

Abstract
To better understand how medication status and task demands affect cognition in Major Depressive Disorder (MDD), we evaluated medication-naïve patients with MDD, medicated patients with MDD receiving the Selective Serotonin Reuptake Inhibitors (SSRI) paroxetine, and healthy controls. All three groups were administered a computer-based cognitive task with two phases, an initial phase in which a sequence is learned through reward-based feedback (which our prior studies suggest is striatal-dependent), followed by a generalization phase that involves a change in the context where learned rules are to be applied (which our prior studies suggest is hippocampal-region dependent). Medication-naïve MDD patients were slow to learn the initial sequence but were normal on subsequent generalization of that learning. In contrast, medicated patients learned the initial sequence normally, but were impaired at the generalization phase. We argue that these data suggest (i) an MDD-related impairment in striatal-dependent sequence learning which can be remediated by SSRIs and (ii) an SSRI-induced impairment in hippocampal-dependent generalization of past learning to novel contexts, not otherwise seen in the medication-naïve MDD group. Thus, SSRIs might have a beneficial effect on striatal function required for sequence learning, but a detrimental effect on the hippocampus and other medial temporal lobe structures critical for generalization.
Keywords: Major Depressive Disorder, Selective Serotonin Reuptake Inhibitor (SSRI), hippocampus, basal ganglia, reward, punishment, sequence learning, context-shift, generalization
1. INTRODUCTION
Major depressive disorder (MDD) is a condition characterized by a long-lasting depressed mood or marked loss of interest or pleasure in all or nearly all activities (Belmaker and Agam, 2008). Relatively little is known about the effects of MDD on striatal-based learning, although it is well known that the striatum is a key brain region disrupted in MDD (Dunlop and Nemeroff, 2007; Nestler and Carlezon, 2006; Nutt, 2006; Perona et al., 2008). Converging evidence from the literature confirms the involvement of the broader basal ganglia dopaminergic system in the pathophysiology and cognitive changes related to MDD. For example, recent imaging studies suggest that patients with MDD show cognitive and neurochemical dysfunction directly related to the nigrostriatal system (Dunlop and Nemeroff, 2007; Robinson et al., 2012; Walter et al., 2007). In addition, the major symptom of MDD, anhedonia, has been linked to dopaminergic dysfunction in the basal ganglia (Bressan and Crippa, 2005; Dhillon et al., 2008; Heinz et al., 1999; Miller et al., 1996; Schmidt et al., 2001). Further, patients with MDD have a three-fold higher than normal risk of developing Parkinson’s disease (PD) (Leentjens et al., 2003; Schuurman et al., 2002), in which nigrostriatal dopaminergic neurons decay (Kish et al., 1988). Finally, studies have reported reductions in the size of the striatum in patients with MDD (Lorenzetti et al., 2009) that has been linked to impairments in motor sequence learning (Naismith et al., 2006).
However, because psychomotor retardation is a common feature of MDD (Buyukdura et al., 2011), it is unclear whether such deficits reflect learning deficits or just motor slowing. The present study addresses this issue by using a computer-based test of sequence learning (Shohamy et al., 2005; Nagy et al., 2007a). In this task, participants learn to execute a chain sequences leading to reward. The chain is gradually lengthened until a complete sequence in learned. Subjects are first trained to learn A→reward, followed by B→A→reward, and so forth until a full sequence is acquired (D→C→B→A→reward). Learning is evaluated by the number of errors that are committed at each stage of the task, and therefore, learning does not dependent on response speed. Converging evidence from the literature suggests that the basal ganglia dopaminergic system is vital for this type of reward-based sequence learning (Haber and Knutson, 2010; Schultz, 1997). For example, a previous study from our group utilizing the same task as in the current study showed that medication-naïve patients with PD were significantly impaired on sequence-learning (Nagy et al., 2007a). A different study showed that dopaminergic medication (L-dopa) remediated this deficit (Shohamy et al., 2005).
The task also contains a subsequent generalization phase, designed to test the generalization of learned stimulus-response associations. In this phase, subjects are presented with a choice between the door that was previously correct in this room, a door that was previously-correct in a different room, and a “distractor” door that was never correct in any room. To successfully pass this phase, subjects are required to apply their previously learned door-room associations from the sequence-learning phase to new contexts with novel distractors. Animal and human work has shown that the medial temporal lobe plays an important role in generalization of learning over multiple contexts (Eichenbaum et al., 1996; Myers et al., 2002; Myers et al., 2003). Specifically, a previous study using this task found that PD patients with nigro-striatal dysfunction (but presumed intact medial temporal lobe function) showed no impairment in generalization, while those with amnestic mild cognitive impairment (aMCI, and presumed medial temporal lobe dysfunction) did show an impairment (Nagy et al., 2007a), consistent with the view that the reward-based sequence learning phase is striatal-dependent while the subsequent generalization phase is hippocampal/medial temporal lobe dependent. This finding is important because patients with MDD are also thought to have a smaller-than-average hippocampus, a key brain region for memory formation located within the medial temporal lobe (Campbell and Macqueen, 2004; MacQueen et al., 2003; Vakili et al., 2000; Vythilingam et al., 2004). Moreover, studies indicate that medication-free patients with MDD are impaired on hippocampal-dependent memory measures such as the delayed paragraph recall of Wechsler Memory Scale and the Selective Reminding Test (Austin et al., 2001; Vythilingam et al., 2004). Thus, we seek to address how MDD influences hippocampal-based generalization, in addition to striatal-based sequence learning.
Of note, a previous study on this task found a sequence-learning deficit but spared generalization in patients with MDD (Polgar et al., 2007). However, this experiment did not control for medication use and so it is still not clear how antidepressants affect cognitive performance. It has been hypothesized that selective serotonin reuptake inhibitors (SSRIs) achieve their therapeutic mood-enhancing effect, in part, by modifying synaptic availability of serotonin, by enhancing dopaminergic function in the brain (Nutt, 2006), and possibly also by enhancing neurogenesis in the hippocampal region (Malberg, 2004). Neuroimaging and animal studies suggest that SSRIs increase the size of the hippocampus by augmenting the rate of neurogenesis in the dentate gyrus, a substructure of the hippocampal region (Boldrini et al., 2009; Malberg and Schechter, 2005; Sahay et al., 2011). Furthermore, it has been shown in animal studies that neurogenesis in the dentate gyrus is key for the mood augmenting effect of antidepressants (David et al., 2009), although the blockage of neurogenesis does not induce depression-like behavior in animals (Santarelli et al., 2003). However, it has not been sufficiently tested what SSRIs would do for medial temporal lobe dependent learning using sensitive measures of cognitive function similar to the task we use in our current study. SSRI-induced neurogenesis could indicate a learning improvement with SSRIs. However, past studies showed ambiguous effects of SSRIs on medial temporal lobe dependent processes (Carlini et al., 2012; Igelstrom and Heyward, 2012; Sass and Wortwein, 2012; Vythilingam et al., 2004),
Thus, it remains unclear how MDD and medication use influences cognitive function on a learning and generalization task. To our knowledge, few studies have conducted thorough assessments of striatal- and hippocampal-dependent learning-and-memory function on patients with MDD both with and without SSRI treatment. In our current study, we investigate the cognitive correlates of striatal and hippocampal function in two groups of patients with MDD, those that are medication-naïve and those that have been treated using SSRIs, as well as healthy matched controls. We predicted that medication-naïve patients with MDD would resemble medication-naïve patients with PD, being impaired at initial sequence learning, whereas SSRI treated patients would not show this impairment. Given past studies showing ambiguous effects of SSRIs on medial temporal lobe dependent processes (Carlini et al., 2012; Igelstrom and Heyward, 2012; Sass and Wortwein, 2012; Vythilingam et al., 2004), it was not clear a priori what, if any, effect SSRIs would have on the generalization phase of this task. As described below, our results indicated that while patients treated with SSRIs did not reveal an MDD-derived sequence-learning deficit, these medications led to an additional and heretofore, novel impairment in the medial temporal lobe dependent generalization of this learning.
2. METHODS
2.1 PARTICIPANTS
We recruited 16 medication-naïve patients with MDD (MDD), 15 SSRI-responding patients with MDD (MDD-T), and 25 HC subjects, from various psychiatric clinics, mental health care centers and primary health care centers throughout the West Bank, Palestinian Territories. All subjects were Caucasians, ranging from 18–60 years of age. Participants were group matched for age, gender and years of education, as shown in Table 1. All subjects underwent screening evaluations that included a medical history and a physical examination. Psychiatric assessment was conducted using an unstructured interview with a psychiatrist using the DSM-IV-TR criteria for the diagnosis of MDD, and the Mini International Neuropsychiatric Interview (MINI) (Amorim et al., 1998). All SSRI-treated patients with MDD received 10–30 mg of paroxetine per day (M=16.67, SD=7.78) as part of their normal ongoing treatment. Inclusion criteria for HC subjects were absence of any psychiatric or other disorders that might affect cognition. MDD-T patient average exposure to SSRIs was 35.35 (SD=43.96) months. MDD-T patients’ response to SSRIs was assessed using subjective reports and scores on the Beck Depression Inventory II. Exclusion criteria for all subjects included psychotropic drug exposure, except for the SSRI paroxetine in the SSRI-treated MDD group; major medical or neurological illness; illicit drug use or alcohol abuse within the past year; lifetime history of alcohol or drug dependence; psychiatric disorders other than major depression (excepting comorbid anxiety symptoms); current pregnancy or breastfeeding. After receiving a complete description of the study, participants provided written informed consent as approved by the Al-Quds University Ethics Committee and the Rutgers Institutional Review Board.
Table 1.
Summary of Demographic and Neuropsychological Results. HC: healthy controls, MDD: medication-naïve patients with MDD, MDD-T: SSRI-treated patients with MDD, Mini-Mental Status Examination (MMSE), the digit span subtest of the Revised Wechsler Adult Intelligence Scale (WAIS-R digit-span), Beck Depression Inventory II (BDI-II), and Beck Anxiety Inventory (BAI).
| Age | Education | MMSE | WAIS-R digit-span | BDI-II | BAI | ||
|---|---|---|---|---|---|---|---|
| HC | Mean | 31.08 | 14.08 | 29.76 | 16.24 | 5.84 | 8.76 |
| SD | 14.01 | 1.87 | 0.44 | 4.93 | 4.89 | 7.01 | |
| MDD | Mean | 30.63 | 12.63 | 29.19 | 11.87 | 31.50 | 24.43 |
| SD | 8.25 | 2.06 | 0.91 | 3.16 | 8.80 | 10.98 | |
| MDD-T | Mean | 34.87 | 13.07 | 28.93 | 11.13 | 8.73 | 11.87 |
| SD | 7.44 | 3.49 | 1.75 | 3.44 | 6.47 | 7.10 | |
2.2 NEUROPSYCHOLOGICAL TEST BATTERY
All subjects completed the Arabic version (Inzelberg et al., 2007) of a battery of neuropsychological test questionnaires: Mini-Mental Status Examination (MMSE (Folstein et al., 1975)), Beck Depression Inventory II (BDI-II) (Beck et al., 1996), Beck Anxiety Inventory (BAI) (Beck et al., 1988) and the digit span subtest of the Revised Wechsler Adult Intelligence Scale (WAIS-R digit span) (Burgess et al., 1992). All results are summarized in Table 1. One-way ANOVAs, with a Bonferroni correction of α=0.005 to protect the level of significance (9 comparisons), revealed a significant effect of group on BDI-II (F(2,53)=79.65, p<0.001, η2=0.75), BAI (F(2,53)=17.90, p<0.001, η2=0.40) and WAIS-R scores (F(2,53)=9.22, p<0.001, η2=0.26). For WAIS-R, there was a significant difference between HCs and both MDD groups (p<0.05). However, BDI-II and BAI scores were significantly correlated. Further, only BDI-II scores significantly correlated to behavioral task results. Hence, we only included BDI-II as a covariate in the analysis of cognitive data (for more details, please see Figure 3-A).
Figure 3.

(A) Correlation between BDI-II scores and the numbers of errors on the sequence-learning phase of the task (chain steps A–D), Pearson’s r=0.360, N=51, p=0.009. (B) Correlation between WAIS-R digit-span scores and the number of errors on the context-shift generalization phase, Pearson’s r=−0.245, N=51, p=0.083.
2.3 COMPUTER-BASED COGNITIVE TASK
2.3.1 Sequence Learning Followed by Generalization with a Context-Shift
The task as previously reported (Shohamy, Bodi). It was run on a Macintosh computer, programmed in the SuperCard language. In this task, participants are instructed to guide an animated character (nicknamed “Kilroy”) through a sequence of four rooms with different colored doors to reach a goal point, the outside world. The rooms have a uniform white background, and are drawn using perspective lines, with three black doors appearing on the far wall. The doors appear about 2″ high, and the colored cards are each 1″ high by 0.5″ wide, and outlined in white for visual clarity. The animated figure (Kilroy) appears about 2″ tall.
On each trial, Kilroy appears in a room with three doors, each of which has a colored card (see Figure 1-A). The three colored cards in each room are consistent for every participant, but no color appears in more than one room during training. For example, room A might have red, green, and purple doors; room B might have yellow, blue, and brown doors, and so on. The colored cards marking the doors in each of six rooms are selected from a set of eighteen unique and highly discriminable colors. Assignment of colors was randomized across subjects. Spatial layout of these three colored cards on the doors (left, center, right) was randomized on each trial, so that the correct answer (left, center, right) varied across trials in a room. Thus, the color of the card, not the location of the door, determines the correct response on each trial.
Figure 1.

Illustration of the Sequence Learning with Context Shift Task.
In each room, the subject uses the computer mouse to move the cursor to click on one of the doors. When the subject selects a door, Kilroy turns, walks to the door, and tries to open it. If the subject’s choice is incorrect, the door is “locked” and Kilroy cannot open it. He puts his hands on his hips and makes a disappointed face, and the word “Locked!” appears on the bottom of the screen (Figure 1-C). Kilroy then moves back to the center of the room, and awaits the subject’s next choice. If the subject’s choice is correct, Kilroy opens the door and steps through. If this room was at the end of the sequence, Kilroy reaches the outside, where he turns and gives a thumbs-up sign (Figure 1-B); if the room was at an earlier stage of the sequence, Kilroy steps through into the next room (Figure 1-D) and, once there, waits for further instructions (as in Figure 1-A). In either case (correct or incorrect response), the outcome appears on the screen for 1 s; there is then a 0.33 s interval before Kilroy appears at the bottom of the screen again, ready for new instructions. There is no limit on response times.
One trial consists of Kilroy traversing a full sequence of rooms until (eventually) reaching the outside. The length of this sequence increases from one to four rooms over the course of training, starting in the room leading directly to the outside world and progressively moving further away. A trial is scored as correct if the subject chooses the correct door on the first opportunity for every room in the sequence. However, a subject may make one or more errors on a trial, by choosing an incorrect door one or more times before choosing the correct door, in each of one or more rooms in the sequence. This means that a subject could make more than one error per trial. Each learning phase continues until the subject completes four consecutive correct trials or to a maximum of fifteen trials. If a subject fails to reach criterion within the maximum number of trials for any phase, that phase is terminated, further training and context-shift phases are skipped, and the subject proceeds directly to the last (retraining) phase of the task.
2.3.2 Procedure
The subject is seated in a quiet testing room at a comfortable viewing distance from the screen. Before the test, the subject is informed that the aim of the game is to help a cartoon figure get out of the house as many times as possible. The following instructions appear: “Welcome to the experiment. In this experiment, you will see a character named Kilroy who is trying to get out of the house. Each room in the house has three doors, and each door has a colored card on it. On each trial, two of the doors are locked, and one door is unlocked. In each room, click on the color card of the door that you think is unlocked. If you are correct, Kilroy will get outside. Good luck!” The task then consisted of the following phases (table 2):
Table 2.
The Sequence Learning with Context-Shift Learning Paradigm
| Phase | Description | Doors shown | Correct response |
|---|---|---|---|
|
| |||
| Practice | Cue-association | P1P2P3 | P1→reward |
|
| |||
| Sequence-Learning | Chain step A | A1A2A3 | A1→ reward |
| Chain step B | B1B2B3 | B1 →A1 → reward | |
| Chain step C | C1C2C3 | C1 →B1 →A1 → reward | |
| Chain step D | D1D2D3 | D1 →C1 →B1 →A1 → reward | |
|
| |||
| Context-Shift Generalization | Example generalization trial | D1B1X1 | D1 →C1 →B1 →A1 → reward |
|
| |||
| Retest | Cue-association | Y1Y2Y3 | Y1→ reward |
Practice. The Practice Room appears, with three colored doors (P1P2P3), and Kilroy in his “waiting-for-instructions” position at the front bottom of the screen. If the subject chooses the correct door (P1), Kilroy makes it outside and the trial is concluded. Every trial terminates with Kilroy (eventually) reaching the outside. The practice phase continues until the subject makes four consecutive correct trials (i.e. chooses the correct door on the first response in each of four trials).
Phase 1: Sequence training. At this point, new instructions appear: “You’ve successfully finished practice! Now Kilroy will be put in some new rooms. Again, in each room, two doors are locked and one door is unlocked. Each time, click on the door that you think is unlocked. Sometimes, Kilroy will have to go through more than one room to reach the outside. Good luck!” Kilroy now appears in his “waiting-for-instructions” position in Room 1. This phase is identical to the practice phase, except that three new colored cards are used (A1A2A3). Here, subjects have to learn to open the correct door (A1, see Table 2). Once this is learned, phase 2 begins, in which Kilroy appears in Room 2, which contains three new colored cards; here, choice of the correct door (B1) leads Kilroy to Room 1, where a correct answer leads him outside. Once this is learned, subjects work through phase 3 (door C1 in Room 3 leads to Room 2 and so on) and phase 4 (door D1 in Room 4 leads to Room 3 and so on) until, by the end of phase 4, subjects should be choosing the correct door in each room: D1→C1→B1→A1→reward.
Phase 2: Generalization with Context-shift. Next comes a generalization with context-shift phase, unsignaled to the subject. At the start of a trial, Kilroy appears in Room 4. Correct responses will, as usual, allow him to progress through the sequence of rooms and reach the outside. However, now the three cards include one that was previously correct in that room, one that was previously correct but in a different room, and a distractor that was never correct in any room. Thus, in Room 2, Kilroy might be presented with a choice between card B1, card A1, and card C3. Card B1 is the correct choice, and should be chosen by a subject who had learned the sequence, that is, what choice to make at each step in the sequence. But a subject who had merely learned non-sequential stimulus-response associations might choose A1, since that is a stimulus that had been directly associated with reward in the past. The generalization with context-shift phase contains six trials, each trial consisting of a trip through the usual four rooms. This phase enables dissociation between subjects who learned the correct sequence (i.e., what choice to make at each step in the sequence) versus subjects who merely learned non-sequential stimulus-response associations (i.e., knowing a stimulus has been associated with a reward regardless of the sequence).
Phase 3: Retraining. The final phase is a retraining phase, in which subjects are required to learn a new room with three new colored cards (Y1Y2Y3), one of which leads directly to the outside. The purpose of this phase is to determine whether any learning deficits observed on the sequence learning or generalization phase are due to fatigue effects or other non-associative factors.
At the end of the test, the subject sees a screen reporting the total number of trials on which Kilroy got out, which is equal to the total number of trials (regardless of intervening errors).
2.4 STATISTICAL ANALYSIS
The normality of data distribution was checked using Kolmogorov–Smirnov tests. All data were normally distributed (p>0.1). Mixed-design ANCOVA using SPSS 20 was used to compare HC subjects, medication-naïve and SSRI-treated patients with MDD, followed by planned one-way ANOVAs and Tukey’s Honestly Significant Difference (HSD) post-hoc tests. Pearson’s product–moment correlation coefficients were calculated between test performance and neuropsychological measures. The level of significance was set at α=0.05.
3. RESULTS
Five participants failed to complete one of the steps of the sequence-learning phase within the maximum allowed trials (2 HC, 2 medication-native MDD, and 1 SSRI-treated MDD). Data from these subjects were excluded from subsequent analyses. The completion rate did not differ among groups, χ2(2) = 0.37, p=0.83.
We conducted mixed-design ANCOVA, with group as the between-subject variable, learning phase (sequence-learning, context-shift generalization and retraining) as the within-subject variable, BDI-II results as a covariate (there was a significant correlation between BDI-II scores, and the number of errors in the sequence-learning phase, see 3.4 Correlational Studies), and number of errors in sequence-learning, context-shift generalization and retraining as the dependent variable.
Box’s test confirmed the equality of covariance matrices of dependent variables across groups (Box’s M=18.371, F(12, 7458.903)=1.38, p=0.167). The ANCOVA revealed a significant effect of group (F(2,47)=6.282, p=0.004, η2=0.211) and learning phase (F(2,94)=16.018, p<0.001, η2=0.254). In addition, there was a significant interaction between group and learning phase (F(4,94)=9.835, p<0.001, η2=0.295) as well as between BDI-II scores and learning phase (F(4,94)=3.633, p=0.030, η2=0.072. Although there was a significant correlation between BDI-II scores and the number of errors in the sequence-learning phase, BDI-II did not have a significant effect on the between-subject effects of the covariance analysis (p>0.05), and was dropped from subsequent analyses.
To further investigate the differences between groups and within phases, we conducted one-way ANOVA on the sequence-learning, generalization and retraining phases separately, with a Bonferroni correction of α=0.017 to protect the level of significance (for three comparisons).
3.1 Phase 1: Sequence Learning
Using one-way ANOVA, with group as the independent variable, and the total number of errors in the sequence-learning phase (A–D, see Table 2) as the dependent variable, we found a significant effect of group (F(2,48)=8.64, p=0.001, η2=0.26). Tukey’s HSD post-hoc test revealed that medication-naïve patients with MDD made significantly more errors than either SSRI-treated patients with MDD or HCs (p<0.05, Figure 2-A).
Figure 2.
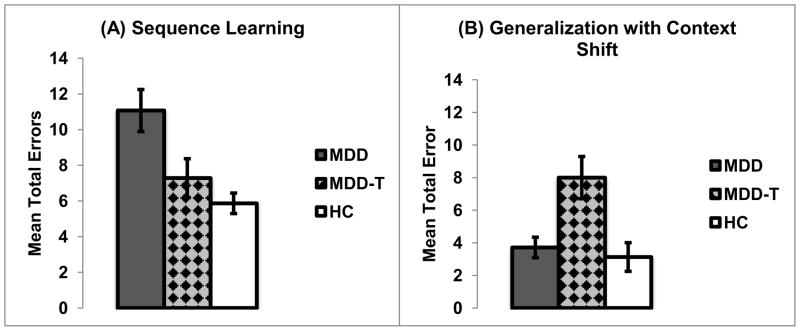
Computer-based cognitive task results. (A) The mean numbers of errors on the sequence-learning phase of the task (chain steps A–D) (±SEM). (B) The mean numbers of errors on the generalization with context-shift phase (±SEM). MDD are medication-naïve patients with MDD, MDD-T are SSRI-treated patients with MDD, HC are healthy controls.
3.2 Phase 2: Generalization with Context-Shift
One-way ANOVA, with group as the independent variable, and the total number of errors in the context-shift generalization phase as the dependent variable, revealed significant effect of group (F(2,48)=10.60, p<0.001, η2=0.31). Follow up post-hoc analysis using Tukey’s HSD showed that SSRI-treated patients with MDD made significantly more errors than either medication-naïve patients with MDD or HCs (p<0.05, Figure 2-B).
3.3 Phase 3: Retraining
One-way ANOVA, with group as the independent variable, and the total number of errors in the retraining phase as the dependent variable, showed no effect of group (F(2,48)=0.43, p=0.64).
3.4 Correlational studies
Pearson’s product–moment correlation coefficients were calculated between test performance on our sequence learning task and neuropsychological measures. There was a significant correlation between BDI-II scores, and the number of errors in the sequence-learning phase. Specifically, the more depressed subjects were, the worse they did at the sequence-learning phase (Pearson’s r=0.360, N=51, p=0.009, Figure 3-A). However, the same correlation did not hold significance when subjects were distributed to their corresponding groups (HC: Pearson’s r=−0.054, N=23, p=0.808; medication-naïve MDD: Pearson’s r=−0.346, N=14, p=0.225; SSRI-treated MDD: Pearson’s r=0.080, N=14, p=0.787). The negative correlation between WAIS-R digit-span results, which represent a measure of short-term memory, and the number of errors on the generalization phase, approached significance; specifically, better working memory scores were associated with better performance (fewer errors) on the generalization phase of the task (Pearson’s r=−0.245, N=51, p=0.083, Figure 3-B). When subjects were split into groups, was that of HCs and SSRI-treated patients with MDD correlation results were approaching significant (HC: Pearson’s r=−0.360, N=23, p=0.092; medication-naïve MDD: Pearson’s r=0.226, N=14, p=0.438; SSRI-treated MDD: Pearson’s r=0.462, N=14, p=0.096).
4. DISCUSSION
The present study investigated the effects of MDD and SSRI administration on sequence learning phase and generalization. As predicted, medication-naïve patients with MDD were impaired on the initial sequence-learning phase of the task. This impairment was not present in the paroxetine-treated MDD group. On the other hand, paroxetine-treated patients with MDD, but not medication-naïve patients with MDD, showed impairment on the generalization phase in which the previously learned rules needed to be applied in a novel context, a function that has been attributed to the medial temporal lobe in a previous study using this task (Nagy et al., 2007a).
The results of our study indicate that medication-naïve patients with MDD show a similar cognitive profile to medication-naïve patients with Parkinson’s disease who were previously tested on this same task (Nagy et al., 2007a), with both patient groups showing impaired learning but spared generalization. This observation might be attributed to the effect of both disorders on striatal dopamine (Kish et al., 1988; McCabe et al., 2010; Walter et al., 2007) as well as a deficit in raphe serotonin (Gervais and Rouillard, 2000; Guiard et al., 2008). Furthermore, there is an overlap between MDD and Parkinson’s disease, where patients with MDD are at a higher risk to develop Parkinson’s disease later in life (Leentjens et al., 2003; Schuurman et al., 2002), whereas 50% of patients with PD develop MDD during the course of the disease (Cummings, 1992; Veiga et al., 2009). However, it is not clear whether this overlap between the two disorders is a consequence of dopaminergic dysfunction alone, or it is a mixture of both serotonergic and dopaminergic effects (Delaville et al., 2012; Kitaichi et al., 2010), which could also have caused the deficit in sequence learning. To dissociate the involvement of serotonergic and dopaminergic systems, studies comparing dopaminergic and serotonergic antidepressants are required and will be an important direction for future research.
SSRIs are the first line of treatment for MDD, due to their well-documented mood-elevating effect (Belmaker, 2008). It is believed that SSRIs achieve their therapeutic outcome by increasing synaptic concentrations of not only serotonin, but also dopamine and norepinephrine (Dunlop and Nemeroff, 2007; Nutt, 2006). SSRI-treated patients in our study received paroxetine mono-therapy to treat their depressive symptoms. Compared to medication-naïve patients with MDD and HC, paroxetine-treated patients with MDD showed no impairment on the sequence-learning phase. This might be a result of paroxetine increasing serotonin and dopamine concentrations, leading to a reduction of reward learning deficit (Cools et al., 2011; Stoy et al., 2011). Minimizing the deficit in reward learning in paroxetine-treated patients with MDD could explain the remediation of learning on the sequence-learning phase, as compared to the medication-naïve patients with MDD who showed impairment on the same phase similar to that seen in medication-naïve patients with Parkinson’s disease (Nagy et al., 2007a; Nagy et al., 2007b; Shohamy et al., 2005). However, the remediation of the sequence-learning deficit in the paroxetine-treated group could be attributed to diminished neural processing of reward and punishment feedback during SSRI treatment, bringing reward and punishment into balance (McCabe et al., 2010). MDD impairs cognitive function at multiple levels, and SSRIs also affect many other neurotransmitter systems that could contribute to the changes in behavioral performance (Belmaker and Agam, 2008),]. It is worth noting that the severity of depressive symptoms, as reflected by BDI-II scores, significantly correlates with the number of errors on the sequence-learning phase of the cognitive task, such that the more depressed people performed most poorly on this learning phase. This is similar to the results we obtained in a previous study using a learning and generalization task with patients with Parkinson’s disease (Herzallah et al., 2010).
LIMITATIONS AND FUTURE DIRECTIONS
An important limitation of the current study is that the different severity of depressive symptoms in SSRI-treated vs. medication-naïve patients might have contributed to the difference between the groups. We did not have access to SSRI-treated patients’ BDI-II scores before they were placed on the SSRI regimen. Therefore, it is impossible to conclude that the observed behavioral effects originate from the medication alone. However, we matched the different groups on almost all measures of neuropsychological and neuropathology tests we used in this study.
On the generalization phase, the performance of medication-naïve patients with MDD was not significantly different from that of HC subjects. In contrast, paroxetine-treated patients with MDD were selectively impaired on the generalization phase, resembling patients with amnestic mild cognitive impairment previously studied using this same task (Nagy et al., 2007a).
Although some previous work has argued that there are “hippocampal-related” cognitive deficits in patients with MDD, and that SSRI administration remediates these deficit (Vythilingam et al., 2004), more recent evidence suggests that there are no volumetric difference at the level of the hippocampus between patients with MDD and normal healthy controls (Kroes et al., 2011; Vythilingam et al., 2004). This finding does not rule out non-volumetric dysfunctions, but we argue that most prior research studies of learning and memory in MDD have used the delayed paragraph recall test (Vythilingam et al., 2004) which is (i) not specific to the medial temporal lobe, as performance can be affected by disrupted frontal function (Loewenstein et al., 2009), and (ii) not sufficiently sensitive to mild degrees of hippocampal atrophy or dysfunction (Loewenstein et al., 2009; Myers et al., 2002).
One possible explanation of our results – and their seeming paradoxical conflict with these past other reports -- might be that SSRI administration results in hippocampal dysfunction via induction of excessive neurogenesis in the dentate gyrus (Meltzer et al., 2005; Ming and Song, 2011). This, of course, is only a conjecture. However, some studies suggest that SSRI-induced neurogenesis produces cells that are characteristically different than cells that are naturally generated in the dentate gyrus (Kobayashi et al., 2010; Liu et al., 2011; O’Leary et al., 2009). These immature newborn cells have different functions than mature cells (Kesner et al., 2004); they are more excitable (Snyder et al., 2001) and tend to inhibit mature neurons in the dentate gyrus (Kobayashi et al., 2010), therefore leading to impaired function of the dentate gyrus in pattern separation of input to the dentate gyrus (Ming and Song, 2011). Moreover, recent evidence in the animal literature also suggests that hippocampal neurogenesis can impair memory retrieval in a radial arm maze (Saxe et al., 2007). Thus, it is possible that the production of immature cells in the hippocampal network as a result of SSRI administration negatively impact memory processes (Saxe et al., 2007). Future research, of course, is needed to evaluate this hypothesis. The near-significant negative correlation between errors on the generalization phase and WAIS-R digit-span scores (a measure of short-term memory) indicates that those who were most impaired on generalization (more errors) had the worst scores on short-term memory (Figure 3-B), which is in line with previous literature that suggests a negative effect of dentate gyrus neurogenesis on some types of memory (Aimone et al., 2009; Saxe et al., 2007; Weisz and Argibay, 2009). A larger study will be required to see if this trend holds consistently and significantly.
Another possible way in which SSRIs could affect hippocampal function via their impact on rapid eye movement (REM) sleep. Studies suggest that SSRI administration suppresses REM sleep (Brooks and Gershon, 1977; Ross et al., 1990), which impairs hippocampal-, but not striatal-, dependent learning {Watts, 2012 #4254;Fogel, 2007 #4255;Hennevin, 1995 #4256;Hennevin, 2007 #4257}. This is in agreement with our findings where SSRI-treated patients with MDD show deficit on the hippocampal-dependent generalization phase, but improved striatal-dependent learning phase of the cognitive task we used. However, it is still unclear how SSRI administration leads to REM suppression and related cognitive deficit. More research is required to disentangle MDD-related from sleep-related cognitive deficits, and further explore the role of SSRIs in subsequent cognitive changes. It will be particularly important in future research to better understand how individual differences in SSRI-related changes in REM correlate with SSRI-mediated changes in both clinical symptoms and cognitive function.
Another possible interpretation of our results is that paroxetine, the SSRI that was used to treat MDD in our sample, has a weak anti-nicotinic anticholinergic effect (Fortin et al., 2011; Mertens and Pintens, 1988). The hippocampus has a high concentration of nicotinic cholinergic receptors (Martin and Aceto, 1981). Previous studies have shown that using multiple SSRIs with anticholinergic properties might impair hippocampal dependent memory functions (Fortin et al., 2011; Herzallah et al., 2010). Thus, this weak anticholinergic effect of paroxetine could have contributed to the deficit we observe in the generalization phase of cognitive task in the SSRI treated MDD patients. However, compared to other anticholinergic agents, paroxetine has been found to have a relatively weak anticholinergic effect (Chew et al., 2008), suggesting that this explanation is unlikely.
We base some of our interpretations of the current study by drawing on findings obtained using the same task in patient populations with amnestic mild cognitive impairment and Parkinson’s disease (Nagy et al., 2007a; Nagy et al., 2007b; Shohamy et al., 2005). However, various studies suggest that amnestic mild cognitive impairment affects many brain systems, other than the medial temporal lobe, and these might contribute to the deficit we found in the generalization phase of the cognitive task (Van Dam et al., 2013). Further, Parkinson’s disease has been shown to affect several systems in the brain other than the nigrostriatal dopamine system (Jellinger, 1991; Mann and Yates, 1983). Subsequently, generalizing earlier results to our findings in MDD might require further research into the overlapping biological correlates between these disorders.
One major limitation to our study is the between-subject design, where the mediation-naïve and the SSRI-treated MDD patients are different individuals. Given the heterogeneity of MDD, and how various subtypes of MDD differ with regards to cognitive function, the current result might be confounded by between-subject variability originating from factors other than MDD and SSRI administration. We did, of course, try to control for that in the current study by recruiting melancholic MDD patients only, and by matching the two groups on various neuropsychological and demographic measures as described earlier. However, future work ought to address this issue through within-subject methodologies, examining the same MDD patients on and off medication.
Another limitation of the current study is the low number of recruited subjects. This becomes evident in the correlational analyses where we collapsed all groups together to investigate correlations within the larger group. However, given that the focus of the current study is task-measured cognitive function, all a priori power analyses indicated the need for 14 subjects per group to achieve power levels higher than 90%, which confirms the sufficiency of the number of subjects in the analysis of our primary cognitive results. Future larger studies, however, should address these imitations and better control for possible confounding variables. Despite these power limitations, we found a double dissociation between medication naïve and SSRI-treated patients with MDD in learning and generalization.
In sum, our results show that SSRIs have both enhancing and deleterious effects on cognition in MDD. To our knowledge, this is the first study to dissociate the effects of MDD and SSRI treatment on cognitive function. Our results examine the effects of chronic (longer than eight weeks) administration of SSRIs. However, there is still much to explore regarding the time course of the effects of SSRIs on cognition, and how this time course might relate to the time course of remediation of depressive symptoms. Moreover, given the heterogeneity of MDD and the wide range of individual differences among people suffering from MDD, further research is needed to better characterize the cognitive correlates of various subcategories of MDD. Additional research is also needed to study other antidepressants in use, such as selective norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants, and dopamine agonists. In addition, the cognitive correlates of other treatment modalities, such as cognitive behavioral therapy, repetitive TMS, and electroconvulsive therapy, and how that might interact with antidepressant treatment regimen also needs further investigation.
Acknowledgments
We would like to thank Al-Quds Cognitive Neuroscience Lab students: Salam Abdellatif, Dana Deeb, Aya Imam, Issa Isaac, Hussain Khdour, Jeries Kort and Mohammad Taha for their excellent technical assistance. Research reported in this publication was supported by the National Institutes of Health Award R21MH095656 from the Fogarty International Center and the National Institute of Mental Health to MAG, the Palestinian American Research Center (PARC), as well as generous donations from Saad N. Mouasher, Daryl Kulok, Dr. Samih Darwazah and Stewart and Lois Gross.
ROLE OF THE FUNDING SOURCE
Authors comply with the NIH Public Access Policy, as per agreements with Elsevier.
Footnotes
CONFLICT OF INTEREST
Authors report no conflict of interest.
CONTRIBUTORS
Mohammad M. Herzallah
Research design, data collection, data analysis, and paper writing.
Ahmed A. Moustafa
Data analysis, and paper writing.
Joman Y. Natsheh
Data collection, data analysis, and paper writing.
Omar A. Danoun
Data collection, data analysis, and paper writing.
Jessica R. Simon
Paper writing.
Yasin I. Tayem
Paper writing.
Mahmud A. Sehwail
Subject recruitment and paper writing.
Ivona Amleh
Subject recruitment and paper writing.
Issam Bannoura
Subject recruitment and paper writing.
Georgios Petrides
Paper writing.
Catherine E. Myers
Research design and paper writing.
Mark A. Gluck
Research design and paper writing.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aimone JB, Wiles J, Gage FH. Computational influence of adult neurogenesis on memory encoding. Neuron. 2009;61:187–202. doi: 10.1016/j.neuron.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amorim P, Lecrubier Y, Weiller E, Hergueta T, Sheehan D. DSM-IH-R Psychotic Disorders: procedural validity of the Mini International Neuropsychiatric Interview (MINI) Concordance and causes for discordance with the CIDI. Eur Psychiatry. 1998;13:26–34. doi: 10.1016/S0924-9338(97)86748-X. [DOI] [PubMed] [Google Scholar]
- Austin MP, Mitchell P, Goodwin GM. Cognitive deficits in depression: possible implications for functional neuropathology. Br J Psychiatry. 2001;178:200–206. doi: 10.1192/bjp.178.3.200. [DOI] [PubMed] [Google Scholar]
- Beck AT, Epstein N, Brown G, Steer RA. An inventory for measuring clinical anxiety: psychometric properties. J Consult Clin Psychol. 1988;56:893–897. doi: 10.1037//0022-006x.56.6.893. [DOI] [PubMed] [Google Scholar]
- Beck AT, Steer RA, Ball R, Ranieri W. Comparison of Beck Depression Inventories -IA and -II in psychiatric outpatients. J Pers Assess. 1996;67:588–597. doi: 10.1207/s15327752jpa6703_13. [DOI] [PubMed] [Google Scholar]
- Belmaker RH. The future of depression psychopharmacology. CNS Spectr. 2008;13:682–687. doi: 10.1017/s1092852900013766. [DOI] [PubMed] [Google Scholar]
- Belmaker RH, Agam G. Major depressive disorder. N Engl J Med. 2008;358:55–68. doi: 10.1056/NEJMra073096. [DOI] [PubMed] [Google Scholar]
- Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, Arango V. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology. 2009;34:2376–2389. doi: 10.1038/npp.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan RA, Crippa JA. The role of dopamine in reward and pleasure behaviour--review of data from preclinical research. Acta Psychiatr Scand Suppl. 2005:14–21. doi: 10.1111/j.1600-0447.2005.00540.x. [DOI] [PubMed] [Google Scholar]
- Brooks DC, Gershon MD. Amine repletion in the reserpinized cat: effect upon PGO waves and REM sleep. Electroencephalography and clinical neurophysiology. 1977;42:35–47. doi: 10.1016/0013-4694(77)90149-3. [DOI] [PubMed] [Google Scholar]
- Burgess A, Flint J, Adshead H. Factor structure of the Wechsler Adult Intelligence Scale-revised (WAIS-R): a clinical sample. Br J Clin Psychol. 1992;31 ( Pt 3):336–338. doi: 10.1111/j.2044-8260.1992.tb01002.x. [DOI] [PubMed] [Google Scholar]
- Buyukdura JS, McClintock SM, Croarkin PE. Psychomotor retardation in depression: biological underpinnings, measurement, and treatment. Progress in neuro-psychopharmacology & biological psychiatry. 2011;35:395–409. doi: 10.1016/j.pnpbp.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Macqueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci. 2004;29:417–426. [PMC free article] [PubMed] [Google Scholar]
- Carlini VP, Poretti MB, Rask-Andersen M, Chavan RA, Ponzio MF, Sawant RS, de Barioglio SR, Schioth HB, de Cuneo MF. Differential effects of fluoxetine and venlafaxine on memory recognition: possible mechanisms of action. Progress in neuro-psychopharmacology & biological psychiatry. 2012;38:159–167. doi: 10.1016/j.pnpbp.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Chew ML, Mulsant BH, Pollock BG, Lehman ME, Greenspan A, Mahmoud RA, Kirshner MA, Sorisio DA, Bies RR, Gharabawi G. Anticholinergic activity of 107 medications commonly used by older adults. J Am Geriatr Soc. 2008;56:1333–1341. doi: 10.1111/j.1532-5415.2008.01737.x. [DOI] [PubMed] [Google Scholar]
- Cools R, Nakamura K, Daw ND. Serotonin and dopamine: unifying affective, activational, and decision functions. Neuropsychopharmacology. 2011;36:98–113. doi: 10.1038/npp.2010.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL. Depression and Parkinson’s disease: a review. Am J Psychiatry. 1992;149:443–454. doi: 10.1176/ajp.149.4.443. [DOI] [PubMed] [Google Scholar]
- David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux JP, Artymyshyn RP, Gardier AM, Gerald C, Antonijevic IA, Leonardo ED, Hen R. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62:479–493. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaville C, Chetrit J, Abdallah K, Morin S, Cardoit L, De Deurwaerdere P, Benazzouz A. Emerging dysfunctions consequent to combined monoaminergic depletions in parkinsonism. Neurobiol Dis. 2012;45:763–773. doi: 10.1016/j.nbd.2011.10.023. [DOI] [PubMed] [Google Scholar]
- Dhillon S, Yang LP, Curran MP. Bupropion: a review of its use in the management of major depressive disorder. Drugs. 2008;68:653–689. doi: 10.2165/00003495-200868050-00011. [DOI] [PubMed] [Google Scholar]
- Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry. 2007;64:327–337. doi: 10.1001/archpsyc.64.3.327. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H, Schoenbaum G, Young B, Bunsey M. Functional organization of the hippocampal memory system. Proc Natl Acad Sci U S A. 1996;93:13500–13507. doi: 10.1073/pnas.93.24.13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel SM, Smith CT, Cote KA. Dissociable learning-dependent changes in REM and non-REM sleep in declarative and procedural memory systems. Behavioural brain research. 2007;180:48–61. doi: 10.1016/j.bbr.2007.02.037. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Fortin MP, Rouch I, Dauphinot V, Gedeon C, Genthon S, Bonnefoy M, Krolak-Salmon P. Effects of anticholinergic drugs on verbal episodic memory function in the elderly: a retrospective, cross-sectional study. Drugs Aging. 2011;28:195–204. doi: 10.2165/11586580-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Gervais J, Rouillard C. Dorsal raphe stimulation differentially modulates dopaminergic neurons in the ventral tegmental area and substantia nigra. Synapse. 2000;35:281–291. doi: 10.1002/(SICI)1098-2396(20000315)35:4<281::AID-SYN6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Guiard BP, El Mansari M, Merali Z, Blier P. Functional interactions between dopamine, serotonin and norepinephrine neurons: an in-vivo electrophysiological study in rats with monoaminergic lesions. Int J Neuropsychopharmacol. 2008;11:625–639. doi: 10.1017/S1461145707008383. [DOI] [PubMed] [Google Scholar]
- Haber SN, Knutson B. The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology. 2010;35:4–26. doi: 10.1038/npp.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz A, Weingartner H, George D, Hommer D, Wolkowitz OM, Linnoila M. Severity of depression in abstinent alcoholics is associated with monoamine metabolites and dehydroepiandrosterone-sulfate concentrations. Psychiatry Res. 1999;89:97–106. doi: 10.1016/s0165-1781(99)00099-2. [DOI] [PubMed] [Google Scholar]
- Herzallah MM, Moustafa AA, Misk AJ, Al-Dweib LH, Abdelrazeq SA, Myers CE, Gluck MA. Depression impairs learning whereas anticholinergics impair transfer generalization in Parkinson patients tested on dopaminergic medications. Cogn Behav Neurol. 2010;23:98–105. doi: 10.1097/WNN.0b013e3181df3048. [DOI] [PubMed] [Google Scholar]
- Igelstrom KM, Heyward PM. Inhibition of hippocampal excitability by citalopram. Epilepsia. 2012;53:2034–2042. doi: 10.1111/j.1528-1167.2012.03660.x. [DOI] [PubMed] [Google Scholar]
- Inzelberg R, Schechtman E, Abuful A, Masarwa M, Mazarib A, Strugatsky R, Farrer LA, Green RC, Friedland RP. Education effects on cognitive function in a healthy aged Arab population. Int Psychogeriatr. 2007;19:593–603. doi: 10.1017/S1041610206004327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol. 1991;14:153–197. doi: 10.1007/BF03159935. [DOI] [PubMed] [Google Scholar]
- Kesner RP, Lee I, Gilbert P. A behavioral assessment of hippocampal function based on a subregional analysis. Rev Neurosci. 2004;15:333–351. doi: 10.1515/revneuro.2004.15.5.333. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N Engl J Med. 1988;318:876–880. doi: 10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- Kitaichi Y, Inoue T, Nakagawa S, Boku S, Kakuta A, Izumi T, Koyama T. Sertraline increases extracellular levels not only of serotonin, but also of dopamine in the nucleus accumbens and striatum of rats. Eur J Pharmacol. 2010;647:90–96. doi: 10.1016/j.ejphar.2010.08.026. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Ikeda Y, Sakai A, Yamasaki N, Haneda E, Miyakawa T, Suzuki H. Reversal of hippocampal neuronal maturation by serotonergic antidepressants. Proc Natl Acad Sci U S A. 2010;107:8434–8439. doi: 10.1073/pnas.0912690107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroes MC, Rugg MD, Whalley MG, Brewin CR. Structural brain abnormalities common to posttraumatic stress disorder and depression. J Psychiatry Neurosci. 2011;36:256–265. doi: 10.1503/jpn.100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leentjens AF, Van den Akker M, Metsemakers JF, Lousberg R, Verhey FR. Higher incidence of depression preceding the onset of Parkinson’s disease: a register study. Mov Disord. 2003;18:414–418. doi: 10.1002/mds.10387. [DOI] [PubMed] [Google Scholar]
- Liu JX, Pinnock SB, Herbert J. Novel control by the CA3 region of the hippocampus on neurogenesis in the dentate gyrus of the adult rat. PLoS One. 2011;6:e17562. doi: 10.1371/journal.pone.0017562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewenstein DA, Acevedo A, Potter E, Schinka JA, Raj A, Greig MT, Agron J, Barker WW, Wu Y, Small B, Schofield E, Duara R. Severity of medial temporal atrophy and amnestic mild cognitive impairment: selecting type and number of memory tests. Am J Geriatr Psychiatry. 2009;17:1050–1058. doi: 10.1097/JGP.0b013e3181b7ef42. [DOI] [PubMed] [Google Scholar]
- Lorenzetti V, Allen NB, Fornito A, Yucel M. Structural brain abnormalities in major depressive disorder: a selective review of recent MRI studies. J Affect Disord. 2009;117:1–17. doi: 10.1016/j.jad.2008.11.021. [DOI] [PubMed] [Google Scholar]
- MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S, Joffe RT, Nahmias C, Young LT. Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci U S A. 2003;100:1387–1392. doi: 10.1073/pnas.0337481100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malberg JE. Implications of adult hippocampal neurogenesis in antidepressant action. J Psychiatry Neurosci. 2004;29:196–205. [PMC free article] [PubMed] [Google Scholar]
- Malberg JE, Schechter LE. Increasing hippocampal neurogenesis: a novel mechanism for antidepressant drugs. Curr Pharm Des. 2005;11:145–155. doi: 10.2174/1381612053382223. [DOI] [PubMed] [Google Scholar]
- Mann DM, Yates PO. Pathological basis for neurotransmitter changes in Parkinson’s disease. Neuropathol Appl Neurobiol. 1983;9:3–19. doi: 10.1111/j.1365-2990.1983.tb00320.x. [DOI] [PubMed] [Google Scholar]
- Martin BR, Aceto MD. Nicotine binding sites and their localization in the central nervous system. Neurosci Biobehav Rev. 1981;5:473–478. doi: 10.1016/0149-7634(81)90017-8. [DOI] [PubMed] [Google Scholar]
- McCabe C, Mishor Z, Cowen PJ, Harmer CJ. Diminished neural processing of aversive and rewarding stimuli during selective serotonin reuptake inhibitor treatment. Biological psychiatry. 2010;67:439–445. doi: 10.1016/j.biopsych.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer LA, Yabaluri R, Deisseroth K. A role for circuit homeostasis in adult neurogenesis. Trends Neurosci. 2005;28:653–660. doi: 10.1016/j.tins.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Mertens C, Pintens H. Paroxetine in the treatment of depression. A double-blind multicenter study versus mianserin. Acta Psychiatr Scand. 1988;77:683–688. doi: 10.1111/j.1600-0447.1988.tb05188.x. [DOI] [PubMed] [Google Scholar]
- Miller HL, Delgado PL, Salomon RM, Berman R, Krystal JH, Heninger GR, Charney DS. Clinical and biochemical effects of catecholamine depletion on antidepressant-induced remission of depression. Arch Gen Psychiatry. 1996;53:117–128. doi: 10.1001/archpsyc.1996.01830020031005. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CE, Kluger A, Golomb J, Ferris S, de Leon MJ, Schnirman G, Gluck MA. Hippocampal atrophy disrupts transfer generalization in nondemented elderly. J Geriatr Psychiatry Neurol. 2002;15:82–90. doi: 10.1177/089198870201500206. [DOI] [PubMed] [Google Scholar]
- Myers CE, Shohamy D, Gluck MA, Grossman S, Onlaor S, Kapur N. Dissociating medial temporal and basal ganglia memory systems with a latent learning task. Neuropsychologia. 2003;41:1919–1928. doi: 10.1016/s0028-3932(03)00127-1. [DOI] [PubMed] [Google Scholar]
- Nagy H, Keri S, Myers CE, Benedek G, Shohamy D, Gluck MA. Cognitive sequence learning in Parkinson’s disease and amnestic mild cognitive impairment: Dissociation between sequential and non-sequential learning of associations. Neuropsychologia. 2007a;45:1386–1392. doi: 10.1016/j.neuropsychologia.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Nagy O, Kelemen O, Benedek G, Myers CE, Shohamy D, Gluck MA, Keri S. Dopaminergic contribution to cognitive sequence learning. J Neural Transm. 2007b;114:607–612. doi: 10.1007/s00702-007-0654-3. [DOI] [PubMed] [Google Scholar]
- Naismith SL, Hickie IB, Ward PB, Scott E, Little C. Impaired implicit sequence learning in depression: a probe for frontostriatal dysfunction? Psychol Med. 2006;36:313–323. doi: 10.1017/S0033291705006835. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biological psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Nutt DJ. The role of dopamine and norepinephrine in depression and antidepressant treatment. J Clin Psychiatry. 2006;67(Suppl 6):3–8. [PubMed] [Google Scholar]
- O’Leary OF, Wu X, Castren E. Chronic fluoxetine treatment increases expression of synaptic proteins in the hippocampus of the ovariectomized rat: role of BDNF signalling. Psychoneuroendocrinology. 2009;34:367–381. doi: 10.1016/j.psyneuen.2008.09.015. [DOI] [PubMed] [Google Scholar]
- Perona MT, Waters S, Hall FS, Sora I, Lesch KP, Murphy DL, Caron M, Uhl GR. Animal models of depression in dopamine, serotonin, and norepinephrine transporter knockout mice: prominent effects of dopamine transporter deletions. Behav Pharmacol. 2008;19:566–574. doi: 10.1097/FBP.0b013e32830cd80f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgar P, Farkas M, Nagy O, Kelemen O, Rethelyi J, Bitter I, Myers CE, Gluck MA, Keri S. Learning cognitive skills in depression: the effect of context-change. Psychiatr Hung. 2007;22:271–275. [PubMed] [Google Scholar]
- Robinson OJ, Cools R, Carlisi CO, Sahakian BJ, Drevets WC. Ventral striatum response during reward and punishment reversal learning in unmedicated major depressive disorder. Am J Psychiatry. 2012;169:152–159. doi: 10.1176/appi.ajp.2011.11010137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RJ, Ball WA, Gresch PJ, Morrison AR. REM sleep suppression by monoamine reuptake blockade: development of tolerance with repeated drug administration. Biological psychiatry. 1990;28:231–239. doi: 10.1016/0006-3223(90)90578-p. [DOI] [PubMed] [Google Scholar]
- Sahay A, Scobie KN, Hill AS, O’Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, Hen R. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011;472:466–470. doi: 10.1038/nature09817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- Sass A, Wortwein G. The effect of subchronic fluoxetine treatment on learning and memory in adolescent rats. Behavioural brain research. 2012;228:169–175. doi: 10.1016/j.bbr.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Saxe MD, Malleret G, Vronskaya S, Mendez I, Garcia AD, Sofroniew MV, Kandel ER, Hen R. Paradoxical influence of hippocampal neurogenesis on working memory. Proc Natl Acad Sci U S A. 2007;104:4642–4646. doi: 10.1073/pnas.0611718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Nolte-Zenker B, Patzer J, Bauer M, Schmidt LG, Heinz A. Psychopathological correlates of reduced dopamine receptor sensitivity in depression, schizophrenia, and opiate and alcohol dependence. Pharmacopsychiatry. 2001;34:66–72. doi: 10.1055/s-2001-15184. [DOI] [PubMed] [Google Scholar]
- Schultz W. Dopamine neurons and their role in reward mechanisms. Curr Opin Neurobiol. 1997;7:191–197. doi: 10.1016/s0959-4388(97)80007-4. [DOI] [PubMed] [Google Scholar]
- Schuurman AG, van den Akker M, Ensinck KT, Metsemakers JF, Knottnerus JA, Leentjens AF, Buntinx F. Increased risk of Parkinson’s disease after depression: a retrospective cohort study. Neurology. 2002;58:1501–1504. doi: 10.1212/wnl.58.10.1501. [DOI] [PubMed] [Google Scholar]
- Shohamy D, Myers CE, Grossman S, Sage J, Gluck MA. The role of dopamine in cognitive sequence learning: evidence from Parkinson’s disease. Behavioural brain research. 2005;156:191–199. doi: 10.1016/j.bbr.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Snyder JS, Kee N, Wojtowicz JM. Effects of adult neurogenesis on synaptic plasticity in the rat dentate gyrus. J Neurophysiol. 2001;85:2423–2431. doi: 10.1152/jn.2001.85.6.2423. [DOI] [PubMed] [Google Scholar]
- Stoy M, Schlagenhauf F, Sterzer P, Bermpohl F, Hagele C, Suchotzki K, Schmack K, Wrase J, Ricken R, Knutson B, Adli M, Bauer M, Heinz A, Strohle A. Hyporeactivity of ventral striatum towards incentive stimuli in unmedicated depressed patients normalizes after treatment with escitalopram. J Psychopharmacol. 2011 doi: 10.1177/0269881111416686. [DOI] [PubMed] [Google Scholar]
- Vakili K, Pillay SS, Lafer B, Fava M, Renshaw PF, Bonello-Cintron CM, Yurgelun-Todd DA. Hippocampal volume in primary unipolar major depression: a magnetic resonance imaging study. Biological psychiatry. 2000;47:1087–1090. doi: 10.1016/s0006-3223(99)00296-6. [DOI] [PubMed] [Google Scholar]
- Van Dam NT, Sano M, Mitsis EM, Grossman HT, Gu X, Park Y, Hof PR, Fan J. Functional neural correlates of attentional deficits in amnestic mild cognitive impairment. PLoS One. 2013;8:e54035. doi: 10.1371/journal.pone.0054035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga BA, Borges V, Silva SM, de Goulart FO, Cendoroglo MS, Ferraz HB. Depression in Parkinson’s disease: clinical-epidemiological correlates and comparison with a controlled group of non-parkinsonian geriatric patients. Rev Bras Psiquiatr. 2009;31:39–42. doi: 10.1590/s1516-44462009000100010. [DOI] [PubMed] [Google Scholar]
- Vythilingam M, Vermetten E, Anderson GM, Luckenbaugh D, Anderson ER, Snow J, Staib LH, Charney DS, Bremner JD. Hippocampal volume, memory, and cortisol status in major depressive disorder: effects of treatment. Biological psychiatry. 2004;56:101–112. doi: 10.1016/j.biopsych.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Walter U, Hoeppner J, Prudente-Morrissey L, Horowski S, Herpertz SC, Benecke R. Parkinson’s disease-like midbrain sonography abnormalities are frequent in depressive disorders. Brain. 2007;130:1799–1807. doi: 10.1093/brain/awm017. [DOI] [PubMed] [Google Scholar]
- Watts A, Gritton HJ, Sweigart J, Poe GR. Antidepressant suppression of non-REM sleep spindles and REM sleep impairs hippocampus-dependent learning while augmenting striatum-dependent learning. J Neurosci. 2012;32:13411–13420. doi: 10.1523/JNEUROSCI.0170-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz VI, Argibay PF. A putative role for neurogenesis in neuro-computational terms: inferences from a hippocampal model. Cognition. 2009;112:229–240. doi: 10.1016/j.cognition.2009.05.001. [DOI] [PubMed] [Google Scholar]