

Abstract
In investigations aimed at exploring the potential of disubstituted allenes in stereoselective synthesis, we report studies that explore the reductive cross-coupling reaction of vinylsilanes with a range of substituted allenes. Regiochemical control is attained by employing allenic alkoxides, where the proximal heteroatom dictates the site-selectivity in a process that proceeds by net formal metallo-[3,3] rearrangement (directed carbometalation/elimination). Stereoselectivity in these reactions is complex, with both the nature of allene substitution and relative stereochemistry of the substrate impacting the stereoselective generation of each alkene of a substituted 1,3-diene. 2009 Elsevier Ltd. All rights reserved.
Keywords: Allenes, Titanium, Reductive Cross-Coupling, Z-dienes
1. Introduction
Reductive cross-coupling is emerging as a useful strategy for bimolecular C–C bond formation.1 While a great many reports continue to appear for the coupling of substituted alkynes with carbonyl-based reaction partners,2 studies that describe the related bimolecular union of coupling partners that diverge from these well-studied processes are rare.3 In a program aimed at the elucidation of reductive cross-coupling reactions between alkynes, allenes and alkenes, we have put forth a variety of heteroatom-directed reaction designs to accomplish selective cross-coupling.4 These designs, which vary by the sequence of steps that result in encapsulation of the metal during the metal-centered [2+2+1] process, have proven to be useful for: 1) control of hetero- vs. homo-coupling, 2) control of regioselectivity, 3) control of relative stereochemistry, and in some cases, 4) provide a means of overcoming the typical sluggish reactivity of highly substituted substrates in reductive cross-coupling chemistry (Figure 1).5
Figure 1.

Recent examples of heteroatom-directed reductive cross-coupling reactions of alkynes and alkenes.
In the course of a recent campaign in total synthesis, we experienced a great deal of difficulty associated with the efficient preparation of a trisubstituted (Z,E)-1,3-diene (Figure 2).6 While we were able to forge the C7-C8 sigma bond of callystatin A by application of well-known palladium-catalyzed coupling chemistry,7 the synthesis of the stereodefined trisubstituted vinyl iodide 1 was quite challenging. Initial explorations relied on multistep functionalization of a dibromo-olefin that proceeded through conversion to a TMS-alkyne, hydrozirconation, iodination, and Pd-catalyzed ethylation.8 In an effort to increase step-economy for the synthesis of 1, we ultimately pursued Wittig chemistry between an α-chrial aldehyde and an iodoethyl-substituted ylid. While this revised route provided a concise entry to the desired vinyliodide, stereoselectivity for the Wittig olefination was 1.7:1, favoring the formation of the undesired isomer.6, 9 Fortunately, chromatographic separation of these isomers was possible and a total synthesis of callystatin A was accomplished in 11 steps from a commercially available chiral alcohol. The difficulty in establishing the C6-C9 1,3-(Z,E)-diene in callystatin prompted us to develop an alternative pathway to establish this type of stereodefined motif. Previous studies in our laboratory had revealed that alkoxide-directed reductive cross-coupling of disubstituted allenes with internal alkynes defined a regio- and stereoselective pathway to cross-conjugated trienes (4 + 5 → 6; Figure 3).4i We speculated that a related reductive cross-coupling reaction between substituted allenes and terminal alkenes could provide a useful pathway to trisubstituted (Z,E)-dienes (7 + 8 → 9) of the substitution pattern seen in C6-C9 of callystatin A. Here, we discuss our studies of this proposed process, and present the scope and limitations of a stereoselective reductive cross-coupling reaction between disubstituted allenes and vinylsilanes for the synthesis of (Z)-olefin-containing 1,3-dienes.
Figure 2.
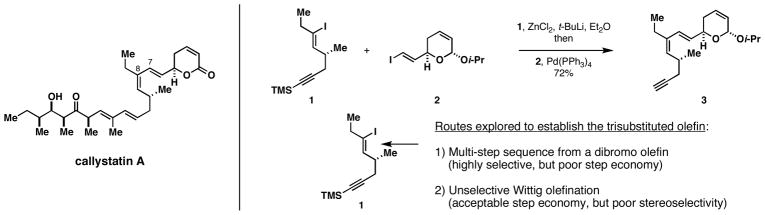
Difficulties experienced in establishing the C6-C9-(Z,E)-diene of callystatin A.
Figure 3.

Allene-alkene reductive cross-coupling for the synthesis of Z-dienes.
2. Results and Discussion
Exploring the utility of a reaction design based on stepwise encapsulation of the metal center in a reductive cross-coupling reaction of allenes with alkenes, we initiated studies of this process with vinyltrimethylsilane – a simple monosubstituted alkene that is well known to be a substrate for Ti(IV) alkoxide-mediated coupling reactions.10 As illustrated in equations 1 and 2 of Figure 4, the basic bond construction proved feasible. Reaction of the isomeric allenic alcohols 10 and 12 with a preformed Ti-complex of vinyltrimethylsilane resulted in formation of the stereo-undefined trisubstituted 1,3-dienes 11 and 13 in 51 and 53% yield. In addition to validating the proposed coupling process, equation 2 demonstrated that this allene-alkene union is effective for establishing a tetrasubstituted alkene.
Figure 4.

Exploring the basic bond construction: Reductive cross-coupling between substituted allenes and vinyltrimethylsilane.
Next, in a brief study, the structural requirements for (Z)-selectivity were examined. Reductive cross-coupling of disubstituted allenes that contain an unbranched substituent distal to the hydroxymethyl group (14 and 16; eqs 3 and 4) proved to be relatively poor substrates for this process. While these coupling reactions proceeded with acceptable conversions (51-56% yield), the resulting dienes (15 and 17) were formed with only moderate levels of selectivity (Z:E = 3:1). In accord with previous observations made in our study of allene–imine4k and allene–alkyne4h–i reductive cross-coupling reactions, (Z)-selectivity is markedly enhanced with substrates that contain branched alkyl substituents distal to the hydroxymethyl group. As illustrated in eqs 5 and 6, the isopropyl- and cyclohexyl-substituted allenes 18 and 20 were converted to the dienes 19 and 21 in 50 and 53% yield, each with ≥ 20:1 selectivity for the formation of the (Z)-diene product.
While we were pleased with the success of our initial investigation, we recognized that the tetraalkylsilane products derived from this coupling process are of limited general synthetic utility. As such, we briefly explored the potential of employing this (Z)-selective coupling reaction for the union of allenes with other coupling partners. Coupling of allene 16 with styrene was successful, but led to the production of the (Z)-diene 23 in 52% yield (Figure 5).11 As would be anticipated from our earlier investigation of allene 16, this coupling reaction proceeded with low levels of stereoselectivity (Z:E = 2:1). Unfortunately, all attempts to accomplish this reductive cross-coupling with simple terminal alkenes was met with failure; in all cases resulting in the formation of complex product mixtures.
Figure 5.

Reductive cross-coupling reactions of allenyl alcohols with styrene and chlorodimethylvinylsilane.
Inspired by the Kulinkovich reaction,12 we hoped that we could employ EtMgBr as a coupling partner in this (Z)-diene-forming coupling reaction. Based on the presumed mechanistic course of the Kulinkovich reaction, that is thought to proceed by the conversion of EtMgBr to a Ti–ethylene complex,12b coupling with allenyl alcohols should result in a net stereoselective ethylation.13 Such a process would define a direct approach to the synthesis of the (Z,E)-diene of callystatin A. Unfortunately, all attempts to accomplish this transformation were also unsuccessful and led to the formation of complex product mixtures.14
Moving on, we refocused our attention on an inexpensive and readily available vinylsilane that, when coupled to substituted allenes would provide a product that could be easily functionalized. This concept was validated as described in equation 8 (Figure 5). Here, coupling of the tertiary alcohol 10 with vinyldimethylchlorosilane 24, followed by oxidation of the σC–Si, provided the primary carbinol 25 in 51% yield (over two steps). Illustrated in equations 9 and 10, a two-step process of reductive cross-coupling and oxidation delivered the stereodefined (Z)-diene-containing primary alcohols 26 and 28 from the racemic allenes 20 and 27. In each case, the (Z)-diene product was formed with ≥ 20:1 stereoelectivity.
Returning to our initial problem, defined by the goal of designing a reductive cross-coupling reaction for the establishment of each stereodefined alkene of a (Z,E)-diene, we questioned whether the present allene–vinylsilane coupling reaction proceeds in a stereospecific fashion (Figure 6). If ligand exchange is a prerequisite for carbometalation (A → B → C) in this reductive cross-coupling reaction, the conversion of preformed metallacyclopropanes (A) to 1,3-diene products was anticipated to follow from a precise sequence of steps whereby the (Z)-trisubstituted alkene would derive from stereoselective carbometalation anti to R2 (B → C), and the (E)-disubstituted alkene would result from stereospecific elimination (C → D).
Figure 6.
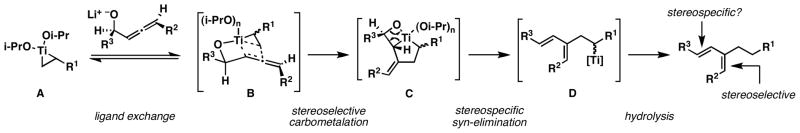
Allene-alkene reductive cross-coupling for the synthesis of (Z)-dienes.
In an effort to explore this proposal, two stereodefined allenes were prepared as described in Figure 7. The isomerically pure diols 29 and 31 were accessed from syn-dihydroxylation of the corresponding (Z)- and (E)-enynes, each prepared from Sonogashira coupling with TMS-acetylene.15 Stereoselective conversion to the isomerically defined allenes 30 and 32 followed from a two-step sequence composed of carbonate formation and SN2’ addition.16
Figure 7.

Preparation of isomeric allenes.
The coupling of allenes 30 and 32 with vinyldimethylchlorosilane is depicted in Figure 8. These reactions, performed as previously discussed in Figure 5, provided substituted 1,3-dienes 33 and 34 with varying levels of selectivity. While in each case, the (Z)-trisubstituted alkene of the products was established with high levels of selectivity (Z:E ≥ 20:1), the disubstituted alkenes of these products was accessed with only moderate levels of selectivity (E:Z = 5:1 and 1:3). This observation is consistent with a mechanistic picture composed of: 1) stereoselective syn-carbometalation, and 2) competition between syn- and anti-elimination of the organometallic intermediate.
Figure 8.

Stereoselective access to (Z,E)- and (Z,Z)-1,3-dienes by reductive cross-coupling.
If ligand exchange was a prerequisite to C–C bond formation, we would expect that the coupling reaction between allene 30 and vinyldimethylchlorosilane would deliver the (Z,E)-1,3-diene 33 with very high levels of stereochemical purity. This expectation follows from a rationale based on facile syn-elimination of the resulting bicyclic metallacyclopentane (C; Figure 8) in preference to ring opening and anti-elimination to deliver the (Z)-disubstituted alkene product.17 The appreciable quantity of (Z,Z)-diene observed (17% of the 1,3-diene product isolated) is consistent with anti-elimination by way of a stereodefined monocyclic metallacyclopentane – itself, likely derived from a competing reaction pathway that does not proceed through alkoxide-directed carbometalation. While such stereochemical erosion has not been observed in related coupling reactions of substituted allylic alcohols,4j, 4l, 4m–p the current observation may relate to the increased reactivity of allenes in bimolecular metal-mediated reductive cross-coupling.18 Overall, this enhanced reactivity (and associated competing non-directed carbometalation) may represent an inherent limitation to the use of allenic alkoxides in stereoselective reductive cross-coupling chemistry. That said, the examples described here demonstrate the potential to employ reductive cross-coupling for the stereoselective synthesis of (Z,E)- and (Z,Z)-trisubstituted 1,3-dienes from isomerically pure disubstituted allenyl alcohols.
3. Conclusion
While reductive cross-coupling chemistry is well known for the union of alkynes with polarized π-bonds (carbonyl electrophiles), it is only beginning to emerge as a useful strategy for union of more substituted and electronically unactivated π-systems (i.e. alkenes, alkynes, and allenes). In efforts directed at the application of an alkyne–alkyne reductive cross-coupling reaction in natural product synthesis, we identified a significant challenge in the establishment of the C6-C9 (Z,E)-1,3-diene of callystatin A. Here, we describe the design and investigation of a new allene–alkene reductive cross-coupling to address this problem in stereoselective synthesis. Our results lead us to conclude that the present coupling reaction provides a highly stereoselective route to the generation of 1,3-dienes that contain a (Z)-trisubstituted alkene. While our data reveals that this coupling process does not proceed in a stereospecific fashion, we have discovered a reductive cross-coupling reaction that forges a central C–C bond in concert with the establishment of two stereodefined alkenes.
4. Experimental section
4.1. General experimental information
All reactions were conducted in flame-dried glassware under nitrogen or argon using anhydrous solvents. Toluene, tetrahydrofuran, and diethyl ether were used after passing through an activated alumina column. Ti(Oi-Pr)4 was used after distillation of the commercially available reagent. All other commercially available reagents were used as received (Aldrich). 1H NMR data were recorded at 400 MHz on a Bruker AM-400 in CDCl3. 13C NMR data were recorded at 100 MHz on a Bruker AM-400. Infrared spectra were recorded on a PerkinElmer SpectrumOne FT-IR instrument. Low resolution mass spectra were acquired on a Varian 500-MS mass spectrometer under soft ionization mode. HRMS data (ESITOF-MS) were obtained by the University of Florida Mass Spectrometry lab. 1H NMR chemical shifts are reported relative to residual CHCl3 (7.26ppm) 13C chemical shifts are reported relative to the central line of CDCl3 (77.16 ppm). Chromatographic purification was performed using 60Å, 35–75μ particle size silica gel from Silicycle. All compounds purified by chromatography were sufficiently pure for use in further experiments, unless indicated otherwise. Semi-preparative HPLC normal phase separations were performed using an HPLC system composed of two Dynamax SD-1 pumps, a Rheodyne injector and a Dynamax UV-1 absorbance detector.
4.2. Cross-coupling between substituted allenes and vinyltrimethylsilane
4.2.1. General synthesis of diene 6
To a −78 °C solution of alkene 5 (1.1 eq) in diethyl ether (0.1 M) was added ClTi(Oi-Pr)3 (1.1 eq) and c-C5H9MgCl (2.2 eq) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 4 (1 eq) in THF (0.5 M) was added n-BuLi (1.1 eq) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature before the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. The crude material was purified by column chromatography on silica gel (ethyl acetate : hexanes) to yield diene 6 as a clear oil.
4.2.2. 1-trimethylsilyl-3-methylene-5-methyl-hex-4-ene (11)
To a −78 °C solution of vinyltrimethylsilane (0.161 mL, 1.1 mmol) in 11 mL of diethyl ether (0.1 M) was added 1.1 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 1.1 mmol) and 1.1 mL of c-C5H9MgCl (2.00 M in ether, 2 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 10 (0.119 mL, 1 mmol) in 2 mL THF was added 0.440 mL of n-BuLi (2.54 M in hexanes, 1.1 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature before the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. The crude material was purified by column chromatography on silica gel (hexanes) to yield diene 11 as a clear oil (93 mg, 51%). 1H NMR (400 MHz, CDCl3) δ 5.61 (s, 1H), 4.97 (d, J = 4.8 Hz, 1H), 4.72 (d, 1H), 2.06-2.02 (m, 2H), 1.80 (s, 3H), 1.79 (s, 3H), 0.65-0.60 (m, 2H), 0.001 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 149.2, 134.9, 126.3, 111.0, 31.9, 26.8, 19.7, 15.5, −1.6; IR (thin film, NaCl) 3443, 2953, 1731, 1377, 1248, 1178, 1064, 838, 758, 691 cm−1; HRMS (EI, M+) calcd for C11H22Si, 182.1491 m/z (M); observed 183.1560 (M + H)+ m/z.
4.2.3. 1-trimethylsilyl-4-methyl-3-vinyl-pent-3-ene (13)
To a −78 °C solution of vinyltrimethylsilane (0.352 mL, 2.4 mmol) in 24 mL of diethyl ether (0.1 M) was added 2.4 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 2.4 mmol) and 2.4 mL of c-C5H9MgCl (2.00 M in ether, 4.8 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 12 (0.235 g, 2 mmol) in 2 mL of THF was added 0.880 mL of n-BuLi (2.50 M in hexanes, 2.2 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature before the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. The crude material was purified by column chromatography on silica gel (hexanes) to yield diene 13 as a clear oil (196.5 mg, 54%). 1H NMR (400 MHz, CDCl3) δ 6.71 (dd, J = 11.0, 17.4 Hz, 1H), 5.06 (d, J = 17.4 Hz, 1H), 4.96 (d, J = 11.0 Hz, 1H), 2.22-2.18 (m, 2H),1.80 (s, 3H), 1.77 (s, 3H), 0.62-0.58 (m, 2H) 0.03 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 134.4, 134.4, 130.3, 110.1, 22.8, 21.8, 21.3, 16.5, −1.7; IR (thin film, NaCl) 3444, 2954, 1731, 1464, 1383, 1248, 861, 757, 691, 466 cm−1; HRMS (EI, M+) calcd for C11H22Si, 182.1491 m/z (M); observed 183.1566 (M + H)+ m/z.
4.2.4. (Z)-1-trimethylsilyl-3-vinyl-non-3-ene (15)
To a −78 °C solution of vinyltrimethylsilane (0.190 mL, 1.3 mmol) in 15 mL of diethyl ether (0.1 M) was added 1.3 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 1.3 mmol) and 1.3 mL of c-C5H9MgCl (2.00 M in ether, 2.6 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 14 (0.165 g, 1.17 mmol) in 2 mL THF was added 0.518 mL of n-BuLi (2.50 M in hexanes, 1.3 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature before the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. Z:E selectivity was determined by 1H NMR of the crude mixture after work-up. The crude material was purified by column chromatography on silica gel (hexanes) to yield diene 15 as a clear oil (147.3 mg, 56%, Z:E 3:1). The stereochemistry of the major product was assigned by analogy to previous examples. (peaks correlating to the major Z-isomer were extracted from the spectrum of the 3:1 mixture) 1H NMR (400 MHz, CDCl3) δ 6.67 (dd, J = 10.8, 17.2 Hz, 1H), 5.43-5.37 (m, 1H), 5.17 (d, J = 17.6 Hz, 1H), 5.06 (d, J = 17.6 Hz, 1H), 2.20-2.12 (m, 2H + 2H), 1.37-1.27 (m, 2H + 2H + 2H), 0.89 (t, 3H), 0.70 (m, 2H), 0.01 (s, 9H); 13C NMR (15 Z + E) (100 MHz, CDCl3) δ 141.4, 140.2, 138.9, 133.0, 132.3, 129.8, 112.8, 110.1, 31.8, 29.8, 29.5, 28.1, 27.5, 22.8, 22.7, 16.8, 16.1, 14.3, −1.6, −1.8; IR (thin film, NaCl) 3444, 2954, 2860, 1755, 1714, 1463, 1248, 837, 757, 692 cm−1; HRMS (EI, M+) calcd for C14H28Si, 224.1960 m/z (M); observed 225.2028 (M + H)+ m/z.
4.2.5. Synthesis of allene 16
To a mixture of n-BuLi (2.51 M in hexanes, 1.1 eq) in THF (0.16 M) was added dropwise tetrahydro-2(2-propynyloxy)-2H-2-pyran (1 eq) at −78 °C under Ar. After the mixture was stirred for 1h, 5-(tert-butyldimethylsiloxy)-pentanal (1.11 eq) was added dropwise. The reaction mixture was stirred for 1 h at −78 °C, warmed to room temperature, stirred for 24 h, and added 20 mL of sat. aq. NH4Cl. The mixture was extracted with 3 × 50 mL EtOAc, dried over MgSO4, filtered, and concentrated. The crude material was purified by column chromatography on silica gel (10–15% ethyl acetate : hexanes) to yield alcohol of 16 as a clear oil. (1.451 g, 61%). To a solution of triphenylphosphine (1.3 eq) in THF (0.16 M) in a flame-dried round bottom flask at −15 °C under Ar was added diethyl azodicarboxylate (1.3 eq) and stirred for 10 min. To the above solution was then added alcohol (1 eq) and stirred for 10 min. Then o-nitrobenzenesulfonylhydrazine (1.3 eq) was added, stirred for 1 h at −15 °C, warmed to room temperature, and stirred for 17 h. Subsequently, the solution was concentrated in vacuo and was purified by column chromatography on silica gel (10% ethyl acetate : hexanes) to yield THP-protected allene of 16 as a clear oil. (1.407 g, 62%). To a solution of THP-protected allene of 16 (1 eq) in Et2O (0.1 M) was added MgBr2-Et2O (3 eq). The resulting solution was stirred at room temperature for 17 h and then added 10 mL of Et2O. The resulting mixture was washed with brine, dried over MgSO4, filtered, and concentrated. The crude material was purified by column chromatography on silica gel (10% ethyl acetate : hexanes) to yield allene 16 as a clear oil (0.424 g, 54%). 1H NMR (400 MHz, CDCl3) δ 5.34-5.29 (m, 1H + 1H), 4.13-4.09 (m, 2H), 3.64-3.60 (m, 2H), 2.07-2.03 (m, 2H), 1.60-1.58 (m, 2H), 1.57-1.49 (m, 2H), 0.90 (s, 9H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 203.3, 93.9, 92.2, 63.2, 61.0, 32.2, 28.4, 26.1, 25.4, 18.5, −5.1; IR (thin film, NaCl) 3380, 2857, 1964, 1727, 1655, 1471, 1360, 1255, 1105, 1024, 836, 775, 663, 565 cm−1; LRMS (EI, M+) calcd for C14H28O2Si, 256.1859 m/z (M); observed 255.10 (M − H)+ m/z.
4.2.6. (Z)-6-(2-(trimethylsilyl)ethyl)octa-5,7-dien-1-ol (17)
To a −78 °C solution of vinyltrimethylsilane (0.431 mL, 2.94 mmol, 1 eq) in 29 mL of diethyl ether (0.1 M) was added 2.94 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 2.94 mmol, 1 eq) and 2.93 mL of c-C5H9MgCl (2.01 M in ether, 5.88 mmol, 3 eq) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 16 (0.256 g, 1 mmol, 0.34 eq) in 2 mL THF (0.5 M) was added 0.465 mL of n-BuLi (2.54 M in hexanes, 1.18 mmol, 0.4 eq) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature before the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. After concentration, the crude material was subjected to TBAF (1 mmol) in 10 mL THF. After stirred for 2 h, 10 mL of H2O was added, and the product was extracted with 50 mL of EtOAc, dried over Na2SO4, and concentrated. Z:E selectivity was determined by 1H NMR of the crude mixture after work-up. The crude material was purified by column chromatography on silica gel (15% ethyl acetate : hexanes) to yield diene 17 as a clear oil (114.8 mg, 51%, Z:E 3:1). The stereochemistry of the major isomer was determined by nOe analysis. The major alkene isomer was separated by HPLC [EtOAc/hexanes: 15%-30 % (0–30 min, 20 mL/min), on a Microsorb (Si 80-120 C5 H410119) column] to yield pure 17. 1H NMR (400 MHz, CDCl3) δ 6.67 (dd, J = 11.2, 17.6 Hz, 1H), 5.41 (t, J = 7.6 Hz, 1H), 5.22 (d, J = 3.0 Hz, 1H), 5.08 (d, J = 11.0 Hz, 1H), 3.65 (t, J = 5.6 Hz, 2H), 2.20-2.15 (m, 2H + 2H), 1.59-1.57 (m, 2H), 1.47-1.46 (m, 2H), 1.26 (brs, 1H), 0.70-0.65 (m, 2H), 0.01 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 139.4, 132.8, 129.0, 113.2, 63.1, 32.5, 27.4, 27.2, 26.2, 16.1, −1.6; IR (thin film, NaCl) 3334, 3088, 2952, 1639, 1455, 1248, 1059, 990, 862, 837, 758, 690 cm−1; HRMS (EI, Na) calcd for C13H26OSi, 226.1753 m/z (M); observed 226.1752 (M) m/z.
4.2.7. (Z)-3-trimethylsilylethyl-4-isopropyl-1,3-butadiene (19)
To a −78 °C solution of vinyltrimethylsilane (0.161 mL, 1.1 mmol) in 11 mL of diethyl ether (0.1 M) was added 1.1 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 1.1 mmol) and 1.1 mL of c-C5H9MgCl (2.06 M in ether, 2.2 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 18 (0.134 mL, 1.0 mmol) in 2 mL THF was added 0.440 mL of n-BuLi (2.50 M in hexanes, 1.1 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature before the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. The crude material was purified by column chromatography on silica gel (hexanes) to yield diene 19 as a clear oil (96.2 mg, 50%, 20:1 Z:E). Z:E selectivity was determined by 1H NMR of the crude mixture after work-up. The stereochemistry of the major product was assigned by analogy to previous examples. The major alkene isomer was separated by HPLC [hexanes: 100 % (0–30 min, 20 mL/min), on a Microsorb (Si 80-120 C5 H410119) column] to yield pure 19. 1H NMR (400 MHz, CDCl3) δ 6.68 (dd, J = 10.8, 17.3 Hz, 1H), 5.23-5.16 (m, 1H + 1H), 5.06 (dd, J = 1.6, 3.2 Hz, 1H), 2.75 (septet, J = 6.6, 9.4, 13.3 Hz, 1H), 2.18-2.13 (m, 2H), 0.97 (d, J = 8.0 Hz, 6H), 0.70-0.65 (m, 2H), 0.01 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 137.1, 136.7, 133.2, 112.9, 27.3, 26.5, 23.4, 15.9, −1.6; IR (thin film, NaCl) 3444, 2956, 1731, 1248, 1177, 837, 758, 692, 468 cm−1; HRMS (EI, M+) calcd for C12H24Si, 196.1647 m/z (M); observed 197.1726 (M + H)+ m/z.
4.2.8. (Z)-1-trimethylsilyl-3(cyclohexymethylene)pent-4-ene (21)
To a −78 °C solution of vinyltrimethylsilane (0.161 mL, 1.1 mmol) in 11 mL of diethyl ether (0.1 M) was added 1.1 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 1.1 mmol) and 1.1 mL of c-C5H9MgCl (2.00 M in ether, 2.2 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 20 (0.155 mL, 1 mmol) in 2 mL THF was added 0.440 mL of n-BuLi (2.50 M in hexanes, 1.1 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature before the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. Z:E selectivity was determined by 1H NMR of the crude mixture after work-up. The crude material was purified by column chromatography on silica gel (hexanes) to yield diene 21 as a clear oil (125.7 mg, 53%, 20:1 Z:E). The stereochemistry of the major product was assigned by analogy to previous examples. The major alkene isomer was separated by HPLC [hexanes: 100 % (0–30 min, 20 mL/min), on a Microsorb (Si 80-120 C5 H410119) column] to yield pure 21. 1H NMR (400 MHz, CDCl3) δ 6.68 (dd, J = 11.6, 17.2 Hz, 1H), 5.25-5.16 (m, 1H + 1H), 5.06 (d, J = 14.8 Hz, 1H), 2.44-2.36 (m, 1H), 2.18-2.14 (m, 2H), 1.69-1.54 (m, 2H +2H), 1.31-1.27 (m, 4H), 1.08-0.90 (m, 2H), 0.70-0.65 (m, 2H), 0.01 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 137.1, 135.7, 133.3, 112.8, 36.4, 33.6, 33.0, 27.3, 26.1, 16.0, −1.5; IR (thin film, NaCl) 3417, 2854, 1714, 1450, 1248, 860, 758, 692 cm−1; HRMS (EI, M+) calcd for C15H28Si, 236.1960 m/z (M); observed 237.2042 (M + H) m/z.
4.3. Cross-coupling reactions of allenyl alcohols with styrene and chlorodimethyvinylsilane
4.3.1. (Z)-6-(2-phenylethyl)octa-5,7-dien-1-ol (23)
To a −78 °C solution of alkene 22 (0.182 mL, 1.92 mmol, 1 eq) in 19.1 mL of diethyl ether (0.1 M) was added 1.92 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 1.92 mmol, 1 eq) and 1.91 mL of c-C5H9MgCl (2.01 M in ether, 3.84 mmol, 3 eq) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 16 (0.168 g, 0.65 mmol, 0.34 eq) in 2 mL THF (0.5 M) was added 0.306 mL of n-BuLi (2.54 M in hexanes, 0.77 mmol, 1.1 eq) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature, and the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. After concentration, the crude material was subjected to TBAF (0.65 mmol) in 6.5 mL THF. After stirred for 2 h, 10 mL of H2O was added, and the product was extracted with 50 mL of EtOAc, dried over Na2SO4, and concentrated. Z:E selectivity was determined by 1H NMR of the crude mixture after work-up. The crude material was purified by column chromatography on silica gel (15% ethyl acetate : hexanes) to yield diene 23 as a clear oil (120.4 mg, 52%, Z:E 2:1). The stereochemistry of the major isomer was determined by nOe analysis. The major alkene isomer was separated by HPLC [EtOAc/hexanes: 15%-30 % (0–30 min, 20 mL/min), on a Microsorb (Si 80-120 C5 H410119) column] to yield pure 23. 1H NMR (400 MHz, CDCl3) δ 7.28-7.17 (m, 5H), 6.69 (dd, J = 8.0, 17.6 Hz, 1H), 5.33-5.28 (m, 1H + 1H), 5.14 (d, J = 12.8 Hz, 1H), 3.62 (m, 2H), 2.78-2.74 (m, 2H), 2.51-2.47 (m, 2H), 2.21-2.15 (m, 2H), 1.55-1.51 (m, 2H), 1.42-1.40 (m, 2H), 1.17 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ 142.6, 136.1, 132.7, 131.1, 128.6, 128.4, 125.9, 113.4, 63.0, 35.5, 35.4, 32.4, 27.1, 26.0; IR (thin film, NaCl) 3351, 2932, 2860, 1635, 1603, 1495, 1454, 1059, 899, 747, 698 cm−1; HRMS (EI, Na) calcd for C16H22O, 231.1671 m/z (M + H); observed 231.1755 (M + H)+ m/z.
4.3.2. 5-methyl-3-methylenehex-4-en-1-ol (25)
To a −78 °C solution of alkene 24 (0.870 mL, 6 mmol) in 60 mL of diethyl ether (0.1 M) was added 6 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 6 mmol) and 6 mL of c-C5H9MgCl (2.01 M in ether, 12 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 10 (0.238 mL, 2 mmol) in 2 mL THF was added 0.880 mL of n-BuLi (2.54 M in hexanes, 2.2 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature before the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. After concentrated in vacuo, the crude material was purified by column chromatography on silica gel (hexanes) to yield diene as an impure, clear oil. This impure product was carried on to the subsequent step without further purification. The impure diene was dissolved in 10 mL 1:1 MeOH : THF, and added KHCO3 (8 mmol) and KF (16 mmol) was added. The reaction mixture was stirred for 5 additional min, 30% H2O2 (10.0 mmol) was added, and the reaction mixture was stirred for 17 h at room temperature. To the reaction was added aqueous Na2S2O3 dropwise, and the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. The crude material was purified by column chromatography on silica gel (15% ethyl acetate : hexanes) to yield diene 25 as a clear oil (128.4 mg, 53%). 1H NMR (400 MHz, CDCl3) δ 5.60 (s, 1H), 5.06 (s, 1H), 4.91 (s, 1H), 3.67 (dt, J = 6.0, 12.0, 18.0 Hz, 2H), 2.35 (t, J = 6.2 Hz, 2H), 1.80 (s, 6H), 1.38 (t, O-H, 1H); 13C NMR (100 MHz, CDCl3) δ 142.7, 136.3, 125.3, 115.4, 61.2, 41.1, 26.8, 19.7; IR (thin film, NaCl) 3417, 2924, 1257, 1042, 799 cm−1; HRMS (EI, M+) calcd for C8H14O, 126.1045 m/z (M); observed 127.1127 (M + H)+ m/z.
4.3.3. (Z)-3-(cyclohexylmethylene)pent-4-en-1-ol (26)
To a −78 °C solution of alkene 24 (0.416 mL, 3 mmol) in 30 mL of diethyl ether (0.1 M) was added 3 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 3 mmol) and 3 mL of c-C5H9MgCl (2.00 M in ether, 6 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 20 (0.115 mL, 1 mmol) in 2 mL THF was added 0.440 mL of n-BuLi (2.50 M in hexanes, 1.1 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature before running through a silica plug and washing with 100mL EtOAc. After concentrated in vacuo, the crude material was purified by column chromatography on silica gel (hexanes) to yield diene as an impure, clear oil. This impure product was carried on to the subsequent step without further purification. The impure diene was dissolved in 10 mL 1:1 MeOH : THF, and added KHCO3 (4 mmol) and KF (8 mmol) was added. The reaction mixture was stirred for 5 additional min, 30% H2O2 (5 mmol) was added, and the reaction mixture was stirred for 17 h at room temperature. To the reaction was added aqueous Na2S2O3 dropwise, and the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. Z:E selectivity was determined by 1H NMR of the crude mixture after work-up. The crude material was purified by column chromatography on silica gel (15% ethyl acetate : hexanes) to yield diene 26 as a clear oil (100.3 mg, 55%, 20:1 Z:E). The stereochemistry of the major product was assigned by analogy to previous examples. The major alkene isomer was separated by HPLC [EtOAc/hexanes: 15%-30 % (0–30 min, 20 mL/min), on a Microsorb (Si 80-120 C5 H410119) column] to yield analytically pure 26. 1H NMR (400 MHz, CDCl3) δ 6.69 (dd, J = 11.0, 17.5 Hz, 1H), 5.31 (d, J = 9.4 Hz, 1H), 5.24 (d, J = 16.0 Hz, 1H), 5.11 (d, J = 11.0 Hz, 1H), 3.68 (t, J = 6.1 Hz, 2H), 2.47-2.43 (dt + m, J = 1.0, 6.0, 12.7 Hz, 2H + 1H), 1.73-1.62 (m, 4H), 1.31-1.01 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 139.8, 132.8, 130.7, 113.8, 61.5, 36.7, 36.6, 33.6, 26.1, 26.0; IR (thin film, NaCl) 3418, 2852, 1727, 1449, 1267, 1122, 1042, 891, 737 cm−1; HRMS (EI, M+ + H) calcd for C12H20O, 181.1514 m/z (M + H); observed 181.1590 (M + H)+ m/z.
4.3.4. (Z)-3-(cyclopentylmethylene)pent-4-en-1-ol (28)
To a −78 °C solution of alkene 24 (0.208 mL, 1.5 mmol) in 15 mL of diethyl ether (0.1 M) was added 1.5 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 1.5 mmol) and 1.5 mL of c-C5H9MgCl (2.00 M in ether, 3 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 27 (0.069 g, 0.5 mmol) in 2 mL THF was added 0.220 mL of n-BuLi (2.54 M in hexanes, 0.55 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, the reaction was quenched with 5 mL of sat. aq. NH4Cl solution. The mixture was warmed to room temperature before running through a silica plug and washing with 100mL EtOAc. After concentrated in vacuo, the crude material was purified by column chromatography on silica gel (hexanes) to yield diene as an impure, clear oil. This impure product was carried on to the subsequent step without further purification. The impure diene was dissolved in 10 mL 1:1 MeOH : THF, and added KHCO3 (2 mmol) and KF (4 mmol) was added. The reaction mixture was stirred for 5 additional min, 30% H2O2 (2.5 mmol) was added, and the reaction mixture was stirred for 17 h at room temperature. To the reaction was added aqueous Na2S2O3 dropwise, and the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. Z:E selectivity was determined by 1H NMR of the crude mixture after work-up. The crude material was purified by column chromatography on silica gel (15% ethyl acetate : hexanes) to yield diene 28 as a clear oil (44.1 mg, 53%, 20:1 Z:E). The stereochemistry of the major product was assigned by analogy to previous examples. The major alkene isomer was separated by HPLC [EtOAc/hexanes: 15%-30 % (0–30 min, 20 mL/min), on a Microsorb (Si 80-120 C5 H410119) column] to yield pure 28. 1H NMR (400 MHz, CDCl3) δ 6.72 (dd, J = 11.0, 17.6 Hz, 1H), 5.41 (d, J = 9.0 Hz, 1H), 5.24 (dd, J = 3.0, 17.5 Hz, 1H), 5.11 (d, J = 11.0 Hz, 1H), 3.69 (t, J = 6.4 Hz, 2H), 2.91-2.85 (m, 1H), 2.49 (t, J = 8.8 Hz, 2H), 1.83-1.70 (m, 2H), 1.60-1.58 (m, 2H), 1.58-1.57 (m, 2H), 1.27-1.24 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 139.2, 132.9, 131.3, 113.7, 61.5, 38.5, 36.7, 34.2, 25.5; IR (thin film, NaCl) 3417, 2869, 1727, 1451, 1258, 1043, 842, 798, 471 cm−1; HRMS (EI, M+) calcd for C11H18O, 167.1358 m/z (M + H); observed 167.1437 (M)+ m/z.
4.4. Preparation of isomeric allenes
4.4.1. Synthesis of allene 30
To a solution of carbonyl diimidazole (1 eq) in benzene (0.1 M) in a flame-dried round bottom flask, diol 29 (1 eq) was added and the solution was refluxed under Ar for 17 h. Subsequently, the solution was cooled to room temperature, and water (10 mL) was added. The resulting mixture was extracted with ether, dried over MgSO4, filtered, and concentrated by vacuum. The crude material was purified by column chromatography on silica gel (25% ethyl acetate : hexanes) to yield carbonate as a clear oil (87% yield). To a −78 °C solution of CuBr-Me2S (2.381 g, 11.58 mmol) in 50 mL of THF was added 6.08 mL of i-PrMgCl 2.00 M (12.16 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution was stirred at −78 °C for 1 h, then the carbonate of 29 (5.79 mmol, 1.339 g) in 10 mL THF was added dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, at −78 °C, then 5 mL of sat. aq. NH4Cl was added. The mixture was warmed to room temperature before aqueous workup and extraction with 3 × 30 mL EtOAc. The crude material was purified by column chromatography on silica gel (10–30% ethyl acetate : hexanes) to yield allene 30 as a clear oil. (0.966 g, 83%). 1H NMR (400 MHz, CDCl3) δ 7.36-7.26 (m, 5H), 5.35-5.31 (m, 1H), 5.28-5.24 (m, 1H), 4.58 (s, 2H), 4.36-4.35 (m, 1H), 3.56 (dd, J = 3.6, 9.6 Hz, 1H), 3.45 (dd, J = 7.6, 9.6 Hz, 1H), 2.36-2.29 (m, 1H +1H), 1.00 (d, J = 8.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 201.8, 138.1, 128.6, 128.4, 127.9, 101.6, 93.0, 74.5, 73.6, 69.1, 27.9, 22.5; IR (thin film, NaCl) 3422, 2925, 1962, 1454, 1363, 1105, 873, 737, 698, 610 cm−1; HRMS (EI, M+) calcd for C15H20O2, 232.1463 m/z (M); observed 231.1393 (M − H)+ m/z.
4.4.2. Synthesis of allene 32
To a solution of carbonyl diimidazole (1 eq) in benzene (0.1 M) in a flame-dried round bottom flask, diol 31 (1 eq) was added and the solution was refluxed under Ar for 17 h. Subsequently, the solution was cooled to room temperature and water (10 mL) was added. The resulting mixture was extracted with ether, dried over MgSO4, filtered, and concentrated by vacuum. The crude material was purified by column chromatography on silica gel (25% ethyl acetate : hexanes) to yield carbonate as a clear oil (84% yield). To a −78 °C solution of CuBr-Me2S (1.99 g, 9.66 mmol) in 50 mL of THF was added 5.07 mL of i-PrMgCl 2.00 M (10.14 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution was stirred at −78 °C for 1 h, then the carbonate of 31 (4.83 mmol, 1.117 g) in 10 mL THF was added dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, at −78 °C, then 5 mL of sat. aq. NH4Cl was added. The mixture was warmed to room temperature before aqueous workup and extraction with 3 × 30 mL EtOAc. The crude material was purified by column chromatography on silica gel (10–30% ethyl acetate : hexanes) to yield allene 32 as a clear oil. (0.698 g, 63%). 1H NMR (400 MHz, CDCl3) δ 7.36-7.26 (m, 5H), 5.35-5.33 (m, 1H), 5.28-5.26 (m, 1H), 4.56 (s, 2H), 4.36 (m, 1H), 3.56 (dd, J = 3.6 Hz, 9.6 Hz, 1H), 3.50 (dd, J = 7.6, 9.6 Hz, 1H), 2.33-2.32 (m, 1H + 1H), 1.01 (d, J = 4.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 201.7, 138.1, 128.6, 128.0, 127.9, 101.9, 93.2, 74.4, 73.5, 68.8, 27.9, 22.6; IR (thin film, NaCl) 3392, 2960, 1962, 1725, 1454, 1364, 1076, 875, 743, 698 cm−1; HRMS (EI, M+) calcd for C15H20O2, 232.1463 m/z (M); observed 231.1755 (M − H)+ m/z.
4.5. Stereoselective access to (Z,E)- and (Z,Z)-1,3-dienes
4.5.1. (2E,4Z)-1-benzyloxy-4-hydroxyethyl-5-isopropyl-2,4-pentadiene (33)
To a −78 °C solution of alkene 24 (0.215 mL, 1.56 mmol) in 16 mL of diethyl ether (0.1 M) was added 1.56 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 1.56 mmol) and 1.56 mL of c-C5H9MgCl (2.01 M in ether, 3.12 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 30 (60.0 mg, 0.26 mmol) in 2 mL THF was added 0.112 mL of n-BuLi (2.54 M in hexanes, 0.286 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath, and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2 h, 5 mL of sat. aq. NH4Cl solution was added. The mixture was warmed to room temperature before the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. The crude material was purified by column chromatography on silica gel (5% ethyl acetate : hexanes) to yield an impure diene as a clear oil. This impure sample was dissolved in 10 mL 1:1 MeOH : THF and KHCO3 (1.04 mmol) and KF (2.08 mmol) was added. The reaction mixture was stirred for 5 additional min, 30% H2O2 (1.3 mmol) was added, and the resulting solution was stirred for 17 h at room temperature. To the reaction was added aqueous Na2S2O3 dropwise, and the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. Z:E selectivity was determined by 1H NMR of the crude mixture after work-up. The crude material was purified by column chromatography on silica gel (20% ethyl acetate : hexanes) to yield diene 33 as a clear oil (38.8 mg, 52%, 5:1 Z,E : Z,Z). The major alkene isomer was separated by HPLC [EtOAc/hexanes: 15%-30 % (0–30 min, 20 mL/min), on a Microsorb (Si 80-120 C5 H410119) column] to yield pure 33. The stereochemistry of the major isomer was determined by nOe (see the analysis following the synthesis of 34). 1H NMR (400 MHz, CDCl3) δ 7.36-7.26 (m, 5H), 6.58 (d, J = 15.9 Hz, 1H), 5.83 (dt, J = 6.2, 12.3, 15.9 Hz, 1H), 5.29 (d, J = 9.4 Hz, 1H), 4.54 (s, 2H), 4.11 (d, J = 8.0 Hz, 2H), 3.68 (m, 2H), 2.81-2.75 (m, 1H), 2.45 (t, J = 6.4 Hz, 2H), 1.29 (O-H, 1H), 0.99 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 141.4, 138.4, 137.2, 129.5, 128.6, 128.0, 127.8, 126.0, 72.5, 71.3, 61.4, 37.4, 26.9, 23.4; IR (thin film, NaCl) 3391, 2958, 2866, 1454, 1360, 1043, 966, 736, 697, 470 cm−1; HRMS (EI, Na) calcd for C17H24O2, 261.1776 m/z (M + H); observed 261.1843 (M + H)+ m/z.
4.5.2. (2Z,4Z)-1-benzyloxy-4-hydroxyethyl-5-isopropyl-2,4-pentadiene (34)
To a −78 °C solution of alkene 24 (0.142 mL, 1.03 mmol) in 11 mL of diethyl ether (0.1 M) was added 1.03 mL of ClTi(Oi-Pr)3 (1.00 M in hexanes, 1.03 mmol) and 1.03 mL of c-C5H9MgCl (2.01 M in ether, 2.05 mmol) dropwise via gas-tight syringe. The resulting clear, yellow solution turned dark red brown while warming to −50 °C over 1 h. The reaction mixture was stirred at −50 °C for 1 h and then cooled to −78 °C. To a separate −78 °C solution of allene 32 (79.5 mg, 0.34 mmol) in 2 mL THF was added 0.150 mL of n-BuLi (2.54M in hexanes, 0.38 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath and added to the −78 °C solution of alkene dropwise via cannula. After warming slowly to 0 °C over 2.5 h, 5 mL of sat. aq. NH4Cl solution was added. The mixture was warmed to room temperature before the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. The crude material was purified by column chromatography on silica gel (5% ethyl acetate : hexanes) to yield an impure diene as a clear oil. This impure sample was dissolved in 10 mL 1:1 MeOH : THF and KHCO3 (1.37 mmol) and KF (2.73 mmol) was added. The reaction mixture was stirred for 5 additional min, 30% H2O2 (1.71 mmol) was added, and the resulting solution was stirred for 17 h at room temperature. To the reaction was added aqueous Na2S2O3 dropwise, and the resulting solution was filtered through a plug of silica, eluting with 100 mL EtOAc. Z:E selectivity was determined by 1H NMR of the crude mixture after work-up. Z:E selectivity was determined by 1H NMR of the crude mixture after work-up. The crude material was purified by column chromatography on silica gel (20% ethyl acetate : hexanes) to yield diene as a clear oil (44.8 mg, 53%, 3:1 Z,Z : Z,E). The major alkene isomer was separated by HPLC [EtOAc/hexanes: 15%-30 % (0–30 min, 20 mL/min), on a Microsorb (Si 80-120 C5 H410119) column] to yield pure 34. 1H NMR (400 MHz, CDCl3) δ 7.33-7.26 (m, 5H), 6.00 (d, J = 11.7 Hz, 1H), 5.77 (dt, J = 6.6, 11.7, 13.1 Hz, 1H), 5.18 (d, J = 9.7 Hz, 1H), 4.51 (s, 2H), 4.01 (d, J = 8.0 Hz, 2H), 3.61 (t, J = 11.7 Hz, 2H), 2.40-2.36 (m, 1H), 2.27 (t, J = 6.2 Hz, 2H), 1.62 (O-H, 1H), 0.90 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 138.9, 138.2, 130.6, 130.0, 128.6, 128.4, 128.1, 127.9, 72.9, 67.2, 60.1, 41.1, 28.2, 22.9; IR (thin film, NaCl) 3401, 3030, 2900, 2867, 1454, 1360, 1094, 1071, 736, 697, 608 cm−1; HRMS (EI, Na) calcd for C17H24O2, 261.1776 m/z (M + H); observed 261.1858 (M + H)+ m/z.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support of this work by the American Cancer Society (RSG-06-117-01), the Arnold and Mabel Beckman Foundation, Boehringer Ingelheim, Eli Lilly & Co., and the National Institutes of Health (GM080266).
Footnotes
Complete experimental details for preparative procedures along with spectral data for all products are provided. Supplementary data associated with this article can be found in the online version at doi:
References and notes
- 1.For recent reviews, see: Montgomery J, Sormunen GJ. Top Curr Chem. 2007;279:1–23.Ng SS, Ho CY, Schleicher KD, Jamison TF. Pure Appl Chem. 2008:929–939. doi: 10.1351/pac200880050929.Moslin RM, Miller-Moslin K, Jamison TF. Chem Commun. 2007:4441–4449. doi: 10.1039/b707737h.Patman RL, Bower JF, Kim IS, Krische MJ. Aldrichemica Acta. 2008;41:95–104.Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394–1401. doi: 10.1021/ar7001123.Jeganmohan M, Chen CH. Chem Eur J. 2008;14:10876–10886. doi: 10.1002/chem.200800904.Reichard HA, McLaughlin M, Chen MZ, Micalizio GC. Eur J Org Chem. 2009 doi: 10.1002/ejoc.200901094.
- 2.a) Harada K, Urabe H, Sato F. Tetrahedron Lett. 1995;36:3203–3206. [Google Scholar]; b) Gao Y, Harada K, Sato F. Tetrahedron Lett. 1995;36:5913–5916. [Google Scholar]; c) Qi X, Montgomery J. J Org Chem. 1999;64:9310–9313. [Google Scholar]; d) Mahandru GM, Liu G, Montgomery J. J Am Chem Soc. 2004;126:3698–3699. doi: 10.1021/ja049644n. [DOI] [PubMed] [Google Scholar]; e) Sa-ei K, Montgomery J. Org Lett. 2006;8:4441–4443. doi: 10.1021/ol061579u. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Chaulagain MR, Sormunen GJ, Montgomery F. J Am Chem Soc. 2007;129:9568–9569. doi: 10.1021/ja072992f. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Baxter RD, Montgomery J. J Am Chem Soc. 2008;130:9662–9663. doi: 10.1021/ja803774s. [DOI] [PubMed] [Google Scholar]; h) Huang WS, Chan J, Jamison TF. Org Lett. 2000;2:4221–4223. doi: 10.1021/ol006781q. [DOI] [PubMed] [Google Scholar]; i) Colby EA, Jamison TF. J Org Chem. 2003;68:156–166. doi: 10.1021/jo0264123. [DOI] [PubMed] [Google Scholar]; j) Patel SJ, Jamison TF. Angew Chem Int Ed. 2003;42:1364–1367. doi: 10.1002/anie.200390349. [DOI] [PubMed] [Google Scholar]; k) Miller KM, Huang WS, Jamison TF. J Am Chem Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y. [DOI] [PubMed] [Google Scholar]; l) Miller KM, Jamison TF. J Am Chem Soc. 2004;126:15342–15343. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]; m) Luanphaisarnnont T, Ndubaku CO, Jamison TF. Org Lett. 2005;7:2937–2940. doi: 10.1021/ol050881k. [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Miller KM, Jamison TF. Org Lett. 2005;7:3077–3080. doi: 10.1021/ol051075g. [DOI] [PubMed] [Google Scholar]; o) Moslin RM, Miller KM, Jamison TF, Tetrahedron 2006;62:7598–7610. [Google Scholar]; p) Jang HY, Huddleston RR, Krische MJ. J Am Chem Soc. 2004;126:4664–4668. doi: 10.1021/ja0316566. [DOI] [PubMed] [Google Scholar]; q) Kong J-R, Cho C-W, Krische MJ. J Am Chem Soc. 2005;127:11269–11276. doi: 10.1021/ja051104i. [DOI] [PMC free article] [PubMed] [Google Scholar]; r) Komanduri V, Krische MJ. J Am Chem Soc. 2006;128:16448–16449. doi: 10.1021/ja0673027. [DOI] [PubMed] [Google Scholar]; s) Cho CW, Krische MJ. Org Lett. 2006;8:3873–3876. doi: 10.1021/ol061485k. [DOI] [PubMed] [Google Scholar]; t) Barchuk A, Ngai MY, Krische MJ. J Am Chem Soc. 2007;129:8432–8433. doi: 10.1021/ja073018j. [DOI] [PubMed] [Google Scholar]; u) Ngai MY, Barchuk A, Krische MJ. J Am Chem Soc. 2007;129:280–281. doi: 10.1021/ja0670815. [DOI] [PubMed] [Google Scholar]; v) Ngai MY, Barchuk A, Krische MJ. J Am Chem Soc. 2007;129:12644–12645. doi: 10.1021/ja075438e. [DOI] [PubMed] [Google Scholar]; w) Skucas E, Kong JR, Krische MJ. J Am Chem Soc. 2007;129:7242–7243. doi: 10.1021/ja0715896. [DOI] [PubMed] [Google Scholar]; x) Cho CW, Skucas E, Krische MJ. Organomet. 2007;26:3860–3867. [Google Scholar]; y) Hong YT, Cho CW, Skucas E, Krische MJ. Org Lett. 2007;9:3745–3748. doi: 10.1021/ol7015548. [DOI] [PubMed] [Google Scholar]; z) Han SB, Kong JR, Krische MJ. Org Lett. 2008;10:4133–4135. doi: 10.1021/ol8018874. [DOI] [PMC free article] [PubMed] [Google Scholar]; aa) Patman RL, Chaulagain MR, Williams VM, Krische MJ. J Am Chem Soc. 2009;131:2066–2067. doi: 10.1021/ja809456u. [DOI] [PMC free article] [PubMed] [Google Scholar]; ab) Nevárez Z, Woerpel KA. Org Lett. 2007;9:3773–3776. doi: 10.1021/ol701424a. [DOI] [PubMed] [Google Scholar]; ac) Bourque LE, Woerpel KA. Org Lett. 2008;10:5257–5260. doi: 10.1021/ol8018538. [DOI] [PMC free article] [PubMed] [Google Scholar]; ad) Anderson LL, Woerpel KA. Org Lett. 2009;11:425–428. doi: 10.1021/ol802412b. [DOI] [PMC free article] [PubMed] [Google Scholar]; ae) Bahadoor AB, Flyer A, Micalizio GC. J Am Chem Soc. 2005;127:3694–3695. doi: 10.1021/ja050039+. [DOI] [PubMed] [Google Scholar]; af) Bahadoor AB, Micalizio GC. Org Lett. 2006;8:1181–1184. doi: 10.1021/ol0600786. [DOI] [PubMed] [Google Scholar]
- 3.For the coupling of two alkynes and an enone for the formation of a substituted cyclohexene, see: Lozanov M, Montgomery J. J Am Chem Soc. 2002;124:2106–2107. doi: 10.1021/ja0175845.For the coupling of cyclopropyl ketones to enones, see: Liu L, Montgomery J. J Am Chem Soc. 2006;128:5348–5349. doi: 10.1021/ja0602187.For the coupling of an imine with an enone, see: Liu L, Montgomery J. Org Lett. 2007;9:3885–3887. doi: 10.1021/ol071376l.For the coupling of an alkyne with an enone, see: Herath A, Thompson BB, Montgomery J. J Am Chem Soc. 2007;129:8712–8713. doi: 10.1021/ja073300q.For aldehyde–epoxide coupling, see: Molinaro C, Jamison TF. Angew Chem Int Ed. 2005;44:129–132. doi: 10.1002/anie.200461705.For enyne–epoxide coupling, see: Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J Am Chem Soc. 2004;126:4130–4131. doi: 10.1021/ja0491735.For the coupling of terminal alkenes to enones, see: Ho CY, Ohmiya H, Jamison TF. Angew Chem Int Ed. 2008;47:1893–1895. doi: 10.1002/anie.200705163.For the coupling of allenes with aldehydes, see: Ng SS, Jamison TF. J Am Chem Soc. 2005;127:7320–7321. doi: 10.1021/ja0521831.Ng SS, Jamison TF. Tetrahedron. 2006;62:11350–11359.For the coupling of aldehydes to terminal alkenes, see: Ng SS, Jamison TF. J Am Chem Soc. 2005;127:14194–14195. doi: 10.1021/ja055363j.Ng SS, Ho CY, Jamison TF. J Am Chem Soc. 2006;128:11513–11528. doi: 10.1021/ja062866w.Ho C-Y, Ng S-S, Jamison TF. J Am Chem Soc. 2006;128:5362–5363. doi: 10.1021/ja061471+.Ho CY, Jamison TF. Angew Chem Int Ed. 2007;46:782–785. doi: 10.1002/anie.200603907.For coupling of a terminal alkene with an isocyanate, see: Schleicher KD, Jamison TF. Org Lett. 2007;9:875–878. doi: 10.1021/ol063111x.For the coupling of N-sulfonyl imines with 2-vinylpyridines, see: Komanduri V, Grant CD, Krische MJ. J Am Chem Soc. 2008;130:12592–12593. doi: 10.1021/ja805056g.For alkene–alkyne coupling, see: Trost BM. Acc Chem Res. 2002;35:695–705. doi: 10.1021/ar010068z.(and references cited therein). Wang CC, Lin PS, Cheng CH. J Am Chem Soc. 2002;124:9696–9697. doi: 10.1021/ja026543l.Chang HT, Jayanth TT, Wang CC, Cheng CH. J Am Chem Soc. 2007;129:12032–12041. doi: 10.1021/ja073604c.For the coupling of alkynes with enols, see: Kuninobu Y, Kawata A, Takai K. J Am Chem Soc. 2006;128:11368–11369. doi: 10.1021/ja064022i.For the coupling of allenes with enones, see: Chang HT, Jayanth TT, Cheng CH. J Am Chem Soc. 2007;129:4166–4167. doi: 10.1021/ja0710196.Trost BM, Pinkerton AB, Seidel M. J Am Chem Soc. 2001;123:12466–12476. doi: 10.1021/ja011428g.Trost BM, McClory A. Org Lett. 2006;8:3627–3629. doi: 10.1021/ol0610136.For the coupling of 1,3-dienes to aldehydes or imines, see: Kimura M, Ezoe A, Shibata K, Tamaru Y. J Am Chem Soc. 1998;120:4033–4034.Kimura M, Miyachi A, Kojima K, Tanaka S, Tamaru Y. J Am Chem Soc. 2004;126:14360–14361. doi: 10.1021/ja0450354.Kojima K, Kimura M, Ueda S, Tamaru Y. Tetrahedron. 2006;62:7512–7520.For the coupling of homoallylic alcohols to acylpyrroles, see: Epstein OL, Seo JM, Masalov N, Cha JK. Org Lett. 2005;7:2105–2108. doi: 10.1021/ol050352g.For coupling of internal alkynes with allylic halides or allylic alcohols, see: Suzuki N, Kondakov DY, Kageyama M, Kotora M, Hara R, Takahashi T. Tetrahedron. 1995:514519–4540.Takai K, Yamada M, Odaka G, Utimoto K, Fujii T, Furukawa I. Chem Lett. 1995:315–316.
- 4.For a discussion of the general design strategy, see ref: 1g. For the coupling of an internal alkyne with a terminal alkyne, see: Shimp HL, Micalizio GC. Org Lett. 2005;7:5111–5114. doi: 10.1021/ol052241n.Perez LJ, Shimp HL, Micalizio GC. J Org Chem. 2009;74:7211–7219. doi: 10.1021/jo901451c.For the cross-coupling of two different internal alkynes, see: Ryan J, Micalizio GC. J Am Chem Soc. 2006;128:2764–2765. doi: 10.1021/ja057352w.For the coupling of internal alkynes with alkenes, see: Reichard HA, Micalizio GC. Angew Chem Int Ed. 2007;46:1440–1443. doi: 10.1002/anie.200603515.For the coupling of internal alkynes with imines, see: McLaughlin M, Takahashi M, Micalizio GC. Angew Chem Int Ed. 2007;46:3912–3914. doi: 10.1002/anie.200605060.Chen MZ, Micalizio GC. Org Lett. 2009;11:4982–4985. doi: 10.1021/ol902169k.For the coupling of alkenes with imines, see: Takahashi M, Micalizio GC. J Am Chem Soc. 2007;129:7514–7516. doi: 10.1021/ja071974v.For the coupling of allenes with alkynes, see: Shimp HL, Micalizio GC. Chem Commun. 2007:4531–4533. doi: 10.1039/b708256h.Shimp HL, Hare A, McLaughlin M, Micalizio GC. Tetrahedron. 2008;64:6831–6837. doi: 10.1016/j.tet.2008.02.015.For allylic alcohol–alkyne coupling, see: Kolundzic F, Micalizio GC. J Am Chem Soc. 2007;129:15112–15113. doi: 10.1021/ja075678u.For allene–imine coupling, see: McLaughlin M, Shimp HL, Navarro R, Micalizio GC. Synlett. 2008:735–738. doi: 10.1055/s-2008-1042808.For allylic alcohol–vinylsilane coupling, see: Belardi JK, Micalizio GC. J Am Chem Soc. 2008;130:16870–16872. doi: 10.1021/ja8074242.For allylic alcohol–imine coupling, see: Takashi M, McLaughlin M, Micalizio GC. Angew Chem Int Ed. 2009;48:3648–3652. doi: 10.1002/anie.200900236.Tarselli MA, Micalizio GC. Org Lett. 2009;11:4596–4599. doi: 10.1021/ol901870n.Umemura S, McLaughlin M, Micalizio GC. Org Lett. 2009;11:5402–5405. doi: 10.1021/ol9022134.Yang D, Micalizio GC. J Am Chem Soc. 2009;131:17548–17549. doi: 10.1021/ja908504z.
- 5.Internal alkynes and substituted alkenes are notoriously unreactive substrates in a variety of bimolecular metal-centered [2+2+1] chemistry. Examples that highlight the ability to overcome this characteristic include refs: 4c, d, g, j, l, m–p.
- 6.Reichard HA, Rieger JC, Micalizio GC. Angew Chem Int Ed. 2008;47:7837–7840. doi: 10.1002/anie.200803031. [DOI] [PubMed] [Google Scholar]
- 7.Negishi EI. Acc Chem Res. 1982;15:340–348. [Google Scholar]
- 8.Langille NF, Panek JS. Org Lett. 2004;6:3203–3206. doi: 10.1021/ol048664r. [DOI] [PubMed] [Google Scholar]
- 9.For study and application of related ylids, see the following selected references: Chen J, Wang T, Zhao K. Tetrahedron Lett. 1994;35:2827–2828.Arimoto H, Kaufman MD, Kobayashi K, Qiu Y, Smith AB., III Synlett. 1998:765–767.Roethle PA, Chen IT, Trauner D. J Am Chem Soc. 2007;129:8960–8961. doi: 10.1021/ja0733033.
- 10.For examples of reductive cross-coupling chemistry with vinylsilanes, see: Mizojiri R, Urabe H, Sato F. J Org Chem. 2000;65:6217–6222. doi: 10.1021/jo000925x.b) ref 4l.
- 11.For the use of styrene in related Ti-mediated coupling reactions, see: Lysenko IL, Kim K, Lee HG, Cha JK. J Am Chem Soc. 2008;130:15997–16002. doi: 10.1021/ja806440m.For an early example of olefin exchange in the context of the Kulinkovich reaction, see: Lee J, Kim H, Cha JK. J Am Chem Soc. 1996;118:4198–4199.
- 12.a) Kulinkovich OG, Sviridov SV, Vasilevski DA, Pritytskaya TS. J Org Chem USSR (Engl Transl) 1990;25:2027. [Google Scholar]; b) Kulinkovich OG, de Meijere A. Chem Rev. 2000;100:2789–2834. doi: 10.1021/cr980046z. [DOI] [PubMed] [Google Scholar]
- 13.Reductive ethylation processes have been reported in Ti-mediated reactions of EtMgBr with allylic ethers and allylic alcohols, see: Kulinkovich OG, Epstein OL, Isakov VE, Khmel’nitskaya EA. Synlett. 2001:49–52.Matyushenkov EA, Churikov DG, Sokolov NA, Kulinkovich OG. Russ J Org Chem. 2003;39:478–485.Also, the reductive ethylation of homoallylic alcohols has been reported: Kulinkovich OG, Shevchuk TA, Isakov VE, Prokhorevich KN. Russ J Org Chem. 2006;42:659–664.See also, ref 11a.
- 14.In no case could a clean sample of (Z)-diene be obtained in any of attempted ethylation of an allenyl alcohol with EtMgBr.
- 15.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467–4470. [Google Scholar]
- 16.Kang SK, Kim SG, Cho DG. Tetrahedron: Asymmetry. 1992;3:1509–1510. [Google Scholar]
- 17.This mechanistic proposal is consistent with the high levels of selectivity observed in related reductive cross-coupling reactions of allylic alkoxides with alkynes, vinylsilanes and imines (see refs: 4j, 4l and 4m–o).
- 18.In comparison to the poor levels of reactivity associated with substituted alkenes in bimolecular metal-centered [2+2+1], Ti-mediated reactions of allenes are known: Hideura D, Urabe H, Sato F. Chem Commun. 1998:271–272.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.