

Abstract
Epithelial-to-mesenchymal transitions (EMTs) underlie cell plasticity required in embryonic development and frequently observed in advanced carcinogenesis. Transforming growth factor-β (TGF-β) induces EMT phenotypes in epithelial cells in vitro and has been associated with EMT in vivo. Here we report that expression of the hairy/enhancer-of-split-related transcriptional repressor Hey1, and the Notch-ligand Jagged1 (Jag1), was induced by TGF-β at the onset of EMT in epithelial cells from mammary gland, kidney tubules, and epidermis. The HEY1 expression profile was biphasic, consisting of immediate-early Smad3-dependent, Jagged1/Notch-independent activation, followed by delayed, indirect Jagged1/Notch-dependent activation. TGF-β-induced EMT was blocked by RNA silencing of HEY1 or JAG1, and by chemical inactivation of Notch. The EMT phenotype, biphasic activation of Hey1, and delayed expression of Jag1 were induced by TGF-β in wild-type, but not in Smad3-deficient, primary mouse kidney tubular epithelial cells. Our findings identify a new mechanism for functional integration of Jagged1/Notch signalling and coordinated activation of the Hey1 transcriptional repressor controlled by TGF-β/Smad3, and demonstrate functional roles for Smad3, Hey1, and Jagged1/Notch in mediating TGF-β-induced EMT.
Keywords: development, differentiation, epithelial, neoplasm, signal transduction, transforming growth factor-β
Introduction
During embryogenesis, epithelial cell layers undergo epithelial-to-mesenchymal transitions (EMTs) characterized by disassembly of cell–cell contacts, reorganization of actin cytoskeleton, and cell–cell separation resulting in fibroblast-like cells with mesenchymal marker expression and migratory properties (Hay, 1995). Extracellular cues, including cell adhesion molecules and growth factors, have been shown to induce various phenotypic aspects of EMT in vitro and in vivo (reviewed in Thiery, 2002). In addition, members of the transforming growth factor-β (TGF-β) superfamily have been identified as important inducers of EMT in cardiac, palatal and hair follicle development, and in cutaneous wound repair (Sanford et al, 1997; Sun et al, 1998; Romano and Runyan, 2000; Camenisch et al, 2002). Importantly, the metastatic potential of certain carcinomas is associated with cooperative signalling by activated Ras and autocrine TGF-β inducing an irreversible EMT phenotype (Janda et al, 2002).
Members of the TGF-β superfamily are multifunctional cytokines that control cell fates, including cell cycle arrest, differentiation, and apoptosis (Roberts and Sporn, 1990). TGF-β signals through complexes of heteromeric transmembrane type I (TbRI) and type II (TbRII) receptors (Massague, 2000), activating Smad2 and/or Smad3, two signalling mediators of the SMAD protein family (Heldin et al, 1997; Hoodless and Wrana, 1998), which associate with the shared partner Smad4 and translocate to the nucleus. Nuclear SMAD protein complexes bind specific DNA sequence motifs and participate in transcriptional regulation of target genes (Derynck et al, 1998; Yang et al, 2003). TGF-β controls a context-dependent spectrum of epithelial cell plasticity, ranging from cell scattering phenotypes to partial and reversible EMT, or complete and persistent EMT, in nontransformed epithelial cells, indicating an important role for TGF-β-dependent signals in inducing EMT phenotypes. Extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinases (MAPKs), phosphatidylinositol 3-kinase (PI3K), RhoA and Rac1 small GTPases, and Smads have all been implicated as mediators of some or all phenotypic aspects of TGF-β-induced EMT (Miettinen et al, 1994; Piek et al, 1999; Bakin et al, 2000; Bhowmick et al, 2001; Zavadil et al, 2001). Zinc-finger transcriptional repressors Snail and Slug, and the two-handed E-box-binding zinc-finger protein SIP1 are targets of TGF-β signalling (Romano and Runyan, 2000; Comijn et al, 2001; Zavadil et al, 2001) with putative roles in EMT (Nieto, 2002). These findings together emphasize our currently incomplete understanding of the complexity of TGF-β signalling in orchestrating EMT phenotypes.
We have previously reported that TGF-β induction of an EMT phenotype in cultured human keratinocytes was associated with early upregulation of components and target genes of Notch receptor signalling, including transcriptional repressors related to Drosophila hairy and enhancer-of-split (H/E(spl)) proteins (Zavadil et al, 2001). Binding of transmembrane Notch receptors with membrane-bound ligands of the Delta, Serrate/Jagged, or Caenorhabditis elegans Lag-2 (DSL) family initiates proteolytic processing of Notch receptors mediated by accessory protein complexes and γ-secretase activity, followed by nuclear translocation of the intracellular domain of Notch (NICD) (Mumm and Kopan, 2000). Activation (derepression) of Notch target genes involves NICD binding with constitutive repressor complexes anchored at CBF-1-binding elements, converting them into transcriptional activator complexes. The basic helix–loop–helix (bHLH) DNA-binding transcriptional repressors of the H/E(Spl) protein family are direct targets of Notch. To date, H/E(Spl) proteins have not been implicated in EMT and/or TGF-β signalling. Although both TGF-β/Smad and Notch pathways are widely involved in cell fate decisions in development and pathobiology, functional and molecular interactions of these pathways have not been reported to date.
Here we report a new signalling mechanism involved in EMT activated by TGF-β. We show that TGF-β stimulates two kinetically coordinated waves of expression of Hey1. Immediate-early activation of Hey1 by TGF-β is mediated directly by Smad3/4 binding at distinct Smad-binding element core repeats (SCRs) in their distal promoter regions and does not require Notch signalling. The immediate-early activation cycle is followed by a second cycle of gene activation that requires classical Notch-dependent signalling, which is activated by TGF-β through Smad3- and ERK MAPK-dependent synthesis of Jagged1. When either Hey1, Jagged1, Notch, or Smad3 is inactivated, TGF-β fails to induce key EMT phenotypes, including disassembly of E-cadherin adherens junctions, disassembly of cortical actin, and cell–cell separation. Our results suggest that functional integration of TGF-β and Notch pathways is mediated by Smad3/ERK MAPK-dependent synthesis of Jagged1 and has important roles in regulation of epithelial cell plasticity by TGF-β.
Results
The Notch-regulated transcriptional repressor Hey1 is a new direct target gene of TGF-β/Smad signalling in epithelial cells
Our previous work indicated a potential role for Notch and Notch target genes of the H/E(spl) family of bHLH transcriptional repressors in TGF-β-induced EMT (Zavadil et al, 2001). Here we found that TGF-β induced expression of the H/E(spl) gene Hey1 in established cell culture models of TGF-β-induced EMT (Figure 1A), including normal murine mammary gland epithelial cells (NMuMGs), kidney proximal tubular epithelial (HK-2), and distal tubular epithelial cell lines Madin–Darby Canine Kidney (MDCK). Induction was RNA synthesis dependent and protein synthesis independent (data not shown), indicating that Hey1 is a new immediate-early target gene of TGF-β. Consistent with mRNA expression, Hey1 protein synthesis was induced upon stimulation by TGF-β (Figure 1B).
Figure 1.
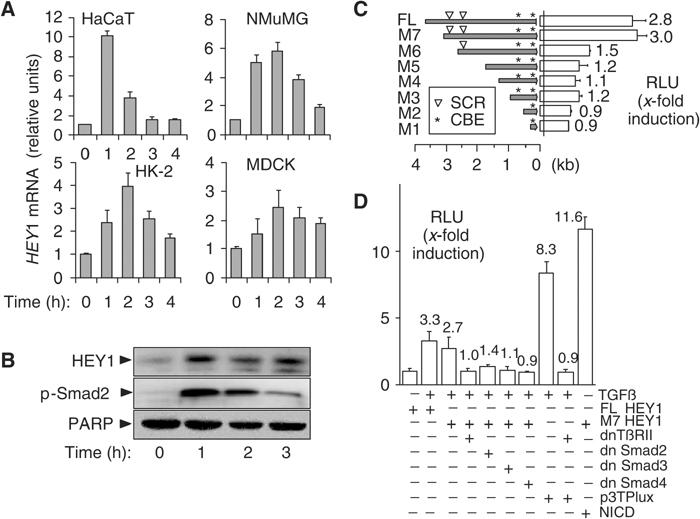
HEY1 is a direct target gene of TGF-β/Smad signalling. (A) Bar graphs show averages of relative HEY1 mRNA abundance determined by quantitative RT-PCR analysis in keratinocytes (HaCaT), mouse mammary gland epithelial cells (NMuMG), human kidney proximal tubular epithelial cells (HK-2), and canine kidney distal tubular epithelial cell line (MDCK). (B) Immunoblots show HEY1 and C-terminally phosphorylated Smad2 p-Smad2 proteins in keratinocytes stimulated with TGF-β. PARP, loading control. (C) Bars show average (N=3) fold induction by TGF-β of relative luciferase units (RLUs) in keratinocytes transfected with reporter constructs of human HEY1 promoter deletions (full-length FL, M7 to M1). (▿) positions of SCRs; (*) positions of NICD-responsive CBEs. (D) Bars indicate average (N=3) fold induction by TGF-β of luciferase activity (RLU) in keratinocytes cotransfected with luciferase reporter constructs (FL-HEY1, M7 HEY1, or 3TPLux) and plasmids expressing dominant-negative mutants of type II TGF-β receptor (dnTbRII), Smad2, 3 and 4 (dn Smad2, 3 and 4), or NICD. p3TPlux, positive control for TGF-β/Smad; NICD, positive control for activation of CBE sites.
To investigate molecular mechanisms of HEY1 activation by TGF-β, we isolated a 3.9 kb fragment of the 5′ flanking region of human HEY1 and tested its response to TGF-β in transiently transfected keratinocytes using a luciferase reporter construct. We observed a significant ∼3-fold increase in luciferase activity upon TGF-β treatment (Figure 1C). Sequence analysis of the HEY1 promoter identified five putative consensus binding elements for Smad3/Smad4 (Dennler et al, 1998). Four of the Smad-binding elements (SBE) were arranged in two GTCTnGTCT repeats at positions −2681 (proximal) and −3005 (distal) relative to the translation initiation codon (Figure 1C). Such SCRs have been shown to mediate high-affinity binding of Smad3/Smad4 protein complexes (von Gersdorff et al, 2000), and are characteristic elements of TGF-β-responsive immediate-early genes (Yang et al, 2003). We used a series of HEY1 promoter deletion constructs to confirm that promoter fragments containing both proximal and distal SCRs were required for the activation by TGF-β signals, while constructs lacking the SCRs (M1–M5) were unresponsive (Figure 1C). Dominant-negative mutant interference with TGF-β signals by cotransfection with expression plasmids for either TβRII-DN-K277R, Smad3Δexon8, or Smad4ΔM4 reduced the TGF-β-inducible activity of HEY1 full-length (FL) or M7 reporter constructs to baseline levels, while Smad2 3S → A had a partial effect (Figure 1D). These results indicate that key components of the TGF-β/Smad pathway were required for induction of the HEY1 promoter. Two Notch-responsive CBEs are located in the proximal part of the promoter in constructs FL, M2–M7 while construct M1 contains only a proximal CBE (Figure 1C). When the reporter constructs were cotransfected with a construct expressing active Notch intracellular domain (NICD), all promoter fragments except for M1 were sufficient to mediate induction of luciferase activity by NICD (data not shown). TGF-β/Smad and Notch signals thus use physically distinct promoter regions to activate HEY1.
TGF-β induces rapid and transient binding of endogenous Smad3/Smad4 complexes at SCRs in the HEY1 promoter
In vivo binding of endogenous Smad3- and Smad4-containing complexes with both HEY1 promoter SCRs at positions −2681 and −3005, respectively, was analyzed by chromatin immunoprecipitation with anti-Smad3 and anti-Smad4 antibodies, followed by quantitative PCR analysis of the SCR loci (ChIP–QPCR). TGF-β induced rapid and transient increases of SCR binding with Smad3/Smad4-containing complexes (Figure 2A). In contrast, we could not detect TGF-β-inducible SCR binding of endogenous Smad2 (data not shown). To confirm our in vivo results, conventional electrophoretic mobility shift assays (EMSAs) and antibody interference studies also demonstrated TGF-β-induced binding of Smad3-containing protein complexes to synthetic oligonucleotide probes containing either the distal or the proximal HEY1 SCR (Figure 2B). Together, these experiments define two separate TGF-β-inducible, Smad3/4 response elements at positions −3005 and −2681 bp relative to the translation initiation site of the human HEY1 gene.
Figure 2.
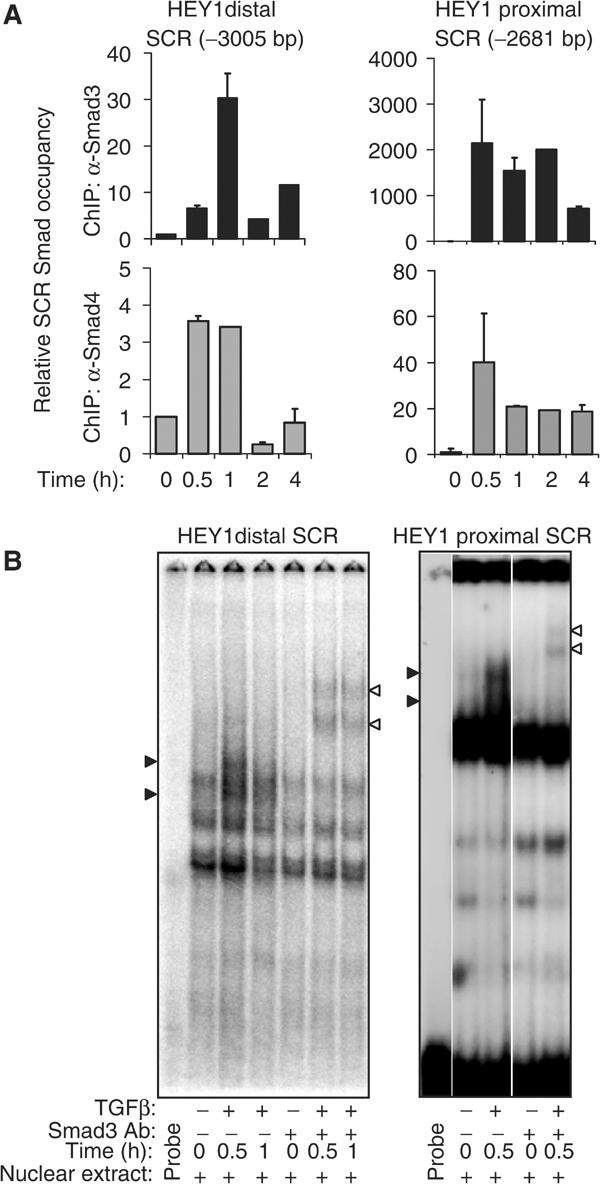
TGF-β induces rapid in vivo binding of endogenous Smad3/Smad4 protein complexes with SCR elements in the HEY1 promoter. (A) Bar graphs show the relative amount of distal HEY1 SCR (−3005 bp), or proximal SCR (−2681 bp), respectively, determined by quantitative, sequence-specific PCR that is bound with immunoprecipitated Smad3 (black bars) or Smad4 (gray bars) in untreated keratinocytes (time 0 h; baseline unit set at 1) and TGF-β-treated keratinocytes. (B) EMSAs demonstrate binding of oligonucleotide probes containing the distal (−3005) SCR or proximal (−2681) SCR, respectively, and nuclear protein extracts from keratinocytes left untreated (−) or treated with TGF-β (+) for 0.5 and 1 h. Specific DNA-binding protein complexes are depicted by block arrowheads, and are supershifted by preincubation with anti-Smad3 antibody (open arrowhead).
Notch receptor activation and function is not required for Smad3/4-dependent, immediate-early induction of HEY1 by TGF-β
To investigate whether TGF-β/Smad3 and Notch pathways may cooperate in the immediate-early induction of HEY1, we examined classical features of Notch receptor signalling. Chemical inhibition (GSI) of γ-secretase activity, typically required for proteolytic activation of Notch (Mumm and Kopan, 2000), did not block induction of HEY1 by TGF-β (Figure 3A). We did not detect nuclear accumulation of transcriptionally active NICD within 60 min of TGF-β treatment (Figures 3B and 5B). TGF-β had no effect on the constitutive CBF-1 protein complex binding with oligonucleotide probes containing proximal (Figure 3C) or distal CBE sites of the HEY1 promoter, respectively. Furthermore, the level of 300 kDa transmembrane form of Notch1, as assessed by immunoblotting, was unchanged within the first hour of TGF-β treatment (see Figure 5B). Together, our results indicate that Notch receptor function was not required for the immediate-early activation of HEY1 mediated by Smad3/4 in TGF-β-treated HaCaT cells.
Figure 3.
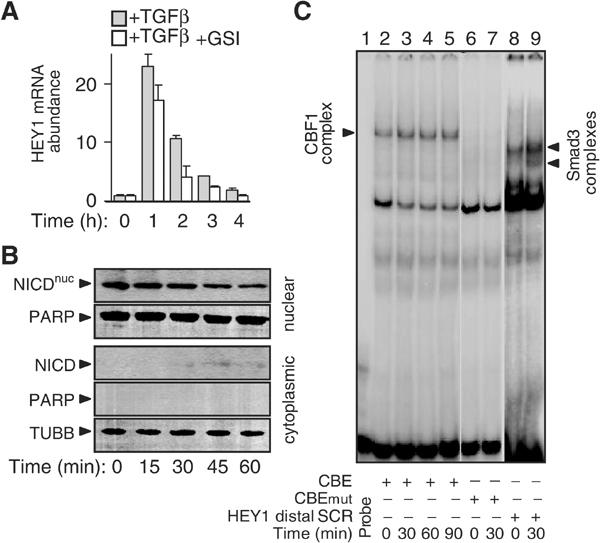
Activation of the Notch receptor is not involved in TGF-β induction of HEY1. (A) Histograms show the relative abundance of HEY1 mRNA expression as determined by quantitative RT-PCR in keratinocytes treated with TGF-β in the absence or presence of γ-secretase inhibitor GSI. (B) Immunoblot showing NICD in nuclear and cytoplasmic protein fractions of keratinocytes treated with TGF-β. PARP (poly-α-ADP-ribosyltransferase) and TUBB (tubulin β) demonstrate purity of cytoplasmic and nuclear protein lysate fractions, respectively. (C) EMSA demonstrates binding of CBF-1 protein complex with oligonucleotide probe containing the distal CBE of the HEY1 promoter (lanes 1–5). Lanes 6 and 7: EMSA using mutated CBE oligonucleotide probe (CBEmut) with single bp exchange (cgtggGaaa → cgtggCaaa). Lanes 8 and 9: EMSA using probe containing the distal (−3005 bp) HEY1 SCR sequence (HEY1 distal SCR) demonstrates TGF-β-inducible Smad protein complexes.
Figure 5.
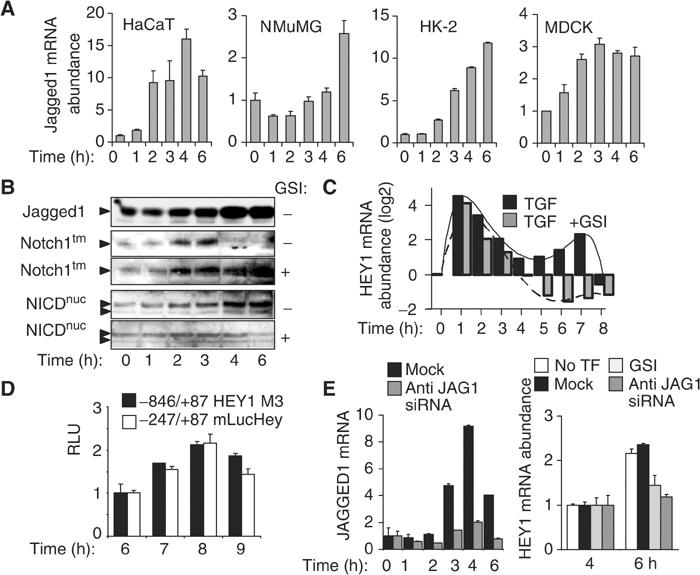
Jagged1-induced Notch receptor activation initiates a second, delayed activation of HEY1. (A) Histograms show the mean of relative mRNA abundance determined by quantitative RT-PCR of Notch ligand JAGGED1 in response to TGF-β in epithelial cells (N=3). (B) Immunoblotting depicts JAGGED1 protein, intact 300 kDa transmembrane form (Notch1tm) of Notch in the absence or presence of γ-secretase inhibitor GSI, and nuclear accumulation of NICD (NICDnuc) in the absence or presence of GSI in TGF-β-treated keratinocytes. (C) Bar graphs indicate the relative abundance of HEY1 mRNA as determined by quantitative RT-PCR analysis in keratinocytes treated with TGF-β in the absence or presence of GSI. (D) Histogram shows RLUs in keratinocytes transfected with reporter constructs containing proximal human and mouse HEY1 promoter fragments, as indicated, after TGF-β stimulation (N=3). (E) Left histogram: Bars indicate a representative experiment showing the relative abundance of JAGGED1 mRNA determined by quantitative RT-PCR in keratinocytes, either control transfected (mock) or transfected with anti-JAGGED1 siRNA (anti-JAG1 siRNA). Right histogram: Bars show the mean relative abundance of HEY1 mRNA at 4 and 6 h after TGF-β treatment in untransfected keratinocytes (no TF), control-transfected keratinocytes (mock), control-transfected keratinocytes pretreated with GSI (GSI), or in anti-JAGGED1 siRNA-transfected keratinocytes (anti-JAG1 siRNA).
Disassembly of E-cadherin adherens junctions, cell–cell separation, and cell motility induced by TGF-β in keratinocytes require HEY1 function
We used antisense oligonucleotides to inactivate HEY1 function. HEY1 induction by TGF-β at 1 h was significantly reduced in antisense oligotransfected, but not in sense oligotransfected, cells (Figure 4A). In confluent keratinocyte cultures transfected with control sense oligonucleotide, TGF-β treatment for 24 h induced typical features of EMT, including disassembly and disappearance of E-cadherin adherens junctions, cell–cell separation, and conversion to fibroblastoid cell morphology (Figure 4B). In contrast, transfection of HEY1 antisense oligonucleotide blocked these manifestations of TGF-β-induced EMT as monitored after 24 h (Figure 4B). In addition, transfection of HEY1 antisense, but not sense oligonucleotide, significantly reduced TGF-β-induced cell motility associated with EMT at 24 and 48 h (Figure 4C), as measured by in vitro scratch-wound assays. These results demonstrate that activation of HEY1 by TGF-β/Smad3 is required for TGF-β-induced EMT phenotype manifestations and cell motility in human keratinocytes.
Figure 4.
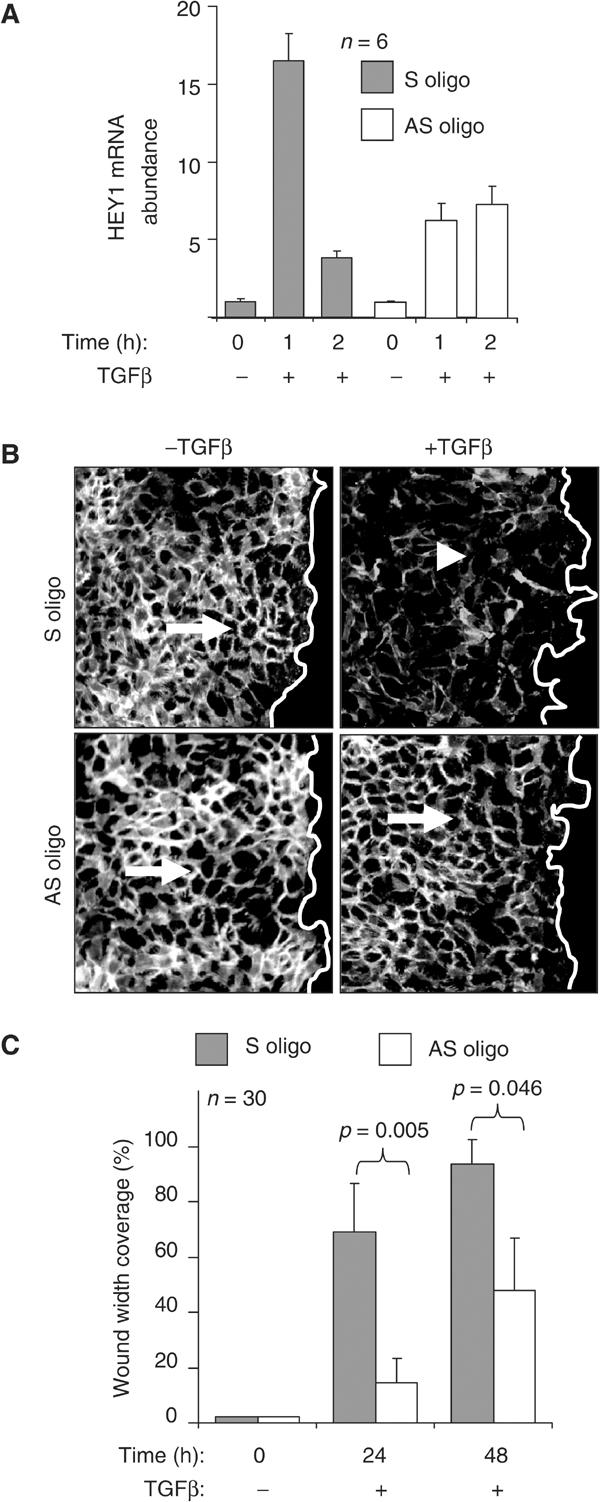
HEY1 is required for EMT and cell motility induced by TGF-β in keratinocytes. (A) Histograms show the relative abundance of HEY1 mRNA after treatment with TGF-β in keratinocytes transfected with the control S (sense) and AS (antisense) oligonucleotides, respectively. (B) In vitro scratch-wound assay of the cells transfected with S and AS oligonucleotide and stimulated by TGF-β for 24 h and immunostained for E-cadherin (white staining). The empty space right of the white line indicates the wound area. The arrow depicts E-cadherin-positive cell adherens junctions in untreated cells, and the arrowhead indicates loss of E-cadherin-positive adherens junctions in TGF-β-treated cells. (C) Histograms indicate the average coverage of scratch wound widths in % relative to baseline wound width (0 h) at 24 and 48 h after TGF-β treatment in keratinocyte cultures transfected with S (gray bars) or AS (open bars) oligonucleotides. Significance of the motility change was determined by Student's t-test. Wound width coverage at 24 and 48 h in untreated control cells was not significantly different from baseline (not shown).
TGF-β induces delayed Jagged1 synthesis to activate kinetically coordinated Notch signalling required for a second, delayed phase of HEY1 induction
We reported previously that stimulation of HaCaT keratinocytes with TGF-β indirectly activates ERK MAPK between 1 and 2 h, and that ERK function was required for delayed induction of the Notch ligand JAG1 after 2 h of TGF-β treatment (Zavadil et al, 2001). Here we show that TGF-β increased the expression of Jagged1 mRNA between 2 and 6 h in all four epithelial cell lines examined (Figure 5A). Moreover, TGF-β treatment strongly induced Jagged1 protein levels beginning at 2 h in keratinocytes. Jagged1 expression was associated with a dramatic decrease of the levels of intact 300 kDa transmembrane Notch protein (Notchtm) and accumulation of NICD in the nucleus (NICDnuc) by 4 h (Figure 5B). The reduction of transmembrane Notch protein and the accumulation of NICD in the nucleus were prevented by chemical inhibitor of γ-secretase activity, suggesting that classical Notch receptor activation was associated with increased Jagged1 synthesis induced by TGF-β. As Notch is known to activate transcription of several H/E(spl) genes directly, including Hey1 (Nakagawa et al, 2000), we examined the expression of HEY1 mRNA over an extended time period (0–8 h) of TGF-β stimulation. The immediate-early, TGF-β/Smad-dependent wave of activation (1–3 h) was followed by a second, delayed induction wave of HEY1 (5–8 h) (Figure 4C). Inhibition of γ-secretase had no effect on the immediate-early activation, but completely blocked the delayed induction of HEY1 mRNA (Figure 4C), indicating that it was mediated by Notch receptor activation. Interestingly, we observed that TGF-β induced biphasic activation profiles of additional H/E(spl) genes HEY2, HES1, and HES5, similar to HEY1 (Supplementary Data Figure S1). Immediate-early regulation of all four TGF-β-induced H/E(spl) genes was Notch independent and associated with inducible Smad3/Smad4 protein complex binding at SCR Smad complex response elements conserved in their promoters (Supplementary Data Figure S2). Similar to HEY1, a delayed activation phase of HEY2, HES1, and HES5 with peak activation between 5 and 8 h was either completely blocked (HEY2, HES5) or diminished (HES1) by GSI γ-secretase inhibitor (Supplementary Data Figure S1). In addition, the delayed Jagged1-mediated activation of Notch was associated with activation of Notch/NICD-responsive and TGF-β/Smad-unresponsive HEY1 promoter regions, as assayed by luciferase reporter assays in cells transfected with luciferase reporter constructs M3 HEY1 (human) and mLucHey (mouse) (Maier and Gessler, 2000) (Figure 5D). Together, these results indicate that delayed activation of Notch by TGF-β leads to classical NICD signalling and transcriptional activation of a broader complement of classical Notch target genes.
To examine directly whether Jagged1 was required for the delayed, Notch-dependent induction wave of HEY1, we used RNA interference to silence TGF-β-inducible Jagged1 expression. We confirmed that anti-Jagged1 short-interfering RNA (siRNA) blocked induction of Jagged1 mRNA synthesis by TGF-β (Figure 5E). The delayed increase of HEY1 mRNA observed at 6 h after TGF-β treatment in untransfected and control oligo (Mock)-transfected cells was completely blocked in cells transfected with anti-Jagged1 siRNA (Figure 5E), similar to cells pretreated with γ-secretase inhibitor (Figure 5E). Taken together, our results demonstrate that TGF-β stimulates two kinetically coordinated waves of activation of HEY1. Immediate-early activation of HEY1 by TGF-β is mediated directly by Smad3/4 binding at two distinct SCR sequences in the distal HEY1 promoter region and does not require Notch signalling. The Smad-dependent, Notch-independent immediate-early activation is followed by a second wave of HEY1 activation mediated by classical Notch-dependent signalling induced through indirect and delayed induction of Jagged1 synthesis by TGF-β.
Functional inactivation of Jagged1 or Notch inhibits disassembly of E-cadherin adherens junctions and cell–cell separation induced by TGF-β
Next, we examined the effects of Jagged1 silencing on EMT phenotypes. In cells transfected with control siRNA, disassembly of E-cadherin adherens junctions and cortical actin bundles, cell–cell separations, and stress fiber formation were induced by TGF-β within 24 h (Figure 6A). In contrast, cells transfected with anti-Jagged1 siRNA retained epithelial morphology and cells failed to separate, although E-cadherin adherens junctions appeared widened, and limited stress fiber formation was detectable (bottom row panels) when compared with untreated, control-transfected cells (upper row panels) (Figure 6A). To further delineate the timeframe for functional requirement of Jagged1-induced Notch signalling in EMT, we used timed pharmacological inhibition of γ-secretase activity (Figure 5). When γ-secretase inhibitor GSI was applied before or, up to 7 h, after treatment with TGF-β, the cells retained epithelial morphology during TGF-β exposure while some stress fiber formation was observed (Figure 6B), similar to cells transfected with anti-Jagged1 siRNA (Figure 6A). In contrast, addition of the inhibitor at 8 h or later time points failed to prevent TGF-β-induced EMT (not shown). Thus, the timeframe for functional requirement of Notch signalling in TGF-β-induced EMT phenotypes coincided with the delayed phase of Jagged1- and γ-secretase-dependent synthesis of HEY1. These results demonstrate that the TGF-β-induced Jagged1-dependent activation of Notch signalling is required for disassembly of E-cadherin adherens junctions and cortical actin bundles, and for cell–cell separation induced by TGF-β in keratinocytes.
Figure 6.
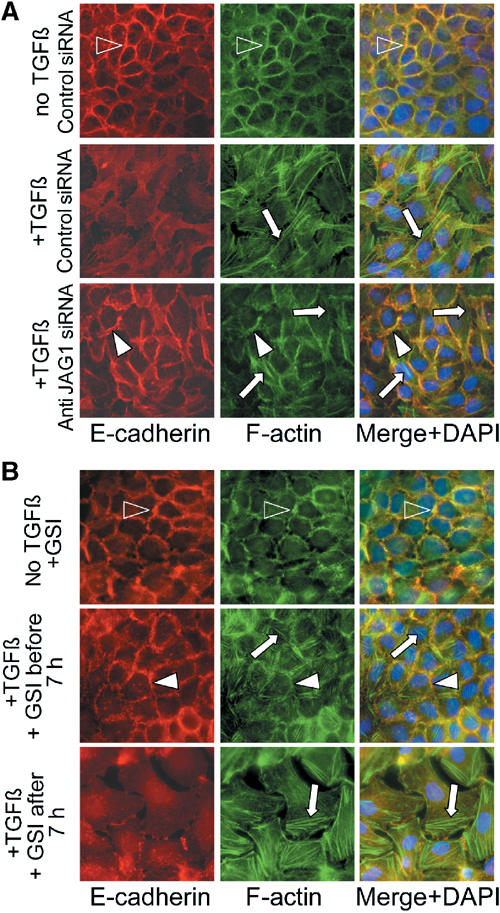
Jagged1 and Notch activation is required for TGF-β-induced EMT. (A) Immunofluorescence labelling for E-cadherin (red), F-actin (green), or both merged with nuclear DAPI staining, as indicated, of control-transfected, untreated (no TGF-β, control siRNA), or TGF-β-treated (+TGF-β, control siRNA) keratinocytes, and keratinocytes transfected with anti-JAGGED1 siRNA and treated with TGF-β (+TGF-β, anti-JAG1 siRNA). Immunostaining assays were performed 24 h after TGF-β induction. Open arrowheads show E-cadherin-positive adherens junctions and cortical actin bundles, respectively. White arrowheads depict widened E-cadherin-positive adherens junctions. Arrows denote actin stress fibers. (B) Experiments shown as in (A) in untransfected keratinocytes treated with TGF-β in the absence or presence of γ-secretase inhibitor (GSI) added either within 0–7 h of TGF-β treatment (+GSI before 7 h) or after 7 h (+GSI after 7 h).
Smad3 is required for induction of Snail family repressors, Hey1, and Jagged1 expression by TGF-β, and is essential for TGF-β-induced EMT phenotypes in primary mouse kidney tubular epithelial cells
To examine the role of Smad proteins in TGF-β signalling in EMT, we established primary tubular epithelial cell cultures (mTEC) from mice with homozygous deletion of Smad3 (Smad3Δex8/Δex8) and wild-type control littermates (Yang et al, 1999). Quantitative real-time PCR (RT-PCR) analysis confirmed that Smad3 mRNA was present in wild-type (mTEC WT) control cells, but not detectable in Smad3 knockout (mTEC S3KO) (Figure 7A). Please note that mTEC S3KO appeared on average larger than mTEC WT. TGF-β strongly induced a biphasic pattern of Hey1 mRNA expression in mTEC WT, including an immediate-early induction (1–3 h) and a delayed induction (5–7 h) (Figure 7B). In contrast, both phases of Hey1 induction were dramatically reduced in mTEC S3KO (Figure 7B). Compared with a progressive and sustained increase of Jagged1 mRNA in mTEC WT after 2 h of TGF-β stimulation, induction of Jagged1 mRNA levels was significantly lower and not sustained in mTEC S3KO (Figure 7C). Next, we analyzed patterns of E-cadherin and actin cytoskeleton in mTEC WT and mTEC S3KO in response to TGF-β. E-cadherin and cortical actin patterns were characteristic of epithelial cell phenotype in untreated mTEC WT and mTEC S3KO cultures (Figure 7D). TGF-β treatment induced disassembly of E-cadherin adherens junctions and cortical actin bundles, cell separation, and actin stress fiber formation in mTEC WT (Figure 7D). In contrast, E-cadherin cell junctions and cortical actin remained intact but appeared wider and cells did not separate in TGF-β-treated mTEC S3KO cultures, while some stress formation was observed (Figure 7D). Together, these findings suggest that Smad3 is required for biphasic induction of HEY1 expression, for progressive increase of Jagged1 expression, and for manifestation of EMT phenotypes induced by TGF-β in primary mouse tubular epithelial cells.
Figure 7.
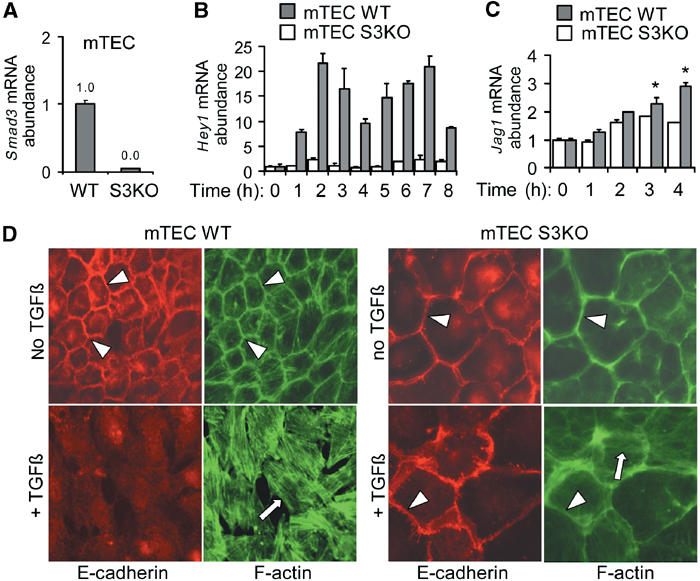
Smad3 is required for induction of Hey1 and Jagged1, and for EMT, in primary mouse kidney tubular epithelial cells (mTEC). (A) Quantitative RT-PCR analysis of Smad3 in wild-type (mTEC WT) and Smad3-deficient (mTEC S3KO) primary tubular epithelial cells of the mouse kidney. (B, C) Quantitative RT-PCR analysis of Hey1 and Jagged1 inductions by TGF-β in wild-type (mTEC WT) and Smad3-deficient (mTEC S3KO) primary mTEC cultures. (D) Immunofluorescence labelling for E-cadherin (red), and phalloidin labelling for F-actin (green) in wild-type (mTEC WT) and Smad3-deficient (mTEC S3KO) primary, left untreated or treated with TGF-β for 24 h. Arrowheads indicate E-cadherin adherens junctions and cortical actin, respectively. Arrows depict actin stress fibers.
To characterize further the role of Smad3 in EMT induced by TGF-β, we examined responsiveness of previously identified TGF-β-regulated genes with functional roles in EMT and repression of E-cadherin, including Slug (Romano and Runyan, 2000), Snail (Peinado et al, 2003), and Sip1 (Comijn et al, 2001). Strong, immediate-early gene inductions by TGF-β were observed for Hey1, Snail, and Sip1 in mTEC WT, but not in mTEC S3KO (Figure 8). Induction of Slug was more modest and persistent in mTEC WT, but also absent in mTEC S3KO (Figure 8). Interestingly, TGF-β strongly induced HEY1 and SLUG, but not SNAIL and SIP1, in human HaCaT keratinocytes; however, in HK-2 tubular epithelial cells, TGF-β induced HEY1 and SNAIL, but not SLUG and SIP1, expression. These findings indicate that TGF-β may induce the bHLH transcriptional repressor HEY1 in a broad spectrum of epithelial cell types, while regulation of zinc-finger transcriptional repressors Snail, Slug, and Sip1 may be more restricted, perhaps depending on the tissue origin of the epithelial cell type.
Figure 8.
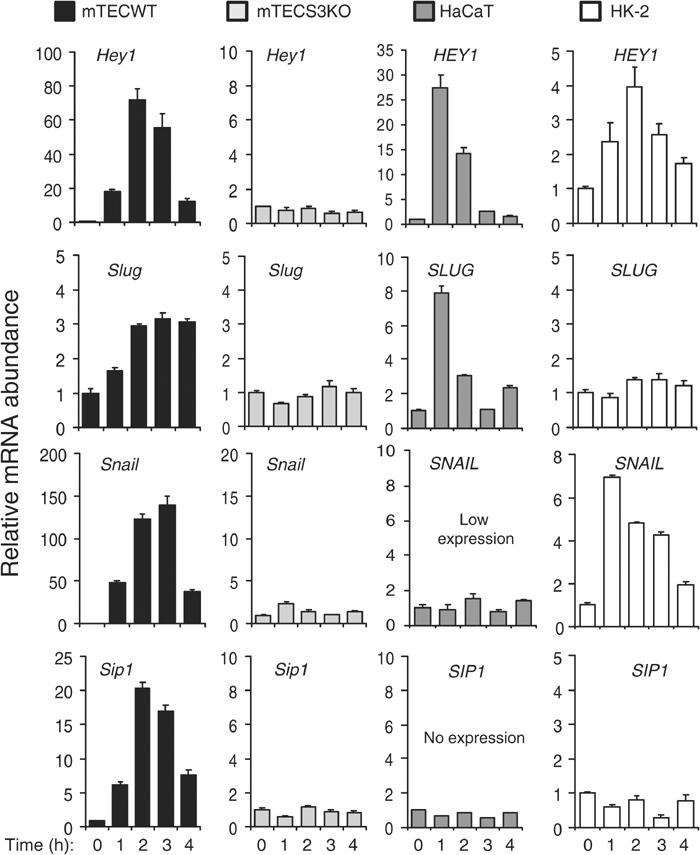
Histograms demonstrate the effects of TGF-β treatments, as indicated, on the relative abundance of Hey1/HEY1, Slug/SLUG, Snail/SNAIL, and Sip1/SIP1 mRNAs, respectively, in primary mouse kidney tubular epithelial cultures derived from wild-type control (mTEC WT) and Smad3 knockout (mTEC S3KO) mice, and in human keratinocytes (HaCaT) and human kidney tubular epithelial cells (HK-2). Graphs show representative results from triplicate experiments.
Discussion
We report a new mechanism of functional integration between TGF-β and Notch signalling leading to coordinated, biphasic activation of bHLH transcriptional repressor Hey1 as an essential mediator for initiation of EMT by TGF-β. First, TGF-β directly activates HEY1 transcription through inducible interaction of Smad3/Smad4 transcriptional activators with two distinct SCRs. Immediate-early activation of HEY1 is observed across a spectrum of epithelial cells known to undergo EMT in response to TGF-β, requires Smad3, and is independent of Notch signalling. Second, a delayed wave of HEY1 activation between 4 and 7 h of TGF-β stimulation is distinct from the immediate-early wave and is also observed in all epithelial cells examined. It is mediated indirectly through delayed synthesis of Jagged1, activating classical Notch signalling. The delayed and sustained activation of Jagged1 by TGF-β requires ERK MAPK, as previously shown (Zavadil et al, 2001), and is significantly reduced in Smad3-deficient tubular epithelial cells. Thus, Smad3 and ERK MAPK may cooperate to integrate the Jagged1/Notch module as part of the TGF-β signalling network. Third, selective inactivations of either Smad3, Hey1, Jagged1, or Notch1, respectively, consistently prevent disassembly of E-cadherin adherens junctions and of cortical actin bundles, and cell–cell separation, but appear to permit some widening of cell–cell contacts and limited stress fiber formation in keratinocytes and kidney tubular epithelial cells treated with TGF-β. These findings are consistent with our previous gene expression profiling study where inhibition of ERK MAPK prevented activation of genes with roles in cell adhesion and motility, but had little effect on genes involved in actin cytoskeleton remodelling (Zavadil et al, 2001). Finally, in primary kidney tubular epithelial cells, Smad3 is not only required for immediate-early gene induction of the bHLH gene Hey1, but also of zinc-finger transcriptional repressors (Slug, Snail, Sip1) with known important roles in disassembly of various cell–cell contacts and/or repression of E-cadherin by TGF-β.
Roles of Smad-dependent signalling mechanisms in TGF-β-induced EMT have been examined with conflicting results by overexpressing Smads (Piek et al, 1999), or dominant-negative mutant Smads (Bhowmick et al, 2001). Our findings presented here, and recent reports by others (Yu et al, 2002; Itoh et al, 2003), strongly support an important role for Smads. For example, TGF-β type I receptor (TbRI) mutants with intact kinase function sufficient to activate MAPK pathways, but inability to bind and activate Smads due to mutations in the critical L45 loop, were not sufficient to induce stress fiber formation and/or EMT (Yu et al, 2002; Itoh et al, 2003). Our own results demonstrate that Smad3 function is at a minimum required for key aspects of EMT, including disassembly of E-cadherin adherens junctions and separation of cells induced by TGF-β. This conclusion is consistent with our finding that Smad3 is essential for immediate-early regulation of Snail family transcriptional repressors known to be required for downregulation of E-cadherin and disassembly of various cell–cell junctions. However, TGF-β was able to induce a widening of cell–cell contacts and cortical actin bundles, and to stimulate a limited degree of stress fiber formation in Smad3-deficient tubular epithelial cells, indicating that either Smad2- and/or Smad-independent pathways may be sufficient to mediate these aspects of EMT, which remain to be defined at a molecular level. For example, RhoA and its target p160ROCK are rapidly activated by TGF-β: RhoA has been shown to be necessary, but not sufficient for cell contact disassembly, while p160ROCK may signal specifically to activate programs leading to formation of stress fibers (Bhowmick et al, 2001). Activation of p38 MAPK appears at least to some extent Smad independent (Yu et al, 2002; Itoh et al, 2003) and contributes important signals to EMT, stress fiber formation, and cell migration induced by TGF-β in NMuMG (Bakin et al, 2002; Yu et al, 2002). We (Zavadil et al, 2001) and others (Peinado et al, 2003) have shown that delayed, indirect activation of ERK MAPK by TGF-β provides important signals for cell migration and turnover of cell–matrix adhesion complexes. However, in contrast with Smad-independent p38 activation, the relationship and hierarchy of Smad and ERK MAPK activation is more complicated and will require further study. Since the outcomes of Jagged1/Notch inhibition overlap largely with the effects of Smad3 deficiency and inactivation of Hey1 in our study, and include the effects of ERK inhibition previously shown (Zavadil et al, 2001), we conclude that Smad3/ERK-dependent coactivation of Jagged1/Notch constitutes an important module of the increasingly complex and differentiated network of signalling pathways that cooperate to mediate the full spectrum of EMT phenotypes induced by TGF-β in epithelial cells.
Our model entails a number of new observations with implications for several active fields of research. First, we identify the H/E(spl) gene Hey1 as a new immediate-early target of TGF-β and define molecular elements of the transcriptional activation of Hey1 by Smad3/Smad4. Interestingly, activation of Notch signalling targets by TGF-β/Smad may be conserved across species as indicated by recent observations on Drosophila organogenesis, which identified decapentaplegic (Dpp) and Mothers-against-decapentaplegic (Mad) (respective fly orthologs of TGF-β/BMP and Smads) as positive regulators of hairy expression in the developing eye and leg sensory organ patterning (Greenwood and Struhl, 1999; Hays et al, 1999). Furthermore, gene knockout experiments in mice indicate that Tgfb2 and Hey2 may be components of a functional pathway directing EMT in endothelial cushion remodelling during cardiogenesis (Camenisch et al, 2002; Donovan et al, 2002; Sakata et al, 2002). In both vertebrates and invertebrates, the members of the H/E(spl) subfamily of bHLH proteins restrict the differentiation of precursor cells in a variety of tissues (Davis and Turner, 2001). Therefore, direct induction of Hey1 by TGF-β/Smad provides a new potential mechanism for TGF-β to regulate and/or switch cellular differentiation programs. In addition, the expression patterns of H/E(spl) genes observed in the developing nervous system, muscle, lymphocyte, blood vessels, and presomitic mesoderm may now be explained by TGF-β/Smad signalling in addition to, or instead of, Notch signalling.
Finally, functional integration of TGF-β/Smad and Notch pathways mediating biphasic activation of Hey1, and likely other yet unknown target genes, may have implications for the disease pathogenesis of multiple sclerosis and injury of kidney tubular epithelium (John et al, 2002; Morrissey et al, 2002). Recent reports indicate that Notch1 may exert a dual role in skin carcinogenesis, similar to TGF-β. While Tgfb1 and Notch1 may function as tumor suppressors in skin by maintaining epidermal homeostasis (Cui et al, 1996; Nicolas et al, 2003), both may also exert overlapping oncogenic activities in skin cancer progression that is characterized by EMT (Cui et al, 1996; Weijzen et al, 2002). In light of these findings, and our observations reported here, in vivo co-analyses of TGF-β/Smad, Jagged/Notch, and H/E(spl) transcriptional repressors in chronic degenerative and malignant diseases seem warranted. Further molecular investigations will be required to differentiate target genes of TGF-β/Smad-induced immediate-early activation of Hey1, and of the Jagged1/Notch pathway in epithelial cell plasticity.
Materials and methods
Cell culture
HaCaT keratinocytes, NMuMG normal mouse mammary gland epithelium, HK-2 human kidney proximal tubule epithelium, and MDCK canine kidney distal tubule epithelium were expanded to 90% confluency followed by serum deprivation in 0.2% FBS for 24 or 48 h prior to stimulation with 10 pM rhTGF-β1 (R&D Systems) in the absence or presence of 5 μM γ-secretase inhibitor X or L-685, 458 (Calbiochem) added 15 min before TGF-β.
Primary cell culture of mouse tubular epithelium
Kidneys were collected from 8- to 12-week-old C57BL/6 wild-type and C57BL/6 Smad3 knockout (Smad3ex8/ex8) mice (Yang et al, 1999). Kidneys were minced and passed through serial sieves (180, 106, 75 μM) in PBS. Tubular fragments were digested for 30 min with 1 mg/ml of collagenase I (Worthington Biochemical) at 37°C in an incubator shaker. Cells were seeded in collagen I-coated plates maintained in culture at 37°C with 5% CO2 in DMEM/F12 medium (Gibco, cat# 11320-033) containing 5% fetal calf serum, penicillin/streptomycin antibiotic, the ITS supplement of 0.5% insulin, transferrin, selenium (Sigma-Aldrich), and 5 nM dexamethasone. Cells were analyzed at passages one to three.
Transfections and gene silencing
Human HEY1 promoter fragment (full-length FL, 3.9 kb) isolated from human BAC clone RP11-27N21 was subcloned into pGL3 luciferase construct (Promega). Deletion mutants M1–M7 were generated by sequential restriction digests of FL. The NICD expression construct was a kind gift of Dr R Kopan. Dominant-negative clones of TbRII (TbRII-DN K277R), Smad2 (Smad2 3S → A, serine residues of SSXS motif changed to alanine), Smad3 (Smad3 Δexon8), and Smad4 (Smad4 ΔM4) were provided by Dr E Piek, and the 3TPlux construct was a gift of Dr J Wrana. Antisense oligonucleotides against human HEY1 were designed as previously described (Henderson et al, 2001). Transfections were carried out in triplicate for using Superfect reagent (Qiagen) and 3 μg of total plasmid DNA per 4 × 105 cells seeded. TGF-β-induced versus uninduced (baseline control) triplicate cultures of identical transfection background were compared in all transfection experiments. The target sequence of the JAG1 siRNA duplex (Dharmacon Research) was AAGGUGUGUGGGGCCUCGGGU. High-efficiency (>90% cells) oligonucleotide and siRNA transfections were carried out in growing cells at 50–60% confluence using the recommended protocol for Oligofectamine (Life Technologies, Invitrogen). In all, 4 μM anti-HEY1 oligonucleotide (AS) or 200 nM anti-JAG1 siRNA concentrations were used. The cells were recovered post-transfection by overnight culture in 10% serum, then serum deprived for 24 h (0.2% serum) and stimulated with TGF-β. The efficiency of transfection in RNAi experiments was monitored by using Cy3-labelled siRNA duplex of GL2 luciferase sequence (Dharmacon Research).
Quantitative RT-PCR
Total RNA was prepared using TRIZOL reagent, and reverse transcribed with SuperScript II reverse transcriptase (Life Technologies, Invitrogen). The cDNA was amplified using SYBR-Green PCR Master Mix (Applied Biosystems) and gene-specific exon–exon junction spanning primers (sequences available upon request) in ABI 7900HT sequence detection system (Applied Biosystems) or iCycler (BioRad). Normalizations across samples were performed using geometric average of constitutive gene expression of GLUD1, HPRT1, and B2M.
Protein extraction
Whole-cell protein extracts from keratinocyte cultures were prepared as described (Zavadil et al, 2001). For cytoplasmic/nuclear cell fractionation, the cells were lysed in 300 μl of sucrose buffer per 100 mm dish (0.32 M sucrose, 10 mM Tris–HCl pH 8.0, 3 mM CaCl2, 2 mM magnesium acetate, 0.1 mM EDTA, 0.5% NP-40), and cytoplasmic fraction was cleared by centrifugation (2500 rpm, 5 min). The nuclei were washed twice with sucrose buffer and resuspended in 40 μl of low-salt buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 20 mM KCl, 0.2 mM EDTA, 25% glycerol, 1% NP-4 (v/v)) and mixed with 40 μl of high-salt buffer (low-salt buffer with 800 mM KCl) and incubated for 40 min at 4°C. Nuclear protein extracts were cleared by centrifugation (13 000 rpm, 20 min).
Quantitative chromatin immunoprecipitation
The formaldehyde-crosslinked protein–DNA complexes in the cell protein lysates were sonicated to the size range of 30–500 bp using Fisher Sonic Dismembrator 500 with a Branson 450 microtip, submerged in the mixture of dry ice in methanol. Protein–DNA complexes were precleared and immunoprecipitated using protein A/G agarose beads (Santa Cruz) and specific antibodies or isotype controls, reverse crosslinked by proteinase K treatment and overnight incubation at 65°C. The enrichment of specific promoter DNA contents was measured by quantitative RT-PCR (primer sequences available upon request) and adjusted relative to background contents of unrelated promoter sequences, to immunoprecipitation by isotype IgG controls and to the relative contents of target sequences in the initial sonicated chromatin input.
Electrophoretic mobility shift assay
In all, 2.5 pmol of 32P-labelled oligonucleotides were incubated with 15 μg of nuclear proteins in binding buffer (4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, 10 mM Tris–HCl pH 7.5, 0.1 mg/ml dI-dC) for 30 min at room temperature. For supershift analysis, the nuclear proteins were preincubated for 2 h at 4°C with 3 μg of anti-Smad3 antibody (Santa Cruz, sc-6202X) or 3 μg of nonrelated goat IgG. Protein–DNA complexes were resolved on 5% polyacrylamide gel in Tris-borate buffer.
Immunodetection of proteins
Immunohistochemical and immunoblot analyses were performed as described (Zavadil et al, 2001), using anti-poly (ADP-ribose) polymerase (anti-PARP) (Enzyme systems, C2.10), anti-β tubulin (Sigma, T-4026), anti-Notch1 (Santa Cruz, sc-6014), anti-HEY1 (Santa Cruz, sc-16424), anti-Jagged1 (Santa Cruz, sc-6011), anti-Smad2 (Santa Cruz, sc-6200), and anti-Smad3 (Santa Cruz, sc-6202X and ZYMED, 51-1500). Phosphospecific anti-Smad2 serum was kindly provided by Dr P ten Dijke. The Q-ChIP immunoprecipitations were performed using anti-Smad3 (FL-425, Santa Cruz), anti-Smad2/3 (Upstate Biotechnology), anti-Smad4 (C-20, Santa Cruz), and anti-Smad4 (Upstate Biotechnology).
Cell motility
Confluent serum-deprived monolayer cultures of HaCaT keratinocytes transfected with AS or S oligonucleotides were ‘wounded' by scratching the layer using 200 μl pipet tip in the presence or absence of TGF-β and monitored by phase contrast microscope photography as described (Cano et al, 2000). Motility of the cells was quantified by 30 measurements in each experimental category (AS versus S) and expressed as a percentage of distance coverage by cells moving into the scratch wound area 24 and 48 h after wounding. Student's t-test was applied to assess the significance of the AS versus S effect on cell motility.
Supplementary Material
Supplementary Data Figure S1
Supplementary Data Figure S2
Acknowledgments
We are grateful to Drs R Kopan, M Gessler, E Piek, CC Hughes, and J Wrana for kindly providing us with gene expression plasmids and reporter constructs. We thank Dr T Stopka for expert assistance with ChIP assays. We thank the Albert Einstein Biotechnology Center for support with quantitative gene expression studies. JZ is a Fellow of the AACR-Sidney Kimmel Foundation for Cancer Research. This work was supported by NIH grants RO1DK56077 and RO1DK60043 to EPB.
References
- Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL (2002) p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci 115: 3193–3206 [DOI] [PubMed] [Google Scholar]
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL (2000) Phosphatidylinositol-3 kinase function is required for TGFbeta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem 275: 36803–36810 [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL (2001) Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell 12: 27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger-De Groot AC, Mcdonald JA, Klewer SE (2002) Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol 248: 170–181 [DOI] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, Del Barrio MG, Portillo F, Nieto MA (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2: 76–83 [DOI] [PubMed] [Google Scholar]
- Comijn J, Berx G, Vermassen P, Verschueren K, Van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, Van Roy F (2001) The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7: 1267–1278 [DOI] [PubMed] [Google Scholar]
- Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, Akhurst RJ (1996) TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 86: 531–542 [DOI] [PubMed] [Google Scholar]
- Davis RL, Turner DL (2001) Vertebrate hairy and enhancer of split related proteins: transcriptional repressors regulating cellular differentiation and embryonic patterning. Oncogene 20: 8342–8357 [DOI] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, Ten Dijke P, Huet S, Gauthier JM (1998) Direct binding of Smad3 and Smad4 to critical TGFb-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 17: 3091–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang Y, Feng XH (1998) Smads: transcriptional activators of TGF-beta responses. Cell 95: 737–740 [DOI] [PubMed] [Google Scholar]
- Donovan J, Kordylewska A, Jan YN, Utset MF (2002) Tetralogy of fallot and other congenital heart defects in Hey2 mutant mice. Curr Biol 12: 1605–1610 [DOI] [PubMed] [Google Scholar]
- Greenwood S, Struhl G (1999) Progression of the morphogenetic furrow in the Drosophila eye: the roles of Hedgehog, Decapentaplegic and the Raf pathway. Development 126: 5795–5808 [DOI] [PubMed] [Google Scholar]
- Hay ED (1995) An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 154: 8–20 [DOI] [PubMed] [Google Scholar]
- Hays R, Buchanan KT, Neff C, Orenic TV (1999) Patterning of Drosophila leg sensory organs through combinatorial signaling by hedgehog, decapentaplegic and wingless. Development 126: 2891–2899 [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, Ten Dijke P (1997) TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390: 465–471 [DOI] [PubMed] [Google Scholar]
- Henderson AM, Wang SJ, Taylor AC, Aitkenhead M, Hughes CC (2001) The basic helix–loop–helix transcription factor HESR1 regulates endothelial cell tube formation. J Biol Chem 276: 6169–6176 [DOI] [PubMed] [Google Scholar]
- Hoodless PA, Wrana JL (1998) Mechanism and function of signaling by the TGF beta superfamily. [Review] [160 Refs]. Curr Top Microbiol Immunol 228: 235–272 [DOI] [PubMed] [Google Scholar]
- Itoh S, Thorikay M, Kowanetz M, Moustakas A, Itoh F, Heldin CH, Ten Dijke P (2003) Elucidation of Smad requirement in transforming growth factor-beta Type I receptor-induced responses. J Biol Chem 278: 3751–3761 [DOI] [PubMed] [Google Scholar]
- Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, Grunert S (2002) Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol 156: 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GR, Shankar SL, Shafit-Zagardo B, Massimi A, Lee SC, Raine CS, Brosnan CF (2002) Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med 8: 1115–1121 [DOI] [PubMed] [Google Scholar]
- Maier MM, Gessler M (2000) Comparative analysis of the human and mouse Hey1 promoter: Hey genes are new Notch target genes. Biochem Biophys Res Commun 275: 652–660 [DOI] [PubMed] [Google Scholar]
- Massague J (2000) How cells read TGF-beta signals. Nat Rev Mol Cell Biol 1: 169–178 [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Ebner R, Lopez AR, Derynck R (1994) TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol 127: 2021–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey J, Guo G, Moridaira K, Fitzgerald M, Mccracken R, Tolley T, Klahr S (2002) Transforming growth factor-beta induces renal epithelial Jagged-1 expression in fibrotic disease. J Am Soc Nephrol 13: 1499–1508 [DOI] [PubMed] [Google Scholar]
- Mumm JS, Kopan R (2000) Notch signaling: from the outside in. Dev Biol 228: 151–165 [DOI] [PubMed] [Google Scholar]
- Nakagawa O, McFadden DG, Nakagawa M, Yanagisawa H, Hu T, Srivastava D, Olson EN (2000) Members of the HRT family of basic helix–loop–helix proteins act as transcriptional repressors downstream of Notch signaling. Proc Natl Acad Sci USA 97: 13655–13660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, Van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F (2003) Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 33: 416–421 [DOI] [PubMed] [Google Scholar]
- Nieto MA (2002) The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol 3: 155–166 [DOI] [PubMed] [Google Scholar]
- Peinado H, Quintanilla M, Cano A (2003) Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem 278: 21113–21123 [DOI] [PubMed] [Google Scholar]
- Piek E, Moustakas A, Kurisaki A, Heldin CH, Ten Dijke P (1999) TGF-(beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci 112 (Part 24): 4557–4568 [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB (1990) The transforming growth factors-β. In Handbook of Experimental Pharmacology. Peptide Growth Factors and their Receptors, Sporn MB, Roberts AB (eds) pp 419–472. Berlin, Heidelberg, New York, London, Paris, Tokyo, Hong Kong: Springer-Verlag [Google Scholar]
- Romano LA, Runyan RB (2000) Slug is an essential target of TGFbeta2 signaling in the developing chicken heart. Dev Biol 223: 91–102 [DOI] [PubMed] [Google Scholar]
- Sakata Y, Kamei CN, Nakagami H, Bronson R, Liao JK, Chin MT (2002) Ventricular septal defect and cardiomyopathy in mice lacking the transcription factor CHF1/Hey2. Proc Natl Acad Sci USA 99: 16197–16202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-De Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T (1997) TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124: 2659–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Vanderburg CR, Odierna GS, Hay ED (1998) TGFbeta3 promotes transformation of chicken palate medial edge epithelium to mesenchyme in vitro. Development 125: 95–105 [DOI] [PubMed] [Google Scholar]
- Thiery JP (2002) Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2: 442–454 [DOI] [PubMed] [Google Scholar]
- Von Gersdorff G, Susztak K, Rezvani F, Bitzer M, Liang D, Böttinger EP (2000) Smad3 And Smad4 mediate transcriptional activation of the human Smad7 promoter by transforming growth factor beta. J Biol Chem 275: 11320–11326 [DOI] [PubMed] [Google Scholar]
- Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, Osborne BA, Gottipati S, Aster JC, Hahn WC, Rudolf M, Siziopikou K, Kast WM, Miele L (2002) Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med 8: 979–986 [DOI] [PubMed] [Google Scholar]
- Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C (1999) Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T Cell responsiveness to TGF-beta. EMBO J 18: 1280–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YC, Piek E, Zavadil J, Liang D, Xie D, Heyer J, Pavlidis P, Kucherlapati R, Roberts AB, Böttinger EP (2003) Hierarchical model of gene regulation by transforming growth factor beta. Proc Natl Acad Sci USA 100: 10269–10274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Hebert MC, Zhang YE (2002) TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J 21: 3749–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, Piek E, Böttinger EP (2001) Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci USA 98: 6686–6691 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data Figure S1
Supplementary Data Figure S2