

Abstract
Hydrocarbon-degrading bacterial communities from freshwater, marine, and hypersaline Brazilian aquatic ecosystems (with water salinities corresponding to 0.2%, 4%, and 5%, respectively) were enriched with different hydrocarbons (heptadecane, naphthalene, or crude oil). Changes within the different microcosms of bacterial communities were analyzed using cultivation approaches and molecular methods (DNA and RNA extraction, followed by genetic fingerprinting and analyses of clone libraries based on the 16S rRNA-coding gene). A redundancy analysis (RDA) of the genetic fingerprint data and a principal component analysis (PCA) of the clone libraries revealed hydrocarbon-enriched bacterial communities specific for each ecosystem studied. However, within the same ecosystem, different bacterial communities were selected according to the petroleum hydrocarbon used. In general, the results demonstrated that Acinetobacter and Cloacibacterium were the dominant genera in freshwater microcosms; the Oceanospirillales order and the Marinobacter, Pseudomonas, and Cycloclasticus genera predominated in marine microcosms; and the Oceanospirillales order and the Marinobacter genus were selected in the different hydrocarbon-containing microcosms in hypersaline water. Determination of total petroleum hydrocarbons (TPHs) in all microcosms after 32 days of incubation showed a decrease in the hydrocarbon concentration compared to that for the controls. A total of 50 (41.3%) isolates from the different hydrocarbon-contaminated microcosms were associated with the dominant operational taxonomic units (OTUs) obtained from the clone libraries, and their growth in the hydrocarbon contaminating the microcosm from which they were isolated as the sole carbon source was observed. These data provide insight into the general response of bacterial communities from freshwater, marine, and hypersaline aquatic ecosystems to petroleum hydrocarbon contamination.
INTRODUCTION
The extraction of crude oil from oil reservoirs and all activities associated with petroleum production, including crude oil transportation and the storage and use of petroleum-derived fuels, are potential sources of environmental contamination (1). Moreover, increased oil extraction activities at offshore platforms contribute to the increased susceptibility of marine environments to crude oil contamination (2). Crude oil is a complex mixture primarily composed of aliphatic and aromatic hydrocarbons, which affect indigenous marine life due to their toxic and carcinogenic effects (3).
Although hundreds of microbial species utilize petroleum-derived hydrocarbons as carbon and energy sources, these species generally comprise only 1 to 10% of the expected biodiversity of a native biome (4). These oil-degrading microorganisms are generally more easily detected in hydrocarbon-contaminated environments (2). Studies performed after the marine BP Deepwater Horizon oil spill in 2010 in the Gulf of Mexico in the United States showed that the oil leak resulted in the enrichment of bacteria belonging to the Oceanospirillales order and Colwellia and Cycloclasticus genera (5–7). In addition, members of the Oceanospirillales were detected as the major constituents of the bacterial community in different studies on oil-polluted marine ecosystems (8, 9). However, microbial succession in petroleum-contaminated environments depends on the hydrocarbon fractions available to the microbial community, as suggested previously by McKew et al. (10). Studies in marine environments have described the predominance of bacteria belonging to the Alcanivorax species (Oceanospirillales order) as the primary aliphatic hydrocarbon degraders (10, 11) and those belonging to the Cycloclasticus species as the primary polycyclic aromatic hydrocarbon (PAH) degraders (12, 13).
Although many studies have shown the significant impact of petroleum hydrocarbons on marine microbial structure, studies on the effects of hydrocarbon contaminations on freshwater and hypersaline ecosystems are underrepresented. Bacterial hydrocarbon degradation in hypersaline environments has been considered a difficult process because the solubility of hydrocarbons and oxygen is reduced when salinity increases (14). To date, members of the Oceanospirillales order and Marinobacter and Martelella genera (14–18) have been detected in petroleum hydrocarbon-contaminated hypersaline waters. Hydrocarbon-degrading bacteria have already been identified in freshwater (19), but the precise functions of bacterial strains in this environment remain unknown.
Brazil is one of the largest petroleum-producing countries. The process of petroleum extraction in offshore platforms on the coast of the Brazilian state of Rio de Janeiro severely impacts the Brazilian coastline. Nevertheless, to our knowledge, studies associated with hydrocarbon-degrading bacterial communities have been performed only in sediments of mangrove ecosystems on the Brazilian coastline (20, 21). Therefore, to provide further insight into the presence and activity of oil-degrading bacterial communities, we collected water samples from three different Brazilian aquatic ecosystems on the coast of Rio de Janeiro. Different experimental enrichments with petroleum hydrocarbons were generated; heptadecane and naphthalene were used as models for the aliphatic and aromatic fractions of petroleum hydrocarbons, respectively, and crude oil was used as a complex mixture of hydrocarbons.
Throughout the experimental procedure, the structure-function relationships of bacterial communities were determined on the basis of DNA and RNA analyses using genetic fingerprinting and clone library techniques. In addition, using culture-based methods, bacterial strains were obtained from the different microcosms. These methods enabled us to describe and isolate hydrocarbon-enriched bacteria from different aquatic ecosystems and to highlight the differences in bacterial responses to petroleum-derived hydrocarbon contamination based on the type of hydrocarbon and the aquatic environment (freshwater, marine, and hypersaline) in which the bacteria were found.
MATERIALS AND METHODS
Sample sites.
The water samples used in this study were collected from three different ecosystems in Massambaba's Environmental Protection Area in Saquarema, Rio de Janeiro, Brazil. Jacarepiá Lagoon (22°91′66″S, 42°42′40″W), Vermelha Lagoon (22°93′30″S, 42°39′28″W), and Massambaba Beach (22°93′61″S, 42°39′90″W) represented freshwater (nonsaline, 0.2% salinity), hypersaline (5% salinity), and marine (saline, 4% salinity) aquatic ecosystems, respectively (see Fig. S1 in the supplemental material). The characteristics of these aquatic ecosystems are presented in Table S1 in the supplemental material. This area is a typical tropical environment with semiarid features (22). To our knowledge, these ecosystems have never been affected by crude oil contamination. Three samples of approximately 500 ml of water (depth, 10 to 50 cm) were collected on 24 January 2011 from each ecosystem (n = 3) at a maximum distance of 100 m between each sampling point and 5 to 10 m from the lagoon margins and/or from the beach sand. The samples were immediately transported to the laboratory in the dark to perform the experiments described below.
Determination of physicochemical parameters.
The water temperature was measured in situ using a field thermometer. The protocols described in Standard Methods for the Examination of Water and Wastewater (SMEWW) of the American Public Health Association (23) were used to determine the pH (electrometric method; SMEWW protocol 4500 H+ B) and water salinity (density method; SMEWW protocol 2520 C).
Experimental microcosms.
The microcosms were established in triplicate, as follows: each water sample (25 ml; freshwater and marine and hypersaline water) was enriched with 1% of the contaminant (corresponding to 280, 60, and 100 μg ml−1 crude oil, heptadecane, and naphthalene, respectively) in 50-ml glass flasks covered with aluminum foil. The contaminants used were heptadecane (a model of aliphatic hydrocarbons; Sigma-Aldrich, St. Louis, MO), naphthalene (a model of aromatic hydrocarbons; Sigma-Aldrich), and crude oil (Baiano oil, a light crude oil with low viscosity [American Petroleum Institute gravity, greater than 30] and a complex mixture of different aliphatic and aromatic hydrocarbons). On the basis of contaminant input, the C/N/K/P proportion was corrected to 100:10:1:1 to facilitate bacterial growth, as described by Niepceron et al. (13). Two controls were used: (i) different water samples with the addition of C, N, K, and P but without any contaminants and (ii) water samples amended with C, N, K, and P, sterilized by autoclaving (121°C, 15 min), and subsequently supplemented with each contaminant. All microcosms were incubated in the dark for up to 32 days at 28°C.
TPH analysis.
Measurement of total petroleum hydrocarbons (TPHs) in the contaminated microcosms (those corresponding to the 32nd day of incubation) and in the controls (autoclaved water samples contaminated with the different hydrocarbons and maintained under the same conditions as the contaminated microcosms) was performed by gas chromatography (GC) to quantify the total concentration of the hydrocarbons extracted using a modified EPA 8015 technique (24). The TPH values obtained for the contaminated microcosms were statistically compared to those obtained for the controls using the Student t test.
DNA and RNA extraction for the analysis of bacterial communities.
Bacterial communities were analyzed using molecular methods before and at 4, 12, and 32 days after initiating the experiments. Microbial DNA was extracted at all sampling points, and the RNA was extracted at 12 and 32 days after the experiments were initiated. For each DNA and RNA extraction, 30-ml aliquots of each noncontaminated water sample and 1.5 ml of the contaminated microcosms were collected and centrifuged to pellet the microbial cells. The microbial cells were subsequently lysed using a combination of lysozyme (1 mg ml−1) and alkaline lysis buffer from a FastDNA spin kit for soil (QBIOgene, Carlsbad, CA). The total RNA extracted from lysates was purified using an RNeasy kit (Qiagen, São Paulo, SP, Brazil) according to the manufacturer's protocol. The DNA was purified according to the methods of Pitcher et al. (25) and subsequently eluted in 50 μl of TE (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0). The amount of DNA and RNA extracted from each sample from sampling point was determined using a NanoDrop 1000 apparatus (Thermo Scientific, Suwanee, GA). Then, the extracted RNAs were treated with RNase-free DNase I (Fermentas International Inc., Burlington, ON, Canada), and the cDNA was synthesized using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions.
Genetic fingerprinting and clone library construction.
Fragments of the 16S rRNA gene from environmental DNA/cDNA were PCR amplified using the primers U968F (5′-GAACGCGAAGAACCTTAC-3′) and L1401R (5′-CGGTGTGTACAAGACCC-3′) in a 25-μl reaction mixture as previously described (26). For genetic fingerprinting, a G+C clamp was attached to the U968F primer. Denaturing gradient gel electrophoresis (DGGE) analysis was performed using a Bio-Rad DCode universal mutation detection system (Bio-Rad Laboratories, Munich, Germany) under the same conditions described previously (27). The DGGE profiles were analyzed using GelCompar II software (version 4.06; Applied Maths, Kortrijk, Belgium). The Pearson correlation coefficient was calculated on the basis of the densitometric curves of the profile lanes as described previously (28). Dendrograms were constructed through a Ward-based coefficient using BioNumerics software (Applied Maths). In addition, the binary matrices generated from the DGGE lanes were exported to PC-ORD software (29) to perform a redundancy analysis (RDA).
The clone libraries were constructed with PCR-generated 16S rRNA amplicons, as described above, using an InsTAclone PCR cloning kit (Fermentas) according to the manufacturer's instructions. The insert-containing clones were sequenced using the forward primer M13F (5′-GTAAAACGACGGCCAGT-3′) of the vector pTZ57R/T in an ABI Prism 3100 automatic sequencer (Applied Biosystems Inc.) at Macrogen (South Korea).
Sequence analysis.
The generated electropherogram files were analyzed using the Phred program (30) for base calling and trimming of vector and low-quality (<20) sequences. The clone sequences were analyzed using the QIIME pipeline (31). Briefly, 16S rRNA gene sequences were aligned using the Greengenes core set (32) and assigned to operational taxonomic units (OTUs) with 97% similarity. Representative sequences for each OTU were taxonomically assigned using the Naive Bayesian classifier from the Ribosomal Database Project (33) with 80% confidence.
Statistical analyses.
Statistical analyses of the clone libraries were performed using the QIIME pipeline (31). OTU-generated matrices were compared using environmental cluster analysis on the basis of weighted and unweighted UniFrac beta diversity (34). OTU-generated matrices were also used to generate network profiles with Cytoscape software (35).
Bacterial isolation and identification.
Microcosm samples corresponding to the 32nd day of incubation were selected for the isolation of enriched bacterial strains. Tryptic soy broth (TSB) and marine broth (MB) agar-containing plates were used to isolate hydrocarbon-enriched bacteria from microcosms containing freshwater (TSB) and marine and hypersaline (MB) waters. Mineral medium (Bushnell Haas [BH]) agar containing the contaminant (heptadecane, naphthalene, or crude oil) as the sole carbon source for bacterial growth was also used to isolate bacterial strains. NaCl (3.5%) was added to BH agar for plating marine and hypersaline waters. After 7 days of incubation at 28°C, the colonies with different morphologies (from each medium and contaminant used) were selected for purification using the streak plate method. The isolates were stored at −80°C in TSB or MB medium at pH 7.0 to 7.2 with 10% glycerol.
The DNA from the isolated strains was extracted using an AxyPrep bacterial genomic DNA miniprep kit (Axygen Bioscience, Union City, CA), and BOX-PCR was used to cluster the bacterial strains according to the methods of Versalovic et al. (36). One representative strain from each BOX-PCR group was selected for 16S rRNA-based molecular identification. The PCR amplification of the 16S rRNA-coding gene and the molecular sequencing methodologies were performed according to the methods of Alvarez et al. (37).
The partial 16S rRNA gene sequences (∼800 bp for the majority of the strains and ∼1,370 bp for five selected isolates) were identified using the BLAST-N tool (www.ncbi.nlm.nih.gov/blast) on the National Center for Biotechnology Information (NCBI) website using the GenBank nonredundant database. Phylogenetic trees were constructed using the unweighted-pair group method with arithmetic mean (UPGMA) (38) and a bootstrap test (1,000 replicates) (39). These analyses were performed using the MEGA program, version 4 (40).
Bacterial growth using petroleum hydrocarbons as a carbon source.
The growth of the different isolates was determined in BH medium containing petroleum hydrocarbons (naphthalene, heptadecane, or crude oil) as the sole carbon source, in accordance with the work of Alvarez et al. (37). Either turbidity or visual observation of any alteration in the oil film compared to the appearance of the controls (without any hydrocarbon source or without a bacterial inoculum) was used to demonstrate bacterial growth.
Bacterial growth in different salt concentrations.
Five isolates representing the ecosystems studied (two strains from freshwater [strains F02-115.1_JO and D06-95_JO], one from marine water [strain MOM4_MO], and two strains from hypersaline water [strains G06-163_VO and H02-159_VO]) were grown in BH medium containing heptadecane as the sole carbon source and different concentrations of NaCl (from 0 to 20%) for 4 days. The number of cells was determined by serial dilutions and plating onto MB or TSB agar (the same medium amended with the equivalent amount of NaCl in which the strains were cultivated) for the calculation of the number of CFU ml−1.
Nucleotide sequence accession numbers.
The sequences were deposited in the GenBank database with the following accession numbers: KC307962 to KC309393 (clone libraries) and KC330685 to KC330734 and KC900371 to KC900375 (isolated strains).
RESULTS
Microcosms and TPH analysis.
Bacterial communities in freshwater and marine and hypersaline water samples and those enriched after contamination with different hydrocarbons (heptadecane, naphthalene, and crude oil) in the microcosms were first characterized using molecular methods. On the 4th day after initiating the experiment, no considerable DNA yields were obtained from naphthalene-contaminated microcosms. However, DNA and RNA were efficiently extracted from all contaminated microcosms after 12 and 32 days of incubation. Neither turbidity nor substantial DNA yields were observed in the controls without hydrocarbon contamination and in the sterilized water containing hydrocarbons.
TPH measurement in the contaminated microcosms (those corresponding to the 32nd day of incubation), performed by GC-mass spectrometry (MS) analysis, resulted in a decrease in hydrocarbon concentration compared to the concentration of hydrocarbon initially introduced into the microcosms. This observation can be explained either by the occurrence of biodegradation or abiotic degradation or by losses during hydrocarbon extraction steps before GC-MS analysis. However, comparing the TPH content in all microcosms after 32 days of incubation with the values obtained for the controls (water samples amended with C, N, K, and P, sterilized by autoclaving, and supplemented with each hydrocarbon contaminant) maintained under the same conditions for the entire incubation period (32 days), the results showed a decrease in hydrocarbon concentration varying from 18% (freshwater microcosms contaminated with naphthalene) to 54% (hypersaline microcosms contaminated with crude oil) (Fig. 1). The degradation values obtained for the contaminated microcosms were significantly different (P = 0.05) from those obtained for the controls. Therefore, the decrease in hydrocarbon concentration suggests the presence of an active hydrocarbon-degrading bacterial community in the microcosms (Fig. 1).
Fig 1.
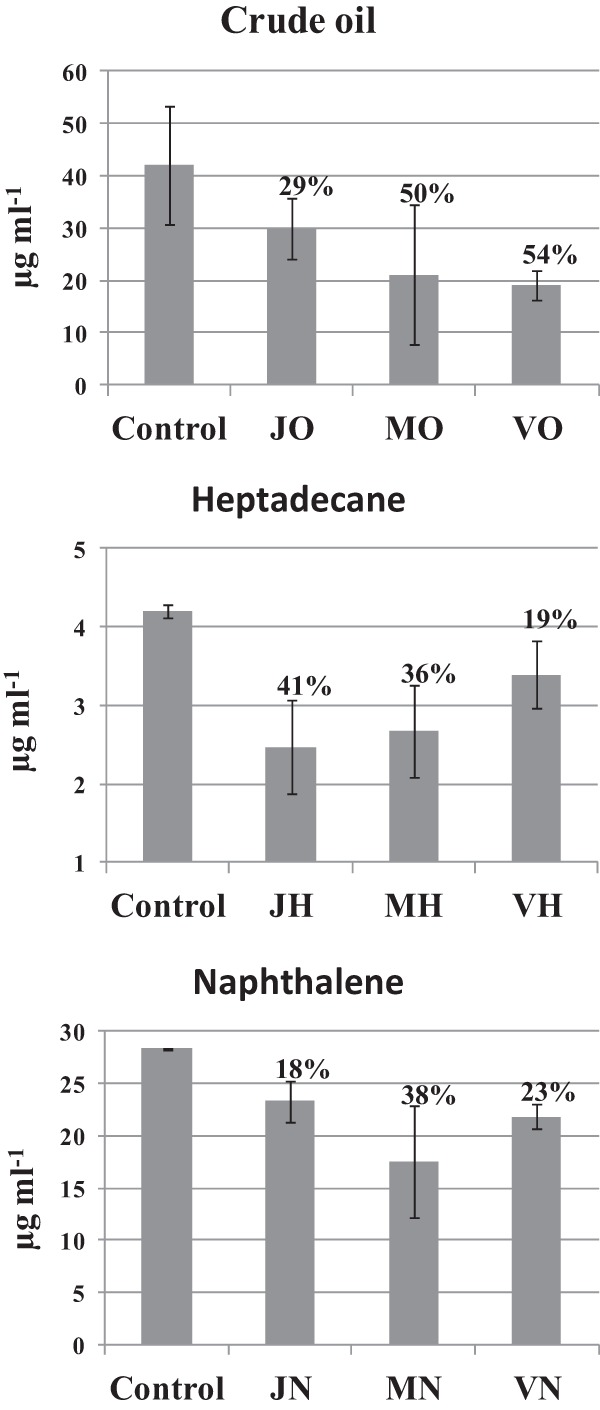
TPH quantification after the 32nd day of incubation of the microcosms. The degradation values (above the bars) were determined as the ratio between the average values obtained for the contaminated microcosms and those obtained for the negative controls used in this study. The samples are denoted as follows: the first letter represents the origin of the sample (J, Jacarepiá Lagoon; M, Massambaba Beach; V, Vermelha Lagoon), followed by the hydrocarbon used in the different microcosms (H, heptadecane; N, naphthalene; O, crude oil).
Genetic fingerprinting analysis.
The DNA- and RNA-based genetic fingerprinting analyses showed that each sample collected (n = 3 each for freshwater and marine and hypersaline water samples) and contaminated with one type of hydrocarbon (naphthalene, heptadecane, or crude oil) had similar bacterial community patterns, generating representative replicates (see Fig. S2A in the supplemental material). To simplify the analyses, the following experiments were performed using the pool of PCR-amplified 16S rRNA genes. A dendrogram analysis of the genetic fingerprinting (DGGE) data (see Fig. S2B in the supplemental material) revealed the formation of three main groups with less than 10% similarity (each group formed by freshwater and marine and hypersaline water samples). Moreover, the following observations were obtained from the DGGE analysis: (i) bacterial changes primarily occurred during the first 4 days after bacterial exposure to crude oil and heptadecane; (ii) in all cases, no strong differences between DNA- and RNA-based bacterial community analyses were observed; (iii) small variations in the enriched bacterial communities were observed throughout the experimental period after the primary change of the bacterial taxa; (iv) bacterial communities enriched through hydrocarbon exposure were different for each ecosystem studied, as was also demonstrated by a redundancy analysis (Fig. 2); and (v) considering the same ecosystem, the hydrocarbon used was responsible for the selection of specific bacterial communities (see Fig. S2B in the supplemental material).
Fig 2.
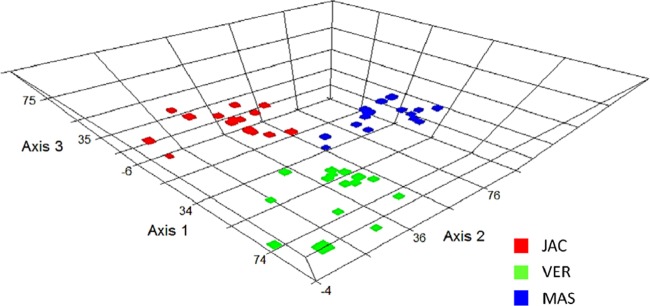
An RDA ordination diagram based on the genetic fingerprint data obtained from hydrocarbon-enriched bacterial communities. The data were plotted using each aquatic ecosystem type, and the different colors represent the microcosm samples obtained from Vermelha Lagoon (VER; green), Massambaba Beach (MAS; blue), and Jacarepiá Lagoon (JAC; red). The detailed genetic fingerprint pattern is presented in Fig. S2B in the supplemental material.
Enriched bacterial communities.
The RNA and DNA extracted from the samples after 12 days of incubation were used to construct the 16S rRNA gene clone libraries, and the dominant hydrocarbon-enriched bacterial communities were compared with those in the noncontaminated samples. More than 1,450 high-quality sequences were obtained, and these sequences (approximately 410 bp without the primer sequences) were further clustered into OTUs with 97% nucleotide similarity.
As observed using genetic fingerprint analysis, the hydrocarbon-enriched bacterial communities were quite different from those in the samples before contamination (Fig. 3A). The profiles of the hydrocarbon-enriched bacterial communities in the various contaminated microcosms, obtained through DNA- and RNA-based analyses, were very similar. Moreover, hydrocarbon contamination resulted in an increase in the relative abundance of two main phyla: Proteobacteria and Bacteroidetes (Fig. 3A). Although the Gammaproteobacteria class was minimally detectable in the original communities of the hypersaline and freshwater ecosystems, the results showed an increase in the relative abundance of this class after hydrocarbon contamination in both microcosm samples (Fig. 3A). In the marine ecosystem, Gammaproteobacteria comprised 55.7% of the original community; however, the relative abundance of this class increased after hydrocarbon contamination, reaching 100% in naphthalene-contaminated microcosms (Fig. 3A).
Fig 3.

Relative abundance of the OTUs in 16S rRNA gene clone libraries. (A) Bacterial classes enriched by petroleum hydrocarbons. (B) Hydrocarbon-enriched bacterial genera detected in contaminated microcosms. To simplify the interpretation of the scheme, only the hydrocarbon-enriched bacterial communities are shown, and bacterial groups with low abundance were clustered in unclassified bacteria and other groups. The taxonomy was assigned using the Greengenes (32) 16S rRNA gene database, and an average of 67 sequences per contaminated sample was obtained. The samples are denoted as follows: the first letter corresponds to the origin of each sample (J, Jacarepiá Lagoon; M, Massambaba Beach; V, Vermelha Lagoon), the second letter indicates the hydrocarbon used in the different microcosms (H, heptadecane; N, naphthalene; O, crude oil), and the third letter represents either the DNA (D)- or RNA (R)-based library.
Hydrocarbon-enriched bacterial communities in freshwater samples.
The clone library results showed that the OTUs associated with the Cloacibacterium genus of the Flavobacteriaceae family represented the majority of the naphthalene-enriched bacterial community (22% of the relative abundance of the DNA-based clone library). The RNA-based analysis showed that Cloacibacterium represented 58% of the relative abundance of the RNA-based sequences. Other enriched members of the bacterial community also detected among the RNA-based sequences obtained from naphthalene-contaminated freshwater included Achromobacter (11% of the relative abundance of the sequences obtained from the DNA-based analysis and 21% from the RNA-based analysis) and Pseudomonas (8% of the DNA- and RNA-based libraries) (Fig. 3B). The crude oil- and heptadecane-enriched OTUs in freshwater microcosms were 72% and 42%, respectively, of the relative abundance of the sequences obtained from the DNA-based libraries of the genus Acinetobacter (and 32% and 70% of the relative abundance of the sequences from the RNA-based libraries, respectively). The relative abundance of Aquabacterium-related OTUs increased in heptadecane-containing (10% and 17% of the DNA- and RNA-based libraries, respectively) and in crude oil-containing (6% and 15% of DNA- and RNA-based libraries, respectively) microcosms, whereas Parvibaculum-related OTUs were detected in the DNA- and RNA-based libraries (20% and 22%, respectively) of crude oil-contaminated freshwater microcosms (Fig. 3B).
Hydrocarbon-enriched bacterial communities in marine samples.
The Cycloclasticus genus, belonging to the Thiotrichales, was enriched by the presence of naphthalene in marine waters, comprising 59% of the relative abundance of the DNA-based sequences and 58% of the RNA-based sequences. Pseudomonas-related OTUs were also increased in naphthalene-contaminated marine microcosms (15% and 32% of the relative abundance of the sequences of the DNA- and RNA-based libraries, respectively). The Oceanospirillales order and the Marinobacter and Mesoflavibacter genera were predominately enriched in heptadecane- and crude oil-contaminated marine microcosms. Oceanospirillales comprised more than 55% of the relative abundance of the DNA- and RNA-based sequences obtained from heptadecane-contaminated microcosms and 31% (DNA-based) and 27% (RNA-based) of the sequences from crude oil-contaminated marine microcosms. The OTUs associated with Mesoflavibacter represented 7% and 12% of the relative abundance of the DNA- and RNA-based sequences obtained from heptadecane-contaminated microcosms, respectively. Similar results were observed in crude oil-contaminated marine microcosms (9% and 17% of the relative abundance of the DNA- and RNA-based sequences, respectively). In addition, Marinobacter-related OTUs represented 28% and 30% of the relative abundance of the DNA- and RNA-based sequences, respectively, obtained from crude oil-contaminated microcosms (Fig. 3B).
Hydrocarbon-enriched bacterial communities in hypersaline samples.
No strong bacterial selection occurred after naphthalene contamination in hypersaline water samples. However, the OTUs associated with the Marinobacter genus were primarily enriched after hypersaline water contamination with naphthalene, heptadecane, and crude oil. Marinobacter-related OTUs comprised 10% and 15% of the relative abundance of the sequences in the DNA- and RNA-based libraries derived from naphthalene-enriched microcosms, respectively. In addition, Marinobacter accounted for more than 80% of the relative abundance of the DNA-based sequences (81% of the RNA-based sequences) in heptadecane-contaminated hypersaline microcosms and up to 41% and 30% of the relative abundance of the DNA- and RNA-based sequences in crude oil-contaminated microcosms, respectively. The genus Oceanobacter (Oceanospirillales order) was also enriched in the bacterial community in crude oil-contaminated hypersaline microcosms (16% of the relative abundance of the sequences from the DNA-based analysis and 25% of the sequences from the RNA-based analysis) (Fig. 3B).
OTUs shared among the contaminated samples.
A network analysis demonstrated the relationship of the OTUs obtained from the hydrocarbon-contaminated environmental samples (see Fig. S3 in the supplemental material). In addition, principal component analyses, constructed using the environmental distance matrix based on weighted and unweighted UniFrac distance (at OTU levels of 97%), grouped each of the microcosms from the different ecosystems into separate groups (see Fig. S4 in the supplemental material).
A more comprehensive phylogenetic analysis based on OTU classification showed the shared distribution patterns of the enriched bacterial communities from different contaminated microcosms (Fig. 4). For example, although Acinetobacter species comprised the dominant OTUs from the freshwater ecosystem contaminated with heptadecane and crude oil, a more detailed analysis of the Acinetobacter communities showed that the Acinetobacter species from crude oil-enriched samples differed from those obtained from heptadecane-enriched samples. The same bacterial distribution pattern was also observed in marine and hypersaline enrichments (e.g., Oceanospirillales- and Marinobacter-related OTUs in heptadecane- and crude oil-contaminated marine and hypersaline microcosms) (Fig. 4).
Fig 4.
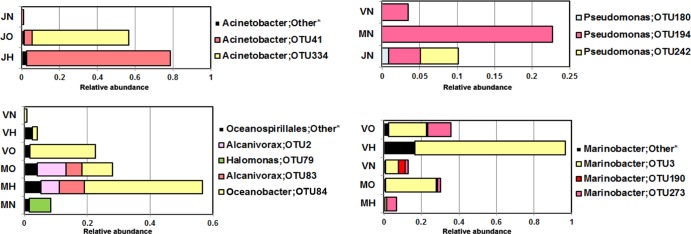
Relative abundance of the OTUs shared among the different enriched microcosms. The samples are denoted as follows: the first letter represents the origin of the samples (J, Jacarepiá Lagoon; M, Massambaba Beach; V, Vermelha Lagoon), followed by the hydrocarbon used in the different microcosms (H, heptadecane; N, naphthalene; O, crude oil). *, low-abundance OTUs are grouped as “other.”
Isolation of bacterial strains from hydrocarbon-contaminated microcosms.
A total of 199 isolates was obtained from all enriched microcosms. On the basis of the DNA genotyping analyses (BOX-PCR), 121 different strains were further identified through 16S rRNA gene sequencing. The results showed that 50 (41.3%) bacterial strains could be associated with the dominant OTUs in the clone libraries (see Table S2 in the supplemental material). All of the strains were able to grow with the hydrocarbon contaminating the microcosms from which they were isolated (see Table S2 in the supplemental material). Five strains related to the dominant bacterial taxa enriched in heptadecane- and crude oil-contaminated marine, hypersaline, and freshwater microcosms (as observed in clone library analysis; Fig. 5A) were further selected to test their growth capacity in different concentrations of salt (NaCl). The selected strains were closely related to Alcanivorax dieselolei, Alcanivorax balearicus, Marinobacter hydrocarbonoclasticus, Acinetobacter junii, and Acinetobacter calcoaceticus (Fig. 5A; see Table S2 in the supplemental material). The Acinetobacter-related strains were able to grow in mineral medium using heptadecane as the sole carbon source in the presence of NaCl over a range of concentrations from 0 to 5% (maximum growth was without NaCl; Fig. 5B). The Marinobacter and Alcanivorax-related strains were able to grow in a broader range of salinity (from 0 to 15% NaCl), although the highest number of cells was obtained at 7% and 8% NaCl for Marinobacter and Alcanivorax, respectively (Fig. 5B).
Fig 5.

(A) Phylogenetic tree based on the 16S rRNA gene sequences of five selected bacterial isolates and the closest related species obtained from the GenBank database (NCBI). The tree was constructed on the basis of the neighbor-joining method. The GenBank accession number of each bacterial species is presented between vertical lines. (B) Number of CFU ml−1 after bacterial growth in different concentrations of NaCl for 4 days using heptadecane as the sole carbon source. a, similar results were obtained for strain F02-115.1_JO; b, similar results were obtained for strain MOM4_MO.
DISCUSSION
The primary question addressed in this study is whether the structure and composition of the hydrocarbon-degrading bacterial communities observed in freshwater, marine, and hypersaline aquatic ecosystems are determined by the properties of the ecosystems in which they are found or by the type of hydrocarbon (naphthalene, heptadecane, and crude oil) contaminating the microcosms. Lozupone and Knight (41) have demonstrated that salinity significantly affects the global pattern of the distribution of microbes in aquatic environments. Moreover, many studies have shown that petroleum hydrocarbons have a large impact on marine microbial structure (8, 42, 43). Although the analyses of the data obtained in this study showed some resemblance among the bacterial taxa in samples from hydrocarbon-contaminated freshwater, marine, and hypersaline ecosystems, the bacterial selection depended more on the environmental system than on the hydrocarbons used in the microcosms.
The bacterial profiles generated from DNA- and RNA-based analyses showed strong similarity, suggesting the role of the enriched bacterial communities in the hydrocarbon degradation activity observed in the microcosms. TPH results corroborated this observation. Although certain amounts of TPH were lost by evaporation, by abiotic degradation, or during hydrocarbon extraction steps before GC-MS analysis, as also observed by Niepceron et al. (13), the occurrence of biodegradation in all contaminated microcosms can be assumed when the TPH results are compared to those obtained in the microcosms used as controls. Moreover, different bacterial strains were also isolated from each contaminated microcosm, and the hydrocarbon-degrading potential of these bacteria was detected. Therefore, in agreement with the results described by McKew et al. (10), different members of the bacterial communities may be responsible for the degradation of petroleum-derived hydrocarbons in each ecosystem.
Most of the hydrocarbon-enriched bacteria from freshwater, marine, and hypersaline microcosms were particularly associated with well-known bacterial hydrocarbon degraders, which have previously been detected in different studies (7, 43–45). The enrichment of OTUs related to the Cloacibacterium genus in freshwater microcosms contaminated with naphthalene is a new finding. No other studies have described PAH degradation by bacteria within this group, although this genus has frequently been found in freshwater ecosystems (46). Furthermore, the high relative abundance of OTUs related to the Cycloclasticus and Pseudomonas genera after naphthalene contamination in marine water-containing microcosms highlights the importance of these genera in these marine ecosystems, as previously suggested (10, 12, 44). Marinobacter should have a role in PAH degradation in high salinity. Bacteria belonging to the Marinobacter genus degrade both aliphatic and aromatic fractions of petroleum hydrocarbons (47) and have been found to be part of a PAH-degrading halophilic microbial consortium in saline soil (18). Considering alkane- and crude oil-contaminated microcosms, a predominance of Acinetobacter in freshwater microcosms and of Oceanospirillales and Marinobacter in marine and hypersaline microcosms was observed. Although Oceanospirillales and Marinobacter equally dominated both crude oil-contaminated marine and hypersaline microcosms, Oceanospirillales prevailed in alkane-contaminated marine microcosms, and Marinobacter dominated in alkane-contaminated hypersaline microcosms.
Because we believe that the prevalence of Acinetobacter, Oceanospirillales, and Marinobacter in alkane-contaminated freshwater, marine, and hypersaline ecosystems, respectively, occurred because of the salinity of each ecosystem, we focused on culture-dependent methods and tested the optimal saline concentration for the growth of these bacteria. Previous studies have shown that bacteria from these genera grow over a broad range of salinities (17, 48). Although the results obtained here explained the predominance of alkane-degrading Acinetobacter in contaminated freshwater microcosms (optimal growth was observed in medium without NaCl), this predominance was not true for both Alcanivorax and Marinobacter strains, which grew over similar ranges of salinity (from 0 to 15% NaCl) (Fig. 5B). Therefore, other environmental factors (not only salinity) may be involved in Alcanivorax and Marinobacter competition for alkanes in marine- and hypersaline-contaminated ecosystems.
Based on the results obtained in this study, we conclude that the general patterns of hydrocarbon-enriched bacterial communities are ecosystem dependent, although within the same ecosystem, the communities are selected by the hydrocarbon contaminating the microcosms. Moreover, the distribution of alkane- and PAH-enriched bacterial communities within a particular ecosystem may be used to determine the most abundant hydrocarbon present in the environment. The establishment of these biogeographic patterns of hydrocarbon-specific and hydrocarbon-enriched bacterial communities might be important for many industrial activities, such as environmental biomonitoring and bioremediation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ricardo B. Pinto for providing local coordinates, Diogo F. Rosas for assistance with sampling, and Armando Franco for critically reading the manuscript.
This study was financially supported through grants from the National Research Council of Brazil (CNPq), CAPES, and FAPERJ.
Footnotes
Published ahead of print 19 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02251-13.
REFERENCES
- 1.Head IM, Jones DM, Röling WF. 2006. Marine microorganisms make a meal of oil. Nat. Rev. Microbiol. 4:173–182 [DOI] [PubMed] [Google Scholar]
- 2.Atlas RM, Hazen TC. 2011. Oil biodegradation and bioremediation: a tale of the two worst spills in US history. Environ. Sci. Technol. 45:6709–6715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGenity TJ, Folwell BD, McKew BA, Sanni GO. 2012. Marine crude-oil biodegradation: a central role for interspecies interactions. Aquat. Biosyst. 8:10. 10.1186/2046-9063-8-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atlas RM. 1981. Microbial degradation of petroleum hydrocarbons: an environmental perspective. Microbiol. Rev. 45:180–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hazen TC, Dubinsky EA, DeSantis TZ, Andersen GL, Piceno YM, Singh N, Jansson JK, Probst A, Borglin SE, Fortney JL, Stringfellow WT, Bill M, Conrad ME, Tom LM, Chavarria KL, Alusi TR, Lamendella R, Joyner DC, Spier C, Baelum J, Auer M, Zemla ML, Chakraborty R, Sonnenthal EL, D'haeseleer P, Holman HY, Osman S, Lu Z, Van Nostrand JD, Deng Y, Zhou J, Mason OU. 2010. Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science 330:204–208 [DOI] [PubMed] [Google Scholar]
- 6.Chakraborty R, Borglin SE, Dubinsky EA, Andersen GL, Hazen TC. 2012. Microbial response to the MC-252 oil and Corexit 9500 in the Gulf of Mexico. Front. Microbiol. 3:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mason OU, Hazen TC, Borglin S, Chain PS, Dubinsky EA, Fortney JL, Han J, Holman HY, Hultman J, Lamendella R, Mackelprang R, Malfatti S, Tom LM, Tringe SG, Woyke T, Zhou J, Rubin EM, Jansson JK. 2012. Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 6:1715–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teramoto M, Suzuki M, Okazaki F, Hatmanti A, Harayama S. 2009. Oceanobacter-related bacteria are important for the degradation of petroleum aliphatic hydrocarbons in the tropical marine environment. Microbiology 155:3362–3370 [DOI] [PubMed] [Google Scholar]
- 9.Gertler C, Näther DJ, Cappello S, Gerdts G, Quilliam RS, Yakimov MM, Golyshin PN. 2012. Composition and dynamics of biostimulated indigenous oil-degrading microbial consortia from the Irish, North and Mediterranean Seas: a mesocosm study. FEMS Microbiol. Ecol. 81:520–536 [DOI] [PubMed] [Google Scholar]
- 10.McKew BA, Coulon F, Osborn AM, Timmis KN, McGenity TJ. 2007. Determining the identity and roles of oil-metabolizing marine bacteria from the Thames estuary, UK. Environ. Microbiol. 9:165–176 [DOI] [PubMed] [Google Scholar]
- 11.Wang W, Shao Z. 2012. Genes involved in alkane degradation in the Alcanivorax hongdengensis strain A-11-3. Appl. Microbiol. Biotechnol. 94:437–448 [DOI] [PubMed] [Google Scholar]
- 12.Wang B, Lai Q, Cui Z, Tan T, Shao Z. 2008. A pyrene-degrading consortium from deep-sea sediment of the West Pacific and its key member Cycloclasticus sp. P1. Environ. Microbiol. 10:1948–1963 [DOI] [PubMed] [Google Scholar]
- 13.Niepceron M, Portet-Koltalo F, Merlin C, Motelay-Massei A, Sylvie Barray S, Bodilis J. 2010. Both Cycloclasticus spp. and Pseudomonas spp. as PAH-degrading bacteria in the Seine estuary (France). FEMS Microbiol. Ecol. 71:137–147 [DOI] [PubMed] [Google Scholar]
- 14.McGenity TJ. 2010. Halophilic hydrocarbon degraders, p 1940–1951 In Timmis KN. (ed), Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, Germany [Google Scholar]
- 15.Mnif S, Chamkha M, Sayadi S. 2009. Isolation and characterization of Halomonas sp. strain C2SS100, a hydrocarbon-degrading bacterium under hypersaline conditions. J. Appl. Microbiol. 107:785–794 [DOI] [PubMed] [Google Scholar]
- 16.Feng TC, Cui CZ, Dong F, Feng YY, Liu YD, Yang XM. 2012. Phenanthrene biodegradation by halophilic Martelella sp. AD-3. J. Appl. Microbiol. 113:779–789 [DOI] [PubMed] [Google Scholar]
- 17.Dastgheib SM, Amoozegar MA, Khajeh K, Ventosa A. 2011. A halotolerant Alcanivorax sp. strain with potential application in saline soil remediation. Appl. Microbiol. Biotechnol. 90:305–312 [DOI] [PubMed] [Google Scholar]
- 18.Dastgheib SM, Amoozegar MA, Khajeh K, Shavandi M, Ventosa A. 2012. Biodegradation of polycyclic aromatic hydrocarbons by a halophilic microbial consortium. Appl. Microbiol. Biotechnol. 95:789–798 [DOI] [PubMed] [Google Scholar]
- 19.Atlas RM, Bartha R. 1973. Abundance, distribution and oil biodegradation potential of microorganisms in Raritan Bay. Environ. Pollut. 4:291–300 [Google Scholar]
- 20.dos Santos HF, Cury JC, do Carmo FL, dos Santos AL, Tiedje J, van Elsas JD, Rosado AS, Peixoto RS. 2011. Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: bacterial proxies for oil pollution. PLoS One 6:e16943. 10.1371/journal.pone.0016943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andreote FD, Jiménez DJ, Chaves D, Dias AC, Luvizotto DM, Dini-Andreote F, Fasanella CC, Lopez MV, Baena S, Taketani RG, de Melo IS. 2012. The microbiome of Brazilian mangrove sediments as revealed by metagenomics. PLoS One 7:e38600. 10.1371/journal.pone.0038600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbiére EB. 1985. Condições climáticas dominantes na porção oriental da lagoa de Araruama (RJ) e suas implicações na diversidade do teor de salinidade. In Caderno de ciências da terra, vol 59 Universidade de São Paulo, SP, Brazil [Google Scholar]
- 23.Clesceri LS, Greenberg AE, Eaton AD. (ed). 1998. Standard methods for examination of water and wastewater, 20th ed. American Public Health Association, Washington, DC [Google Scholar]
- 24.EPA 2000. Nonhalogenated organics using GC/FID. EPA 8015C, revision 3. EPA, Washington, DC [Google Scholar]
- 25.Pitcher DG, Saunders NA, Owen RJ. 1989. Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett. Appl. Microbiol. 8:151–156 [Google Scholar]
- 26.Heuer H, Krsek M, Baker P, Smalla K, Wellington EM. 1997. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microbiol. 63:3233–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jurelevicius D, Korenblum E, Casella R, Vital RL, Seldin L. 2010. Polyphasic analysis of the bacterial community in the rhizosphere and roots of Cyperus rotundus L. grown in a petroleum-contaminated soil. J. Microbiol. Biotechnol. 20:862–870 [DOI] [PubMed] [Google Scholar]
- 28.Hardoim CC, Costa R, Araújo FV, Hajdu E, Peixoto R, Lins U, Rosado AS, van Elsas JD. 2009. Diversity of bacteria in the marine sponge Aplysina fulva in Brazilian coastal waters. Appl. Environ. Microbiol. 75:3331–3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCune B, Mefford MJ. 2011. PC-ORD. Multivariate analysis of ecological data, version 6.0. MjM Software, Gleneden Beach, OR [Google Scholar]
- 30.Ewing B, Green P. 1998. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 8:186–194 [PubMed] [Google Scholar]
- 31.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lozupone CA, Hamady M, Kelley ST, Knight R. 2007. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73:1576–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13:2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Versalovic J, Schneider M, De Bruijn FJ, Lupski JR. 1994. Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Methods Mol. Cell. Biol. 5:25–40 [Google Scholar]
- 37.Alvarez VM, Santos SC, da costa Casella R, Vital RL, Sebástian GV, Seldin L. 2008. Bioremediation potential of a tropical soil contaminated with a mixture of crude oil and production water. J. Microbiol. Biotechnol. 18:1966–1974 [PubMed] [Google Scholar]
- 38.Sokal RR, Michener CD. 1958. A statistical method for evaluating systematic relationships. Univ. Kans. Sci. Bull. 38:1409–1438 [Google Scholar]
- 39.Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791 [DOI] [PubMed] [Google Scholar]
- 40.Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599 [DOI] [PubMed] [Google Scholar]
- 41.Lozupone CA, Knight R. 2007. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U. S. A. 104:11436–11440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bælum J, Borglin S, Chakraborty R, Fortney JL, Lamendella R, Mason OU, Auer M, Zemla M, Bill M, Conrad ME, Malfatti SA, Tringe SG, Holman HY, Hazen TC, Jansson JK. 2012. Deep-sea bacteria enriched by oil and dispersant from the Deepwater Horizon spill. Environ. Microbiol. 14:2405–2416 [DOI] [PubMed] [Google Scholar]
- 43.Kostka JE, Prakash O, Overholt WA, Green SJ, Freyer G, Canion A, Delgardio J, Norton N, Hazen TC, Huettel M. 2011. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the Deepwater Horizon oil spill. Appl. Environ. Microbiol. 77:7962–7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kasai Y, Kishira H, Harayama S. 2002. Bacteria belonging to the genus Cycloclasticus play a primary role in the degradation of aromatic hydrocarbons released in a marine environment. Appl. Environ. Microbiol. 68:5625–5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alonso-Gutiérrez J, Figueras A, Albaigés J, Jiménez N, Viñas M, Solanas AM, Novoa B. 2009. Bacterial communities from shoreline environments (Costa da Morte, northwestern Spain) affected by the Prestige oil spill. Appl. Environ. Microbiol. 75:3407–3418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao SJ, Deng CP, Li BZ, Dong XQ, Yuan HL. 2010. Cloacibacterium rupense sp. nov., isolated from freshwater lake sediment. Int. J. Syst. Evol. Microbiol. 60:2023–2026 [DOI] [PubMed] [Google Scholar]
- 47.Vila J, María Nieto J, Mertens J, Springael D, Grifoll M. 2010. Microbial community structure of a heavy fuel oil-degrading marine consortium: linking microbial dynamics with polycyclic aromatic hydrocarbon utilization. FEMS Microbiol. Ecol. 73:349–362 [DOI] [PubMed] [Google Scholar]
- 48.Bouvet PJM, Grimont PAD. 1986. Taxonomy of the genus Acinetobacter with the recognition of Acinetobacter baumannii sp. nov., Acinetobacter haemolyticus sp. nov., Acinetobacter johnsonii sp. nov., and Acinetobacter junii sp. nov. and emended descriptions of Acinetobacter calcoaceticus and Acinetobacter lwoffii. Int. J. Syst. Evol. Microbiol. 36:228–240 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.