

Abstract
Troglitazone, a first-generation thiazolidinedione of antihyperglycaemic properties, was withdrawn from the market due to unacceptable idiosyncratic hepatotoxicity. Despite intensive research, the underlying mechanism of troglitazone-induced liver toxicity remains unknown. Here we report the use of the Sod2+/– mouse model of silent mitochondrial oxidative-stress-based and quantitative mass spectrometry-based proteomics to track the mitochondrial proteome changes induced by physiologically relevant troglitazone doses. By quantitative untargeted proteomics, we first globally profiled the Sod2+/– hepatic mitochondria proteome and found perturbations including GSH metabolism that enhanced the toxicity of the normally nontoxic troglitazone. Short- and long-term troglitazone administration in Sod2+/– mouse led to a mitochondrial proteome shift from an early compensatory response to an eventual phase of intolerable oxidative stress, due to decreased mitochondrial glutathione (mGSH) import protein, decreased dicarboxylate ion carrier (DIC), and the specific activation of ASK1-JNK and FOXO3a with prolonged troglitazone exposure. Furthermore, mapping of the detected proteins onto mouse specific protein-centered networks revealed lipid-associated proteins as contributors to overt mitochondrial and liver injury when under prolonged exposure to the lipid-normalizing troglitazone. By integrative toxicoproteomics, we demonstrated a powerful systems approach in identifying the collapse of specific fragile nodes and activation of crucial proteome reconfiguration regulators when targeted by an exogenous toxicant.
Keywords: mitochondria, proteomics, toxicology, drug-induced liver injury, SOD2
Introduction
Drug-induced liver injury (DILI) is a leading cause of acute liver failure,1 thus constituting a major reason for drug candidates failing during development or withdrawal from the market. Because of drug-related toxicity, many drug candidates that may otherwise be potentially efficacious in the treatment of disorders or diseases have been discontinued; this precipitous discontinuation represents a major setback to a larger population which may benefit from further development of these drug candidates. In addition, from the pharmaceutical industry’s perspective, the resultant regulatory actions have driven up the development cost to meet acceptable safety requirements. Drugs that cause DILI rarely do so at doses tolerated well by most patients at frequencies of less than 1 in 10,000 patients, and generally do not exhibit dose-dependency. Consequently, they often escape detection preclinically and even in Phase II/III trials due to its limited patient pool versus the much larger population exposed to the drug.1,2 Establishing definitive prediction of drugs that exhibit DILI can be difficult and signals an urgent need for innovative approaches that can rigorously detect early signs of potential hepatotoxicity in drug candidates.3 Current toxicological studies using normally healthy animals are often insufficient in addressing the mechanisms behind human diseases and are relatively insensitive in detecting drug-induced toxicity effects.4 Therefore, considerations of animal models with underlying genetic abnormalities relevant to human diseases may be a useful strategy in revealing and evaluating potential drug-induced adverse effects. Recently, transgenic mice of heterozygous genetic knockouts are proposed as alternative animal models in a diverse range of research fields including toxicity assessment due to their intermediate phenotypes.4
Troglitazone, a once-marketed first-generation thiazolidinedione used in the treatment of Type-II Diabetes Mellitus was withdrawn from the market due to unacceptable idiosyncratic hepatotoxicity risks.5 In early drug safety assessments and even in long-term studies, troglitazone did not cause hepatotoxicity in normal healthy rodents and monkeys.6−8 This highlights the shortcomings and remarkable difficulty in predicting idiosyncratic DILI at the standard preclinical animal testing phase.4,9 Numerous attempts to explain the mechanisms underlying troglitazone-induced liver toxicity (TILI) or to recapitulate the human scenario have been unsuccessful, and in vitro results and ensuing hypotheses provided little mechanistic relevance to address clinical TILI.10,11 We and others have demonstrated that the Sod2+/– mouse exhibits higher sensitivity toward the mitochondrial damaging effects of drugs, including troglitazone.12−17 The Sod2+/– mouse model presents an interesting phenotype that is clinically silent yet amenable to unmasking potential drug-induced adverse reactions of normally mild drugs, thereby representing a useful model in drawing correlations between increased mitochondrial oxidative stress and drug-induced adverse effects. Two-dimensional liquid chromatography–difference gel electrophoresis on the Sod2+/– hepatic mitochondrial proteome (henceforth referred to as mitoproteome) revealed molecular changes that recapitulate the clinical features of TILI in a time-dependent fashion.18
Encouraged by the involvement of specific mitochondrial proteins in troglitazone-induced hepatotoxicity and the superior comprehensiveness of mass spectrometry-based proteomics,19 we sought to deepen the coverage of mitochondrial protein changes between Sod2+/+ and Sod2+/– mice, as well as to track Sod2+/– mitoproteome changes with troglitazone administration (Figure 1). Integrating quantitative proteomics, toxicological end points, and topological changes, we found fragilities in mitochondrial glutathione (mGSH) transport and oxidative-stress-induced dysregulation of lipid-associated proteins as crucial nodes that underlie the transition from early compensatory responses to late hepatic injury in the Sod2+/– mouse. Furthermore, we show that deficiency in mGSH transport by dicarboxylate ion carrier (DIC) accelerates troglitazone-induced cytotoxicity. This quantitative systems approach represents a new and powerful way toward understanding DILI with major implications for its early prediction.
Figure 1.

Flow-chart summary of the iTRAQ experimental designs of 4-plex and 8-plex systems. (A) Quantification of proteins differentially expressed in the Sod2+/+ and Sod2+/– hepatic mitochondria using the 4-plex iTRAQ channels. The proteomics experiment was performed in technical replicates. (B) Quantitative shotgun proteomics using the 8-plex iTRAQ labels to elucidate and identify mitoproteome dynamics over two periods of daily vehicle (VEH) or troglitazone (TRG) dosing. The experiment was performed in technical replicates. See text for further details to experimental design. NC, no change; NS, not significant; S, significant; SCX, strong cation exchange; RP, reverse phase.
Experimental Procedures
Animals and Drug Administration
All protocols involving animals were in compliance with the Institutional Animal Care and Use Committee and in accordance with the guidelines of the National Advisory Committee for Laboratory Animal Care and Research. Heterozygous Sod2tm1Leb/J mice, congenic in the C57BL/6 background, were obtained from Jackson Laboratory (Bar Harbor, ME). A breeding colony was established by crossing male Sod2+/– with female wild type Sod2+/+ mice.
Female Sod2+/– mice were randomly divided into four groups (n = 3–6) and injected daily intraperitoneally with 9% solutol HS-15 (10 μL/g body weight) or troglitazone (30 mg/kg body weight; Cayman Chemical, Ann Arbor, MI) for 14 or 28 days. After 14 or 28 days of treatment, the mice were anesthetized with pentobarbital (60 mg/kg, intraperitoneally), and immediately after necropsy, livers were excised; one portion of liver sample for use in histopathological evaluation was fixed in 4% neutral buffered formalin while the remaining portion was used to prepare mitochondrial fractions. Blood was drawn via cardiac puncture; serum was prepared by allowing blood to clot for 30 min and centrifuging at 2000g at 4 °C for 10 min; and the supernatant was decanted for analysis. All mice were food-deprived the night before sacrifice.
Sample Preparation
The mitochondrial fraction was obtained by centrifugation of liver homogenates first at 800g and then twice at 8000g. The enriched mitochondrial pellet was resuspended in wash buffer (210 mM mannitol, 70 mM sucrose, 5 mM Hepes, 1 mM EGTA, pH 7.4) and gently layered onto a modified discontinuous Percoll gradient. A gradient was generated by layering 1.5 mL of 60%, 4 mL of 32%, 1.5 mL of 23%, and 1.5 mL of 15% (v/v) Percoll in wash buffer. After centrifugation at 134000g at 4 °C for 1 h, purified mitochondria were recovered from the 60/32% interface, which was then diluted with 4 volumes of wash buffer before being centrifuged at 12000g at 4 °C. The purified mitochondrial proteins were resuspended in lysis buffer (7 M urea, 2 M thiourea, 4% (w/v) CHAPS) with Halt protease inhibitor cocktail, PhosStop phosphatase inhibitor (Pierce, Rockford, IL), DNase 1, and RNase A (Roche Applied Science, Mannheim, Germany), with protein content being determined and then with storage as described previously.18
Proteomics
Two iTRAQ experiments were performed: (i) 4-plex system for the Sod2+/– versus Sod2+/+ comparative study and (ii) 8-plex system for the troglitazone study. Schematics of the experimental designs can be found in Figure 1. 4-plex and 8-plex iTRAQ labeling was performed according to manufacturers’ protocols (AB SCIEX, CA). The labeled samples were combined and passed through a strong cation exchange cartridge (AB SCIEX) and SepPak (Millipore), vacuum-dried, and stored at −80 °C until further use. Each of the iTRAQ-labeled peptide mixtures was separated by 2D-LC using an Ultimate dual gradient LC system (Dionex-LC Packings) equipped with a Probot MALDI spotting device. The LC fractions were mixed directly with MALDI matrix solution (7 mg/mL α-cyano-4-hydroxycinnamic acid and 130 μg/mL ammonium citrate in 75% ACN) at a flow rate of 5.4 μL/min via a 25 nL mixing tee (Upchurch Scientific) before they were spotted onto a 192-well stainless steel MALDI target plate (AB SCIEX) using a Probot Micro Precision Fraction Collector (Dionex-LC Packings) at a speed of 5 s/well.
The samples on the MALDI target plates were analyzed using a 4700 Proteomics Analyzer mass spectrometer (AB SCIEX). MS/MS analyses were performed using nitrogen at a collision energy of 1 kV and a collision gas pressure of 1 × 10–6 Torr. One thousand shots were accumulated for each MS spectrum. For MS/MS, 6000 shots were combined for each precursor ion with a signal-to-noise (S/N) ratio greater than or equal to 100. For precursors with a S/N ratio between 50 and 100, 10000 shots were acquired. The resolution used to select the parent ion was 200. No smoothing was applied before peak detection for both MS and MS/MS, and the peaks were deisotoped. For MS/MS, only the peaks from 60 to 20 Da below each precursor mass and with S/N ≥ 10 were selected. Peak density was limited to 30 peaks per 200 Da, and the maximum number of peaks was set to 125. Cysteine methanethiolation, N-terminal iTRAQ labeling, and iTRAQ-labeled lysine were selected as fixed modifications; methionine oxidation was considered as a variable modification. One missed cleavage was allowed. Precursor error tolerance was set to 100 ppm; MS/MS fragment error tolerance was set to 0.4 Da. Maximum peptide rank was set to 2.
iTRAQ Data Analysis
iTRAQ data analyses were performed using ProteinPilot (AB SCIEX) by searching the spectra against the International Protein Index (IPI) mouse database (version 3.48) concatenated with a randomized “decoy” version of itself, restricted to tryptic peptides. “Instantaneous” FDR estimation was set at 5% (Supporting Information Figure S1A). As a further refinement, proteins have to meet the criteria of (i) ProteinPilot Unused ProtScore of ≥2.0 (99% C.I.) and (ii) two or more tryptic peptides with at least one unique peptide of high confidence (>99%). The iTRAQ fold change (R) is a relative ratio of either (i) Sod2+/– over Sod2+/+ or (ii) TRG over VEH. For quantification purposes, MS and MS/MS spectra were manually examined and detected sequences rejected on the basis of one or more of the following criteria: (i) C.I. < 99%, (ii) partially cleaved tryptic sequences or internal miscleavages, except when proline is the C-terminus of lysine or arginine, (iii) “crowding” of one or more adjacent precursor peaks whose intensity is half or more than the matched monoisotopic precursor peak, within a precursor ion tolerance window of ±5 Da, (iv) unlabeled iTRAQ, (v) nonspecific iTRAQ labeling, (vi) plausible PTMs, and (vii) weak iTRAQ reporter ion intensity (threshold at fifth percentile, <3500 relative intensity for 4-plex and <1500 for 8-plex; Supporting Information Figure S1B). To achieve accurate and reliable quantification, cutoff was tailed at the fifth percentile to restrict peptides of low reporter ion signals because they tend to result in less robust quantification. Proteins that have P-values ≤0.05 in at least one data set and showed consistent changes in the replicates were considered as significantly altered in the expression level. Keratin was disregarded.
Validation by Immunoblotting and MRM
As a confirmatory step to our mass spectrometric results, we selected a number of candidate targets and analyzed them via immunoblotting. Antibodies against NDUFS3 (MitoSciences, Invitrogen, Carlsbad, CA), SDHB, COX IV (Molecular Probes, Invitrogen), ACADM, DIC, MSRA, SOD2, mt-COX1, TOMM20, UQCRFS1 (Abcam), and cytochrome c (BD Pharmingen, San Jose, CA) were used. Bands were visualized by utilizing peroxidase-conjugated secondary antibodies and advanced chemiluminescence (GE Healthcare, Uppsala, Sweden). Total cytochrome c levels were determined from liver extracts. To detect activation of the mitogen-activated protein kinase (MAPK) signaling cascade, equal amounts of liver lysates were separated and subjected to immunblotting with antibodies specific for ASK1, phospho-ASK1Serine83, phospho-ASK1Threonine845, JNK, phospho-JNKThreonine183/Tyrosine185, p38 MAPK, and phospho-p38 MAPKThreonine180/Tyrosine182 (Cell Signaling Technology, Beverly, MA). To assess the transcriptional regulatory circuits leading to expression of mitochondria gene products, liver lysates were immunoblotted using antibodies against NRF-1, ERRα (Abcam), PGC-1β (Santa Cruz Biotechnology), PGC-1α (Aviva Systems Biology, San Diego, CA), proliferator-activated receptor gamma (PPARγ), pFOXO3aSerine253, and FOXO3a (Cell Signaling Technology). Quantification of band densities was obtained by scanning the blots with a GS-800 densitometer (Bio-Rad).
MRM was employed as an alternative validation platform where commercial antibodies were not available. In addition, MRM provided the opportunity to detect selected proteins missed in the iTRAQ assay, especially lower abundant proteins. Individually, 80 μg of peptides from Sod2+/+ and Sod2+/– mice liver were labeled with light and heavy mTRAQ channels (AB SCIEX) and subsequently mixed in a 1:1 ratio. The peptides were analyzed using the MRM technique on a Tempo nano-LC (AB SCIEX) coupled to a Q TRAP 4000 configuration (AB SCIEX). Five microliters of sample was injected and desalted online using a Reprosil C18-Aq trap column (5 μm × 0.3 mm i.d.; SGE Analytical Science, Victoria, Australia), running at 0.1% formic acid in water for 5 min at 20 μL/min. The peptides were then eluted from a Chromolith CapRod C18 column (0.1 mm i.d. × 150 mm; EMD Darmstadt, Germany) with a linear gradient starting from 5% B to 30% B (A, 0.1% formic acid in water; B, 0.1% formic acid in ACN) for 30 min at 0.3 μL/min. The ion source parameters were optimized at an ion spray voltage of 2800 V, with ion source gas 1 at 40 V, and with the interface heater temperature at 150 °C. The MRM transitions of the peptides were generated from TIQAM software (www.proteomecenter.org). Peptides were excluded where there were cysteines and methionines and peptides with two and more consecutive basic amino acids on either the C- or N-terminus. In addition, the molecular masses of the peptides selected were between 400 and 1200 Da. The masses of the precursor and product ions were then modified on the basis of the mTRAQ labeling technique. A total of 430 MRM transitions were used to detect and quantify 25 proteins. The MRM transitions, the dwell time and collision energy of the mTRAQ-labeled peptides are illustrated in Supporting Information Table S1. In order to confirm the presence of the protein, at least 2 MRM transitions from each of its 2 peptides must be detected.
Serum ALT Analysis, Histopathology, and Immunohistochemistry
Serum ALT activity was measured using a Cobas c111 analyzer (Roche). For histopathological analysis, livers from the same set of mice used in the iTRAQ experiments were formalin-fixed and paraffin-embedded. The fixed tissues were subsequently processed with an automatic tissue processor (Leica TP 1020, Germany) and embedded in paraffin blocks. Two micrometer tissue sections were stained with hematoxylin and eosin and analyzed by light microscopy.
For IHC, the formalin-fixed, paraffin-embedded sections were dewaxed and rehydrated, and antigen retrieval was performed by heating in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 20 min. Endogenous peroxidase activity was blocked using 0.3% hydrogen peroxide for 10 min. Sections were then blocked and immunostained using an Envision + System-HRP Labeled Polymer Kit (Dako, Denmark). Anti-phospho-FOXO3aSerine 253 antibodies (Cell Signaling Technology) were incubated overnight at 4 °C, and detection of antigens was performed using 3,3-diaminobenzidine. The sections were counterstained using Lillie’s modified Mayers Hematoxylin, dehydrated, mounted, and viewed under a light microscope (Carl Zeiss, Oberkochen, Germany).
Determination of Mitochondrial Glutathione, Nitrotyrosine, and Cytochrome c Levels
mGSH was determined in isolated mitochondria using monochlorobimane as fluorochrome. Monochlorobimane features a high selectivity for GSH and is conjugated to glutathione (GSH) by a glutathione-S-transferase-catalyzed reaction. Mitochondrial samples were incubated with monochlorobimane for 1 h at 37 °C. The fluorescence was determined at 380/460 nm, and GSH levels were calculated from a standard curve formed from glutathione positive controls. Cytochrome c (R & D Systems, Minneapolis, MN) and nitrotyrosine (Kamiya biomedical, Seattle, WA) ELISA was performed according to the manufacturer’s instructions.
Determination of ACO2 Activity and Modification
Mitochondrial ACO2 activity was determined from isolated mitochondria using an aconitase activity assay (Cayman Chemicals), which measures the formation of NADPH from NADP+ concomitant with the conversion of citrate to isocitrate and of isocitrate to α-ketoglutarate. The reaction was monitored at 340 nm.
8% nondenaturing gels were used to monitor ACO2 higher molecular weight aggregates (>250 kDa) from mitochondrial extracts. 12.5% denaturing gels were used to monitor intact (83 kDa) and cleaved ACO2 (∼40 kDa). Antibodies specifically targeting residues 767–780 of the carboxy terminus of ACO2 (a generous gift from Dr. Bi Xuezi)20 were used for immunoblotting analysis. Bands were visualized by utilizing peroxidase-conjugated secondary antibodies and advanced chemiluminescence (GE Healthcare).
RNAi
HepG2 cells were cultured in the presence of Dulbecco’s modified Eagle’s medium (DMEM; Sigma), 100 IU penicillin/μg streptomycin, and 10% fetal bovine serum at 37 °C in a humidified incubator with 5% CO2. The cells were washed with PBS and treated with various concentrations of troglitazone, dissolved in 0.1% DMSO, at different time-points in serum-free DMEM (1, 2, and 5 h). At 5 h, 100 μM troglitazone was determined as the cytotoxic dose, which is consistent with previous studies.21 HepG2 cells were seeded (0.25 × 106/well; 24-wells), and at 70–80% confluency, they were transfected with a pool of four predesigned siRNA targeting Slc25a10 (50 nM, ON-TARGETplus SMARTpool siRNA, Dharmacon, Thermo Scientific, Waltham, MA) for 18 h using 2.5 μL of TransIT-TKO as the transfection reagent (Mirus Bio, Madison, WI) in the absence of streptomycin. Sequences of Slc25a10 siRNA are shown in the Table 1. GAPD siRNA (catalog no. D-001830-20, Thermo Scientific), scrambled nontargeting siRNA pool (catalog no. D-001810-10, Thermo Scientific), DMEM + 0.1% DMSO, and DMEM + TransIT-TKO (mock) served as controls. Small molecule inhibition via 20 mM butylmalonate (Sigma) or antioxidant GSH-ethyl ester (Sigma) was added to HepG2 cells for 1 h prior to the addition of troglitazone. Cell viability was measured by the formation of formazan using the MTS (Promega, Madison, WI) assay at 490 nm.
Table 1.
| siRNA pool | target sequence |
|---|---|
| DIC (Slc25a10) siRNA 1 | CGGCGGAUGUGCCACGUUU |
| DIC (Slc25a10) siRNA 2 | GAGGUGAAGCUUCGAAUGA |
| DIC (Slc25a10) siRNA 3 | GUGCUGAAGACUCGCCUGA |
| DIC (Slc25a10) siRNA 4 | CGGCAUCAGUGGUUUAACU |
Data Integration and PEP
We performed our network-based profiling utilizing the Proteomics Expansion Pipeline or PEP as described previously.22 Briefly, the mitoproteome data set identified formed the basis of our seed selection. A seed here is a high-confidence identified protein defined by the criteria as stated above. This selection process reduced the original 277 identified proteins to a set of 113 seeds. We built an expanded mouse PPIN by merging data from two data sources: MppDB23 and IntNetDB.24 MppDB is a mouse protein–protein interaction (PPI) database, and we used the reference set of mouse PPI data collected over five PPI databases: DIP, BIND, MIPS, MINT, and IntAct. This network was rather sparse, with limited information. Hence, we used IntNetDB to obtain interologs from human PPIN to mouse, and we merged this with the MppDB data set. The resultant network of 10307 nodes and 124866 edges was then subfiltered for edges between mitochondrial and its associated proteins from MitoCarta25 (http://www.broadinstitute.org/pubs/MitoCarta/). This subfiltered network, which we henceforth refer to as the Mitonetwork, was utilized for functional analysis of our proteomics data.
Statistical and In Silico Analysis
Ingenuity Pathway analysis (IPA; version 7.1) was used for the analysis of over-represented biological pathways. Right-tailed Fisher’s Exact test (P < 0.05) is used for multiple testing of over-represented pathways. To detect differences in distribution of fold changes, we performed Student’s t-test on the wild type versus Sod2+/– mouse (P < 0.05) and 1-way ANOVA with Bonferroni posthoc test for the toxicological study (14 and 28 days; P < 0.05). Detected proteins were clustered using HCL to detect patterns in protein expressions. Distances between samples were determined using Euclidean distance, and cluster distances were determined using Ward’s method. Gene ontology (GO) enrichment analysis was performed using GO-Term Finder.26 Statistically enriched GO “biological process” terms were identified using the standard hypergeometric test; significance was defined at Padjusted < 0.01 using the Bonferroni multiple testing correction. The GO terms “metabolic process, cellular metabolic process, and primary metabolic process” were considered broad terms and were not included in the analysis. Unique annotation terms were extracted by pairwise comparisons between all identified expression clusters.
For the prediction of troglitazone induction on PPAR target genes, corresponding matched DNA genomic sequences were obtained and screened using Matinspector (version 8.0) for PPAR response elements (PPREs). Genomic sequences and transcription start sites were obtained from the Ensembl database (release 49), and regions spanning 5000 bp upstream and downstream of the transcriptional starting sites were extracted to accommodate for genes with unusual PPRE locations.27 Only the V$PERO matrix family was included in the search. To increase the stringency, core similarity and matrix similarity scores for each prediction were assigned cut-offs at 0.8 each. This gave rise to 325 potential PPRE sites in 76 genes/proteins (14 days, 136 genes with 507 predicted sites; 28 days, 148 genes with 584 sites; noting several common genes between the two time courses). To test if the presence of a predicted PPRE site gives rise to larger fold changes, we split the expression data into two categories on the basis of bespoke PPRE analyses, and we compared their fold distributions. As the fold distributions are non-normal, significant differences were determined using the nonparametric Mann–Whitney test.
Results
Sod2 Haplodeficiency Moderately Affects the Mitoproteome
The monoallelic loss of Sod2 provided the opportunity to examine how the mitoproteome copes with the damaging effects of free radicals under diminished protection.28 Liver mitochondria from Sod2+/– and Sod2+/+ mice (n = 4 per group) were compared, and 321 mitochondrial proteins were identified with high confidence (Figure 2A). We confirmed the quantitative specificity of MS to detect Sod2 haplodeficiency (R+/–:+/+ ≈ 0.53; Figure 2B). iTRAQ has been reported to suppress quantification, in particular with the larger fold changes.29 Therefore, while the observed iTRAQ ratio of SOD2 follows that of the expected ratio, we cannot rule out potential underestimation of the larger fold changes. The expression of several selected proteins was confirmed by immunoblotting and MRM, demonstrating the accuracy of our MS quantification (Supporting Information Figure 2A and B). Silver staining of whole proteomes run on 1D SDS-PAGE gel acted as loading controls (Supporting Information Figure 2C).
Figure 2.

Sod2 haplodeficiency and mitoproteome. (A) Volcano plot of liver mitochondrial proteins. Vertical dotted lines denote significant fold change cut-offs, defined as R+/–:+/+ ≥ 1.2 for up-regulation and ≤0.833 for down-regulation, and horizontal dotted lines denoted P ≤ 0.05. The inset shows the proportion of the Sod2+/– mitoproteome that was unchanged, downregulated, or upregulated relative to Sod2+/+. (B) MS/MS spectrum of the m/z region of reporter ions generated from iTRAQ labeled SOD2 tryptic peptide. Immunoblotting against SOD2 showed consistency with MS quantification. Note that channels 114/115 and 116/117 form a pair of duplicates.
Compensatory responses to maintain mitochondrial functionality28 were evidenced by the up-regulation of a larger proportion of the mitochondrial proteins (25%) compared to down-regulated proteins (13%; Supporting Information Table S2). ROS levels are modulated in young Sod2+/– mice,30 and we asked if this ROS homeostatic maintenance is maintained in part by the antioxidant enzymes housed within the mitochondria. GSH, a major low-molecular-weight antioxidant is regulated by a network of enzymes and oxidoreductases to maintain its homeostasis. Depletion of mGSH is a hallmark of endogenous and chemically induced oxidant stress, and in the absence of de novo GSH synthesis in the mitochondria, maintenance of the mGSH pool is crucial to ensure mitochondrial redox balance.31 The up-regulation of GSTK1, MGST1 (Supporting Information Table S2), and GPX132 suggests increased recycling of mGSH to restore redox equilibrium with the partial ablation of Sod2. Proteins with iron–sulfur ([Fe–S]) clusters are known for their exquisite sensitivity to oxidative stress and used if these mitochondrial proteins are affected. Because of their low abundance, we measured FXN and FDX1 levels by MRM and found lower levels in Sod2+/– liver (Supporting Information Figure 2B). Under oxidative stress, [Fe–S] proteins may be damaged and degraded, or their biogenesis interrupted.33 Given the many roles of [Fe–S] proteins, which include stabilizing structures and acting as electron carriers and regulatory sensors,34 it is crucial to maintain a steady pool of [Fe–S] proteins with nondisrupted [Fe–S] clusters. Concordantly, we found enrichment of TST (R+/–:+/+ ≈ 1.46), whose role includes the formation of [Fe–S] clusters. Similar levels of endogenous mGSH, mitochondrial protein carbonyl, and nitrotyrosyl adducts in Sod2+/+ and Sod2+/– mice were observed (Student’s t-test, P = 0.19, P = 0.11, and P = 0.92 respectively; Supporting Information Figure 3A), reflecting GSH-induced protection from oxidative stress.
GO enrichment analysis of the up- and down-regulated proteins revealed statistically significant alteration of several “biological processes” associated with the loss of a single Sod2 allele (Supporting Information Figure 3B). Expectedly, electron transport chain and redox homeostasis were implicated (Padj = 5.24 × 10–06 and 3.61 × 10–06, respectively), consistent with increased mitochondrial oxidative stress and respiration.28,35 Many biochemical functions were also perturbed, including fatty acid metabolism (Padj = 1.95 × 10–08). Glutamine is a precursor of GSH,36 and the overrepresentation of glutamine metabolism (Padj = × 10–05) and GSH metabolism (Padj = 8.66 × 10–05) provided further evidence of GSH perturbation. Taken together, our data suggest that there is attenuation of ROS due to an upregulated antioxidant defense, and also discrete molecular aberrations that are present that may sensitize the Sod2+/– mouse to clinically silent drug toxicities.37
Troglitazone Administration Leads to Delayed Hepatic Sod2+/– Mitoproteome Damage
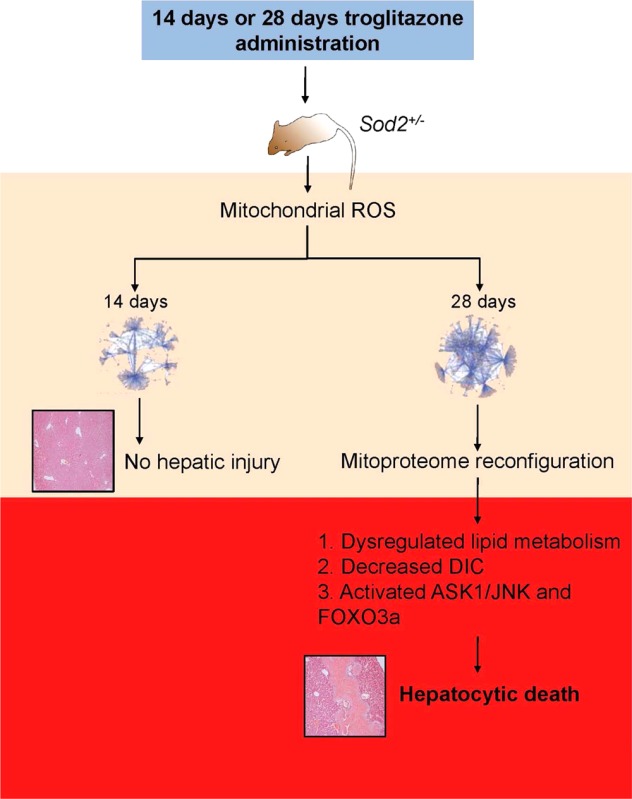
A hallmark of TILI is the delayed onset of liver injury, which could abruptly progress to life-threatening irreversible liver failure.5 Troglitazone administration in Sod2+/+ (wild type) mice did not result in hepatic injuries,16 and because Sod2+/– mice below the age of 20 weeks accumulate clinically silent oxidative stress,28 we focused on studying troglitazone effects on the Sod2+/– hepatic mitochondria. Histological examination of Sod2+/– liver displayed a two-phase response that mirrors the delayed onset of liver injury characteristic of idiosyncratic TILI in humans—an early nontoxic phase and a later sustained phase of liver injury, exemplified by confluent areas of cytoplasmic vacuolation and hepatocytic death (Figure 3A). Serum alanine aminotransferase levels significantly increased (187%) in Sod2+/– treated with 28 days troglitazone relative to solutol administered Sod2+/– mice (Figure 3B).
Figure 3.

Sod2 haplodeficiency delays troglitazone hepatotoxicity, as revealed by quantitative proteomics. (A) Representative hematoxylin and eosin liver sections prepared from solutol, and troglitazone-treated Sod2+/– mice dosed over either 14 days or 28 days at 100× magnification. Insets at the top left are observations made at 400×. Black arrows indicate vacuolation of hepatocytes. (B) Serum alanine aminotransferase levels of Sod2+/– mice administered with 14 days or 28 days troglitazone. (C) Hierarchical clustering with heatmap of mitochondrial proteins. The different colors on the side bars represent the three major clusters. (D) Significantly perturbed pathways in Sod2+/– liver mitochondria with troglitazone administration. The total number of proteins that make up a pathway is displayed on the top of columns. The ratio is defined as the number of proteins analyzed against the total number of proteins in a pathway. Mean ± SD values are shown. n = 3 per group; *P < 0.05. VEH, vehicle-administered; TRG, troglitazone-administered.
To understand the associated mitoproteome changes, we performed 8-plex iTRAQ to interrogate the mitochondrial profiles across 14 and 28 days drug administration (30 mg/kg daily, i.p.). A total of 314 proteins were identified (Supporting Information Table S3), and hierarchical clustering (HCL) showed that temporal-defined proteins were classified into three major clusters (Figure 3C). By immunoblotting, we validated the differential expression of selected mitochondrial proteins and found good correspondence with MS-quantification (Supporting Information Figure 4A). We evaluated the attributes of detected proteins against MitoCarta,25 and they followed the distribution of annotated mitochondrial proteins (Supporting Information Figure 4B). The detected proteins have the tendency to describe their functional clusters substantially, covering 44 out of 48 GO Slim terms annotated to the mitochondrial “Biological Process” (Supporting Information Figure 4C). Pathway analysis revealed that these perturbed clusters include mitochondrial dysfunction, fatty acid metabolism, and oxidative stress (Figure 3C, Supporting Information Figure 5 and Table S4A), concordant with biochemical end points supporting the association of mitochondrial oxidative stress in troglitazone hepatotoxicity.11
Protein networks can reflect interdependencies between proteins and potentially identify functionally important groups of proteins and/or important proteins (hubs).38 To better understand the emergent modular properties of the mitoproteome with troglitazone exposure, we applied a network-cleaned clique-enrichment approach22,39 on our proteomics data set and derived an extended mouse specific mitochondrial network of 798 nodes, and 1145 edges from the single largest connected component and 1206 nodes, 2103 edges (Supporting Information Figure 6A). The well-defined topological interconnectivity of the 14 and 28 days PPINs suggests a transitory response from an early adaptor phase to a stressed phase with prolonged troglitazone administration. At 14 days, there were few major changes in the mitoproteome, of which we observed slight perturbation in lipid metabolism, specific to the troglitazone’s intended pharmacological action to normalize glucose and lipid levels. This is also consistent with little hepatic damage observed at this point. In contrast, at 28 days, as a consequence of prolonged troglitazone treatment, there were widespread mitoproteome changes, including cell death, fatty acid metabolism perturbation, and responses to oxidative-stress clusters (Supporting Information Figure 6B and Table S4B and C).
Troglitazone induces the expression of PPAR-responsive lipid metabolism genes,40 and this motivated us to investigate any inadvertent “off-target” troglitazone effects on fatty acid metabolism. To test this, a position-weight matrix was applied to predict PPRE sequences among the differentially regulated proteins. Within expectations, at 14 days, proteins with PPRE motifs (Supporting Information Table 5) were up-regulated relative to PPRE-absent proteins (Wilcoxon P < 0.01). In contrast, at 28 days, the protein expressions of both groups were similar (Wilcoxon P = 0.196; Supporting Information Figure 6C), suggesting a lesser degree of troglitazone-induced regulation on PPRE protein expressions and also off-target PPAR-dependent toxicity. These data suggest that perturbations in lipid metabolism partially contribute to the TILI, together with other perturbed clusters. However, we do not preclude the possibility that certain determinants of toxicity may interact and overlap with pharmacologically mediated changes.
Troglitazone Causes ROS-Induced Mitochondrial Stress and Cytotoxicity
We hypothesized that prolonged troglitazone exposure leads to accumulated oxidative stress that results in a transition from redox equilibrium to disequilibrium breaches. Therefore, we assessed the ramifications of troglitazone administration on the mitochondrial redox proteins. We found SOD2 up-regulation (R28days:14days ≈ 1.29) despite intrinsic heterozygosity of Sod2 (Figure 4A), concomitant with the up-regulation of PRDX3, GLRX5, and GPX1, proteins that were involved in maintaining redox homeostasis (R28days:14days ≈ 1.44, 1.21, and 1.23, respectively). To further investigate the impact of ROS-induced mitochondrial stress caused by troglitazone, we examined ACO2, a mitochondrial matrix [Fe–S] tricarboxylic acid cycle enzyme that displays exquisite sensitivity to ROS. ACO2 activity decreased by 30% in 14 days and >55% with 28 days of troglitazone treatment, while ACO2 levels remain basal (R14days ≈ 0.98 and R28days ≈ 1.08; Supporting Information Figure 7A). By immunoblotting, ∼40 kDa ACO2 fragments suggest ROS-induced cleavage of ACO2 with 14 days of troglitazone administration, consistent with proteolytic degradation by LONP1 under moderate stress.41 Continued troglitazone administration caused the aggregation of ACO2 (Supporting Information Figure 7B), which is reflective of increased, chronic O2•– in the Sod2+/– mouse liver mitochondria.20 Other indices of oxidative damage, such as mitochondrial nitrotyrosine adducts, NO levels (1-way ANOVA, P < 0.05, P < 0.001, respectively; Supporting Information Figure 7C and D), and protein carbonyls,15,18 increased significantly after 28 days of troglitazone administration, with a concomitant mGSH depletion (1-way ANOVA, P < 0.05; Figure 4B) and increase of cytochrome c release into the cytosol (1-way ANOVA, P < 0.01; Supporting Information Figure 7F), a key initial step in the apoptosis process.
Figure 4.

Troglitazone induces oxidative stress through impaired mitochondrial GSH transport. (A) MS/MS spectrum of a SOD2 tryptic peptide. iTRAQ channels 116 and 114 constitute 14 days of troglitazone administration, channels 115 and 113 constitute 14 days of control treatment, while channels 121 and 118 constitute 28 days of troglitazone administration, and channels 119 and 117 constitute 28 days of control treatment. The proteomics experiment was performed in technical replicates. (B) Measurements of mitochondrial GSH levels of Sod2+/– mice after 14 and 28 days troglitazone administration. Mean ± SD values are shown. n = 3–6 per group; 1-way ANOVA with Bonferroni post-test; VEH, vehicle-administered; TRG, troglitazone-administered. #P < 0.05 for 28 days treated compared with 14 days vehicle; %P < 0.05 for 28 days treated compared with 14 days treated; *P < 0.05 and ***P < 0.001 for 28 days treated compared with 28 days vehicle. (C) Immunoblot analysis after DIC knockdown in HepG2 cells. β-actin served as normalization control. (D) HepG2 cells were transfected with a pool of siRNAs, and cell viability was measured by MTT assay. Nontargeting siRNA acted as negative control, and GAPD siRNA acted as positive control. Dose response assays were normalized to 0.1% DMSO, and troglitazone-treated siRNA assays were normalized to mock. The graph is a representation of three independent experiments. Mean ± SD values are shown. ###P < 0.001 relative to DMEM + 0.1% DMSO; ***P < 0.0001 relative to 100 μM troglitazone (2 h) after normalizing to 0.1% DMSO; %%%P < 0.0001 relative to DIC siRNA. BM, butylmalonate; Ctrl, nontargeting siRNA; Mock, transfection reagent; GSH-EE, GSH-ethyl ester; TRG, troglitazone.
Depletion of mGSH may be due to the down-regulation of dicarboxylate carrier (DIC; Slc25a10; R28days:14days ≈ 0.71), one of the two mGSH import proteins.31,42 Because of the importance in maintaining mGSH homeostatic levels through active transport, we investigated the functional relevance of troglitazone-induced DIC down-regulation via genetic and pharmacological assays. Attenuation of Slc25a10 (Figure 4B) by RNAi in HepG2 cells treated with 100 μM troglitazone accelerated troglitazone-induced cytotoxicity from 5 to 2 h (Figure 4C; 1-way ANOVA, P < 0.0001 relative to 2 h troglitazone treatment). Treatment with GAPD-siRNA or nontargeting siRNA in the presence of troglitazone confirmed the “toxic-enhancement” effect of DIC knockdown. Similarly, incubation with butylmalonate, a DIC inhibitor, in the presence of troglitazone resulted in increased cytotoxicity (1-way ANOVA, P < 0.0001 relative to 2 h troglitazone incubation). When cells were pretreated with GSH-ethyl ester, troglitazone-induced cytotoxicity was blunted (1-way ANOVA, P < 0.001). Therefore, DIC is necessary for the maintenance of mGSH transport and attenuates the cytotoxic effects of troglitazone. Taken together, these data suggest that chronic troglitazone exposure in Sod2+/– mouse liver leads to decreased mGSH import and a buildup of ROS, potentially leading to intolerable mitochondrial damage and triggering the abrupt progression of hepatocytic death.
Prolonged Troglitazone Treatment Activates FOXO3a in an Akt-Independent Manner, Possibly by Antagonistic Action of ASK1-JNK
ROS are potent stressors to JNK and p38 activation, and their involvement in troglitazone-induced cytotoxicity has been noted previously.43,44 We observed ASK1 activation, the upstream activator of JNK (phosphorylation of Thr845 and dephosphoryation of inhibitory residue Ser83) and JNK1/2 activation but not p38 after 28 days of troglitazone administration (Figure 5A). By contrast, JNK1/2 were minimally phosphorylated in control and 14 days treated groups (Figure 5A). The phosphorylated form of p38 was minimal in all study groups. Therefore, accumulated mitochondrial oxidative stress enhanced with troglitazone led to ASK1-JNK activation in vivo but not p38 in the Sod2+/– mice.
Figure 5.

ASK1-JNK mediates troglitazone-potentiated mitochondrial oxidative stress through FOXO3a. (A) Liver homogenates were subjected to immunoblotting with pASK1Thr845, pASK1Ser83, ASK1, pJNK, JNK1/2, pp38, and p38 antibodies. Elevated oxidative stress from 4 weeks of troglitazone administration induced ASK1 and JNK1/2 activation. The right panel shows mean normalized density values relative to the corresponding nonphosphorylated form. (B) Immunoblot analysis of transcriptional regulators and coactivators involved in regulating nuclear-encoded mitochondrial proteins in Sod2+/+ and Sod2+/– mice. (C) Liver homogenates were subjected to immunoblotting with FOXO3a, pFOXO3aSer253, PGC-1α, PGC-1β, ERRα, NRF-1, and PPARγ antibodies. (D) Liver sections were subjected to immunohistochemistry by incubating with pFOXO3aSer253 antibody and counterstained. Extended administration of Sod2+/– mice with troglitazone induced the nuclear translocation of FOXO3a in areas of hepatocytic degeneration but not the vehicle (solutol). Original magnification ×100. The inset illustrates the cytosolic localization of pFOXO3a (original magnification ×400). (E) Proposed schematic diagram showing that extended troglitazone administration coupled to increased mitochondrial oxidative stress in Sod2+/– mice primed ASK1-JNK-FOXO3a activation, which in turn resulted in mitoproteome reconfiguration and dysregulated lipid metabolism, enhancing mitochondrial ROS regulation in a feedback loop. TRG, troglitazone.
JNK has been implicated in the activation of FOXO3a under sustained oxidative stress in antagonism to Akt.45 Notably, in vertebrates and C. elegans, FOXO3a acts as a transcriptional factor in the up-regulation of SOD2 as a form of oxidative-stress defense.46,47 As shown in Figure 5B, endogenous FOXO3a was inherently activated in the Sod2+/– mouse liver (indicated by moderate dephosphorylation at Ser253), possibly in response to elevated ROS. In Sod2+/+ mouse, however, FOXO3a remained inactivated (phosphorylated). Administering Sod2+/– mice with troglitazone for 28 days resulted in markedly reduced p-FOXO3aSer253 and its retention in the cytosol (Figure 5C and D). Using immunohistochemistry, we showed that, in areas of confluent hepatocytic degeneration, activation of FOXO3a occurred in a dephosphorylation-dependent manner. In contrast, in the neighboring areas denoted by surviving hepatocytes, FOXO3a remained phosphorylated at Ser253 and was retained in the cytoplasm (Figure 5D).
Besides FOXO3a, several crucial transcriptional factors or activators of nuclear-encoded gene regulatory programs48 were evaluated in their contributions in governing the observed differential expression of the Sod2+/– mitochondrial proteins in the absence and presence of troglitazone. Given that in certain cell types PGC-1α drives the gene expression of Sod2 under pro-oxidant status49,50 and that troglitazone restores PGC-1α levels,51 we investigated if troglitazone-treated Sod2+/– mitoproteome changes were PGC-1α and PGC-1β-dependent. Consistent with the lack of increase of LRPPRC levels (R28days:14days ≈ 0.96), which up-regulates PGC-1α and PGC-1β,52 PGC-1α and PGC-1β remained at similar levels throughout the study and were independent of the form of treatment (Figure 5C). Hepatocyte PPARγ expression has been reported to increase with troglitazone,40,53 but our immunoblot analysis revealed no significant differences, consistent with another pharmacoproteomic study.54 In addition, no significant difference was observed with duration or drug-treatment for two other well-established transcriptional regulators of nuclear-encoded mitochondrial proteins, NRF-1 and ERR-α (Figure 5C). Taken together, our results demonstrate that a compromised intrinsic mitochondrial antioxidant defense combined with prolonged troglitazone exposure leads to the specific activation of ASK1-JNK and FOXO3a, driving a reconfiguration of the liver mitoproteome. Despite compensatory measures such as the upregulation of important antioxidant mitochondrial proteins, oxidative damage due to the lack of sustained mGSH import can no longer be managed and hepatic injury ensues (Figure 5E).
Discussion
In this study we described an integrative toxicoproteomics approach on a mouse model of compromised mitochondrial antioxidant defense for the study of the idiosyncratic hepatotoxicant, troglitazone. By combining high throughput MS-based mitoproteome-wide profiling, biochemical end points, and network biology, we demonstrated that the Sod2+/– hepatic mitoproteome followed a two-phase response to repeated troglitazone administration that cumulated in liver injuries by the fourth week. This integrative approach identified the combined deterioration of key fragile nodes including attenuated DIC levels and a dysfunctional mGSH transport system that lead to the eventual toxicity of troglitazone. Inhibition of DIC through pharmacological or genetic means sensitized cells to troglitazone-induced cytotoxicity and was reversed when pretreated with a GSH donor.
The mGSH node is intrinsically perturbed by a compromised mitochondrial ROS defense in the Sod2+/– mouse, and this fragile node conferred a point of sensitization for troglitazone to expose its toxic effect. Data documented by us and others suggest that DIC may be a key protein in exemplifying TILI: (i) its temporal inverse differential expression parallels the two-phase change observed at tissue level; (ii) DIC knockdown in C. elegans increases ROS production, which could result in further oxidative damage;55 and (iii) the unaffected levels of OGC, together with DIC, account for GSH transport into liver mitochondria.56 Association studies on troglitazone-prescribed Type II Diabetes Mellitus in Japanese patients with elevated serum liver transaminases revealed gene-polymorphisms with combined Gstt1–/– and Gstm1–/– genotypes,57 and such a correlation suggested that these acquired genetic factors at least partially predisposed certain groups of patients to TILI. Importantly, this is consistent with our results and implicated clinical associations of silent genetic abnormalities in ROS detoxification and mGSH transport with TILI. It would be interesting to test for the potential convergence in mGSH cluster perturbation of other DILI toxicants implicated with mitochondrial dysfunction. However, to more accurately reflect mGSH status, determination of coenzyme A and coenzyme A–glutathione mixed disulfide may be employed because measurements of mGSH may not entirely account for intramitochondrial thiol/disulfide exchanges with mGSH.58
Activation of p38 and JNK has been previously implicated in troglitazone-induced cytotoxicity.43,44 Here we demonstrated in vivo JNK activation in mediating ROS-induced cell death and that mGSH depletion could trigger a signaling crosstalk between ROS-mediated ASK1-JNK and FOXO3a, plausibly through JNK-dependent phosphorylation of 14-3-3.45,59 This led to the activation of transcriptional programs to reconfigure the mitoproteome, including increased SOD2 expression to attenuate troglitazone-induced oxidative stress. In addition, the recurring themes of lipid metabolism suggest that continual troglitazone exposure might perturb lipid metabolism as collateral damage. This is crucial because free fatty acids can enhance mitochondrial-generated ROS by inhibiting oxidative phosphorylation and by interfering with electron flow,60 further exacerbating oxidative damage.
Application of proteomic studies will help to elucidate the complexity behind idiosyncratic-DILI as a probable multifactorial disease and perhaps offer better therapeutic design. Deeper coverage by MS-based proteomics strengthens such efforts by unbiasedly capturing the complexities and dynamics of proteome changes at a global scale. It has been proposed that differences in the proportion of abnormal mitochondria and Sod2+/+ mitochondria in individual cells (heteroplasmy) have different consequences on the viability of a cell or the ability to function optimally, also known as the threshold effect.61 Undoubtedly, single cell proteomics can be applied to assess if heteroplasmic cells harboring more mitochondrial mutations have a higher propensity to initiate drug-induced toxic cascades.62 At a time when drug-induced toxicities are a growing concern, platforms have been initiated63 to modernize the drug development process and drug safety evaluation, including to improve our understanding of the mechanisms underlying DILI and for its better prediction. Broadly, this present study may represent a powerful step forward in using a systems approach in a mouse model of underlying genetic abnormality to advance our understanding of the risk factors in other idiosyncratic toxic drugs and tests of hepatotoxic signals.
Acknowledgments
We would like to thank Teck Kwang Lim for his LC-MS/MS technical expertise. This work was supported by grants from the NUS Environmental Toxicology Programme and Biomedical Research Council Singapore.
Glossary
Abbreviations
- [Fe–S]
iron–sulfur
- two-dimensional liquid chromatography
- 2D-LC
two-dimensional liquid chromatography
- DILI
drug-induced liver injury
- HCL
hierarchical clustering
- iTRAQ
isobaric tag for relative and absolute quantitation
- mGSH
mitochondrial glutathione
- MRM
multiple reaction monitoring
- NO
nitric oxide
- PEP
proteomics expansion pipeline
- PPRE
PPAR response element
- PPIN
protein–protein interaction network
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- TILI
troglitazone-induced liver injury
Supporting Information Available
Summary of MRM transitions of 25 mitochondrial proteins; list of differentially expressed hepatic mitochondrial proteins detected by MS with the partial loss of Sod2; summary of mitochondrial proteins and their fold change detected by MS with troglitazone administration; summary of functionally enriched terms based on Mitonetwork and IPA; list of murine genes matching identified proteins with potential PPRE sites; iTRAQ analysis of mouse hepatic mitoproteome; validation of MS detected mitochondrial proteins; Sod2+/– versus wild type mouse hepatic mitoproteome and biochemical end points; proteomics of troglitazone treated Sod2+/– mitochondria; functional and toxicological analysis of troglitazone-treated Sod2+/– mitoproteome; topology dynamics of Sod2+/– hepatic mitoproteome with troglitazone exposure; and Sod2 genotype and ACO2 activity and damage with troglitazone. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
○ Y.H.L.: Biosystems and Micromechanics, and Infectious Diseases Interdisciplinary Research Groups, Singapore MIT Alliance for Research and Technology, Singapore 138602.
Author Present Address
□ M.R.: Singapore Lipidomics Incubator, Center for Life Sciences, National University of Singapore, 28 Medical Drive, Level #03-03A, Singapore 117456.
Author Contributions
UAB, MCMC and YHL conceived and designed experiments; YHL, QL and TKL performed LC-MS/MS assays. WG and LW performed bioinformatic analyses. CKN and MR performed MRM assays. LYH analyzed the data. YHL wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat. Rev. Drug Discovery 2005, 46489–99. [DOI] [PubMed] [Google Scholar]
- Dykens J. A.; Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discovery Today 2007, 1217–18777–85. [DOI] [PubMed] [Google Scholar]
- Opar A. Overtaking the DILI Model-T. Nat. Rev. Drug Discovery 2012, 118585–6. [DOI] [PubMed] [Google Scholar]
- Dixit R.; Boelsterli U. A. Healthy animals and animal models of human disease(s) in safety assessment of human pharmaceuticals, including therapeutic antibodies. Drug Discovery Today 2007, 127–8336–42. [DOI] [PubMed] [Google Scholar]
- Graham D. J.; Green L.; Senior J. R.; Nourjah P. Troglitazone-induced liver failure: A case study. Am. J. Med. 2002, 114, 299–306. [DOI] [PubMed] [Google Scholar]
- Matsunuma N.; Kimura K.; Yamashita K.; Miyakoshi N.; Manabe S. Chronic toxicity study on CS-045 in rats treated orally for 1 year. J. Clin. Ther. Med. 1993, 9Suppl 3299–315. [Google Scholar]
- Mayfield R.; McLean T.; Crook D.; Buist D. P.; Gopinath C. CS-045 toxicity to cynomolgus monkeys by repeated oral administration for 52 weeks. J. Clin. Ther. Med. 1993, 9Suppl 3317–41. [Google Scholar]
- Rothwell C.; Bleavins M.; McGuire E.; Iglesia F. d. l.; Masuda H. 52-Week oral toxicity study of troglitazone in cynomolgus monkeys [abstract]. Toxicologist 1997, 36, 273. [Google Scholar]
- Ulrich R. G. Idiosyncratic toxicity: a convergence of risk factors. Annu. Rev. Med. 2007, 58, 17–34. [DOI] [PubMed] [Google Scholar]
- Chojkier M. Troglitazone and liver injury: in search of answers. Hepatology 2005, 412237–46. [DOI] [PubMed] [Google Scholar]
- Smith M. T. Mechanisms of troglitazone hepatotoxicity. Chem. Res. Toxicol. 2003, 166679–87. [DOI] [PubMed] [Google Scholar]
- Hsiao C. J.; Younis H.; Boelsterli U. A. Trovafloxacin, a fluoroquinolone antibiotic with hepatotoxic potential, causes mitochondrial peroxynitrite stress in a mouse model of underlying mitochondrial dysfunction. Chem. Biol. Interact. 2010, 1881204–13. [DOI] [PubMed] [Google Scholar]
- Kashimshetty R.; Desai V. G.; Kale V. M.; Lee T.; Moland C. L.; Branham W. S.; New L. S.; Chan E. C.; Younis H.; Boelsterli U. A. Underlying mitochondrial dysfunction triggers flutamide-induced oxidative liver injury in a mouse model of idiosyncratic drug toxicity. Toxicol. Appl. Pharmacol. 2009, 2382150–9. [DOI] [PubMed] [Google Scholar]
- Leitner H.; Goldstein M.; Min E.; Gauthier S.; Day B. Mitochondrial antioxidant status and nucleoside reverse transcriptase inhibitor (NTRI) toxicity. Free Radical Biol. Med. 2005, 39, S153–S154. [Google Scholar]
- Ong M. M.; Latchoumycandane C.; Boelsterli U. A. Troglitazone-induced hepatic necrosis in an animal model of silent genetic mitochondrial abnormalities. Toxicol. Sci. 2007, 971205–13. [DOI] [PubMed] [Google Scholar]
- Ong M. M. K.; Wang A. S.; Leow K. Y.; Khoo Y. M.; Boelsterli U. A. Nimesulide-induced hepatic mitochondrial injury in heterozygous Sod2± mice. Free Radical Biol. Med. 2006, 40, 420–429. [DOI] [PubMed] [Google Scholar]
- Ramachandran A.; Lebofsky M.; Weinman S. A.; Jaeschke H. The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 2011, 2513226–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. H.; Chung M. C.; Lin Q.; Boelsterli U. A. Troglitazone-induced hepatic mitochondrial proteome expression dynamics in heterozygous Sod2(±) mice: two-stage oxidative injury. Toxicol. Appl. Pharmacol. 2008, 231143–51. [DOI] [PubMed] [Google Scholar]
- Mann M.; Kelleher N. L. Precision proteomics: the case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. U. S. A. 2008, 1054718132–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bota D. A.; Davies K. J. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 2002, 49674–80. [DOI] [PubMed] [Google Scholar]
- Tirmenstein M. A.; Hu C. X.; Gales T. L.; Maleeff B. E.; Narayanan P. K.; Kurali E.; Hart T. K.; Thomas H. C.; Schwartz L. W. Effects of troglitazone on HepG2 viability and mitochondrial function. Toxicol. Sci. 2002, 691131–8. [DOI] [PubMed] [Google Scholar]
- Goh W. W.; Lee Y. H.; Zubaidah R. M.; Jin J.; Dong D.; Lin Q.; Chung M. C.; Wong L. Network-Based Pipeline for Analyzing MS Data: An Application toward Liver Cancer. J. Proteome Res. 2011, 10, 2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Cai H.; Xu J.; Ying S.; Zhang Y. A mouse protein interactome through combined literature mining with multiple sources of interaction evidence. Amino Acids 2010, 3841237–52. [DOI] [PubMed] [Google Scholar]
- Xia K.; Dong D.; Han J. D. IntNetDB v1.0: an integrated protein-protein interaction network database generated by a probabilistic model. BMC Bioinform. 2006, 7, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini D. J.; Calvo S. E.; Chang B.; Sheth S. A.; Vafai S. B.; Ong S. E.; Walford G. A.; Sugiana C.; Boneh A.; Chen W. K.; Hill D. E.; Vidal M.; Evans J. G.; Thorburn D. R.; Carr S. A.; Mootha V. K. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 1341112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle E. I.; Weng S.; Gollub J.; Jin H.; Botstein D.; Cherry J. M.; Sherlock G. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20183710–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine C.; Dubois G.; Duguay Y.; Helledie T.; Vu-Dac N.; Gervois P.; Soncin F.; Mandrup S.; Fruchart J. C.; Fruchart-Najib J.; Staels B. The orphan nuclear receptor Rev-Erbalpha is a peroxisome proliferator-activated receptor (PPAR) gamma target gene and promotes PPARgamma-induced adipocyte differentiation. J. Biol. Chem. 2003, 2783937672–80. [DOI] [PubMed] [Google Scholar]
- Kokoszka J. E.; Coskun P.; Esposito L.; Wallace D. C. Increased mitochondrial oxidative stress in the Sod2± mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp N. A.; Huber W.; Sadowski P. G.; Charles P. D.; Hester S. V.; Lilley K. S. Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell Proteomics 2010, 991885–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri A.; Muller F. L.; Liu Y.; Ng R.; Faulkner J.; Hamilton M.; Richardson A.; Huang T. T.; Epstein C. J.; Van Remmen H. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech. Ageing Dev. 2006, 1273298–306. [DOI] [PubMed] [Google Scholar]
- Fernandez-Checa J. C.; Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005, 2043263–73. [DOI] [PubMed] [Google Scholar]
- Lee Y. H.; Boelsterli U. A.; Lin Q.; Chung M. C. Proteomics profiling of hepatic mitochondria in heterozygous Sod2(±) mice, an animal model of discreet mitochondrial oxidative stress. Proteomics 2008, 83555–568. [DOI] [PubMed] [Google Scholar]
- Lill R.; Hoffmann B.; Molik S.; Pierik A. J.; Rietzschel N.; Stehling O.; Uzarska M. A.; Webert H.; Wilbrecht C.; Muhlenhoff U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 2012, 182391491–508. [DOI] [PubMed] [Google Scholar]
- Beinert H.; Holm R. H.; Munck E. Iron-sulfur clusters: nature’s modular, multipurpose structures. Science 1997, 2775326653–9. [DOI] [PubMed] [Google Scholar]
- Williams M. D.; Van Remmen H.; Conrad C. C.; Huang T. T.; Epstein C. J.; Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J. Biol. Chem. 1998, 2734328510–5. [DOI] [PubMed] [Google Scholar]
- Amores-Sanchez M. I.; Medina M. A. Glutamine, as a precursor of glutathione, and oxidative stress. Mol. Genet. Metab. 1999, 672100–5. [DOI] [PubMed] [Google Scholar]
- Lee Y. H.; Lin Q.; Boelsterli U. A.; Chung M. C. The Sod2 mutant mouse as a model for oxidative stress: a functional proteomics perspective. Mass Spectrom. Rev. 2010, 292179–96. [DOI] [PubMed] [Google Scholar]
- Park J.; Lee D. S.; Christakis N. A.; Barabasi A. L. The impact of cellular networks on disease comorbidity. Mol. Syst. Biol. 2009, 5, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Zimmerman L. J.; Park B. H.; Tabb D. L.; Liebler D. C.; Zhang B. Network-assisted protein identification and data interpretation in shotgun proteomics. Mol. Syst. Biol. 2009, 5, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memon R. A.; Tecott L. H.; Nonogaki K.; Beigneux A.; Moser A. H.; Grunfeld C.; Feingold K. R. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 2000, 141114021–31. [DOI] [PubMed] [Google Scholar]
- Bulteau A. L.; Lundberg K. C.; Ikeda-Saito M.; Isaya G.; Szweda L. I. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc. Natl. Acad. Sci. U. S. A. 2005, 102175987–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash L. H. Mitochondrial glutathione transport: physiological, pathological and toxicological implications. Chem. Biol. Interact. 2006, 1631–254–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae M. A.; Song B. J. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Mol. Pharmacol. 2003, 632401–8. [DOI] [PubMed] [Google Scholar]
- Lim P. L.; Liu J.; Go M. L.; Boelsterli U. A. The mitochondrial Superoxide/Thioredoxin-2/Ask1 signaling pathway is critically Involved in troglitazone-induced cell injury to human hepatocytes. Toxicol. Sci. 2008, 1012341–9. [DOI] [PubMed] [Google Scholar]
- Sunayama J.; Tsuruta F.; Masuyama N.; Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J. Cell Biol. 2005, 1702295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops G. J.; Dansen T. B.; Polderman P. E.; Saarloos I.; Wirtz K. W.; Coffer P. J.; Huang T. T.; Bos J. L.; Medema R. H.; Burgering B. M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 4196904316–21. [DOI] [PubMed] [Google Scholar]
- Honda Y.; Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999, 13111385–93. [PubMed] [Google Scholar]
- Scarpulla R. C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 882611–38. [DOI] [PubMed] [Google Scholar]
- St-Pierre J.; Drori S.; Uldry M.; Silvaggi J. M.; Rhee J.; Jager S.; Handschin C.; Zheng K.; Lin J.; Yang W.; Simon D. K.; Bachoo R.; Spiegelman B. M. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 1272397–408. [DOI] [PubMed] [Google Scholar]
- Valle I.; Alvarez-Barrientos A.; Arza E.; Lamas S.; Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 663562–73. [DOI] [PubMed] [Google Scholar]
- Jove M.; Salla J.; Planavila A.; Cabrero A.; Michalik L.; Wahli W.; Laguna J. C.; Vazquez-Carrera M. Impaired expression of NADH dehydrogenase subunit 1 and PPARgamma coactivator-1 in skeletal muscle of ZDF rats: restoration by troglitazone. J. Lipid Res. 2004, 451113–23. [DOI] [PubMed] [Google Scholar]
- Cooper M. P.; Qu L.; Rohas L. M.; Lin J.; Yang W.; Erdjument-Bromage H.; Tempst P.; Spiegelman B. M. Defects in energy homeostasis in Leigh syndrome French Canadian variant through PGC-1alpha/LRP130 complex. Genes Dev. 2006, 20212996–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G. F.; Khandelwal R. L.; Roesler W. J. Troglitazone induces expression of PPARgamma in liver. Mol. Cell Biol. Res. Commun. 1999, 23202–8. [DOI] [PubMed] [Google Scholar]
- Lanne B.; Dahllof B.; Lindahl C.; Ebefors K.; Kanmert I.; von Bahr H.; Miliotis T.; Nystrom A. C.; Arnerup G.; Paulsons I.; Kerb S.; Oakes N. PPARα and PPARγ regulation of liver and adipose proteins in obese and dyslipidemic rodents. J. Proteome Res. 2006, 581850–9. [DOI] [PubMed] [Google Scholar]
- Lee T. H.; Mun J. Y.; Han S. M.; Yoon G.; Han S. S.; Koo H. S. DIC-1 over-expression enhances respiratory activity in Caenorhabditis elegans by promoting mitochondrial cristae formation. Genes Cells 2009, 143319–27. [DOI] [PubMed] [Google Scholar]
- Zhong Q.; Putt D. A.; Xu F.; Lash L. H. Hepatic mitochondrial transport of glutathione: studies in isolated rat liver mitochondria and H4IIE rat hepatoma cells. Arch. Biochem. Biophys. 2008, 4741119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe I.; Tomita A.; Shimizu M.; Sugawara M.; Yasumo H.; Koishi R.; Takahashi T.; Miyoshi K.; Nakamura K.; Izumi T.; Matsushita Y.; Furukawa H.; Haruyama H.; Koga T. A study to survey susceptible genetic factors responsible for troglitazone-associated hepatotoxicity in Japanese patients with type 2 diabetes mellitus. Clin. Pharmacol. Ther. 2003, 735435–55. [DOI] [PubMed] [Google Scholar]
- O’Donovan D. J.; Rogers L. K.; Kelley D. K.; Welty S. E.; Ramsay P. L.; Smith C. V. CoASH and CoASSG levels in lungs of hyperoxic rats as potential biomarkers of intramitochondrial oxidant stresses. Pediatr. Res. 2002, 513346–53. [DOI] [PubMed] [Google Scholar]
- Sunters A.; Madureira P. A.; Pomeranz K. M.; Aubert M.; Brosens J. J.; Cook S. J.; Burgering B. M.; Coombes R. C.; Lam E. W. Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res. 2006, 661212–20. [DOI] [PubMed] [Google Scholar]
- Schonfeld P.; Wojtczak L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radical Biol. Med. 2008, 453231–41. [DOI] [PubMed] [Google Scholar]
- Wallace D. C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutstein H. B.; Morris J. S.; Annangudi S. P.; Sweedler J. V. Microproteomics: analysis of protein diversity in small samples. Mass Spectrom. Rev. 2008, 274316–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key FDA Critical Path Activities Under Way in 2007; U.S. Department of Health and Human Services, U.S. Food and Drug Administration, Silver Spring, MD, 2008.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.