

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen (LANA) is a 1,162-amino-acid protein that mediates the maintenance of episomal viral genomes in latently infected cells. The two central components of episome persistence are DNA replication with each cell division and the segregation of DNA to progeny nuclei. LANA self-associates to bind KSHV terminal-repeat (TR) DNA and to mediate its replication. LANA also simultaneously binds to TR DNA and mitotic chromosomes to mediate the segregation of episomes to daughter nuclei. The N-terminal region of LANA binds histones H2A and H2B to attach to mitotic chromosomes, while the C-terminal region binds TR DNA and also associates with chromosomes. Both the N- and C-terminal regions of LANA are essential for episome persistence. We recently showed that deletion of all internal LANA sequences results in highly deficient episome maintenance. Here we assess independent internal LANA regions for effects on episome persistence. We generated a panel of LANA mutants that included deletions in the large internal repeat region and in the unique internal sequence. All mutants contained the essential N- and C-terminal regions, and as expected, all maintained the ability to associate with mitotic chromosomes in a wild-type fashion and to bind TR DNA, as assessed by electrophoretic mobility shift assays (EMSA). Deletion of the internal regions did not reduce the half-life of LANA. Notably, deletions within either the repeat elements or the unique sequence resulted in deficiencies in DNA replication. However, only the unique internal sequence exerted effects on the ability of LANA to retain green fluorescent protein (GFP) expression from TR-containing episomes deficient in DNA replication, consistent with a role in episome segregation; this region did not independently associate with mitotic chromosomes. All mutants were deficient in episome persistence, and the deficiencies ranged from minor to severe. Mutants deficient in DNA replication that contained deletions within the unique internal sequence had the most-severe deficits. These data suggest that internal LANA regions exert critical roles in LANA-mediated DNA replication, segregation, and episome persistence, likely through interactions with key host cell factors.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV-8), is a gamma-2-herpesvirus. KSHV is tightly associated with Kaposi's sarcoma, primary effusion lymphoma (PEL), and multicentric Castleman's disease (1–4). In tumor cells, KSHV infection is predominantly latent, and only a small subset of viral genes are expressed. Cells latently infected with KSHV have multiple copies of the viral genome maintained as extrachromosomal, covalently closed circular (ccc) DNA (episomes) (1, 5). Latency-associated nuclear antigen (LANA) is necessary and sufficient for episome persistence in the absence of other viral genes (6, 7).
LANA is an 1,162-amino-acid protein that contains a proline-rich region, a central acidic repeat region, and a putative leucine zipper (see Fig. 1). For episomes to persist, DNA must replicate with each cell cycle and must then segregate to daughter nuclei during mitosis. LANA fulfills both of these functions. Both the N- and C-terminal regions of LANA are necessary for episome persistence. The C-terminal region binds directly to a specific sequence in the KSHV terminal-repeat (TR) elements, and this binding is required for LANA-mediated replication and episome persistence (6, 8–15). The KSHV genome contains approximately 40 TR copies (16, 17), and each TR has two adjacent LANA binding sites (11, 12, 18). LANA also segregates KSHV DNA to daughter nuclei by tethering episomes to mitotic chromosomes during mitosis. The association of LANA with chromosomes (7, 19–21) is necessary for episome persistence (22). LANA has two independent chromosome binding regions, located in its N- and C-terminal regions (19, 20, 22–26) (see Fig. 1). The N-terminal region is the dominant chromosome association region and binds to mitotic chromosomes by interacting directly with histones H2A and H2B. This interaction is important for DNA replication and is essential for episome maintenance (12, 22, 27).
Fig 1.
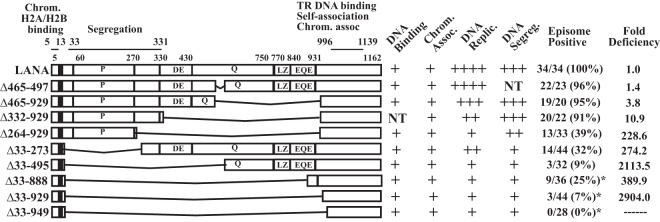
Schematic diagram of KSHV LANA and LANA deletion mutants. Indicated are the proline-rich region (P), the aspartate and glutamate (DE), glutamine (Q), and glutamate and glutamine (EQE) regions, and the putative leucine zipper (LZ). The DE, Q, EQE, and LZ regions all contain repeat elements. The shaded area represents region I of the N-terminal nuclear localization signal (NLS) within amino acids 24 to 30 (20, 69). The C-terminal portion of LANA can also localize to nuclei, but an NLS has not been precisely mapped. Amino acids 5 to 13 mediate chromosome association through interaction with histones H2A/H2B. Amino acids 996 to 1139 contain TR DNA binding, self-association, and chromosome association functions. The capabilities for TR DNA binding, mitotic chromosome association, DNA replication, DNA segregation (as suggested by retention of p2TR-ΔRE-GFP), and episome persistence for each of the constructs are shown on the right. For episome persistence, fractions indicate the number of G418-resistant cell lines with episomes divided by the total number of G418-resistant cell lines assayed by Gardella analysis; percentages are given in parentheses. Fold deficiencies in episome maintenance ability were determined by dividing the value for each mutant in Table 1 by that for WT LANA. NT, not tested. LANA deletion mutants marked with asterisks, and their abilities to bind TR DNA, associate with chromosomes, and maintain p8TR episomes, have been described previously (28), but their TR DNA replication and segregation capabilities were further investigated here.
Although the N- and C-terminal regions of LANA are essential for episome persistence, we showed recently that they are not sufficient for efficient episome maintenance. Fusion of the N-and C-terminal regions of LANA resulted in mutants that retained the ability to associate with chromosomes and could also bind and replicate TR DNA; however, these LANA deletion mutants were highly deficient in episome maintenance (28). This work now investigates the importance of individual internal regions of LANA for episome persistence. DNA replication was reduced by multiple different deletions spanning the internal sequence. Retention of green fluorescent protein (GFP) expression from replication-deficient TR-containing episomes localized to an N-terminal internal region, consistent with a role for this sequence in episome segregation. Mutants in which both DNA replication and the unique N-terminal region were disrupted had the most-severe episome persistence defects.
MATERIALS AND METHODS
Cell lines.
BJAB and Loukes cells were maintained in RPMI medium containing 10% bovine growth serum (BGS) (HyClone) or 10% FetalPlex (Gemini) and 15 μg/ml gentamicin. Cells stably expressing LANA were grown in a medium containing hygromycin B at 200 U/ml (Calbiochem). KSHV-infected BCBL-1 cells were maintained in RPMI medium containing 20% BGS or 20% FetalPlex and 15 μg/ml gentamicin.
Plasmids.
All T7 epitope-tagged LANA constructs have the first 3 LANA amino acids deleted and were cloned into the pSG5 oligonucleotide (28). pT7 and pT7LANA have been described previously (28). To generate the pSG5 oligonucleotide T7LANAΔ33-273, pT7LANA was digested with BamHI, and the large LANA BamHI fragment was cloned into the pSG5 oligonucleotide, which was then termed pSG5 oligonucleotide LANA 273. To generate the N-terminal portion for pSG5 oligonucleotide T7LANAΔ33-273, T7LANA 4-32 was amplified using primers NotI T7 tag-F (ATAAGAATGCGGCCGCCCACCATGGCATCGATGACAGGTGGC) and LANA273N-R (TTGATATCCTCTTTCCGGAGACCTGTTTCG). Primer LANA273N-R adds an extra nucleotide to correct the open reading frame (ORF) and an EcoRV site at the 5′ end. After amplification, T7LANA 4-32 was digested and was ligated into the NotI/EcoRV sites from pSG5 oligonucleotide LANA 273, resulting in pSG5 oligonucleotide T7LANAΔ33-273, which has amino acids 33 to 273 deleted, encodes amino acids glycine, tyrosine, and glutamine, and includes an EcoRV site in place of the deleted sequence. To generate pSG5 oligonucleotide T7LANAΔ465-929, the SmaI/PstI fragment from pSG5 oligonucleotide T7LANA (28) (LANA 4-463) was cloned into pBluescript KS(−) (pBS), and the resulting construct was termed pBS LANA 4-464 (amino acid 464 is reconstituted after cloning into pBS). The C-terminal LANA portion of T7LANAΔ465-929 was amplified with primers LANA930Pst-F (TCCACTGCAGCAGGAGACGGTGGAAGAGCC) and LANA1162EcoXbaHind-R (TTAAGCTTTCTAGAGATATCTTATGTCATTTCCTGTGGAGAGTCCC). LANA930Pst-F adds a PstI site at the 5′ end, and LANA1162EcoXbaHind-R adds EcoRV, XbaI, and HindIII sites at the 3′ end, of the amplification product. After amplification, the C-terminal region of LANA was cloned into the PstI/HindIII site of pBS LANA 4-464, and the resulting construct was termed pBS LANA Δ465-929. The SmaI/HindIII fragment from pBS LANA Δ465-929 was then cloned into the pT7 vector, and the resulting construct was termed pT7 LANA Δ465-929. pT7 LANA Δ465-929 was digested with KpnI and EcoRV, and the LANA fragment was used to replace the KpnI/EcoRV LANA fragment from pSG5 oligonucleotide T7LANAΔ33-929 (28) to generate pSG5 oligonucleotide T7LANAΔ465-929, which encodes amino acids Leu and Gln and has a PstI restriction site in place of amino acids 465 to 929. To generate pSG5 oligonucleotide T7LANAΔ33-495, the PstI/BamHI fragment from pT7LANA was cloned into pBS, and the resulting construct was termed pBS LANA 496-1162. The N-terminal region of LANA was amplified from pT7LANA using primers NotI T7 tag-F and LANA495N-R (5′ TTGATATCTCTTTCCGGAGACCTGTTTCG 3′). LANA495N-R adds an EcoRV site at the 3′ end of the amplified PCR fragment. The amplified fragment was then cloned into the XhoI/EcoRV site of pBS LANA 496-1162, and the resulting construct was termed pBS LANA Δ33-495. pBS LANA Δ33-495 was then digested with HindIII and NruI, and the LANA fragment was used to replace the HindIII/NruI fragment from a pSG5 oligonucleotide T7LANA N-C fusion mutant, described previously (28), to generate pSG5 oligonucleotide T7LANAΔ33-495, which encodes Asp, Ile, Glu, and Phe and includes EcoRV and EcoRI sites in place of amino acids 33 to 495. To generate pSG5 oligonucleotide T7LANAΔ332-929, the N-terminal portion (T7LANA 4-331) was amplified from pSG5 oligonucleotide T7LANAΔ465-929 using primers NotI T7 tag-F and LANA331Pst-R (5′ GGGGCTGCAGATCATCCTTATTGTCATTGTC 3′). LANA331Pst-R adds a PstI site to the 3′ end of the amplified product. The PCR product was then digested with PstI (a second PstI site is located upstream of the LANA ORF and 3′ to the T7 epitope) and was used to replace the PstI fragment from pSG5 oligonucleotide T7LANAΔ465-929 to generate pSG5 oligonucleotide T7LANAΔ332-929, which encodes amino acids Leu and Gln and has a PstI restriction site in place of amino acids 332 to 929. pSG5 oligonucleotide T7LANAΔ264-929 was generated by PCR. T7LANA 4-263 was amplified from pT7LANA using primers NotI T7 tag-F and LANA263Xho-R (5′ CTCGAGTGCTGCGGGAGATGTAGGCGGTTGGCGTGGCGG 3′). LANA 930-1162 was then amplified with primers LANA1162 EcoRV-R (5′ TGATATCTTATGTCATTTCCTGTGGAGAGTCCC 3′) and LANA930Xho-F (5′ GCAGCACTCGAGCAGGAGACGGTGGAAGAGCC 3′). The products of the N- and C-terminal LANA amplifications were then combined and amplified with primers NotI T7 tag-F and LANA1162 EcoRV-R. The final PCR product, T7LANAΔ264-929, was then cloned into the NotI/EcoRV sites of the pSG5 oligonucleotide to generate pSG5 oligonucleotide T7LANAΔ264-929, which encodes amino acids Leu and Glu and has an XhoI site in place of amino acids 264 to 929. pSG5 oligonucleotide T7LANA and pSG5 oligonucleotide T7LANAΔ465-497 have been described previously (28). pEGFP LANA 1-32 has been described previously (22). To generate pEGFP LANA 1-331, LANA 1-331 was amplified by PCR from pEGFP LANA (22) using forward primer GFP LANA 1-331wt Fwd (CCGGAATTCCGCAATGGCGCCCCCGGGAATGCGCCTGAGGTCGGGA) and reverse primer GFP LANA 331stopKpn Rev (CGGGGTACCCTATTAATCATCCTTATTGTCATTGTC). To generate pEGFP LANA 1-331 GMR, LANA 1-331 GMR was amplified from pEGFP LANA GMR (22) using primer GFP LANA 1-331 GMR Fwd (CCGGAATTCCGCAATGGCGCCCCCGGCGGCCGCGCTGAGG) and primer GFP LANA 331stopKpn Rev. LANA 33-331 was amplified from pEGFP LANA using primer GFP LANA 33-331 Fwd (CCGGAATTCCGCATGTGACCTTGGCGATGACCTACATCTA) and primer GFP LANA 331stopKpn Rev. After amplification, PCR fragments were digested with EcoRI and KpnI and were ligated into the EcoRI/KpnI sites of pEGFP-NLS (29). All constructs were confirmed by sequencing. p8TR contains eight copies of the KSHV terminal-repeat unit (TR) cloned into pRep9 (Invitrogen), which was first modified by deleting the sequence between ClaI and KpnI (7, 22). p8TR-gB is a modified version of p8TR that contains a small target sequence cloned into the HindIII site and is used to measure p8TR replication by real-time PCR (30). p2TR-GFP and p2TR-ΔRE-GFP were kindly provided by Rolf Renne (31). Each contains two copies of the TR and also a GFP expression cassette. p2TR-ΔRE-GFP lacks the 32-bp replication element (RE), which is adjacent to the two LANA binding sites present in each TR (32); this RE is required for LANA-mediated replication but not for the binding of LANA to the TR. Therefore, the plasmid can be used to investigate the ability of LANA to segregate TR-containing DNA in the absence of replication.
Generation of BJAB cells stably expressing LANA proteins.
Ten million BJAB cells were transfected in 400 μl of RPMI medium at 200 V and 960 μF in a 0.4-cm-gap cuvette with a Bio-Rad electroporator (7). pSG5 oligonucleotide plasmids (70 μg) encoding T7LANA or the T7LANA deletion mutants were cotransfected with a plasmid carrying the hygromycin resistance gene downstream of a simian virus 40 promoter (10 μg) into BJAB cells. After 48 h, cells were seeded at 1,000/well into 96-well plates and were selected for hygromycin B resistance (200 U hygromycin B/ml; Calbiochem). Clones resistant to hygromycin B were screened for LANA expression using Western blotting and immunofluorescence.
Fluorescence microscopy.
For metaphase spreads of cells stably expressing LANA or LANA mutants, 0.5 × 106 cells/ml were incubated overnight in 1 μg/ml of colcemid (Calbiochem). Colcemid-treated cells were swollen in hypotonic buffer (1% sodium citrate, 10 mM CaCl2, 10 mM MgCl2) for 5 min, spread onto slides by a Cytospin system (Thermo Scientific Shandon), and fixed for 10 min in 4% paraformaldehyde (Polysciences) in phosphate-buffered saline. To detect LANA or the LANA mutants, cell spreads were incubated with an anti-T7 tag monoclonal antibody (1:1,000; Novagen), which detects a single epitope in T7 LANA, or an anti-LANA monoclonal antibody (1:500; Advanced Biotechnologies), which detects a multicopy epitope located in the LANA repeat region. For the secondary antibody, an Alexa Fluor 488-conjugated anti-mouse antibody or an Alexa Fluor 488-conjugated anti-rat antibody (1:2,000; Molecular Probes) was used. Cells were counterstained with propidium iodide (1 μg/ml; Molecular Probes), and Aqua-Poly/Mount (Polysciences) was applied to coverslips. Microscopy was performed with a Zeiss Axioskop microscope, PCM 2000 hardware, and C-Imaging software (Compix, Inc.).
For GFP fusion proteins, 5 × 106 BJAB cells were transfected with 5 μg of pEGFP-NLS, pEGFP LANA 1-32, pEGFP-NLS LANA 1-331, pEGFP-NLS LANA 1-331 GMR, or pEGFP-NLS LANA 33-331. Twenty-four hours posttransfection, 2 × 106 cells were seeded in 6-well plates at 0.5 × 106/ml and were treated with colcemid at 1.5 μg/ml for 8 h. Cells were then swollen in hypotonic buffer, spread onto slides, fixed, and counterstained with propidium iodide as described above.
Half-life determination.
Due to intrinsic cycloheximide resistance of these BJAB cells, Loukes cells were used to assess the half-life (t1/2) of LANA. Ten million Loukes cells were transfected with 7 μg of the vector and LANA or LANA deletion mutants, using Amaxa Nucleofector program X-005, in 150 μl solution V. Cells were seeded in 6 ml of medium in T25 flasks and were kept in log phase. Two days after transfection, 1 × 106 cells were seeded in 6-well plates at 0.5 × 106/ml and were treated with 100 μg/ml cycloheximide (Calbiochem). Cells were collected at serial time points up to 15 h, because longer incubation resulted in cell death. As a control, BCBL-1 cells were similarly incubated with cycloheximide. LANA was detected with an affinity-purified antibody against the C-terminal region of LANA. As a control for effective arrest of protein synthesis by cycloheximide, c-Myc expression was detected with antibody 9E10 (Santa Cruz Biotechnology). Blots were stripped, and α-tubulin was detected as a loading control.
DNA replication assay.
To assess LANA-mediated DNA replication, cells were transfected as described previously (30). Briefly, 10 million cells were transfected with 5 μg of p8TR-gb by using Amaxa Nucleofactor program O-17 and solution V. After transfection, cells were seeded in 6-well plates in 5 ml of medium. Twenty-four hours after transfection, cells were transferred to 25-cm2 flasks, and ∼ 5 × 106 cells were harvested for the isolation of low-molecular-weight DNA using the Hirt method (33). The remaining cells were kept in log-phase growth by resuspending cells daily at 0.4 million/ml. Seventy-two hours after transfection, low-molecular-weight DNA was again harvested from cells. For Hirt DNA extraction, cells were lysed in 1 ml lysis buffer (0.6% SDS, 10 mM EDTA, 10 mM Tris-HCl [pH 7.5], 50 μg/ml RNase A) per 5 million cells and were incubated at 37°C for 2 h. NaCl was then added to a final concentration of 1 M NaCl, and cells were incubated overnight at 4°C. After a 30-min centrifugation at 11,000 × g and 4°C, DNA was extracted once with phenol-chloroform (1:1) and twice with chloroform–isoamyl alcohol (24:1) and was ethanol precipitated, and the DNA pellet was washed with 70% ethanol, air dried, and resuspended in buffer (10 mM Tris [pH 8], 0.1 mM EDTA).
For real-time PCR, primers gB-F (5′ TCAAGACCACCTCCTCCATGA 3′) and gB-R (5′ CGGCCCAACATATCGTTGAC 3′) and a fluorescent probe (5′ 6-carboxyfluorescein [FAM]-TCGCCCGGCTGATCGCAGTT-6-carboxytetramethylrhodamine [TAMRA] 3′) were used as described previously (30). For the PCR, 300 nM primers gB-F and gB-R, 250 nM fluorescent probe, and TaqMan Universal master mix (Applied Biosystems) were combined in a final volume of 25 μl. PCRs were performed using an ABI 7300 real-time PCR system (Applied Biosystems) (1 cycle at 50°C for 2 min; 1 cycle at 95°C for 10 min; 40 cycles of 95°C for 30 s and 60°C for 1 min). To detect total p8TR-gB, 2 μg of Hirt DNA was digested with 30 U of EcoRV (New England Biolabs [NEB]) in a 50-μl reaction mixture overnight at 37°C, followed by heat inactivation of EcoRV at 80°C for 30 min. PCR amplification was performed in triplicate using 3.75 μl of the EcoRV-digested product as the template. Amplified DNA was quantified using a standard curve from a p8TR-gB plasmid dilution series. To detect replicated p8TR-gB, 6 μg of Hirt DNA was digested with 60 U DpnI in NEB buffer 2 in a 50-μl reaction mixture overnight at 37°C, followed by exonuclease III (ExoIII) (NEB) treatment (120 U) for 30 min at 37°C to reduce the background due to incompletely DpnI digested DNA (30, 34). After heat inactivation of ExoIII at 70°C for 30 min, the volumes of the digested products were increased to 60 μl by using NEB buffer 2, and DNA was digested with 80 U of EcoRV at 37°C for at least 6 h. EcoRV was then heat inactivated at 80°C for 30 min. PCR amplification was performed in triplicate with 4.5 μl of each digested product as the template.
To normalize the replication of each test sample, the total amount of p8TR-gB present at 24 h posttransfection was divided by the amount detected in BJAB control cells and was termed the ratio for normalization. The amount of replicated DNA detected in the sample at 3 days posttransfection was divided by the corresponding ratio for normalization to obtain the normalized replication of the particular test sample. To calculate the fold replication of each sample compared to that of BJAB control cells, the normalized replication of the sample was divided by the normalized replication of BJAB control cells.
EMSA.
For electrophoretic mobility shift assays (EMSA), LANA and LANA mutants were translated in vitro using a TNT Quick Coupled reticulocyte lysate system (Promega). Similar amounts of in vitro-translated proteins, as assessed by Western blotting, were incubated in DNA binding buffer [20 mM Tris (pH 7.5), 10% glycerol, 50 mM KCl, 0.1 mM dithiothreitol, 10 mM MgCl2, 1 mM EDTA, 20 μg/ml of poly(dI-dC)] with 50,000 cpm of 32P-labeled oligonucleotide TR-13 for 30 min at room temperature. TR-13 contains a 20-bp LANA binding sequence (6). Bound complexes were resolved on a 5% nondenaturing polyacrylamide gel, and signals were detected by autoradiography.
Selection of G418-resistant cells and Gardella gel analysis.
BJAB cells alone and BJAB cells stably expressing T7LANA or T7LANA deletion mutants were cotransfected with 4 μg of GFP and 30 μg of p8TR using a Bio-Rad electroporator as described above. GFP transfection efficiencies were assessed at 24 h posttransfection by determining the percentage of fluorescent cells by microscopy or by fluorescence-activated cell sorting (FACS) and were similar for different transfections. Seventy-two hours posttransfection, LANA or LANA mutant protein expression was assayed by Western blotting, and cells were seeded in 96-well plates at 1,000/well in a medium containing G418 (600 μg/ml) (Gibco or Gemini). Alternatively, for limiting-dilution assays, cells were seeded in 96-well plates at 1,000/well, 100/well, 10/well, or 1/well in a medium containing G418. GraphPad Prism was used to perform nonlinear regression analyses with LANA limiting-dilution outgrowth values to compare episome maintenance efficiencies. Gardella analyses were performed on G418-resistant clones by in situ lysis of cells in gel-loading wells with protease (Sigma) and sodium dodecyl sulfate (35), followed by electrophoresis in Tris-borate-EDTA. DNA was transferred to a nylon membrane, and KSHV DNA was detected using a 32P-labeled TR probe. Signals were detected by autoradiography.
GFP retention assay.
Ten million BJAB cells alone or LANA-expressing BJAB cells in log-phase growth were transfected with 5 × 1010 copies (0.08 pmol) of plasmid p2TR-GFP (407 ng DNA) or p2TR-ΔRE-GFP (467 ng DNA), using Amaxa Nucleofactor program O-17, in 150 μl of solution V. After transfection, cells were seeded in 5 ml medium in a six-well plate. Eighteen to 20 h posttransfection, cells were sorted for GFP-positive cells (BD FACSAria sorter) and were suspended in RPMI medium containing hygromycin B and Normocin (50 μg/ml; Invivogen) at a concentration of 150,000 cells/ml (day zero). The cell concentration was measured daily by FACS for the first 5 days after sorting, and the percentages of GFP-positive cells were monitored daily and were plotted over periods of 14 days for p2TR-GFP and 7 days for p2TR-ΔRE-GFP. After reaching a concentration of ∼1 × 106/ml, cells were kept in log-phase growth by reducing the concentration to ∼0.2 × 105/ml. Cell concentrations were measured by FACS using CountBright Absolute Counting Beads (Invitrogen).
Although all LANA mutant cell lines grew similarly, some small differences in growth occurred after sorting. To compare the efficiencies of plasmid segregation of the different LANA mutants while accounting for differences in rates of cell growth, the percentages of GFP-positive cells were compared at a mutant-specific, plasmid-specific time point (denoted t*sp, where s is the mutant [strain] and p is the plasmid) at which the cell concentration reached e1.5 (4.48) times the concentration measured at day 1 after cell seeding. The factor e1.5 was used because this was well within the approximately linear part of the growth pattern for all transfected cells. To estimate the date posttransfection at which cells reached e1.5 times the concentration at day 1, growth curve models were fitted to replicate data for each of the transfections. Repeated observations of GFP retention were made on replicates for each mutant, and quadratic logistic regression was used to model the relationship between the proportion of cells expressing GFP and the time that had elapsed from the initial transfection for each replicate transfection. For each mutant-plasmid combination, the estimated proportion of cells expressing GFP at t*sp for transfected cells was obtained using replicate-specific logistic regression parameter estimates. The distributions of these growth-corrected GFP expression rates are summarized in boxplots. Nonparametric tests of the null hypothesis that different LANA mutants have common medians were performed for 8 families of comparisons. For cell lines transfected with p2TR-GFP, group A, composed of LANA, LANAΔ465-929, and LANAΔ332-929, was compared to group B, composed of LANAΔ33-273, LANAΔ33-495, LANAΔ33-888, LANAΔ33-929, and LANAΔ33-949; group A was also compared to LANAΔ264-929. In addition, LANAΔ264-929 was compared to group B and, individually, to LANAΔ33-495. The same sets of comparisons were made for cell lines transfected with p2TR-ΔRE-GFP. Holm's procedure (36) for adjusting inferences for multiple comparisons was used to calculate P values for the comparisons described above (see Fig. 6). All computations for the GFP retention analyses were performed using R, version 2.15.2.
Fig 6.

The unique internal LANA sequence is important for the retention of GFP expression from episomal TR-containing DNA. (A) Ten million BJAB cells alone or BJAB cells stably expressing LANA or the different LANA deletion mutants were transfected with 5 × 1010 copies (5,000 copies per cell) of p2TR-GFP. Eighteen to 20 h posttransfection, cells were sorted for GFP expression and were seeded at similar densities. GFP expression was monitored daily by FACS for 14 days. To account for some small differences in cell growth after sorting, GFP expression was compared at a time point when cells had reached a concentration e1.5 times that at day 1 of seeding. P values for comparisons between the indicated groups are shown. (B) Cell lines were assessed as for panel A but were initially transfected with p2TR-ΔRE-GFP, which is identical to p2TR-GFP except for deletion of the RE, abolishing LANA-mediated DNA replication. GFP expression was monitored for 7 days. The results in panels A and B are each from six experiments. (C) Western blotting of BJAB cells alone or BJAB cells expressing LANA or LANA mutants at the time of DNA segregation. (Left) (Top) Anti-T7 epitope blot; (bottom) anti-tubulin blot. (Right) Anti-LANA blot. Brightness and contrast were uniformly adjusted within each panel with Adobe Photoshop. Approximately 3.5 × 105 cells were loaded per lane for the anti-T7 and anti-tubulin blots, and ∼1.0 × 105 cells were loaded per lane for the anti-LANA blot. (D) GFP NLS, GFP LANA 1-32, GFP LANA 1-331, GFP LANA 1-331 GMR, and GFP LANA 33-331 were each expressed in BJAB cells. Cells were arrested in metaphase with colcemid. GFP is green, and chromosomes were counterstained with propidium iodide (red). The overlay of green and red generates yellow. Brightness and contrast in individual panels were uniformly adjusted using Adobe Photoshop. Magnification, ×630.
RESULTS
LANA internal-deletion mutants associate with mitotic chromosomes.
To persist stably in proliferating cells, KSHV episomes must replicate with each cell division and must segregate efficiently to daughter cell nuclei. The N- and C-terminal portions of LANA are essential for these processes. In addition, internal LANA sequences are critical for function, since deletion of the internal sequences by fusion of the N- and C-terminal portions of LANA results in highly deficient episome maintenance (28). To investigate the roles of distinct LANA internal regions in episome persistence, we generated a panel of LANA deletion mutants termed LANAΔ465-497, LANAΔ33-495, LANAΔ33-273, LANAΔ465-929, LANAΔ332-929, and LANAΔ264-929 (Fig. 1).
Although all mutants contain the N- and C-terminal chromosome association regions, we wanted to confirm that each associates with mitotic chromosomes, since this function is essential for DNA tethering and episome persistence. BJAB cells alone or BJAB cells stably expressing LANA, LANAΔ465-497, LANAΔ33-495, LANAΔ33-273, LANAΔ465-929, LANAΔ332-929, or LANAΔ264-929 (Fig. 1) were arrested in metaphase by overnight treatment with colcemid, and the subcellular localization of LANA was analyzed by confocal microscopy. LANA (green) was detected using an antibody against LANA or against the T7 tag; mitotic chromosomes were stained with propidium iodide (red). As expected, no LANA staining was detected in BJAB cells (Fig. 2a and f). In agreement with previous results, LANA associated with mitotic chromosomes (yellow, resulting from the colocalization of green and red), with concentrations near telomeres and centromeres (Fig. 2b and g; arrows indicate peritelomeric staining, and arrowheads indicate pericentromeric staining) (28, 37). LANAΔ465-497 (Fig. 2c and h), LANAΔ33-273 (Fig. 2d and i), LANAΔ33-495 (Fig. 2e and j), LANAΔ465-929 (Fig. 2k), LANAΔ332-929 (Fig. 2l), and LANAΔ264-929 (Fig. 2m) associated with mitotic chromosomes and also displayed preferential localization near the centromeres and telomeres, like LANA. Since the anti-LANA antibody detects the central repeat elements that are deleted in LANAΔ465-929, LANAΔ332-929, and LANAΔ264-929, these mutants were detected only with an antibody to the N-terminal T7 epitope tag. These data demonstrate that deletion of the internal domains does not affect the ability of LANA to interact with mitotic chromosomes.
Fig 2.
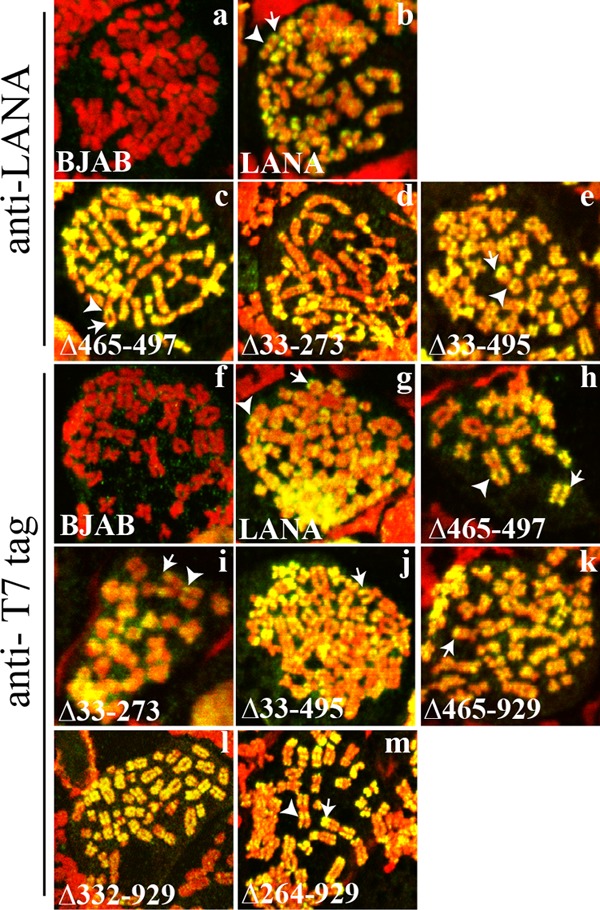
LANA and LANA deletion mutants associate with mitotic chromosomes, with preferential localization to areas near centromeres and telomeres. BJAB cells alone and BJAB cells stably expressing different LANA mutants were arrested in metaphase with colcemid and were analyzed for LANA localization by confocal microscopy. LANA (green) was detected with an antibody against LANA or against the T7 epitope tag. Chromosomes were counterstained with propidium iodide (red). The overlay of green and red results in yellow. White arrowheads and arrows indicate pericentromeric and peritelomeric localization of LANA, respectively. Brightness and contrast were similarly adjusted for the panels, and individual panels were uniformly adjusted using Adobe Photoshop. Magnification, ×630.
LANA internal-deletion mutants bind TR DNA.
Since LANA DNA binding is mediated through the C-terminal portion of LANA, which is present in all mutants, we expected all LANA deletion proteins to bind TR DNA. However, since LANA TR DNA binding is essential for episome persistence (14), we wanted to confirm DNA binding, and we assessed this by electrophoretic mobility shift assays (EMSA). LANA or each deletion mutant was translated in vitro and was incubated with a radiolabeled probe containing the LANA TR binding site. Incubation of rabbit reticulocyte lysate (RRL) with TR DNA in the absence of LANA did not shift the TR probe (Fig. 3, lanes 1 and 7). In agreement with previous results (6, 23, 28, 38), incubation of wild-type (WT) LANA with the TR probe resulted in two major complexes (Fig. 3, lanes 2 and 8, top two arrows). LANAΔ465-497 (Fig. 3, lane 3) and LANAΔ33-273 (Fig. 3, lane 4) also generated two complexes. LANAΔ33-495 (Fig. 3, lane 5) bound TR DNA and formed several detectable complexes. LANAΔ465-929 bound TR DNA, but as a single complex (Fig. 3, lane 6). Previous experiments had indicated that LANA amino acids 900 to 929 are responsible for generating multiple EMSA complexes (28). LANAΔ264-929 (Fig. 3, lane 9) also bound TR DNA in one predominant complex. Therefore, all LANA deletion mutants retained the ability to bind TR DNA.
Fig 3.
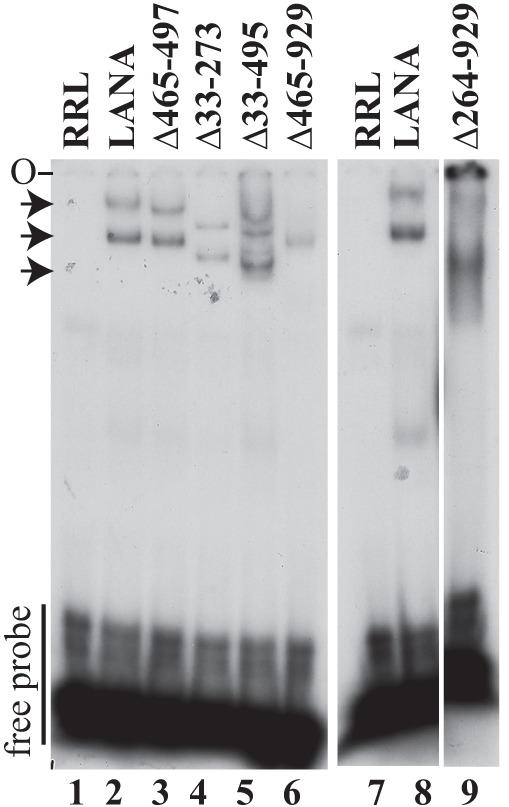
Deletion of internal regions of LANA does not abolish TR DNA binding as assessed by EMSA. A radiolabeled TR DNA probe was incubated with rabbit reticulocyte lysate (RRL) (lanes 1 and 7) or similar amounts (as detected by Western blotting) of in vitro-translated LANA (lanes 2 and 8), LANAΔ465-497 (lane 3), LANAΔ33-273 (lane 4), LANAΔ33-495 (lane 5), LANAΔ465-929 (lane 6), or LANAΔ264-929 (lane 9). After incubation, complexes were resolved on a nondenaturing polyacrylamide gel. Brightness and contrast were uniformly adjusted with Adobe Photoshop. All panels are from the same gel. Arrows indicate LANA-TR or mutated LANA-TR complexes. O, gel origin.
Deletion of the internal repeat elements does not reduce the half-life of LANA.
We investigated whether deletion of internal regions reduces the half-life of LANA, since previous work suggested that the repeat elements enhance the stability of LANA (39). We assessed the half-life of LANA in KSHV-infected BCBL-1 cells or after LANA expression in uninfected Loukes cells. Treatment with cycloheximide and assessment by immunoblotting at serial time points demonstrated a t1/2 of ∼9 to 12 h in both BCBL-1 and Loukes cells (Fig. 4A). LANAΔ33-273, LANAΔ33-495, LANAΔ465-929, LANAΔ332-929, LANAΔ264-929, LANAΔ33-888, LANAΔ33-929, and LANAΔ33-949 each had a half-life similar to that of LANA (Fig. 4A). As a control for effective arrest of protein synthesis by cycloheximide, we also detected c-Myc, which had a t1/2 of ∼1 to 3 h in the presence of LANA expression and a t1/2 of ∼0.5 to 1 h in the absence of LANA (Fig. 4B). LANA is known to enhance the stability of c-Myc (40, 41). Therefore, deletion of the internal LANA regions, including the repeat elements, did not reduce the half-life of LANA.
Fig 4.

Deletion of internal LANA regions does not reduce the half-life of LANA. Cells were treated with cycloheximide to arrest protein synthesis, and LANA levels were assessed by immunoblotting. (A) Results for BCBL-1 cells (∼1.5 × 105 cells loaded per lane) and for Louckes cells (∼3 × 105 cells loaded per lane) transfected with LANA, LANAΔ33-273, LANAΔ33-495, LANAΔ465-929, LANAΔ332-929, LANAΔ264-929, LANAΔ33-888, LANAΔ33-929, or LANAΔ33-949 are shown at time zero and at serial time points after cycloheximide arrest. Untransfected cells (not transf.) show a comigrating cross-reactive band with LANAΔ264-929. LANA was detected with affinity-purified antibody directed against the C-terminal region of LANA. Tubulin immunoblots are shown below each panel. (B) c-Myc was detected in the same cells as those used for panel A at time zero and at serial time points in order to assess the evidence for effective cycloheximide arrest. Results for untransfected cells are also shown. Tubulin blots are shown below each panel.
The LANA deletion mutants are deficient in TR DNA replication.
LANA-mediated DNA replication is essential for KSHV episome persistence. The C-terminal portion of LANA binds directly to KSHV TR DNA to mediate replication (11–15, 42), and the N-terminal chromosome binding region is also critical for replication (12, 15, 42). In addition, we showed previously that deletion of all internal LANA sequences reduced the efficiency of DNA replication (28). Therefore, we wanted to assess the contributions of specific internal LANA regions to DNA replication.
To assay for TR DNA replication, BJAB cells alone and BJAB cells stably expressing LANA, LANAΔ465-497, LANAΔ33-273, LANAΔ33-495, LANAΔ465-929, LANAΔ332-929, LANAΔ264-929, LANAΔ33-929, LANAΔ33-949, or LANAΔ33-888 (Fig. 1) were transfected with p8TR-gB. LANAΔ33-929, LANAΔ33-949, and LANAΔ33-888 are LANA deletion mutants lacking all internal sequences that were previously shown to have reduced TR DNA replication by Southern blot assays for DpnI-resistant DNA (28) and were included for comparison. The transfected plasmid p8TR-gB is Dam-methylated DNA, purified from Dam methylase-positive bacteria, and contains 8 copies of the KSHV TR elements. Dam methylation renders the DNA susceptible to DpnI digestion. Since mammalian cells lack Dam methylase, DNA that undergoes replication after transfection is resistant to DpnI digestion.
Figure 5 shows the normalized DNA replication (Fig. 5A) and fold replication (Fig. 5B) of LANA and LANA mutants over that for BJAB control cells. LANA efficiently mediated DNA replication at 27.06-fold over the control level, and LANAΔ465-497, which contains only a 33-amino-acid deletion within the glutamine-rich repeat region, also efficiently replicated DNA at 44.20-fold over the control level. In agreement with previous results using Southern blot detection of DpnI-resistant DNA (28) that showed reductions in replication, deletion of all internal sequences in LANAΔ33-888, LANAΔ33-929, and LANAΔ33-949 resulted in dramatically reduced DNA replication, at only 3.94-, 5.04-, and 1.52-fold over the control level, ranging from 5.6% to 18.6% of the WT level.
Fig 5.
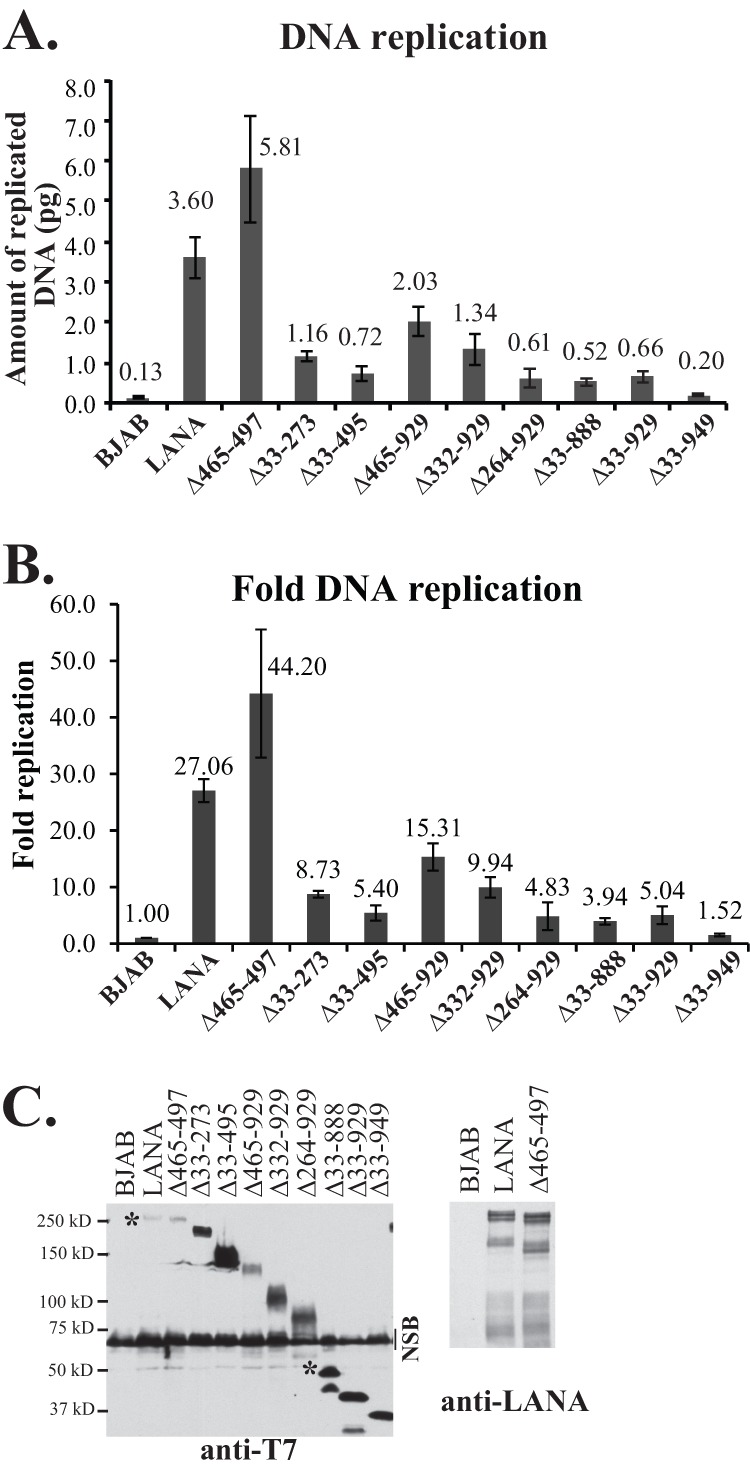
Deletion of internal LANA regions results in decreased DNA replication. (A and B) BJAB cells alone or BJAB cells stably expressing LANA or different LANA deletion mutants were transfected with p8TR-gB, which contains 8 copies of the TR. Hirt DNA was isolated at 24 h posttransfection for the assessment of transfection efficiencies and again at 72 h posttransfection for the assessment of replicated DNA. The amounts of p8TR-gB present in the Hirt DNA samples were quantified by real-time PCR. To normalize for transfection efficiencies, the amount of p8TR-gB detected in the samples at 24 h posttransfection was divided by the amount detected in BJAB control cells, and that result was termed the ratio of normalization. These values were close to 1. To normalize the amount of replicated p8TR-gB, the amount of replicated p8TR DNA detected at 72 h was divided by the corresponding ratio of normalization for each mutant. To calculate the fold p8TR-gB replication relative to the negative control (BJAB cells), the normalized replication of the samples was divided by the normalized replication detected for the BJAB control cells. The results represent averages for 4 experiments. (C) Western blotting of BJAB cells alone or BJAB cells expressing LANA or LANA mutants at the time of DNA replication. (Left) Anti-T7 epitope blot. Approximately 3.5 × 105 cells were loaded per lane. NSB, nonspecific band. Asterisks indicate WT LANA and full-length LANAΔ33-888 bands. (Right) Anti-LANA blot of LANA and LANAΔ465-497. Approximately 1.5 × 105 cells were loaded per lane. Brightness and contrast within each panel were uniformly adjusted with Adobe Photoshop. Deletions of the central repeat regions result in increased LANA expression (28, 39).
Deletion of smaller internal regions resulted in different levels of reduction in DNA replication. LANAΔ465-929, in which most of the glutamine-rich internal repeat regions and all of the predicted leucine zipper are deleted, had 15.31-fold replication over that of the control, a level modestly reduced at 56.7% of WT replication. LANAΔ332-929, which lacks all internal repeat elements, had 9.94-fold replication over the control level, equivalent to 36.7% of the WT level. LANAΔ264-929, which lacks only 68 additional amino acids relative to LANAΔ332-929, replicated DNA at only 4.83-fold over the control level, or 17.8% of the WT level, suggesting that these additional amino acids are important for replication. Deletion of unique sequence, including the proline-rich region, in LANAΔ33-273 resulted in 8.73-fold replication, or 32.3% of WT LANA replication. Notably, LANAΔ33-495, which lacks the unique internal sequence (LANA amino acids 33 to 331) and some of the repeat elements, showed substantially reduced replication at 5.40-fold over the control level, or 20.0% of the WT level. The reductions in replication were not due to reduced protein expression, since all mutants were expressed at levels at least equivalent to that of WT LANA (Fig. 5B). Therefore, deletions within either the unique internal sequence of LANA or the repeat regions resulted in reduced DNA replication.
The unique internal sequence of LANA is critical for the retention of GFP expression from replication-deficient TR episomes.
We wanted to assess the episome segregation of the LANA mutants, since segregation of KSHV DNA to daughter nuclei following mitosis is essential for episome persistence; otherwise, DNA is degraded. The N- and C-terminal portions of LANA, which mediate chromosome attachment and KSHV DNA binding to tether episomes to mitotic chromosomes, are necessary for this process. However, the role of the internal LANA sequence is unknown. Each KSHV TR contains two adjacent LANA binding sites and an adjacent 32-bp replication element that is required for LANA-mediated replication (32). We used GFP expression from TR-containing DNA to monitor for the presence of episomes. To investigate the abilities of the LANA deletion mutants to retain GFP expression from TR DNA, BJAB cells alone or stably expressing LANA, LANAΔ465-929, LANAΔ332-929, LANAΔ264-929, LANAΔ33-273, LANAΔ33-495, LANAΔ33-888, LANAΔ33-929, or LANAΔ33-949 were transfected with equimolar amounts of p2TR-GFP or p2TR-ΔRE-GFP (kindly provided by Rolf Renne) (31). p2TR-GFP contains two TR elements and a GFP expression cassette. p2TR-ΔRE-GFP is identical to p2TR-GFP except that it lacks the replication element (RE). Since p2TR-ΔRE-GFP retains LANA binding sites, this plasmid can be used to assess the ability of LANA to segregate TR episomes in the absence of replication. Eighteen to 20 h after transfection with p2TR-GFP or p2TR-ΔRE-GFP, cells were sorted to collect GFP-positive cells and were seeded at 1.5 × 105 cells/ml. GFP expression and cell concentrations were assessed daily.
Figure 6A is a boxplot showing the percentages of GFP-positive cells after transfection with p2TR-GFP, which is capable of both LANA-mediated DNA replication and segregation. In the absence of selection, TR DNA is expected to be lost from proliferating cells, and LANA reduces the rate of this loss (30, 31, 43). To account for slightly different growth rates after the sorting process, the percentages of GFP-positive cells were compared after a constant amount of cell proliferation (e1.5, or ∼4.48-fold) for each cell line, which generally occurred ∼ 3 to 4 days postsorting and was well within the approximate linear growth range for all cell lines. In the absence of LANA, GFP expression was rapidly lost from nearly all BJAB cells (Fig. 6A). In contrast, >80% of LANA-expressing cells (median value) retained GFP. The abilities of LANAΔ33-273, LANAΔ33-495, LANAΔ33-888, LANAΔ33-929, and LANAΔ33-949 to maintain p2TR-GFP were all substantially reduced from that of LANA; for each of these mutants, fewer than 20% of cells (median value) were GFP positive. Notably, LANAΔ465-929 maintained p2TR-GFP at nearly WT levels. For both LANAΔ332-929 and LANAΔ264-929, the ability to maintain p2TR-GFP was reduced to intermediate levels, with about 45% and 30% of cells expressing GFP. Holm's procedure was used to adjust the P values used to measure the statistical significance of tests comparing the groups; the adjusted P values may be individually compared to a significance threshold of 5%. Comparison of group A, composed of LANA, LANAΔ465-929, and LANAΔ332-929, with group B, which included LANAΔ33-273, LANAΔ33-495, LANAΔ33-888, LANAΔ33-929, and LANAΔ33-949, yielded a P value of 2.68 × 10−16; comparison of group A to LANAΔ264-929 yielded a P value of 1.57 × 10−5. P values for the comparison of LANAΔ264-929 to group B and individually to LANAΔ33-495 were 2.24 × 10−7 and 1.17 × 10−3, respectively (Fig. 6A).
In contrast to p2TR-GFP, transfection with p2TR-ΔRE-GFP assesses the ability of LANA to retain TR DNA in the absence of replication, since loss of the RE abolishes the ability of LANA to replicate DNA. Therefore, GFP retention is expected to directly reflect LANA-mediated segregation. As expected, nearly all BJAB cells, which lack LANA, rapidly lost GFP expression (Fig. 6B). In contrast, about 50% of cells expressing LANA expressed GFP. Notably, both LANAΔ465-929 and LANAΔ332-929 retained p2TR-ΔRE-GFP at wild-type levels. In contrast, as with p2TR-GFP, LANAΔ33-273, LANAΔ33-495, LANAΔ33-888, LANAΔ33-929, and LANAΔ33-949 were all highly deficient in the ability to retain GFP expression (Fig. 6B), whereas LANAΔ264-929 had an intermediate phenotype. Holm's procedure was again used to adjust P values. Comparison of group A (LANA, LANAΔ465-929, and LANAΔ332-929) with group B (LANAΔ33-273, LANAΔ33-495, LANAΔ33-888, LANAΔ33-929, and LANAΔ33-949) resulted in a P value of 6.32 × 10−16; comparison of group A with LANAΔ264-929 yielded a P value of 2.69 × 10−5; comparison of LANAΔ264-929 with group B yielded a P value of 5.60 × 10−6; and comparison of LANAΔ264-929 with LANAΔ33-495 yielded a P value of 1.75 × 10−2 (Fig. 6B). The reductions in LANA-mediated GFP retention were not due to reduced protein expression, because all mutants were expressed at levels higher than that of WT LANA (Fig. 6C). It remains possible that increased expression of LANAΔ465-929 or LANAΔ332-929 may be masking a defect for these mutants, although this is unlikely, since the expression of LANAΔ465-929 was only modestly higher than that of WT LANA. These results indicate that the unique N-terminal LANA internal sequence within amino acids 33 to 331, which are deleted in group B, is critical for the retention of p2TR-ΔRE-GFP, consistent with an important role in episome segregation, while the central repeat elements (deleted in LANAΔ332-929 and LANAΔ465-929) are dispensable for this function.
LANA amino acids 33 to 331 do not independently associate with mitotic chromosomes.
Since LANA residues 33 to 331 were implicated as critical for episome segregation, we assessed whether this region could independently associate with mitotic chromosomes. GFP LANA 1-32, GFP LANA 1-331, and GFP LANA 33-331 each contain the designated LANA residues fused downstream of GFP. GFP LANA 1-32 contains LANA residues 5 to 13, which bind histones H2A/H2B on the nucleosome surface to mediate chromosome association. As expected, GFP (green) did not associate with mitotic chromosomes (red), while GFP LANA 1-32 (green) painted chromosomes (the overlay of green and red generates yellow) (Fig. 6D). GFP 1-331 also associated tightly with chromosomes. GFP LANA 1-331 5GMR7, which contains alanine substitutions at the indicated residues that abolish interactions with histone H2A/H2B and with chromosomes (22, 27), did not associate with chromosomes. GFP LANA 33-331 (green), which lacks the histone-interacting LANA residues 5 to 13, also did not associate with chromosomes (Fig. 6D). Therefore, LANA residues 33 to 331 do not independently associate with mitotic chromosomes.
Both the unique internal sequence and repeat elements have roles in episome persistence.
We investigated the abilities of the internal LANA deletion mutants to mediate episome persistence. BJAB cells alone or expressing either LANA, LANAΔ465-497, LANAΔ33-495, LANAΔ33-273, LANAΔ465-929, LANAΔ332-929, or LANAΔ264-929 were transfected with a plasmid containing 8 copies of the KSHV terminal repeats (p8TR), seeded at 1,000 cells/well into microtiter plates, and selected for G418 resistance (which is encoded on the plasmid vector). After 7 to 8 days of G418 selection, >94% of LANA and LANAΔ465-497 wells had robust, G418-resistant outgrowth that was macroscopically visible and could be expanded to 24-well plates. LANAΔ465-929 also had G418-resistant growth in >94% of the wells, but the cells could not be expanded to 24-well plates until ∼10 to 11 days of selection. LANAΔ332-929 had somewhat less-robust G418-resistant outgrowth, in 90% of wells (average value), and cells could be expanded at ∼11 to 12 days of selection. Like the negative-control BJAB cells, LANAΔ33-495, LANAΔ33-273, and LANAΔ264-929 had G418-resistant outgrowth in fewer than 90% of wells and could not be expanded until ∼12 to 14 days under G418 selection. The lower G418-resistant outgrowth of BJAB cells than of LANA-expressing cells is due to the need for integration of p8TR DNA, which is a rare event compared with episome persistence. These results were not due to lower transfection efficiencies as assessed by GFP cotransfection. Therefore, these G418-resistant outgrowth results were consistent with the notion that LANAΔ465-929 has wild-type or only slightly reduced episome maintenance efficiency, while that of LANAΔ332-929 may be modestly reduced. In contrast, LANAΔ33-495, LANAΔ33-273, and LANAΔ264-929 appeared either not to mediate episome persistence or to do so with greatly reduced efficiency.
To detect the presence of p8TR episomes, Gardella gel analyses were performed on G418-resistant cell lines. In Gardella gels, live cells are loaded into the wells of an agarose gel and are lysed in situ in the presence of SDS and protease. During electrophoresis, large chromosomal DNA remains at the gel origin, while extrachromosomal DNA (as large as several hundred kilobases) migrates into the gel. DNA is then detected by Southern blotting by using a radiolabeled probe. Analysis of BCBL-1 cells, a KSHV-infected primary effusion lymphoma (PEL) cell line, results in a slowly migrating band representing the viral episome and more quickly migrating linear DNA that is due to lytic replication of the virus (Fig. 7A, lane 2, and B and C, lanes 1). As expected, G418-resistant BJAB cells lacking LANA did not have episomal DNA (Fig. 7A through C, lanes 3 and 4). In contrast, and in agreement with previous results, BJAB cells expressing LANA (Fig. 7A, lanes 5 and 6, and B and C, lanes 5 to 7) or LANAΔ465-497 (Fig. 7A, lanes 7 to 10) all contained episomal DNA (28). As observed previously (6, 22, 28), much of the episomal DNA migrated more slowly than the covalently closed circular (ccc) p8TR plasmid DNA. This slower migration is due to duplication of the TR sequence and multimerization of input plasmids (6, 28). Like LANA and LANA Δ465-497, LANAΔ465-929 (Fig. 7A, lanes 11 to 14) and LANA Δ332-929 (Fig. 7B, lanes 8 to 15) also efficiently maintained TR episomes in all lanes. From a total of 11 experiments, LANA had episomes in 34/34 (100%) G418-resistant clones; from 4 experiments, LANAΔ465-497 had episomes in 22/23 (96%); from 5 experiments, LANAΔ465-929 had episomes in 19/20 (95%); and from 3 experiments, LANAΔ332-929 had episomes in 20/22 (91%) (Fig. 1). Therefore, deletion of most of the glutamine-rich internal repeat regions and the predicted leucine zipper in LANAΔ465-929, or deletion of all of the internal repeat sequence in LANA Δ332-929, had only had modest effects on the ability of LANA to maintain episomes in these assays.
Fig 7.

LANA mutants have differing levels of episome maintenance deficiencies as assessed by Gardella gel analyses. BJAB cells alone, or BJAB cells stably expressing LANA or LANA deletion mutants, were transfected with p8TR. Seventy-two hours posttransfection, cells were seeded in 96-well plates and were selected for G418 resistance. Gardella cell analysis was performed to detect p8TR episomes from G418-resistant cell lines expanded from the microtiter plates. Approximately 1 × 106 cells were loaded per lane in Gardella gels. (A) Gardella gel containing a naked p8TR plasmid (lane 1), BCBL-1 cells (a KSHV-infected primary effusion lymphoma cell line) (lane 2), p8TR-transfected, G418-resistant BJAB cells (lanes 3 and 4), or BJAB cells stably expressing LANA (lanes 5 and-6), LANAΔ465-497 (lanes 7 to 10), LANAΔ465-929 (lanes 11 to 14), LANAΔ33-273 (lanes 15 to 18), or LANAΔ33-495 (lanes 19 to 22). Vertical lines (lanes 20 and 22) indicate a faint episomal signal. The gel origin (O), BCBL-1 episomal (E) and linear (L) forms (linear due to lytic replication), and p8TR covalently closed circular (ccc) DNA and nicked (N) DNA are indicated. Gardella gel analysis was performed after 24 days of G418 selection. (B) Gardella gel with BCBL-1 cells (lane 1), naked p8TR plasmid (lane 2), p8TR-transfected, G418-resistant BJAB cells (lanes 3 and 4), or BJAB cells stably expressing LANA (lanes 5 to 7) or LANAΔ332-929 (lanes 8 to 15). Gardella gel analysis was performed after 34 days of G418 selection. (C) Gardella gel with BCBL-1 cells (lane 1), naked p8TR plasmid (lane 2), p8TR-transfected, G418-resistant BJAB cells (lanes 3 and 4), or BJAB cells stably expressing LANA (lanes 5 to 7) or LANAΔ264-929 (lanes 8 to 13). Gardella gel analysis was performed after 36 days of selection. (D to F) Immunoblot analyses for LANA or LANA mutants expressed in G418-resistant cell lines used in Gardella gel analyses. Panels D to F correspond to panels A to C, respectively. LANA was detected using an anti-T7 monoclonal antibody or an anti-LANA antibody. Approximately 3.5 × 105 cells were loaded per lane for anti-T7 blots and ∼1.5 × 105 cells per lane for the anti-LANA blot. Asterisks indicate LANAΔ332-929 or LANA bands. Brightness and contrast in individual panels were uniformly adjusted using Adobe Photoshop. Lowercase letters indicate individually selected G418-resistant cell lines. NSB, nonspecific bands.
In contrast, LANAΔ264-929, LANAΔ33-273, and LANAΔ33-495 were highly compromised for episome persistence. Gardella analysis showed that LANAΔ33-273 (Fig. 7A, lanes 15 to 18) had episomes in only two of four lanes, LANAΔ264-929 (Fig. 7C, lanes 8 to 13) had episomes in only two of the six lanes, and LANAΔ33-495 (Fig. 7A, lanes 19 to 22) had only very faint signals in two of four lanes. Overall, from 7 experiments, LANAΔ264-929 had episomes in 13/33 G418-resistant cell lines (39%); from 8 experiments, LANAΔ33-273 had episomes in 14/44 (32%); and from 4 experiments, LANAΔ33-495 had episomes in 3/32 (9%) (Fig. 1). All mutants were expressed at levels at least as high as that of LANA (Fig. 7D to F), so deficits were not due to reduced protein expression. Protein expression levels were highest for LANAΔ264-929 and LANAΔ332-929 (Fig. 7E and F). This is due to the deletion of the internal repeat regions, which exert inhibitory effects on LANA expression (39). Therefore, the lower levels of episome persistence for LANAΔ264-929, LANAΔ33-273, and LANAΔ33-495 were not due to decreased protein expression. These results show that LANA mutants with deletions that included the unique internal sequence (LANA amino acids 33 to 331) had the largest episome maintenance deficits.
Assessment of the episome maintenance deficiencies of LANAΔ332-929 and LANAΔ465-929 in limiting-dilution assays.
Although both LANAΔ465-929 and LANAΔ332-929 mediated long-term episome maintenance of TR DNA and maintained episomes in 95% and 91% of G418-resistant cell lines (Fig. 1), respectively, the G418-resistant outgrowth of both was delayed by several days relative to that of LANA after plating at 1,000 cells/well, as described above. In contrast, LANAΔ465-497 did not exhibit delayed outgrowth. These differences may be due to fewer G418-resistant LANAΔ465-929 and LANAΔ332-929 episome-containing cells in each well with outgrowth, resulting in delayed G418-resistant outgrowth. To further investigate these differences, we performed limiting-dilution assays with p8TR-transfected cells.
First, we assessed LANAΔ465-497 as a control. BJAB cells alone or BJAB cells stably expressing LANA or LANAΔ465-497 were transfected with p8TR, seeded in 96-well plates at 1,000, 100, 10, and 1 cell/well, and selected for G418 resistance (Fig. 8A). Cells expressing LANA and LANAΔ465-497 had 100% well outgrowth when seeded at 1,000 cells/well and 100 cells/well. When seeded at 10 cells/well or 1 cell/well, LANA had G418-resistant outgrowth in 70 and 10 wells on average, respectively, while LANAΔ465-497 had outgrowth in 51 and 8 wells, respectively. In contrast, BJAB cells lacking LANA had much lower G418-resistant outgrowth, which occurred in 76, 12, 2, and 0 wells after seeding at 1,000, 100, 10, and 1 cell/well, respectively. Similarly, the number of days necessary for cells to expand sufficiently for transfer from 96-well to 24-well plates was the same for LANA and LANAΔ465-497 but was significantly higher for BJAB cells (Fig. 8C). Therefore, LANA and LANAΔ465-497 showed similar abilities to mediate G418-resistant outgrowth after limiting dilution, while BJAB cells, which lack LANA and rely on the integration of p8TR for G418 resistance, were much less efficient.
Fig 8.
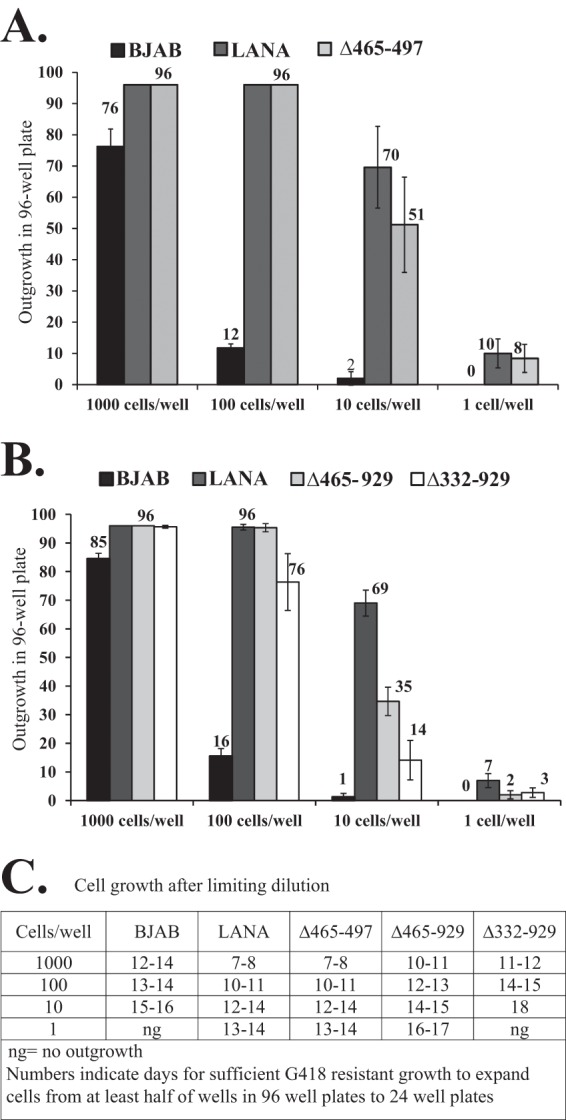
LANAΔ465-929 and LANAΔ332-929 exhibit deficiencies in limiting-dilution outgrowth after TR DNA transfection and G418 selection. (A) BJAB cells alone and BJAB cells stably expressing LANA or LANAΔ465-497 were transfected with plasmid p8TR and were seeded in 96-well plates at different cell concentrations. Well outgrowth was recorded at 20 days under G418 selection. Values are averages for 3 experiments; error bars indicate standard deviations. (B) Limiting-dilution assay for BJAB cells alone and BJAB cells stably expressing LANA, LANAΔ465-929, or LANAΔ332-929 after p8TR transfection and G418 selection. (C) Cells stably expressing LANAΔ465-929 or LANAΔ332-929 were assessed for rates of G418-resistant outgrowth after plating at different cell concentrations. Shown are the numbers of days necessary for sufficient G418-resistant outgrowth to allow the expansion of at least half of the positive wells in 96-well plates to 24-well plates.
We then performed limiting-dilution experiments after transfection of BJAB cells alone or BJAB cells expressing LANA, LANAΔ465-929, or LANAΔ332-929. In results similar to those for the previous experiment, after transfection with p8TR, LANA showed G418-resistant outgrowth in 96, 96, 69, and 7 wells on average after seeding at 1,000, 100, 10, and 1 cell/well (Fig. 8B). LANAΔ465-929 showed slightly reduced G418-resistant outgrowth, in 96, 96, 35, and 2 cells/well after seeding at 1,000, 100, 10, and 1 cell/well. The outgrowth of LANAΔ332-929 was further reduced, occurring in 96, 76, 14, and 3 cells/well after seeding at 1,000, 100, 10, and 1 cell/well (Fig. 8B). The reduced outgrowth of LANAΔ465-929 and LANAΔ332-929 relative to that of LANA was also reflected in the number of days necessary for cells to expand sufficiently for transfer from 96-well plates to 24-well plates (Fig. 8C). The reductions in G418-resistant outgrowth for LANAΔ465-929 and LANAΔ332-929 were not due to differences in transfection efficiencies as assessed by GFP cotransfection. Therefore, LANAΔ465-929 was modestly less efficient than LANA, and LANAΔ332-929 was even more deficient in the ability to mediate G418-resistant outgrowth after limiting dilution.
Based on these results, and to facilitate the comparison of mutants, the predicted numbers of cells seeded/well needed to obtain 63.2% of wells with outgrowth of episome-containing cells were determined using nonlinear regression analyses. By adopting the Poisson distribution for the number of episome-containing cells in a well, when the average number of episome-containing cells seeded per well is 1, an average of 36.8% of wells are expected to be vacant. Therefore, nonlinear regression analyses were used with the limiting-dilution data to estimate the number of cells seeded/well expected to yield 63.2% of wells with at least 1 episome-containing cell. These values were 7.7, 11.1, 28.9, and 84.1 for LANA, LANAΔ465-497, LANAΔ465-929, and LANA Δ332-929, respectively (Table 1). Comparison of the values for the mutants to that for LANA indicated deficiencies of 1.4-fold, 3.8-fold, and 10.9-fold for LANAΔ465-497, LANAΔ465-929, and LANA Δ332-929, respectively (Fig. 1). In contrast, for BJAB cells, which require the integration of transfected DNA for G418 resistance, 577 cells/well were necessary for G418-resistant outgrowth in 63.2% of wells, a 75-fold deficiency relative to the value for LANA.
Table 1.
Efficiency of episome maintenance
| Cell line | Predicted no. of cells/well necessary for 63.2% outgrowth of wells with episome-containing cells |
|---|---|
| LANA | 7.7 |
| LANAΔ465-497 | 11.1 |
| LANAΔ465-929 | 28.9 |
| LANAΔ332-929 | 84.1 |
| LANAΔ264-929 | 1,757.9 |
| LANAΔ33-273 | 2,108.6 |
| LANAΔ33-495 | 16,255.5 |
| LANAΔ33-888 | 2,999.2 |
| LANAΔ33-929 | 22,335.5 |
The numbers of cells seeded per well needed for 63.2% outgrowth with episome-containing cells were also estimated for LANAΔ264-929, LANAΔ33-273, and LANAΔ33-495. These estimates were based on the percentages of cell lines that contained episomes (Fig. 1) and the total numbers of wells with G418-resistant outgrowth after seeding at 1,000 cells/well, which were, on average, 84, 78, and 76 wells for LANAΔ264-929, LANAΔ33-273, and LANAΔ33-495, respectively. The Poisson distribution was then used to estimate episome-containing cell outgrowth at higher cell-seeding numbers prior to nonlinear regression analyses. Similar estimates were made for LANAΔ33-888 and LANAΔ33-929, which have been described previously (28) and had G418-resistant outgrowth after seeding at 1,000 cells/well in 77 and 74 wells on average, respectively. These results indicate deficiencies of 228.6-fold, 274.2-fold, 2,113.5-fold, 389.9-fold, and 2,904.0-fold for LANAΔ264-929, LANAΔ33-273, LANAΔ33-495, LANAΔ33-888, and LANAΔ33-929 relative to LANA, respectively (Fig. 1). Therefore, the efficiency of episome maintenance of each of these mutants was lower than the efficiency of integration, since BJAB cells had a 75-fold deficiency relative to LANA. Accordingly, G418-resistant cell lines from these mutants typically had integrated DNA and lacked episomes (Fig. 1).
DISCUSSION
This work demonstrates that both the unique internal sequence and the repeat elements of LANA play roles in episome maintenance. These findings were not due to an unexpected deficiency in mitotic chromosome association or a loss of DNA binding, because these functions are mediated by N- and C-terminal portions of LANA that were present in all mutants, and preservation of these functions was confirmed. Interestingly, all internal sequences tested were found to contribute to DNA replication. The ability to retain GFP expression from TR episomes deficient in DNA replication localized to the unique internal LANA sequence, consistent with an important role for this region in episome segregation. Deficiencies in the ability to mediate episome persistence ranged from minor to severe, and mutants with compromised replication and with deletions within the unique internal region implicated in segregation had the most-severe deficits (Fig. 1).
The finding that levels of DNA replication were reduced in all mutants suggests that different LANA regions may recruit cellular DNA replication factors. Deleted sequences with effects on replication included both the repeat elements and the unique internal sequence. Two relatively small regions of sequence that were notable for their effects on DNA replication were LANA residues 264 to 331 and 930 to 949. The replication of LANAΔ264-929 was reduced more than 2-fold from that of LANAΔ332-929, demonstrating the importance of amino acids 264 to 331, and the replication of LANAΔ33-949 was reduced more than 3-fold from that of LANAΔ33-929, demonstrating the importance of residues 930 to 949 (Fig. 5A). It is possible that key DNA replication factors bind to these sequences.
Several proteins involved in DNA replication have been associated with LANA. Origin recognition complex (ORC) proteins are central to the formation of the prereplication complex, and ORC1 to ORC6 interact with LANA (15, 44) and are recruited to TR DNA in a LANA-dependent manner (45). However, these factors associate predominantly with the C-terminal portion of LANA, which was included in all mutants, although conflicting results of either no (15) or weak (44) binding of ORC1 to LANA residues 1 to 340 have been reported. Moreover, minichromosome maintenance complex (MCM), a DNA helicase necessary for DNA replication initiation and elongation, and HBO1, a histone acetyltransferase important for DNA replication licensing, are recruited to the TR DNA region in a LANA-dependent manner, and their expression contributes to LANA-mediated TR-DNA replication (45). However, LANA has not been shown to interact physically with these proteins. Topoisomerase IIβ (TopoIIβ) induces double-stranded DNA (dsDNA) breaks during DNA replication (46), and ellipticine, a TopoIIβ inhibitor, reduced KSHV TR DNA replication. TopoIIβ associates with LANA, but the association is with the N-terminal portion of LANA, which was present in all mutants. The ubiquitin-specific protease USP7 associates with LANA, and a deletion of the USP7 binding site enhanced LANA DNA replication (47). However, this interaction was with LANA C-terminal residues 971 to 986, which were present in all mutants. LANA also interacts with structure-specific recognition protein 1 (SSRP1), whose expression is important for LANA-mediated DNA replication; however, the specific region of interaction within LANA has not been mapped (48). SSRP1 is a subunit of the chromatin structure modulator FACT (facilitating chromatin transcription), which disrupts and reassembles nucleosomes through the removal and replacement of an H2A/H2B dimer, thus allowing RNA and DNA polymerases to access DNA (49–52). LANA also associates with replication proteins A1 and A2, which complex together with an additional subunit to bind single-stranded DNA (ssDNA) during replication, although the specific region of interaction in LANA has not been mapped (53). Therefore, it remains possible that internal LANA regions recruit some of these or other DNA replication factors.
LANA mediates segregation to progeny nuclei by tethering KSHV DNA to mitotic chromosomes, allowing episomes to “hitchhike” on chromosomes. The essential components of this tethering mechanism enable LANA to bind mitotic chromosomes and TR DNA simultaneously. LANA N-terminal residues 5 to 13 bind histones H2A/H2B to attach to chromosomes (27), and the C-terminal portion of LANA also plays a role in chromosome association, leading to the concentration of LANA at pericentromeric and peritelomeric regions (24). The C-terminal portion of LANA also has the essential function of binding TR DNA, and both the N- and C-terminal portions of LANA were included in all mutants. Importantly, all mutants interacted with mitotic chromosomes in a WT fashion (Fig. 2), bound TR DNA (Fig. 3), and did not have reduced half-lives (Fig. 4). Since the essential tethering components were present in all mutants, the findings of severe segregation deficits were unexpected. The unique internal sequence, comprising residues 33 to 331, was found to be critical for LANA-mediated retention of GFP expression from p2TR-ΔRE-GFP, while the large central repeat region was not involved. LANAΔ332-929, which lacks all internal repeat elements, was as efficient as wild-type LANA at GFP retention (Fig. 6B), while LANAΔ33-273, which lacks nearly all the N-terminal unique internal sequence, was highly deficient. Notably, LANAΔ264-929 exhibited an intermediate ability to retain GFP, consistent with the location of amino acid 264 near the C-terminal border of the critical unique sequence that exerts the segregation function. LANA residues 33 to 331 did not independently associate with chromosomes (Fig. 6D), suggesting a novel role for this region in segregation.
LANA is associated with chromosomes in both interphase and metaphase (27), but during certain phases of the cell cycle, LANA may dissociate from nucleosomes and then reattach, and host cell proteins may mediate this process. For instance, during S phase, nucleosomes, to which LANA binds, may be transiently removed from DNA to allow DNA replication to occur (54, 55). A host cell protein(s) may be important for ensuring that H2A/H2B histones loaded with LANA are deposited onto newly synthesized DNA or that LANA is reloaded onto newly formed nucleosomes. If temporarily dissociated from nucleosomes, LANA may maintain a distinct association with host cell chromatin or DNA through a cell factor(s). Interactions may also be necessary to stabilize or strengthen the interaction of LANA with histones H2A/H2B. In addition, the precise distribution of LANA, with its cargo of viral DNA, to each sister chromatid is expected to be critical for persistence, and this trafficking may occur through a host cell protein(s).
A number of cell proteins involved in chromatin association, remodeling, or mitosis have been shown to interact with LANA. It is intriguing to speculate that FACT may play a role in segregation during nucleosome disruption through the interaction of LANA with SSRP1. HMGA1 and HMGB1 remodel chromatin and interact with LANA (53), although the interactions within LANA have not been mapped. Both the N- and C-terminal portions of LANA are required for the localization of LANA with methyl-CpG-binding protein (MeCP2) at chromocenters, locations of concentrated pericentric heterochromatin (56), and MeCP2 has been proposed to play a role in the binding of LANA to chromosomes (19). Although results differ as to whether MeCP2 binds the N-terminal (19) or the C-terminal (56) portion of LANA, both these regions were present in all mutants. Interestingly, the kinetochore proteins CENP-F and Bub1 colocalized with LANA at ∼30% or ∼50% of sites, respectively, on mitotic chromosomes in infected cells (57). Knockdown of Bub1, a mitotic spindle checkpoint protein, but not knockdown of CENP-F, reduced episomal DNA levels in infected cells. CENP-F and Bub1 each interact both with LANA amino acids 1 to 340 and with LANA amino acids 842 to 1162 (57), which overlap the unique region shown here to be important for segregation. The nuclear mitotic apparatus protein, NuMA, locates to the spindle poles during mitosis and has been shown to associate with LANA during interphase, but not during mitosis. However, NuMA interacts with LANA residues 762 to 1162 (58), which were not implicated in segregation here. The BET (bromodomain and extraterminal domain) proteins BRD2, BRD3, and BRD4, which interact with acetylated histones, associate with LANA (59–62). Further, BRD4 colocalizes with LANA on mitotic chromosomes (62). The C-terminal portion of LANA is the major interaction region with BRD proteins and was included in all mutants. A minor binding region for BRD4 exists in LANA residues 475 to 777 (62), but this sequence is located in the repeat region, not in the unique internal sequence. Therefore, some of these proteins, or others yet to be identified, may play important roles in LANA-mediated segregation.
It is notable that once episomes are established, even LANA mutants that are highly deficient in episome persistence are able to maintain episomes stably for months, and likely indefinitely (28). As discussed previously (28), it is likely that recombination events within TR DNA provide compensatory changes that allow long-term persistence even in the presence of substantial LANA deficiency. As observed here (Fig. 7) and previously (6, 28), much of the episomal DNA is substantially larger than the input covalently closed circular DNA and comigrates with KSHV episomes from infected BCBL-1 cells, which are ∼200 kb. This increase in size is due to TR duplication and the multimerization of input plasmids (6, 28). It is possible that greatly increased TR numbers, or amplification of certain components of TR elements, such as LANA binding sites, may contribute to efficient persistence.
Despite the ability of LANA mutants to mediate episome persistence at reduced efficiencies, found in this work, it is likely that even relatively modest deficiencies have considerable consequences for KSHV fitness. For instance, LANAΔ332-929, which lacks all internal repeat elements, was 10.9-fold deficient in episome persistence relative to WT LANA (Fig. 1). A recombinant KSHV containing a very similar LANA mutant lacking all the internal repeat elements, LANAΔ329-931, was recently shown to be incapable of establishing stable cell lines containing viral episomes, in contrast to control KSHV (63). The system used here has a large dynamic range, allowing the identification of mutants that are >1,000-fold deficient (Fig. 1). This system uses the transfection of high copy numbers of TR DNA into cell lines stably expressing LANA. In contrast, KSHV expresses LANA in cis, and the generation of recombinant KSHV is much more limited with regard to the number of copies of genomes available for initial transfection or infection. In fact, these lower copy numbers of recombinant KSHV are similar to, or even higher than, the copy number of KSHV in vivo (64–68). Further, while the system used here may allow TR DNA to undergo extensive recombination to select episomes for increased episome maintenance efficiency, the virus does not have this luxury. Therefore, even modest LANA deficiencies, as defined in these assays, are likely highly deleterious to KSHV.
This work suggests that critical functional sequences are distributed within the internal sequence of LANA. As discussed above, it is likely that certain host cell proteins interact with specific LANA sites to play critical roles in LANA function, both in DNA replication and in episome segregation. Future identification of the protein(s) mediating these functions will provide important insight into the mechanisms underlying LANA function.
ACKNOWLEDGMENTS
This work was supported by grants (to K.M.K.) from the National Cancer Institute (CA082036) and from the U.S. Department of Defense (PR093491).
Footnotes
Published ahead of print 4 September 2013
REFERENCES
- 1.Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. 1995. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 86:2708–2714 [PubMed] [Google Scholar]
- 2.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 3.Moore PS, Chang Y. 1995. Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N. Engl. J. Med. 332:1181–1185 [DOI] [PubMed] [Google Scholar]
- 4.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 5.Decker LL, Shankar P, Khan G, Freeman RB, Dezube BJ, Lieberman J, Thorley-Lawson DA. 1996. The Kaposi sarcoma-associated herpesvirus (KSHV) is present as an intact latent genome in KS tissue but replicates in the peripheral blood mononuclear cells of KS patients. J. Exp. Med. 184:283–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ballestas ME, Kaye KM. 2001. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 75:3250–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644 [DOI] [PubMed] [Google Scholar]
- 8.Cotter MA, II, Subramanian C, Robertson ES. 2001. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology 291:241–259 [DOI] [PubMed] [Google Scholar]
- 9.Fejér G, Medveczky MM, Horvath E, Lane B, Chang Y, Medveczky PG. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts preferentially with the terminal repeats of the genome in vivo and this complex is sufficient for episomal DNA replication. J. Gen. Virol. 84:1451–1462 [DOI] [PubMed] [Google Scholar]
- 10.Garber AC, Shu MA, Hu J, Renne R. 2001. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:7882–7892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garber AC, Hu J, Renne R. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 277:27401–27411 [DOI] [PubMed] [Google Scholar]
- 12.Hu J, Garber AC, Renne R. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 76:11677–11687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grundhoff A, Ganem D. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 77:2779–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Komatsu T, Ballestas ME, Barbera AJ, Kelley-Clarke B, Kaye KM. 2004. KSHV LANA1 binds DNA as an oligomer and residues N-terminal to the oligomerization domain are essential for DNA binding, replication, and episome persistence. Virology 319:225–236 [DOI] [PubMed] [Google Scholar]
- 15.Lim C, Sohn H, Lee D, Gwack Y, Choe J. 2002. Functional dissection of latency-associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J. Virol. 76:10320–10331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Judde JG, Lacoste V, Briere J, Kassa-Kelembho E, Clyti E, Couppie P, Buchrieser C, Tulliez M, Morvan J, Gessain A. 2000. Monoclonality or oligoclonality of human herpesvirus 8 terminal repeat sequences in Kaposi's sarcoma and other diseases. J. Natl. Cancer Inst. 92:729–736 [DOI] [PubMed] [Google Scholar]
- 17.Duprez R, Lacoste V, Briere J, Couppie P, Frances C, Sainte-Marie D, Kassa-Kelembho E, Lando MJ, Essame Oyono JL, Nkegoum B, Hbid O, Mahé A, Lebbe C, Tortevoye P, Huerre M, Gessain A. 2007. Evidence for a multiclonal origin of multicentric advanced lesions of Kaposi sarcoma. J. Natl. Cancer Inst. 99:1086–1094 [DOI] [PubMed] [Google Scholar]
- 18.Lagunoff M, Ganem D. 1997. The structure and coding organization of the genomic termini of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). Virology 236:147–154 [DOI] [PubMed] [Google Scholar]
- 19.Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 76:11596–11604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piolot T, Tramier M, Coppey M, Nicolas JC, Marechal V. 2001. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J. Virol. 75:3948–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szekely L, Kiss C, Mattsson K, Kashuba E, Pokrovskaja K, Juhasz A, Holmvall P, Klein G. 1999. Human herpesvirus-8-encoded LNA-1 accumulates in heterochromatin-associated nuclear bodies. J. Gen. Virol. 80:2889–2900 [DOI] [PubMed] [Google Scholar]
- 22.Barbera AJ, Ballestas ME, Kaye KM. 2004. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 N terminus is essential for chromosome association, DNA replication, and episome persistence. J. Virol. 78:294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelley-Clarke B, Ballestas ME, Srinivasan V, Barbera AJ, Komatsu T, Harris TA, Kazanjian M, Kaye KM. 2007. Determination of Kaposi's sarcoma-associated herpesvirus C-terminal latency-associated nuclear antigen residues mediating chromosome association and DNA binding. J. Virol. 81:4348–4356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelley-Clarke B, Ballestas ME, Komatsu T, Kaye KM. 2007. Kaposi's sarcoma herpesvirus C-terminal LANA concentrates at pericentromeric and peri-telomeric regions of a subset of mitotic chromosomes. Virology 357:149–157 [DOI] [PubMed] [Google Scholar]
- 25.Lim C, Seo T, Jung J, Choe J. 2004. Identification of a virus trans-acting regulatory element on the latent DNA replication of Kaposi's sarcoma-associated herpesvirus. J. Gen. Virol. 85:843–855 [DOI] [PubMed] [Google Scholar]
- 26.Wong LY, Matchett GA, Wilson AC. 2004. Transcriptional activation by the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen is facilitated by an N-terminal chromatin-binding motif. J. Virol. 78:10074–10085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861 [DOI] [PubMed] [Google Scholar]
- 28.De León Vázquez E, Kaye KM. 2011. The internal Kaposi's sarcoma-associated herpesvirus LANA regions exert a critical role on episome persistence. J. Virol. 85:7622–7633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hung SC, Kang MS, Kieff E. 2001. Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1. Proc. Natl. Acad. Sci. U. S. A. 98:1865–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De León Vázquez E, Kaye KM. 2011. Rapid and quantitative assessment of KSHV LANA-mediated DNA replication. Arch. Virol. 156:1323–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skalsky RL, Hu J, Renne R. 2007. Analysis of viral cis elements conferring Kaposi's sarcoma-associated herpesvirus episome partitioning and maintenance. J. Virol. 81:9825–9837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu J, Renne R. 2005. Characterization of the minimal replicator of Kaposi's sarcoma-associated herpesvirus latent origin. J. Virol. 79:2637–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365–369 [DOI] [PubMed] [Google Scholar]
- 34.Taylor ER, Morgan IM. 2003. A novel technique with enhanced detection and quantitation of HPV-16 E1- and E2-mediated DNA replication. Virology 315:103–109 [DOI] [PubMed] [Google Scholar]
- 35.Gardella T, Medveczky P, Sairenji T, Mulder C. 1984. Detection of circular and linear herpesvirus DNA molecules in mammalian cells by gel electrophoresis. J. Virol. 50:248–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holm S. 1979. A simple sequentially rejective multiple test procedure. Scand. J. Statist. 6:65–70 [Google Scholar]
- 37.Kelley-Clarke B, De León-Vázquez E, Slain K, Barbera AJ, Kaye KM. 2009. Role of Kaposi's sarcoma-associated herpesvirus C-terminal LANA chromosome binding in episome persistence. J. Virol. 83:4326–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srinivasan V, Komatsu T, Ballestas ME, Kaye KM. 2004. Definition of sequence requirements for latency-associated nuclear antigen 1 binding to Kaposi's sarcoma-associated herpesvirus DNA. J. Virol. 78:14033–14038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwun HJ, da Silva SR, Shah IM, Blake N, Moore PS, Chang Y. 2007. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J. Virol. 81:8225–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bubman D, Guasparri I, Cesarman E. 2007. Deregulation of c-Myc in primary effusion lymphoma by Kaposi's sarcoma herpesvirus latency-associated nuclear antigen. Oncogene 26:4979–4986 [DOI] [PubMed] [Google Scholar]
- 41.Liu J, Martin HJ, Liao G, Hayward SD. 2007. The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J. Virol. 81:10451–10459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim C, Choi C, Choe J. 2004. Mitotic chromosome-binding activity of latency-associated nuclear antigen 1 is required for DNA replication from terminal repeat sequence of Kaposi's sarcoma-associated herpesvirus. J. Virol. 78:7248–7256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Invest. 113:124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verma SC, Choudhuri T, Kaul R, Robertson ES. 2006. Latency-associated nuclear antigen (LANA) of Kaposi's sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 80:2243–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stedman W, Deng Z, Lu F, Lieberman PM. 2004. ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J. Virol. 78:12566–12575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purushothaman P, McDowell ME, McGuinness J, Salas R, Rumjahn SM, Verma SC. 2012. Kaposi's sarcoma-associated herpesvirus-encoded LANA recruits topoisomerase IIβ for latent DNA replication of the terminal repeats. J. Virol. 86:9983–9994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jäger W, Santag S, Weidner-Glunde M, Gellermann E, Kati S, Pietrek M, Viejo-Borbolla A, Schulz TF. 2012. The ubiquitin-specific protease USP7 modulates the replication of Kaposi's sarcoma-associated herpesvirus latent episomal DNA. J. Virol. 86:6745–6757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu J, Liu E, Renne R. 2009. Involvement of SSRP1 in latent replication of Kaposi's sarcoma-associated herpesvirus. J. Virol. 83:11051–11063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan BC, Chien CT, Hirose S, Lee SC. 2006. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J. 25:3975–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. 2003. FACT facilitates transcription-dependent nucleosome alteration. Science 301:1090–1093 [DOI] [PubMed] [Google Scholar]
- 51.Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D. 1998. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92:105–116 [DOI] [PubMed] [Google Scholar]
- 52.Reinberg D, Sims RJ., III 2006. de FACTo nucleosome dynamics. J. Biol. Chem. 281:23297–23301 [DOI] [PubMed] [Google Scholar]
- 53.Shamay M, Liu J, Li R, Liao G, Shen L, Greenway M, Hu S, Zhu J, Xie Z, Ambinder RF, Qian J, Zhu H, Hayward SD. 2012. A protein array screen for Kaposi's sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J. Virol. 86:5179–5191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henikoff S. 2008. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 9:15–26 [DOI] [PubMed] [Google Scholar]
- 55.Li G, Reinberg D. 2011. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 21:175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsumura S, Persson LM, Wong L, Wilson AC. 2010. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J. Virol. 84:2318–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiao B, Verma SC, Cai Q, Kaul R, Lu J, Saha A, Robertson ES. 2010. Bub1 and CENP-F can contribute to Kaposi's sarcoma-associated herpesvirus genome persistence by targeting LANA to kinetochores. J. Virol. 84:9718–9732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Si H, Verma SC, Lampson MA, Cai Q, Robertson ES. 2008. Kaposi's sarcoma-associated herpesvirus-encoded LANA can interact with the nuclear mitotic apparatus protein to regulate genome maintenance and segregation. J. Virol. 82:6734–6746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Platt GM, Simpson GR, Mittnacht S, Schulz TF. 1999. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 73:9789–9795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Viejo-Borbolla A, Ottinger M, Bruning E, Burger A, Konig R, Kati E, Sheldon JA, Schulz TF. 2005. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 79:13618–13629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ottinger M, Christalla T, Nathan K, Brinkmann MM, Viejo-Borbolla A, Schulz TF. 2006. Kaposi's sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 80:10772–10786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.You J, Srinivasan V, Denis GV, Harrington WJ, Jr, Ballestas ME, Kaye KM, Howley PM. 2006. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol. 80:8909–8919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alkharsah KR, Schulz TF. 2012. A role for the internal repeat of the Kaposi's sarcoma-associated herpesvirus latent nuclear antigen in the persistence of an episomal viral genome. J. Virol. 86:1883–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koelle DM, Huang ML, Chandran B, Vieira J, Piepkorn M, Corey L. 1997. Frequent detection of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in saliva of human immunodeficiency virus-infected men: clinical and immunologic correlates. J. Infect. Dis. 176:94–102 [DOI] [PubMed] [Google Scholar]
- 65.Alkharsah KR, Dedicoat M, Blasczyk R, Newton R, Schulz TF. 2007. Influence of HLA alleles on shedding of Kaposi sarcoma-associated herpesvirus in saliva in an African population. J. Infect. Dis. 195:809–816 [DOI] [PubMed] [Google Scholar]
- 66.Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185–6196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 86:9708–9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Budt M, Hristozova T, Hille G, Berger K, Brune W. 2011. Construction of a lytically replicating Kaposi's sarcoma-associated herpesvirus. J. Virol. 85:10415–10420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cherezova L, Burnside KL, Rose TM. 2011. Conservation of complex nuclear localization signals utilizing classical and non-classical nuclear import pathways in LANA homologs of KSHV and RFHV. PLoS One 6:e18920. 10.1371/journal.pone.0018920 [DOI] [PMC free article] [PubMed] [Google Scholar]