

Abstract
Structure-based modeling combined with rational drug design, and high throughput screening approaches offer significant potential for identifying and developing lead compounds with therapeutic potential. The present review focuses on these two approaches using explicit examples based on specific derivatives of Gossypol generated through rational design and applications of a cancer-specific-promoter derived from Progression Elevated Gene-3. The Gossypol derivative Sabutoclax (BI-97C1) displays potent anti-tumor activity against a diverse spectrum of human tumors. The model of the docked structure of Gossypol bound to Bcl-XL provided a virtual structure-activity-relationship where appropriate modifications were predicted on a rational basis. These structure-based studies led to the isolation of Sabutoclax, an optically pure isomer of Apogossypol displaying superior efficacy and reduced toxicity. These studies illustrate the power of combining structure-based modeling with rational design to predict appropriate derivatives of lead compounds to be empirically tested and evaluated for bioactivity. Another approach to cancer drug discovery utilizes a cancer-specific promoter as readouts of the transformed state. The promoter region of Progression Elevated Gene-3 is such a promoter with cancer-specific activity. The specificity of this promoter has been exploited as a means of constructing cancer terminator viruses that selectively kill cancer cells and as a systemic imaging modality that specifically visualizes in vivo cancer growth with no background from normal tissues. Screening of small molecule inhibitors that suppress the Progression Elevated Gene-3-promoter may provide relevant lead compounds for cancer therapy that can be combined with further structure-based approaches leading to the development of novel compounds for cancer therapy.
Keywords: Progression Elevated Gene-3, Sabutoclax, Apogossypol, BI-97C1, Gossypol, AP-1, PEA3, ETV4, E1AF, c-fos, c-jun, Cancer Terminator Virus
STRUCTURE-BASED APPROACHES FOR CANCER DRUG DISCOVERY
Cancer is a progressive disease characterized by sequential changes in gene expression mediated by both genetic and epigenetic alterations in the evolving tumor cell [1–3]. This intrinsic complexity and the heterogeneous nature of cancer, which is often further exacerbated during cancer expansion and progression to metastasis, has proven to be a significant impediment for developing effective therapies [3]. An inherent property of many cancer cells involves resistance to therapeutic agents that promote programmed cell death (apoptosis) in tumors, which is designed to limit growth and destroy tumors. [4]. Apoptosis plays a fundamental role in tissue homeostasis by ensuring a proper balance between cell loss and cell production. Defective regulation of apoptosis is implicated to various extents in virtually every malignancy [4]. Inhibition of apoptosis provides cancer cells with a selective growth advantage promoting survival and chemoresistance [5, 6]. The B-cell lymphoma/leukemia-2 (Bcl-2) family of proteins is a central regulator of apoptosis [7]. The Bcl-2 family of anti-apoptotic proteins comprise Bcl-2, Mcl-1, Bcl-XL, Bfl-1, Bcl-W and Bcl-B. The Bcl-2 protein family also includes pro-apoptotic effector proteins Bak, Bax, Bad, Bid, and Bim. The pro-apoptotic Bcl-2 family proteins heterodimerize with the anti-apoptotic proteins neutralizing their activity and inhibiting apoptosis [5]. Heterodimerization occurs through a hydrophobic crevice on the surface of the anti-apoptotic Bcl-2 family member and the α-helix BH3 domain of pro-apoptotic family members [7]. Because overexpression of Bcl-2 family members is prevalent in many types of cancer, they provide attractive targets for the development of novel therapeutics with anti-cancer properties. Molecules that mimic the BH3 domain of pro-apoptotic Bcl-2 members may effectively induce apoptosis and/or neutralize the ability of anti-apoptotic Bcl-2 family members to promote cancer cell survival [8–10].
A small library of natural products was screened using a combination of nuclear magnetic resonance (NMR)-based binding assays and fluorescence polarization displacement assays (FPA) identifying two polyphenols, Gossypol (NSC19048) and Purpurogallin, that could inhibit a BH3 peptide binding to BCLxL. Gossypol and Purpurogallin displace the BH3 peptide from binding Bcl-XL in the FPA assay with an IC50 of 0.5 μM and 2.2 μM, respectively [11]. Gossypol is a natural product derived from cotton seed extracts which was originally investigated as a male contraceptive agent in China [12]. Gossypol inhibits the growth of a diverse array of human breast, prostate, ovarian and pancreatic cancer cell lines and xenograft models [13–16]. Studies aimed at identifying the target of Gossypol suggested that this compound can induce apoptosis in cells overexpressing Bcl-2 or Bcl-XL [17]. Purpurogallin a less potent inhibitor of Bcl-XL is an anti-oxidant found in edible oils reported to inhibit tyrosine-specific protein kinase and DNA synthesis in glioblastoma cells [18, 19].
Mapping chemical-shift differences induced by Gossypol or Purpurogallin binding to 15N-labeled-Bcl-XL with heteronuclear NMR spectroscopy revealed that Gossypol binds more tightly than Purpurogallin to Bcl-XL, and that the binding of the polyphenols occurs most likely in the BH3 binding pocket of Bcl-XL. Docking studies using the Bcl-XL confirmation found in complex with Bax peptide revealed an optimal location for Gossypol binding in a deep hydrophobic cleft normally occupied by the Bak helical BH3 peptide domain. Furthermore, the docking studies showed that the (−) enantiomer of Gossypol binds more tightly than (+) enantiomer of Gossypol. Purpurogallin produced ambiguous results in the docking studies, however studying the inhibitory effect of Purpurogallin analogues allowed the identification of essential pharmacophoric sub-structures common to these polyphenols [11].
The (−) enantiomer of Gossypol (AT-101) is in clinical trials involving patients with solid tumors, lymphoma and leukemia. Although Gossypol is efficacious in cancer patients’ adverse events are associated with treatment including hepatocytotoxicity and gastrointestinal cytotoxicity. Since Gossypol is only a modest inhibitor of Bcl-XL it is likely that high doses of compound would be required for efficacy in patients, however dose limiting toxicity is apparent in the clinical trials. This issue generated a need to develop a structure-activity relationship (SAR) with Gossypol to identify more potent and selective inhibitors of Bcl-XL. The model of the docked structure of Gossypol into Bcl-XL also allowed for a virtual structure activity relationship where modifications of Gossypol were predicted on a rational basis leading to more potent and selective inhibitors [20]. We hypothesized that the reactive aldehyde residues in Gossypol were undesirable because they participate in Schiff’s base-type reactions, which may react with proteins and nucleic acids indiscriminately causing adverse side effects. We docked a small library of eight Gossypol derivatives modifying the aldehyde residues and found Apogossypol (1,1′,6,6′,7,7′-hexahydroxy-3,3′-dimethyl-5,5′-bis (1-methylethyl)-[2.2′-binaphthalene], NSC736630), a Gossypol derivative with the aldehyde residues eliminated having the lowest binding energy to Bcl-XL [20].
We compared the toxicity profile of Apogossypol and Gossypol to validate our hypothesis that the aldehyde residues contribute to adverse events of the compound. Both compounds are orally administrable, however single dose-pharmacokinetic studies revealed that Apogossypol has superior pharmacokinetic characteristics than Gossypol including greater blood concentrations over time (area under curve) due to reduced clearance of the compound [21, 22]. Mice tolerated two to four times higher doses of Apogossypol than Gossypol with less adverse events of hepatotoxicity and gastrointestinal toxicity in the group receiving Apogossypol. Accompanying reduced toxicity of Apogossypol was its superior efficacy in a model resembling human low-grade follicular lymphoma induced in transgenic mice by over-expression of Bcl-2 in B-cells [23]. B-cells from mice over-expressing Bcl-2 were more sensitive to cytotoxicity caused by in vitro Apogossypol treatment than Gossypol with LD50 values of 3 to 5 μM and 7.5 to 10 μM respectively [23]. Daily dosing of the maximum tolerated doses of these polyphenols illustrated that Apogossypol was superior to Gossypol in reducing splenomegaly and B-cell counts in spleens of Bcl-2 transgenic mice [23]. Taken together these results indicate that Apogossypol is a superior lead compound for anti-cancer therapy to the parent gossypol compound with increased activity and reduced adverse events.
Apogossypol is a racemic compound with the individual isomers of the compound being as effective in inhibiting Bcl-2 family members as the racemic mixture [24]. We synthesized 5, 5′ amide and ketone substituted Apogossypol derivatives and evaluated their activities and found compounds BI-79D10 and 8r with improved in vitro and in vivo efficacy compared to Apogossypol [24]. The structure of BI-97D10, 8r and the polyphenol parent compounds are illustrated in Fig. (1). Compound 8r is a mixture of diastereomers and has three centers of chirality. Because different enantiomers and diastereomers of compounds can have different physical, chemical and pharmacological properties as evident in the example presented above with Gossypol, we synthesized the four optically pure diastereomers of compound 8r and evaluated their activities. Of the four diastereomers, Sabutoclax (BI-97C1) was found to bind most tightly to BCLxL as we had done previously with NMR-spectroscopy. Sabutoclax inhibits binding of BH3 peptides to Bcl-XL, Bcl-2, Mcl-1, and Bfl-1 with IC50 values of 0.13, 0.56, and 0.049 μM respectively.
Fig. 1.
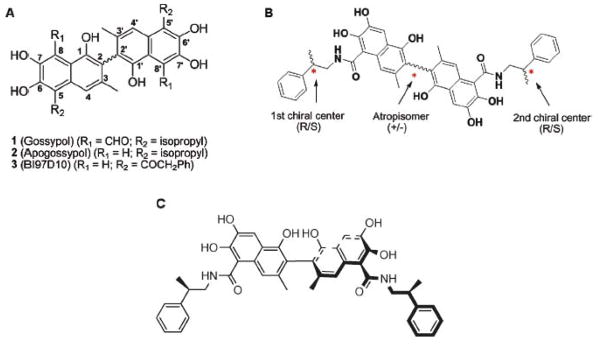
(A) Structure of Gossypol, Apogossypol and BI97D10. (B) Structure of compound 8r. (C) Structure of Sabutoclax (BI-97C1). Adapted from Wei et al. (24), J. Med. Chem., 2009.
Concordant with the observation that Sabutoclax binds most tightly to Bcl-2 family members, it inhibits the growth of human prostate cancer and lung cancer and B-cell lymphoma lines with EC50 values of 0.13, 0.56, and 0.049 μM respectively [24]. Sabutoclax displays a 20-fold better efficacy in inhibiting the growth of PC-3 cells compared to BI-97D10. Furthermore, Sabutoclax show less cytotoxicity in bax/bak double-knockout cells compared to BI-97D10, suggesting that the mechanism of cell death provoked is dependent on Bcl-2 family members. Sabutoclax has superior in vivo efficacy compared to BI-97D10 in a transgenic mouse model of lymphoma where Bcl-2 is over-expressed in B-cells and in a human prostate cancer xenograft model dependent on Mcl-1 for growth [24]. Pharmacological inhibition of Mcl-1 expression with Sabutoclax, is sufficient to sensitize prostate tumors to mda-7/IL-24-induced apoptosis illustrating the power of combining an inhibitor Bcl-2 family proteins with a novel gene delivery modality [25, 26].
We have determined that Sabutoclax is a viable drug candidate due to the critical role of Bcl-2 family proteins in cancer progression, and this compound has been licensed to Oncothyreon Inc. for further development for human trials. From the initial discovery of polyphenols as inhibitors of Bcl-2 family proteins emerged Sabutoclax, a product of integration of structure-based modeling with rational design to predict derivatives of lead compounds to be empirically tested and evaluated for bioactivity.
UTILIZATION OF THE PEG-3-PROMOTER FOR CANCER IMAGING, GENE THERAPY AND DRUG DISCOVERY
Cancer is a progressive disease culminating in the acquisition of metastatic potential by a subset of evolving tumor cells [3]. Although extensively investigated resulting in significant progress in recent years, the precise molecular and biochemical events underlying tumor etiology in many cancers and cancer progression to metastasis remain ambiguous [1, 3]. Progression Elevated Gene-3 (PEG-3) was cloned by subtraction hybridization of cDNA libraries generated from E11, a mutant Ad5-(H5ts125)-transformed rat embryo cell clone that forms small, slow-growing, and compact tumors, and from an E11 isogenic clone developed from a highly aggressive tumorigenic nude mouse, E11-NMT (Fig. 2) [27–30]. The expression of PEG-3 directly correlates with cancer progression and acquisition of oncogenic potential by transformed rodent cells [30]. PEG-3 shares significant nucleotide and amino acid sequence homology with the hamster growth arrest and DNA damage-inducible gene gadd34 and a homologous murine gene, MyD116 that is induced during induction of terminal differentiation by interleukin-6 in murine myeloid leukemia cells [30]. The PEG-3 gene is a mutated and truncated form of gadd34/MyD116 that is commonly mutated in diverse rodent tumors and functions as a dominant-negative mutant of gadd34, promoting instead of inhibiting the cancer phenotype [31]. No similar mutation is observed in human cancers, but the promoter of rodent PEG-3 is selectively active in human cancer cells, as it is in rodent cancer cells [32–36]. The cancer specific activity of the minimal promoter region of PEG-3 has been demonstrated in human cancer cell lines of various tissues such as brain, prostate, breast and pancreas, as well as in metastatic melanoma (Fig. 3 and data not shown) [32–37]. The cancer-specific activity of the PEG-3-promoter is a versatile platform that we have used to successfully create cancer-imaging techniques (Figs. 4 and 5), conditionally replicating cancer terminator viruses (CTV) (Fig. 6), and a novel screening platform that identifies inhibitors of the transformed state as potential lead compounds with anti-cancer properties (Fig. 7).
Fig. 2.

Photograph comparing tumor growth of E11 and its derivatives in nude mice. Pictures were taken 20 days postinjection with the indicated cell type. Cell lines: E11, H5ts125-transformed primary rat embryo cell line showing the non-progressed localized tumor phenotype; E11-NMT, nude mouse tumor-derived E11 clone, showing the progressed aggressive transformed phenotype; E11/PEG-3 S, a clone of E11 cells stably overexpressing a transduced PEG-3 gene in the sense orientation (S), displaying an aggressive progressed transformed phenotype, gain-of-function phenotype; E11-NMT/PEG-3 AS, a clone of E11-NMT cells stably expressing an antisense construct (AS) targeting the PEG-3 gene, displaying a non-progressed transformed phenotype, loss-of-function phenotype. Taken from Su et al. (29), Proc. Natl. Acad. Sci. USA, 1999.
Fig. 3.

PEG-3-promoter drives the expression of green fluorescence protein (GFP) only in cancer cells but not in normal cells. The indicated cells were infected with either Ad.CMV-GFP or Ad.PEG-GFP at a moi of 100 pfu per cell, and GFP expression was analyzed by an immunofluorescence microscope at 2 d postinfection. Cell types: HuPEC, early passage primary human prostate epithelial cells; Du-145, PC-3 and LNCaP: human prostate carcinoma cell lines; P69, SV40 T-antigen immortalized normal human prostate epithelial cell line; M2182, is a P69-derived clone that is tumorigenic but not metastatic in animal models; M12, is a P69-derived clone displaying both tumorigenic and metastatic properties in animal models; HMEC, early passage primary human mammary epithelial cells; MCF7, T47D, MDA-MB-157, MDA-MB-231 and MDA-MB-453, human breast carcinoma cell lines; PHFA, early passage normal human primary fetal astrocytes; U251MG, T98G and U87MG, human glioblastoma multiforme cell lines. Taken from Su et al. (32), Proc. Natl. Acad. Sci. USA, 2005.
Fig. 4.

Cancer-specific PEG-3-promoter activity shown by bioluminescence imaging in experimental metastasis models of human melanoma (Mel) and breast cancer (BCa). (a) Bioluminescence imaging of a representative healthy control mouse (Ctrl-2). (b) Bioluminescence imaging showing firefly luciferase expression observed in a representative melanoma model (Mel-3). Each mouse was imaged from four directions (V, ventral; L, left side; R, right side; D, dorsal views) to cover the entire body. Pseudocolor images from the two groups were adjusted to the same threshold. (c) Quantification of bioluminescence imaging signal intensity in the control group (Ctrl) and melanoma group at 24 and 48 h after injection of pPEG-Luc–PEI polyplex. Quantified values are shown in total flux. ***P < 0.0001. (d,e) CT scans and gross anatomical views of lung from one representative mouse from the control group (d) and the melanoma group (e). (f,g) Bioluminescence imaging of one representative mouse from the control group (f, Ctrl-3) and the experimental breast cancer metastasis group (g, BCa-1). The pseudocolor images were adjusted to the same threshold. (h) Quantification of bioluminescent signal intensity in the Ctrl and breast cancer groups at 24 and 48 h after injection of pPEG-Luc–PEI polyplex. **P = 0.0066. (i) A CT image and a macroscopic view of lung from a representative breast cancer mouse. Displayed bioluminescent images (a,b,f,g) were obtained at 48 h after the systemic delivery of pPEG-Luc–PEI polyplex. Black arrows (e,i) indicate metastatic nodules observed in the lung. Scale bars, 5 mm. From Bhang et al. (37), Nature Medicine, 2011.
Fig. 5.

Detection and localization of metastatic masses by SPECT-CT imaging after the systemic administration of pPEG-HSV1tk. (a–j) Systemic metastatic sites were detected based on the whole body SPECT-CT images in three representative mice, Mel-2 (a,b), Mel-3 (c–h) and BCa-1 (i,j). (a,c,e,g) Transverse, coronal and sagittal views of co-registered SPECT-CT images of Mel-2 (a) and Mel-3 (c,e,g). All images were obtained at 24 h after [125I]FIAU injection. (b,d,f,h) Gross anatomical details of the metastatic masses that were located on the basis of the SPECT-CT images (a,c,e,g). Multiple metastatic sites were detected by imaging in Mel-2 (a, dotted circle). Necropsy of the corresponding area revealed melanoma masses under the brown adipose tissue in the upper dorsal area (b, dotted circle). (c) Accumulated radioactivity was detected adjacent to the thoracic mid-spine (arrow), which corresponded to a tumor at this location (d, arrow). Additional metastatic sites demonstrated by SPECT-CT imaging (e,g, arrow and dotted circle) correlated with melanoma masses uncovered immediately above the diaphragm (f, dotted circle) and in the left inguinal lymph node (h, arrow), respectively. (i,j) Cross-comparison of PEG-3 promoter–mediated imaging and FDG-PET in a breast cancer metastasis model, BCa-1. Two nodules (Tu-1 and Tu-2) were detected by [125I]FIAU-SPECT near the heart (i) and were confirmed by necropsy (j). Although Tu-1 was also detected by [18F]FDG-PET, Tu-2, a smaller nodule attached to the heart, was not obvious in the PET image. SPECT images were acquired 48 h after injection of [125I]FIAU. The PET and SPECT images were acquired on the same day (i). (Tu, tumor; Ht, heart; Bf, brown fat.) Scale bars, 10 mm. From Bhang et al. (37), Nature Medicine, 2011.
Fig. 6.

Conditionally replication competent adenovirus, CTV (Ad.PEG-E1A-mda-7), eradicates primary and distant tumors in athymic nude mice. Subcutaneous tumor xenografts from T47D cells were established in athymic nude mice in both right and left flanks, and only tumors on the left side were injected with PBS (control) or with the indicated Ad for 3 wk (total of seven injections). (A and C) Measurement of tumor volume. The data represent mean ± SD with a minimum of five mice per group. (B and D) Measurement of tumor weight at the end of the study. The data represent mean ± SD with at least five mice per group. Qualitatively similar results were obtained in an additional study. From Sarkar et al. (34) Proc. Natl. Acad. Sci. USA, 2005.
Fig. 7.

Schematic representation of the screening strategy to recover selective inhibitors of the PEG-3-promoter. HeLa cells stably expressing the PEG-3-promoter will be utilized as the primary screen. A hit from the primary screen will be designated as a compound that inhibits the PEG-3-promoter but does not inhibit a non-specific promoter and is not toxic to untransformed CREF cells. Compounds that meet these criteria will be designated probe compounds for follow up studies. Cytotoxicity profiling of hits from the HTS campaign in human cancer and normal counterpart cells will facilitate lead selection. We will begin to dissect the mechanism of action of the PEG-3-promoter-inhibitors through cytotoxicity profiling in oncogenic CREF cells and by studying the activity of the compounds on the PEG-3-promoter mutated at the AP1 or PEA3 binding sites.
PEG-3-promoter activity is regulated by Activating Protein-1 (AP-1) and PEA3/ETV4/E1AF transcription factors that are juxtaposed in the 512-bp region of the minimal PEG-3-promoter [38]. The PEA3 family of transcription factors comprises ETV5/ERM, ETV1/ER81 and PEA3. Elevated PEA3 expression correlates with poor prognosis in a broad array of clinical cancer samples from tissues including lung, stomach, liver, colon and others [39]. Many reports demonstrate the involvement of PEA3 in the transcriptional regulation of matrix metalloproteases (MMPs) involved with degradation of the extracellular matrix, a function required for cancer metastasis [40]. The AP-1 protein family is implicated in transformation and cell proliferation and has also been linked to apoptosis, differentiation, cellular migration, wound healing and inflammation [41–44]. AP1 and PEA3 are primarily regulated by the MAPKinase pathway, including extracellular related kinase (ERK), P38MAPK and SAPK/JNK cascades [45, 46]. There are also reports that link Rho/RACK or Wnt signaling pathway to PEA-3 leading to tumorigenesis [47]. A summary of the network of signaling pathways known to regulate AP-1 and PEA3 transcription are illustrated in Fig. (8). Juxtaposed AP-1 and PEA-3 binding sites are observed in genes that contribute to cancer progression including Her2-mediated activation of the COX2 promoter, expression of urokinase plasminogen activator, induction of pro-angiogenic factor IL-8, and more recently expression of Notch ligands [48–51].
Fig. 8.

Schematic representation of the PEG-3-promoter and the network of signaling pathways which it potentially monitors. ERK, p38, SAPK/JNK, Wnt, and Rho signaling activate transcription of AP-1 and PEA3, which are required for PEG-3-promoter activity. Small molecules that can inhibit PEG-3-promoter activity may be valuable anti-cancer therapeutics as well as probe compounds to study new pathways involved in catalyzing transformation.
Molecular genetic imaging enables the visualization and quantification of the activity of a variety of tumor-specific gene promoters. Because of the cancer specific activity exerted by the PEG-3-promoter we illustrated that the systemic delivery of a PEG-3-promoter-driven imaging construct can be used to visualize melanoma and breast cancer metastases in vivo with two distinct reporters (Figs. 4 and 5) [37]. Previous promoters utilized to visualize cancer in vivo were derived from human telomerase reverse transcriptase, survivin, and carcinoembryonic antigen that employed adenovirus delivery of the promoter for tumor-specific reporter expression [52–54]. Due to the use of adenoviral vectors for delivery, treatment was limited to local delivery with systemic delivery resulting in accumulation of the vector in the liver. The advantage of utilizing the PEG-3-promoter for tumor specific imaging is that it uses a non-viral vector allowing for systemic delivery of the vector. Since the PEG-3-promoter has a high level of constitutive activity it drives reporter genes at level detectable without the requirement of promoter amplification, a common requirement in other promoter-based imaging methods [37]. Further studies are required in other tumor types to determine if a wide variety of tumor types can be imaged with this methodology. Based on previous studies indicating the wide spectrum tumor-specific activity of the PEG-3-promoter [32–37], it is highly likely that this methodology will be applicable to a large number of additional tumor indications. The translational implications of this finding would not only aid tumor detection, but also extend to pre-operative planning, intraoperative management and therapeutic monitoring. Additionally, by combining this unique delivery methodology in combination with both an imaging and therapeutic agent, “theranostics”, it would be possible to both visualize the cancer and destroy it using a single reagent [55].
The cancer selectivity of the PEG-3-promoter has been utilized to successfully construct Cancer Terminator Viruses (CTVs), which successfully eradicate pancreatic, prostate and melanoma tumors (Fig. 6) [33–36]. Cancer-specific replication of the CTV occurs because the E1A gene, required for replication, is driven by the PEG-3 promoter. The CTV simultaneously expresses a second transgene in the E3 region that encodes an apoptosis-inducing or immunomodulatory cytokine, such as interferon (IFN-gamma) or melanoma differentiation associated gene-7/interleukin-24 (mda-7/IL-24) [33–36]. These CTVs then produces large quantities of the transgene protein as a function of adenovirus replication uniquely in cancer cells and eradicates the cancer. The advantages of using mda-7/IL-24 as the second transgene relate to both the direct and indirect anti-tumor effects of this cancer selective apoptosis- and toxic autophagy-inducing cytokine [56–71]. mda-7/IL-24: induces apoptosis or toxic autophagy in a broad spectrum of human cancers without affecting normal cells or tissues; inhibits tumor angiogenesis; is a potent anti-cancer immune modulatory agent; synergizes with current modalities of therapy including radiation, chemotherapy and antibody therapy; induces a potent ‘bystander’ anti-tumor effect as a secreted therapeutic cytokine; and has been shown to be safe, well tolerated and clinically efficacious in a Phase I clinical trial when injected intratumorally in patients with advanced cancers [56–77].
Since a major challenge of gene therapy is to develop an effective means to deliver toxic gene products we then set out to develop an alternative to delivering the CTVs intratumorally. We achieved this through engineering complexes of the CTV virus in Ultrasound (US) contrast agents (microbubbles), which would allow for targeted release through ultrasound following systemic injection of the CTV. This ultrasound-targeted microbubble-destruction (UTMD) approach involving US-guided CTV-microbubbles eradicated prostate cancer tumors in mice, not only in the targeted region, but also non-targeted sites distant from the primary tumor because of secondary viral infection and secreted mda-7/IL-24 [2, 78]. This approach has significant potential for therapeutic applications, since it has been used to successfully deliver a non-replicating tropism modified adenovirus (Ad.5/3-mda-7) into the prostate of Hi-Myc transgenic mice that develop pathogenic changes similar to humans developing prostate cancer [25]. When UTMD with Ad.5/3-mda-7 was combined with Sabutoclax (BI-97C1) in the Hi-Myc mice tumor growth and progression was abrogated [25]. These developments highlight the potential use of this novel image-guided viral gene therapy technology for use in patients with advanced metastatic disease.
Since the PEG-3-promoter provides a relevant unbiased (not based on the genetic change(s) causing the cancer) readout of the transformed state and cancer progression, compounds that inhibit PEG-Prom activity can potentially reverse the cancer phenotype. We have designed a screening paradigm to identify potential inhibitors of the transformed state utilizing the PEG-3-promoter as a primary screen. HeLa cells stably expressing a construct encoding the PEG-3-promoter driving luciferase expression (HeLa-PEG-luc) were screened with the full NIH Molecular Libraries Small Molecule Repository library consisting of >330,000 compounds to identify potential inhibitors of the PEG-3-promoter. A total of 6,143 compounds inhibited the PEG-3-promoter at least 50% and will be subjected to further counterscreens (Pubchem AID: 588405).
Hits from the primary high-throughput-screening (HTS) will be evaluated in a number of secondary assays (Fig. 7). We will determine if hits can inhibit the activity of the GAPDH-promoter driving luciferase expression stably in HeLa cells. Counter-screening with the GAPDH-promoter will allow us to discard general inhibitors of transcription/translation as well as discard compounds that can inhibit luciferase enzymatic activity. Furthermore, compound cytotoxicity will be determined to ensure promoter inhibition observed is not a consequence of generalized cellular cytotoxicity. A direct secondary screen will be developed in which the individual regions of the PEG-3-promoter that contain a functional or mutated AP-1 or PEA-3 site linked to a Luc reporter gene will be engineered into HeLa cells. These cells will be used to determine if specific compounds inhibit PEG-3-promoter function by blocking activity of AP-1 or PEA-3. Identified small molecule inhibitors could, in principle, exert their effects through any of these known pathways or through yet to be discovered pathways (Fig. 8). Our screening paradigm should permit a distinction and identification of the pathways through which any identified inhibitor may function.
We have developed a series of clones derived from CREF (Fischer cloned rat embryo fibroblast) cells [79] that are transformed morphologically and biologically (induce cells to grow anchorage independently and induce tumors in nude mice) by a single oncogene [30, 38, 79–84]. This series contains transformed CREF cells derived from transfection and overexpression of Ha-ras, v-raf, v-src, human papilloma virus type 18, AEG-1, type 5 adenovirus, c-Jun, high molecular weight DNA from human prostate cancer, etc. [79–84]. This panel of cells will permit us to define potential oncogene-regulated pathways modulated by specific small molecules identified in the HTS efforts. Confirmation of specific mode of action could also be ascertained by specifically extinguishing the function of defined oncogenes using known pharmacological or small molecule inhibitors of distinct oncogenic pathways, such as ras or src inhibitors, or by using siRNA approaches [84–86]. These studies will help elucidate potential mechanisms of action of chemical probes identified.
We conducted a pilot screen of ~14,400 compounds using the strategy described above and identified a PEG-3-promoter-inhibitor (unpublished data). This PEG-3-promoter inhibitor selectively kills human cancer cell lines derived from breast, prostate, pancreatic and brain with minimal cytotoxicity to untransformed counterpart cell lines. Treatment with the PEG-3-promoter-inhibitor suppresses PEA3 protein expression in cancer cell lines, which may be the event responsible for inhibiting the PEG-3-promoter. Since PEA3 associates with cancer progression this may also be a mechanism by which this PEG-3-promoter-inhibitor suppresses cancer growth. More follow up studies are required to delineate the target of this PEG-3-promoter-inhibitor, however this study suggests that this screening methodology is a viable strategy to produce lead compound with anti-cancer properties. Once targets of compounds are identified, structure-based modeling and rational design will be applied to yield second-generation compounds with superior anti-cancer activity. Furthermore, the use of the PEG-3-promoter as a systemic imaging agent as described earlier will allow us to determine if molecular probes identified using our HTS approaches can affect tumor expression of the PEG-3-promoter in vivo.
Acknowledgments
The present studies were supported in part by National Institutes of Health grants R03 MH093195, R01 CA097318 and P01 CA104177, and the National Foundation for Cancer Research (P.B.F.); the Samuel Waxman Cancer Research Foundation (D.S. and P.B.F.); USAMRAA W81XWH-10-PCRP-SIDA (P.B.F. and X.-Y.W.); USAMRAA, Army Prostate Cancer DoD Award W81XWH-11-1-0186 (S.S and P.B.F.); National Institutes of Health grant R01 CA149668 (J.C.R., M.P.) and California Institute for Regenerative Medicine grant TR2-01789 (M.P.); National Institutes of Health grants CA092871 and CA151838 (M.G.P.); National Institutes of Health Grant R01 CA138540 and Grants from the James S. McDonnell Foundation (D.S.).
ABBREVIATIONS
- AP-1
Activating Protein-1
- AS
Antisense
- BCL-2
B-cell lymphoma/leukemia-2
- CREF
Fischer cloned rat embryo fibroblast
- CTV
Cancer terminator viruses
- ERK
Extracellular related kinase
- FPA
Fluorescence polarization displacement assays
- GFP
Green fluorescent protein
- HTS
High throughput screening
- IFN
Interferon
- mda-7/IL-24
melanoma differentiation associated gene-7/interleukin-24
- MMP
Matrix metalloprotease
- NMR
Nuclear magnetic resonance
- PEG-3
Progression elevated gene-3
- SAR
Structure activity relationship
- US
Ultrasound
- UTMD
Ultrasound-targeted microbubble-destruction
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Dash R, Azab B, Shen XN, Sokhi UK, Sarkar S, Su ZZ, Wang XY, Claudio PP, Dent P, Dmitriev IP, Curiel DT, Grant S, Sarkar D, Fisher PB. Developing an effective gene therapy for prostate cancer: New technologies with potential to translate from the laboratory into the clinic. Discovery Med. 2011;11(56):46–56. [PMC free article] [PubMed] [Google Scholar]
- 3.Das SK, Bhutia SK, Kegelman TP, Peachy L, Oyesanya RA, Dasgupta S, Sokhi UK, Azab B, Dash R, Quinn BA, Kim K, Barral PM, Su ZZ, Boukerche H, Sarkar D, Fisher PB. MDA-9/syntenin: a positive gatekeeper of melanoma metastasis. Front Biosci. 2012;17:1–15. doi: 10.2741/3911. [DOI] [PubMed] [Google Scholar]
- 4.Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol. 1999;17(9):2941–53. doi: 10.1200/JCO.1999.17.9.2941. [DOI] [PubMed] [Google Scholar]
- 5.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108(2):153–64. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 6.Reed JC. Apoptosis-based therapies. Nat Rev Drug Discov. 2002;1(2):111–21. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 7.Reed JC. Bcl-2 family proteins. Oncogene. 1998;17(25):3225–36. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- 8.Reed JC. Bcl-2 family proteins: strategies for overcoming chemoresistance in cancer. Adv Pharmacol. 1997;41:501–32. doi: 10.1016/s1054-3589(08)61070-4. [DOI] [PubMed] [Google Scholar]
- 9.Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol. 2001;3(2):173–82. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 10.Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, Croce CM, Alnemri ES, Huang Z. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci US A. 2000;97(13):7124–9. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46(20):4259–64. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- 12.Wu D. An overview of the clinical pharmacology and therapeutic potential of gossypol as a male contraceptive agent and in gynaecological disease. Drugs. 1989;38(3):333–341. doi: 10.2165/00003495-198938030-00001. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert NE, O’Reilly JE, Chang CJ, Lin YC, Brueggemeier RW. Antiproliferative activity of gossypol and gossypolone on human breast cancer cells. Life Sci. 1995;57(1):61–67. doi: 10.1016/0024-3205(95)00243-y. [DOI] [PubMed] [Google Scholar]
- 14.Wu YW, Chik CL, Knazek RA. An in vitro and in vivo study of antitumor effects of gossypol on human SW-13 adrenocortical carcinoma. Cancer Res. 1989;49(14):3754–8. [PubMed] [Google Scholar]
- 15.Thomas M, von Hagen V, Moustafa Y, Montmasson MP, Monet JD. Effects of gossypol on the cell cycle phases in T-47D human breast cancer cells. Anticancer Res. 1991;11(4):1469–75. [PubMed] [Google Scholar]
- 16.Le Blanc M, Russo J, Kudelka AP, Smith JA. An in vitro study of inhibitory activity of gossypol, a cottonseed extract, in human carcinoma cell lines. Pharmacol Res. 2002;46(6):551–5. doi: 10.1016/s104366180200230x. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Wang J, Wong SC, Chow LS, Nicholls JM, Wong YC, Liu Y, Kwong DL, Sham JS, Tsa SW. Cytotoxic effect of gossypol on colon carcinoma cells. Life Sci. 2000;67(22):2663–71. doi: 10.1016/s0024-3205(00)00857-2. [DOI] [PubMed] [Google Scholar]
- 18.Abou-Karam M, Shier WT. Inhibition of oncogene product enzyme activity as an approach to cancer chemoprevention. Tyrosine-specific protein kinase inhibition by purpurogallin from Quercus sp. nutgall. Phytother Res. 1999;13(4):337–40. doi: 10.1002/(SICI)1099-1573(199906)13:4<337::AID-PTR451>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 19.Fung KP, Wu TW, Lui CP. Purpurogallin inhibits DNA synthesis of murine fibrosarcoma L-929 and human U-87 MG glioblastoma cells in vitro. Chemotherapy. 1996;42(3):199–205. doi: 10.1159/000239442. [DOI] [PubMed] [Google Scholar]
- 20.Becattini B, Kitada S, Leone M, Monosov E, Chandler S, Zhai D, Kipps TJ, Reed JC, Pellecchia M. Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L) Chem Biol. 2004;11(3):389–95. doi: 10.1016/j.chembiol.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 21.Wei J, Stebbins JL, Kitada S, Dash R, Zhai D, Placzek W, Wu B, Rega MF, Zhang Z, Barile E, Yang L, Dahl R, Fisher PB, Reed JC, Pellecchia M. An optically pure Apogossypolone derivative as pan-active inhibitor of anti-apoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. Frontiers. Oncology. 2012 doi: 10.3389/fonc.2011.00028. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia L, Coward LC, Kerstner-Wood CD, Cork RL, Gorman GS, Noker PE, Kitada S, Pellecchia M, Reed JC. Comparison of pharmacokinetic and metabolic profiling among gossypol, apogossypol and apogossypol hexaacetate. Cancer Chemother Pharmacol. 2008;61(1):63–73. doi: 10.1007/s00280-007-0446-3. [DOI] [PubMed] [Google Scholar]
- 23.Kitada S, Kress CL, Krajewska M, Jia L, Pellecchia M, Reed JC. Bcl-2 antagonist apogossypol (NSC736630) displays single-agent activity in Bcl-2-transgenic mice and has superior efficacy with less toxicity compared with gossypol (NSC19048) Blood. 2008;111(6):3211–9. doi: 10.1182/blood-2007-09-113647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei J, Kitada S, Rega MF, Stebbins JL, Zhai D, Cellitti J, Yuan H, Emdadi A, Dahl R, Zhang Z, Yang L, Reed JC, Pellecchia M. Apogossypol derivatives as pan-active inhibitors of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J Med Chem. 2009;52(14):4511–23. doi: 10.1021/jm900472s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dash R, Azab B, Quinn BA, Shen X, Wang XY, Das SK, Rahmani MJ, Wei M, Dent P, Hedvat, Dmitriev IP, Curiel DT, Grant S, Wu B, Stebbins JL, Pellecchia M, Reed JC, Sarkar D, Fisher PB. Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. Proc Natl Acad Sci USA. 2011;108(21):8785–90. doi: 10.1073/pnas.1100769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S, Oyesanya RA, Dasgupta S, Dent P, Grant S, Rahmani M, Curiel DT, Dmitriev I, Hedvat M, Wei J, Wu B, Stebbins JL, Reed JC, Pellecchia M, Sarkar D, Fisher PB. Targeting Mcl-1 for the therapy of cancer. Expert Opin Invest Drug. 2011;20(10):1397–411. doi: 10.1517/13543784.2011.609167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Babiss LE, Zimmer SG, Fisher PB. Reversibility of progression of the transformed phenotype in Ad5-transformed rat embryo cells. Science. 1985;228(4703):1099–101. doi: 10.1126/science.2581317. [DOI] [PubMed] [Google Scholar]
- 28.Fisher PB, Bozzone JH, Weinstein IB. Tumor promoters and epidermal growth factor stimulate anchorage-independent growth of adenovirus-tranformed rat embryo cells. Cell. 1979;18(3):695–705. doi: 10.1016/0092-8674(79)90124-7. [DOI] [PubMed] [Google Scholar]
- 29.Su ZZ, Goldstein NI, Jiang H, Wang MN, Duigou GJ, Young CS, Fisher PB. PEG-3, a nontransforming cancer progression gene is a positive regulator of cancer aggressiveness and angiogenesis. Proc Natl Acad Sci USA. 1999;96(26):15115–20. doi: 10.1073/pnas.96.26.15115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su ZZ, Shi Y, Fisher PB. Subtraction hybridization identifies a transformation progression-associated gene PEG-3 with sequence homology to a growth arrest and DNA damage-inducible gene. Proc Natl Acad Sci US A. 1997;94(17):9125–30. doi: 10.1073/pnas.94.17.9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su ZZ, Emdad L, Sarkar D, Randolph A, Valerie K, Yacoub A, Dent P, Fisher PB. Potential molecular mechanism for rodent tumorigenesis: mutational generation of Progression Elevated Gene-3 (PEG-3) Oncogene. 2005;24(13):2247–55. doi: 10.1038/sj.onc.1208420. [DOI] [PubMed] [Google Scholar]
- 32.Su ZZ, Sarkar D, Emdad L, Duigou GJ, Young CS, Ware J, Randolph A, Valerie K, Fisher PB. Targeting gene expression selectively in cancer cells by using the progression-elevated gene-3 promoter. Proc Natl Acad Sci US A. 2005;102(4):1059–64. doi: 10.1073/pnas.0409141102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarkar D, Su ZZ, Vozhilla N, Park ES, Gupta P, Fisher PB. Dual cancer-specific targeting strategy cures primary and distant breast carcinomas in nude mice. Proc Natl Acad Sci US A. 2005;102(39):14034–9. doi: 10.1073/pnas.0506837102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarkar D, Su ZZ, Vozhilla N, Park ES, Randolph A, Valerie K, Fisher PB. Targeted virus replication plus immunotherapy eradicates primary and distant pancreatic tumors in nude mice. Cancer Res. 2005;65(19):9056–63. doi: 10.1158/0008-5472.CAN-05-1261. [DOI] [PubMed] [Google Scholar]
- 35.Sarkar D, Lebedeva IV, Su ZZ, Park ES, Chatman L, Vozhilla N, Dent P, Curiel DT, Fisher PB. Eradication of therapy-resistant human prostate tumors using a cancer terminator virus. Cancer Res. 2007;67(11):5434–42. doi: 10.1158/0008-5472.CAN-07-0195. [DOI] [PubMed] [Google Scholar]
- 36.Sarkar D, Su ZZ, Park ES, Vozhilla N, Dent P, Curiel DT, Fisher PB. A cancer terminator virus eradicates both primary and distant human melanomas. Cancer Gene Ther. 2008;15(5):293–302. doi: 10.1038/cgt.2008.14. [DOI] [PubMed] [Google Scholar]
- 37.Bhang HE, Gabrielson KL, Laterra J, Fisher PB, Pomper MG. Tumor-specific imaging through progression elevated gene-3 promoter-driven gene expression. Nat Med. 2011;17(1):123–9. doi: 10.1038/nm.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su ZZ, Shi Y, Fisher PB. Cooperation between AP1 and PEA3 sites within the progression elevated gene-3 (PEG-3) promoter regulate basal and differential expression of PEG-3 during progression of the oncogenic phenotype in transformed rat embryo cells. Oncogene. 2000;19(30):3411–21. doi: 10.1038/sj.onc.1203666. [DOI] [PubMed] [Google Scholar]
- 39.Launoit de Y, Baert JL, Chotteau-Lelievre A, Monte D, Coutte L, Mauen S, Firlej V, Degerny C, Verreman K. The Ets transcription factors of the PEA3 group: transcriptional regulators in metastasis. Biochim Biophys Acta. 2006;1766(1):79–87. doi: 10.1016/j.bbcan.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Higashino F, Yoshida K, Noumi T, Seiki M, Fujinaga K. Ets-related protein E1A-F can activate three different matrix metalloproteinase gene promoters. Oncogene. 1995;10(7):1461–3. [PubMed] [Google Scholar]
- 41.Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117(Pt 25):5965–73. doi: 10.1242/jcs.01589. [DOI] [PubMed] [Google Scholar]
- 42.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4(5):E131–6. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 43.Wagner EF, Eferl R. Fos/AP-1 proteins in bone and the immune system. Immunol Rev. 2005;208:126–40. doi: 10.1111/j.0105-2896.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 44.Kang DC, Motwani M, Fisher PB. Role of the transcription factor AP1 in melanoma differentiation. (review) Intl J Oncology. 1998;13(6):1117–26. doi: 10.3892/ijo.13.6.1117. [DOI] [PubMed] [Google Scholar]
- 45.Vojtek AB, Der CJ. Increasing complexity of the Ras signaling pathway. J Biol Chem. 1998;273(32):19925–8. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 46.O’Hagan RC, Tozer RG, Symons M, McCormick F, Hassell JA. The activity of the Ets transcription factor PEA3 is regulated by two distinct MAPK cascades. Oncogene. 1996;13(6):1323–33. [PubMed] [Google Scholar]
- 47.Hakuma N, Kinoshita I, Shimizu Y, Yamazaki K, Yoshida K, Nishimura M, Dosaka-Akita H. E1AF/PEA3 activates the Rho/Rho-associated kinase pathway to increase the malignancy potential of non-small-cell lung cancer cells. Cancer Res. 2005;65(23):10776–82. doi: 10.1158/0008-5472.CAN-05-0060. [DOI] [PubMed] [Google Scholar]
- 48.Clementz AG, Rogowski A, Pandya K, Miele L, Osipo C. NOTCH-1 and NOTCH-4 are novel gene targets of PEA3 in breast cancer: novel therapeutic implications. Breast Cancer Res. 2011;13(3):R63. doi: 10.1186/bcr2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subbaramaiah K, Norton L, Gerald W, Dannenberg AJ. Cyclooxygenase-2 is overexpressed in HER-2/neu-positive breast cancer: evidence for involvement of AP-1 and PEA3. J Biol Chem. 2002;277(21):18649–57. doi: 10.1074/jbc.M111415200. [DOI] [PubMed] [Google Scholar]
- 50.Gavrilov D, Kenzior O, Evans M, Calaluce R, Folk WR. Expression of urokinase plasminogen activator and receptor in conjunction with the ets family and AP-1 complex transcription factors in high grade prostate cancers. Eur J Cancer. 2001;37(8):1033–40. doi: 10.1016/s0959-8049(01)00077-6. [DOI] [PubMed] [Google Scholar]
- 51.Iguchi A, Kitajima I, Yamakuchi M, Ueno S, Aikou T, Kubo T, Matsushima K, Mukaida N, Maruyama I. PEA3 and AP-1 are required for constitutive IL-8 gene expression in hepatoma cells. Biochem Biophys Res Commun. 2000;279(1):166–71. doi: 10.1006/bbrc.2000.3925. [DOI] [PubMed] [Google Scholar]
- 52.Kishimoto H, Kojima T, Watanabe Y, Kagawa S, Fujiwara T, Uno F, Teraishi F, Kyo S, Mizuguchi H, Hashimoto Y, Urata Y, Tanaka N, Fujiwara T. In vivo imaging of lymph node metastasis with telomerase-specific replication-selective adenovirus. Nat Med. 2006;12(10):1213–9. doi: 10.1038/nm1404. [DOI] [PubMed] [Google Scholar]
- 53.Ray S, Paulmurugan R, Patel MR, Ahn BC, Wu L, Carey M, Gambhir SS. Noninvasive imaging of therapeutic gene expression using a bidirectional transcriptional amplification strategy. Mol Ther. 2008;16(11):1848–56. doi: 10.1038/mt.2008.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiao J, Doubrovin M, Sauter BV, Huang Y, Guo ZS, Balatoni J, Akhurst T, Blasberg RG, Tjuvajev JG, Chen SH, Woo SL. Tumor-specific transcriptional targeting of suicide gene therapy. Gene Ther. 2002;9(3):168–75. doi: 10.1038/sj.gt.3301618. [DOI] [PubMed] [Google Scholar]
- 55.Hingtgen S, Kasmieh R, Elbayly E, Nesterenko I, Figueiredo JL, Dash R, Sarkar D, Hall D, Kozakov D, Vajda S, Fisher PB, Shah K. A first-generation multi-functional cytokine for simultaneous optical tracking and tumor therapy. PLoS One. 2012 doi: 10.1371/journal.pone.0040234. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang H, Lin JJ, Su ZZ, Goldstein NI, Fisher PB. Subtraction hybridization identifies a novel melanoma differentiation associated gene, mda-7, modulated during human melanoma differentiation, growth and progression. Oncogene. 1995;11(12):2477–86. [PubMed] [Google Scholar]
- 57.Jiang H, Su Z-Z, Lin JJ, Goldstein NI, Young CSH, Fisher PB. The melanoma differentiation associated gene mda-7 suppresses cancer cell growth. Proc Natl Acad Sci USA. 1996;93(17):9160–5. doi: 10.1073/pnas.93.17.9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Su ZZ, Madireddi MT, Lin JJ, Young CSH, Kitada S, Reed JC, Goldstein NI, Fisher PB. The cancer growth suppressor gene mda-7 selectively induces apoptosis in human breast cancer cells and inhibits tumor growth in nude mice. Proc Natl Acad Sci USA. 1998;95(24):14400–5. doi: 10.1073/pnas.95.24.14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lebedeva IV, Su ZZ, Chang Y, Kitada S, Reed JC, Fisher PB. The cancer growth suppressing gene mda-7 induces apoptosis selectively in human melanoma cells. Oncogene. 2002;21(5):708–18. doi: 10.1038/sj.onc.1205116. [DOI] [PubMed] [Google Scholar]
- 60.Sauane M, Gopalkrishnan RV, Sarkar D, Su ZZ, Lebedeva IV, Dent P, Pestka S, Fisher PB. Mda-7/IL-24: novel cancer growth suppressing and apoptosis inducing cytokine. Cytokine Growth Factor Rev. 2003;14(1):35–51. doi: 10.1016/s1359-6101(02)00074-6. [DOI] [PubMed] [Google Scholar]
- 61.Lebedeva IV, Su ZZ, Sarkar D, Kitada S, Dent P, Waxman S, Reed JC, Fisher PB. Melanoma differentiation associated gene-7, mda-7/IL-24, promotes apoptosis in prostate cancer cells by promoting mitochondrial dysfunction and inducing reactive oxygen species. Cancer Res. 2003;63(23):8138–44. [PubMed] [Google Scholar]
- 62.Fisher PB. Is mda-7/IL-24 a ‘magic bullet’ for cancer? Cancer Res. 2005;65(22):10128–38. doi: 10.1158/0008-5472.CAN-05-3127. [DOI] [PubMed] [Google Scholar]
- 63.Gupta P, Su ZZ, Lebedeva IV, Sarkar D, Sauane M, Emdad L, Bachelor MA, Grant S, Curiel DT, Dent P, Fisher PB. mda-7/IL-24: Multifunctional cancer-specific apoptosis-inducing cytokine. Pharmacol Ther. 2006;111(3):596–628. doi: 10.1016/j.pharmthera.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gupta P, Walter MR, Su ZZ, Lebedeva IV, Emdad L, Randolph A, Valerie K, Sarkar D, Fisher PB. BiP/GRP78 is an intracellular target for mda-7/IL-24 induction of cancer-specific apoptosis. Cancer Res. 2006;66(16):8182–91. doi: 10.1158/0008-5472.CAN-06-0577. [DOI] [PubMed] [Google Scholar]
- 65.Sarkar D, Lebedeva IV, Gupta P, Emdad L, Sauane M, Dent P, Curiel DT, Fisher PB. Melanoma differentiation associated gene-7 (mda-7)/IL-24: a “magic bullet” for cancer therapy? Expert Opin Biol Ther. 2007;7(5):577–86. doi: 10.1517/14712598.7.5.577. [DOI] [PubMed] [Google Scholar]
- 66.Yacoub A, Park MA, Gupta P, Rahmani M, Zhang G, Hamed H, Hanna D, Sarkar D, Lebedeva IV, Emdad L, Sauane M, Vozhilla N, Spiegel S, Koumenis C, Graf M, Curiel DT, Grant S, Fisher PB, Dent P. Caspase-, cathepsin- and PERK-dependent regulation of MDA-7/IL-24-induced cell killing in primary human glioma cells. Mol Cancer Ther. 2008;7(2):297–313. doi: 10.1158/1535-7163.MCT-07-2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sauane M, Su ZZ, Gupta P, Lebedeva IV, Dent P, Sarkar D, Fisher PB. Autocrine regulation of mda-7/IL-24 mediates cancer-specific apoptosis. Proc Natl Acad Sci US A. 2008;105(28):9763–8. doi: 10.1073/pnas.0804089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao P, Sun X, Chen X, Wang Y, Foster BA, Subjeck J, Fisher PB, Wang XY. Secretable chaperone Grp170 enhances therapeutic activity of a novel tumor suppressor, mda-7/IL-24. Cancer Res. 2008;68(10):3890–8. doi: 10.1158/0008-5472.CAN-08-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emdad L, Lebedeva IV, Su ZZ, Gupta P, Sauane M, Dash R, Grant S, Dent P, Curiel DT, Sarkar D, Fisher PB. Historical perspective and recent insights into our understanding of the molecular and biochemical basis of the antitumor properties of mda-7/IL-24. Cancer Biol Ther. 2009;8(5):391–400. doi: 10.4161/cbt.8.5.7581. [DOI] [PubMed] [Google Scholar]
- 70.Dash R, Bhutia SK, Azab B, Su ZZ, Quinn BA, Kegelmen TP, Das SK, Kim K, Lee SG, Park MA, Yacoub A, Rahmani M, Emdad L, Dmitriev IP, Wang X-Y, Sarkar D, Grant S, Dent P, Curiel DT, Fisher PB. mda-7/IL-24: a unique member of the IL-10 gene family promoting cancer-specific toxicity. Cytokine Growth Factor Rev. 2010;21(5):381–91. doi: 10.1016/j.cytogfr.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dent P, Yacoub A, Hamed HA, Park MA, Dash R, Bhutia SK, Sarkar D, Gupta P, Emdad L, Lebedeva IV, Sauane M, Su ZZ, Rahmani M, Broaddus WC, Young HF, Lesniak M, Grant S, Fisher PB. MDA-7/IL-24 as a cancer therapeutic: from bench to bedside. Anticancer Drugs. 2010;21(8):725–31. doi: 10.1097/CAD.0b013e32833cfbe1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fisher PB, Gopalkrishnan RV, Chada S, Ramesh R, Grimm EA, Rosenfeld MR, Curiel DT, Dent P. mda-7/IL-24, a novel cancer selective apoptosis inducing cytokine gene: from the laboratory into the clinic. Cancer Biol Ther. 2003;2(4 Suppl 1):S23–37. [PubMed] [Google Scholar]
- 73.Cunningham CC, Chada S, Merritt JA, Ton A, Senzer N, Zhang Y, Mhashilkar A, Parker K, Vukelja S, Richards D, Hood J, Coffee K, Nemunaitis J. Clinical and local biological effects of an intratumoral injection of mda-7 (IL24; INGN 241) in patients with advanced carcinoma: a phase I study. Mol Ther. 2005;11(1):149–59. doi: 10.1016/j.ymthe.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 74.Tong AW, Nemunaitis J, Su D, Zhang Y, Cunningham C, Senzer N, Netto G, Rich D, Mhashilkar A, Parker K, Coffee K, Ramesh R, Ekmekcioglu S, Grimm EA, van Wart Hood J, Merritt J, Chada S. Intratumoral injection of INGN 241, a nonreplicating adenovector expressing the melanoma-differentiation associated gene-7 (mda-7/IL24): biologic outcome in advanced cancer patients. Mol Ther. 2005;11(1):160–72. doi: 10.1016/j.ymthe.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 75.Fisher PB, Sarkar D, Lebedeva IV, Emdad L, Gupta P, Sauane M, Su ZZ, Grant S, Dent P, Curiel DT, Senzer N, Nemunaitis J. Melanoma differentiation associated gene-7/interleukin-24 (mda-7/IL-24): novel gene therapeutic for metastatic melanoma. Toxicol Appl Pharmacol. 2007;224(3):300–7. doi: 10.1016/j.taap.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lebedeva IV, Emdad L, Su Z-Z, Gupta P, Sauane M, Sarkar D, Staudt MR, Liu SJ, Taher MM, Xiao R, Barral P, Lee S-G, Wang D, Vozhilla N, Park ES, Chatman L, Boukerche H, Ramesh R, Inoue S, Chada S, Li R, De Pass AL, Mahasreshti PJ, Dmitriev IP, Curiel DT, Yacoub A, Grant S, Dent P, Senzer N, Nemunaitis JJ, Fisher PB. mda-7/IL-24, novel anticancer cytokine: focus on bystander antitumor, radiosensitization and antiangiogenic properties and overview of the phase I clinical experience (Review) Int J Oncol. 2007;31(5):985–1007. [PubMed] [Google Scholar]
- 77.Eager R, Harle L, Nemunaitis J. Ad-MDA-7; INGN 241: a review of preclinical and clinical experience. Expert Opin Biol Ther. 2008;8(10):1633–43. doi: 10.1517/14712598.8.10.1633. [DOI] [PubMed] [Google Scholar]
- 78.Greco A, Di Benedetto A, Howard CM, Kelly S, Nande R, Dementieva Y, Miranda M, Brunetti A, Salvatore M, Claudio L, Sarkar D, Dent P, Curiel DT, Fisher PB, Claudio PP. Eradication of therapy-resistant human prostate tumors using an ultrasound-guided site-specific cancer terminator virus delivery approach. Mol Ther. 2010;18(2):295–306. doi: 10.1038/mt.2009.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fisher PB, Babiss LE, Weinstein IB, Ginsberg HS. Analysis of type 5 adenovirus transformation with a cloned rat embryo cell line (CREF) Proc Natl Acad Sci USA. 1982;79(11):3527–31. doi: 10.1073/pnas.79.11.3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Su ZZ, Austin VN, Zimmer SG, Fisher PB. Defining the critical gene expression changes associated with expression and suppression of the tumorigenic and metastatic phenotype in Ha-ras-transformed cloned rat embryo fibroblast cells. Oncogene. 1993;8(5):1211–9. [PubMed] [Google Scholar]
- 81.Su ZZ, Lin J, Shen R, Fisher PE, Goldstein NI, Fisher PB. Surface-epitope masking and expression cloning identifies the human prostate carcinoma tumor antigen gene PCTA-1 a member of the galectin gene family. Proc Natl Acad Sci USA. 1996;93(14):7252–7. doi: 10.1073/pnas.93.14.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Emdad L, Lee SG, Su ZZ, Jeon HY, Boukerche H, Sarkar D, Fisher PB. Astrocyte elevated gene-1 (AEG-1) functions as an oncogene and regulates angiogenesis. Proc Natl Acad Sci US A. 2009;106(50):21300–5. doi: 10.1073/pnas.0910936106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Su Z, Shi Y, Friedman R, Qiao L, McKinstry R, Hinman D, Dent P, Fisher PB. PEA3 sites within the progression elevated gene-3 (PEG-3) promoter and mitogen-activated protein kinase contribute to differential PEG-3 expression in Ha-ras and v-raf oncogene transformed rat embryo cells. Nucleic Acids Res. 2001;29(8):1661–71. doi: 10.1093/nar/29.8.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Su ZZ, Lee SG, Emdad L, Lebdeva IV, Gupta P, Valerie K, Sarkar D, Fisher PB. Cloning and characterization of SARI (suppressor of AP-1, regulated by IFN) Proc Natl Acad Sci US A. 2008;105(52):20906–11. doi: 10.1073/pnas.0807975106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dash R, Richards JE, Su ZZ, Bhutia SK, Azab B, Rahmani M, Dasmahapatra G, Yacoub A, Dent P, Dmitriev IP, Curiel DT, Grant S, Pellecchia M, Reed JC, Sarkar D, Fisher PB. Mechanism by which Mcl-1 regulates cancer-specific apoptosis triggered by mda-7/IL-24, an IL-10-related cytokine. Cancer Res. 2010;70(12):5034–45. doi: 10.1158/0008-5472.CAN-10-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bhutia SK, Dash R, Das SK, Azab B, Su ZZ, Lee SG, Grant S, Yacoub A, Dent P, Curiel DT, Sarkar D, Fisher PB. Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer Res. 2010;70(9):3667–76. doi: 10.1158/0008-5472.CAN-09-3647. [DOI] [PMC free article] [PubMed] [Google Scholar]