

Abstract
Tunable promoters represent a pivotal genetic tool for a wide range of applications. Here we present such a system for sphingomonads, a phylogenetically diverse group of bacteria that have gained much interest for their potential in bioremediation and their use in industry and for which no dedicated inducible gene expression system has been described so far. A strong, constitutive synthetic promoter was first identified through a genetic screen and subsequently combined with the repressor and the operator sites of the Pseudomonas putida F1 cym/cmt system. The resulting promoter, termed PQ5, responds rapidly to the inducer cumate and shows a maximal induction ratio of 2 to 3 orders of magnitude in the different sphingomonads tested. Moreover, it was also functional in other Alphaproteobacteria, such as the model organisms Caulobacter crescentus, Paracoccus denitrificans, and Methylobacterium extorquens. In the noninduced state, expression from PQ5 is low enough to allow gene depletion analysis, as demonstrated with the essential gene phyP of Sphingomonas sp. strain Fr1. A set of PQ5-based plasmids has been constructed allowing fusions to affinity tags or fluorescent proteins.
INTRODUCTION
Regulated gene expression systems are a powerful tool to study physiology, allowing, for example, dosage-effect studies, conditional expression of toxic alleles, and depletion analysis of essential genes; accordingly, they are well developed for model organisms (1–7) but are missing for many non-model organisms. Most systems rely on a transcriptional repressor that tightly binds to operator sites in the promoter region of target genes in the absence of an inducer, thereby preventing transcription; when an inducer is present, it allosterically binds to and inactivates the transcriptional regulator, leading to derepression of promoters. Despite this simple concept, identification of inducible promoters, control elements, and inducing conditions in a particular organism is not always a trivial task. On the other hand, it is often difficult to exploit a particular system for use in organisms other than the original host because of the need for dedicated transporters for the inducer or a different promoter specificity of the RNA polymerase holoenzyme or the requirement of a coactivator for full promoter activity (8). Some of these obstacles can be circumvented by engineering artificial inducible promoters by placing a constitutive minimal promoter known to be active in a particular organism with operator sequences and the repressor of a heterologous system (2, 9, 10). We here describe such a system for sphingomonads, a phylogenetically diverse group of environmentally abundant bacteria comprising the genera Sphingomonas, Sphingobium, Novosphingobium, and Sphingopyxis (11). Members of this group are well known for their unusual ability to degrade a wide range of compounds, including pesticides, herbicides, xenobiotics, and many other aromatics. Due to this property, they are prospective candidates for bioremediation; in addition, several strains are used in industrial applications, and others have been described as potential candidates for biocontrol (12–15). Despite this interest in many different aspects of their physiology, no dedicated inducible gene expression system for sphingomonads has existed so far. Here, by combining the operator sequences (CuO) and the cumate-responsive CymR repressor from the Pseudomonas putida F1 cym/cmt system (16, 17) with an engineered minimal housekeeping promoter, Psyn2, derived from Sphingomonas sp. strain Fr1 (henceforth Sphingomonas Fr1), we have developed a highly inducible and rapidly responding conditional gene expression system that shows low basal expression without induction (i.e., is tight). We demonstrate its functionality in a number of different sphingomonads as well as in more distantly related Alphaproteobacteria not belonging to the Sphingomonadales. Due to the modular design of the system, core promoter sequences can be readily exchanged, opening the possibility to adapt the system to other organisms as well.
MATERIALS AND METHODS
Strains, growth conditions, and plasmid delivery.
Escherichia coli TOP10 (Invitrogen), “ccdB survival” (Invitrogen), and “dam− dcm−” (NEB; catalog no. C2925I) were used for cloning. Sphingomonas Fr1 wild-type (JVZ857) and ΔphyR ΔphyP::loxP mutant (JVZ1357) strains were described previously (18). Other Sphingomonas, Sphingobium, and Novosphingobium strains used in this work were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ). Methylobacterium extorquens AM1 rif (19), Caulobacter crescentus CB15 (20), and Agrobacterium tumefaciens C58 (21) were laboratory stocks. Paracoccus denitrificans JVZ2585 is a spontaneous rifampin-resistant derivative of P. denitrificans DSM 413 (DSMZ) obtained by selection on LB-Lennox containing 50 μg rifampin/ml; it carries the A1610T mutation in “cluster I” of rpoB, resulting in amino acid change Q537L (equivalent to Q513L in E. coli), which likely accounts for rifampin resistance (22). E. coli strains were routinely grown on LB-Lennox at 37°C. Other strains were grown at 28°C on solidified medium (1.5% agar) or as 20-ml cultures in 100-ml baffled flasks (with shaking at 160 rpm) in LB-Lennox (Sphingomonas Fr1, P. denitrificans, and A. tumefaciens), PYE (0.2% peptone, 0.1% yeast extract, 0.02% MgSO4·7H2O, 0.01% CaCl2·H2O; C. crescentus), Nutrient broth (Fluka Analytical, catalog no. 17181) (all other sphingomonads), or minimal medium (23) with methanol (M. extorquens). When appropriate, concentrations (in μg/ml) of antibiotics for E. coli, sphingomonads, and M. extorquens were used as follows: tetracycline, 10; kanamycin, 50; carbenicillin, 50; streptomycin, 100; chloramphenicol, 34. For other strains, tetracycline to maintain plasmids was added to the media in the following concentrations (in μg/ml): 2 (C. crescentus); 3 (A. tumefaciens); and 1 (P. denitrificans).
Plasmids were delivered into sphingomonads by conjugal transfer or electroporation as described previously (24). For A. tumefaciens and C. crescentus, electroporation was performed on a GenePulser Xcell system (Bio-Rad) using the standard preset E. coli program; for M. extorquens, electroporation was performed as described before (24). For P. denitrificans, plasmids were delivered by conjugal transfer from E. coli S17-1λpir (25) with mating on minimal medium without a carbon source as previously described (24). Mating mixtures were plated on LB-Lennox containing tetracycline (10 μg/ml) and rifampin (50 μg/ml) for plasmid selection and E. coli counterselection, respectively.
Measurement of promoter activity.
Cumate (4-isopropylbenzoic acid) was purchased from Sigma-Aldrich (catalog no. 269402-5G) and dissolved in 100% ethanol to result in a 100 mM stock solution. Dilutions were prepared in ethanol to give a 1,000-fold stock. For “no cumate” controls, an equal volume of ethanol was added to cultures.
For measuring the steady-state activity of PQ5 in Sphingomonas Fr1, precultures of the wild-type strain harboring pQF-lacZ were grown for 8 to 10 h to an optical density at 600 nm (OD600) of 1.0, diluted into fresh medium at an OD600 of 0.0005 to 0.001 with different cumate concentrations (see Results and Discussion), and grown for 20 to 24 h. For measuring induction kinetics in Sphingomonas Fr1, cultures were grown to the exponential phase (OD600 of 0.5 to 1.5) and cumate was added to reach a final concentration of 50 μM. For depletion analysis, cultures were grown in LB containing 50 μM cumate to the exponential phase (OD600 of 1 to 2), washed 5 times with 20 ml of LB, and resuspended in 20 ml of LB (OD600 of 0.025) with (50 μM) or without cumate. Cultures were grown for 20 to 24 h before measuring nepRp-lacZ+ activity.
Measurements of steady-state activity of PQ5 in other sphingomonads were performed as described for Sphingomonas Fr1 using 25 μM cumate. For A. tumefaciens and P. denitrificans, cultures inoculated from a single colony were grown overnight (16 to 20 h) with (25 μM) or without cumate. For C. crescentus, cultures in the exponential phase (OD600 of 0.6 to 0.8) were split in two and induced with 50 μM cumate or subjected to mock treatment with ethanol for 4 h before measurement of PQ5 activity. For M. extorquens, the wild-type strain harboring the lux reporter plasmid (pQ5-lux or pQ2148-lux) was grown to the exponential phase (OD600 of 0.3 to 0.6) and PQ5 or PQ2148 was induced by adding 90 μl of culture to wells containing 10 μl of a 10-fold aqueous stock solution of cumate at different concentrations. Measurements were started immediately (<2 min after induction) and followed for approximately 3 h in 2-min intervals.
β-Galactosidase activity was determined using the chromogenic substrate 2-nitrophenyl-β-d-galactopyranoside (ONPG; Fluka) according to the method of Miller (26). For M. extorquens, luciferase activity was measured in 96-well black/white isoplates (PerkinElmer) on a Victor3 multilabel plate reader (PerkinElmer) essentially as described previously (27) except that the measured values were corrected for the initial OD600.
SDS-PAGE and Western blots.
Aliquots (1 ml) were taken from the same cultures used to measure nepRp-lacZ+ activity, cells were pelleted by centrifugation (6,000 × g, 5 min) and resuspended to an OD600 of 10 in SDS-PAGE sample buffer (28), and 5-μl samples were resolved on SDS-PAGE gels (12.5%) followed by transfer to nitrocellulose membranes. Western blotting was performed with primary mouse monoclonal anti-FLAG M2 antibodies (Invitrogen) and secondary goat anti-mouse horseradish peroxidase-coupled IgG antibodies (Bio-Rad) and ECL Western blotting detection reagents (GE Healthcare).
Plasmid construction.
Standard molecular biology protocols were followed (29). PCR was done with Phusion DNA polymerase (Thermo Scientific) in GC buffer containing 10% dimethyl sulfoxide (DMSO), and oligonucleotides were purchased from Microsynth (Balgach, Switzerland). Restriction enzymes were from Thermo Scientific, and T4 DNA ligase was from New England BioLabs. For oligonucleotide annealing, the two complementary oligonucleotides (10 μM each) were mixed in 100 μl of oligonucleotide annealing buffer (8 mM Tris-HCl [pH 7.5], 40 mM NaCl) in a microcentrifuge tube; the tube was placed in 1 liter of boiling water, which was then allowed to cool to room temperature for several hours. Relevant plasmids are listed in Table 1 and oligonucleotides in Table S1 in the supplemental material.
Table 1.
Plasmids used in the study
| Plasmid category and name | Description/relevant featuresa | Source or reference |
|---|---|---|
| Basic cloning plasmids | ||
| pAK200 | pBBR oriV; Kmr | 18 |
| pAK206 | pBBR oriV; Gmr | 18 |
| pAK200Cm | pAK200 derivative; Cmr | This study |
| pBBR1MCS5 | pBBR oriV; Gmr | 32 |
| pCM62 | IncP oriV; ColE1 oriV; RP4 oriT; Tcr | 33 |
| pME6010 | pVS1 oriV; p15A oriV; Tcr | 34 |
| Standard PQ5-based plasmids | ||
| pQF | pCM62 with cymR*, PQ5, and MCS for N- and C-terminal fusions to 3×FLAG tag; Tcr | This study |
| pQH | pQF derivative for C-terminal fusion to 3×FLAG and N-terminal fusion to HA tag; Tcr | This study |
| pQM | pQF derivative for C-terminal fusion to 3×FLAG and N-terminal fusion to myc tag; Tcr | This study |
| pQG | pQF derivative for C-terminal fusion to 3×FLAG and N-terminal fusion to EGFP; Tcr | This study |
| pQY | pQF derivative for C-terminal fusion to 3×FLAG and N-terminal fusion to SYFP2; Tcr | This study |
| pQR | pQF derivative for C-terminal fusion to 3×FLAG and N-terminal fusion to mCherry; Tcr | This study |
| pQC | pQF derivative for C-terminal fusion to 3×FLAG and N-terminal fusion to ECFP; Tcr | This study |
| pQH2 | pQH derivative with pBBR oriV; Tcr | This study |
| pQFD | pQF derivative with Gateway cassette; contains ccdB; Cmr Tcr | This study |
| pQHD | pQH derivative with Gateway cassette; contains ccdB; Cmr Tcr | This study |
| pQMD | pQM derivative with Gateway cassette; contains ccdB; Cmr Tcr | This study |
| pQGD | pQG derivative with Gateway cassette; contains ccdB; Cmr Tcr | This study |
| pQYD | pQY derivative with Gateway cassette; contains ccdB; Cmr Tcr | This study |
| pQRD | pQR derivative with Gateway cassette; contains ccdB; Cmr Tcr | This study |
| Reporter plasmids | ||
| pTOPO-TERM193 | pCR2.1-TOPO with TERM193; Kmr | This study |
| pAK127 | pCM62 with TERM193; Tcr | This study |
| pAK127lacZ | Promoter-probe plasmid (intermediate construct); pAK127 with E. coli lacZ; Tcr | This study |
| pAK127lacZ(MCS) | Promoter-probe plasmid (intermediate construct); pAK127lacZ with new MCS; Tcr | This study |
| pAK200Cm-lacZ | Promoter-probe plasmid (intermediate construct); pAK200Cm with TERM193-MCS-lacZ fragment of pAK127lacZ(MCS); Cmr | This study |
| pAK501 | Promoter-probe plasmid; pME6010 with TERM193-MCS-lacZ-cat fragment of pAK200Cm-lacZ; Cmr | This study |
| pAK501-nepRp | nepR promoter (from −42 to −2) in pAK501; Cmr | This study |
| pAK128 | Promoter-probe plasmid; pAK127 with SYFP2; Tcr | This study |
| pQF-lacZ | pCM62-based reporter plasmid with cymR* and PQ5-lacZ+ fusion; Tcr | This study |
| pLM05 | Promoter-probe plasmid carrying luxCDABE from Photorhabdus luminescens; Kmr | 27 |
| pQ5-lux | pLM05 with cymR* and PQ5-luxCDABE fusion; Kmr | This study |
| pQ2148 | pQH derivative in which Psyn2 is replaced by the META1_2148 promoter from M. extorquens AM1; Tcr | This study |
| pQ2148-lacZ | Reporter plasmid; pQ2148 with lacZ; Tcr | This study |
| pQ2148-lux | pLM05 with cymR* and PQ2148-luxCDABE fusion; Kmr | This study |
| Other plasmids | ||
| pQH-PhyP | pQH derivative encoding PhyP-3×FLAG fusion; Tcr | This study |
| pAK200-QPhyP | pAK200 derivative with PQ5 driving PhyP-3×FLAG expression; Kmr | This study |
| pCM62-PhyR | pCM62 harboring phyR under the control of its native promoter and RBS; Tcr | 18 |
| pCM62-PhyR(D194A) | pCM62 harboring phyR(D194A) under the control of its native promoter and RBS; Tcr | 18 |
Cmr, chloramphenicol resistance; Gmr, gentamicin resistance; Kmr, kanamycin resistance; Tcr, tetracycline resistance.
PQ5-based plasmids.
pQF was assembled in multiple steps, mostly from synthetic DNA (Invitrogen and MWG Operon Eurofins) and short double-stranded DNA (dsDNA) (generated by oligonucleotide annealing), and details are outlined in the supplemental material. All other PQ5-based plasmids were derivatives of pQF, except pQF-lacZ, from which pQF itself was derived (see the supplemental material). The multiple-cloning site (MCS) of pQF is flanked by sequences encoding triple-FLAG (3×FLAG) tags. Derivatives of pQF were obtained by exchanging the N-terminal 3×FLAG tag for other affinity tags or fluorescent proteins while maintaining the reading frame of the original MCS and the C-terminal 3×FLAG in all constructs. The N-terminal tags in the different plasmids are as follows: pQF, 3×FLAG tag; pQH, hemagglutinin (HA) tag; pQM, myc tag; pQG, enhanced green fluorescent protein (EGFP); pQY, “super” yellow fluorescent protein 2 (SYFP2); pQR, mCherry; pQC, enhanced cyan fluorescent protein (ECFP). Their construction is detailed in the following explanation. pQH was generated by replacing the HindIII-VspI fragment of pQF by a fragment generated by annealing oligonucleotides oJVZ1088 and oJVZ1089. Similarly, pQM was generated by replacing the HindIII-VspI fragment of pQF with a fragment generated by annealing oligonucleotides oJVZ1090 and oJVZ1091. pQF derivatives for C-terminal fusions to fluorescence proteins were constructed by replacing the HindIII-VspI fragment of pQF with PCR fragments encoding fluorescence proteins amplified with oligonucleotides oJVZ1107 and oJVZ1108. PCR templates for genes encoding fluorescent proteins were as follows: pBBRBB-egfp (30) (Addgene plasmid 32549) for EGFP; pLM-sYFP2 (18) for SYFP2; pMP4516 (31) for ECFP; and pHC119 (a gift from David H. H. Chou) for mCherry. Destination vectors based on PQ5 compatible with Gateway cloning (Invitrogen) were obtained by PCR amplification of the Gateway cassette (including att sites, ccdB, and cat) from pDEST-565 (Addgene plasmid 11520) with oligonucleotides oJVZ740 and oJVZ739, digestion with XbaI and Acc65I, and ligation into the same sites of pQF and its derivatives. Destination plasmids were given the parent's name followed by a “D” (e.g., pQHD was derived from pQH by insertion of the Gateway cassette and allows fusion to the N-terminal HA tag). The pQH derivative pQH2 contains the pBBR1 origin of replication (oriV) and was constructed by PCR amplification of a fragment containing rep, mob, and oriT from pBBR1MCS5 (32) using oligonucleotides oJVZ1436 and oJVZ1437, digestion with PscI and Eco147I, and cloning between the same sites of pQH.
lacZ reporter constructs.
The lacZ transcriptional reporter plasmid pAK501 was constructed in several steps that involved cloning of a putative transcriptional terminator, a “low-background” multiple-cloning site (MCS), and the reporter gene lacZ in plasmid pCM62 (33) followed by exchange of the antibiotic resistance cassette and the origin of replication. Individual steps are described in the following explanation. First, TERM193, a fragment encompassing tandem copies of a putative bidirectional transcriptional terminator encoded in the Sphingomonas Fr1 genome, was amplified with oligonucleotide pair oJVZ745 and oJVZ746 from pTOPO-TERM193 (see Fig. S1 in the supplemental material) and cloned into pCM62 via PscI and HindIII sites, generating plasmid pAK127. Next, lacZ was amplified from E. coli BTH101 (Euromedex) using oligonucleotide pair oJVZ1082 and oJVZ1083 and cloned between the EcoRI and Acc65I sites of pAK127, resulting in the lacZ reporter plasmid pAK127lacZ. Because pAK127lacZ had a high level of background lacZ activity despite the presence of terminators, the MCS of pAK127lacZ was replaced by another MCS generated by cloning annealed oligonucleotides (oJVZ1086 and oJVZ1087) between the HindIII and Acc65I sites of pAK127lacZ, giving pAK127lacZ(MCS). This plasmid showed an approximately 10-fold-reduced background compared to its parent. To construct pAK501, the “lacZ reporter cassette” of pAK127lacZ(MCS) comprising TERM193-MCS-lacZ was combined in several steps with a different broad-host-range oriV (pSV1) and a different antibiotic-resistant marker (cat). To this end, pAK200Cm was first constructed by PCR amplification of cat from pBAD33 (1) using oligonucleotide pair oJVZ1103 and oJVZ1104 and cloning the BamHI-BcuI-digested PCR product between the BglII and BcuI sites of pAK200 (18). pAK200Cm-lacZ was then constructed by cloning a NcoI-NheI-digested PCR product containing the lacZ reporter cassette (TERM193-MCS-lacZ) from pAK127lacZ(MCS) amplified with oligonucleotides oJVZ1105 and oJVZ1106 into the PscI and NheI sites of pAK200Cm. Finally, pAK501 was generated by ligating a Eco147I-BglII-digested fragment amplified by PCR with oligonucleotides oJVZ1102 and oJVZ1103 containing TERM193, the MCS, lacZ, and cat from pAK200Cm-lacZ with the 5.1-kb fragment of pME6010 (34) released by Eco147I-BamHI digestion (pME6010 was purified from the E. coli “dam− dcm−” strain since Eco147I cleavage is methylation impaired in the context of pME6010). Plasmid pAK501 carries the broad-host-range pSV1 origin of replication and is thus compatible with other commonly used origins of replication, such as IncP and pBBR1.
Plasmid pAK501-nepRp reporting general stress response (GSR) activity was generated by cloning annealed oligonucleotides oJVZ1291 and oJVZ1292 harboring the mapped Sphingomonas Fr1 nepR promoter (18) between the HindIII and BamHI sites of pAK501.
syfp2 reporter constructs.
The syfp2 reporter plasmid pAK128 used for screening for strong promoters was constructed by PCR amplifcation of syfp2 from pLM-sYFP2 (18) with oligonucleotides oJVZ743 and oJVZ744 and cloning into pAK127 (see above) via Acc65I and EcoRI sites. Construction of the rpmBp mutant promoter library is described in a separate section (see below).
luxCDABE reporter constructs.
To construct pQ5-lux, a fragment containing PQ5 and cymR* was subcloned from pQF-lacZ into the luciferase reporter plasmid pLM05 (27) using restriction sites NotI and Acc65I. pQ2148-lux was constructed in several steps. First, Psyn2 of plasmid pQH was replaced by a fragment containing the META1_2148 promoter generated by oligonucleotide annealing of oJVZ1434 and oJVZ1435 using HpaI and BsrGI restriction sites, giving plasmid pQ2148. Then, as an intermediary step, a XbaI-EcoRI fragment of pAK127lacZ(MCS) containing lacZ was inserted between the SpeI and EcoRI sites of pQ2148, generating pQ2148-lacZ. Finally, a fragment containing cymR* and PQ2148 was subcloned from pQ2148-lacZ into pLM05 using NotI and Acc65I sites, giving pQ2148-lux.
Other plasmids.
pQH-PhyP was constructed by amplifying phyP with primers oJVZ534 and oJVZ1093, digestion with HindIII and Acc65I, and ligation into pQH digested with the same enzymes; in this construct, PhyP is fused to a 3×FLAG tag at its C terminus and the expression of the PhyP-3×FLAG fusion is driven by PQ5 and the natural phyP ribosome binding site (RBS). pAK200-QPhyP was derived by subcloning a PscI-EcoRI fragment of pQH-PhyP into pAK200 digested with the same enzymes. This plasmid carries the pBBR oriV and is thus compatible with the pAK501 reporter plasmid (pSV1 oriV) and pCM62-based plasmids (IncP oriV). pCM62-PhyR and pCM62-PhyR(D194A) have been described previously (18).
In silico analysis of putative promoters of housekeeping genes.
A permanent draft of the Sphingomonas Fr1 genome (genome name “Sphingomonas melonis FR1”) is available through the Integrated Microbial Genomes (IMG) system (35) on the DOE Joint Genome Institute website (http://img.jgi.doe.gov/cgi-bin/w/main.cgi). Upstream regions of putative housekeeping genes (see Table S2 in the supplemental material) were subjected to a motif search by MEME (36) with the following parameters: distribution of motif occurrences, 0 or 1 per sequence; number of different motifs, 15; minimum number of sites, 5; maximum number of sites, 30; minimum motif width, 20; maximum motif width, 50; searching, “given strand only.” Putative rpmB promoter sequences of sphingomonads were retrieved from the IMG website and aligned using MultAlin (37) using symbol comparison table “DNA” and default parameters.
Screening for and identification of Psyn2.
A 119-bp region upstream of the rpmB (Sphme2DRAFT_0682) open reading frame with randomized nucleotides in the putative −10 and −35 promoter regions was generated by oligonucleotide annealing and extension with Phusion DNA polymerase using oligonucleotides PL28pNTGACN_s and PL28pTANNNGC_as. Products were purified, digested with HindIII and BamHI, cloned into the same sites of pAK128 (a syfp2 reporter plasmid), and transformed into E. coli TOP10. Plasmids were purified from a pool of approximately 10,000 colonies to generate the rpmB promoter library. This library was subsequently transformed into Sphingomonas Fr1, and approximately 20,000 colonies were screened for high fluorescence using an IVIS Spectrum system (Caliper LifeSciences). A total of 14 colonies showing high fluorescence were picked, and the rpmB promoter regions were amplified by colony PCR and sequenced (Microsynth, Switzerland).
Nucleotide sequence accession numbers.
Sequences of pAK501 and pQF have been deposited in GenBank (http://www.ncbi.nlm.nih.gov/GenBank/index.html) under accession numbers KF536587 and KF536588, respectively.
RESULTS AND DISCUSSION
Screening for a strong housekeeping promoter.
Since high gene expression at full induction for the inducible gene expression system was desired, we first sought to identify strong housekeeping promoters in Sphingomonas Fr1. This promoter should constitute a minimal (or core) promoter sequence in order to minimize the risk of additional factors influencing promoter activity. Regions of 99 to 120 bp upstream of 23 putative Sphingomonas Fr1 housekeeping genes (see Table S2 in the supplemental material) were subjected to a motif search using the MEME suite (36). The conserved motif TTGACN-N17-TANNNGC resembling the σ70-dependent promoter consensus of E. coli (TTGACA-N17-TATAAT) was identified in 11 of 23 putative promoter regions by MEME (Fig. 1A), and 7 additional putative promoters from the original set could be manually aligned to this consensus (Fig. 1B). We focused in the following on one of those promoters, rpmBp, which drives expression of the gene encoding ribosomal protein L28 (Sphme2DRAFT_0682).
Fig 1.
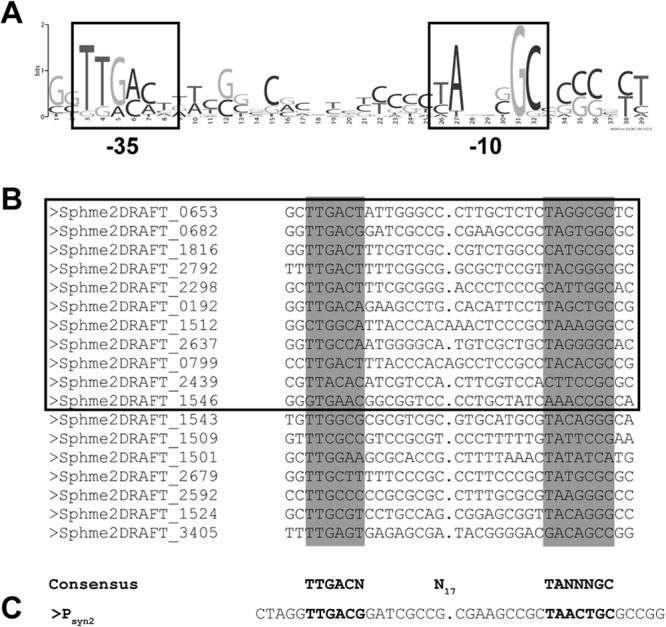
(A) Weblogo output of the motif found by MEME in 11 of 23 query sequences. (B) Alignment of the 11 sequences found by MEME harboring the putative promoter motif (boxed) and additional putative promoters that were aligned manually after reinspection of the promoter regions of the remaining housekeeping genes. Note that the linker length had been adjusted manually by inserting a 1-bp gap in most promoters to better align the −35 and −10 boxes of two promoters. Labels refer to the gene locus tags of the permanent genome draft (http://img.jgi.doe.gov/cgi-bin/w/main.cgi). (C) Sequence of Psyn2 corresponding to positions −43 to +1 in the original rpmB (Sphme2DRAFT_0682) promoter. Note that 1 bp had been inserted in the linker region to align it to the sequences shown in panel B.
Alignment of the putative rpmBp regions of different sphingomonads showed that the presumed promoter motif is the most conserved part in the rpmB upstream region with the putative −35 box TTGACN consensus sequence and the −10 box TANNNGC consensus sequence (see Fig. S3 in the supplemental material), suggesting that the identified motif is the true promoter of rpmB. However, the wild-type rpmB promoter was rather weak when tested as a transcriptional fusion to E. coli lacZ (data not shown). We reasoned that changing nonconserved nucleotides in the −10 and −35 boxes (see above) could generate a stronger promoter and thus screened a library of mutant rpmBp promoters in which those nucleotides had been randomized for increased activity (see Materials and Methods). Among the 14 promoters identified in this screen (see Fig. S4 in the supplemental material), five were identical with the core promoter motif TTGACG-N17-TAACTGC, and the corresponding 44-bp fragment ranging from position −43 to position +1 is referred to as Psyn2 in the following (Fig. 1C).
Construction of a cumate-inducible promoter.
In order to render Psyn2 regulated, we combined it with the control elements of the Pseudomonas putida F1 cym/cmt system, i.e., the transcriptional repressor CymR and its corresponding operator sites (CuO) (17). This system was chosen because the inexpensive inducer cumate is membrane permeative, i.e., does not require any dedicated transporter, and Sphingomonas Fr1 apparently cannot metabolize it since its genome lacks homologs of cumate utilization genes. These features should make induction stable with minimal interference with physiology.
Because it is difficult to predict the behavior of a promoter when it is placed under the control of heterologous regulatory elements (38), several arrangements of Psyn2 with CuO operator sequences and different promoters for cymR were tested (data not shown). In the final configuration shown in Fig. 2, the cumate-inducible promoter, referred to as PQ5, consists of Psyn2 flanked by two CuO operator sequences; expression of a high-GC-content codon-optimized gene for the CymR repressor (cymR*) is driven divergently by a modified bla promoter (Pbla-mut1T) in which the −35 box has been changed to TTGACA and the −10 box to TACAAT. This unit has been assembled together with a multiple cloning site flanked by sequences encoding triple FLAG (3×FLAG) tags into plasmid pCM62 (33), generating plasmid pQF. Details on construction of pQF are given in Fig. S2 and Materials and Methods in the supplemental material.
Fig 2.
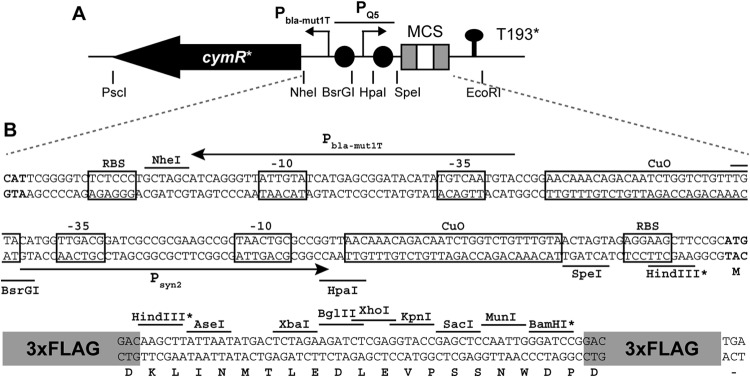
(A) Organization of cymR*, PQ5, and the multiple-cloning site (MCS) on plasmid pQF (not to scale). Restriction sites flanking important features are indicated. T193* denotes a putative transcriptional terminator derived from Sphingomonas Fr1 (see the supplemental material). Gray boxes in the MCS indicate 3×FLAG tags and black circles CuO sites. (B) Nucleotide sequence of the PQ5 promoter region and the MCS. cymR* and MCS start codons are in bold. Ribosome binding sites (RBS), operator sequences (CuO), and −35 and −10 promoter elements are boxed. Core promoter sequences of PQ5 (Psyn2) and the promoter driving cymR* expression (Pbla-mut1T) are indicated by arrows. Translation of the MCS is indicated below the nucleotide sequence. All restriction sites shown are unique in pQF, except those marked with an asterisk. The 3×FLAG tag (peptide sequence DYKDHDGDYKDHDIDYKDDDDK) is shown schematically.
Characterization of PQ5.
To test whether E. coli lacZ could be used as a reporter gene for measuring cumate-dependent gene expression, we first determined the endogenous β-galactosidase activity of the Sphingomonas Fr1 wild type grown with or without cumate. Irrespective of the cumate concentration, no significant β-galactosidase activity (<5 Miller units) was observed, indicating that the strain had no endogenous β-galactosidase activity under the conditions tested and that lacZ is an appropriate reporter gene. Thus, to characterize PQ5-dependent gene expression, a transcriptional fusion of PQ5 with lacZ (plasmid pQF-lacZ) was used and promoter activity was followed by measuring the β-galactosidase activity of Sphingomonas Fr1/pQF-lacZ cultures grown with different cumate concentrations. As seen from the dose-response curve in Fig. 3A, PQ5 steady-state activity was dependent on the inducer concentration and showed a maximal induction ratio of >250-fold at 25 and 50 μM cumate (see also Table 2). We noted that at cumate concentrations of >10 μM, growth was somewhat impaired. However, this was likely due to the high expression level of lacZ and not to the cumate concentration itself because Sphingomonas Fr1 without plasmid pQF-lacZ or carrying another gene, phyP, under PQ5 control (see below) did not show any growth defect even at 100 μM cumate. In time course experiments, addition of 50 μM cumate to an exponentially growing noninduced culture of the same strain (Sphingomonas Fr1/pQF-lacZ) led to a rapid and sustained increase in β-galactosidase activity that was apparent within 10 min after induction and peaked after 3 h (Fig. 3B). The differences in levels of absolute lacZ expression between the experiments whose results are shown in Fig. 3A and B were likely due to the different experimental setups (see Materials and Methods). In summary, these results demonstrate that PQ5 provides a rapidly responding and tunable system for gene expression in Sphingomonas Fr1.
Fig 3.
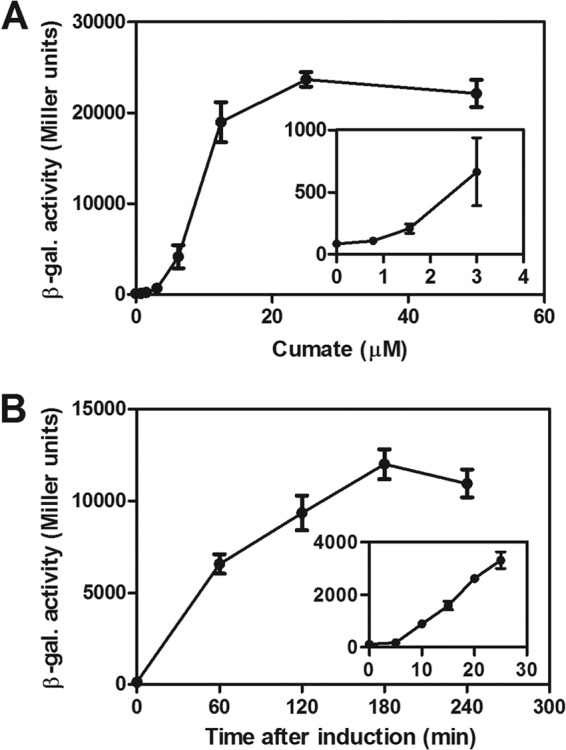
(A) Dose-response curve of PQ5 assayed as β-galactosidase (β-gal.) activity of the PQ5-lacZ+ transcriptional fusion in the Sphingomonas Fr1 wild type (strain JVZ857/pQF-lacZ) grown with different cumate concentrations (no cumate and 2-fold increments from 0.78 to 50 μM cumate). The inset is a closeup of the same dose-response curve illustrating responsiveness to low cumate concentrations. (B) Induction kinetics of PQ5 was assayed by adding 50 μM cumate to a noninduced culture of strain JVZ857/pQF-lacZ in the mid-exponential phase and following β-galactosidase activity of PQ5-lacZ+ over time. The inset represents a set of independent experiments focusing on the first 25 min after inducer addition. Data represent the means ± standard deviations of the results of three independent biological replicate experiments, each performed with two technical replicates.
Table 2.
PQ5 activity in different Alphaproteobacteria
| Strain | β-Galactosidase activity (Miller units)a,c |
Ratiob,c | |
|---|---|---|---|
| Without cumate | With cumate | ||
| Sphingomonas sp. strain Fr1 (JVZ857) | 87 ± 6.9 | 24,000 ± 800 | 270 |
| Sphingomonas aerolata DSM 14751 | 61 ± 7.0 | 16,000 ± 1,300 | 260 |
| Sphingomonas melonis DSM 14444 | 75 ± 3.6 | 6,800 ± 1,400 | 90 |
| Sphingobium indicum DSM 16412 | 1.2 ± 2.1 | 1,700 ± 36 | 1,400 |
| Novosphingobium rosa DSM 7285 | 28 ± 24 | 13,000 ± 3,000 | 480 |
| Caulobacter crescentus CB15 | 10 ± 3.1 | 11,000 ± 1,200 | 1,000 |
| Paracoccus denitrificans JVZ2585 | 1.8 ± 0.6 | 8,700 ± 1,300 | 4,700 |
Data represent means ± standard deviations of the results of three independent biological replicates.
Data represent ratios of unrounded mean values (induced results/uninduced results).
Values are given with two significant digits.
PQ5 allows gene depletion analysis.
One of the most informative applications of conditional gene expression systems is the study of essential genes by depletion analysis. To test whether this was possible with PQ5, we chose the essential gene phyP of Sphingomonas Fr1 (18). phyP encodes a putative phosphatase of the response regulator PhyR that is a master regulator of the general stress response (GSR) in Alphaproteobacteria (39). phyP cannot be deleted unless phyR is also deleted or replaced by the phyRD194A allele encoding a nonphosphorylatable version of PhyR, suggesting that phosphorylation of PhyR is the cause of lethality. Accordingly, it has been proposed that phyP is essential because its deletion would lead to overactivation of the GSR (18).
To test this hypothesis, we used a strain deleted for chromosomal phyP and phyR (strain JVZ1357) (18) complemented in trans with a functional PhyP-3×FLAG fusion expressed under the control of PQ5 (plasmid pAK200-QPhyP) and phyR under the control of its own promoter (plasmid pCM62-PhyR). As a readout for activation of the GSR, this strain also harbored a plasmid carrying the nepRp-lacZ+ transcriptional fusion (plasmid pAK501-nepRp) in which lacZ expression is driven by the previously mapped, PhyR-dependent nepR promoter (18). Growth of this strain on LB agar or in liquid LB required the addition of cumate (data not shown), suggesting that in the absence of cumate, PhyP-3×FLAG was depleted below the level required for viability. When grown in liquid culture containing cumate, removal of the inducer by extensive washing and dilution into fresh medium lacking cumate (OD600 of 0.025) resulted in growth arrest in the exponential phase (OD600 of ca. 2) after 6 to 7 generations; in contrast, when the same culture was diluted into fresh medium containing cumate, growth was normal and continued until it reached the stationary phase (OD600 of >10). Western blots confirmed that PhyP-3×FLAG levels were dramatically decreased in the depleted strain compared to the nondepleted strain (Fig. 4A). Decreased levels of PhyP-3×FLAG correlated with increased nepRp-lacZ+ activity as measured with β-galactosidase assays (Fig. 4B), demonstrating that, indeed, loss of PhyP leads to GSR overactivation. As expected, a control strain expressing nonphosphorylatable PhyRD194A [plasmid pCM62-PhyR(D194A)] instead of wild-type PhyR displayed reduced nepRp-lacZ+ activity and showed no dependence on cumate with respect to growth (data not shown) and GSR activation (Fig. 4). Similar results were obtained for otherwise isogenic strains lacking pAK501-nepRp (data not shown), indicating that the observed phenotypes were not an artifact of lacZ expression. Altogether, these results verify that phyR is epistatic to phyP (18) and support the idea that phyP is essential because it prevents detrimental overactivation of the general stress response through prevention of constitutive PhyR phosphorylation. More generally, they demonstrate the utility of PQ5 for studying essential gene function.
Fig 4.
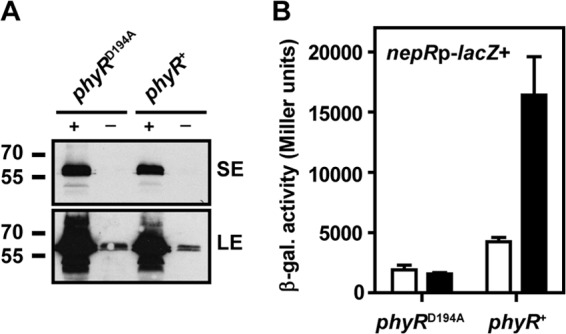
(A) Western blot of the PhyP-3×FLAG-depletion strain harboring the phyR wild-type (phyR+) or D194A mutant (phyRD194A) allele grown with (+) or without (−) cumate (see the main text for details). PhyP-3×FLAG was detected using primary mouse monoclonal anti-FLAG M2 antibodies and secondary goat anti-mouse horseradish peroxidase-coupled IgG antibodies and ECL Western blotting detection reagent. Membranes were exposed to X-ray films for a short exposure of 20 s (SE) as well as a long exposure of 15 min (LE) to visualize residual PhyP-3×FLAG in the depleted strains. Numbers on the left indicate molecular mass markers (in kDa). (B) nepRp-lacZ+ β-galactosidase activity assayed in the same strains as represented in panel A grown with cumate (white bars) or without cumate (black bars). Three independent biological replicate experiments were performed, and data are displayed as means ± standard deviations. Results in panel A are representative of the results of three replicate experiments.
Use of PQ5 in other Alphaproteobacteria.
To broaden the potential use of our system, we first tested it in a number of other sphingomonads using plasmid pQF-lacZ. As seen from Table 1, in all cases high induction ratios of approximately 100- to 1,000-fold between noninduced and cumate-induced cultures were obtained as measured by β-galactosidase assays. In the cases where the induction ratio was even higher than in Sphingomonas Fr1, this stemmed from a tighter repression rather than from higher promoter activity. Indeed, Sphingomonas Fr1 displayed the highest absolute β-galactosidase activity of all strains tested. In general, we speculate that strain-specific differences in induction ratios and absolute β-galactosidase activity can be attributed to different strengths of PQ5 and/or the promoter driving expression of CymR, Pbla-mut1T, although we cannot exclude the possibility that there are other factors involved.
We next tested PQ5 in other alphaproteobacterial species representing the orders Caulobacterales (C. crescentus), Rhodobacterales (P. denitrificans), and Rhizobiales (M. extorquens and A. tumefaciens). For all species except M. extorquens, PQ5 activity was monitored using plasmid pQF-lacZ; for M. extorquens, a fragment containing PQ5 and cymR* was subcloned in a previously described bacterial luciferase (luxCDABE) transcriptional reporter plasmid (27). Whereas PQ5 was essentially inactive in A. tumefaciens (<2 Miller units with or without cumate; data not shown), the promoter was inducible in the other species tested with induction ratios of approximately 1,000- to 5,000-fold (Table 2 and Fig. 5B). As discussed above for sphingomonads, these high induction ratios are due to tighter repression rather than to higher activity of PQ5. Similar to results obtained from Sphingomonas Fr1, in M. extorquens the response to inducer addition was rapid (apparent after approximately 10 min) and expression was tunable over a wide range of inducer concentrations. We also tested a derivative of PQ5, PQ2148, in which the Psyn2 core promoter had been replaced by a minimal promoter from M. extorquens AM1 (Fig. 5A) naturally driving expression of rpsL (META1_2148). This promoter was chosen because it shows a clear single transcriptional start site and is active in both methanol- and succinate-grown bacteria according to transcriptome sequencing (RNA-Seq) data (A. Francez-Charlot and J. A. Vorholt, unpublished data). Similar to PQ5, PQ2148 was rapidly and highly inducible and showed approximately 2-fold-higher activity than PQ5 at the highest cumate concentration tested (Fig. 5C).
Fig 5.
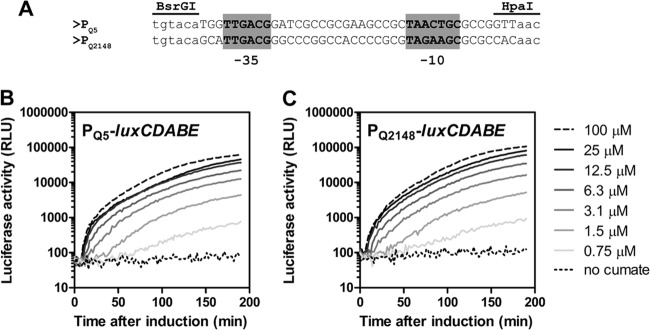
(A) Alignment of core promoter regions in PQ5 and PQ2148. The sequence between BsrGI and HpaI is the only difference between the two reporter plasmids pQ5-lux and pQ2148-lux. (B and C) Time courses of cumate-dependent induction of PQ5-luxCDABE and PQ2148-luxCDABE reporter constructs. Cumate concentrations used are indicated on the right. Luciferase activity is given in arbitrary units (RLU [relative luminescence units]). Data are from a single experiment that is representative of three independent biological replicates.
Altogether, these results indicate that PQ5 can be used as a versatile inducible expression system in Sphingomonadales and also in members of other orders of the Alphaproteobacteria. In addition, results with PQ2148 suggest that Psyn2 may be easily exchanged for other species-specific promoters to achieve, for example, tighter repression or higher expression levels in cases where Psyn2 does not provide the desired characteristics.
A comprehensive set of cumate-inducible plasmids.
In addition to pQF, a number of PQ5-based plasmids have been constructed (see Table 1) that carry, in addition to the carboxy-terminal 3×FLAG tag, different amino-terminal epitope tags (HA, myc, fluorescent proteins) commonly used for immunodetection or protein localization studies. In all constructs, the original reading frame displayed in Fig. 2B is maintained. Besides conventional cloning vectors, for most plasmids derivatives compatible with the Gateway cloning system (Invitrogen) have also been constructed. Last, we have constructed a PQ5-based plasmid carrying the broad-host-range pBBR origins of replication and transfer that may be used in bacteria for which pCM62-based plasmids are not suitable, such as Bradyrhizobium japonicum (H.-M. Fischer, personal communication). Plasmids are available through the Addgene plasmid repository (www.addgene.org).
Conclusions.
We developed a tight and highly inducible gene expression system for sphingomonads, for which this is the first such system described, and demonstrated its successful application for depletion analysis. This system adds a valuable part to the genetic toolbox of sphingomonads, and we anticipate that it will be of broad use for both basic and applied research. In addition, the system was demonstrated to work in a number of other Alphaproteobacteria in which it may be used to complement already existing inducible gene expression systems (3, 6, 40–42).
Supplementary Material
ACKNOWLEDGMENTS
We thank James Tauber for performing initial experiments on PhyP depletion. We are grateful to David H. H. Chou, Denis Faure, Dieter Haas, Dominic Esposito, Hans-Martin Fischer, Tobias J. Erb, and Urs Jenal for providing strains and/or plasmids.
This work was supported by the Swiss National Science Foundation (SNF) through research grant 31003A-135623.
Footnotes
Published ahead of print 30 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02296-13.
REFERENCES
- 1. Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iniesta AA, Garcia-Heras F, Abellon-Ruiz J, Gallego-Garcia A, Elias-Arnanz M. 2012. Two systems for conditional gene expression in Myxococcus xanthus inducible by isopropyl-beta-D-thiogalactopyranoside or vanillate. J. Bacteriol. 194:5875–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meisenzahl AC, Shapiro L, Jenal U. 1997. Isolation and characterization of a xylose-dependent promoter from Caulobacter crescentus. J. Bacteriol. 179:592–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Quisel JD, Burkholder WF, Grossman AD. 2001. In vivo effects of sporulation kinases on mutant Spo0A proteins in Bacillus subtilis. J. Bacteriol. 183:6573–6578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stephens C, Christen B, Watanabe K, Fuchs T, Jenal U. 2007. Regulation of D-xylose metabolism in Caulobacter crescentus by a LacI-type repressor. J. Bacteriol. 189:8828–8834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thanbichler M, Iniesta AA, Shapiro L. 2007. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res. 35:e137. 10.1093/nar/gkm818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yansura DG, Henner DJ. 1984. Use of the Escherichia coli lac repressor and operator to control gene expression in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 81:439–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Browning DF, Busby SJ. 2004. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2:57–65 [DOI] [PubMed] [Google Scholar]
- 9. Choi YJ, Morel L, Bourque D, Mullick A, Massie B, Miguez CB. 2006. Bestowing inducibility on the cloned methanol dehydrogenase promoter (PmxaF) of Methylobacterium extorquens by a applying regulatory elements of Pseudomonas putida F1. Appl. Environ. Microbiol. 72:7723–7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choi YJ, Morel L, Le Francois T, Bourque D, Bourget L, Groleau D, Massie B, Miguez CB. 2010. Novel, versatile, and tightly regulated expression system for Escherichia coli strains. Appl. Environ. Microbiol. 76:5058–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takeuchi M, Hamana K, Hiraishi A. 2001. Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int. J. Syst. Evol. Microbiol. 51:1405–1417 [DOI] [PubMed] [Google Scholar]
- 12. Balkwill DL, Fredrickson JK, Romine MF. 2006. Sphingomonas and related genera, p 605–629 In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E. (ed), The prokaryotes: a handbook on the biology of bacteria, vol 7, proteobacteria, delta and epsilon subclasses. Deeply rooting bacteria Springer-SBM, New York, NY [Google Scholar]
- 13. Fialho AM, Moreira LM, Granja AT, Popescu AO, Hoffmann K, Sa-Correia I. 2008. Occurrence, production, and applications of gellan: current state and perspectives. Appl. Microbiol. Biotechnol. 79:889–900 [DOI] [PubMed] [Google Scholar]
- 14. Innerebner G, Knief C, Vorholt JA. 2011. Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl. Environ. Microbiol. 77:3202–3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. White DC, Sutton SD, Ringelberg DB. 1996. The genus Sphingomonas: physiology and ecology. Curr. Opin. Biotechnol. 7:301–306 [DOI] [PubMed] [Google Scholar]
- 16. Eaton RW. 1996. p-Cumate catabolic pathway in Pseudomonas putida Fl: cloning and characterization of DNA carrying the cmt operon. J. Bacteriol. 178:1351–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eaton RW. 1997. p-Cymene catabolic pathway in Pseudomonas putida F1: cloning and characterization of DNA encoding conversion of p-cymene to p-cumate. J. Bacteriol. 179:3171–3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaczmarczyk A, Campagne S, Danza F, Metzger LC, Vorholt JA, Francez-Charlot A. 2011. Role of Sphingomonas sp. strain Fr1 PhyR-NepR-σEcfG cascade in general stress response and identification of a negative regulator of PhyR. J. Bacteriol. 193:6629–6638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nunn DN, Lidstrom ME. 1986. Isolation and complementation analysis of 10 methanol oxidation mutant classes and identification of the methanol dehydrogenase structural gene of Methylobacterium sp. strain AM1. J. Bacteriol. 166:581–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poindexter JS. 1964. Biological properties and classification of the Caulobacter group. Bacteriol. Rev. 28:231–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smith EF, Townsend CO. 1907. A plant-tumor of bacterial origin. Science 25:671–673 [DOI] [PubMed] [Google Scholar]
- 22. Jin DJ, Gross CA. 1988. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J. Mol. Biol. 202:45–58 [DOI] [PubMed] [Google Scholar]
- 23. Peyraud R, Kiefer P, Christen P, Massou S, Portais JC, Vorholt JA. 2009. Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. Proc. Natl. Acad. Sci. U. S. A. 106:4846–4851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaczmarczyk A, Vorholt JA, Francez-Charlot A. 2012. Markerless gene deletion system for sphingomonads. Appl. Environ. Microbiol. 78:3774–3777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simon R, Priefer U, Pühler A. 1983. A broad-host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Nat. Biotechnol. 1:784–791 [Google Scholar]
- 26. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 27. Metzger LC, Francez-Charlot A, Vorholt JA. 2013. Single-domain response regulator involved in the general stress response of Methylobacterium extorquens. Microbiology 159:1067–1076 [DOI] [PubMed] [Google Scholar]
- 28. Laemmli UK. 1970. Cleavage of structural proteins during assembly of head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 29. Sambrook J, Russel D. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 30. Vick JE, Johnson ET, Choudhary S, Bloch SE, Lopez-Gallego F, Srivastava P, Tikh IB, Wawrzyn GT, Schmidt-Dannert C. 2011. Optimized compatible set of BioBrick vectors for metabolic pathway engineering. Appl. Microbiol. Biotechnol. 92:1275–1286 [DOI] [PubMed] [Google Scholar]
- 31. Bloemberg GV, Wijfjes AH, Lamers GE, Stuurman N, Lugtenberg BJ. 2000. Simultaneous imaging of Pseudomonas fluorescens WCS365 populations expressing three different autofluorescent proteins in the rhizosphere: new perspectives for studying microbial communities. Mol. Plant Microbe Interact. 13:1170–1176 [DOI] [PubMed] [Google Scholar]
- 32. Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, II, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176 [DOI] [PubMed] [Google Scholar]
- 33. Marx CJ, Lidstrom ME. 2001. Development of improved versatile broad-host-range vectors for use in methylotrophs and other Gram-negative bacteria. Microbiology 147:2065–2075 [DOI] [PubMed] [Google Scholar]
- 34. Heeb S, Itoh Y, Nishijyo T, Schnider U, Keel C, Wade J, Walsh U, O'Gara F, Haas D. 2000. Small, stable shuttle vectors based on the minimal pVS1 replicon for use in gram-negative, plant-associated bacteria. Mol. Plant Microbe Interact. 13:232–237 [DOI] [PubMed] [Google Scholar]
- 35. Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang JH, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 40:D115–D122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers, p 28–36 In Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. AAAI Press, Menlo Park, CA [PubMed] [Google Scholar]
- 37. Corpet F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16:10881–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lanzer M, Bujard H. 1988. Promoters largely determine the efficiency of repressor action. Proc. Natl. Acad. Sci. U. S. A. 85:8973–8977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Francez-Charlot A, Frunzke J, Reichen C, Ebneter JZ, Gourion B, Vorholt JA. 2009. Sigma factor mimicry involved in regulation of general stress response. Proc. Natl. Acad. Sci. U. S. A. 106:3467–3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chubiz LM, Purswani J, Carroll SM, Marx CJ. 2013. A novel pair of inducible expression vectors for use in Methylobacterium extorquens. BMC Res. Notes 6:183. 10.1186/1756-0500-6-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ind AC, Porter SL, Brown MT, Byles ED, de Beyer JA, Godfrey SA, Armitage JP. 2009. Inducible-expression plasmid for Rhodobacter sphaeroides and Paracoccus denitrificans. Appl. Environ. Microbiol. 75:6613–6615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tett AJ, Rudder SJ, Bourdes A, Karunakaran R, Poole PS. 2012. Regulatable vectors for environmental gene expression in Alphaproteobacteria. Appl. Environ. Microbiol. 78:7137–7140 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.