

Background: Bile acids cause ryanodine receptor (RyR) Ca2+ release and lead to injury in pancreatic acinar cells, yet the mechanism is unknown.
Results: Inhibition of the RyR activator cADPR reduces bile acid-induced acinar cell Ca2+ release, cell injury, and pancreatitis.
Conclusion: CD38-cADPR facilitates bile-induced Ca2+ release, cell injury, and pancreatitis.
Significance: The CD38-cADPR pathway may serve as a target for the treatment of some forms of pancreatitis.
Keywords: Bile Acid, Calcium Signaling, CD38, Cyclic ADP-ribose, NAADP, Pancreas, Ryanodine Receptor
Abstract
Aberrant Ca2+ signals within pancreatic acinar cells are an early and critical feature in acute pancreatitis, yet it is unclear how these signals are generated. An important mediator of the aberrant Ca2+ signals due to bile acid exposure is the intracellular Ca2+ channel ryanodine receptor. One putative activator of the ryanodine receptor is the nucleotide second messenger cyclic ADP-ribose (cADPR), which is generated by an ectoenzyme ADP-ribosyl cyclase, CD38. In this study, we examined the role of CD38 and cADPR in acinar cell Ca2+ signals and acinar injury due to bile acids using pharmacologic inhibitors of CD38 and cADPR as well as mice deficient in Cd38 (Cd38−/−). Cytosolic Ca2+ signals were imaged using live time-lapse confocal microscopy in freshly isolated mouse acinar cells during perifusion with the bile acid taurolithocholic acid 3-sulfate (TLCS; 500 μm). To focus on intracellular Ca2+ release and to specifically exclude Ca2+ influx, cells were perifused in Ca2+-free medium. Cell injury was assessed by lactate dehydrogenase leakage and propidium iodide uptake. Pretreatment with either nicotinamide (20 mm) or the cADPR antagonist 8-Br-cADPR (30 μm) abrogated TLCS-induced Ca2+ signals and cell injury. TLCS-induced Ca2+ release and cell injury were reduced by 30 and 95%, respectively, in Cd38-deficient acinar cells compared with wild-type cells (p < 0.05). Cd38-deficient mice were protected against a model of bile acid infusion pancreatitis. In summary, these data indicate that CD38-cADPR mediates bile acid-induced pancreatitis and acinar cell injury through aberrant intracellular Ca2+ signaling.
Introduction
Ca2+ signals within the pancreatic acinar cell play a critical role in both physiology and disease. In the physiologic state, localized Ca2+ transients are initiated by hormones and neurotransmitters (1). These Ca2+ signals are tightly linked to the secretion of zymogens into the acinar lumen (2). In the disease state, however, the localized Ca2+ signals are converted to sustained high-amplitude global signals, which are associated with early events in pancreatitis (3–7). They include premature activation of digestive enzymes within the acinar cell (8–10), cytokine expression (11), vacuolization (8, 12, 13), mitochondrial depolarization (14), loss of plasma membrane integrity (15), and cell death (16–18).
Although aberrant Ca2+ signals are known to play an important role in acinar cell injury, the mechanism by which pancreatitis-inducing insults lead to the disease is unclear. We and others have previously identified a selective intracellular Ca2+ channel, the ryanodine receptor (RyR),3 as an important contributor to these Ca2+ signals (10, 15, 18, 19). The RyR mediates intra-acinar zymogen activation and pancreatitis (15, 18) and is one of two major Ca2+ release channels localized to the endoplasmic reticulum, the other being the inositol 1,4,5-trisphosphate receptor. The RyR is a large homotetramer of 2.3 MDa and is found primarily in the basal region of the acinar cell (10, 20–22). RyR Ca2+ release is regulated by a host of factors, with cytosolic Ca2+ being the most potent activator, making the RyR a prototypic Ca2+-induced Ca2+ release channel. Other activators of the RyR include calmodulin, ATP, and the cyclic adenine nucleotide cyclic ADP-ribose (cADPR).
cADPR is a metabolite of NAD+ and is produced in mammalian cells by the ADP-ribosyl cyclase CD38 (cluster of differentiation 38). cADPR sensitizes the RyR to Ca2+ in a manner similar to that of caffeine, yet with higher potency (23). The action of cADPR on the RyR requires additional protein factors, including calmodulin and FK506-binding protein (24–26).
In this study, we investigated the role of bile acids in mediating intracellular Ca2+ release through a CD38-cADPR-dependent pathway. Bile acid exposure is known to cause acinar cell injury, and it may account for biliary pancreatitis, the most common etiology of acute pancreatitis in both children and adults (27). Bile acids exert their injurious effects on the acinar cell through Ca2+-dependent pathways. Specifically, bile acids trigger Ca2+ release from endoplasmic reticulum and vesicular Ca2+ stores. This occurs through activation of RyRs and inositol 1,4,5-trisphosphate receptors (5, 18, 28) and subsequent opening of store-operated Ca2+ entry channels (7, 16, 29).
In a recent study by Cosker et al. (30), CD38 was shown to play a role in pancreatic acinar cell Ca2+ signaling. Although another enzymatic product of CD38, nicotinic acid adenine dinucleotide phosphate (NAADP), was the focus of this study, cADPR levels were also reduced at base line in Cd38−/− acinar cells compared with wild-type cells, and importantly, cholecystokinin stimulation did not induce an increase in cADPR levels, as it did in the wild-type cells. To exclude the effects of Ca2+ influx, Cd38−/− acini were stimulated in the absence of extracellular Ca2+ and failed to evoke a Ca2+ transient. On the basis of this work and the previous studies implicating RyR Ca2+ signaling in bile acid-mediated pathology, in this study, we examined the role of CD38 and cADPR in intracellular Ca2+ release, acinar cell injury, and pancreatitis due to bile acid exposure.
EXPERIMENTAL PROCEDURES
Reagents and Animals
All reagents were purchased from Sigma-Aldrich unless stated otherwise. Male Swiss Webster mice (weighing 20–25 g; Charles River Laboratories, Wilmington, MA) were fed standard laboratory chow, given free access to water, and randomly assigned to control or experimental groups. Cd38−/− mice were generated by one of us (F. E. L.) (31). Age-, sex-, and strain-matched control mice (C57BL/6 mice, The Jackson Laboratory) were used as wild-type controls. All animal experiments were performed using a protocol approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Preparation of Pancreatic Acini for Ca2+ Imaging
Groups of pancreatic acinar cells were isolated as described previously (10) with minor modifications. Briefly, the pancreas was removed and then minced for 5 min in buffer containing 20 mm HEPES (pH 7.4), 95 mm NaCl, 4.7 mm KCl, 0.6 mm MgCl2, 1.3 mm CaCl2, 10 mm glucose, and 2 mm glutamine plus 1% BSA, 1× Gibco® minimum Eagle's medium nonessential amino acids (Invitrogen), 200 units/ml type 4 collagenase (Worthington), and 1 mg/ml soybean trypsin inhibitor. The tissue was incubated for 30 min at 37 °C with shaking at 90 rpm. The digest was transferred to a 15-ml conical tube and washed with collagenase-free buffer. The suspension was vigorously shaken for 15–20 s to separate the cells into smaller clusters.
Detection and Analysis of Cellular Ca2+ Signals
Acinar cells were loaded at room temperature with the high-affinity Ca2+-sensing dye Fluo-4/AM (Kd = 300 nm; Invitrogen). Acinar cells were plated on acid-washed glass coverslips and then mounted on a perifusion chamber. They were stimulated at room temperature with the bile acid taurolithocholic acid 3-sulfate (TLCS) at the concentrations indicated. A Zeiss LSM 710 laser scanning confocal microscope was used with a 40× 1.4-numerical aperture objective. The dye was excited at a wavelength of 488 nm, and emission signals of >515 nm were collected every 2.5 s. Fluorescence from individual acinar cells was recorded. Recordings were analyzed using NIH ImageJ software, and mean fluorescence over time in each region was graphed.
Preparation of Pancreatic Acini
Groups of pancreatic acinar cells were isolated as described previously (32) with minor modifications. Briefly, the pancreas was removed and minced for 5 min in 1× DMEM/nutrient mixture F-12 without phenol red (Invitrogen) plus 0.1% BSA and 2 mg/ml type-4 collagenase. The suspension was briefly incubated for 5 min at 37 °C with shaking at 90 rpm. The buffer was removed, replaced with new collagenase buffer, and then incubated for 35 min. The suspension was filtered through a 300-μm mesh (Sefar American, Depew, NY) and washed three times with collagenase-free buffer. Acinar cells were allowed to equilibrate for 5 min at 37 °C prior to use.
Cell Injury Assays
Acinar cell injury was measured using a cytotoxicity assay for lactate dehydrogenase (LDH) leakage (Promega). Absorbance was measured at 492 nm. Results are expressed as percent LDH released into the medium. For propidium iodide uptake, acinar cells were incubated in a 48-well plate with 50 μg/ml propidium iodide (Sigma) for 30 min prior to addition of 500 μm TLCS. Fluorescence was measured at 536-nm excitation and 617-nm emission wavelengths. Data were normalized to total DNA by again measuring fluorescence after cell lysis with 0.5% Triton X-100.
Preparation of Human Acinar Cells
Pancreatic tissue was harvested from cadaveric donors as described by Bottino et al. (33). Briefly, specimens were transported in cold preservation fluid (histidine/tryptophan/ketoglutarate) with a cold ischemia time of 13 h. Fat, connective tissue, and blood vessels were trimmed away. The pancreas was washed with a mixture of antibiotics and then cut at the level of the neck to reveal the pancreatic duct. Catheters were placed in both sides of the transected duct, and a blend of exogenous enzymes, including collagenases and neutral proteases (GMP-grade, Serva, Heidelberg, Germany), freshly dissolved in Hanks' balanced salt solution was prewarmed to 28–30 °C and introduced intraductally. The pancreatic organ was then transferred to a Ricordi digestion chamber, and the pancreatic tissue was mechanically disrupted as described by Ricordi (34). Pancreatic cells were washed several times with cold RPMI 1640 medium supplemented with human serum albumin (2.5% total volume). Endocrine cell contamination was <1%. Acinar cells were kept in calcium- and magnesium-free Hanks' buffer, and cell injury assays were performed as described above.
Enzyme Activity Assays
Protease activity assays were performed at room temperature using fluorogenic substrates as described previously (35) with modifications. Briefly, 50 μl of 400 μm enzyme substrate were added to each homogenized sample, and the accumulation of fluorescence was measured over 12 min using a Synergy H1 fluorescence plate reader (BioTek, Winooski, VT) at 380-nm excitation and 440-nm emission wavelengths. The trypsin substrate was supplied by Peptides International (Louisville, KY) and had the amino acid sequence t-butoxycarbonyl-Gln-Ala-Arg-7-amino-4-methylcoumarin. The chymotrypsin substrate was supplied by Calbiochem and had the amino acid sequence succinyl-Ala-Ala-Pro-Phe-7-amino-4-methylcoumarin. Zymogen activity was normalized to total protein content.
Intraductal Bile Acid Infusion Model of Pancreatitis
Pancreatitis was induced by retrograde infusion of the bile acid TLCS (3 mm) into the distal common bile duct and pancreatic duct as described recently (36). Briefly, C57BL/6 mice between 8 and 12 weeks of age were anesthetized with isoflurane. A ventral incision was made to reveal the abdominal cavity. The duodenum was flipped to reveal its distal side and held in place by ligatures. The bile duct was identified, and a 30-gauge needle was inserted through the antimesenteric aspect of the duodenum to cannulate the biliopancreatic duct. TLCS was infused at 10 μl/min for 5 min using a P33 perfusion pump (Harvard Apparatus, Holliston, MA). The exterior wound was closed using 7-mm wound clips, and a single injection of buprenorphine (0.075 mg/kg) was given immediately after the surgery. Normal saline-infused animals served as sham controls. Animals were allowed to recover on a heating pad for 90 min after the procedure. Mice were euthanized 24 h after induction.
Tissue Preparation and Histological Grading
The pancreas, duodenum, and spleen were fixed at room temperature for 24 h in 4% paraformaldehyde solution and transferred to 70% ethanol. Paraffin-embedded sections were stained with hematoxylin and eosin and graded using a 40× objective over 10 separate fields in a blinded fashion. Pancreatic tissue was graded for edema, acinar cell vacuole formation, inflammation, and necrosis as described by Wildi et al. (37).
RESULTS
Pharmacologic Inhibition of CD38 Attenuates Bile Acid-induced Ca2+ Signals
We used TLCS to examine the effects of bile acids on acinar cell Ca2+ release for two primary reasons. First, TLCS induces Ca2+ signals at submillimolar concentrations below the critical micellar concentration. Second, it is the least hydrophilic and thus most potent of the naturally occurring bile acids (38).
To examine intracellular Ca2+ release and to exclude the influence of extracellular Ca2+, acini were loaded with the high-affinity Ca2+ dye Fluo-4/AM (Fig. 1A) and perifused in a nominally Ca2+-free medium. Using time-lapse laser scanning confocal microscopy, we observed that perifusion with 500 μm TLCS caused Ca2+ oscillations in over 90% of the acini (Fig. 1B). Lower concentrations of TLCS (50 μm) in the absence of Ca2+ did not induce a Ca2+ transient (data not shown).
FIGURE 1.

Pharmacologic inhibition of CD38 attenuates bile acid-induced Ca2+ signals. A, pseudo-colored images of acinar cells loaded with the Ca2+ dye Fluo-4/AM and then stimulated with TLCS. Images are shown at base line (1), during peak fluorescence (2), and upon returning to base line (3). Est., estimated. B, representative Ca2+ signals observed in cells perifused with TLCS (500 μm) in the absence of extracellular Ca2+. C, representative trace of an experiment in which TLCS (500 μm) was perifused for 2 min, followed by the addition of nicotinamide (20 mm) to the solution. After 3.5 min, nicotinamide was washed off, and TLCS remained in the solution. D, amplitude of the Ca2+ signal shown in C, represented as normalized fluorescence. E, representative trace of an experiment in which cells were pretreated with nicotinamide (20 mm) for 30 min. Subsequently, the cells were perifused with TLCS (500 μm) and nicotinamide for 4 min. After 4 min, nicotinamide was washed off, and TLCS remained in the solution. F, amplitude of the Ca2+ signal shown in D, represented as normalized fluorescence. Data were obtained from three separate days of experimentation (n = 50–60 cells/condition). #, p < 0.05 compared with TLCS alone.
To determine whether TLCS-induced Ca2+ release is dependent on CD38, acinar cells were first perifused with TLCS (500 μm) and then co-treated with nicotinamide (20 mm). We found that nicotinamide abolished the Ca2+ response to TLCS (Fig. 1, C and D). To determine whether the effect was reversible, TLCS was administered after washing off nicotinamide, and a second peak was observed. Similar results were observed when acinar cells were pretreated with nicotinamide prior to TLCS stimulation (Fig. 1, E and F). The Ca2+ transient observed following withdrawal of nicotinamide was a global surge, which appears as a single Ca2+ spike.
Genetic Deletion of Cd38 Attenuates Bile Acid-induced Ca2+ Signals
In addition to inhibiting CD38, nicotinamide has several nonspecific effects. It can inhibit poly(ADP-ribose) polymerase (39) and sirtuins (40) and can scavenge reactive oxygen species (41). Therefore, to complement the pharmacologic data, we isolated acinar cells from Cd38-deficient mice (Cd38−/−) and stimulated them with TLCS (500 μm). These mice have no pancreatic defect at base line and no gross phenotypic differences compared with wild-type mice (30, 31). We observed that compared with wild-type cells, Cd38−/− acinar cells exhibited a 30% reduction in Ca2+ release (p < 0.05) (Fig. 2, A and B). Both the pharmacologic and genetic inhibition of the CD38 pathway attenuates the amount of intracellular Ca2+ release observed with TLCS. To examine the nonspecific effects of nicotinamide, Cd38-deficient acinar cells were stimulated with TLCS in the presence or absence of the inhibitor. We observed that nicotinamide caused reductions in the TLCS-induced Ca2+ signal (Fig. 2, C and D), suggesting that this inhibitor has nonspecific targets that are in addition to inhibiting CD38.
FIGURE 2.

Genetic deletion of Cd38 attenuates bile acid-induced Ca2+ signals. A, intracellular Ca2+ measurements were taken from wild-type or Cd38-deficient acinar cells stimulated with TLCS (500 μm). B, amplitude of the Ca2+ signal represented as normalized fluorescence. Data were obtained from three separate days of experimentation. C, Cd38-deficient acinar cells were stimulated with TLCS in the presence or absence of the inhibitor nicotinamide. D, amplitude of the Ca2+ signal represented as normalized fluorescence (n = 50–60 cells/condition). #, p < 0.05 compared with TLCS alone.
Bile Acid-induced Ca2+ Signals Are Dependent on cADPR
Bile acids target the RyR to induce aberrant acinar cell Ca2+ signals and cell injury (18). Because CD38 drives the synthesis of the RyR activator cADPR, we next asked whether bile acid-induced Ca2+ release is mediated by cADPR.
We found that pretreatment with the cADPR inhibitor 8-Br-cADPR (30 μm) reduced the TLCS-stimulated Ca2+ transient by 75% (p < 0.05) (Fig. 3). The effect was reversible because a return peak was observed after the inhibitor was washed out. These data suggest that the mechanism for CD38-mediated Ca2+ release following TLCS stimulation is through cADPR.
FIGURE 3.
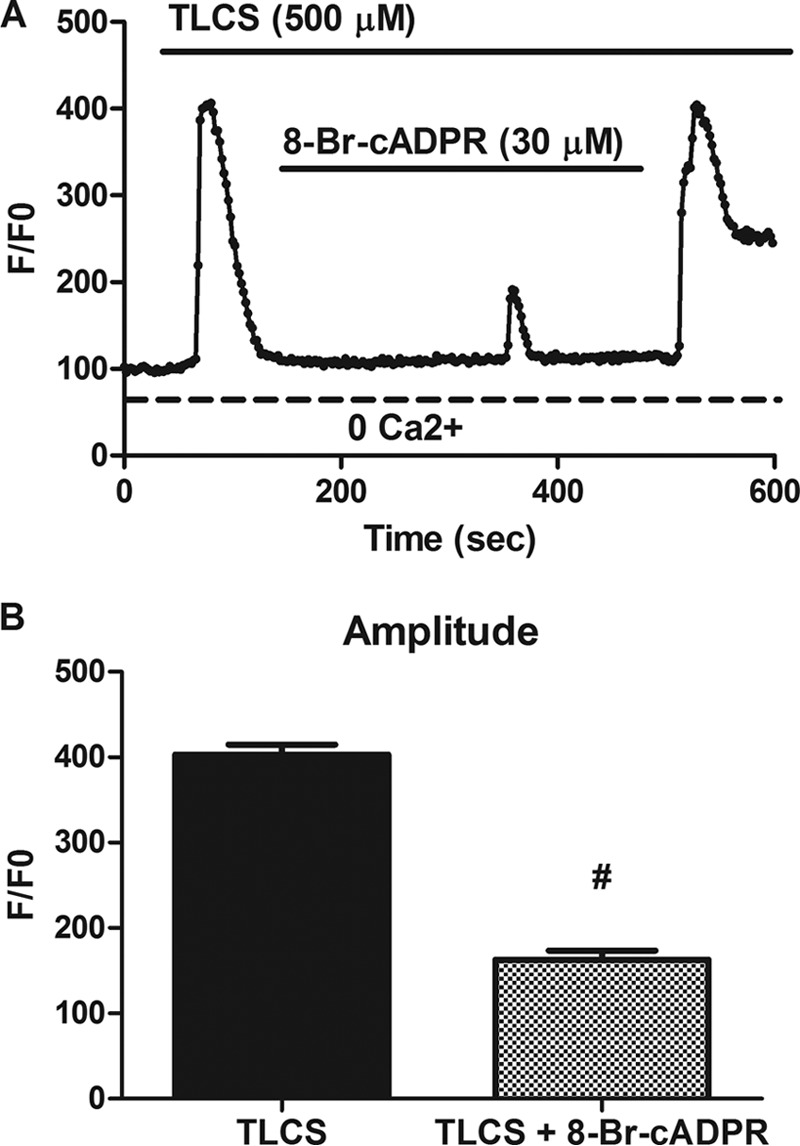
Bile acid-induced Ca2+ signals are dependent on cADPR. A, representative Ca2+ signals observed in cells perifused with TLCS (500 μm) with or without the cADPR inhibitor 8-Br-cADPR (30 μm). B, amplitude of the Ca2+ signal represented as normalized fluorescence. Data were obtained from three separate days of experimentation (n = 50–60 cells/condition). #, p < 0.05 compared with TLCS alone.
Bile Acid-induced Cell Injury Is Dependent on CD38-cADPR
Bile acids cause injury to pancreatic acinar cells (18, 42, 43). The injury is dependent on an aberrant rise in cytosolic Ca2+ release. To examine whether CD38 is involved in the pathogenesis of acinar injury, we pretreated isolated acinar cells with varying concentrations of nicotinamide prior to TLCS stimulation. Higher concentrations of nicotinamide abrogated TLCS-induced LDH release (p < 0.05) (Fig. 4A). In addition, nicotinamide significantly reduced propidium iodide uptake (p < 0.05) (Fig. 4B).
FIGURE 4.

Bile acid-induced cell injury is dependent on CD38. Acinar cells were pretreated with increasing concentrations of nicotinamide for 30 min prior to a 4-h incubation with TLCS (500 μm). Cell injury was measured by percent LDH release (A) and propidium iodide uptake (B). C, human acinar cells were obtained from a cadaveric specimen and stimulated with TLCS (500 μm) with or without nicotinamide. Cell injury was measured as percent LDH release. Wild-type or Cd38-deficient acinar cells were stimulated with TLCS (500 μm) for 6 h, and cell injury was assessed by LDH release (D) and propidium iodide uptake (E) (n = 3). * and #, p < 0.05 compared with the control and TLCS alone, respectively. RLU, relative light units.
To determine the relevance of the current findings to the human condition, we obtained live human pancreatic acinar cells from a 9-year-old female donor who died of anoxia. The cold ischemia time was ∼13 h. The cells were stimulated with TLCS in the presence or absence of nicotinamide. Similar to what was seen in rodent acini, TLCS (500 μm) caused an increase in LDH leakage (p < 0.05) (Fig. 4C). In addition, nicotinamide prevented cell injury (p < 0.05). Although the data are limited due to the availability of fresh human samples and nonspecific effects of the CD38 inhibitor, they provide relevance to the experimental finding that CD38 plays a critical role in mediating bile acid-induced acinar cell injury.
To further examine the role of CD38 and cADPR in cell injury, we isolated acinar cells from Cd38−/− mice and stimulated them for 6 h with TLCS (500 μm). Cell injury measurements revealed that Cd38-deficient acinar cells were protected against TLCS-induced cell injury over the duration of the 6-h time course (Fig. 4, D and E). We also observed reductions in TLCS-induced cell injury (down to the base line) following pretreatment with 8-Br-cADPR (Fig. 5). To understand whether premature activation of digestive enzymes contributes to this injury, we measured the activity of the protease chymotrypsin from wild-type and Cd38-deficient mice simulated with TLCS. Fig. 6 shows that 1 h after stimulation, TLCS induced a 6-fold increase in chymotrypsin activity. The activity was reduced by 20% relative to control levels in Cd38-deficient acinar cells (p < 0.05). Taken together, these data demonstrate the importance of CD38 and cADPR in bile acid-induced cell injury and protease activation.
FIGURE 5.

Bile acid-induced cell injury is dependent on cADPR. Acinar cells were pretreated with the cADPR inhibitor 8-Br-cADPR (30 μm) for 30 min prior to a 2-h incubation with TLCS (500 μm). Cell injury was measured by percent LDH release (A) and propidium iodide uptake (B) (n = 3). * and #, p < 0.05 compared with the control and TLCS alone, respectively.
FIGURE 6.

Bile acid-induced chymotrypsin activity is dependent on CD38. Acinar cells from wild-type or Cd38-deficient mice were stimulated with TLCS (500 μm). Chymotrypsin activity was measured 1 h later. Data are expressed as -fold increase versus control (n = 3). * and #, p < 0.05 compared with the control and TLCS alone, respectively.
Cd38−/− Mice Are Protected against Bile Acid Infusion Pancreatitis
To examine the clinical relevance of CD38-cADPR in the intact animal, we employed an in vivo model of bile acid infusion in which wild-type or Cd38−/− mice received a brief retrograde duct infusion of TLCS (3 mm). We evaluated pancreatic sections for early indices of acute pancreatitis, including edema, inflammation, vacuolization, and necrosis. Remarkably, the Cd38-deficient mice were protected against pancreatitis. Each of the histological parameters of pancreatic injury was reduced to control levels (Fig. 7).
FIGURE 7.

Cd38−/− mice are protected against bile acid infusion pancreatitis. TLCS (3 mm) was infused into the pancreatic duct of wild-type or Cd38−/− mice. Tissues were collected 24 h after infusion. A, representative H&E sections from the pancreatic head. B and C, overall severity scores and subscores, respectively (n = five to seven animals/group). * and #, p < 0.05 compared with the wild-type control and TLCS, respectively.
DISCUSSION
The key findings of this study are that CD38 and cADPR mediate TLCS-induced Ca2+ release, acinar cell injury, and bile acid infusion pancreatitis in vivo. Although it is known that bile acids trigger several Ca2+-mediated injurious pathways within the acinar cell, neither the origin of the aberrant Ca2+ signals nor their targets have been fully clarified. Bile acids may work through ligand binding of a G protein-coupled bile acid receptor (Gpbar1; also known as Tgr5) or through bile transport into the cell (7). They can cause sustained Ca2+ release, which then triggers opening of store-operated Ca2+ entry channels (7). In addition, several targets of aberrant acinar cell Ca2+ following bile acid exposure have been suggested and include mitochondria (44, 45), sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps (7), and a host of Ca2+-mediated proteins (28, 42). We have shown that the serine/threonine phosphatase calcineurin is activated in response to Ca2+ generated by bile acid exposure (42) and that calcineurin plays an important role in regulating bile acid-induced NF-κB activation (46).
In the acinar cell, bile acids potentiate Ca2+ release from the endoplasmic reticulum and vesicular Ca2+ stores through opening of both inositol 1,4,5-trisphosphate receptors and RyRs (5, 28). TLCS-induced Ca2+ transients were reduced by the inositol 1,4,5-trisphosphate receptor inhibitor caffeine (5) and in two-photon permeabilized acinar cells by the RyR inhibitor ruthenium red (28). The latter result is of particular interest because RyR-dependent Ca2+ release in the pancreatic acinar cell is a potential target for blocking aberrant Ca2+ signals. The RyR was initially shown to mediate pancreatitis in a secretagogue hyperstimulation model of pancreatitis (10, 15) and, subsequently, during bile acid exposure (18, 28).
cADPR was first identified as a critical Ca2+ second messenger in sea urchins (47, 48). Okamoto and co-workers (49) demonstrated that cADPR modulates stimulus-secretion coupling in the pancreas using permeabilized mouse islets. Several groups have demonstrated a role for cADPR in the generation of cytosolic Ca2+ signals in the pancreatic acinar cell. First, either intracellular application of cADPR (50) or localized uncaging of cADPR (51, 52) can induce Ca2+ signals or cause depletion of the intracellular Ca2+ pool (53–55). Second, cADPR levels within the acinar cell rise during both acetylcholine and cholecystokinin stimulation (30, 56, 57). Third, the cADPR antagonist 8-amino-cADPR inhibits Ca2+ transients induced by cholecystokinin (58), acetylcholine (51), or bombesin (59). To our knowledge, this work is the first to show that cADPR also mediates acinar cell Ca2+ signals and injury following bile acid exposure.
cADPR is synthesized from NAD+ by ADP-ribosyl cyclases. The main ADP-ribosyl cyclase that generates cADPR in the acinar cell appears to be CD38 because Cd38−/− acini have reduced cADPR levels and fail to increase cADPR levels following secretagogue stimulation (30, 56). In contrast, non-CD38 ribosyl cyclases are active in brain (60) and may be contributed by another member of the superfamily of ribosyl cyclases, CD157, also known as BST-1 (61–63). Several important physiologic roles have been associated with CD38, including the control of insulin secretion (64), clearance of bacterial infections (60), and altered social behavior (65). Our findings using primarily Cd38−/− mice demonstrate that CD38 regulates pancreatic acinar cell injury and in vivo pancreatitis following exposure to bile acids.
Our data raise a few unresolved questions in the CD38 field (47). First, it is unclear how a stimulus, e.g. a bile acid, regulates CD38. The regulation of CD38 by cGMP (66) or cAMP-mediated phosphorylation (67) has been suggested. Second, because CD38 is an ectoenzyme, there is the topological paradox of how a cytosolic substrate such as NAD+ comes into contact with the active site of CD38, which faces the extracellular space, and then how its product cADPR is shuttled back into the cell (68). Third, CD38 synthesizes NAADP and NAD+, two other nucleotides besides cADPR that can affect Ca2+ signals. NAADP is derived from NADP and is a potent activator of the two pore channels in endolysosomes (69). The other nucleotide made by CD38 from NAD+ is the non-cyclic compound ADPR, which activates the Ca2+ influx channel TRPM2 (transient receptor potential cation channel member 2) (60, 70). For this reason, our Ca2+ signaling studies were designed to exclude the influence of Ca2+ influx by using Ca2+-free medium. Nonetheless, we cannot exclude the possibility that CD38-generated ADPR contributes to acinar cell injury or pancreatitis due to bile acids. In summary, we offer the first demonstration that bile acids cause acinar cell Ca2+ release, injury, and pancreatitis through CD38. These data provide further insight into the Ca2+-dependent mechanisms of bile acid-induced pathology and suggest that the CD38-cADPR pathway may serve as a target for the treatment of some forms of pancreatitis.
Acknowledgments
We acknowledge Drs. Mark Lowe and George Perides for helpful discussion and Taimur Ahmad and Iman Benbourenane for assistance in editing the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants DK083327 and DK093491 (to S. Z. H.).
- RyR
- ryanodine receptor
- cADPR
- cyclic ADP-ribose
- NAADP
- nicotinic acid adenine dinucleotide phosphate
- TLCS
- taurolithocholic acid 3-sulfate
- LDH
- lactate dehydrogenase.
REFERENCES
- 1. Petersen O. H. (2005) Ca2+ signalling and Ca2+-activated ion channels in exocrine acinar cells. Cell Calcium 38, 171–200 [DOI] [PubMed] [Google Scholar]
- 2. Williams J. A. (2001) Intracellular signaling mechanisms activated by cholecystokinin-regulating synthesis and secretion of digestive enzymes in pancreatic acinar cells. Annu. Rev. Physiol. 63, 77–97 [DOI] [PubMed] [Google Scholar]
- 3. Parekh A. B. (2000) Calcium signaling and acute pancreatitis: specific response to a promiscuous messenger. Proc. Natl. Acad. Sci. U.S.A. 97, 12933–12934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mooren F. Ch., Hlouschek V., Finkes T., Turi S., Weber I. A., Singh J., Domschke W., Schnekenburger J., Krüger B., Lerch M. M. (2003) Early changes in pancreatic acinar cell calcium signaling after pancreatic duct obstruction. J. Biol. Chem. 278, 9361–9369 [DOI] [PubMed] [Google Scholar]
- 5. Voronina S., Longbottom R., Sutton R., Petersen O. H., Tepikin A. (2002) Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J. Physiol. 540, 49–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Criddle D. N., Raraty M. G., Neoptolemos J. P., Tepikin A. V., Petersen O. H., Sutton R. (2004) Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc. Natl. Acad. Sci. U.S.A. 101, 10738–10743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim J. Y., Kim K. H., Lee J. A., Namkung W., Sun A. Q., Ananthanarayanan M., Suchy F. J., Shin D. M., Muallem S., Lee M. G. (2002) Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology 122, 1941–1953 [DOI] [PubMed] [Google Scholar]
- 8. Raraty M., Ward J., Erdemli G., Vaillant C., Neoptolemos J. P., Sutton R., Petersen O. H. (2000) Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 97, 13126–13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krüger B., Albrecht E., Lerch M. M. (2000) The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am. J. Pathol. 157, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Husain S. Z., Prasad P., Grant W. M., Kolodecik T. R., Nathanson M. H., Gorelick F. S. (2005) The ryanodine receptor mediates early zymogen activation in pancreatitis. Proc. Natl. Acad. Sci. U.S.A. 102, 14386–14391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han B., Logsdon C. D. (2000) CCK stimulates mob-1 expression and NF-κB activation via protein kinase C and intracellular Ca2+. Am. J. Physiol. Cell Physiol. 278, C344–C351 [DOI] [PubMed] [Google Scholar]
- 12. Mareninova O. A., Hermann K., French S. W., O'Konski M. S., Pandol S. J., Webster P., Erickson A. H., Katunuma N., Gorelick F. S., Gukovsky I., Gukovskaya A. S. (2009) Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J. Clin. Invest. 119, 3340–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sherwood M. W., Prior I. A., Voronina S. G., Barrow S. L., Woodsmith J. D., Gerasimenko O. V., Petersen O. H., Tepikin A. V. (2007) Activation of trypsinogen in large endocytic vacuoles of pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 104, 5674–5679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Criddle D. N., Murphy J., Fistetto G., Barrow S., Tepikin A. V., Neoptolemos J. P., Sutton R., Petersen O. H. (2006) Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology 130, 781–793 [DOI] [PubMed] [Google Scholar]
- 15. Orabi A. I., Shah A. U., Ahmad M. U., Choo-Wing R., Parness J., Jain D., Bhandari V., Husain S. Z. (2010) Dantrolene mitigates caerulein-induced pancreatitis in vivo in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G196–G204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim M. S., Hong J. H., Li Q., Shin D. M., Abramowitz J., Birnbaumer L., Muallem S. (2009) Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim M. S., Lee K. P., Yang D., Shin D. M., Abramowitz J., Kiyonaka S., Birnbaumer L., Mori Y., Muallem S. (2011) Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology 140, 2107–2115.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Husain S. Z., Orabi A. I., Muili K. A., Luo Y., Sarwar S., Mahmood S. M., Wang D., Choo-Wing R., Singh V. P., Parness J., Ananthanaravanan M., Bhandari V., Perides G. (2012) Ryanodine receptors contribute to bile acid-induced pathological calcium signaling and pancreatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G1423–G1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gerasimenko J. V., Lur G., Sherwood M. W., Ebisui E., Tepikin A. V., Mikoshiba K., Gerasimenko O. V., Petersen O. H. (2009) Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc. Natl. Acad. Sci. U.S.A. 106, 10758–10763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pandol S. J., Periskic S., Gukovsky I., Zaninovic V., Jung Y., Zong Y., Solomon T. E., Gukovskaya A. S., Tsukamoto H. (1999) Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology 117, 706–716 [DOI] [PubMed] [Google Scholar]
- 21. Straub S. V., Giovannucci D. R., Yule D. I. (2000) Calcium wave propagation in pancreatic acinar cells: functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J. Gen. Physiol. 116, 547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leite M. F., Dranoff J. A., Gao L., Nathanson M. H. (1999) Expression and subcellular localization of the ryanodine receptor in rat pancreatic acinar cells. Biochem. J. 337, 305–309 [PMC free article] [PubMed] [Google Scholar]
- 23. Lee H. C. (1993) Potentiation of calcium- and caffeine-induced calcium release by cyclic ADP-ribose. J. Biol. Chem. 268, 293–299 [PubMed] [Google Scholar]
- 24. Lee H. C., Aarhus R., Graeff R., Gurnack M. E., Walseth T. F. (1994) Cyclic ADP-ribose activation of the ryanodine receptor is mediated by calmodulin. Nature 370, 307–309 [DOI] [PubMed] [Google Scholar]
- 25. Lee H. C., Aarhus R., Graeff R. M. (1995) Sensitization of calcium-induced calcium release by cyclic ADP-ribose and calmodulin. J. Biol. Chem. 270, 9060–9066 [DOI] [PubMed] [Google Scholar]
- 26. Tanaka Y., Tashjian A. H., Jr. (1995) Calmodulin is a selective mediator of Ca2+-induced Ca2+ release via the ryanodine receptor-like Ca2+ channel triggered by cyclic ADP-ribose. Proc. Natl. Acad. Sci. U.S.A. 92, 3244–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bai H. X., Lowe M. E., Husain S. Z. (2011) What have we learned about acute pancreatitis in children? J. Pediatr. Gastroenterol. Nutr. 52, 262–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gerasimenko J. V., Flowerdew S. E., Voronina S. G., Sukhomlin T. K., Tepikin A. V., Petersen O. H., Gerasimenko O. V. (2006) Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J. Biol. Chem. 281, 40154–40163 [DOI] [PubMed] [Google Scholar]
- 29. Gerasimenko J. V., Lur G., Ferdek P., Sherwood M. W., Ebisui E., Tepikin A. V., Mikoshiba K., Petersen O. H., Gerasimenko O. V. (2011) Calmodulin protects against alcohol-induced pancreatic trypsinogen activation elicited via Ca2+ release through IP3 receptors. Proc. Natl. Acad. Sci. U.S.A. 108, 5873–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cosker F., Cheviron N., Yamasaki M., Menteyne A., Lund F. E., Moutin M. J., Galione A., Cancela J. M. (2010) The ecto-enzyme CD38 is a nicotinic acid adenine dinucleotide phosphate (NAADP) synthase that couples receptor activation to Ca2+ mobilization from lysosomes in pancreatic acinar cells. J. Biol. Chem. 285, 38251–38259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cockayne D. A., Muchamuel T., Grimaldi J. C., Muller-Steffner H., Randall T. D., Lund F. E., Murray R., Schuber F., Howard M. C. (1998) Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood 92, 1324–1333 [PubMed] [Google Scholar]
- 32. Orabi A. I., Shah A. U., Muili K., Luo Y., Mahmood S. M., Ahmad A., Reed A., Husain S. Z. (2011) Ethanol enhances carbachol-induced protease activation and accelerates Ca2+ waves in isolated rat pancreatic acini. J. Biol. Chem. 286, 14090–14097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bottino R., Bertera S., Grupillo M., Melvin P. R., Humar A., Mazariegos G., Moser A. J., Walsh R. M., Fung J., Gelrud A., Slivka A., Soltys K., Wijkstrom M., Trucco M. (2012) Isolation of human islets for autologous islet transplantation in children and adolescents with chronic pancreatitis. J. Transplant. 2012, 642787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ricordi C. (ed) (1995) Methods in Cell Transplantation, R. G. Landes, Austin, TX [Google Scholar]
- 35. Chaudhuri A., Husain S. Z., Kolodecik T. R., Grant W. M., Gorelick F. S. (2007) Cyclic AMP-dependent protein kinase and Epac mediate cyclic AMP responses in pancreatic acini. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1403–G1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perides G., van Acker G. J., Laukkarinen J. M., Steer M. L. (2010) Experimental acute biliary pancreatitis induced by retrograde infusion of bile acids into the mouse pancreatic duct. Nat. Protoc. 5, 335–341 [DOI] [PubMed] [Google Scholar]
- 37. Wildi S., Kleeff J., Mayerle J., Zimmermann A., Böttinger E. P., Wakefield L., Büchler M. W., Friess H., Korc M. (2007) Suppression of transforming growth factor β signalling aborts caerulein induced pancreatitis and eliminates restricted stimulation at high caerulein concentrations. Gut 56, 685–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hofmann A. F., Roda A. (1984) Physicochemical properties of bile acids and their relationship to biological properties: an overview of the problem. J. Lipid Res. 25, 1477–1489 [PubMed] [Google Scholar]
- 39. Siegel C., McCullough L. D. (2011) NAD+ depletion or PAR polymer formation: which plays the role of executioner in ischaemic cell death? Acta Physiol. 203, 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu D., Gharavi R., Pitta M., Gleichmann M., Mattson M. P. (2009) Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromol. Med. 11, 28–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gale E. A. (1996) Molecular mechanisms of β-cell destruction in IDDM: the role of nicotinamide. Horm. Res. 45, 39–43 [PubMed] [Google Scholar]
- 42. Muili K. A., Wang D., Orabi A. I., Sarwar S., Luo Y., Javed T. A., Eisses J. F., Mahmood S. M., Jin S., Singh V. P., Ananthanaravanan M., Perides G., Williams J. A., Molkentin J. D., Husain S. Z. (2013) Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J. Biol. Chem. 288, 570–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Perides G., Laukkarinen J. M., Vassileva G., Steer M. L. (2010) Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology 138, 715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Voronina S. G., Barrow S. L., Gerasimenko O. V., Petersen O. H., Tepikin A. V. (2004) Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating Δψm. J. Biol. Chem. 279, 27327–27338 [DOI] [PubMed] [Google Scholar]
- 45. Voronina S. G., Barrow S. L., Simpson A. W., Gerasimenko O. V., da Silva Xavier G., Rutter G. A., Petersen O. H., Tepikin A. V. (2010) Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology 138, 1976–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Muili K. A., Jin S., Orabi A. I., Eisses J. F., Javed T. A., Le T., Bottino R., Jayaraman T., Husain S. Z. (2013) Pancreatic acinar cell nuclear factor κB activation because of bile acid exposure is dependent on calcineurin. J. Biol. Chem. 288, 21065–21073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee H. C. (2012) Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 287, 31633–31640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Galione A., Lee H. C., Busa W. B. (1991) Ca2+-induced Ca2+ release in sea urchin egg homogenates: modulation by cyclic ADP-ribose. Science 253, 1143–1146 [DOI] [PubMed] [Google Scholar]
- 49. Takasawa S., Nata K., Yonekura H., Okamoto H. (1993) Cyclic ADP-ribose in insulin secretion from pancreatic β cells. Science 259, 370–373 [DOI] [PubMed] [Google Scholar]
- 50. Thorn P., Gerasimenko O., Petersen O. H. (1994) Cyclic ADP-ribose regulation of ryanodine receptors involved in agonist evoked cytosolic Ca2+ oscillations in pancreatic acinar cells. EMBO J. 13, 2038–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leite M. F., Burgstahler A. D., Nathanson M. H. (2002) Ca2+ waves require sequential activation of inositol trisphosphate receptors and ryanodine receptors in pancreatic acini. Gastroenterology 122, 415–427 [DOI] [PubMed] [Google Scholar]
- 52. Yamasaki M., Masgrau R., Morgan A. J., Churchill G. C., Patel S., Ashcroft S. J., Galione A. (2004) Organelle selection determines agonist-specific Ca2+ signals in pancreatic acinar and β cells. J. Biol. Chem. 279, 7234–7240 [DOI] [PubMed] [Google Scholar]
- 53. Gerasimenko J. V., Sherwood M., Tepikin A. V., Petersen O. H., Gerasimenko O. V. (2006) NAADP, cADPR and IP3 all release Ca2+ from the endoplasmic reticulum and an acidic store in the secretory granule area. J. Cell Sci. 119, 226–238 [DOI] [PubMed] [Google Scholar]
- 54. Göbel A., Krause E., Feick P., Schulz I. (2001) IP3 and cyclic ADP-ribose induced Ca2+ release from intracellular stores of pancreatic acinar cells from rat in primary culture. Cell Calcium 29, 29–37 [DOI] [PubMed] [Google Scholar]
- 55. Krause E., Göbel A., Schulz I. (2002) Cell side-specific sensitivities of intracellular Ca2+ stores for inositol 1,4,5-trisphosphate, cyclic ADP-ribose, and nicotinic acid adenine dinucleotide phosphate in permeabilized pancreatic acinar cells from mouse. J. Biol. Chem. 277, 11696–11702 [DOI] [PubMed] [Google Scholar]
- 56. Fukushi Y., Kato I., Takasawa S., Sasaki T., Ong B. H., Sato M., Ohsaga A., Sato K., Shirato K., Okamoto H., Maruyama Y. (2001) Identification of cyclic ADP-ribose-dependent mechanisms in pancreatic muscarinic Ca2+ signaling using CD38 knockout mice. J. Biol. Chem. 276, 649–655 [DOI] [PubMed] [Google Scholar]
- 57. Yamasaki M., Thomas J. M., Churchill G. C., Garnham C., Lewis A. M., Cancela J. M., Patel S., Galione A. (2005) Role of NAADP and cADPR in the induction and maintenance of agonist-evoked Ca2+ spiking in mouse pancreatic acinar cells. Curr. Biol. 15, 874–878 [DOI] [PubMed] [Google Scholar]
- 58. Cancela J. M., Petersen O. H. (1998) The cyclic ADP ribose antagonist 8-NH2-cADP-ribose blocks cholecystokinin-evoked cytosolic Ca2+ spiking in pancreatic acinar cells. Pflugers Arch. 435, 746–748 [DOI] [PubMed] [Google Scholar]
- 59. Burdakov D., Cancela J. M., Petersen O. H. (2001) Bombesin-induced cytosolic Ca2+ spiking in pancreatic acinar cells depends on cyclic ADP-ribose and ryanodine receptors. Cell Calcium 29, 211–216 [DOI] [PubMed] [Google Scholar]
- 60. Partida-Sánchez S., Cockayne D. A., Monard S., Jacobson E. L., Oppenheimer N., Garvy B., Kusser K., Goodrich S., Howard M., Harmsen A., Randall T. D., Lund F. E. (2001) Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med. 7, 1209–1216 [DOI] [PubMed] [Google Scholar]
- 61. Kaisho T., Ishikawa J., Oritani K., Inazawa J., Tomizawa H., Muraoka O., Ochi T., Hirano T. (1994) BST-1, a surface molecule of bone marrow stromal cell lines that facilitates pre-B-cell growth. Proc. Natl. Acad. Sci. U.S.A. 91, 5325–5329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Itoh M., Ishihara K., Tomizawa H., Tanaka H., Kobune Y., Ishikawa J., Kaisho T., Hirano T. (1994) Molecular cloning of murine BST-1 having homology with CD38 and Aplysia ADP-ribosyl cyclase. Biochem. Biophys. Res. Commun. 203, 1309–1317 [DOI] [PubMed] [Google Scholar]
- 63. Kajimoto Y., Miyagawa J., Ishihara K., Okuyama Y., Fujitani Y., Itoh M., Yoshida H., Kaisho T., Matsuoka T., Watada H., Hanafusa T., Yamasaki Y., Kamada T., Matsuzawa Y., Hirano T. (1996) Pancreatic islet cells express BST-1, a CD38-like surface molecule having ADP-ribosyl cyclase activity. Biochem. Biophys. Res. Commun. 219, 941–946 [DOI] [PubMed] [Google Scholar]
- 64. Kato I., Yamamoto Y., Fujimura M., Noguchi N., Takasawa S., Okamoto H. (1999) CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J. Biol. Chem. 274, 1869–1872 [DOI] [PubMed] [Google Scholar]
- 65. Higashida H., Salmina A. B., Olovyannikova R. Y., Hashii M., Yokoyama S., Koizumi K., Jin D., Liu H.-X., Lopatina O., Amina S., Islam M. S., Huang J.-J., Noda M. (2007) Cyclic ADP-ribose as a universal calcium signal molecule in the nervous system. Neurochem. Int. 51, 192–199 [DOI] [PubMed] [Google Scholar]
- 66. Galione A., White A., Willmott N., Turner M., Potter B. V., Watson S. P. (1993) cGMP mobilizes intracellular Ca2+ in sea urchin eggs by stimulating cyclic ADP-ribose synthesis. Nature 365, 456–459 [DOI] [PubMed] [Google Scholar]
- 67. Bruzzone S., Moreschi I., Usai C., Guida L., Damonte G., Salis A., Scarfì S., Millo E., De Flora A., Zocchi E. (2007) Abscisic acid is an endogenous cytokine in human granulocytes with cyclic ADP-ribose as second messenger. Proc. Natl. Acad. Sci. U.S.A. 104, 5759–5764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. De Flora A., Guida L., Franco L., Zocchi E. (1997) The CD38/cyclic ADP-ribose system: a topological paradox. Int. J. Biochem. Cell Biol. 29, 1149–1166 [DOI] [PubMed] [Google Scholar]
- 69. Lee H. C., Aarhus R. (1995) A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J. Biol. Chem. 270, 2152–2157 [DOI] [PubMed] [Google Scholar]
- 70. Togashi K., Hara Y., Tominaga T., Higashi T., Konishi Y., Mori Y., Tominaga M. (2006) TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 25, 1804–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]