raises the interesting possibility that the Dam1 complex does not simply contribute an additional microtubule-binding domain when it interacts with the Ndc80 complex, but changes how the Ndc80 complex binds to the microtubule lattice. The ins839 mutation could partially mimic this effect, which explains why the mutant Ndc80 complex is not further influenced by the Dam1 complex.
An alternative explanation for our observations is that the ins839 mutation disrupts the interaction interface between Ndc80 and the Dam1 complex. Recent studies have proposed that the Dam1 complex binds directly to Ndc80 within the Ndc80 complex, but did not agree on the location of this interaction interface (Maure et al. 2011; Lampert et al. 2013). One of these proposed regions, a stretch of Ndc80 between the CH domain and the hairpin (residues 256–273; Figure 4A), lies in close proximity to the ins839 mutation. When this region was deleted, the Dam1 complex did not enhance the cosedimentation of the mutant complex with microtubules in vitro (Lampert et al. 2013). The authors concluded that residues 256–273 represent an interaction interface between Ndc80 and the Dam1 complex. However, like the ins839 mutation, a deletion of this region alters the microtubule-binding behavior of the Ndc80 complex alone (Lampert et al. 2013). Therefore, it remains unclear whether these mutations directly disrupt the Dam1 binding interface, or if microtubule attachments made by the mutant complexes are simply unable to be strengthened by the Dam1 complex. Future studies will be aimed at determining whether this region of Ndc80 is in direct contact with the Dam1 complex.
Identification of novel domains in Ndc80 and defining the tetramerization domain
Our linker-scanning mutagenesis screen successfully identified three previously uncharacterized segments of Ndc80 as regions essential for its function in vivo. These three lethal insertion clusters lie in the last ∼300 amino acid residues of Ndc80, predicted to form the “rod-shaped” part of the complex as seen by electron microscopy (Wei et al. 2005; Wang et al. 2008). The first cluster is centered on residue K380 of Ndc80, and coincides with a change in predicted coiled-coil character that may represent a transition point where the coiled-coil begins. Consistent with this prediction, limited proteolysis experiments identified K380 as a cleavage site that is likely unprotected by coiled-coil (Wei et al. 2005). The representative insertion from this cluster, ins1148, is predicted to abolish the formation of coiled coil after the insertion (Figure S5). Nevertheless it forms a stable tetramer both in vivo and in vitro, suggesting the coiled coil in this region is not driving the interaction between Ndc80 and Nuf2, Spc24, or Spc25. Similarly the cluster represented by ins1687 abolishes the probability of coiled-coil formation after this insertion, but again has no effect on tetramer formation and represents a domain with an, as yet, unknown function.
The cluster represented by ins1957 is in a region predicted to overlap with the N termini of Spc24/Spc25 by crosslinking experiments (Maiolica et al. 2007). While this general region is proposed to mediate tetramerization of the complex (Ciferri et al. 2005; Wei et al. 2005; Maiolica et al. 2007), it is not clear which complex components are directly involved and which segments of these components are required. We found that the ins1957 mutation disrupts formation of the Ndc80 complex in vivo and that the mutant Ndc80 was consequently defective at incorporating into kinetochores. These results suggest that the lethal insertion cluster represented by the ins1957 mutation defines a region in Ndc80 important for tetramerization of the Ndc80 complex.
Using the linker-scanning mutagenesis screen, we have identified lethal insertions in known structural elements of Ndc80 and, additionally, defined new functional domains in the protein. Future experiments will examine individual domains with the goal of defining binding interfaces with known partners, searching for novel protein interactions, and uncovering new roles for Ndc80. This approach is generally applicable to other genes in S. cerevisiae and is particularly useful in revealing functional properties and domains in proteins that are not yet amenable to structural characterization.
Supplementary Material
Acknowledgments
We thank members of the Davis Lab and the Seattle Mitosis group for helpful discussions. We also thank I. Onn, D. Koshland, C. Lee, and J. Shendure for technical assistance and A. Desai for the α-Ndc80 antibody. This work was supported by a National Sciences and Engineering Research Council of Canada scholarship (to J.F.T.), National Institutes of Health Grants T32 GM008268 (to N.T.U.) and T32 GM007270 (to K.K.F.), Searle Scholar Award Grant 06-L-111 (to C.L.A.), Packard Fellowship for Science and Engineering Grant 2006-30521 (to C.L.A), National Center for Research Resources Grant S10 RR26406 (to C.L.A.), and National Institute of General Medical Sciences Grants R01 GM40506 (to T.N.D.) and R01 GM079373 (to C.L.A.). M.J.D. is funded by the National Institute of General Medical Sciences Grant P41 GM103533 to T.N.D. M.J.D. is a Rita Allen Scholar.
Footnotes
Communicating editor: O. Cohen-Fix
Literature Cited
- Akiyoshi B., Nelson C. R., Ranish J. A., Biggins S., 2009. Analysis of Ipl1-mediated phosphorylation of the Ndc80 kinetochore protein in Saccharomyces cerevisiae. Genetics 183: 1591–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alushin G. M., Ramey V. H., Pasqualato S., Ball D. A., Grigorieff N., et al. , 2010. The Ndc80 kinetochore complex forms oligomeric arrays along microtubules. Nature 467: 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asbury C. L., Gestaut D. R., Powers A. F., Franck A. D., Davis T. N., 2006. The Dam1 kinetochore complex harnesses microtubule dynamics to produce force and movement. Proc. Natl. Acad. Sci. USA 103: 9873–9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A., Pringle J. R., 1991. Use of a screen for synthetic lethal and multicopy suppresser mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 11: 1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackshields G., Sievers F., Shi W., Wilm A., Higgins D. G., 2010. Sequence embedding for fast construction of guide trees for multiple sequence alignment. Algorithms Mol. Biol. 5: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke D., Dawson D., Stearns T., 2000. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Ciferri C., De Luca J., Monzani S., Ferrari K. J., Ristic D., et al. , 2005. Architecture of the human ndc80-hec1 complex, a critical constituent of the outer kinetochore. J. Biol. Chem. 280: 29088–29095. [DOI] [PubMed] [Google Scholar]
- Ciferri C., Pasqualato S., Screpanti E., Varetti G., Santaguida S., et al. , 2008. Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell 133: 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis T. N., 1992. Mutational analysis of calmodulin in Saccharomyces cerevisiae. Cell Calcium 13: 435–444. [DOI] [PubMed] [Google Scholar]
- DeLuca K. F., Lens S. M., DeLuca J. G., 2011. Temporal changes in Hec1 phosphorylation control kinetochore-microtubule attachment stability during mitosis. J. Cell Sci. 124: 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wulf P., McAinsh A. D., Sorger P. K., 2003. Hierarchical assembly of the budding yeast kinetochore from multiple subcomplexes. Genes Dev. 17: 2902–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimona M., Djinovic-Carugo K., Kranewitter W. J., Winder S. J., 2002. Functional plasticity of CH domains. FEBS Lett. 513: 98–106. [DOI] [PubMed] [Google Scholar]
- Guimaraes G. J., Dong Y., McEwen B. F., Deluca J. G., 2008. Kinetochore-microtubule attachment relies on the disordered N-terminal tail domain of Hec1. Curr. Biol. 18: 1778–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hach F., Hormozdiari F., Alkan C., Birol I., Eichler E. E., et al. , 2010. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods 7: 576–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi I., Ikura M., 2003. Crystal structure of the amino-terminal microtubule-binding domain of end-binding protein 1 (EB1). J. Biol. Chem. 278: 36430–36434. [DOI] [PubMed] [Google Scholar]
- He X., Rines D. R., Espelin C. W., Sorger P. K., 2001. Molecular analysis of kinetochore-microtubule attachment in budding yeast. Cell 106: 195–206. [DOI] [PubMed] [Google Scholar]
- Hornung P., Maier M., Alushin G. M., Lander G. C., Nogales E., et al. , 2011. Molecular architecture and connectivity of the budding yeast Mtw1 kinetochore complex. J. Mol. Biol. 405: 548–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C., Ortiz J., Lechner J., Shevchenko A., Magiera M. M., et al. , 2001. The budding yeast proteins Spc24p and Spc25p interact with Ndc80p and Nuf2p at the kinetochore and are important for kinetochore clustering and checkpoint control. EMBO J. 20: 777–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C., Ortiz J., Tanaka T. U., Lechner J., Schiebel E., 2002. Four new subunits of the Dam1–Duo1 complex reveal novel functions in sister kinetochore biorientation. EMBO J. 21: 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmler S., Stach M., Knapp M., Ortiz J., Pfannstiel J., et al. , 2009. Mimicking Ndc80 phosphorylation triggers spindle assembly checkpoint signalling. EMBO J. 28: 1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline-Smith S. L., Sandall S., Desai A., 2005. Kinetochore-spindle microtubule interactions during mitosis. Curr. Opin. Cell Biol. 17: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenbaum E., Rivero F., 2002. Calponin homology domains at a glance. J. Cell Sci. 115: 3543–3545. [DOI] [PubMed] [Google Scholar]
- Lampert F., Hornung P., Westermann S., 2010. The Dam1 complex confers microtubule plus end-tracking activity to the Ndc80 kinetochore complex. J. Cell Biol. 189: 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampert F., Mieck C., Alushin G. M., Nogales E., Westermann S., 2013. Molecular requirements for the formation of a kinetochore-microtubule interface by Dam1 and Ndc80 complexes. J. Cell Biol. 200: 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiolica A., Cittaro D., Borsotti D., Sennels L., Ciferri C., et al. , 2007. Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol. Cell. Proteomics 6: 2200–2211. [DOI] [PubMed] [Google Scholar]
- Malvezzi F., Litos G., Schleiffer A., Heuck A., Mechtler K., et al. , 2013. A structural basis for kinetochore recruitment of the Ndc80 complex via two distinct centromere receptors. EMBO J. 32: 409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskell D. P., Hu X. W., Singleton M. R., 2010. Molecular architecture and assembly of the yeast kinetochore MIND complex. J. Cell Biol. 190: 823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maure J. F., Komoto S., Oku Y., Mino A., Pasqualato S., et al. , 2011. The Ndc80 loop region facilitates formation of kinetochore attachment to the dynamic microtubule plus end. Curr. Biol. 21: 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell A. V., Jiang T., Keating A. E., Berger B., 2006. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 22: 356–358. [DOI] [PubMed] [Google Scholar]
- Miller S. A., Johnson M. L., Stukenberg P. T., 2008. Kinetochore attachments require an interaction between unstructured tails on microtubules and Ndc80(Hec1). Curr. Biol. 18: 1785–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milutinovich M., Unal E., Ward C., Skibbens R. V., Koshland D., 2007. A multi-step pathway for the establishment of sister chromatid cohesion. PLoS Genet. 3: e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda J. J., De Wulf P., Sorger P. K., Harrison S. C., 2005. The yeast DASH complex forms closed rings on microtubules. Nat. Struct. Mol. Biol. 12: 138–143. [DOI] [PubMed] [Google Scholar]
- Muller E. G., 1996. A glutathione reductase mutant of yeast accumulates high levels of oxidized glutathione and requires thioredoxin for growth. Mol. Biol. Cell 7: 1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller E. G. D., Snydsman B. E., Novik I., Hailey D. W., Gestaut D. R., et al. , 2005. The organization of the core proteins of the yeast spindle pole body. Mol. Biol. Cell 16: 3341–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T., Vinh D. B. N., Crawford D. K., Davis T. N., 1998. A genetic analysis of interactions with Spc110p reveals distinct functions of Spc97p and Spc98p, components of the yeast gamma-tubulin complex. Mol. Biol. Cell 9: 2201–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino T., Rago F., Hori T., Tomii K., Cheeseman I. M., et al. , 2013. CENP-T provides a structural platform for outer kinetochore assembly. EMBO J. 32: 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne M. A., Schlenstedt G., Jinks T., Silver P. A., 1994. Nuf2, a spindle pole body-associated protein required for nuclear division in yeast. J. Cell Biol. 125: 853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajunen M., Turakainen H., Poussu E., Peranen J., Vihinen M., et al. , 2007. High-precision mapping of protein protein interfaces: an integrated genetic strategy combining en masse mutagenesis and DNA-level parallel analysis on a yeast two-hybrid platform. Nucleic Acids Res. 35: e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic A., Pasqualato S., Dube P., Krenn V., Santaguida S., et al. , 2010. The MIS12 complex is a protein interaction hub for outer kinetochore assembly. J. Cell Biol. 190: 835–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., et al. , 2004. UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25: 1605–1612. [DOI] [PubMed] [Google Scholar]
- Petyuk V., McDermott J., Cook M., Sauer B., 2004. Functional mapping of Cre recombinase by pentapeptide insertional mutagenesis. J. Biol. Chem. 279: 37040–37048. [DOI] [PubMed] [Google Scholar]
- Powers A. F., Franck A. D., Gestaut D. R., Cooper J., Gracyzk B., et al. , 2009. The Ndc80 kinetochore complex forms load-bearing attachments to dynamic microtubule tips via biased diffusion. Cell 136: 865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P., Longden I., Bleasby A., 2000. EMBOSS: the European Molecular Biology Open Software Suite.. Trends Genet 16: 276–277. [DOI] [PubMed] [Google Scholar]
- Scannell D. R., Zill O. A., Rokas A., Payen C., Dunham M. J., et al. , 2011. The awesome power of yeast evolutionary genetics: new genome sequences and strain resources for the Saccharomyces sensu stricto genus. G3: Genes, Genomes, Genetics 1: 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J. C., Arthanari H., Boeszoermenyi A., Dashkevich N. M., Wilson-Kubalek E. M., et al. , 2012. The kinetochore-bound Ska1 complex tracks depolymerizing microtubules and binds to curved protofilaments. Dev. Cell 23: 968–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimogawa M. M., Wargacki M. M., Muller E. G., Davis T. N., 2010. Laterally attached kinetochores recruit the checkpoint protein Bub1, but satisfy the spindle checkpoint. Cell Cycle 9: 3619–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien J. F., Umbreit N. T., Gestaut D. R., Franck A. D., Cooper J., et al. , 2010. Cooperation of the Dam1 and Ndc80 kinetochore complexes enhances microtubule coupling and is regulated by aurora B. J. Cell Biol. 189: 713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbreit N. T., Gestaut D. R., Tien J. F., Vollmar B. S., Gonen T., et al. , 2012. The Ndc80 kinetochore complex directly modulates microtubule dynamics. Proc. Natl. Acad. Sci. USA 109: 16113–16118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. W., Long S., Ciferri C., Westermann S., Drubin D., et al. , 2008. Architecture and flexibility of the yeast Ndc80 kinetochore complex. J. Mol. Biol. 383: 894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R. R., Sorger P. K., Harrison S. C., 2005. Molecular organization of the Ndc80 complex, an essential kinetochore component. Proc. Natl. Acad. Sci. USA 102: 5363–5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R. R., Schnell J. R., Larsen N. A., Sorger P. K., Chou J. J., et al. , 2006. Structure of a central component of the yeast kinetochore: the Spc24p/Spc25p globular domain. Structure 14: 1003–1009. [DOI] [PubMed] [Google Scholar]
- Wei R. R., Al-Bassam J., Harrison S. C., 2007. The Ndc80/HEC1 complex is a contact point for kinetochore-microtubule attachment. Nat. Struct. Mol. Biol. 14: 54–59. [DOI] [PubMed] [Google Scholar]
- Wigge P. A., Kilmartin J. V., 2001. The Ndc80p complex from Saccharomyces cerevisiae contains conserved centromere components and has a function in chromosome segregation. J. Cell Biol. 152: 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigge P. A., Jensen O. N., Holmes S., Soues S., Mann M., et al. , 1998. Analysis of the Saccharomyces spindle pole by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. J. Cell Biol. 141: 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.