

Abstract
Ligand affinities can be optimized by interfacial cavity filling. A hollow (Phe43 cavity) between HIV-1 surface protein (gp120) and cluster of differentiation 4 (CD4) receptor, extends beyond residue phenylalanine 43 of CD4 and cannot be fully accessed by natural amino acids. To increase HIV-1 gp120 affinity for a family of CD4-mimetic miniproteins (miniCD4s), we targeted the gp120 Phe43 cavity with eleven non-natural phenylalanine derivatives, introduced into a miniCD4 named M48 (1). The best derivative named M48U12 (13) binds HIV-1 YU2 gp120 with 8 pM affinity, and shows potent HIV-1 neutralization. It contained a methylcyclohexyl derivative of 4-aminophenylalanine and its co-crystal structure with gp120 revealed the cyclohexane ring buried within the gp120 hydrophobic core but able to assume multiple orientations in the binding pocket, and an aniline nitrogen potentially providing a focus for further improvement. Altogether, the results provide a framework for filling the interfacial Phe43 cavity to enhance miniCD4 affinity.
Keywords: miniCD4, gp120, HIV, protein interaction, HIV-1 neutralization, X-ray crystallography
Introduction
Cell entry of the human immunodeficiency virus type 1 (HIV-1) is a multi-step process initiated by the binding of the HIV-1 surface protein (gp120) envelope glycoprotein on the viral spike to the cluster of differentiation 4 (CD4) receptor on appropriate target cells. This association stabilizes a conformation of gp120 that enables it to bind to a chemokine co-receptor, CCR5 or CXCR4.1-3 Co-receptor binding in turn activates the fusogenic properties of the noncovalently associated transmembrane component (gp41) of the viral spike, and subsequent spikeconformational rearrangements and virus-host membrane fusion lead to entry of the HIV-1 genetic material into the cell.4 Each of these steps represents a potential target for therapeutic intervention.5-8
For over a decade, we have been developing peptide CD4 mimics, called miniCD4s, which are based on the transplantation of the gp120-binding surface of CD4 into the context of various scorpion-toxin scaffolds.9 These disulfide rich peptides belong to the knottin family, which has been widely utilized as scaffolds for developing stable peptide ligands through loop grafting of bioactive peptides.10 and references therein The 31-residue scyllatoxin, from the scorpion Leiurus quinquestriatus hebraeus, was selected based on the similarity of the gp120-binding portion of CD4 with the scyllatoxin structure, which consists of a β-hairpin linked to a short helix. The scyllatoxin β-hairpin between residues 18-29scyllatoxin superimposes with a root-mean-square deviation (RMSD) of 1.1 Å with CD4 residues 36-47CD4, which are involved in binding gp1209 (to aid in clarity, residues are identified by a subscript of the parent macromolecule). Moreover, the scyllatoxin fold, stabilized by three disulphide bridges (Figure S1),11 is highly permissive to mutation,12 stable to acidic pH and high temperatures,12 relatively resistant towards proteolytic degradation,13 and poorly immunogenic.14 Optimization of the scyllatoxin peptide structure led to miniCD4s of only 27 residues and affinities for gp120 comparable to that of CD4 itself.15 These miniproteins stabilize the CD4-bound conformation of gp120 as observed by X-ray crystallography15,16 as well as by immunogenic elicitation: thus, cross-linking of a miniCD4 to HIV-1 gp120 or HIV-1 gp140 (gp120 coupled to the extracellular part of gp41) enhances its binding to the CCR5 receptor, and injection of this cross-linked complex into rabbits significantly increases the production of CD4-induced antibodies that recognize the site of CCR5 binding on gp120.17,18 MiniCD4s are also able to neutralize HIV entry in vitro15,16,19 and in vivo, as shown recently in a macaque vaginal simian-human immunodeficiency virus (SHIV) challenge.20
An interfacial cavity, called the Phe43 cavity, extends from the tip of the gp120-interactive β-hairpin of CD4 into the heart of HIV-1 gp120. This mostly hydrophobic cavity is capped by the phenyl side chain of residue 43CD4. The miniCD4, named M48 (1),15 recognizes the entrance of the Phe43 cavity in a manner similar to that of CD4.15 To enhance the interaction between gp120 and miniCD4, we synthesized peptide CD4 mimics, in which the side chain of residue 23miniCD4 – the miniCD4 residue positioned at the mouth of the Phe43 cavity– was replaced by a number of unnatural phenylalanine derivatives. The antiviral activity of four such peptides (2-4 and 14) was determined previously,15,19 with 2 being the most potent. In a related study, we determine the structural basis for 2 recognition of HIV-1 gp120 and show that optimal filling of the Phe43 cavity is a mechanism for enhancing affinity and neutralization potency of a CD4-binding site ligand.21 To provide a fuller understanding of how interactions with the Phe43 cavity might contribute to HIV-1 gp120 affinity, we synthesized miniCD4 derivatives of 1, each with a unique 23miniCD4 extension. This paper describes the synthesis of eleven novel unnatural amino acids, which were used to replace residue 23M48. Their incorporation, along with three commercially available unnatural amino acids, into 1, allowed us to compare the relative affinity for 15 miniCD4 (including 1) ligands to HIV-1, and we did so with two gp120s, one belonging to clade B and the other to clade C. The antiviral activity of the six tightest binding derivatives of 1 was assessed against two clade B lab strains, two clade C primary isolates and two transmitter/founder (T/F) viruses, one belonging to clade B and the other to clade C. The tightest binder and most potent neutralizer M48U12 (13) was analyzed for its gp120-binding affinity by surface plasmon resonance (SPR) and crystallized in complex with HIV-1 gp120 from the clade B YU2 isolate. The co-crystal structure at 1.9 Å-resolution provides atomic-level details for 13, which displays both extraordinary affinity and potent neutralization. Together, these results show how appropriately filling of an interfacial cavity through a combination of rational design, chemical synthesis, and functional/structural feedback allows for potent near-pan neutralization of HIV-1 by miniCD4 mimetics.
Results
Peptide Synthesis
MiniCD4s were obtained by standard solid phase peptide synthesis using Fmoc chemistry and by introducing various commercial or homemade amino acid derivatives at position 23miniCD4. After deprotection, the miniCD4s were folded by adding the crude peptide mixtures to Tris buffer (pH 8) containing the glutathione redox couple. Addition of two molar guanidinium hydrochloride in the folding buffer was required to increase the solubility of the various linear peptides. MiniCD4s were purified by reverse phase HPLC. Two series of miniCD4s were synthesized. The first one was obtained on the M3313 (16) family (TpaNLHFCQLRCKSLGLLGKCAGSXCACV-NH2, where Tpa stands for thiopropionyl, and X for Phe, Trp or unnatural amino acid) with commercially available amino acids. The second was built with three commercially available and eleven homemade unnatural amino acids on the more recently developed M48 (1) family (TpaNLHFCQLRCKSLGLLGRCA-DPro-TXCACV-NH2, where X stands for Phe or unnatural amino acid). We thus obtained a panel of twenty-four miniCD4s, some of which have been partially described previously.13
Competitive Binding Assays on a M33 (16) Series
Relative apparent gp120-binding affinities of the various miniCD4s, modified at position 23, were determined by competitive ELISA, using soluble CD4 (sCD4) as competitor. The miniCD4s were tested on clade B gp120SF162 and on clade C gp120CN54. A preliminary study done on the M33 (16) family, which contains commercially available, natural or unnatural amino acids at position 23, showed systematically higher potency with various miniCD4s for clade B virus compared to the clade C virus (Table S1; IC50CN54/IC50SF162 > 10). Most of the miniCD4s showed inhibition constants in the nanomolar range for clade B gp120SF162, but apart from the biphenyl- (16) or phenylalanine (17) derivatives, they all have micromolar IC50s for clade C gp120CN54. While the biphenyl derivative marginally improves the apparent affinity over the phenyl derivative, neither tryptophan nor the other six unnatural amino acids tested showed an improvement in affinity (see ratio A and B in Table S1). The modifications, whether extending the linker between the alpha carbon and the phenyl moiety (18 and 19) or using bicyclic residues (20 and 21), or introducing a methoxy (23), a hydrophobic methyl (22) or tert-butyl (24) group at the para position of the phenylalanine, all led to a loss in gp120-binding affinity of the miniCD4s. While both tryptophan and 2-naphtylalanine were deleterious, the latter was less deleterious, since 20 showed low nanomolar affinity for the SF162 surface protein. The most deleterious effect was observed on 24 and 18, where the tert-butyl or the phenyl moiety, respectively, probably clash with the narrowest part of the pocket (this narrowing can be seen in Figure 1). Lengthening the linker between the alpha carbon and the aromatic ring, as was done for 19, enhanced the affinity compared to 18 suggesting that the aromatic ring probably lies in the widest and deepest part of the cavity. The presence of a p-methyl group (22) on the phenylalanine resulted in a small loss of affinity for SF162 gp120, which could be reversed by replacing the p-methyl by a p-phenyl moiety (16), which again probably increases hydrophobic contacts with the deepest part of the pocket,16 while the indole entity of 21 appears to create a steric hindrance at the inlet of the cavity.
Figure 1.

HIV-1 YU2 gp120 binding to sCD4 and CD4 mimetic miniproteins. (A) gp120 binding to the CD4-mimetic miniproteins and to sCD4. gp120 is shown in cartoon representation. CD4 (shown as a transparent yellow surface) binds at the interface of three gp120 domains- inner domain (pale cyan), outer domain (pale green) and bridging sheet (light pink). Phe43CD4 (yellow stick representation) inserts into a gp120 cavity, known as the Phe43 cavity, formed at the intersection of these domains. The miniCD4s are shown in red cartoon representation with side chain of position 23 shown as sticks. Residue 23miniCD4 inserts into the gp120 Phe43 cavity; (B) 90° rotated view of (A). For clarity, the CD4 surface is not shown in this figure. (C) Zoomed-in cross-section of gp120 surface showing the Phe43 cavity with the bound inserts: Phe43 for CD4 (yellow, PDB ID: 1RZK), Phe23 for 1 (wheat, PDB ID: 2I60), biphenylalanine 23 for 14 (slate, PDB ID: 2I5Y), Phe23 for 17 (brown, PDB ID: 1YYM), biphenylalanine 23 for 16 (marine, PDB ID: 1YYL). (D) 180° rotated view of C. For clarity, gp120 surface from only one complex (PDB ID: 2I60) is shown in C and D.
While these results provided useful structure activity relationship of ligand occupation of the gp120 Phe43 cavity, no improvements in affinity of 16 could be achieved using commercially available amino acids. We, therefore, decided to synthesize novel, unnatural amino acids to introduce into the more recently developed M48 (1) sequence.15
Chemistry
Novel unnatural amino acids suitable for Fmoc-SPPS were obtained using a synthetic route described earlier,22 producing a series of ethers derived from the tyrosine amino acid, using the Mitsunobu reaction on solid phase synthesis (Scheme 1). The tyrosine (1′) phenol and amino groups were first protected by an allyloxycarbonyl group. The obtained compound (2′) was immobilized onto a solid support (Wang resin) via its carboxylic moiety. Loading was estimated by cleavage and weight analysis of small samples of resin. 1H NMR of crude aliquots confirmed the reaction progress. An average 87% loading was obtained for the bis-allyloxycarbonyl protected compound (3′), which was close to the earlier 91% obtained by Morley.23 Piperidine treatment led to selective removal of the carbonate group in mild conditions thereby, avoiding detachment of the template from the solid support and deprotection of the allylcarbamate. With a 95% reaction yield, estimated by weighing the resin, an average loading of 1.03 mmol/g was obtained for the resin harboring the monoprotected allyloxycarbonyl derivative (4′). This selective removal of the phenolic protecting group generated an intermediate suitable for the synthesis of an ether library by using the Mitsunobu reaction. Commercially available alcohols were used to prepare resins 5′-12′. Monoprotected diols 44′-46′, used for the synthesis of resins 10′-12′, were prepared by one protecting step from commercially available compounds (Scheme 2). To obtain resin 13′, the N-Cbz-L-proline (47′) carboxylic acid had first to be transformed into an alcohol function. This was done by formation of a mixed anhydride with isobutyl chloroformate, followed by a reduction with sodium borohydride, yielding N-Cbz-(S)-(+)-2-pyrrolidinemethanol 48′ (Scheme 3). The Mitsunobu reaction was carried out in a 1/1 mixture of DCM/THF with PPh3, diisopropylazodicarboxylate (DIAD) and the various alcohols, in a double coupling strategy, as proton NMR showed that some phenol remained after the first coupling. Loading yield of the resins 5′-13′ obtained after the Mitsunobu reaction, as well as the next two steps yields were not determined. Instead, a global yield for these three steps was calculated at the end of the synthesis. The next step was the allyloxycarbonyl moiety removal on the nitrogen atom to reveal the primary amine, by activation with Pd(PPh3)4 in mild conditions where other functional groups remained inert. Then, the resulting amino-resins 14′-22′ were protected by a Fmoc moiety to give Fmoc-amino-acid resins 23′-31′. The last step, i.e. cleavage from the solid support, was carried out under acidic conditions in a mixture of TFA/DCM. After extraction and filtration, the final Fmoc-protected amino acid compounds 32′ to 40′ were obtained with overall yields ranging between 26% and 63% for the last three steps.
Scheme 1. Synthesis of unnatural alkoxy-derivatives of phenylalanine protected for Fmoc-peptide synthesisa.

aReagents: (a) ClCOOCH2CHCH2, NaOH 4 N; (b) Wang resin, diisopropylcarbodiimide, DMAP, NMP; (c) 20% piperidine, DMF; (d) ROH, PPh3, DIAD, DCM/THF; (e) Pd(PPh3)4, Ar, THF/DMSO/HCl/morpholine; (f) Fmoc-Cl, i-Pr2NEt, DCM; (g) TFA/DCM
Scheme 2. Monoprotection of diol derivativesa.

aReagents: (a) BnBr, NaH, dry THF
Scheme 3. Synthesis of alcohol 48′a.

a Reagents: (a) 4-Methylmorpholine, (CH3)2CHCH2OCOCl, DME; (b) NaBH4, DME/H2O
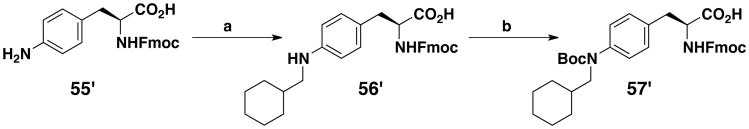
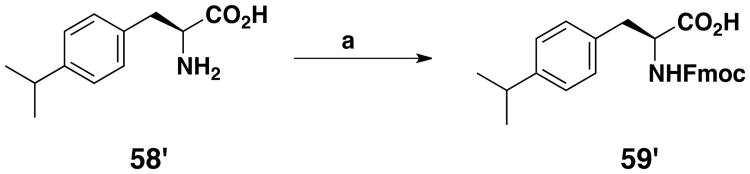
Two Fmoc-protected amino acids 54′ and 57′ were obtained by solution synthesis. For 54′ (Scheme 4), one of the two alcohol functions of 2,2′-thiodiethanol (49′) was first protected with a tert-butyldimethylsilyl moiety. The derivative that was obtained (50′) was coupled to Cbz-Tyr(OH)-OBn using the Mitsunobu reaction. The carboxylic and amine functions were then deprotected by catalytic reduction on Pd black, just before a standard Fmoc protection of the amine moiety. Finally, the sulfoxide (54′) was obtained in a 33% overall yield by oxidation with a tert-butyl hydroperoxide and thiourea dioxide mixture. The last unnatural amino acid that we developed was a methylcyclohexyl derivative of Fmoc-p-aminophenylalanine (57′). It is an analog of 32′, with a nitrogen atom leading to a phenylalkyl amine replacing the oxygen atom of the phenyl ether moiety. It was synthesized by reductive amination on Fmoc-p-amino-L-phenylalanine (55′) using sodium triacetoxyborohydride in CH3CN/AcOH with cyclohexanecarboxaldehyde (Scheme 5). The secondary amine that was obtained was then protected with the tert-butyloxycarbonyl moiety compatible with Fmoc peptide chemistry. The overall yield of 57′ was 73%. Fmoc-protected amino acid 59′ was obtained by reaction of the commercially available amino acid 58′ with Fmoc-chloroformate (Scheme 6).
Scheme 4. Synthesis of the sulfoxide Fmoc-protected amino acid 54′a.

aReagents: (a) TBSCl, NaH, dry THF; (b) Z-Tyr-OBn, PPh3, DEAD, DMF/toluene; (c) Pd black, H2, MeOH; (d) Fmoc-O-succinimide, NaHCO3, DMF, CH3CN, H2O; (e) TBHP, thiourea dioxide, DCM
Scheme 5. Synthesis of Fmoc-protected amino acid 57′a.
aReagents: (a) C6H11CHO, NaBH(OAc)3, CH3CN/AcOH; (b) (Boc)2O, i-Pr2NEt, THF
Scheme 6. Synthesis of Fmoc-protected amino acid 59′a.
aReagents: (a) Fmoc-Cl, Na2CO3, dioxane/H2O
Competitive Binding Assays on a M48 (1) Series
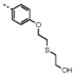
Competitive ELISA assay showed the global superiority of the M48 (1) series over the M33 (16) series (Table 1 compared to Table S1). Eight derivatives shared subnanomolar and five low nanomolar apparent affinities for the clade B SF162 strain. Moreover, two derivatives had subnanomolar and five low nanomolar affinities for the clade C strain. Four derivatives (1, 11, 12, 14) were made with commercially available amino acids, and as was observed for the M33 (16) series, no improvement in binding affinity to gp120, over that of 1, could be achieved for 11, 12 or 14 (previously called M4715). Moreover, the results obtained with 12 confirmed the data obtained for 24, as these p-tert-butylphenylalanine derivatives share the worst IC50 values for both gp120s. However, the affinity loss, on changing the phenylalanine residue for a p-tert-butylphenylalanine, is much higher in the M48 (1) family versus the M33 (16) one (26-fold higher for gp120SF162 and about 13-fold higher for gp120CN54). Interestingly, replacing the p-tert-butyl group by a slightly less bulky isopropyl one (11) enhanced the apparent affinity on SF162 by a factor of seven, while no difference was observed on the C strain gp120, suggesting differences in the Phe43 cavity shape between these two gp120 proteins. Notably, this phenomenon is also present for 2, 4, 11, 13 and 15, with their B/A ratio (impact of the miniCD4 amino acid modification on position 23 on the affinity for gp120 on clade C compared to clade B; see Table 1) deviating significantly from 1. While for the M33 (16) series, the biphenyl moiety marginally increased the apparent binding affinity to gp120 compared to the phenyl group (Table S1; A = 0.9, B = 0.6), the effect was opposite in the M48 (1) family (Table 1; A = 2.6, B = 5.3), with the biphenyl substitution resulting in loss in gp120 binding affinity. X-ray crystallographic studies have already been published for the complexes between the clade B YU2 gp120 and 16, 17 (earlier called F23),16 1 (earlier called [Phe23M47]) and 14 (earlier called M47).15 These data are consistent with the overall small differences between these representative miniCD4s observed in our binding assays, especially on the clade B strain, as the maximum root-mean-square deviation of gp120 is within 0.5 Å for the biphenyl group and 0.3 Å for the phenyl moiety, when comparing the two families (Figure 1). Of the miniCD4s built with the eleven novel unnatural amino acids synthesized in our lab, six showed enhanced affinity for gp120SF162, compared to 1 and four also showed an affinity increase for the clade C gp120CN54. Interestingly, extending the linker between the phenyl ether and the cyclohexyl moiety one carbon atom at a time, going from 2 to 5 and then to 6, demonstrated that shorter the linker, better the affinity for the HIV-1 surface protein. A small loss of affinity was observed at each step, and the effect, once again, was greater on the clade C strain. Exchanging the cyclohexyl moiety in 5 for a phenyl group gave opposite results on the two strains, with an increase in affinity for gp120SF162 but slightly decreased affinity for gp120CN54. On the other hand, exchanging the cyclohexyl moiety for a less bulky cyclopentyl group (3) resulted in enhanced affinity for both strains. The difference between 2 and 10 is the replacement of the cyclohexyl group for a pyrrolidine, which again engendered a higher loss in affinity for clade C CN54 than for clade B SF162 gp120 surface proteins. Filling the Phe43 cavity with linear flexible ethers gave miniCD4s with high affinity for gp120, notably 8 and 9, while a shorter linker between the alcohol function and the phenyl group, as for 7, led to a small loss of affinity. Oxidation of the thioether in 9 to the racemic sulfoxide in 15 was dramatically deleterious for the interaction between the miniCD4 and gp120CN54 and dropped the affinity by a factor of over 300-fold, whereas, affinity for the SF162 strain was reduced by a factor of 29. This large difference in affinities between the two gp120 proteins due to this small chemical modification in the miniCD4 was unexpected (Table 1; B/A = 28). The two best miniCD4s identified in this study were 13 and the previously published miniprotein 2,19 both methylcyclohexyl derivatives of either an aniline (13) or a phenol (2) entity. The NHCH2 or OCH2 linkers appear to have the right length to place the cyclohexyl moiety in the largest part of the Phe43 cavity. The non-planar shape of the cyclohexane ring possibly allows better hydrophobic contacts with the deepest part of the pocket than the planar phenyl group in 14. Interestingly, these two miniCD4s are the only ones that share almost the same subnanomolar affinities for both gp120 (see Table 1, IC50CN54/IC50SF162 ≈ 1) suggesting that the methylcyclohexyl moiety contacts the regions of the cavity that are conserved between the two gp120s.
Table 1. ELISA IC50 values, obtained by competition against sCD4 on gp120SF162 and gp120CN54, of the miniCD4s belonging to the M48 series.
| NO |
|
R | gp120SF162 IC50 (nM ± SEM) | gp120CN54 IC50 (nM ± SEM) | A: gp120SF162 |
B: gp120SF162 |
|
B/A | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M48c |
|
0.52 ± 0.04 | 1.9 ± 0.1 | 1 | 1 | 4 | 1 | |
| 2 | M48U1c |
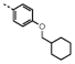
|
0.35 ± 0.07 | 0.37 ± 0.04 | 0.7 | 0.13 | 1 | 0.2 | |
| 3 | M48U2c |
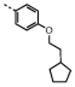
|
0.50 ± 0.09 | 1.7 ± 0.3 | 0.9 | 0.59 | 3 | 0.7 | |
| 4 | M48U3c |
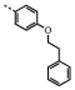
|
0.32 ± 0.06 | 11.7 ± 0.1 | 0.6 | 4.0 | 37 | 6.7 | |
| 5 | M48U4* |
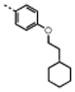
|
1.13 ± 0.18 | 6.6 ± 1 | 2.1 | 2.3 | 6 | 1.1 | |
| 6 | M48U5* |
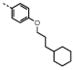
|
5.17 ± 1.26 | 50.2 ± 5.7 | 9.8 | 17 | 10 | 1.7 | |
| 7 | M48U6* |
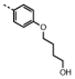
|
0.86 ± 0.07 | 10.1 ± 1.6 | 1.6 | 3.5 | 12 | 2.2 | |
| 8 | M48U7* |
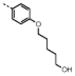
|
0.30 ± 0.06 | 2.4 ± 0.4 | 0.6 | 0.83 | 8 | 1.4 | |
| 9 | M48U8* |
|
0.28 ± 0.05 | 3.4 ± 1.1 | 0.5 | 1.2 | 12 | 2.4 | |
| 10 | M48U9* |
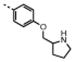
|
5.28 ± 0.46 | 38.2 ± 4.6 | 10 | 13 | 7 | 1.3 | |
| 11 | M48U10* |
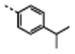
|
284 ± 21 | 13076 ± 415a | 536 | 4509 | 46 | 8.4 | |
| 12 | M48U11* |
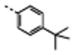
|
2026 ± 277 | 12061 ± 27a | 3823 | 4159 | 6 | 1.1 | |
| 13 | M48U12* |
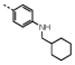
|
0.26 ± 0.05 | 0.31 ± 0.05 | 0.5 | 0.11 | 1 | 0.2 | |
| 14 | M48U13b,c |
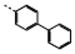
|
1.40 ± 0.46 | 15.5 ± 1.1 | 2.6 | 5.3 | 11 | 2 | |
| 15 | M48U14* |
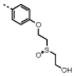
|
8.1 ± 1.6 | 1230 ± 180 | 15 | 424 | 152 | 28 |
Extrapolated values (maximum concentration tested was 10 mM).
Mentioned in reference (19).
New compounds. IC50 values are presented as the mean ± SEM values from three independent experiments.
Antiviral Activity and Cytotoxicity
In addition to competitive binding assays, the antiviral activity of those CD4-mimetics with highest binding affinities was determined against two subtype B and two subtype C viruses. In an earlier paper19 we have described the antiviral activity of 2, 3 and 4 in pseudoviruses using GHOST target cells expressing CD4 and either CCR5 or CXCR4 HIV co-receptor. Nevertheless, we tested these molecules again in this study using a TZM-bl and replication-competent HIV and showed that the results are similar in both assays (Table 2), particularly for Bal, IIIB and VI829 viruses. Minor differences were however observed for VI1358 as the EC50s are about one order of magnitude smaller. Nanomolar EC50s were found for all six M48 miniCD4s tested on lab strain clade B viruses (Table 2). Overall, highest activity was associated with miniproteins 2, 3 and 13 against subtype B HIV-1. Of note, all tested M48 miniCD4s were more active against the CXCR4-tropic strain IIIB, with low nanomolar or picomolar EC50s. While 4, 8 and 9 lost potency against subtype C viruses, 2, 3 and 13 could still inhibit the entry of the HIV-1 subtype C virus. These miniCD4s were also tested against two transmitted/founder viruses (T/F, subtype B and C) (Table 2). Interestingly, while 4, 8 and 9 were highly active against the subtype B lab strains, their antiviral activity was dramatically reduced against the T/F subtype B strain. Although the EC50 of 2, 3 and 13 against the T/F virus was higher than the EC50 values against other viruses, submicromolar activity was retained. The most notable difference between 2 and 13 was against the T/F clade C strain, where 13 with an EC50 value of 32 nM was 8.5-fold more active than 2.
Table 2. Antiviral activity of CD4 miniproteins against HIV-1 lab strains (Bal, IIIB), primary isolates (VI829, VI1358) and transmitted/founder viruses (pREJO.c/2864, p246F10) and cytotoxicity of the miniCD4s.
| EC50 (nM) | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| MiniCD4 | Bal (B, R5) | IIIB (B, X4) | pREJO.c/2864 (T/F, B, R5) | VI829 (C, R5) | VI1358 (C, R5) | p246F10 (T/F, C, R5) | CC50 (nM) |
| 2 | 1 (2)a | 0.1 (0.2)a | 14 | 17 (11)a | 130 (5)a | 272 | >50000 |
| 3 | 3 (4)a | 0,3 (0.5)a | 68 | 103 (98)a | 826 (42)a | 898 | >15000 |
| 4 | 22 (67)a | 1.2 (3)a | >1000 | >1000 (951)a | >1000 (364)a | >1000 | >15000 |
| 8 | 17 | 1 | 682 | >1000 | >1000 | >1000 | >50000 |
| 9 | 48 | 2.5 | >1000 | >1000 | >1000 | >1000 | >50000 |
| 13 | 0.4 | 0.1 | 9.8 | 12 | 37 | 32 | >40000 |
In parentheses: results obtained with GHOST cells in an earlier paper.19
Cytotoxicity of the same series of miniCD4s was evaluated using the water-soluble tetrazolium-1 (WST-1) cell proliferation assay on TZM-bl cells. With minimum CC50 values of 15 μM, no significant cytotoxicity was associated with the six CD4 miniproteins tested (Table 2).
Surface Plasmon Resonance (SPR) Analysis
Results from the competitive binding and virological assays described above establish knottin 13 as the most potent miniCD4 synthesized to date, surpassing the in vitro biological properties of the closely related miniprotein 2, which has demonstrated efficacy as a microbicide in macaque challenge models.20 To determine the affinity of interaction of 13 with HIV-1 gp120, we performed SPR analysis with three Tier 2 HIV-1 viruses namely, clade B isolates YU2 and TRO.11, and clade C isolate ZM135. We compared 13 affinity for HIV-1 gp120 with that of the closely related miniCD4 2 (Table 3 and Figure S2). In a recently published manuscript21 we have reported an affinity for peptide 2 of 15.4 ± 0.8 pM for the clade B YU2 isolate. In this study we performed the SPR assays with extended dissociation time (30 min) to obtain more accurate estimates of affinity of these tight binding complexes. We obtained an affinity of 18.1 ± 0.1 pM for 2 binding to YU2 gp120, comparable to the value obtained in the related study.21 CD4-mimetic 13 showed an affinity of 8.4 ± 0.2 pM for YU2 gp120 with the increase in affinity primarily a result of an enhanced association rate. For TRO.11, the other clade B isolate tested, a ∼14-fold enhancement in affinity for 13 relative to 2, derived from both an increased on-rate as well as a reduced off-rate of binding, the latter suggesting the formation of a more stable complex with 13. At 6.20 × 106 M-1.s-1, the on-rate of 13 binding to TRO.11 gp120 is ∼3-fold enhanced compared to that of 2, and at 1.10 × 10-4 s-1, its off-rate is ∼5-fold slower than 2. For the clade C ZM135 isolate, 16 ± 1 pM binding affinity of 13 to gp120 is a ∼3-fold enhancement over 2, with the enhancement coming primarily as a result of a 3-fold increase in the on-rate of binding.
Table 3.
Binding parameters of miniCD4s 2 and 13 to full-length gp120 from HIV-1 clade B isolates YU2 and TRO.11, and clade C isolate ZM135.
| On-rate (M-1s-1) | Off-rate (s1) | KD (pM) | |
|---|---|---|---|
| YU2 (clade B) | |||
| 2 | 3.82 × 106±2.0 × 104 | 6.87 × 10-5 ± 2.0 × 10-6 | 18.1 ± 0.1 |
| 13 | 2.70 × 107± 2.0 × 104 | 2.25 × 10-4± 1.0 × 10-6 | 8.4 ±0.2 |
| TRO.11 (clade B) | |||
| 2 | 2.04 × 106± 2.0 × 104 | 5.26 × 10-4± 4.0 × 10-6 | 257 ±11 |
| 13 | 6.20 × 106± 3.0 × 104 | 1.10 × 10-4± 5.0 × 10-6 | 17.7 ± 0.3 |
| ZM135 (clade C) | |||
| 2 | 2.34 × 106± 1.0 × 104 | 1.12 × 10-4± 4.0 × 10-6 | 47 ±2 |
| 13 | 6.84 × 106± 3.0 × 104 | 1.08 × 10-4± 5.0 × 10-5 | 16 ±1 |
The SPR results show that, for all three HIV-1 strains tested, peptide 13 binds gp120 with greater affinity than 2, although the degree of enhancement was isolate dependent. Such isolate dependence is consistent with observations of isolate-specific differences observed with the relative binding affinities obtained by CD4 competition ELISA. With all three isolates tested, an increase in rate of association of 13 with gp120 was observed, and with the clade B TRO.11 gp120 we also observed a ∼5-fold reduction in the off-rate.
Co-crystal Structure of 13 in Complex with HIV-1 gp120
To obtain an atomic-level understanding of the extraordinary affinity and neutralization breadth of 13, we crystallized miniCD4 13 with an extended core version of gp120 from the HIV-1 clade B YU2 strain.24 Crystals diffracted to 1.9 Å and the structure was solved by molecular replacement and refined to an Rfree/Rwork of 22.4%/16.8% (Table S4).
Overall, and as expected, the conformation of gp120 bound to 13 resembled the conformation of gp120 bound to CD4 or other previously structurally characterized miniCD4s15,16 with an RMSD variation ranging from 0.3-0.8 Å. Miniprotein 13 bound to the CD4 binding site of gp120 (Figure 2A), with the non-natural amino acid at position 23 inserting its side chain into the gp120 Phe43 cavity (Figure 2B) at the intersection of three gp120 domains: the inner domain, the outer domain, and the bridging sheet. In a related study, we structurally characterized gp120 binding of the closely related miniCD4 2, which contains a methylcyclohexyl derivative of tyrosine at position 23.21 Miniprotein 13 contains a methylcyclohexyl derivative of 4-aminophenylalanine at position 23. Superposing the gp120 molecules in the 2-gp120 and 13-gp120 complexes revealed close overall similarity in the environment around the Phe43 cavity in the two complexes, including the near-identical placement of the water molecules in the conserved gp120 solvent channel (Figure S3). The orientation of the Phe43 insert side chain in 2 and 13 revealed however subtle differences in the two complexes.
Figure 2.

MiniCD4 13 binds at the CD4-binding surface and extends into the gp120 Phe43 cavity. (A) gp120 is shown in grey surface representation with the CD4 binding footprint colored yellow. Knottin 13 is depicted in cartoon representation with the miniprotein colored red except residue 23, which is colored by element type with the nitrogen atoms colored blue and the carbon atoms of one conformation colored orange and the other magenta. (B) A cross-section of gp120 showing the side chain of residue 23 of peptide 13 penetrating the gp120 Phe43 cavity, packing coefficients and surface complementarity for each alternate conformation. Packing Coefficient (%) = (Volume of ligand/Volume of binding pocket) × 100. The colored dots span the region of the ligand used for each calculation.
The overall orientation of the side chain in the Phe43 cavity is unambiguously defined by the electron density in the miniprotein-13-bound cavity (Figure 3A-C and Figure S3), with the anilino group at position 23 appropriately positioned for interacting with the conserved gp120 solvent channel by hydrogen bonding with the proximal water molecule (Figure 3D and 3E). While the overall definition of the insert in the Phe43 cavity is clear for 13, its cyclohexane ring displayed asymmetric electron density, indicative of local ligand disorder in the bound state (Figures 3A-C, Figure S3 and Figure S4). Interestingly, the electron density indicates mobility or disorder at similar positions as was observed in different crystal forms of gp120-bound 2 (Figure S3), suggesting a precise and conserved recognition mode in the Phe43 cavity that allows these Phe43 cavity inserts to retain conformational mobility. The mono-substituted cyclohexane ring was modeled in the chair conformation with the sole substitution that connects the ring to the rest of the miniprotein placed in an equatorial orientation, thereby defining the cyclohexane conformation. The electron density in the cavity appeared to be well modeled by rotations about the flexible linker that connects the cyclohexane ring to the phenyl ring of 13 (Figure 3F).
Figure 3.

MiniCD4 13 binds at the intersection of gp120 inner domain, outer domain and bridging sheet. (A-C) Zoomed-in views of 13 bound to the gp120 Phe43 cavity. gp120 is shown in cartoon representation and colored by domain. Inner domain, outer domain and bridging sheet are colored cyan, green, and pink, respectively. MiniCD4 13 is similarly represented as in Figure 2. A grey mesh surrounding the residue depicts a 2Fo-Fc map at 2.0 σ defining the position of residue 23. In (A-C) the cartoon is made transparent to highlight the placement of residue 23 side chain in the electron density. (D) MiniCD4 13 interacting with a conserved gp120 solvent channel. The red spheres are water molecules located in the solvent channel that starts from inside the Phe43 cavity, traverses under the bridging sheet, and emerges at the CD4-induced coreceptor binding site of gp120. (E) LIGPLOT diagram illustrating the environment around 13 in the gp120 Phe43 cavity in 2-dimensional representation, and showing the 13 hydrogen bonding with the conserved gp120 solvent channel. Only one side chain conformation is shown for clarity. (F) 2D structure of the side chain of 13 with arrows showing the bonds that rotate and result in multiple conformations bound to the gp120 Phe43 cavity. Riding hydrogen atoms used during refinement are shown in white.
The cyclohexane ring was packed against a hydrophobic patch in the Phe43 cavity comprised of atoms from residues Val255, Trp112, Phe382 and Ile424. The observation of residual ligand entropy in the gp120 Phe43 cavity was reminiscent of synthetic host-guest systems, where an enthalpic-entropic balance was found to be critical to achieve optimal binding.25 For complexes in confined apolar cavities, it has been surmised that optimal stability is attained when the guest (or ligand) occupies 55 ± 9% of the cavity volume.26 This rule, first observed in synthetic host-guest systems, was later found to also apply to enzymatic systems.27,28 To determine whether this rule also applies to miniCD4 13 binding to the predominantly apolar gp120 Phe43 cavity, we quantified the packing efficiency of its insert in the Phe43 cavity by calculating the packing coefficient, which is the fraction of the pocket volume occupied by the ligand. We also calculated shape complementarity, which related to the goodness of fit between protein and ligand surfaces. We observed that the insert moiety occupies 80% of the available volume in the Phe43 cavity (Figure 2B and Figure S5). The cyclohexane ring, on the other hand, only fills 66% of the volume of the cavity to which it binds. The observed packing coefficient for the cyclohexane ring of 66% is just above the conjectured optimum of 55 ± 9%. Taken together these calculations show that, while the overall occupation of the Phe43 cavity by the insert of 13 is enthalpy driven and suggestive of tight interactions of the ligand with the protein, local regions of looser packing allow retention of entropy in the bound state. In addition, results from packing coefficient calculations appear to be consistent with shape complementarity analysis: the cyclohexane ring shows lower shape complementarity compared to the overall insert moiety, where tight packing results in high shape complementarity.
Discussion
Although crystal structures of full-length gp120 or trimeric HIV envelope protein gp160 are not yet available, the three-dimensional structures of HIV gp120 core variants have been determined in the CD4-bound conformation29-33 as well as in the unliganded state.24 CD4 binding stabilizes a cavity of roughly 150 Å3, which penetrates about 14 Å into the gp120 hydrophobic core at the intersection of three gp120 domains. The Phe43CD4 side chain caps the entrance of this pocket, hence the Phe43 cavity name. However, this pocket is induced not only by the CD4 receptor, but also by CD4-binding-site antibodies34 and by synthetic entry inhibitors24,35 including miniaturized CD4-mimetics,15,16 called miniCD4s. To increase the affinity of the miniCD4s for the gp120 surface protein and to improve their ability to neutralize HIV-1, we targeted the gp120 Phe43 cavity with various unnatural amino acid side chains. While the effect of modifying position 23 on the miniCD4 was often not identical on the two HIV-1 isolates tested, SF162 and CN54 (see ratio B/A in Tables 1 and S1), the overall trends were similar. For most derivatives, the variation of affinity compared to knottins 17, in the M33 series, or 1, in the M48 series, is more pronounced on the CN54 strain than on SF162. In both strains, all the cavity-lining residues are identical, except for Arg426 (HXBc2 numbering) from strain SF162 and Met426 from strain CN54 (Figure S6). With the non-conserved residue 426, the Phe43 cavity was lined by its invariant main chain component, with the non-conserved side chain facing away from the cavity. The differences in recognition of SF162 and CN54 thus likely relates to isolate-specific differences in Phe43 cavity shape, due to either the variation in residue 426 or the effect of residue variation outside the Phe43 cavity.
Since commercially available amino acids proved to be of limited utility for improving the activity of the miniCD4s, we developed unnatural residues designed to fill the gp120 Phe43 cavity. Of the miniCD4s synthesized and tested thus far, knottin 13 showed the highest affinity for gp120s tested as well as the most potent antiviral activity on laboratory-adapted strains, primary isolates and transmitter/founder viruses.
A cavity-filling analysis was previously performed on the CD4 mutant, called D1D2F43C,36 in which various chemical moieties were attached to the free cysteine at residue 43CD4 (analogous to residue 23miniCD4). One of the best derivatives of D1D2F43C contained a cyclohexyl group at a distance from residue 23miniCD4 Cα similar to that of 2 and 13. Introduction of the cyclohexyl group to D1D2F43C increased affinity ∼30-fold, to a level similar to the unmodified D1D2 (sCD4) case.
In the current study, introduction of a cyclohexyl moiety enhanced the relative affinity of 13 by a factor six for gp120CN54 and two for gp120SF162 compared to 1, resulting in a CD4-binding-site ligand with subnanomolar apparent affinity, and about one order of magnitude better IC50 of sCD4 on the SF162 receptor (4 nM, personal communication). SPR analyses on gp120 from three different strains from clades B and C show an increased affinity of 13 for gp120 compared to 2. The SPR results from this study and from a related study,21 together show that the affinity of 13 for YU2 gp120 surpasses that of 1, 2, 14 and sCD4. The in vitro antiviral activity of 13 improved more than 100-fold versus 1, and between 5 to 25-fold versus sCD4,19 on the laboratory adapted and primary isolates tested.
The crystal structure of 13 bound to YU2 gp120 shows that the introduction of the aniline nitrogen results in a hydrogen bond interaction with the gp120 solvent channel. Conjugation of this nitrogen may provide a means to improve affinity even further, by utilizing interaction with a conserved gp120 solvent channel, which runs adjacent and roughly perpendicular to the Phe43 cavity. Our results further suggest that binding of the insert moiety of 13 to the Phe43 cavity is primarily enthalpy driven while at the same time torsional movements about the flexible linker bonds allow different orientations of the cyclohexane ring to be accommodated in the wider, innermost recess of the cavity, thereby preserving conformational entropy of the ligand in its bound state. It remains to be tested whether modifications of the cyclohexane ring via structureguided substitution will result in better ring packing and higher affinity. While peptides 8 and 9 showed IC50 values similar to miniCD4 13 in the gp120SF162 (clade B) assay, their antiviral activities were not comparable to that of 13 even for clade B isolates. The differences in their pharmacological activity may result from the differences in their cavity-filling properties, described by X-ray crystallographic analyses in this study and in a related one.21 All these inserts displayed flexibility and hydrophobic interactions, but the inserts of 221 and 13 (this study) showed better shape complementarity with the Phe43 cavity than the insert of 821 and probably also of peptide 9. Taken together our results show that a subtle change in the chemistry of Phe43 cavity insert can result in improved chemical complementarity and substantially improved biological activity.
Conclusion
The miniCD4s built on the scyllatoxin scaffold are stable to acidic pH and high temperatures, relatively resistant towards proteolytic degradation, poorly immunogenic and are not cytotoxic. They stabilize the CD4-bound conformation of gp120 and one miniCD4 representative (2) was also shown to neutralize HIV entry in vitro and in vivo. Cost is a key consideration for the potential use of such compounds as microbicides especially in developing countries. One way to achieve low cost is to enhance the potency of the miniCD4s, thereby reducing the amount of drug that needs to be administered. In this paper, we showed that the synthesis of novel unnatural phenylalanine derivatives allowed us to design the most effective miniCD4 ever developed. It is able to block the viral activity of T/F viruses in the nanomolar range not only for clade B but also for clade C, being therefore more potent than the previously described miniprotein 2. From the perspective of synthesis of future miniCD4 variants, 13 has a distinct advantage over 2. Replacement of the phenol moiety by an aniline group, i.e. the presence of a secondary amine in place of ether, allows further modification in direction of the gp120 solvent channel. Such modifications may lead to development of miniCD4s with even greater potency and therefore lower cost in a potential use as a microbicide. It will also allow exploration of a very highly conserved but relatively poorly understood feature in the HIV-1 envelope structure.
Experimental Section
Synthesis Procedures
All reagents and solvents used in the synthesis and purification steps were purchased from Sigma-Aldrich, Fluka, Novabiochem, SDS or Lancaster and were of the highest purity available. THF was distilled from sodium/benzophenone immediately prior to use. Wang resin (p-benzyl-oxybenzyl alcohol, polymer bound resin), used for amino acids synthesis, was purchased from Aldrich (100-200 mesh, loading level 1.82 mmol OH/g). Fmoc-PAL-PEG-PS, used for solid-phase peptide synthesis, was purchased from Applied Biosystems (100-200 mesh, loading level 0.2 mmol/g). Standard Fmoc-protected amino acids were obtained from Novabiochem. Unnatural amino acid Fmoc-L-4-tert-butyl-Phe was purchased from PepTech Corp., Fmoc-L-Phe(4-i-Pr)-OH from Carbone Scientific, Fmoc-L-2-naphtylalanine-OH from Senn Chemicals, Fmoc-L-styrylalanine from PolyPeptide, Fmoc-L-Phe(4-Ph)-OH and Fmoc-homoPhe-OH from Bachem, Fmoc-L-Tyr(Me)-OH and Fmoc-L-Phe(4-Me)-OH from Advanced ChemTech. Flash chromatography was carried out with Merck silica gel Si 60 (40-63 μm) as stationary phase. Melting points of the crystallized compounds were determined on a Kofler melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on Bruker AVANCE 250 and 400 NMR spectrometers. The molecular weights of the amino acids and peptides were determined by electrospray mass spectrometry (ES/MS), performed on a Quattro micro mass spectrometer (Micromass, Altricham, U.K.). The purity of Fmoc-amino acids, denoted in parentheses, was evaluated by analytical-HPLC, at 214 nm, on a thermo HPLC apparatus (32′-40′, 56′, 57′ and 59′) or a Waters 600E HPLC (54′) with a Chromolith SpeedROD RP-18e column (VWR, 50 × 4.6 mm, 2 μm) with a linear gradient from 10% to 90% solvent B over 6 min (0.1% TFA in water (solvent A), 90% CH3CN with 0.09% TFA in water (solvent B) at 3 mL/min constant flow rate (these conditions will be noted HPLC-A). The purity of the various tested peptides (see tables S2 and S3 in the supporting information) was checked on a Waters 600E HPLC, at 214 nm, by reverse phase chromatography at a constant 1 mL/min flow rate, on an analytical Discovery BIO Wide Pore C18, 5 μM (15 cm × 4.6 mm) column with a gradient going from 10% to 50% B in 30 min. Even if we could not always reach a purity ≥95% for the newly synthesized Fmoc-amino acids, all the tested compounds, i.e. the various miniCD4s, have a purity >95%. Retention times (Rt) are reported in minutes.
N,O-Bis-allyloxycarbonyl-L-tyrosine (2′)
Tyrosine (1′) (5.44 g, 30 mmol) was dissolved in 15 mL of 4 N sodium hydroxide (2 equiv). The solution was cooled in an ice-bath and treated with 7 mL (2.2 equiv) of allyl chloroformate and additional 15 mL of 4 N sodium hydroxide, each added in five equal portions, with vigorous shaking for a few minutes after each addition. After the last addition, the mixture was shaken vigorously for 1.5 h, letting it warming up to rt. The water layer was extracted twice with ether, and then acidified to pH 4-5 with concentrated hydrochloric acid. After cooling overnight, crystals were filtered, washed with cold water and dissolved in DCM. The organic layer was washed with H2O and brine, dried over Na2SO4 and evaporated to yield 9.35 g (89%) of 2′ as a white solid: Rf (DCM/MeOH/AcOH: 90/8/2) = 0.75; mp 106-107 °C (105-106 °C);37 1H NMR (CDCl3) δ 3.14 (dt, J = 13.9, 5.2 Hz, 2H), 4.56 (d, J = 5.4 Hz, 2H), 4.73 (d, J = 5.8 Hz, 2H), 4.72-4.75 (m, 1H), 5.21 (d, J = 11.1 Hz, 1H), 5.34 (d, J = 10.4 Hz, 1H), 5.35 (d, J = 18.8 Hz, 1H), 5.43 (d, J = 18.6 Hz, 1H), 5.92-6.26 (m, 2H), 6.36 (br s, 1H), 7.12 (d, J = 8.6 Hz, 2H), 7.20 (d, J = 8.6 Hz, 2H); 13C NMR (CDCl3) δ 37.1, 55.1, 66, 69.2, 118, 119.6, 121.1, 130.4, 131, 132.4, 134.1, 150, 153.6, 156, 175.6.
N,O-Bis-allyloxycarbonyl-L-tyrosine immobilized on Wang resin (3′)
Wang resin (8.5 g; 1.82 mmol OH/g) was swelled in NMP (50 mL) for 15 min in a reactor with external shaking. The resin was filtered and resuspended in NMP (50 mL). 7.27 mL of diisopropylcarbodiimide (3 equiv) and 189 mg of DMAP (0.1 equiv) were added and the mixture was stirred for 15 min. Compound 2′ (16.2 g, 3 equiv) in solution in NMP (50 mL) was added and shaking was continued for 48 h at rt. The resin was filtered, rinsed with DCM (3 × 100 mL) and finally dried in vacuo to afford 12.95 g of a 0.99 mmol/g (87% theoretical) resin-bound N,O-bis-allyloxycarbonyl-L-tyrosine 3′.
Estimated resin loading by cleavage. A small weighed sample of resin (between 50 and 100 mg) was cleaved by 5 mL of TFA/DCM: 3/7 during 1 h. The mixture was filtered, the resin rinsed with DCM (2 × 3 mL) and the filtrate evaporated. The oil was mixed with a few mL of water, frozen and lyophilized. The obtained crude compound was weighed and analyzed by 1H NMR. Loading was estimated by comparison with the theoretical obtained quantity.
N-allyloxycarbonyl-L-tyrosine immobilized on Wang resin (4′)
12 g (11.92 mmol) of bis-protected tyrosine resin 3′ (0.99 mmol/g) were treated with a solution (150 mL) of 20% piperidine in DMF (v/v) for 24 h at rt. The resin was filtered, rinsed with DCM (3 × 100 mL) and finally dried in vacuo to afford 11.05 g of a 1.03 mmol/g (95% theoretical) resin-bound N-allyloxycarbonyl-L-tyrosine 4′.
N-allyloxycarbonyl-O-(c-alkyl or c-aryl-alkyl chain)-L-tyrosine immobilized on Wang resin (5′ to 13′)
General procedure (G1). PPh3 and the corresponding alcohol (4 to 8 equiv depending on the nature of the alcohol, see below) were added to resin 4′ (4.5 mmol, 4.37 g), suspended in an anhydrous 22 mL 1:1 (v/v) DCM/THF mixture. After 15 min, DIAD (4 to 8 equiv depending on the nature of the alcohol, see below), in solution in a few mL of DCM, was added portionwise, with shaking for a few minutes after each addition. The reaction continued for 24 h, at rt. The resin was filtered and rinsed with DCM (3 × 100 mL). This procedure was repeated a second time. After 24 hours shaking, the resin was filtered and rinsed with DMF (2 × 100 mL), DCM (1 × 100 mL), MeOH (1 × 100 mL) and DCM (2 × 100 mL). A small weighed sample of resin was cleaved in the same conditions as described above. Crude compound was used for 1H NMR analysis to check the disappearance of the phenol moiety. Loading was not estimated.
N-allyloxycarbonyl-O-(cyclohexylmethyl)-L-tyrosine immobilized on Wang resin (5′) was synthesized according to the procedure G1 using 3.9 mL of cyclohexylmethanol (7 equiv, 31.5 mmol) with 7 equiv of PPh3 and DIAD.
For 6′ to 13′: see supporting information
O-(c-alkyl or c-aryl-alkyl chain)-L-tyrosine immobilized on Wang resin (14′ to 22′)
General procedure (G2). Under argon atmosphere, 2.6 g of tetrakistriphenylphosphine palladium (0.5 equiv, 2.25 mmol) were added to the resin 5′-13′ (loadings were estimated at 4.5 mmol) in suspension in 150 mL of THF/DMSO/0.5 M HCl/morpholine: 20/20/10/1 ratio. The reactor was protected against light, as Pd(Ph3)4 is light sensitive, and stirred for 24 h. The resin was filtered and rinsed with DMF (2 × 100 mL), a 0.1 M solution of diethyldithiocarbamic acid in DMF (5 × 100 mL), DMF (2 × 100 mL) and DCM (3 × 100 mL). A 100% yield was estimated whatever the 5′-13′ resins used.
Washings with 0.1 M solution of diethyldithiocarbamic acid in DMF were essential in order to remove all traces of catalyst from the resin.
O-(cyclohexylmethyl)-L-tyrosine immobilized on Wang resin (14′) was synthesized according to the procedure G2 using resin 5′.
For 15′ to 22′: see supporting information
N-(9-fluorenylmethoxycarbonyl)-O-(c-alkyl or c-aryl-alkyl chain)-L-tyrosine immobilized on Wang resin (23′ to 31′)
General procedure (G3). To the resin 14′-22′ (loadings not calculated, estimated at 4.5 mmol), in suspension in DCM (90 mL), were added 3.14 mL of diisopropylethylamine (4 equiv, 18 mmol). The mixture was stirred for 15 min. 3.5 g of Fmoc-Cl (3 equiv, 13.5 mmol), in solution in 30 mL of DCM, were added to this suspension and the reaction continued for 24 h at rt. The resin was filtered, rinsed with DCM (3 × 100 mL) and dried in vacuo.
N-(9-fluorenylmethoxycarbonyl)-O-(cyclohexylmethyl)-L-tyrosine immobilized on Wang resin (23′) was synthesized according to the procedure G3 using resin 14′.
For 24′ to 31′: see supporting information
N-(9-fluorenylmethoxycarbonyl)-O-(c-alkyl or c-aryl-alkyl chain)-L-tyrosine (32′ to 40′)
General procedure for resin cleavage (G4). The resin 23′-31′ were treated with 100 mL of TFA/DCM: 3/7 for 1 h at rt. The mixture was filtered and the obtained organic layer was washed with H2O (2 × 50 mL), dried over MgSO4 and evaporated. The crude residue was applied to silica gel flash chromatography with DCM and DCM/MeOH: 97/3 (flash chromatography condition 1: FC1) or DCM and DCM/AcOH: 99/1 (flash chromatography condition 2: FC2) as eluents to give compound 32′ to 40′ with yields comprised between 26 and 63%.
N-(9-fluorenylmethoxycarbonyl)-O-(cyclohexylmethyl)-L-tyrosine (32′)
Procedure G4 was applied on 23′ and the crude residue was purified according to FC2 to yield 1.32 g of 32′ (59%) as a white solid: Rf (DCM/AcOH: 99/1) = 0.47; mp 136-137 °C; 1H NMR (CDCl3) δ 0.96-1.10 and 1.14-1.37 (2m, 6H), 1.63-1.91 (m, 5H), 3.11 (ddt, J = 14.0, 16.6, 5.8 Hz, 2H), 3.70 (d, J = 6.2 Hz, 2H), 4.21 (t, J = 7.1 Hz, 1H), 4.37 (dd, J = 9.8, 6.6 Hz, 1H), 4.45 (dd, J = 10.5, 7.1 Hz, 1H), 4.67 (dt, J = 6.0, 5.9 Hz, 1H), 5.17 (d, J = 8.1 Hz, 1H), 6.81 (d, J = 8.1 Hz, 2H), 7.04 (d, J = 8.1 Hz, 2H), 7.31 (t, J = 7.4 Hz, 2H), 7.41 (t, J = 7.4 Hz, 2H), 7.55 (t, J = 6.2 Hz, 2H), 7.76 (d, J = 7.3 Hz, 2H); 13C NMR (CDCl3) δ 25.79, 26.50, 30.30, 36.38, 37.66, 47.10, 54.57, 66.99, 73.43, 114.63, 119.98, 125.03, 127.05, 127.72, 130.32, 141.26, 143.76, 155.72, 158.51, 175.78; ES/MS for C31H33NO5 (negative ionization): Mol.wt calcd: 499.2, found: 499.1; Rt (HPLC-A): 5.11 (94%).
For compounds 33′ to 40′: see supporting information
4-benzyloxy-1-butanol (44′)
To sodium hydride (4.45 g, 111.2 mmol, 60% in mineral oil) in suspension in dry THF (200 mL), under argon, cooled to 0 °C, were added slowly 50 g of 1,4-butanediol (41′) (5 equiv, 555 mmol). The reaction was stirred for 1 h. Benzyl bromide (13 mL, 111.2 mmol), in 200 mL of dry THF, was added dropwise and the reaction mixture was stirred 5 more hours. After addition of 300 mL H2O, the solution was extracted twice with Et2O. The organic layer was washed with water and brine, dried over MgSO4 and the solvent was evaporated under reduced pressure. After purification by column chromatography on silica gel (eluted with DCM and DCM/MeOH: 97/3), 13.2 g (66%) of 44′ were obtained as a slightly yellow oil: Rf (DCM/MeOH: 97/3) = 0.39; 1H NMR (CDCl3) δ 1.57-1.71 (m, 4H), 3.48 (t, J = 6.1 Hz, 2H), 3.53 (s, 1H), 3.56 (t, J = 6.3 Hz, 2H), 4.49 (s, 2H), 7.24-7.30 (m, 1H), 7.31-7.36 (m, 4H); 13C NMR (CDCl3) δ 26.33, 29.58, 62.10, 70.24, 72.86, 127.67, 128.35, 138.19.
For compounds 45′ and 46′: see supporting information
N-Cbz-(S)-(+)-2-pyrrolidinemethanol (48′)
To 9 g of (-) Cbz-L-proline (47′) (36.1 mmol) in DME (55 mL) was added 4-methylmorpholine (1.1 equiv, 39.7 mmol). The reaction mixture was cooled to -15 °C and isobutylchloroformate (1.3 equiv, 46.9 mmol) was added slowly. After 20 min of stirring at this temperature, the white precipitate was quickly filtered off and washed with DME. To the obtained solution, cooled again to -15 °C, was added sodium borohydride (1.5 equiv, 54.2 mmol), in 23 mL of water, and the reaction mixture stirred during 1 h, letting it warming up to rt. After addition of 65 mL of water and evaporation of DME under reduced pressure, the solution was extracted twice with AcOEt. The organic layer was washed with 1 N aqueous KHSO4 solution, 1 N aqueous NaHCO3 solution and finally with brine. It was dried over MgSO4 and the solvent was evaporated under reduced pressure. 7.4 g (87%) of 48′ were obtained as a colorless thick oil: Rf (DCM/MeOH: 95/5) = 0.55; 1H NMR (CDCl3) δ 1.52-1.93 (m, 4H), 3.32 (t, J = 6.8 Hz, 1H), 3.42 (t, J = 6.8 Hz, 1H), 3.55 (d, J = 5.4 Hz, 2H), 3.86-3.96 (m, 1H), 5.05 (s, 2H), 7.20-7.29 (m, 5H); 13C NMR (CDCl3) δ 24.53, 29.01, 47.82, 61.10, 67.00, 67.72, 128.43, 128.97, 129.04, 137.06, 157.48;
2-((2-((tert-butyldimethylsilyl)oxy)ethyl)thio)ethanol (50′)
5.8 mL (57.6 mmol) of 2,2′-thiodiethanol (49′) were dissolved in dry THF containing 3.2 g (63.4 mmol) of NaH (60% in oil), and stirred for 1.5 h, under inert atmosphere. The solution was cooled to 0 °C and 8.6 g (57.6 mmol) of tert-butyldimethylsilyl chloride were added slowly. The mixture was stirred overnight at rt. 10% K2CO3 was then added, THF was evaporated and diethyl ether was added and extracted with water and brine. The organic phase was dried over Na2SO4 and the solvent was evaporated under reduced pressure. Purification by column chromatography on silica gel (eluted with hexane/AcOEt: 8/2) afforded 8.1 g of 50′, as an almost colorless oil (59%). 1H NMR (CDCl3) δ 0.86 (s, 6H), 0.91 (s, 9H), 2.68 (t, J = 6.5 Hz, 2H), 2.76 (t, J = 6.0 Hz, 2H), 2.88 (br s, 1H), 3.74 (t, J = 6.0 Hz, 2H), 3.79 (t, J = 6.75 Hz, 2H); 13C NMR (CDCl3) δ -5.3, 18.3, 25.9, 34.2, 35.8, 60.9, 63.4.
Benzyl 2-(((benzyloxy)carbonyl)amino)-3-(4-(2-((2-((tert-butyldimethylsilyl)oxy)ethyl)-thio)ethoxy)phenyl)propanoate (51′)
4.1 g (17.4 mmol) of 50′ were dissolved in toluene and cooled to 0 °C. 8.8 g (1.2 equiv) of Z-Tyr-OBn dissolved in 10 mL of dry DMF were added together with 5.7 g (1.2 equiv) of PPh3. After dissolution, DEAD (1.2 equiv) was added dropwise. The reaction was then stirred overnight at rt. Sodium phosphate buffer (0.5 M, pH 7) was added to the mixture, which was then extracted with AcOEt. The organic phase was washed with a saturated solution of NH4Cl, water and brine, dried over Na2SO4 and the solvent was evaporated under reduced pressure. Purification by column chromatography on silica gel (eluted with hexane/AcOEt: 9/1 to 85/15) afforded 10.45 g of 51′ as a colorless oil (96%). 1H NMR (CDCl3) δ 0.08 (s, 6H), 0.9 (s, 9H), 2.74 (t, J = 6.75 Hz, 2H), 2.92 (t, J = 6.75 Hz, 2H), 3.03 (d, J = 5.75 Hz, 2H), 3.81 (t, J = 7.0 Hz, 2H), 4.06 (t, J = 7.0 Hz, 2H), 4.65 (m, 1H), 5.11 (m, 4H), 5.27 (d, J = 8.25 Hz, 1H), 6.71 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 7.30 (m, 10H); 13C NMR (CDCl3) δ -5.2, 18.3, 25.9, 31.4, 34.9, 37.2, 54.9, 63.4, 66.9, 67.2, 67.7, 114.6, 127.7, 128.1, 28.2, 128.4, 128.5, 130.3, 135.1, 136.2, 155.6, 157.6, 171.4.
2-amino-3-(4-(2-((2-((tert-butyldimethylsilyl)oxy)ethyl)thio)ethoxy)phenyl)propanoic acid (52′)
10.45 g (16.8 mmol) of 51′ were dissolved in 20 mL of MeOH and reduced overnight with palladium black under 3.4 bars of H2. The slurry was filtrated. The catalyst was washed five times with MeOH and the filtrate was evaporated yielding 6.62 g of compound 52′ (quantitative). ES/MS for C19H33NO2SSi (positive ionization): Mol.wt calcd: 399.1, found: 399.1
2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-(2-((2-((tert-butyldimethylsilyl)-oxy)ethyl)thio)ethoxy)phenyl)propanoic acid (53′)
Compound 52′, dissolved in 50 mL DMF, was added to 5.9 g (17.5 mmol) of Fmoc N-hydroxysuccinimide ester in a mixture of 750 mL of CH3CN, 370 mL H2O and 5.3 g of NaHCO3 and stirred overnight at rt. The solution was concentrated and extracted with AcOEt. The organic layer was washed with 10% citric acid, a saturated solution of NH4Cl, water and brine, dried over Na2SO4 and the solvent was evaporated under reduced pressure. Purification by column chromatography on silica gel (eluted with DCM/AcOH: 99.5/0.5 to DCM/MeOH/AcOH: 95/5/0.5) afforded 8 g of 53′ as a colorless oil (78%). 1H NMR (CDCl3) δ 0.09 (s, 6H), 0.9 (s, 9H), 2.74 (t, J = 6.75 Hz, 2H), 2.92 (t, J = 6.75 Hz, 2H), 3.09 (m, 2H), 3.81 (t, J = 6.75 Hz, 2H), 4.08 (t, J = 6.75 Hz, 2H), 4.19 (t, J = 6.85 Hz, 1H), 4.41 (m, 2H), 4.65 (m, 1H), 5.28 (t, J = 10.5 Hz, 1H), 6.80 (d, J = 8.5 Hz, 2H), 7.05 (d, J = 8.5 Hz, 2H), 7.16-7.43 (m, 4H), 7.53-7.58 (m, 2H), 7.76 (d, J = 7.5 Hz, 2H), 8.94 (br s, 1H); 13C NMR (CDCl3) δ -5.2, 18.4, 26.0, 31.5, 34.9, 36.9, 47.2, 54.8, 63.5, 67.1, 67.8, 114.8, 120.1, 125.2, 27.2, 127.8, 128.2, 130.5, 141.4, 143.8, 155.9, 157.8, 176.2. ES/MS for C34H43NO6SSi (positive ionization): Mol.wt calcd: 622.2, found: 622.2
2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-(2-((2-((tert-butyldimethylsilyl)-oxy)ethylsulfinyl)ethoxy)phenyl)propanoic acid (54′)
1.24 g (2 mmol) of 53′ were dissolved in 6 mL of dry DCM and treated with 534 mL (1.5 equiv) of tert-butyl hydroperoxide (5.5 M in decane) and 10 mg (6% in mol) of thiourea dioxide for 3 days. The mixture was diluted with DCM and washed with water, brine, and dried over anhydrous Na2SO4. Purification by column chromatography on silica gel (eluted with DCM/MeOH/AcOH: 95/5/0.1) afforded 1.89 g of 54′ as a white foam (74%): mp not measurable, behaves like a wax; 1H NMR (CDCl3) δ 0.1 (s, 6H), 0.9 (s, 9H), 3.06-3.24 (m, 6H), 4.00-4.62 (m, 8H), 5.55 (d, J = 7.75 Hz, 1H), 6.78 (d, J = 8.25 Hz, 2H), 7.05 (d, J = 8.25 Hz, 2H), 7.25-7.41 (m, 4H), 7.54-7.59 (m, 2H), 7.75 (d, J = 7.5 Hz, 2H), 11.19 (br s, 1H); 13C NMR (CDCl3) δ -5.5, 18.2, 25.8, 36.9, 47.1, 51.43, 54.7, 55.2, 56.1, 60.3, 66.8, 114.5, 119.9, 125.1, 127.0, 127.7, 128.0, 130.6, 141.2, 143.8, 155.7, 156.9, 173.7. ES/MS for C34H43NO7SSi (positive ionization): Mol.wt calcd: 637.3, found: 637.3; Rt (HPLC-A on a Waters 600E HPLC): 5.23 (94%).
N-(9-fluorenylmethoxycarbonyl)-p-(N-cyclohexylmethylamino)-L-phenylalanine (56′)
Cyclohexanecarboxaldehyde (900 mL, 1.5 equiv, 7.45 mmol) was added to a suspension of Fmoc-p-amino-L-phenylalanine (55′) (2 g, 4.98 mmol) in 50 mL CH3CN/AcOH (1% v/v). The reaction mixture was stirred for 2 h at rt. Solid NaBH(OAc)3 (2 equiv, 9.94 mmol) was then added portionwise and stirring was continued overnight, at rt. The mixture was poured into 250 mL of a 0.1 N aqueous HCl solution and the solution was extracted twice with AcOEt. The organic layer was washed twice with brine, dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. After purification by column chromatography on silica gel (eluted with DCM/AcOH: 100/0.5 to DCM/MeOH/AcOH: 99/1/0.5), 1.9 g (77%) of 56′ were obtained as a white solid: Rf (DCM/MeOH/AcOH: 99/1/0.5) = 0.18; mp 95 °C; 1H NMR (CDCl3) δ 0.86-0.98 (m, 2H), 1.02-1.34 (m, 5H), 1.42-1.83 (m, 6H), 2.88 (d, J = 6.6 Hz, 2H), 3.07 (d, J = 5.4 z, 2H), 4.21 (t, J = 6.9 Hz, 1H), 4.35 (dd, J = 10.3, 6.8 Hz, 1H), 4.43 (dd, J = 10.3, 7.3 Hz,1H), 4.63 (dt, J = 7.9, 5.3 Hz, 1H), 5.28 (d, J = 8.4 Hz, 1H), 6.60 (d, J = 8.4 Hz, 2H), 6.97 (d, J =8.3 Hz, 2H), 7.31 (t, J = 7.4 Hz, 2H), 7.40 (t, J = 7.2 Hz, 2H), 7.57 (d, J = 7.1 Hz, 2H), 7.76 (d, J = 7.4 Hz, 2H); 13C NMR (acetone-d 6) δ 26.4, 27.0, 37.0, 37.8, 47.6, 50.7, 56.8, 66.6, 112.5,120.5, 125.1, 126.1, 127.7, 128.2, 130.4, 141.6, 144.7, 148.7, 156.6, 174.3; ES/MS forC31H34N2O4 (positive ionization): Mol.wt calcd: 498.3, found: 498.3; Rt (HPLC-A): 3.25 (98.5%).
N-(9-fluorenylmethoxycarbonyl)-p-(N-cyclohexylmethyl-N’-tert-butyloxycarbonyl-amino)-L-phenylalanine (57′)
To 1.07 g (2.15 mmol) of 56′, dissolved in 45 mL of THF, were added 750 mL of diisopropylethylamine (2 equiv) and 1.64 g (3.5 equiv) of di-tert-butyldicarbonate. The reaction mixture was stirred at 45-50 °C for 3 days. The mixture was concentrated in vacuo. After addition of 100 mL AcOEt, the organic layer was washed with 10% citric acid, brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure. After purification by column chromatography on silica gel (eluted with DCM/MeOH/AcOH: 97/3/1.5), 1.22 g (94.8%) of 57′ were obtained as a pale yellow foam: Rf (DCM/MeOH/AcOH: 97/3/1.5) = 0.45; mp not measurable, behaves like a wax; 1H NMR (CDCl3) δ 0.85-0.90 (m, 2H), 1.04-1.14 (m, 3H), 1.45 (s, 9H), 1.53-1.70 (m, 6H), 3.15 (d, J = 4.5 Hz, 2H), 3.44 (d, J = 7.2 Hz, 2H), 4.19 (t, J = 7.0 Hz, 1H), 4.27-4.44 (m, 3H), 4.71 (m, 1H), 5.64 (br s, 1H), 6.98-7.19 (m, 4H), 7.29 (dt, J = 7.2, 1.2 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.57 (t, J = 6.5 Hz, 2H), 7.75 (d, J = 7.2 Hz, 2H); 13C NMR (CDCl3) δ 25.8, 26.4, 28.3, 30.6, 36.7, 37.1,47.1, 54. 3, 56.1, 67.1, 80.36, 119.9, 125.2, 127.1, 127.4, 127.7, 130.0, 133.8, 141.3, 143.7,155.3, 173.6; ES/MS for C36H42N2O6 (positive ionization): Mol.wt calcd: 598.3, found: 598.4; Rt(HPLC-A): 5.29 (96%).
N-(9-fluorenylmethoxycarbonyl)-p-(isopropyl)-L-phenylalanine (59′)
350 mg (1.69 mmol) of p-(isopropyl)-L-phenylalanine (58′) were dissolved in 6.8 mL (4 equiv, 6.76 mmol) of 1 N Na2CO3 aqueous solution and 5 mL of dioxane. Half of the solvent volume was removed under reduced pressure. Then, the residual was diluted in 7.5 mL of water (pH was controlled at 9-10) and 5 mL of dioxane, and cooled with an ice bath. 547 mg (1.25 equiv, 2.11 mmol) of Fmoc-chloroformate, dissolved in 5 mL of dioxane, were added slowly. The reaction mixture was stirred 2 h at 0 °C and overnight at rt (additional 2 mL of 1 N Na2CO3 aqueous solution were added in order to keep pH around 9). After addition of 40 mL H2O, the solution was acidified with a 2 N HCl aqueous solution until the product precipitated. The aqueous layer was quickly extracted twice with Et2O. The organic layer was washed twice with H2O to eliminate all traces of HCl, dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude residue was applied to silica gel flash chromatography (eluted with DCM and DCM/MeOH: 97/3) to yield 0.34 g of 59′ (46%) as a white powder: Rf (DCM/MeOH: 95/5) = 0.33; mp 113 °C; 1H NMR (CDCl3) δ 1.21 and 1.24 (2s, 6H), 2.88 (sept, J = 6.9 Hz, 1H), 3.15 (ddt, J = 14.5, 10.6, 5.1 Hz, 2H), 4.21 (t, J = 6.8 Hz, 1H), 4.36 (dd, J = 10.3, 7.2 Hz, 1H), 4.45 (dd, J = 10.6, 7.1 Hz, 1H), 4.67-4.72 (m, 1H), 5.21 (d, J = 8.1 Hz, 1H), 7.07 (d, J = 8.1 Hz, 2H), 7.16 (d, J = 7.4 Hz, 2H), 7.30 (t, J = 7.4 Hz, 2H), 7.41 (t, J = 7.4 Hz, 2H), 7.54-7.55 (m, 2H), 7.77 (d, J = 7.4 Hz, 2H); 13C NMR (CDCl3) δ 23.92, 23.95, 33.70, 37.22, 47.11, 54.50, 67.08, 119.98, 119.99, 126.76, 127.05, 127.73, 129.28, 132.63, 141.29, 143.67, 155.77, 175.58; ES/MS for C27H27NO4 (negative ionization): Mol.wt calcd: 429.1, found: 429.1; Rt (HPLC-A): 4.25 (100%).
Peptides Synthesis, Refolding and Purification
The sequences of the various synthesized peptides were TpaNLHFCQLRCKSLGLLGKCAGSXCACV-NH2 for the M33 (16) series and TpaNLHFCQLRCKSLGLLGRCA-DPro-TXCACV-NH2 for the M48 (1) series. Tpa stands for thiopropionyl and × for phenylalanine or a phenylalanine derivative. Peptides were synthesized on an ABI-433A (Applied Biosystems) automated peptide synthesizer, using the stepwise solid-phase method and standard Fmoc chemistry. Synthesis was performed on a 0.1 mmol scale with 10 equiv Fmoc-protected amino acids, 20% piperidine in NMP for Fmoc-deprotection, DCC and Cl-HOBt for activation, and acetic anhydride for capping. For the M48 (1) series, N-terminal thiopropionyl group (Tpa) was introduced in its disulphide form. The miniprotein was cleaved from the resin with simultaneous removal of side-chain protecting groups by treatment with reagent K′ [TFA/H2O/phenol/thioanisole/1,2-ethanedithiol/triisopropylsilane: 81.5/5/5/5/2.5/1 (all v/v)], or a mixture of TFA/H2O/triisopropylsilane: 9.5/0.25/0.25, for 2.5 h at rt. The resin was then filtered off and the fully deprotected peptide was precipitated in methyl-tert-butyl ether at 4 °C. After centrifugation and washing with methyl-tert-butyl ether, peptides were dissolved in acetic acid (20% v/v) and freeze-dried. Refolding was done on the crude reduced peptide dissolved at 0.1 mg/mL in Tris/HCl buffer (0.1 M, pH 8.0), containing 2 M guanidinium chloride, 5.0 mM glutathione and 0.5 mM oxidized glutathione. Glutathione was added 15 min before oxidized glutathione in order to deprotect the Tpa moiety, when present, before refolding. The refolded peptides were purified by reverse phase chromatography on a Discovery BIO Wide Pore C18, 5 μM (25 cm × 10 mm), using H2O/5%CH3CN/0.1% TFA as solvent A and CH3CN/10%H2O/0.09%TFA as solvent B. A classical gradient was 10% to 50% B in 90 min. Peptides were lyophilized and dissolved in water. Their concentration was determined by total hydrolysis under highly acidic conditions. Molecular characteristics and purity of the various miniCD4s are described in the supporting information (Tables S2 and S3).
Competitive Binding Assays
Competition binding assays in ELISA were performed in 96-well plates (Maxisorb; Nunc). Competition assay on gp120SF162: 50 ng/well antibody D7324 (Aalto Bio Reagents, Dublin, Ireland) was coated overnight at 4 °C. Wells were then saturated with phosphate-buffered saline containing 3% bovine serum albumin buffer and washed three times, and 15 ng/well gp120SF162 were added, followed by addition of 1.25 ng of sCD4 (Progenics) and different concentrations of soluble competitors. After one night at 4 °C, we successively added anti-CD4 monoclonal antibody L120.3 (Centralized Facility for AIDS Reagents, NIBSC, UK), goat anti-mouse peroxidase-conjugated antibody (Jackson ImmunoResearch, West Grove, PA, USA), and the 3,3′,5,5′-tetramethylbenzidine substrate (Sigma). After acidification, optical density was measured at 450 nm and expressed as the mean of experiments performed in duplicate. The conditions used for competition assay on gp120CN54 were the same as for gp120SF162, except that (i) D7324 was replaced by 500 ng of concanavalin A (Sigma-Aldrich), (ii) 5 ng/well of gp120CN54 were added and (iii) 1 ng of sCD4 was used.
Antiviral Activity Assays
The antiviral activity of peptides 2-4, 8, 9 and 13 was determined by pre-incubating 104 TZM-bl cells/well in a 96-well plate for 30 min at 37 °C and 5% CO2 with or without a serial dilution of compound. Next, 200 TCID50 of Bal (subtype B, CCR5), IIIB (subtype B, CXCR4), VI829 (subtype C, CCR5), VI1358 (subtype C, CCR5), pREJO.c/2864 (transmitted/founder, subtype B, CCR5) or p246F10 (transmitted/founder, subtype C, CCR5) viruses were added to each well, and cultures were incubated for 48 h before luciferase activity was quantified. Each compound was tested in triplicate in a single experiment. Antiviral activity was expressed as the percentage of viral inhibition compared to the untreated control and plotted against the compound concentration. Next, non-linear regression analysis was used to calculate the 50% effective concentration (EC50).
Cellular Toxicity Assays
Cytotoxicity was determined using the water soluble tetrazolium-1 (WST-1) cell proliferation assay, which is based on the cleavage of the tetrazolium salt WST-1 to a formazan dye by cellular dehydrogenases. Because this bioreduction is dependent on the glycolytic production of NAD(P)H in viable cells, the amount of formazan dye formed is correlated directly to the number of viable cells in a culture. Quantification is done by measuring absorbance at 450 nm in a multiwell plate reader. 104 cells were plated per well in a 96-well plate and a serial dilution of compound was added. 48 h later, cell proliferation reagent was added and cell viability was measured compared to untreated control cultures. Cell viability was plotted against the compound concentration and non-linear regression analysis was performed to evaluate the 50% cytotoxic concentration (CC50).
SPR Assays
Experiments were carried out on a Biacore 3000 or Biacore T200 instrument (GE Healthcare). gp120 was covalently coupled to a CM5 chip at 1000-2000 RU, and a blank surface with no antigen was created under identical coupling conditions for use as a reference. MiniCD4s 2 and 13 were serially diluted 2-fold, into 10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA and 0.05% polysorbate 20 (HBS-EP) and injected over the immobilized gp120 and reference cells at 50 μL/min. The data were processed with SCRUBBER-2 and double referenced by subtraction of the blank surface and a blank injection (no analyte). Binding curves were globally fit to a 1:1 binding model.
Complex Preparation, Crystallization, Data Collection, Structure Determination, Refinement, and Analyses
HIV-1 clade B YU2 gp120 coree was expressed, purified and deglycosylated as previously described.24 The 13-gp120 complex was formed by mixing deglycosylated YU2 gp120 and the CD4-mimetic miniprotein (1:2 molar ratio) at room temperature and purified by size exclusion chromatography (Hiload 26/60 Superdex S200 prep grade, GE Healthcare) with buffer containing 0.35 M NaCl, 2.5 mM Tris pH 7.5, 0.02% NaN3. Fractions with gp120-miniprotein complexes were concentrated to ∼10 mg/ml, flash frozen with liquid nitrogen before storing at -80 °C and used for crystallization experiments.
Vapor-diffusion hanging drops were set up by mixing 0.5 μL of protein with an equal volume of reservoir solution composed of 9.0% PEG 4000, 14.0% isopropanol, 100 mM sodium citrate, pH 5.6.38 Droplets were allowed to equilibrate at 20 °C, and diffraction-quality crystals were obtained in 1-5 days.
Diffraction data were collected under cryogenic conditions using 15% (2R,3R)-butanediol as cryoprotectant. X-ray diffraction data were collected at ID-22 beamline (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory, with 1.0000 Å radiation, processed and reduced with HKL2000.39
The crystal structure of the 13-gp120 complex was solved by molecular replacement using Phaser40 in the CCP4 Program Suite.41 The structure of YU2 gp120 bound to CD4-mimetic miniprotein 221 was used as search model. Refinement was carried out with PHENIX.42 Starting with torsion-angle simulated annealing with slow cooling, iterative manual model building was carried out on COOT43 with maps generated from combinations of standard positional, individual B-factor, TLS refinement algorithms and non-crystallographic symmetry (NCS) restraints. Ordered solvents were added during each macro cycle. Throughout the refinement processes, a cross validation (Rfree) test set consisting of 5% of the data was used and hydrogens were included as riding model. Structure validations were performed periodically during the model building/refinement process with MolProbity44 and pdb-care.45 X-ray crystallographic data and refinement statistics are summarized in Table S4. For reporting resolution, the data were scaled to a highest resolution of 1.89 Å. For refinement, all data collected to a highest resolution of 1.79 Å were used.
Superpositions were performed within the CCP4 program suite. Figures were made using Pymol. LIGPLOT46 was used to generate the 2-dimensional representation of 13 environment within the gp120 Phe43 cavity. Pocket-Finder (http://www.modelling.leeds.ac.uk/pocketfinder/), the web interface based on the pocket detection algorithm LIGSITE47 was used to define the volume of the Phe43 cavity and the region of the Phe43 cavity that binds to the cyclohexane ring of 13. The volume of ligands was calculated within the ChemBio3D Ultra 12.0 module within ChemBioOffice 2010 (http://www.cambridgesoft.com/Ensemble_for_Biology/ChemBio3D).
Supplementary Material
Acknowledgments
This work was supported by the 6th European Union Framework Program Microbicide Project (EMPRO), by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health, and Seventh Framework Program (FP7/2007-2013) under grant agreement no. 242135 (CHAARM). L120.3 Ab was provided by the Centralised Facility for AIDS Reagents supported by EU program EVA/MRC (contract QLKZ-CT-1999-00609 and GP828102) and the UK Medical Research Council. We also thank Novartis Vaccine for providing gp120SF162.
Abbreviations Used
- gp120
HIV-1 surface protein
- miniCD4
CD4-mimetic miniprotein
- CD4
cluster of differentiation 4
- DIAD
diisopropylazodicarboxylate
- SPR
surface plasmon resonance
Footnotes
Supporting information: miniCD4s characterization data, gp120 sequence alignment, competition ELISA data, SPR profiles, crystallographic statistics and supplemental figures are included in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession codes: Structure factors and coordinates for 13-HIV-1-gp120 complex structure have been deposited in the Protein Data Bank with PDB ID 4KA2
Author Contributions: L.M.C., A.D. and P.K. synthesized the molecules. ELISA experiments were run by O.C., and J.M., K.K.A and G.V. obtained the neutralization data. P.A., Y.Y., P.K., L.M. and P.D.K. are related to structure analysis. O.R., L.M., K.K.A., P.A., P.D.K. and P.K. wrote the manuscript. All authors have given approval to the final version of the manuscript.
Notes: The authors declare no competing financial interest
References
- 1.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 2.Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature. 1996;384:184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 3.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 4.Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: Fusogens, antigens, and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 5.Blair WS, Lin PF, Meanwell NA, Wallace OB. HIV-1 entry - an expanding portal for drug discovery. Drug Discov Today. 2000;5:183–194. doi: 10.1016/s1359-6446(00)01484-7. [DOI] [PubMed] [Google Scholar]
- 6.Moore JP, Doms RW. The entry of entry inhibitors: A fusion of science and medicine. Proc Natl Acad Sci U S A. 2003;100:10598–10602. doi: 10.1073/pnas.1932511100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vermeire K, Schols D. Anti-HIV agents targeting the interaction of gp120 with the cellular CD4 receptor. Expert Opin Investig Drugs. 2005;14:1199–1212. doi: 10.1517/13543784.14.10.1199. [DOI] [PubMed] [Google Scholar]
- 8.Ryser HJ, Fluckiger R. Progress in targeting HIV-1 entry. Drug Discov Today. 2005;10:1085–1094. doi: 10.1016/S1359-6446(05)03550-6. [DOI] [PubMed] [Google Scholar]
- 9.Vita C, Drakopoulou E, Vizzavona J, Rochette S, Martin L, Ménez A, Roumestand C, Yang YS, Ylisastigui L, Benjouad A, Gluckman JC. Rational engineering of a miniprotein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein. Proc Natl Acad Sci USA. 1999;96:13091–13096. doi: 10.1073/pnas.96.23.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Getz JA, Rice JJ, Daugherty PS. Protease-resistant peptide ligands from a knottin scaffold library. ACS Chem Biol. 2011;6:837–844. doi: 10.1021/cb200039s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martins JC, Zhang WG, Tartar A, Lazdunski M, Borremans FAM. Solution Conformation of Leiurotoxin-I (Scyllatoxin) by H-1 Nuclear Magnetic-Resonance - Resonance Assignment and Secondary Structure. Febs Lett. 1990;260:249–253. doi: 10.1016/0014-5793(90)80115-y. [DOI] [PubMed] [Google Scholar]
- 12.Drakopoulou E, Zinn-Justin S, Guenneugues M, Gilqin B, Ménez A, Vita C. Changing the structural context of a functional beta-hairpin. Synthesis and characterization of a chimera containing the curaremimetic loop of a snake toxin in the scorpion alpha/beta scaffold. J Biol Chem. 1996;271:11979–11987. doi: 10.1074/jbc.271.20.11979. [DOI] [PubMed] [Google Scholar]
- 13.Martin L, Stricher F, Misse D, Sironi F, Pugniere M, Barthe P, Prado-Gotor R, Freulon I, Magne X, Roumestand C, Ménez A, Lusso P, Veas F, Vita C. Rational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes. Nat Biotechnol. 2003;21:71–76. doi: 10.1038/nbt768. [DOI] [PubMed] [Google Scholar]
- 14.Martin L, Heyd B, Sun Y, Martin G, Kan E, Joly P, Kessler P, Ménez A, Ulmer JB, Vita C, Barnett BW, Srivastava IK. Development of covalent HIV-1 gp120-CD4 mimic complexes for vaccine application. Antiviral Therapy. 2006;11(Suppl 2):P09d10. [Google Scholar]
- 15.Stricher F, Huang CC, Descours A, Duquesnoy S, Combes O, Decker JM, Kwon YD, Lusso P, Shaw GM, Vita C, Kwong PD, Martin L. Combinatorial optimization of a CD4-mimetic miniprotein and cocrystal structures with HIV-1 gp120 envelope glycoprotein. J Mol Biol. 2008;382:510–524. doi: 10.1016/j.jmb.2008.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang CC, Stricher F, Martin L, Decker JM, Majeed S, Barthe P, Hendrickson WA, Robinson J, Roumestand C, Sodroski J, Wyatt R, Shaw GM, Vita C, Kwong PD. Scorpion-toxin mimics of CD4 in complex with human immunodeficiency virus gp120 crystal structures, molecular mimicry, and neutralization breadth. Structure. 2005;13:755–768. doi: 10.1016/j.str.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Martin G, Burke B, Thai R, Dey AK, Combes O, Heyd B, Geonnotti AR, Montefiori DC, Kan E, Lian Y, Sun YD, Abache T, Ulmer JB, Madaoui H, Guerois R, Barnett SW, Srivastava IK, Kessler P, Martin L. Stabilization of HIV-1 Envelope in the CD4-bound Conformation through Specific Cross-linking of a CD4 Mimetic. J Biol Chem. 2011;286:21706–21716. doi: 10.1074/jbc.M111.232272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dey AK, Burke B, Sun YD, Sirokman K, Nandi A, Hartog K, Lian Y, Geonnotti AR, Montefiori D, Franti M, Martin G, Carfi A, Kessler P, Martin L, Srivastava IK, Barnett SW. Elicitation of Neutralizing Antibodies Directed against CD4-Induced Epitope(s) Using a CD4 Mimetic Cross-Linked to a HIV-1 Envelope Glycoprotein. PLoS One. 2012;7:e30233. doi: 10.1371/journal.pone.0030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Herrewege Y, Morellato L, Descours A, Aerts L, Michiels J, Heyndrickx L, Martin L, Vanham G. CD4 mimetic miniproteins: potent anti-HIV compounds with promising activity as microbicides. J Antimicrob Chemother. 2008;61:818–826. doi: 10.1093/jac/dkn042. [DOI] [PubMed] [Google Scholar]
- 20.Dereuddre-Bosquet N, Morellato-Castillo L, Brouwers J, Augustijns P, Bouchemal K, Ponchel G, Ramos OH, Herrera C, Stefanidou M, Shattock R, Heyndrickx L, Vanham G, Kessler P, Le Grand R, Martin L. MiniCD4 Microbicide Prevents HIV Infection of Human Mucosal Explants and Vaginal Transmission of SHIV(162P3) in Cynomolgus Macaques. PLoS Pathog. 2012;8:e1003071. doi: 10.1371/journal.ppat.1003071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acharya P, Luongo T, Louder MK, McKee K, Yang Y, Kwon YD, Mascola JR, Kessler P, Martin L, Kwong PD. Structural basis for highly effective HIV-1 neutralization by CD4-mimetic miniproteins revealed by 1.5 Å co-crystal structure of gp120 and M48U1. Structure. 2013 doi: 10.1016/j.str.2013.04.015. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morley AD. Optimisation and synthesis of libraries derived from phenolic amino acid scaffolds. Tetrahedron Lett. 2000;41:7405–7408. [Google Scholar]
- 23.Morley AD. Allyloxycarbonyl - a useful protecting group for phenolic amino acids and applications on solid support. Tetrahedron Lett. 2000;41:7401–7404. [Google Scholar]
- 24.Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc Natl Acad Sci USA. 2012;109:5663–5668. doi: 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cromwell WC, Bystrom K, Eftink MR. Cyclodextrin Adamantanecarboxylate Inclusion Complexes - Studies of the Variation in Cavity Size. J Phys Chem. 1985;89:326–332. [Google Scholar]
- 26.Mecozzi S, Rebek J. The 55% solution: A formula for molecular recognition in the liquid state. Chem Eur J. 1998;4:1016–1022. [Google Scholar]
- 27.Zürcher M, Diederich F. Structure-based drug design: exploring the proper filling of apolar pockets at enzyme active sites. J Org Chem. 2008;73:4345–4361. doi: 10.1021/jo800527n. [DOI] [PubMed] [Google Scholar]
- 28.Kawasaki Y, Chufan EE, Lafont V, Hidaka K, Kiso Y, Mario Amzel L, Freire E. How much binding affinity can be gained by filling a cavity? Chem Biol Drug Des. 2010;75:143–151. doi: 10.1111/j.1747-0285.2009.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwong PD, Wyatt R, Majeed S, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure. 2000;8:1329–1339. doi: 10.1016/s0969-2126(00)00547-5. [DOI] [PubMed] [Google Scholar]
- 31.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pancera M, Majeed S, Ban YEA, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou TQ, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci USA. 2010;107:1166–1171. doi: 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diskin R, Marcovecchio PM, Bjorkman PJ. Structure of a clade C HIV-1 gp120 bound to CD4 and CD4-induced antibody reveals anti-CD4 polyreactivity. Nat Struct Mol Biol. 2010;17:608–613. doi: 10.1038/nsmb.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu XL, Zhou TQ, Zhu J, Zhang BS, Georgiev I, Wang C, Chen XJ, Longo NS, Louder M, McKee K, O'Dell S, Perfetto S, Schmidt SD, Shi W, Wu L, Yang YP, Yang ZY, Yang ZJ, Zhang ZH, Bonsignori M, Crump JA, Kapiga SH, Sam NE, Haynes BF, Simek M, Burton DR, Koff WC, Doria-Rose NA, Connors M, Mullikin JC, Nabel GJ, Roederer M, Shapiro L, Kwong PD, Mascola JR, Progra NCS. Focused Evolution of HIV-1 Neutralizing Antibodies Revealed by Structures and Deep Sequencing. Science. 2011;333:1593–1602. doi: 10.1126/science.1207532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LaLonde JM, Kwon YD, Jones DM, Sun AW, Courter JR, Soeta T, Kobayashi T, Princiotto AM, Wu XL, Schon A, Freire E, Kwong PD, Mascola JR, Sodroski J, Madani N, Smith AB. Structure-Based Design, Synthesis, and Characterization of Dual Hotspot Small-Molecule HIV-1 Entry Inhibitors. J Med Chem. 2012;55:4382–4396. doi: 10.1021/jm300265j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie H, Ng D, Savinov SN, Dey B, Kwong PD, Wyatt R, Smith AB, 3rd, Hendrickson WA. Structure-activity relationships in the binding of chemically derivatized CD4 to gp120 from human immunodeficiency virus. J Med Chem. 2007;50:4898–4908. doi: 10.1021/jm070564e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens CM, Watanabe R. Amino Acid Derivatives .1. Carboallyloxy Derivatives of Alpha-Amino Acids. J Am Chem Soc. 1950;72:725–727. [Google Scholar]
- 38.Majeed S, Ofek G, Belachew A, Huang CC, Zhou T, Kwong PD. Enhancing protein crystallization through precipitant synergy. Structure. 2003;11:1061–1070. doi: 10.1016/s0969-2126(03)00185-0. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 40.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKinnon LR, Nyanga B, Chege D, Izulla P, Kimani M, Huibner S, Gelmon L, Block KE, Cicala C, Anzala AO, Arthos J, Kimani J, Kaul R. Characterization of a Human Cervical CD4(+) T Cell Subset Coexpressing Multiple Markers of HIV Susceptibility. J Immunol. 2011;187:6032–6042. doi: 10.4049/jimmunol.1101836. [DOI] [PubMed] [Google Scholar]
- 42.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lutteke T, von der Lieth CW. pdb-care (PDB CArbohydrate REsidue check): a program to support annotation of complex carbohydrate structures in PDB files. BMC Bioinformatics. 2004;5:69. doi: 10.1186/1471-2105-5-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1996;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 47.Hendlich M, Rippmann F, Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model. 1997;15(389):359–363. doi: 10.1016/s1093-3263(98)00002-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.