

Abstract
Airway epithelia absorb Na+ through the epithelial Na+ channel (ENaC) and secrete Cl− through the cystic fibrosis transmembrane conductance regulator (CFTR) anion channel. This balance maintains sufficient airway surface liquid hydration to permit efficient mucus clearance, which is needed to maintain sterility of the lung. Cystic fibrosis (CF) is a common autosomal recessive inherited disease caused by mutations in the CFTR gene that lead to the reduction or elimination of the CFTR protein. CF is a multi-organ disease that affects epithelia lining the intestines, lungs, pancreas, sweat ducts and vas deferens, among others. CF lungs are characterized by viscous, dehydrated mucus, persistent neutrophilia and chronic infections. ENaC is negatively regulated by CFTR and, in patients with CF, the absence of CFTR results in a double hit of reduced Cl−/HCO3− and H2O secretion as well as ENaC hyperactivity and increased Na+ and H2O absorption. Together, these effects are hypothesized to trigger mucus dehydration, resulting in a failure to clear mucus. Rehydrating CF mucus has become a recent clinical focus and yields important end-points for clinical trials. However, while ENaC hyperactivity in CF airways has been detected in vivo and in vitro, recent data have brought the role of ENaC in CF lung disease pathogenesis into question. This review will focus on our current understanding of the contribution of ENaC to CF pathogenesis.
Key points
Lung hydration and mucus clearance rates are set by a balance between CFTR-mediated Cl− secretion and ENaC-led Na+ absorption. In CF airways, CFTR is diminished, and ENaC is upregulated, leading to mucus dehydration and increased chance of infection.
Evidence for ENaC upregulation in CF airways includes electrophysiological evidence, increased ASL absorption rates, increased cleavage of CF ENaC and increased basolateral Na+/K+ ATPase activity in CF airways.
The mechanism of Na+ hyperabsorption in CF airways is unknown and it may be due to altered protein-protein interactions and/or increased proteolysis of ENaC in CF airways. However, inhibition of ENaC is predicted to increase CF mucus hydration/clearance and thus, ENaC remains an important therapeutic target for the treatment of CF lung disease.
 |
Carey Hobbs (left), Chong Da Tan (centre) and Robert Tarran (right) all performed studies on the epithelial sodium channel (ENaC) in the Cystic Fibrosis/Pulmonary Research and Treatment Centre in the University of North Carolina at Chapel Hill. Carey is a structural biologist/biochemist, while Chong and Robert are physiologists with backgrounds in electrophysiology and imaging. One of the major research efforts in the lab is in understanding how SPLUNC1 proteins can regulate ENaC in healthy and diseased airways. This present paper discusses the relevance of ENaC hyperactivity to cystic fibrosis lung disease.
Introduction
The epithelial Na+ channel (ENaC) constitutes the rate-limiting step for Na+ absorption in the airways and is postulated to play a significant role in influencing mucus hydration levels (Zhou et al. 2011; Fig. 1A and B). ENaC is a heterotrimer that is typically composed of α,β and γ subunits (Stockand et al. 2008). However, in some tissues, a fourth δ-ENaC subunit may be expressed, resulting in the formation of δαβγ-ENaCs with altered biophysical characteristics (Ji et al. 2006; Bangel-Ruland et al. 2010). The extracellular loops of α- and γ-ENaC subunits can be proteolytically cleaved at multiple sites, leading to activation of ENaC and increased Na+ absorption, whilst β-ENaC may serve as a regulatory subunit (Gaillard et al. 2010). Protein–protein interactions, shear stress, cellular trafficking and intracellular 2nd messengers including Na+, cAMP and PIP2 also play roles in regulating ENaC, and may have different effects depending on where ENaC is expressed (Kashlan & Kleyman, 2012; Palmer et al. 2012; Soundararajan et al. 2012; Thibodeau & Butterworth, 2013). As such, ENaC is highly sensitive to both the intracellular and extracellular environments, and can rapidly alter its activity depending on the needs of the lungs. In cystic fibrosis (CF) airways, Na+ absorption is elevated, either as a direct or indirect consequence of the lack of CF transmembrane conductance regulator (CFTR). This increase in Na+ absorption may be due to a constitutive increase in ENaC activity, excessive proteolytic cleavage of ENaC and/or abnormal activation of ENaC by cAMP, all of which may contribute to a depletion of airway surface liquid (ASL) volume (Fig. 1A and B; Gaillard et al. 2010).
Figure 1. Na+ hyperabsorption leads to increased O2 consumption and airway surface liquid (ASL) volume depletion.
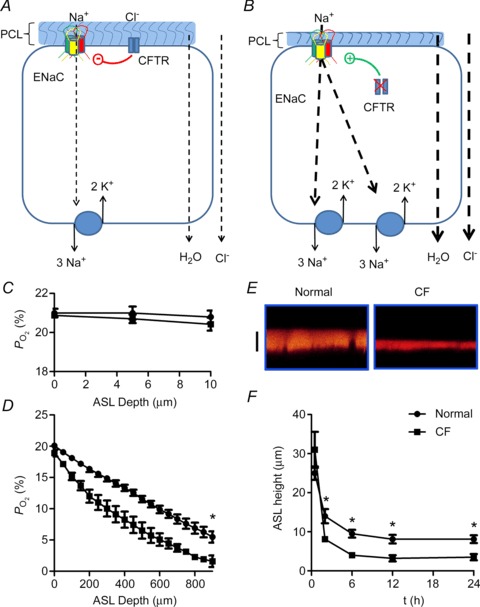
A and B, schematic diagrams of Na+ absorption in normal and cystic fibrosis (CF) airway epithelia, respectively. Na+ is transported down its electrochemical gradient through epithelial Na+ channel (ENaC) in the apical membrane and is pumped out across the basolateral membrane by the Na+/K+-ATPase. Because airway epithelia are highly water permeable and have NaCl-permeable paracellular pathways, Cl− and H2O follow Na+. C, under thin-film conditions (in this case, ASL height was set at ∼10 μm in both normal and CF), the near apical membrane 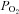 is ∼21%. D, when cultures are flooded with 500 μl Ringer,
is ∼21%. D, when cultures are flooded with 500 μl Ringer, 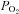 declines with depth. Due to upregulated Na+/K+-ATPase activity, CF HBECs consume more O2 than normal cultures. E, representative ASL images obtained by XZ-confocal microscopy of Texas red-dextran (10 kDa) on normal and CF HBECs. F, mean ASL height with time in normal and CF HBECs taken from E, following addition of 20 μl Ringer solution at t= 0. *P < 0.05 difference between normal and CF. CFTR, cystic fibrosis transmembrane conductance regulator; periciliary liquid layer (PCL).
declines with depth. Due to upregulated Na+/K+-ATPase activity, CF HBECs consume more O2 than normal cultures. E, representative ASL images obtained by XZ-confocal microscopy of Texas red-dextran (10 kDa) on normal and CF HBECs. F, mean ASL height with time in normal and CF HBECs taken from E, following addition of 20 μl Ringer solution at t= 0. *P < 0.05 difference between normal and CF. CFTR, cystic fibrosis transmembrane conductance regulator; periciliary liquid layer (PCL).
ENaC-led dehydration is thought to induce mucus stasis and increase the incidence of airway infections, leading to pulmonary destruction and patient morbidity (Zhou et al. 2011; Thibodeau & Butterworth, 2013). Genes encoding ENaC subunits may further affect the severity of CF by causing gain of function type mutations in ENaC. For example, in patients with atypical CF, a heterozygous mutation in the α-ENaC subunit (W493R) reduces the inhibitory effect of extracellular Na+ leading to increased ENaC activity, thus contributing to the pathophysiology of patients with CF carrying this mutation (Rauh et al. 2010). Similarly, a mutation in the β subunit, V348M, was also identified, which increased ENaC open probability by destabilizing the closed channel state (Mutesa et al. 2009; Rauh et al. 2013). ENaC has also been associated with other diseases. For instance, Liddle's syndrome (pseudoaldosteronism) is caused by mutations in β- or γ-ENaC PY motifs that prevent ENaC from being degraded by NEDD4–2 and the ubiquitin ligase system. This leads to an accumulation of surface ENaC in the kidney, Na+ retention and serious hypertension (Schild et al. 1996). However, ENaC in the lungs of patients with Liddle's syndrome is unaffected as it can still be functionally inhibited by CFTR (Hopf et al. 1999; Mall et al. 2010). As well as gain of function ENaC mutations, loss of function ENaC mutations associated with pseudohypoaldosteronism type 1 (PHA-1) have been detected (Grunder et al. 1997). Here, ENaC activity is severely diminished, resulting in significantly increased fluid volume in the lung (Kerem et al. 1999). Surprisingly, whilst too much ENaC is detrimental to lung health, too little ENaC is not, and patients with PHA-1 clear the excess liquid by an increase in mucociliary transport rates above and beyond those seen in normal subjects without adverse effects (Kerem et al. 1999).
The role of ENaC in CF lung disease has recently been reviewed elsewhere (Mall, 2009; Collawn et al. 2012; Althaus, 2013). In this review, we shall discuss ENaC activity in the context of epithelial transport under thick-film versus thin-film conditions, and try to integrate this with what is known of ENaC in animal models and in human lung disease.
Electrophysiological evidence for Na+ hyperabsorption
Boucher and colleagues found that the nasal potential difference and its amiloride-sensitive component were elevated in patients with CF as compared with normal controls (Knowles et al. 1981, 1983a). These experiments were performed in vivo using a flowing electrode placed under the inferior nasal turbinate that was linked to a subcutaneous reference electrode. Due to the paucity of channels/transporters in the apical membrane of airway epithelia, amiloride is essentially specific for ENaC in this tissue (Qadri et al. 2012) and abolishes all Na+ transport (Boucher et al. 1986). In vitro, the amiloride-sensitive short circuit current (IscAmil) and transepithelial voltage (VtAmil) were also elevated in CF airway epithelia as compared with normal and disease controls (Knowles et al. 1983b; Boucher et al. 1986). In contrast, transepithelial resistance (Rt) and conductance (Gt, the reciprocal of resistance) were unchanged (Boucher et al. 1986). These experiments were performed ex vivo on tissues from 41 normal, 25 CF and nine disease control patients (Boucher et al. 1986). In freshly-excised tissues, the conductance of the shunt/paracellular pathway is much larger than that of the transcellular pathway. That is, Rt is very low (∼100 Ω cm−2) and Gt is very high (∼10 mS cm−2). Due to this, the changes in apical membrane Na+ or Cl− permeability seen in normal versus CF airways do not affect the overall shunt-dominated conductance. Because Rt and Gt were unchanged, according to Ohm's law (V=IR), V and I were directly proportional, suggesting that the enhanced Vt seen in vivo was due to increased Isc. Consistent with these observations, the radioactive Na+ flux was elevated in CF tissues in the absorptive direction under both short circuit and open circuit conditions (Boucher et al. 1986; Boucher, 1994).
Consistent with the increase in mucosal to submucosal Na+ flux, basolateral Na+/K+-ATPase activity was also elevated in airway epithelia, leading to increased O2 consumption (Stutts et al. 1986; Worlitzsch et al. 2002; Fig. 1C and D). This increase in Na+/K+-ATPase activity was required to remove excess cellular Na+ due to hyperactive ENaC and served as an independent marker of increased Na+ absorption in CF airways. With the advent of reliable tissue culture techniques, these bioelectric measurements were reprised in polarized airway epithelia, and elevated Na+ currents, potential differences and absorptive fluxes were identified in CF as compared with normal airway cultures, while Rt and Gt remained unchanged (Boucher et al. 1988).
It has been proposed that the elevated Vt and Isc seen in CF airway epithelia were artifacts caused by: (i) increased driving force for Cl− in CF airway epithelia due to the lack of CFTR; and (ii) amiloride-induced hyperpolarization of the apical plasma membrane and subsequent increased Cl− secretion due to this increased driving force (Chen et al. 2010). However, driving forces have been directly measured in excised nasal epithelia using intracellular microelectrodes, and the apical membrane potential (Va) was found to be reduced in CF tissues (−11 ± 5 mV) as opposed to normal tissues (−29 ± 4 mV) due to the dominance of EK/ENa in the absence of ECl (Cotton et al. 1987), suggesting that raised Isc/Vt is not due to increased driving force. Furthermore, microelectrode studies have been performed under thin-film conditions in normal versus CF bronchial epithelia (Tarran et al. 2005, 2006). These studies revealed that the magnitude of Vt was not different between normal and CF bronchial epithelial cultures (both were ∼11 mV), which is similar to levels reported in vivo (∼12 mV; Alton et al. 1991). However, the bumetanide-sensitive component (i.e. Cl− secretion) was significantly greater in normal than CF cultures, and the amiloride-sensitive component (i.e. Na+ absorption) was significantly greater in CF than normal cultures. These data indicate that increased ENaC activity is preserved in primary cultures. Importantly, in these experiments, Cl− secretion was inhibited before the amiloride-sensitive Vt was measured, thus avoiding any confounding effects of apical membrane hyperpolarization due to the effects of amiloride on Cl− secretion (Tarran et al. 2005, 2006).
Beyond electrophysiology: ENaC and ASL regulation
ENaC activity is frequently assayed using Ussing chambers, which have been a mainstay of ion transport studies for over 50 years, but were originally designed by Hans Ussing to study frog skin (Ussing & Zerhan, 1951; Li et al. 2004). To allow for sufficient clamping, epithelial surfaces must be flooded in several milliliters of Ringer solution. While this flooding is appropriate for many types of epithelia, the airways are normally bathed by only a thin-film of ASL (as low as 1 μl cm−2). Over 100 proteins, as well as nucleotides and nucleosides, have been detected in the ASL, which are likely involved in various aspects of innate defense (Kesimer et al. 2009). We have proposed that some of these molecules act as soluble ‘reporter molecules’ that sense ASL height/volume and signal to the underlying epithelia to turn on or off CaCC, CFTR and ENaC as required. Known reporter molecules include ATP, adenosine and SPLUNC1, which primarily regulate CaCC, CFTR and ENaC, respectively. However, ATP and ADO may also regulate ENaC (reviewed elsewhere; Chambers et al. 2007; Gaillard et al. 2010). Thus, the flooding method of experimentation: (i) washes away endogenous ATP and ADO to deactivate spontaneous Cl− secretion; (ii) washes away SPLUNC1, inducing cleavage of ENaC; and (iii) triggers trafficking of ENaC into the plasma membrane through an unknown mechanism that also increases ENaC activity (Myerburg et al. 2006, 2010; Tarran et al. 2006; Tan et al. 2011). As such, airway epithelia mounted in Ussing chambers strongly suffer from the ‘Observer Effect’. That is, the mounting/flooding of airway cultures itself changes ENaC activity. Thus, any experiments performed in Ussing chambers with airway epithelia must take flooding-induced ENaC activation/insertion into account, and should be interpreted accordingly.
In airway epithelia, up to 40% of all O2 is consumed by the Na+/K+ATPase in order to pump Na+ absorbed by ENaC out of the cell. The cardiac glycoside oubain inhibits the Na+/K+ATPase and reduces O2 consumption by airway epithelia. As such, oubain-sensitive O2 consumption is a marker of Na+ absorption and is increased two–threefold in CF airway epithelia, which is consistent with increased ENaC activity (Stutts et al. 1986). Airway epithelia are also sensitive to flooding with regards to changes in ASL O2 levels. O2 has a very low solubility level in ASL, which is ∼98% H2O. However, the diffusion rate of O2 across the ASL is not outpaced by the metabolic demands of the epithelia, and under thin-film conditions, 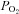 in the ASL is close to atmospheric O2 levels (∼20%; Fig. 1C; Worlitzsch et al. 2002). Tissues bathed in Ussing chambers are typically circulated with 95% O2/5% CO2 gas mix and are not hypoxic. However, addition of Ringer solution with out the gas mix causes the
in the ASL is close to atmospheric O2 levels (∼20%; Fig. 1C; Worlitzsch et al. 2002). Tissues bathed in Ussing chambers are typically circulated with 95% O2/5% CO2 gas mix and are not hypoxic. However, addition of Ringer solution with out the gas mix causes the 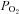 to drastically drop to ∼3% (Fig. 1D; Worlitzsch et al. 2002). Consistent with this finding, application of 100 μl of media to the mucosal side of polarized airway epithelia induced hypoxic stress and resulted in the activation of AMP-activated protein kinase, a cellular energy sensor (Tan et al. 2012). Other groups found no difference in 22Na+ flux rates between normal and CF airway cultures (Chen et al. 2010). However, they performed their 22Na+ flux experiments with 500 μl Ringer solution flooding the apical surface and with atmospheric O2. Thus, Na+ transport would have been inhibited in these hypoxic conditions due to the lack of Na+/K+ATPase activity, which likely accounted for their failure to detect increased mucosal-serosal 22Na+ flux in CF epithelia.
to drastically drop to ∼3% (Fig. 1D; Worlitzsch et al. 2002). Consistent with this finding, application of 100 μl of media to the mucosal side of polarized airway epithelia induced hypoxic stress and resulted in the activation of AMP-activated protein kinase, a cellular energy sensor (Tan et al. 2012). Other groups found no difference in 22Na+ flux rates between normal and CF airway cultures (Chen et al. 2010). However, they performed their 22Na+ flux experiments with 500 μl Ringer solution flooding the apical surface and with atmospheric O2. Thus, Na+ transport would have been inhibited in these hypoxic conditions due to the lack of Na+/K+ATPase activity, which likely accounted for their failure to detect increased mucosal-serosal 22Na+ flux in CF epithelia.
New techniques for indirectly measuring ENaC activity by gravitational methods and XZ-confocal microscopy emerged in the 1990s, which interfaced with well-differentiated human bronchial epithelial cultures (HBECs; Jiang et al. 1993; Matsui et al. 1998) and were suited to measuring ASL height/volume under thin-film conditions without inducing flooding-type effects seen in Ussing chambers (Fig. 1E and F). Using these methods, it was demonstrated that normal HBECs simultaneously absorb and secrete ions/water, and vary the relative contribution of each process to bulk fluid movement (Tarran et al. 2001a, 2005, 2006). While this is inefficient, it allows for a rapid reversal of net flux and the ability to switch between absorptive and secretive modes. CF HBECs were found to consistently hyperabsorb ASL as compared with normal cultures (Jiang et al. 1993; Matsui et al. 1998; Tarran et al. 2006), which was consistent with the elevated Na+ fluxes and IscAmil/VtAmil seen elsewhere (Boucher et al. 1986; Mall et al. 1998). Thus, the decrease in ASL volume is most likely due to both Cl− hyposecretion and Na+ hyperabsorption.
Animal model systems for studying ENaC in the context of CF lung disease
The trachea and pancreas of the CFTR knockout mouse (cftr−/−) do not develop a CF disease phenotype (Snouwaert et al. 1992; Colledge et al. 1996; Grubb & Boucher, 1999), which may be due to the predominance of Ca2+-activated Cl− currents in these tissues (Clarke et al. 1994). However, cftr−/− murine nasal epithelia do exhibit signs of pathology (Snouwaert et al. 1992) and also display reduced ASL volume (Tarran et al. 2001b). cftr−/− murine nasal epithelia had elevated Na+ currents as compared with wild-type controls, which persisted in the presence of bilateral Cl−-free media and precluded the possibility that altered driving force due to Cl− could influence the results (Grubb, 1995). Thus, the appearance of disease correlates well with the appearance of Na+ hyperabsorption in mice.
Because the lower airways of cftr−/− mice failed to display CF-like pathology, mice overexpressing β-ENaC on the Clara cell-specific promoter were developed. These mice displayed constitutive increases in Na+ absorption, though CFTR expression and function were not altered in these mice (Mall et al. 2004; Zhou et al. 2011). As a result, the β-ENaC mouse provided a model to test the effect of ENaC hyperactivity on ASL volume and mucus dehydration in vivo. The β-ENaC mice displayed mucus dehydration that was broadly similar to that seen in patients with CF. In turn, this dehydration resulted in a decrease in mucociliary clearance, confirming that ENaC-led Na+ hyperabsorption can initiate mucus stasis (Mall et al. 2004). Similar to patients with CF, the β-ENaC mouse also exhibited mucus plugging, goblet cell metaplasia, mucus hypersecretion and chronic airway inflammation. CF-like lung disease was also seen in mice lacking NEDD4–2, which leads to an abundance of ENaC in the apical membrane (Kimura et al. 2011). Taken together, these data strongly suggest that Na+ hyperabsorption-induced mucus dehydration is sufficient to trigger the inflammation and pathology seen in CF airways.
Crossing the β-ENaC mouse with either a CFTR−/− or a ΔF508−/− mouse enhanced the spontaneous lung disease seen in the β-ENaC mice (Johannesson et al. 2012; Livraghi-Butrico et al. 2013), suggesting that existing CFTR is moderately protective against unrestrained Na+ hyperabsorption. The β-ENaC mouse also was backcrossed onto C57/Bl6 or BalbC backgrounds. C57/Bl6 β-ENaC mice had both more endogenous Cl− secretion and a significantly increased survival as compared with BalbC β-ENaC mice (Johannesson et al. 2012). However, the β-ENaC mice were also crossbred with a human CFTR-overexpressing mouse, which resulted in increased CFTR-mediated Cl− currents but no reduction in either ENaC-mediated currents or pathology (Grubb et al. 2012). There are several possibilities as to why this cross did not affect disease severity, while cross-breeding onto the C57/Bl6 background did. The increased survival in C57/Bl6 mice may have been due to an as-yet unidentified difference that had nothing to do with Cl− secretion. Alternatively, human-CFTR and mouse-ENaC may not have been expressed in the same cells. Furthermore, while CFTR currents were increased ∼fivefold, ENaC currents were increased up to 100-fold. Thus, there may not have been enough CFTR to regulate ENaC. Finally, enhanced β-ENaC expression may cause the formation αβ-ENaCs, which could be differentially regulated than αβγ-ENaCs. While neither biochemical nor biophysical evidence for αβ-ENaC has been produced, Mall et al. (2010) demonstrated altered proteolytic regulation of amiloride-sensitive Isc in β-ENaC mice, which they speculated was due to the existence of αβ-ENaCs. These data highlight a deficit in the field, namely that ENaC stoichiometry and regulation is not well understood in native airway epithelia.
CFTR−/− pigs that share characteristics with human CF neonates, including pancreatitis, meconium ileus, early focal biliary cirrhosis and microgallbladder, have recently been developed (Stoltz et al. 2013). Like newborn humans, CF piglets lack airway inflammation at birth, and their lungs appear relatively normal (Rogers et al. 2008). One hurdle to the widespread use of the CF pig is that 100% of the pigs develop meconium ileus, which is fatal if not treated with surgery soon after birth. Accordingly, many studies have been performed on ≤24 h old pigs, or on tissues derived therein. Mammalian neonates use Cl− secretion to help expand their lungs in utero and then transition from having fluid-filled lungs to air-filled lungs post-partum. Much of this excess fluid is rapidly absorbed via an ENaC-led process (Hummler et al. 1996). Importantly, ENaC activity significantly declines after birth and takes about 6 weeks to reach levels seen in the fetus or in adult animals (Egan et al. 1975). Chen et al. (2010) did not see Na+ hyperabsorption in tissues from 24 h old CF piglets. However, as with other mammals, 24 h old piglets may not have much absorbing capacity due to post-partum downregulation of ENaC, which could explain why there is a discrepancy between the piglet studies (Chen et al. 2010) and the results obtained from adults human and adult human tissues (Boucher et al. 1986). Clearly, detailed studies need to be performed to better understand the post-partum changes in ion transport in piglets as well as in older CF pigs once the meconium ileus issue is resolved.
CFTR–ENaC Interactions and Na+ hyperabsorption
The effect of cAMP on Na+ transport is CFTR-dependent. Isoprenaline/forskolin inhibited IscAmil and VtAmil in normal nasal epithelia, while it activated Na+ absorption in CF nasal epithelia (Boucher et al. 1986; Mall et al. 1998). Subsequently, using patch-clamping, Stutts et al. demonstrated that, when transfected into fibroblasts, αβγ-ENaC was activated by agonists that elevate cAMP/PKA. However, when CFTR was cotransfected into the cells, the regulation reversed, and ENaC became inhibited by cAMP/PKA (Stutts et al. 1995). More recently, Blouquit-Laye et al. (2012) demonstrated that cGMP/NO regulation of ENaC was defective in CF airways. In H441 cells, which do not express CFTR, forskolin induced α-ENaC translocation to the apical membrane and increased amiloride-sensitive Na+ transport (Woollhead & Baines, 2006). This variable sensitivity to cAMP/PKA appears to be tissue specific, and in sweat glands and alveolar epithelia, the sensitivity of ENaC to cAMP is unaffected by CFTR (Reddy et al. 1999; Bove et al. 2010).
While the mechanism that underlies altered cAMP regulation of ENaC in CF airways is not well understood, CFTR and ENaC do co-immunoprecipitate and undergo fluorescence resonance energy transfer (Berdiev et al. 2007; Gentzsch et al. 2010), suggesting that they can be functionally linked. Direct binding between CFTR and ENaC has not been demonstrated using purified proteins. However, functional regulation of ENaC by CFTR has been demonstrated in planar lipid bilayers, suggesting that minimal accessory proteins are needed for this interaction (Berdiev et al. 2000). Co-expression of ENaC and CFTR also decreased the open probability (Po) without affecting the surface expression of ENaC (Konstas et al. 2003). It has been suggested that the first nucleotide binding fold of CFTR may regulate ENaC (Schreiber et al. 1999). Inhibition of ENaC by intracellular Cl− has also been proposed as an indirect mechanism for the regulation of ENaC by CFTR (Konig et al. 2001; Bachhuber et al. 2005). While this cannot be excluded as a physiological mechanism to regulate ENaC, CFTR mutants that can still conduct Cl−, such as N1303K, also fail to inhibit ENaC (Suaud et al. 2007), suggesting that the exact mechanism whereby CFTR regulates ENaC remains unknown.
Protease-antiprotease imbalance and chronic Na+ hyperabsorption
In Ussing chambers, ENaC can be inhibited equally well by inhibitors of trypsin-like serine proteases in normal and CF HBECs (Bridges et al. 2001). However, under thin-film conditions (i.e. in the presence of native ASL), ENaC was inhibited by the serine protease inhibitor aprotinin in CF HBECs but not in normal HBECs. This indicates that ENaC was spontaneously inhibited in normal but not CF HBECs (Tarran et al. 2006; Gaillard et al. 2010). Subsequently, under thin-film conditions, Gentzsch et al. noted an increase in proteolytic cleavage of α-ENaC in CF HBECs as compared with normal HBECs (Gentzsch et al. 2010). This could be the result of the upregulation of channel-activating proteases in the absence of CFTR (Tarran et al. 2006; Myerburg et al. 2006) or the accumulation of soluble ENaC inhibitors such as SPLUNC1, which are hypothesized to be functional in normal but not CF ASL (Garcia-Caballero et al. 2009). These data suggest that different results may be returned regarding ENaC proteolysis under thick- versus thin-film conditions, especially in normal airways. Importantly, there may be an inherent protease/inhibitor imbalance in CF airways that is extenuated under thin-film conditions and contributes to ENaC hyperactivity in CF airways.
We have identified short palate lung and nasal epithelial clone 1 (SPLUNC1, now renamed BPFIA1, but also known as LUNX and SPURT) as a soluble ENaC inhibitor that is contained in the ASL and acts as an ASL height reporter molecule (Garcia-Caballero et al. 2009). SPLUNC1 is primarily expressed in the upper airways and nasopharyngeal regions (Bingle & Bingle, 2011). SPLUNC1 is also expressed in the auditory canal, and knockdown of SPLUNC1 in this tissue led to a failure of mucus clearance and dehydration in this organ, suggesting that ENaC activity was enhanced following SPLUNC1 knockdown (McGillivary & Bakaletz, 2010). SPLUNC1 binds to ENaC, causing ENaC removal from the plasma membrane, which prevents it from being proteolytically cleaved (Garcia-Caballero et al. 2009). Indeed, stable knockdown of SPLUNC1 in normal airway cultures caused ASL volume to become unregulated and to drop to CF-like levels (Garcia-Caballero et al. 2009). SPLUNC1 is also present in CF airways (Bingle et al. 2007), but because CF airway cultures cannot regulate ENaC activity or ASL height (Tarran et al. 2005, 2006), we postulate that CF SPLUNC1 is non-functional, although the underlying cause of this dysfunction remains unknown.
Although the sequence of events at the onset of inflammation remains the subject of debate, the response to infection and inflammation response may further contribute to CF Na+ hyperabsorption. Human neutrophil elastase is present in CF airways at high concentrations (1 μm; Konstan et al. 1994) and apical application of neutrophil elastase to human bronchial cells increased ENaC activity (Caldwell et al. 2005), suggesting that neutrophil elastase could contribute to Na+ hyperabsorption. Pseudomonas aeruginosa’s alkaline protease can also activate ENaC and increase Na+ transport (Butterworth et al. 2012). Thus, the loss of CFTR may contribute to Na+ hyperabsorption in CF airways either directly, through protein interactions, or indirectly, subsequent to the acquisition of airway disease and the conversion to a protease-rich environment (Fig. 2). It is possible that other as-yet undiscovered host and bacterial proteases are upregulated in diseased CF lungs that can further exacerbate Na+ hyperabsorption.
Figure 2. The contribution of excess protease activity to Na+ hyperabsorption and CF lung disease progression.
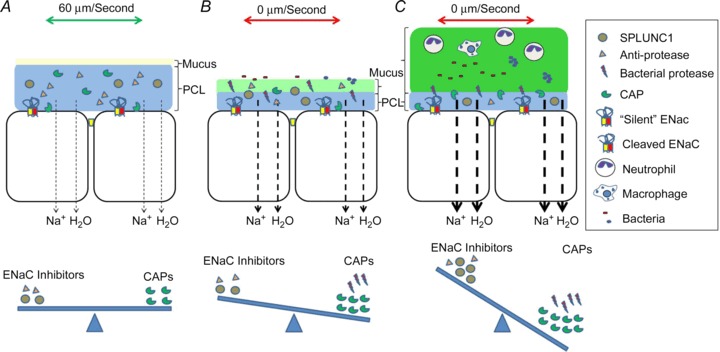
Channel-activating proteases (CAPs) can be either secreted or cell attached, and their action is offset by either anti-proteases or soluble inhibitors such as SPLUNC1. A, normally, the balance between CAPs and inhibitors allows for controlled Na+ absorption, which removes excess ASL without depleting PCL volume. B, CF airways are less able to manage the protease-inhibitor ratio, and either basally or following bacterial infection, excess protease activity contributes to Na+ hyperabsorption, which leads to ASL depletion and mucus stasis. C, mucin secretion (not shown) continues in the absence of PCL leading to increased height of the mucus layer. Chronic neutrophilia exacerbates this condition and leads to further protease release. ENaC, epithelial Na+ channel.
Whilst many researchers have focused on decreased ASL height that is concomitant with Cl− hyposecretion and Na+ hyperabsorption, we predict that the decrease in ASL height in an affected airway is only evident for a short period of time and will then transition into an airway with a greater ASL height than normal airways (Fig. 2A–C). Our reasoning is thus: low volume will induce mucus stasis. However, mucus secretion will continue in the face of Na+ hyperabsorption and the ASL/mucus layer will increase in height, albeit with decreased water availability and increased mucus dehydration as compared with normal ASL. Indeed, the height of the ASL in a dehydrated CF culture or CF airway can be ≥100 μm (Kesimer et al. 2013). This continued secretion of mucus in the face of ASL hyposecretion and mucus stasis may explain how mucus plugs form in CF airways. Thus, while ASL depletion studies are extremely useful for studying the early consequences of altered epithelial ion transport, under the chronic conditions seen in diseased lungs, measurements of mucus dehydration, mucus clearance rates or the simple presence/absence of a periciliary liquid layer (PCL) may also be useful to assay ASL status.
Conclusions
In conclusion, IscAmil and VtAmil are elevated in freshly isolated CF versus normal airway epithelia despite no change in Rt or Gt and despite a reduced apical membrane driving force (Boucher et al. 1986; Cotton et al. 1987). These observations are consistent with biochemical measurements, confocal measurements, Na+ fluxes, patch-clamp studies, optical gravitational measurements, increased ouabain-sensitive O2 consumption and Na+ net transport measurements in CF airways (Stutts et al. 1986, 1995; Jiang et al. 1993; Matsui et al. 1998; Tarran et al. 2005; Berdiev et al. 2007; Gentzsch et al. 2010). Furthermore, due to the protease-rich environment in diseased lungs, ENaC is likely to become further activated following chronic lung infections. However, it is the opinion of these authors that regardless of whether ENaC is more or less active in CF than in normal airways, any level of ENaC activity will further exacerbate Cl− hyposecretion and drive the mucus dehydration seen in CF airways. Importantly, rehydrating the ASL with hypertonic saline has been shown to improve lung function and decrease the rate of exacerbations by ∼50% (Donaldson et al. 2006). Thus, understanding the role that ENaC plays in CF pathogenesis is essential for devising novel therapeutics for the treatment of CF lung disease. While ENaC antagonists such as amiloride failed in the clinic due to their short retention time in the lung (Knowles et al. 1991), alternate approaches to inhibiting ENaC activity, such as adjusting the protease/anti-protease imbalance, may yet prove beneficial.
Acknowledgments
The authors thank Syanne Olson for expert editing assistance.
Glossary
- ASL
airway surface liquid
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- ENaC
epithelial Na+ channel
- HBEC
human bronchial epithelial culture
- PHA-1
pseudohypoaldosteronism type 1
Additional information
Competing interests
None declared.
Funding
The authors thank the NIH/NHLBI (R01 HL108927) for funding.
References
- Althaus M. ENaC inhibitors and airway re-hydration in cystic fibrosis: state of the art. Curr Mol Pharmacol. 2013;6:3–12. doi: 10.2174/18744672112059990025. [DOI] [PubMed] [Google Scholar]
- Alton EW, Khagani A, Taylor RFH, Logan-Sinclair R, Yacoub M, Geddes DM. Effect of heart-lung transplantation on airway potential difference in patients with and without cystic fibrosis. Eur Respir J. 1991;4:5–9. [PubMed] [Google Scholar]
- Bachhuber T, Konig J, Voelcker T, Murle B, Schreiber R, Kunzelmann K. Cl− interference with the epithelial Na+ channel ENaC. J Biol Chem. 2005;280:31587–31594. doi: 10.1074/jbc.M504347200. [DOI] [PubMed] [Google Scholar]
- Bangel-Ruland N, Sobczak K, Christmann T, Kentrup D, Langhorst H, Kusche-Vihrog K, Weber WM. Characterization of the epithelial sodium channel delta-subunit in human nasal epithelium. Am J Respir Cell Mol Biol. 2010;42:498–505. doi: 10.1165/rcmb.2009-0053OC. [DOI] [PubMed] [Google Scholar]
- Berdiev BK, Cormet-Boyaka E, Tousson A, Qadri YJ, Oosterveld-Hut HM, Hong JS, Gonzales PA, Fuller CM, Sorscher EJ, Lukacs GL, Benos DJ. Molecular proximity of cystic fibrosis transmembrane conductance regulator and epithelial sodium channel assessed by fluorescence resonance energy transfer. J Biol Chem. 2007;282:36481–36488. doi: 10.1074/jbc.M708089200. [DOI] [PubMed] [Google Scholar]
- Berdiev BK, Shlyonsky VG, Karlson KH, Stanton BA, Ismailov II. Gating of amiloride-sensitive Na(+) channels: subunit-subunit interactions and inhibition by the cystic fibrosis transmembrane conductance regulator. Biophys J. 2000;78:1881–1894. doi: 10.1016/S0006-3495(00)76737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingle L, Barnes FA, Cross SS, Rassl D, Wallace WA, Campos MA, Bingle CD. Differential epithelial expression of the putative innate immune molecule SPLUNC1 in cystic fibrosis. Respir Res. 2007;8:79. doi: 10.1186/1465-9921-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingle L, Bingle CD. Distribution of human PLUNC/BPI fold-containing (BPIF) proteins. Biochem Soc Trans. 2011;39:1023–1027. doi: 10.1042/BST0391023. [DOI] [PubMed] [Google Scholar]
- Blouquit-Laye S, Dannhoffer L, Braun C, Dinh-Xuan AT, Sage E, Chinet T. Effect of nitric oxide on epithelial ion transports in noncystic fibrosis and cystic fibrosis human proximal and distal airways. Am J Physiol Lung Cell Mol Physiol. 2012;303:L617–L625. doi: 10.1152/ajplung.00368.2011. [DOI] [PubMed] [Google Scholar]
- Boucher RC. Human airway ion transport (Part 1) Am J Respir Crit Care Med. 1994;150:271–281. doi: 10.1164/ajrccm.150.1.8025763. [DOI] [PubMed] [Google Scholar]
- Boucher RC, Cotton CU, Gatzy JT, Knowles MR, Yankaskas JR. Evidence for reduced Cl− and increased Na+ permeability in cystic fibrosis human primary cell cultures. J Physiol. 1988;405:77–103. doi: 10.1113/jphysiol.1988.sp017322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher RC, Stutts MJ, Knowles MR, Cantley L, Gatzy JT. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J Clin Invest. 1986;78:1245–1252. doi: 10.1172/JCI112708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove PF, Grubb BR, Okada SF, Ribeiro CM, Rogers TD, Randell SH, O’Neal WK, Boucher RC. Human alveolar type II cells secrete and absorb liquid in response to local nucleotide signalling. J Biol Chem. 2010;285:34939–34949. doi: 10.1074/jbc.M110.162933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges RJ, Newton BB, Pilewski JM, Devor DC, Poll CT, Hall RL. Na+ transport in normal and CF human bronchial epithelial cells is inhibited by BAY 39–9437. Am J Physiol Lung Cell Mol Physiol. 2001;281:L16–L23. doi: 10.1152/ajplung.2001.281.1.L16. [DOI] [PubMed] [Google Scholar]
- Butterworth MB, Zhang L, Heidrich EM, Myerburg MM, Thibodeau PH. Activation of the epithelial sodium channel (ENaC) by the alkaline protease from Pseudomonas aeruginosa. J Biol Chem. 2012;287:32556–32565. doi: 10.1074/jbc.M112.369520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol. 2005;288:L813–L819. doi: 10.1152/ajplung.00435.2004. [DOI] [PubMed] [Google Scholar]
- Chambers LA, Rollins BM, Tarran R. Liquid movement across the surface epithelium of large airways. Respir Physiol Neurobiol. 2007;159:256–270. doi: 10.1016/j.resp.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JH, Stoltz DA, Karp PH, Ernst SE, Pezzulo AA, Moninger TO, Rector MV, Reznikov LR, Launspach JL, Chaloner K, Zabner J, Welsh MJ. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143:911–923. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LL, Grubb BR, Yankaskas JR, Cotton CU, McKenzie A, Boucher RC. Relationship of a non-CFTR mediated chloride conductance to organ-level disease in cftr(-/-) mice. Proc Natl Acad Sci U S A. 1994;91:479–483. doi: 10.1073/pnas.91.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collawn JF, Lazrak A, Bebok Z, Matalon S. The CFTR and ENaC debate: how important is ENaC in CF lung disease. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1141–L1146. doi: 10.1152/ajplung.00036.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge WH, Abella BS, Southern KW, Ratcliff R, Jiang C, Cheng SH, MacVinish LJ, Anderson JR, Cuthbert AW, Evans MJ. Generation and characterization of a deltaF508 cystic fibrosis mouse model. Nat Genet. 1996;10:445–452. doi: 10.1038/ng0895-445. [DOI] [PubMed] [Google Scholar]
- Cotton CU, Stutts MJ, Knowles MR, Gatzy JT, Boucher RC. Abnormal apical cell membrane in cystic fibrosis respiratory epithelium. An in vitro electrophysiologic analysis. J Clin Invest. 1987;79:80–85. doi: 10.1172/JCI112812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354:241–250. doi: 10.1056/NEJMoa043891. [DOI] [PubMed] [Google Scholar]
- Egan EA, Olver RE, Strang LB. Changes in non-electrolyte permeability of alveoli and the absorption of lung liquid at the start of breathing in the lamb. J Physiol. 1975;244:161–179. doi: 10.1113/jphysiol.1975.sp010789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard EA, Kota P, Gentzsch M, Dokholyan NV, Stutts MJ, Tarran R. Regulation of the epithelial Na+ channel and airway surface liquid volume by serine proteases. Pflugers Arch. 2010;460:1–17. doi: 10.1007/s00424-010-0827-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Caballero A, Rasmussen JE, Gaillard E, Watson MJ, Olsen JC, Donaldson SH, Stutts MJ, Tarran R. SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc Natl Acad Sci U S A. 2009;106:11412–11417. doi: 10.1073/pnas.0903609106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch M, Dang H, Dang Y, Garcia-Caballero A, Suchindran H, Boucher RC, Stutts MJ. The cystic fibrosis transmembrane conductance regulator impedes proteolytic stimulation of the epithelial Na+ channel. J Biol Chem. 2010;285:32227–32232. doi: 10.1074/jbc.M110.155259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb BR. Ion transport across the jejunum in normal and cystic fibrosis mice. Am J Physiol Gastrointest Liver Physiol. 1995;268:G505–G513. doi: 10.1152/ajpgi.1995.268.3.G505. [DOI] [PubMed] [Google Scholar]
- Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev. 1999;79:S193–S214. doi: 10.1152/physrev.1999.79.1.S193. [DOI] [PubMed] [Google Scholar]
- Grubb BR, O’Neal WK, Ostrowski LE, Kreda SM, Button B, Boucher RC. Transgenic hCFTR expression fails to correct beta-ENaC mouse lung disease. Am J Physiol Lung Cell Mol Physiol. 2012;302:L238–L247. doi: 10.1152/ajplung.00083.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunder S, Firsov D, Chang SS, Jaeger NF, Gautschi I, Schild L, Lifton RP, Rossier BC. A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. EMBO J. 1997;16:899–907. doi: 10.1093/emboj/16.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf A, Schreiber R, Mall M, Greger R, Kunzelmann K. Cystic fibrosis transmembrane conductance regulator inhibits epithelial Na+ channels carrying Liddle's syndrome mutations. J Biol Chem. 1999;274:13894–13899. doi: 10.1074/jbc.274.20.13894. [DOI] [PubMed] [Google Scholar]
- Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC. Early death due to defective neonatal lung liquid clearance in alpha ENaC-deficient mice. Nat Genet. 1996;12:325–328. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- Ji HL, Su XF, Kedar S, Li J, Barbry P, Smith PR, Matalon S, Benos DJ. Delta-subunit confers novel biophysical features to alpha beta gamma-human epithelial sodium channel (ENaC) via a physical interaction. J Biol Chem. 2006;281:8233–8241. doi: 10.1074/jbc.M512293200. [DOI] [PubMed] [Google Scholar]
- Jiang C, Finkbeiner WE, Widdicombe JH, McCray PB, Jr, Miller SS. Altered fluid transport across airway epithelium in cystic fibrosis. Science. 1993;262:424–427. doi: 10.1126/science.8211164. [DOI] [PubMed] [Google Scholar]
- Johannesson B, Hirtz S, Schatterny J, Schultz C, Mall MA. CFTR regulates early pathogenesis of chronic obstructive lung disease in betaENaC-overexpressing mice. PLoS ONE. 2012;7:e44059. doi: 10.1371/journal.pone.0044059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashlan OB, Kleyman TR. Epithelial Na(+) channel regulation by cytoplasmic and extracellular factors. Exp Cell Res. 2012;318:1011–1019. doi: 10.1016/j.yexcr.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerem E, Bistritzer T, Hanukoglu A, Hofmann T, Zhou Z, Bennett W, MacLaughlin E, Barker P, Nash M, Quittell L, Boucher R, Knowles MR. Pulmonary epithelial sodium channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N Engl J Med. 1999;341:156–162. doi: 10.1056/NEJM199907153410304. [DOI] [PubMed] [Google Scholar]
- Kesimer M, Ehre C, Burns KA, Davis CW, Sheehan JK, Pickles RJ. Molecular organization of the mucins and glycocalyx underlying mucus transport over mucosal surfaces of the airways. Mucosal Immunol. 2013;6:379–392. doi: 10.1038/mi.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesimer M, Kirkham S, Pickles RJ, Henderson AG, Alexis NE, DeMaria G, Knight D, Thornton DJ, Sheehan JK. Tracheobronchial air-liquid interface cell culture: a model for innate mucosal defense of the upper airways. Am J Physiol Lung Cell Mol Physiol. 2009;296:L92–L100. doi: 10.1152/ajplung.90388.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Kawabe H, Jiang C, Zhang W, Xiang YY, Lu C, Salter MW, Brose N, Lu WY, Rotin D. Deletion of the ubiquitin ligase Nedd4L in lung epithelia causes cystic fibrosis-like disease. Proc Natl Acad Sci U S A. 2011;108:3216–3221. doi: 10.1073/pnas.1010334108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles M, Gatzy J, Boucher R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med. 1981;305:1489–1495. doi: 10.1056/NEJM198112173052502. [DOI] [PubMed] [Google Scholar]
- Knowles M, Gatzy J, Boucher R. Relative ion permeability of normal and cystic fibrosis nasal epithelium. J Clin Invest. 1983a;71:1410–1417. doi: 10.1172/JCI110894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles MR, Church NL, Waltner WE, Yankaskas JR, Gilligan P, King M, Edwards LJ, Helms RW, Boucher RC. Aerosolized amiloride as a treatment of cystic fibrosis lung disease: a pilot study. Adv Exp Med Biol. 1991;290:119–132. doi: 10.1007/978-1-4684-5934-0_14. [DOI] [PubMed] [Google Scholar]
- Knowles MR, Stutts MJ, Spock A, Fischer N, Gatzy JT, Boucher RC. Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science. 1983b;221:1067–1070. doi: 10.1126/science.6308769. [DOI] [PubMed] [Google Scholar]
- Konig J, Schreiber R, Voelcker T, Mall M, Kunzelmann K. The cystic fibrosis transmembrane conductance regulator (CFTR) inhibits ENaC through an increase in the intracellular Cl− concentration. EMBO Rep. 2001;2:1047–1051. doi: 10.1093/embo-reports/kve232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstan MW, Hilliard KA, Norvell TM, Berger M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinical mild lung disease suggest ongoing infection and inflammation. Am J Respir Crit Care Med. 1994;150:448–454. doi: 10.1164/ajrccm.150.2.8049828. [DOI] [PubMed] [Google Scholar]
- Konstas AA, Koch JP, Korbmacher C. cAMP-dependent activation of CFTR inhibits the epithelial sodium channel (ENaC) without affecting its surface expression. Pflugers Arch. 2003;445:513–521. doi: 10.1007/s00424-002-0957-z. [DOI] [PubMed] [Google Scholar]
- Li H, Sheppard DN, Hug MJ. Transepithelial electrical measurements with the Ussing chamber. J Cyst Fibros. 2004;3(Suppl 2):123–126. doi: 10.1016/j.jcf.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Livraghi-Butrico A, Kelly EJ, Wilkinson KJ, Rogers TD, Gilmore RC, Harkema JR, Randell SH, Boucher RC, O’Neal WK, Grubb BR. Loss of Cftr function exacerbates the phenotype of Na+ hyperabsorption in murine airways. Am J Physiol Lung Cell Mol Physiol. 2013;304:L469–L480. doi: 10.1152/ajplung.00150.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall M, Bleich M, Greger R, Schreiber R, Kunzelmann K. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J Clin Invest. 1998;102:15–21. doi: 10.1172/JCI2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall M, Grubb BR, Harkema JR, O’Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- Mall MA. Role of the amiloride-sensitive epithelial Na+ channel in the pathogenesis and as a therapeutic target for cystic fibrosis lung disease. Exp Physiol. 2009;94:171–174. doi: 10.1113/expphysiol.2008.042994. [DOI] [PubMed] [Google Scholar]
- Mall MA, Button B, Johannesson B, Zhou Z, Livraghi A, Caldwell RA, Schubert SC, Schultz C, O’Neal WK, Pradervand S, Hummler E, Rossier BC, Grubb BR, Boucher RC. Airway surface liquid volume regulation determines different airway phenotypes in liddle compared with betaENaC-overexpressing mice. J Biol Chem. 2010;285:26945–26955. doi: 10.1074/jbc.M110.151803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- McGillivary G, Bakaletz LO. The multifunctional host defense peptide SPLUNC1 is critical for homeostasis of the mammalian upper airway. PLoS ONE. 2010;5:e13224. doi: 10.1371/journal.pone.0013224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutesa L, Azad AK, Verhaeghe C, Segers K, Vanbellinghen JF, Ngendahayo L, Rusingiza EK, Mutwa PR, Rulisa S, Koulischer L, Cassiman JJ, Cuppens H, Bours V. Genetic analysis of Rwandan patients with cystic fibrosis-like symptoms: identification of novel cystic fibrosis transmembrane conductance regulator and epithelial sodium channel gene variants. Chest. 2009;135:1233–1242. doi: 10.1378/chest.08-2246. [DOI] [PubMed] [Google Scholar]
- Myerburg MM, Butterworth MB, McKenna EE, Peters KW, Frizzell RA, Kleyman TR, Pilewski JM. Airway surface liquid volume regulates ENaC by altering the serine protease-protease inhibitor balance: a mechanism for sodium hyperabsorption in cystic fibrosis. J Biol Chem. 2006;281:27942–27949. doi: 10.1074/jbc.M606449200. [DOI] [PubMed] [Google Scholar]
- Myerburg MM, Harvey PR, Heidrich EM, Pilewski JM, Butterworth MB. Acute regulation of the epithelial sodium channel in airway epithelia by proteases and trafficking. Am J Respir Cell Mol Biol. 2010;43:712–719. doi: 10.1165/rcmb.2009-0348OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LG, Patel A, Frindt G. Regulation and dysregulation of epithelial Na+ channels. Clin Exp Nephrol. 2012;16:35–43. doi: 10.1007/s10157-011-0496-z. [DOI] [PubMed] [Google Scholar]
- Qadri YJ, Rooj AK, Fuller CM. ENaCs and ASICs as therapeutic targets. Am J Physiol Cell Physiol. 2012;302:C943–C965. doi: 10.1152/ajpcell.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh R, Diakov A, Tzschoppe A, Korbmacher J, Azad AK, Cuppens H, Cassiman JJ, Dotsch J, Sticht H, Korbmacher C. A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self inhibition. J Physiol. 2010;588:1211–1225. doi: 10.1113/jphysiol.2009.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh R, Soell D, Haerteis S, Diakov A, Nesterov V, Krueger B, Sticht H, Korbmacher C. A mutation in the beta-subunit of ENaC identified in a patient with cystic fibrosis-like symptoms has a gain-of-function effect. Am J Physiol Lung Cell Mol Physiol. 2013;304:L43–L55. doi: 10.1152/ajplung.00093.2012. [DOI] [PubMed] [Google Scholar]
- Reddy MM, Light MJ, Quinton PM. Activation of the epithelial Na+ channel (ENaC) requires CFTR Cl− channel function. Nature. 1999;402:301–304. doi: 10.1038/46297. [DOI] [PubMed] [Google Scholar]
- Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, Rogan MP, Pezzulo AA, Karp PH, Itani OA, Kabel AC, Wohlford-Lenane CL, Davis GJ, Hanfland RA, Smith TL, Samuel M, Wax D, Murphy CN, Rieke A, Whitworth K, Uc A, Starner TD, Brogden KA, Shilyansky J, McCray PB, Jr, Zabner J, Prather RS, Welsh MJ. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837–1841. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC. Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. EMBO J. 1996;15:2381–2387. [PMC free article] [PubMed] [Google Scholar]
- Schreiber R, Hopf A, Mall M, Greger R, Kunzelmann K. The first-nucleotide binding domain of the cystic-fibrosis transmembrane conductance regulator is important for inhibition of the epithelial Na+ channel. Proc Natl Acad Sci U S A. 1999;96:5310–5315. doi: 10.1073/pnas.96.9.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- Soundararajan R, Lu M, Pearce D. Organization of the ENaC-regulatory machinery. Crit Rev Biochem Mol Biol. 2012;47:349–359. doi: 10.3109/10409238.2012.678285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockand JD, Staruschenko A, Pochynyuk O, Booth RE, Silverthorn DU. Insight toward epithelial Na+ channel mechanism revealed by the acid-sensing ion channel 1 structure. IUBMB Life. 2008;60:620–628. doi: 10.1002/iub.89. [DOI] [PubMed] [Google Scholar]
- Stoltz DA, Rokhlina T, Ernst SE, Pezzulo AA, Ostedgaard LS, Karp PH, Samuel MS, Reznikov LR, Rector MV, Gansemer ND, Bouzek DC, Alaiwa MH, Hoegger MJ, Ludwig PS, Taft PJ, Wallen TJ, Wohlford-Lenane C, McMenimen JD, Chen JH, Bogan KL, Adam RJ, Hornick EE, Nelson GA, Hoffman EA, Chang EH, Zabner J, McCray PB, Jr, Prather RS, Meyerholz DK, Welsh MJ. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest. 2013;123:2685–2693. doi: 10.1172/JCI68867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- Stutts MJ, Knowles MR, Gatzy JT, Boucher RC. Oxygen consumption and ouabain binding sites in cystic fibrosis nasal epithelium. Pediatr Res. 1986;20:1316–1320. doi: 10.1203/00006450-198612000-00026. [DOI] [PubMed] [Google Scholar]
- Suaud L, Yan W, Carattino MD, Robay A, Kleyman TR, Rubenstein RC. Regulatory interactions of N1303K-CFTR and ENaC in Xenopus oocytes: evidence that chloride transport is not necessary for inhibition of ENaC. Am J Physiol Cell Physiol. 2007;292:C1553–C1561. doi: 10.1152/ajpcell.00064.2006. [DOI] [PubMed] [Google Scholar]
- Tan CD, Selvanathar IA, Baines DL. Cleavage of endogenous gammaENaC and elevated abundance of alphaENaC are associated with increased Na(+) transport in response to apical fluid volume expansion in human H441 airway epithelial cells. Pflugers Arch. 2011;462:431–441. doi: 10.1007/s00424-011-0982-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CD, Smolenski RT, Harhun MI, Patel HK, Ahmed SG, Wanisch K, Yáñez-Muñoz RJ, Baines DL. Br J Pharmacol. 2012;167:368–382. doi: 10.1111/j.1476-5381.2012.01993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarran R, Button B, Picher M, Paradiso AM, Ribeiro CM, Lazarowski ER, Zhang L, Collins PL, Pickles RJ, Fredberg JJ, Boucher RC. Normal and cystic fibrosis airway surface liquid homeostasis: the effects of phasic shear stress and viral infections. J Biol Chem. 2005;280:35751–35759. doi: 10.1074/jbc.M505832200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarran R, Grubb BR, Gatzy JT, Davis CW, Boucher RC. The relative roles of passive surface forces and active ion transport in the modulation of airway surface liquid volume and composition. J Gen Physiol. 2001a;118:223–236. doi: 10.1085/jgp.118.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarran R, Grubb BR, Parsons D, Picher M, Hirsh AJ, Davis CW, Boucher RC. The CF salt controversy: in vivo observations and therapeutic approaches. Mol Cell. 2001b;8:149–158. doi: 10.1016/s1097-2765(01)00286-6. [DOI] [PubMed] [Google Scholar]
- Tarran R, Trout L, Donaldson SH, Boucher RC. Soluble mediators, not cilia, determine airway surface liquid volume in normal and cystic fibrosis superficial airway epithelia. J Gen Physiol. 2006;127:591–604. doi: 10.1085/jgp.200509468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeau PH, Butterworth MB. Proteases, cystic fibrosis and the epithelial sodium channel (ENaC) Cell Tissue Res. 2013;351:309–323. doi: 10.1007/s00441-012-1439-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussing HH, Zerhan K. Active transport of sodium as the source of electric current in the short-circuited isolated frogskin. Acta Physiol Scand. 1951;23:110–127. doi: 10.1111/j.1748-1716.1951.tb00800.x. [DOI] [PubMed] [Google Scholar]
- Woollhead AM, Baines DL. Forskolin-induced cell shrinkage and apical translocation of functional enhanced green fluorescent protein-human alphaENaC in H441 lung epithelial cell monolayers. J Biol Chem. 2006;281:5158–5168. doi: 10.1074/jbc.M509947200. [DOI] [PubMed] [Google Scholar]
- Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiss T, Botzenhart K, Yankaskas JR, Randell S, Boucher RC, Doering G. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest. 2002;109:317–325. doi: 10.1172/JCI13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Duerr J, Johannesson B, Schubert SC, Treis D, Harm M, Graeber SY, Dalpke A, Schultz C, Mall MA. The ENaC-overexpressing mouse as a model of cystic fibrosis lung disease. J Cyst Fibros. 2011;10(Suppl 2):S172–S182. doi: 10.1016/S1569-1993(11)60021-0. [DOI] [PubMed] [Google Scholar]