

Abstract
While accurate measures of heritability are needed to understand the pharmacogenetic basis of drug treatment response, these are generally not available, since it is unfeasible to give medications to individuals for which treatment is not indicated. Using a polygenic linear mixed modeling approach, we estimated lower-bounds on asthma heritability and the heritability of two related drug-response phenotypes, bronchodilator response and airway hyperreactivity, using genome-wide SNP data from existing asthma cohorts. Our estimate of the heritability for bronchodilator response is 28.5% (se 16%, p = 0.043) and airway hyperresponsiveness is 51.1% (se 34%, p = 0.064), while we estimate asthma genetic liability at 61.5% (se 16%, p < 0.001). Our results agree with previously published estimates of the heritability of these traits, suggesting that the LMM method is useful for computing the heritability of other pharmacogenetic traits. Furthermore, our results indicate that multiple SNP main-effects, including SNPs as yet unidentified by GWAS methods, together explain a sizable portion of the heritability of these traits.
Keywords: Asthma, Pharmacogenetics, Heritability, Bronchodilator Response, Airway Hyperresponsiveness
Main Text
The extent to which genomic medicine is applicable to a disease or drug-response phenotype is limited by the genetic architecture and heritability of the traits, and is thus of paramount importance to pharmacogenetics. Such heritability estimates are generally not available for pharmacological responses, since it is unfeasible to give medications to individuals for which treatment is not indicated in family studies. Herein, we employed a recent method for estimating heritability from genome-wide SNP data, polygenic linear mixed modeling (LMM) using genome-wide complex trait analysis (GCTA),1, 2 and we applied these approaches to analysis of asthma and two asthma-related pharmacological phenotypes for which there are existing heritability estimates: bronchodilator response (BDR) and airway hyperresponsiveness (AHR).
Bronchodilators, of which short-acting beta2-agonists are most common, are the most frequently used asthma drug.3 However, it has long been observed that patients have variable responses to these medications.4 BDR, measured by the change in forced expiratory volume in 1 second (FEV1) before and after beta2-agonist administration, has a significant genetic component contributing to the variable bronchodilator response in different patients.5
AHR refers to the characteristic airway obstruction produced by a variety of stimuli in asthmatics.6 AHR is assessed by provocative concentration of methacholine producing a 20% decline in forced expiratory volume in one second (PC20). AHR is the phenotype that changes the most with steroid (anti-inflammatory) treatment7 and has also been shown to be highly heritable across a number of studies and populations.8, 9
Using LMM, estimates of the proportion of phenotypic variance attributable to additive genetic effects,2 or narrow-sense heritability (h2), can be established independent of familial data. Narrow-sense heritability is a subset of the total, or broad-sense, heritability (h2). Heritability measured by LMM (h2) does not include other types of genetic effects such as dominance effects, gene-environment interaction, and gene-gene or variant-variant effects.10 The LMM method looks for variance explained by SNPs in aggregate assuming that many SNPs may be associated with the trait of interest, albeit with effect sizes small enough that they would not be detected in traditional association studies. It is, in effect, answering the question: if we could find every associated SNP, how much heritability will all those genetic effects explain?
This method has been used previously to argue that the heritability of height is almost entirely due to additive effects.2 It has further been applied to metabolic syndrome traits,11 where lower-bounds were given for BMI (15%), and systolic blood pressure (24%). The method has also shown one third of the individual variation in liability to schizophrenia (23% of 70%) is due to additive SNP effects.12 To date, however, this method has not been applied to the study of drug treatment responses, which, in contrast to studies of baseline quantitative traits or affection status, are not typically amenable to classic familial studies of heritability. We hypothesized that LMM would provide reliable heritability estimates of pharmacogenetic and affection traits in asthma. We tested our hypothesis on the BDR, AHR, and affection phenotypes, which permitted correlation of the LMM results with previously published heritability estimates for these traits.
Methods
We utilized subjects from various asthma clinical trials: ACRN,13 CAMP,14 CARE,15 LOCCS,16 LODO,17 and Sepracor.5 These have been described in detail in the provided references. To assess the heritability of asthma, we utilized Caucasian participants of ACRN, CAMP, and CARE, together with publicly available Caucasian controls from iControlDB (Illumina Inc, San Diego, CA). For BDR, we utilized participants from ACRN, CAMP, CARE, LOCCS, LODO, and Sepracor. The AHR cohort was composed of patients from ACRN, CAMP, and CARE. These cohorts are described in more detail in Table 1. These cohorts have elsewhere been used successfully in traditional genome-wide association analyses for asthma18, BDR19, and AHR (forthcoming).
Table 1.
Asthma trials combined for heritability analysis. Sample size is given per-phenotype, per-trial. Samples sizes are post-quality control. Genotyping for the LOCCS, LODO, and Sepracor studies was performed at Riken (Kanagawa, Japan) on Infinium HD Human610-Quad BeadChip chips (Illumina). Genotyping for CAMP was performed at the Channing Division of Network Medicine (Boston, MA) on Infinium Human610 and HumanHap550 BeadChip (Illumina). Genotyping of ACRN and CARE on Affymetrix 1M human hap chips was performed by Affymetrix Inc. (Santa Clara, CA). Quality control was performed at the Channing. Imputation for the BDR cohort was done vs. the 1000 Genomes Project (June 2010) using the Markov Chain Haplotyping (MaCH) software program.20
| Trial | Full Name | Criteria | Genotyping | Cohort (n) | Reference |
|---|---|---|---|---|---|
| ACRN | Asthma Clinical Research Network | Adult | Affymetrix 6.0, Affymetrix. | BDR (217) AHR (355) | Denlinger LC, Sorkness CA, Chinchilli VM, Lemanske RF, Jr. Guideline-defining asthma clinical trials of the National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network and Childhood Asthma Research and Education Network. J Allergy Clin Immunol 2007;119:3–11; quiz 2–3. |
| CAMP | Childhood Asthma Management Program | Children, 5–12 yr old. | Illumina HumanHap550v3 or Infinium HD Human610-Quad BeadChip, Channing. | BDR (521) AHR (513) Asthma (581) | The Childhood Asthma Management Program (CAMP): design, rationale, and methods. Childhood Asthma Management Program Research Group. Control Clin Trials 1999;20:91–120. |
| CARE | Childhood Asthma Research and Education Network | Children, 2–17 yr old. | Affymetrix 6.0, Affymetrix. | BDR (166) AHR (186) | Guilbert TW, Morgan WJ, Krawiec M, et al. The Prevention of Early Asthma in Kids study: design, rationale and methods for the Childhood Asthma Research and Education network. Control Clin Trials 2004;25:286–310. |
| LOCCS | Leukotriene Modifier or Corticosteroid or Corticosteroid-Salmeterol Trial | Adult | Illumina Infinium HD Human610-Quad BeadChip, RIKEN. | BDR (114) Asthma (132) | Peters SP, Anthonisen N, Castro M, et al. Randomized comparison of strategies for reducing treatment in mild persistent asthma. N Engl J Med 2007;356:2027–39. |
| LODO | Effectiveness of Low Dose Theophylline as an Add-on Treatment in Asthma | Adult | BDR (105) Asthma (121) | American Lung Association Asthma Clinical Research Centers. Clinical trial of low-dose theophylline and montelukast in patients with poorly controlled asthma. Am J Respir Crit Care Med 2007;175:235–42. | |
| Sepracor | Sepracor | BDR > 15%, Adult | BDR (375) Asthma (432) | Baron RM, Palmer LJ, Tantisira K, et al. DNA sequence variants in epithelium-specific ETS-2 and ETS-3 are not associated with asthma. American journal of respiratory and critical care medicine 2002;166:927–32. | |
| Illumina | Illumina Controls | Illumina HumanHap550v3, Illumina. | Asthma (1367) | Illumina Inc, San Diego, CA |
BDR was measured by FEV1 change before and after the administration of a bronchodilator (albuterol); where BDR was defined as the change in FEV1 as a percentage of the pre-bronchodilator FEV1. AHR was assessed by methacholine challenge and measured by the log provocative concentration of methacholine resulting in a 20% decrease in airway capacity (measured by FEV1). In the asthma case-control study, asthma cases were defined as any participant in an asthma trial and controls were defined as any of the patients from iControlDB.
Heritability computations with the LMM method were performed using the GCTA software tool.1 Following the guidelines of Yang et al.,2 we excluded SNPs with minor allele frequency < 0.05, missing rate > 0.005, p-value for being outside Hardy-Weinberg equilibrium < 0.001, MaCH20 imputation r2 quality scores < 0.9, and a genomic relatedness score > 0.025. The genomic relatedness filter removed one of each pair of participants with more than 2.5% genomic similarity, which corresponds to removing anyone related as closely as (approximately) 3rd or 4th cousins.21 We included age, sex, height, study and the top 20 genotype principle components as covariates in the LMM model. Although genotyping was performed on each cohort separately, in many cases on separate chips and in separate laboratories (table 1), the inclusion of study and genotype principal components resulted in stratification-free analysis, as evidenced by a genomic inflation factor of 1.017 in a qq-plot of association with asthma affection status in the case-control analysis. Including the top 20 principal components as covariates removes any variance explained by the population structure they might represent from the estimate of heritability.21 Since heritability of case-control traits depends on the prevalence, GCTA measures heritability on an underlying liability scale, where attributable genetic risk has a normal distribution.22 To adjust the estimate of asthma heritability to the liability scale, we assumed a prevalence of 10% for asthma. P-values for statistical significance are reported from GCTA output, and for heritability measurements represent probability of difference from zero.
Results
The cohorts utilized in this analysis are summarized in Table 1. To increase the quality of our LMM estimates, we combined our study cohorts for each phenotype, yielding a total sample size of 2633 for asthma affection status, 1497 for bronchodilator response, and 1054 for AHR.
We computed narrow-sense heritability estimates for asthma and two pharmacogenetic phenotypes: BDR and AHR.1 The estimated heritability of asthma was 61.5% (se 16%, p < 0.001). BDR was 28.6% (se 16%, p = 0.043) and AHR was 51.1% (se 34%, p = 0.064). Results are summarized in Table 2. We next compared our results to published heritability results for each of these three traits (Table 2). Overall, the GTCA results reflected a minimum of 67–76% of the published heritability estimates.
Table 2.
Estimated contributions to heritability of SNP variants in Asthma and two related drug-response phenotypes. Heritability estimated using polygenic linear mixed modeling. Standard error is the error associated with the heritability estimate. Percent explained is the percentage of the published heritability estimates represented by the heritability.
| Sample Size (n) | Heritability (h2) | Standard Error | p – value | Published Heritability | % Explained | |
|---|---|---|---|---|---|---|
| Asthma | 2633 | 61.5% | 16% | < .001 | 70%–90% | 68%–88% |
| BDR | 1497 | 28.6% | 16% | 0.043 | 10%–40% | 72%–286% |
| AHR | 1054 | 51.1% | 34% | 0.064 | 40%–67% | 76%–128% |
Even though asthma is known to have different prevalences among child and adult cases, we were not able to conduct separate heritability estimates without reducing our sample size below the point of usable results. For purposes of comparison, we did alter the estimate of asthma prevalence used in the GCTA method (10%), and arrived at somewhat different results: assuming a 5% prevalence gives a heritability estimate of 49.6% (se 13%), and assuming a 15% prevalence gives 69.9% (se 18%). We also compared heritability estimates for each phenotype when including differing numbers of genotype principal components as covariates (figure 1). This largely resulted in similar estimates of heritability, although including too few (less than 10) or too many PCs (more than 35) could lead to poorer results: as either stratification and batch effects are not sufficiently adjusted for, or too many PCs siphon away increasing amounts of the true genetic variance. That the estimates for AHR seem to be most variable for different numbers of PC covariates is probably due to that phenotype having the smallest sample size and consequently the largest error associated with its heritability estimates.
Figure 1.
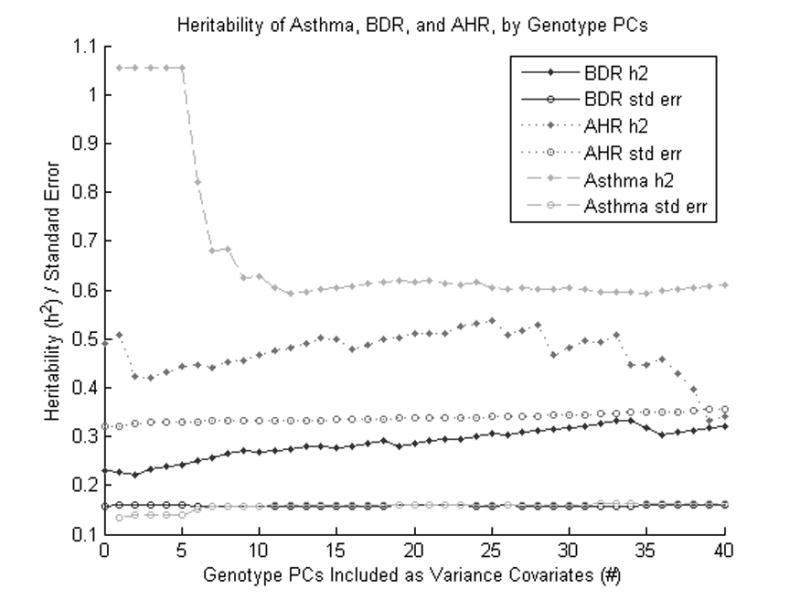
Heritability of Asthma, BDR, and AHR by different numbers of genotype principal components included. Heritability values above 1.0 represent model convergence failures, and do not represent stable estimates of true heritability. This figure shows that the heritability estimates of each phenotype can vary with the number of included genotype PCs, but remain mostly stable for a range of PCs between 10 and 35.
Discussion
Overall, the heritability estimates we obtained using LMM were very consistent with estimates established by pedigree studies, with each of the results reflecting a minimum of 67–76% of the published heritability estimates. The estimates are based on the coverage of the genome provided by the SNP array employed, and thus represent a lower-bound on the narrow-sense heritability (h2). As such, this method is a valid approach for estimating heritability from GWAS data in studies in which familial data is unavailable, or unfeasible, such as pharmacogenetic studies. However, it does not include other types of additive genetic effects which may not be in linkage disequilibrium with the SNP array used, including rare alleles and alleles with very-low minor allele frequency.21
The heritability of asthma has been assessed in a variety of twin studies, the majority of which have estimated heritability between 70% and 90%,23 a value approached by our estimate of 61%. This indicates that a substantial portion of the total variance in individual liability to asthma is due to the additive effects of common variants, a result consistent with the theory of asthma as a complex polygenic disorder. Some researchers have included as covariates in the LMM models SNPs representing loci known to account for significant portions of the heritability of the phenotype, and thus measured the heritability of genomic variants uncorrelated with these loci.24 In our analysis, we included 13 SNPs robustly associated with asthma from recent GWAS meta-studies: GABRIEL25 and EVE.26 This did not result in a significant reduction in heritability of asthma (p = 0.5, compared with 13 random SNPs), indicating that these 13 variants do not by themselves constitute a significant fraction of the genomic liability of asthma.
There have been a few previous studies that measured the heritability of AHR or BDR. Two variance component analyses looked at the heritability of AHR in pedigree studies, obtaining estimates between 50% and 65%,8, 9 while two similar studies of BDR found estimates of heritability of 10% and 10.5%.27, 28 Others have computed the intraclass correlation coefficient (ICC) of AHR and BDR, where the ICC is a ratio of the within-subject variance to the total variance, and represents an upper-bound on the broad-sense heritability (H2). ICCs of 67%29 and 40%30 for AHR_ENREF_24 have been reported, as well as 40%29 for BDR. Another study measured the familial correlation of BDR at between 9% and 16%,31 corresponding to a heritability estimation between 12.7% and 22.6%.
Since the heritability of BDR and AHR are much lower than that of asthma itself, and certain ranges of BDR are frequently used as inclusion criteria for clinical trials and genetic studies, it is possible that important sources of genetic determinants of asthma are missed in such studies. Similarly, since the heritabilities of AHR and BDR are substantially different, this suggests that the biological underpinnings of airway constriction and dilation may be quite different, even though they are sometimes conflated.
Furthermore, our estimates include only heritability due to additive SNP effects. The fact that these numbers are quite close to the heritability estimates established through other means indicates that a significant portion of the genetic liability of these conditions is due to additive effects of simple, common variants. Thus future interrogation of GWAS data related to BDR, AHR, and asthma itself should turn up increasingly more variants associated with these conditions as those GWAS become increasingly well-powered to detect variants with small effects. However, it is likely that foreseeable increases in GWAS sample size will not be sufficient to find all the associated variants, particularly those of smallest effect, and that greater methodological advancements will be required to move the field forward.
In conclusion, our work further establishes narrow-sense heritability estimates for BDR and AHR, two drug-response phenotypes of interest in asthma treatment and diagnosis, as well as for heritability of asthma itself. Application of this method to pharmacogenetic phenotypes will enhance the ability to better appraise the relative value of individual pharmacogenetic variants to the overall response profile and subsequently improve the ability to formulate predictive pharmacogenomic models.
Acknowledgments
We thank all CAMP subjects for their ongoing participation in this study. We acknowledge the CAMP investigators and research team, supported by the National Heart, Lung and Blood Institute (NHLBI), for collection of CAMP Genetic Ancillary Study data. All work on data collected from the CAMP Genetic Ancillary Study was conducted at the Channing Division of Network Medicine of the Brigham and Women’s Hospital under appropriate CAMP policies and human subject’s protections. We thank the American Lung Association Asthma Clinical Research Centers (ALA-ACRC) for use of LOCCS and LODO study samples. We acknowledge and thank the participants and executors of the asthma trials: ACRN, CARE, LOCCS, LODO, and Sepracor. The help of lab members at RIKEN and Channing Division of Network Medicine in providing accurate genotyping across these trials is very much appreciated.
Funding
This research was funded by NIH grants NIH U01 HL065899, R01 NR013391, R01 HL090197, K99 HL105663 and by P-STAR, a component of NIH U19 HL065926, the Pharmacogenomics Research Network (PGRN). Additional funding provided by the Pharmacogenomics Research Network (PGRN) – RIKEN Center for Genomic Medicine (CGM) Global Alliance and by funding from the BioBank Japan project that was supported by the Ministry of Education, Culture, Sports, Sciences and Technology of the Japanese government. The Childhood Asthma Management Program (CAMP) Genetics Ancillary Study is supported by U01 HL075419, P01 HL083069, R01 HL086601, and T32 HL07427 from the NHLBI, NIH. Dr Himes was supported by NIH K99 HL105663.
Footnotes
Conflict of Interest
Dr. Peters has served as a consultant for AstraZeneca, Aerocrine, Airsonett AB, Delmedica, GlaxoSmithKline, Merck, and TEVA, and is a member of Speakers’ Bureaus sponsored by Integrity Continuing Education and Merck, and is a consultant to the ALA-ACRC’s DCC and was PI of its LOCCS trial. Dr. Lima has received a $75000 research grant from Merck. Drs Himes, Irvin, McGeachie, Pendergrass, Plenge, Ritchie, Stahl, and Tantisira declare no competing financial interests.
References
- 1.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. American journal of human genetics. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nature genetics. 2010;42:565–9. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raby BA, Weiss ST. Beta2-adrenergic receptor genetics. Current opinion in molecular therapeutics. 2001;3:554–66. [PubMed] [Google Scholar]
- 4.Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baron RM, Palmer LJ, Tantisira K, et al. DNA sequence variants in epithelium-specific ETS-2 and ETS-3 are not associated with asthma. American journal of respiratory and critical care medicine. 2002;166:927–32. doi: 10.1164/rccm.200201-048OC. [DOI] [PubMed] [Google Scholar]
- 6.Bauer S, Park HN, Seo HS, et al. Assessment of bronchodilator responsiveness following methacholine-induced bronchoconstriction in children with asthma. Allergy Asthma Immunol Res. 2011;3:245–50. doi: 10.4168/aair.2011.3.4.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long-term effects of budesonide or nedocromil in children with asthma. The Childhood Asthma Management Program Research Group. N Engl J Med. 2000;343:1054–63. doi: 10.1056/NEJM200010123431501. [DOI] [PubMed] [Google Scholar]
- 8.Celedon JC, Soto-Quiros ME, Avila L, et al. Significant linkage to airway responsiveness on chromosome 12q24 in families of children with asthma in Costa Rica. Human genetics. 2007;120:691–9. doi: 10.1007/s00439-006-0255-5. [DOI] [PubMed] [Google Scholar]
- 9.Ferreira MA, O’Gorman L, Le Souef P, et al. Variance components analyses of multiple asthma traits in a large sample of Australian families ascertained through a twin proband. Allergy. 2006;61:245–53. doi: 10.1111/j.1398-9995.2005.00954.x. [DOI] [PubMed] [Google Scholar]
- 10.Visscher PM, Hill WG, Wray NR. Heritability in the genomics era--concepts and misconceptions. Nature reviews Genetics. 2008;9:255–66. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- 11.Vattikuti S, Guo J, Chow CC. Heritability and Genetic Correlations Explained by Common SNPs for Metabolic Syndrome Traits. PLoS genetics. 2012;8:e1002637. doi: 10.1371/journal.pgen.1002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SH, DeCandia TR, Ripke S, et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nature genetics. 2012;44:247–50. doi: 10.1038/ng.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denlinger LC, Sorkness CA, Chinchilli VM, Lemanske RF., Jr Guideline-defining asthma clinical trials of the National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network and Childhood Asthma Research and Education Network. J Allergy Clin Immunol. 2007;119:3–11. doi: 10.1016/j.jaci.2006.10.015. quiz 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Childhood Asthma Management Program (CAMP): design, rationale, and methods. Childhood Asthma Management Program Research Group. Control Clin Trials. 1999;20:91–120. [PubMed] [Google Scholar]
- 15.Guilbert TW, Morgan WJ, Krawiec M, et al. The Prevention of Early Asthma in Kids study: design, rationale and methods for the Childhood Asthma Research and Education network. Control Clin Trials. 2004;25:286–310. doi: 10.1016/j.cct.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Peters SP, Anthonisen N, Castro M, et al. Randomized comparison of strategies for reducing treatment in mild persistent asthma. N Engl J Med. 2007;356:2027–39. doi: 10.1056/NEJMoa070013. [DOI] [PubMed] [Google Scholar]
- 17.American Lung Association Asthma Clinical Research Centers. Clinical trial of low-dose theophylline and montelukast in patients with poorly controlled asthma. Am J Respir Crit Care Med. 2007;175:235–42. doi: 10.1164/rccm.200603-416OC. [DOI] [PubMed] [Google Scholar]
- 18.Himes BE, Hunninghake GM, Baurley JW, et al. Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am J Hum Genet. 2009;84:581–93. doi: 10.1016/j.ajhg.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Himes BE, Jiang X, Hu R, et al. Genome-wide association analysis in asthma subjects identifies SPATS2L as a novel bronchodilator response gene. PLoS genetics. 2012;8:e1002824. doi: 10.1371/journal.pgen.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Visscher PM, Yang J, Goddard ME. A commentary on ‘common SNPs explain a large proportion of the heritability for human height’ by Yang et al. (2010) Twin research and human genetics : the official journal of the International Society for Twin Studies. 2010;13:517–24. doi: 10.1375/twin.13.6.517. [DOI] [PubMed] [Google Scholar]
- 22.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. American journal of human genetics. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomsen SF, van der Sluis S, Kyvik KO, Skytthe A, Backer V. Estimates of asthma heritability in a large twin sample. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2010;40:1054–61. doi: 10.1111/j.1365-2222.2010.03525.x. [DOI] [PubMed] [Google Scholar]
- 24.Stahl EA, Wegmann D, Trynka G, et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nature genetics. 2012;44:483–9. doi: 10.1038/ng.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moffatt MF, Gut IG, Demenais F, et al. A large-scale, consortium-based genomewide association study of asthma. The New England journal of medicine. 2010;363:1211–21. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torgerson DG, Ampleford EJ, Chiu GY, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nature genetics. 2011;43:887–92. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palmer LJ, Celedon JC, Chapman HA, Speizer FE, Weiss ST, Silverman EK. Genome-wide linkage analysis of bronchodilator responsiveness and post-bronchodilator spirometric phenotypes in chronic obstructive pulmonary disease. Human molecular genetics. 2003;12:1199–210. doi: 10.1093/hmg/ddg125. [DOI] [PubMed] [Google Scholar]
- 28.Hersh CP, Soto-Quiros ME, Avila L, et al. Genome-wide linkage analysis of pulmonary function in families of children with asthma in Costa Rica. Thorax. 2007;62:224–30. doi: 10.1136/thx.2006.067934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu AC, Tantisira K, Li L, Schuemann B, Weiss S. Repeatability of response to asthma medications. The Journal of allergy and clinical immunology. 2009;123:385–90. doi: 10.1016/j.jaci.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rijcken B, Schouten JP, Weiss ST, Rosner B, De Vries K, Van der Lende R. Long-term variability of bronchial responsiveness to histamine in a random population sample of adults. The American review of respiratory disease. 1993;148:944–9. doi: 10.1164/ajrccm/148.4_Pt_1.944. [DOI] [PubMed] [Google Scholar]
- 31.Niu T, Rogus JJ, Chen C, et al. Familial aggregation of bronchodilator response: a community-based study. American journal of respiratory and critical care medicine. 2000;162:1833–7. doi: 10.1164/ajrccm.162.5.9908127. [DOI] [PubMed] [Google Scholar]