

Abstract
Carotenoids and their metabolic derivatives serve critical functions in both prokaryotic and eukaryotic cells, including pigmentation, photoprotection and photosynthesis as well as cell signaling. These organic compounds are also important for visual function in vertebrate and non-vertebrate organisms. Enzymatic transformations of carotenoids to various apocarotenoid products are catalyzed by a family of evolutionarily conserved, non-heme iron-containing enzymes named carotenoid cleavage oxygenases (CCOs). Studies have revealed that CCOs are critically involved in carotenoid homeostasis and essential for the health of organisms including humans. These enzymes typically display a high degree of regio- and stereo-selectivity, acting on specific positions of the polyene backbone located in their substrates. By oxidatively cleaving or isomerizing specific double bonds, CCOs generate a variety of apocarotenoid isomer products. Recent structural studies have helped illuminate the mechanisms by which CCOs mobilize their lipophilic substrates from biological membranes to perform their characteristic double bond cleavage and/or isomerization reactions. In this review, we aim to integrate structural and biochemical information about CCOs to provide insights into their catalytic mechanisms.
Keywords: carotenoid oxygenase, carotenoid, apocarotenoid, ACO, VP14, RPE65
Introduction
The most widespread color pigments found in nature, carotenoids comprise a >600 member class of fat-soluble isoprenoid compounds with up to fifteen conjugated double-bonds. These compounds are synthesized by many types of organisms ranging from archaea and eubacteria to eukaryotes (algae, fungi and plants) (for review, see [1]) and perform a host of functions in living organisms that can be related to either the light-absorbing and anti-oxidant properties of their polyene backbone chains or their ability to act as signaling molecules. For instance, their well-known pigmentation allows them to impart colors to plants and various animals (birds, marine organisms, etc.). Such coloring improves the chance of reproductive success by attracting insects to disperse pollen in plants or by increasing the sexual attractiveness in animals [2]. A second highly important physiological function pertains to their light capturing, photoprotective and anti-oxidant properties. Carotenoids are important accessory pigments for light capture by the light harvesting complex in photosynthetic organisms. The conjugated double bond system of β,β-carotene can absorb light over a broad range of wavelengths in the blue region of the visible light spectrum and subsequently transfer that energy to chlorophyll (reviewed in [3]). Their polyene structure also allows them to react with free radical products and thereby limit damage by excessive light exposure or free radical metabolites (recently reviewed in [4, 5]). Carotenoids and their derivatives are also essential for human health playing key roles in ontogeny, immune function and light perception by the eye. However, animals (including humans) cannot synthesize carotenoids de novo but instead obtain these compounds from their diet.
In living organisms, carotenoids can be enzymatically converted to a wide array of products. One such group, termed apocarotenoids, is generated by cleavage of a double bond within the polyene backbone by molecular oxygen forming aldehyde or ketone groups at the scissile double bond position. The first evidence suggesting the existence of a specific carotenoid cleavage enzyme can be traced back to 1965, when two groups reported an enzyme from rat liver and intestine that centrally cleaved β,β-carotene to form retinal [6, 7]. However, it was not until more than thirty years later, that the first member of this group, named Viviparous 14 (vp14), was cloned and molecularly identified in a screen for viviparous maize seed that showed a decreased level of abscisic acid (ABA) resulting from the vp14 mutation [8]. This breakthrough facilitated the identification and biochemical characterization of several additional carotenoid cleavage enzymes, not only in plants but also in animals, fungi and bacteria [9-12].
These cleavage enzymes belong to a family of non-heme iron enzymes named carotenoid cleavage oxygenases (CCOs). The overall amino acid sequence identity among family members is variable and can approach random levels. However, the family possesses consensus regions of absolute sequence conservation including the four fully conserved, iron-coordinating His residues. These enzymes exist in all kingdoms of life except Archaea and play important roles in maintaining carotenoid and retinoid homeostasis. CCOs typically display a surprisingly high degree of regio- and stereo- specificity for various carotenoid substrates [13]. All conjugated double bonds in carotenoid rigid backbones are potential cleavage sites, and their cleavage by CCOs requires dioxygen resulting in a large variety of apocarotenoids involved in various physiological processes.
In recent years, many efforts have been focused on elucidating the CCO-catalyzed reaction mechanism(s). Recently obtained structural information on CCOs has provided valuable insights into these processes. Since 2005, crystal structures have been solved for three different CCOs members: apocarotenoid oxygenase (ACO) from cyanobacteria Synechocystis [12], RPE65 from Bos Taurus [14], and VP14 from Zea mays [15]. These structural data together with well-documented biochemical and functional properties of these enzymes provide unprecedented insights into the structural basis for the functional diversity of this protein family.
CCOs display high substrate and cleavage site specificities
VP14 was the first CCO member found to be involved in ABA synthesis, an important hormone that regulates seed maturation and responses to various stresses in plants [16, 17]. Subsequent study of this recombinant enzyme confirmed that VP14 cleaves 9-cis-violaxanthin at its C11-C12 double bond to generate xanthoxin, the immediate precursor for ABA biosynthesis [18]. Homology-based analysis with the vp14 sequence helped to identify many other CCOs in plants. Two functionally different groups have been documented to date. The first group is represented by CCDs (carotenoid cleavage dioxygenases). Members in this group cleave the polyene backbone either symmetrically or asymmetrically. CCD7 from Arabidopsis cleaves β,β-carotene asymmetrically at the 9, 10 position, producing β-ionone and apo-10’-β-carotenal; this C27 product can be further cleaved by CCD8 at the 13, 14 position generating the C18 product, 13’-apo-β-carotenal [19]. Recently, CCDs and β,β-carotene have been shown to play critical roles in the synthesis of the plant hormone strigolactone [20]. Whereas some members recognize and accept specific carotenoids or apocarotenoids, others are more promiscuous in their substrate specificity. For instance, CCD1 from maize specifically cleaves at the 9, 10 position of both cyclic and acyclic carotenoids (e.g. lycopene, β,β-carotene, and zeaxanthin) [21]. Subsequent research found the C5-C6 double bond also is a cleavage site for CCD1 [22, 23]. Another group, named NCEDs (9-cis-epoxy-carotenoid dioxygenases), shares a cluster with VP14 and all are implicated in ABA biosynthesis. All NCEDs cleave 9-cis-epoxycarotenoids at the 11, 12 position to yield the ABA precursor xanthoxin [8, 18]. A kinetic study of VP14 revealed that the 9-cis configuration is strictly required for cleavage activity, but also showed that some flexibility is permitted in the ring structure both distal and adjacent to 9-cis double bond including the presence or absence of an epoxide group [18, 24].
The human genome encodes three CCO members, all of which have been biochemically characterized. Two members, β,β-carotene oxygenase 1 (BCO1) and β,β-carotene oxygenase 2 (BCO2), play critical roles in dietary carotene metabolism, catalyzing the oxidative cleavage of β,β-carotene at distinct double bonds in the polyene backbone (Fig 1). BCO1 cleaves at the central 15, 15’ site and thus converts β,β-carotene into two molecules of retinal, which is a precursor for visual chromophore (11-cis-retinal) and other signaling molecules such as all-trans-retinoic acid. Therefore, BCO1 is an essential participant in the mammalian visual cycle, embryonic development and regulation of gene transcription, especially in the absence of dietary sources of preformed vitamin A (e.g. retinyl esters). Analysis of substrate specificity revealed that two β-ionone rings of the carotenoid substrate are specifically required for the oxidative cleavage reaction, indicating a limited substrate spectrum for BCO1. By contrast, BCO2 cleaves primarily at the 9, 10 site of a broad range of carotenoid substrates, including β,β-carotene, 5-cis and 13-cis lycopene isomers [25, 26]. Additionally, the enzyme can cleave xanthophylls and 4-oxo-carotenoids [27, 28]. The third CCO member, known as retinal pigment epithelium protein with an apparent molecular mass of 65 kDa (hence the name, RPE65), shares significant sequence homology with BCO1/2 (Fig 2) but was identified as an isomerase rather than a carotenoid oxygenase [9, 29-31]. RPE65 specifically cleaves and isomerizes all-trans-retinyl esters to generate 11-cis-retinol and a fatty acid (Fig 1), a key step for regeneration of the visual chromophore, 11-cis-retinal, in vertebrates [29, 30].
Figure 1. Enzymatic reactions mediated by five selected carotenoid cleavage oxygenases.
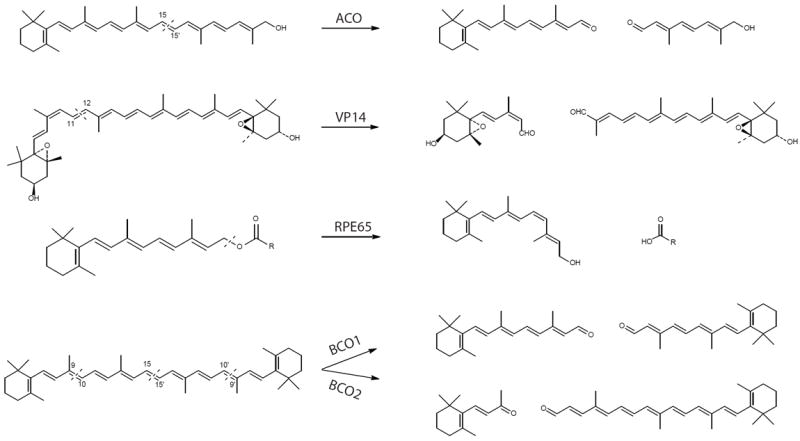
Dashed lines in substrates indicate cleavage sites. ACO, apocarotenoid oxygenase; VP14, viviparous 14; RPE65, retinal pigment epithelium-specific 65 kDa protein; BCO1, β,β-carotene-15, 15’-oxygenases; BCO2, β,β-carotene-9, 10-oxygenases.
Figure 2. Structure-based sequence alignment of selected carotenoid cleavage oxygenases.

The red background indicates sequence identity and red letters stand for sequence similarity. All structural elements of VP14, ACO and RPE65 are shown over the sequence alignment. Structural elements of α-helices and β-strands are displayed as blue squiggles and arrows, respectively. The strictly conserved iron-coordinating His residues (
 ) and their fixating Glu residues (
) and their fixating Glu residues (
 ) are labeled. Dots mark every tenth residue. The sequences were aligned with with T-coffee [75] and the figure was generated with ESPript [76].
) are labeled. Dots mark every tenth residue. The sequences were aligned with with T-coffee [75] and the figure was generated with ESPript [76].
Interestingly, CCOs in microorganisms display relatively broad substrate specificities. One of the best examples is ACO from cyanobacteria, which specifically cleaves apocarotenoids of various polyene chain lengths from C20 to C27 (C4’). This enzyme accepts either terminal aldehydes or alcohol distal to the ionone ring end as well as apocarotenoids with and without a 3-hydroxy group on their β-ionone rings, but it selectively cleaves at the 15, 15’ double bond position within these diverse apocarotenoids to generate C20 retinal and a second aldehyde product [12, 13].
Conserved architecture of CCOs
The available crystal structural data reveal a striking architecture consisting of a rigid seven-bladed β-propeller among CCOs from eubacteria to plants and mammals (Fig 3). In all three solved structures, blades I, II, IV and V consist of four antiparallel β-strands; blade VI and VII have a single-strand extensions and therefore contain five antiparallel strands. The third blade of RPE65 contains a two-strand extension not present in ACO and VP14 structures. Notably the rigid seven β-propeller scaffold can be viewed as the key structural signature of all CCO family members (Fig 2 and 3).
Figure 3. Crystal structure and topology diagram of Synechocystis ACO (left), maize VP14 (center) and bovine RPE65 (right).
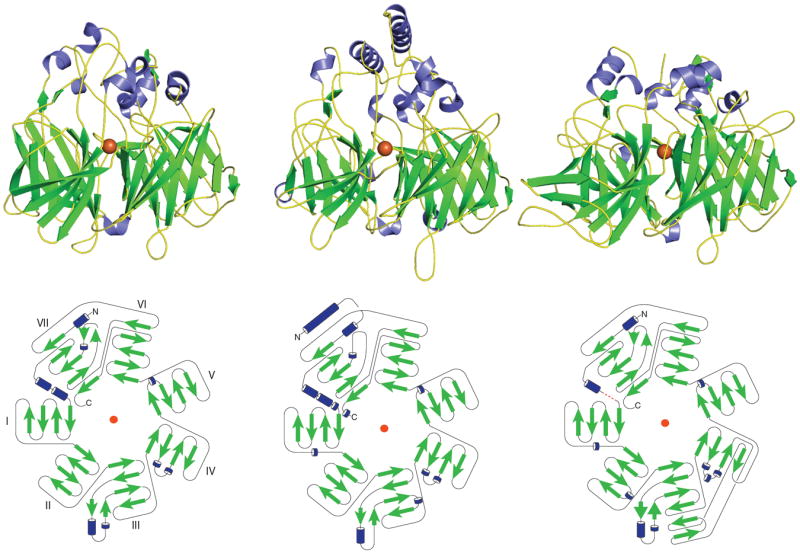
(PDB accession codes: 2BIW, 3NPE and 3FSN). The ferrous catalytic iron is colored in orange. Secondary structural elements consisting of α-helices and β-sheets are colored in blue and green, respectively. The red dashed line in the RPE65 diagram represents the unmodeled loop. The blade labeling shown for the ACO topology diagram is the same for the other two topology diagrams.
All three CCO secondary structures start with α-helices. Moreover, α-helices, short, two-strand β-sheets and various loop regions serve as transitional elements connecting β-strands within each blade, thus bringing separate propeller motifs together (Fig 3). The connectivity of the core propeller fold is identical amongst the three enzymes. Interestingly, although the bottom face of the propeller has some short α-helices bridging the strands, most helical elements are crowded together on the top face of the propeller domain (Fig 3). The α-helix elements, along with strands and extended loops, come together and form a large dome region above the previously mentioned β-propeller domain. In contrast to the propeller portion, the structure of the dome region exhibits more diversity with respect to both sequence homology and tertiary structure (Figs 2 and 3). Covered by the dome, the ferrous iron cofactor strictly required for cleavage activity is bound near the top face of the propeller on its central axis. This Fe2+ ion is coordinated by the Nε atoms of four absolutely conserved His residues (Figs 2 and 4). Notably, in all three crystal structures the innermost strand and immediately surrounding loop regions of blades II, III, IV and VII each contribute a single iron-coordinating His residue. In addition, three of the four His residues hydrogen bond to a set of three conserved Glu residues that are found in the innermost strand or surrounding loop regions from three remaining propeller blades. These Glu residues form a second coordination sphere with average hydrogen bonding distances of about ~2.9 Å. Of note, although the side chain of one of the conserved Glu residues in the VP14 structure (Glu477, PDB accession code 3NPE) points away from its presumed His hydrogen bonding partner (Fig 4B), inspection of electron density map in this region indicates that the Glu side chain is mis-modeled in the structure.
Figure 4. Catalytic centers of ACO (A), VP14 (B) and RPE65 (C).
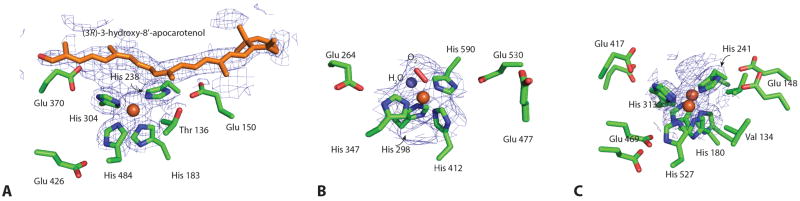
The iron ion is shown as an orange sphere, with the six proposed coordination sites arranged in an octahedral geometry. Four sites are occupied by the strictly conserved His residues. The second coordination sphere formed by three conserved Glu residues most likely helps orient the direct His ligands and may modulate the iron redox potential. The di-cis apocarotenoid substrate modeled in the ACO structure is displayed as orange sticks. Dioxygen and water (red stick and blue sphere, respectively) are modeled in the two remaining coordination sites of the VP14 iron center. Glu477 in VP14 points away from its putative coordinating His residue.
Catalytic center for the cleavage reaction
The requirement for divalent iron in CCOs is well documented by previous studies [18, 26, 31-33]. The putative role of Fe2+ is to activate oxygen for cleavage of carotenoid/apocarotenoid substrates [13, 34]. The ferrous iron center in these enzymes is invariably coordinated by four strictly conserved His residues, with three Glu residues forming the second coordination sphere (Fig 4). The moderate resolution crystal structures of ACO and RPE65 show average Fe-Nε bond lengths of ~2.1-2.2 Å, a distance consistent with the 2.15 Å Fe-Nε bond length measured for RPE65 by X-ray absorption spectroscopy (XAS) [35]. As indicated in the structure of iron-free ACO, this platform for iron-binding is rigid and does not change upon iron binding [12]. Mutagenesis studies of the key metal binding first and second sphere His and Glu residues, respectively, indicate that iron is absolutely required for CCOs to perform their catalytic roles and that both first and second sphere ligands contribute to catalytic function and iron binding [36-38].
Several excellent reviews provide detailed discussions about oxygen-activation by mononuclear non-heme iron containing oxygenases [39, 40]. As within other ferrous iron containing oxygenase enzymes, the iron cofactor in CCOs is used to activate triplet oxygen for reaction with singlet organic molecules, a process otherwise “spin-forbidden” [41]. Besides the four His residues two remaining cis-oriented coordination sites of the iron that face the active site cavity are vacant and available for exogenous ligands. In the VP14 structure, the electron density surrounding the catalytic Fe2+ center most likely implies a bound water trans to His412 plus a dioxygen molecule trans to His298 at the two vacant sites (Fig 4B). Although a similar dioxygen or water molecule trans to His183 and His304 is also implied in the ACO structure, it still remains unclear as to whether a water molecule binds to the sixth coordinating site of Fe2+ due to the hydrophobic microenvironment resulting from the methyl group of Thr136 located 4.4 Å away (Fig 4A). Similarly, one of the two open Fe2+ coordination sites in the octahedral geometry is partially blocked by the Cγ methyl group of Val134 in RPE65 (Fig 4C). A triangular-shaped electron density feature is in close proximity to the fifth and sixth coordination sites of RPE65’s iron center. The shape and position of this density, which is maintained in RPE65 crystals obtained from protein purified from different detergents [35], indicates the presence of a bound fatty acid potentially derived from the retinyl ester cleavage reaction (Figs 1 and 4). This RPE65 substrate-interaction model is further supported by XAS data, which suggests the presence of a bound carboxylate group at the open coordination site(s) of the iron [35].
Often the iron at the active site of mononuclear non-heme oxygenases is coordinated by two or three protein ligands, usually a combination of His and other residues (mostly Asp, Glu, and Tyr). The four His coordination system utilized by CCOs is rare in nature and to date has been observed in only a few other protein families [42-44]. The imidazole-rich coordination and absence of strongly charged directly donating ligands are likely to stabilize the ferrous form of the enzyme. Indeed, chemical and spectroscopic analyses indicate that CCOs maintain iron in the ferrous form even following their aerobic purification in the absence of strong reducing agents [15, 35].
Interestingly, Kloer et al claimed that isomerization activity was embedded in a cyanobacteria ACO because an additional crooked, rod-shaped electron density feature appeared in the active site cavity after soaking ACO crystals with the substrate [12]. The apocarotenoid substrate could only be modeled into the crooked rod density by changing the C13-C14 and C17-C18 double bonds from a trans to a cis configuration. This study provided a novel model for a CCO-mediated carotenoid cleavage reaction that is accompanied by a potential isomerization process. However, electron densities for both the β-ionone ring and alcohol tail were invisible and only the central part of the substrate density was observed. From a chemical perspective, an evident energy source is lacking for the enzyme to overcome the unfavorable energy barrier needed for generation of this double-cis isomer. In addition, the crystals were obtained in the presence of polyethylene glycol and the detergent octylpolyoxyethylene. Those molecules also bear elongated carbon chains resembling the isoprene chain in the apocarotenoid substrate. So the possibility that the observed electron density at the active site might simply be a detergent or PEG molecule certainly cannot be ruled out. Furthermore, an analysis of ACO-catalyzed reaction products revealed that only low levels of isomerized products were detected by liquid chromatography [45], which are most likely attributable to thermal isomerization. Therefore, the mechanisms for CCO-substrate interactions and their catalytic processes require further clarification.
A hydrophobic patch for membrane penetration
Unlike other soluble compounds, most substrates for CCOs show high lipophilic features and thus prefer to reside in a water-free environment. Indeed, large amounts of apocarotenoids are found in the thylakoid membrane of plants and microorganisms [46]. And in mammalian cells, carotenoids and their derivatives are stored in lipid droplets called liposomes as well as in other lipid-enriched organelles [47-49]. Thus, these soluble CCO proteins must adopt a mechanism to extract their lipid-soluble substrates. Biochemical studies revealed that CCO enzymes display variable levels of aqueous solubility. For example, BCO1 behaves largely like a soluble enzyme [50], whereas RPE65 behaves like an integral monotopic membrane protein [51]. ACO can be expressed in a soluble form and purified without detergent [12], but it requires detergent for optimal activity and binds to synthetic liposomes via hydrophobic interactions [52].
Surface analysis of all three CCO structures reveals similar nonpolar patches consisting largely of protruding hydrophobic residues (Fig 5). The proposed role of the hydrophobic patch is that it is used by CCOs to dip into a membrane and extract hydrophobic substrates. In VP14, two antiparallel α-helices mainly consisting of hydrophobic residues (α1-helix from 88 to 108 and α2-helix from 222 to 237) form a large putative membrane penetrating patch covering a surface area of ~2200 Å2 (Fig 5B). In addition, a few positively charged residues (e.g. Arg89) are found in the α1 helix and nearby loop region (e.g. Arg373). These residues may be involved in the interaction of VP14 with negatively charged lipid in the interior membrane. Unlike VP14, ACO bears a smaller patch with its ~1000 Å2 accessible surface area consisting mostly of Leu and Phe residues, as well as a few positively charged residues (Lys123 and Arg129, 266) located in the nearby loop region (Fig 5A). Similarly, three groups of residues have been noted in the lipid-depleted RPE65 structure (residues 196-202, 234-236, and 261-271) where abundant hydrophobic residues reside including Phe196, 200, 235, 262, 264, Leu261, 265, 270 and the aromatic Trp268, 271 (Fig 5C). Nonpolar residue protruding from surrounding loop region (e.g. Phe108) may also contribute to the overall hydrophobicity of RPE65 and thus mediate the interaction between the nonpolar patch region and membrane. Also, a number of positively charged residues are found in those regions as well (e.g. Lys236). These large, nonpolar hydrophobic patches seem to contribute to the overall hydrophobicity of CCO enzymes, explaining the requirement for detergent for their purification, crystallization and/or enzymatic activities. Previous studies suggested that RPE65 is reversibly palmitoylated by LRAT at Cys231, 329, 330, providing a potential strategy to regulate the membrane affinity and alter substrate-binding activity of this enzyme [53-55]. However, both mass spectrometry and mutagenesis studies failed to support a palmitoylation modification of these Cys residues, and no electron density has been found in crystal structures of RPE65 extracted from its native environment [14, 37, 53, 56]. Therefore, reversible Cys residue palmitoylation is unlikely to regulate RPE65 membrane association and catalytic activity.
Figure 5. Surface views of the three crystal structures of ACO (A), VP14 (B) and RPE65 (C) with their hydrophobic patches for putative membrane binding.
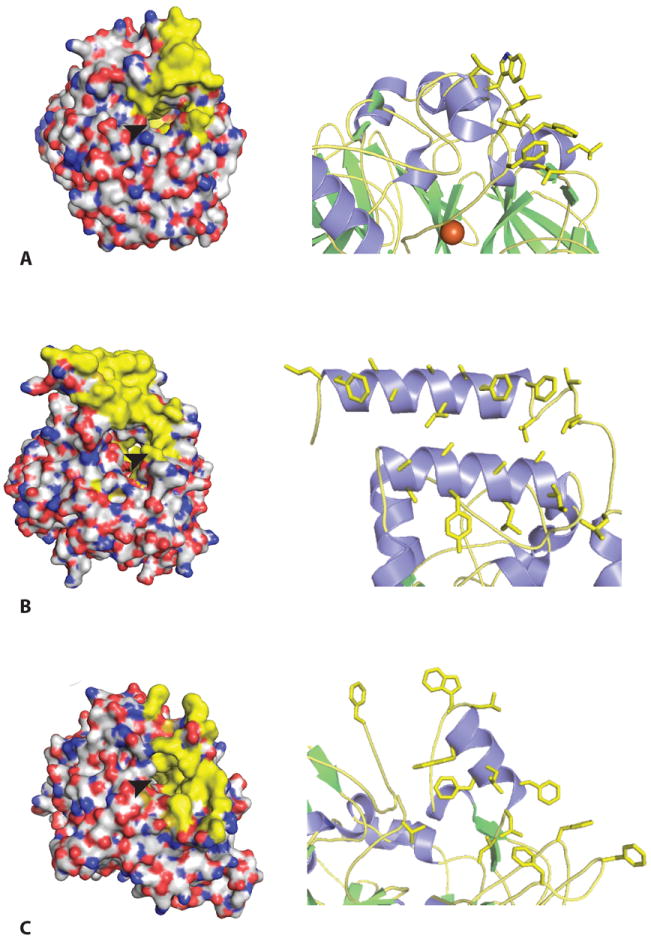
Left, hydrophobic surface portions of each enzyme are colored in yellow. Right, hydrophobic residues colored in yellow for membrane penetration are shown in each structure. The arrowhead indicates the opening of cavities that lead to the active site iron.
Interestingly, a subsequent study revealed a potential role for phospholipids in modifying RPE65 structure [35]. When the protein was extracted from microsomes and crystallized in a membrane-like environment, residues involved in the membrane interaction region adopted a substantially different and more ordered conformation as indicated by lower B-factors and a clearer electron density. A distinct change was the loss of the α5-helix (residues 263-271) accompanied by movement of side chain Phe264 and Trp268 towards the active center. Several other large movements of side residues including Trp271, Phe200, 196, Lys198 and Asn199 were also observed. Notably, the completely disordered and invisible chain containing residues 110-126 in the detergent-solubilized RPE65 displayed a weak but continuous electron density which is orientated parallel and close to the membrane surface. This segment was previously predicted to anchor RPE65 to the membrane [57]. And later studies found that an S-palmitoylated Cys112 in the segment played an important role in mediating membrane association [14, 58]. However, more biochemical and structural studies are needed to determine whether this native membrane-induced conformational change also applies to other CCO enzymes.
Hydrophobic tunnels leading to the active site
Another prominent feature of CCOs is a tunnel extending from outside of the protein and entering its active center relatively perpendicular to the propeller axis (Fig 6, red mesh). All tunnels in the three CCO structures pass by the iron metal and end with an interior Leu residue (Leu446 in VP14, Leu400 in ACO, and Leu439 in RPE65). These long hydrophobic residue-constituted tunnels act as a conduit for the passage of lipophilic substrates. The mouths of these tunnels are surrounded by a large hydrophobic patch for membrane insertion as mentioned above, thereby providing an ideal lipophilic environment to accommodate the substrate. The tunnel hydrophobic residues (mainly Phe, Val, Leu), together with few aromatic and hydrophilic residues (Tyr, Trp and His), work in concert to interact with their hydrophobic carotenoids/apocarotenoids/retinyl ester substrates via van der Waals (hydrophobic) forces to guarantee both the specificity and correct orientation of substrate for the cleavage or isomerization reactions. Docking experiments by modeling 9-cis-violaxanthin into VP14 hydrophobic tunnels revealed that hydrophobic residues around the methylenecyclohexane group and isoprene chain of 9-cis-violaxanthin interact with this substrate via hydrophobic interactions and also hold it in register for cleavage. Notably, three Phe residues (Phe171, 411, 589) surrounding the cleavage site and catalytic iron as well as a Val residue (Val478) at the proximal methylenecyclohexane group are key players for substrate and cleavage site specificity [15]. Mutagenesis studies also demonstrated the importance of those hydrophobic and aromatic residues in determining RPE65 isomerase activity, and recent studies indicates that aromatic residues in the proposed substrate tunnel of RPE65 determine the retinol isomerization process [59, 60]. Interestingly, most residues determining the shape of hydrophobic conduit are located in the extended loop and α-helical regions, with only a slight contribution from the well-conserved rigid β-propeller scaffold domain. Consistently, sequence alignment clearly reveals that regions of the greatest diversity are within the loop and helix sections (Fig 2). The propeller domain in CCOs is highly conserved among evolutionarily separated species, whereas the dome region above the propeller demonstrates the most versatile structural properties. Therefore, residues comprising the dome regions appear to be the major determinants of substrate regio- and stereospecificity.
Figure 6. Tunnels lead to the active center of ACO (A), VP14 (B) and RPE65 (C).
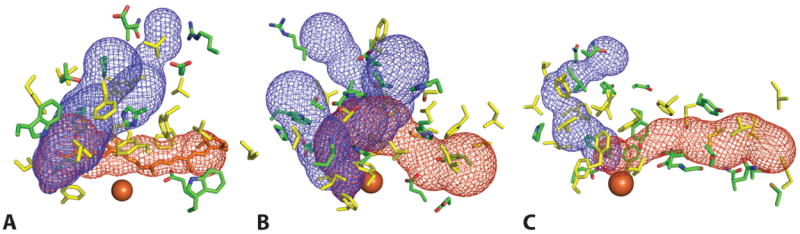
The red and blue mesh represent tunnels connecting the membrane binding region of the protein to the active site and the active site to cytosolic-facing regions, respectively. The location of rightmost portion of the red mesh corresponds to the sites indicated by arrowheads in Fig. 5. Retinoid active site entry presumably occurs via the channel delineated by red mesh. Residues lining the tunnels are shown as sticks. Hydrophobic residues are colored in yellow, and both charged and polar residues are colored green. The catalytic iron is shown as an orange sphere.
Curiously, extra tunnels have been found in all three CCOs structures (Fig 6, blue mesh). Similarly, the amino residue components of those tunnels are mainly hydrophobic, with some hydrophilic residues facing the cytosol at the mouth. Three tunnels in VP14 and two in ACO are connected with their substrate tunnels at the catalytic center, so these could function as exit conduits for the aldehyde products (Fig 6, blue mesh). Though a substantially narrower secondary tunnel of unknown functional importance was identified in the RPE65 structure, a constriction in this tunnel would prevent the passage of retinoid substrate or product. Moreover, an improved resolution RPE65 structure revealed well-ordered water molecules in this tunnel [51]. Therefore, unlike VP14 and ACO, the RPE65 retinoid substrate entry and product exit most likely occur in the same tunnel. This difference in structure is due to an extension of the α-helical segment located between the inner strands of blade IV of the RPE65 structure, a region referred to as a “metazoan loop” [59]. The extension results in the formation of a three helix motif that packs tightly against other portions of the dome region, in contrast to the loose packing of the structurally analogous region in the ACO and VP14 structures. Although the conformation of this region is essentially invariant in the RPE65 structures reported to date, Redmond has proposed that this region could open to allow the entry/exit of compounds including RPE65 inhibitors [61]. However, direct structural evidence to support this hypothesis has yet to be obtained. Of note, the three CCO structures feature a hydrophilic cavity that runs along the propeller axis and terminate just before reaching the iron center. The exact function of these tunnels is not clear but they may allow passage of water or other small molecules/ions e.g. O2 or Fe2+, to the active center. However, the possibility that such molecules could also enter the active center via substrate/product tunnels cannot be ruled out.
Thus when CCOs associate with membrane, interior hydrophobic substrates are extracted and channeled into the nearby substrate tunnel where key residues promote the accurate orientation of the substrate mainly through hydrophobic interactions prior to substrate cleavage. In ACO and VP14, reaction products exit the active center via product exit tunnels, whereas in RPE65 both substrate and product most likely share the same tunnel to enter and exit the active site.
Reaction mechanisms
Despite numerous biochemical studies demonstrating the strict requirement of ferrous state iron and oxygen for the reaction, the actual CCOs oxidative mechanisms still remain controversial. In the monooxygenase reaction, the double bond at the cleavage site forms an epoxide intermediate with a reactive oxygen species, and only one of the two oxygen molecules participates in the reaction (Fig 7, left panel). In an in vitro study using purified BCO1 from chicken intestinal mucosa, the cleavage reaction was initiated in the presence of both 17O2 and H218O. Subsequent GC-MS analysis of retinol products found almost equal quantities of oxygen derived from O2 and H2O, providing evidence interpreted as support for a monooxygenase mechanism [50]. Therefore, BCO1 is also named as BCMO1 (β,β-carotene-15, 15’-mono-oxygenase) and the latter term is now preferred.
Figure 7. Monooxygenase and dioxygenase catalytic mechanisms proposed for carotenoid cleavage enzymes.
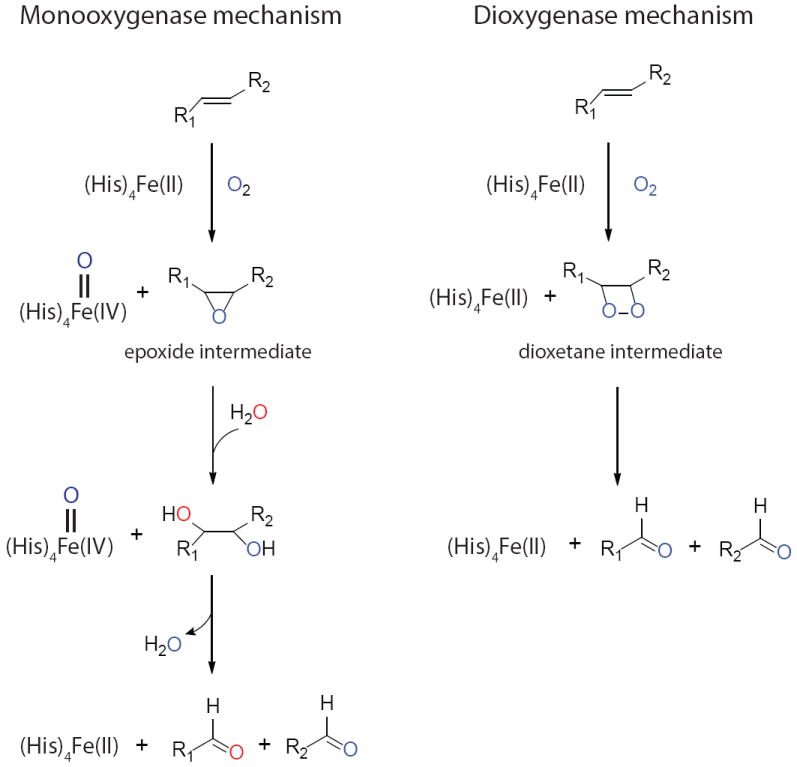
Except for two vacant sites, the catalytic metal irons are occupied by imidazole rings from conserved His residues. Dioxygen binding to the iron activates it for attack of the double bond in the substrate. In the monooxygenase reaction, an epoxide is formed with involvement of one O2-derived oxygen. Only one oxygen remains in the aldehyde products with the other derived from water. In the dioxygenase reaction, an unstable dioxetane intermediate is formed, and both dioxygen atoms remain in the aldehyde products.
In the dioxygenase reaction mechanism, however, two molecules of oxygen are involved in attacking the double bond in the substrate and forming a dioxetane intermediate. This unstable dioxetane species rapidly decays into two aldehyde products, and water molecules are not required for the reaction (Fig 7, right panel). An isotope labeling experiment involving plant CCD1 from Arabidopsis thaliana shows that, when performed in an 18O2 atmosphere, 96% of the β-ionone keto-group and 27% of the aldehyde product were labeled with 18O2 derived oxygen [62]. Because aldehyde oxygens in reaction products exchange rapidly with those of bulk water, this could account for the decreased isotope signal in the aldehyde product. Therefore, this study favors a dioxygen mechanism. In another study that took advantage of available CCO structural information, a pure computational approach suggested that the dioxygenase mechanism is also preferred for the ACO enzyme [34].
However, due to the nature of oxygen chemical exchange among different species, isotope-labeling experiments cannot be considered conclusive. Thus there has been criticism about the long incubation periods used for the reaction supporting the monooxygen mechanism [50]. Oxygen exchange between retinal, molecular oxygen and water during long incubation periods could cause inaccurate interpretations for the equal amounts of formed isolabeled products. Indeed, research already has shown that most of the oxygen in retinal exchanges with H218O during 14 days [63]. In addition, the alcohol dehydrogenase used to convert retinal to retinol could potentially scramble the oxygens during the reaction with NADH, and the increased level of NAD+ could exacerbate this process. More data are required to prove that the aldehyde oxygen in retinal does not exchange with the medium during this reaction. And in experiments reportedly supporting the dioxygen mechanism [62], the small fraction of iso-labeled aldehyde product formed constitutes weak evidence for the proposed dioxygen mechanism, even though isotopic signals were detected in almost all keto-products. Therefore, although growing bodies of data have been accumulated in this area, the question of whether CCOs employ a mono- or dioxygenase mechanism requires further investigation.
CCOs and human health
So far, three CCO members (BCO1, BCO2 and RPE65) have been identified and characterized in humans. These are key players in regulating carotenoid metabolism and thus are critical for human health. BCO1 and BCO2 act on different double bonds of the β,β-carotene backbone to generate different products. Vitamin A, generated via central cleavage of β,β-carotene by BCO1, is the critical precursor for retinoic acid (RA). RA is known be involved in gene regulation and also participates in a wide range of important physiological processes, including embryonic and fetal development, cell differentiation and metabolic control. Genetic variations of BCO1 caused by mutations or polymorphisms and their effects on carotenoid metabolism in humans have already been described [64, 65]. Although less is known about BCO2, its protective role against carotenoid-caused oxidative stress was implied by a recent study [28]. The physiological functions of BCO1 and BCO2 have been reviewed in detail [66, 67].
RPE65 has gained much more attention because a number of mutations within the RPE65 gene are associated with a severe autosomal recessive early onset retinal dystrophy known as Leber’s congenital amaurosis (LCA) as well as other milder retinal dystrophies [68, 69]. To date, over 60 different pathogenic mutations have been found in the RPE65 gene spread over all 14 exon as well as intron regions. Although the structure of human RPE65 is currently unavailable, the bovine RPE65 structure provides us with much valuable information about how RPE65 mutations can affect visual function. Because bovine RPE65 shares 99% sequence identity with human RPE65, it appears also highly probable that both enzymes adopt virtually the same chain folds and share the same three dimensional structures. This close relationship enables us to analyze and evaluate mutational effects at a structural level.
Mutations in strictly conserved iron-binding His residues or second coordination sphere Glu residues produce type II LCA or retinitis pigmentosa (RP). Because the seven-bladed β-propeller domain is the most conserved portion among CCOs and it serves as a core scaffold supporting the active center and dome of the molecule, any residue substitution causing significant conformational changes in this region should also affect function. Indeed, most disease-associated mutations occur within or adjacent to the β-propeller region, especially in blades V and VII, and quite few exist in the helical or loop regions (Fig 8). Beside the propeller domain, Arg91, one of the most frequently affected positions in patients with RP or LCA [70, 71] forms a salt bridge with Glu127 which is located on the C-terminal end of a potential membrane binding region. Recently, a position (Arg118) within this putative membrane binding region has been shown to be substituted in a patient with RP/LCA (Fig 8) [72].
Figure 8. LCA or RP-associated amino acid substitutions in RPE65.
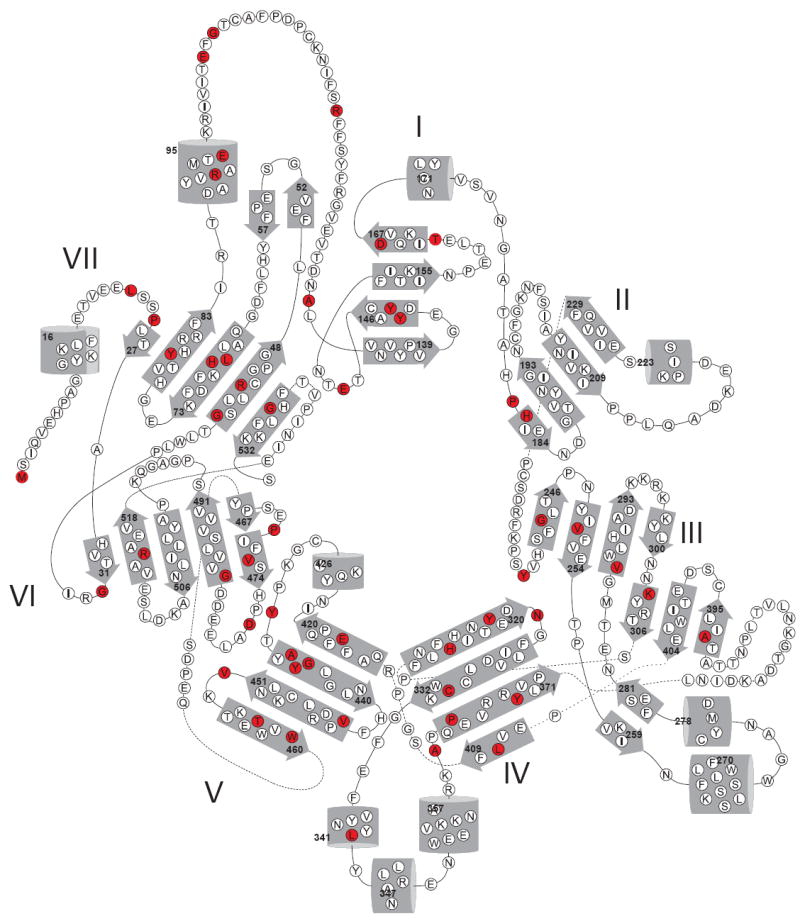
An RPE65 topology diagram reveals amino acid positions (colored in red) found substituted in patients with LCA or RP. Numbers indicate positions in the RPE65 amino acid sequence of residues in each secondary structural element. The figure is adapted from [14].
Conclusions and perspectives
Since the first CCO member VP14 was discovered, more and more members in this family from different species have been identified and characterized, and tremendous progress has been made in defining their roles in carotenoid metabolism of living organisms. Efforts made by structural biologists and breakthroughs in structural studies are vital to further our understanding of CCOs. Well conserved and specialized structures, together with a unique iron-coordination system are all key properties that distinguish CCOs from other iron-requiring protein families. The hydrophobic tunnels found in all known CCO structures have improved our comprehension of their substrate specificity, and the hydrophobic patch also explains how CCOs extract their carotenoid/apocarotenoid substrates from a hydrophobic membrane environment.
However, many issues regarding this enzyme family remain unresolved. For example, although two isotope labeling experiments have been carried out, it is still controversial whether CCOs employ a mono or dioxygen mechanism. Detailed reaction mechanisms and the involvement of oxygen species need to be clarified. More interestingly, another emerging member named NinaB (denoting neither inactivation nor afterpotential mutant B) is of great current interest. Discovered in insect cells, NinaB combines both cleavage and isomerase activities in single CCO protein [9, 73, 74]. Therefore, in contrast to the traditional double bond cleavage activity of CCOs, the isomeroxygenase activity of NinaB and isomerase activity of RPE65 separate these specialized members from other canonical CCO family members. One promising way to study catalytic mechanisms by different CCOs is to obtain the crystal structures of definitive enzyme-substrate complexes, but this still remains a formidable experimental challenge because their native substrate(s) show a high degree of hydrophobicity and feature a relatively large size.
Though challenging, understanding the substrate specificity and catalytic mechanism of these specialized CCO members is important. Because the roles of RPE65 and BCO1/2 in human health are significant, such studies would definitely help us cope with CCO-related diseases. Moreover, greater efforts are still needed to uncover additional CCOs genes involved in the synthesis and metabolic conversions of both carotenoids and apocarotenoids. And because a growing body of evidence indicates a variety of important biological roles for these hydrophobic isoprenoid compounds, it would not be surprising if novel psychological functions for carotenoids and apocarotenoids as well as new CCO members, are discovered in the near future.
Highlights.
After showing and comparing all known CCO structures, we unravel universal properties shared by these enzymes.
Based on available biochemical and structural data, we propose working mechanisms for CCO action.
Catalytic mechanisms of non-heme iron mediated reactions by CCOs are discussed.
Some human hereditary diseases caused by genetic alternations in RPE65 and other CCO members are analyzed at the structural level.
Acknowledgments
We thank Dr. Leslie T. Webster Jr. (Case Western Reserve University) for proof reading and valuable comments on the manuscript. This work was supported by grants from the National Institutes of Health (NIH), National Eye Institute (NEI) R01EY009339, and R01EY020551. K.P. is John H. Hord Professor of Pharmacology.
Abbreviations
- ABA
abscisic acid
- ACO
apocarotenoid oxygenase
- BCO1
β,β-carotene-15, 15’-oxygenases
- BCO2
β,β-carotene-9,10-oxygenases
- CCDs
carotenoid cleavage dioxygenases
- CCOs
carotenoid cleavage oxygenases
- LCA
Leber’s congenital amaurosis
- RP
Retinitis Pigmentosa
- NADH/NAD+
nicotinamide adenine dinucleotides
- NCEDs
9-cis-epoxy-carotenoid dioxygenases
- NinaB
denoting neither inactivation nor afterpotential mutant B
- RA
retinoic acid
- RPE65
retinal pigment epithelium-specific 65 kDa protein
- VP14
viviparous14
- XAS
X-ray absorption spectroscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walter MH, Strack D. Natural product reports. 2011;28:663–692. doi: 10.1039/c0np00036a. [DOI] [PubMed] [Google Scholar]
- 2.Blount JD. Archives of biochemistry and biophysics. 2004;430:10–15. doi: 10.1016/j.abb.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 3.Fraser NJ, Hashimoto H, Cogdell RJ. Photosynthesis research. 2001;70:249–256. doi: 10.1023/A:1014715114520. [DOI] [PubMed] [Google Scholar]
- 4.Stahl W, Sies H. The American journal of clinical nutrition. 2012;96:1179S–1184S. doi: 10.3945/ajcn.112.034819. [DOI] [PubMed] [Google Scholar]
- 5.Carranco Jauregui ME, Calvo Carrillo L, Mde, Romo FP. Archivos latinoamericanos de nutricion. 2011;61:233–241. [PubMed] [Google Scholar]
- 6.Olson JA, Hayaishi O. Proceedings of the National Academy of Sciences of the United States of America. 1965;54:1364–1370. doi: 10.1073/pnas.54.5.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodman DS, Huang HS. Science. 1965;149:879–880. doi: 10.1126/science.149.3686.879. [DOI] [PubMed] [Google Scholar]
- 8.Tan BC, Schwartz SH, Zeevaart JA, McCarty DR. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:12235–12240. doi: 10.1073/pnas.94.22.12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Lintig J, Vogt K. The Journal of biological chemistry. 2000;275:11915–11920. doi: 10.1074/jbc.275.16.11915. [DOI] [PubMed] [Google Scholar]
- 10.Wyss A, Wirtz G, Woggon W, Brugger R, Wyss M, Friedlein A, Bachmann H, Hunziker W. Biochemical and biophysical research communications. 2000;271:334–336. doi: 10.1006/bbrc.2000.2619. [DOI] [PubMed] [Google Scholar]
- 11.Prado-Cabrero A, Scherzinger D, Avalos J, Al-Babili S. Eukaryotic cell. 2007;6:650–657. doi: 10.1128/EC.00392-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kloer DP, Ruch S, Al-Babili S, Beyer P, Schulz GE. Science. 2005;308:267–269. doi: 10.1126/science.1108965. [DOI] [PubMed] [Google Scholar]
- 13.Kloer DP, Schulz GE. Cellular and molecular life sciences : CMLS. 2006;63:2291–2303. doi: 10.1007/s00018-006-6176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiser PD, Golczak M, Lodowski DT, Chance MR, Palczewski K. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:17325–17330. doi: 10.1073/pnas.0906600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Messing SA, Gabelli SB, Echeverria I, Vogel JT, Guan JC, Tan BC, Klee HJ, McCarty DR, Amzel LM. The Plant cell. 2010;22:2970–2980. doi: 10.1105/tpc.110.074815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoecker U, Vasil IK, McCarty DR. Genes & development. 1995;9:2459–2469. doi: 10.1101/gad.9.20.2459. [DOI] [PubMed] [Google Scholar]
- 17.Giraudat J. Current opinion in cell biology. 1995;7:232–238. doi: 10.1016/0955-0674(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz SH, Tan BC, Gage DA, Zeevaart JA, McCarty DR. Science. 1997;276:1872–1874. doi: 10.1126/science.276.5320.1872. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz SH, Qin X, Loewen MC. The Journal of biological chemistry. 2004;279:46940–46945. doi: 10.1074/jbc.M409004200. [DOI] [PubMed] [Google Scholar]
- 20.Alder A, Jamil M, Marzorati M, Bruno M, Vermathen M, Bigler P, Ghisla S, Bouwmeester H, Beyer P, Al-Babili S. Science. 2012;335:1348–1351. doi: 10.1126/science.1218094. [DOI] [PubMed] [Google Scholar]
- 21.Sun Z, Hans J, Walter MH, Matusova R, Beekwilder J, Verstappen FW, Ming Z, van Echtelt E, Strack D, Bisseling T, Bouwmeester HJ. Planta. 2008;228:789–801. doi: 10.1007/s00425-008-0781-6. [DOI] [PubMed] [Google Scholar]
- 22.Ilg A, Beyer P, Al-Babili S. The FEBS journal. 2009;276:736–747. doi: 10.1111/j.1742-4658.2008.06820.x. [DOI] [PubMed] [Google Scholar]
- 23.Vogel JT, Tan BC, McCarty DR, Klee HJ. The Journal of biological chemistry. 2008;283:11364–11373. doi: 10.1074/jbc.M710106200. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz SH, Tan BC, McCarty DR, Welch W, Zeevaart JA. Biochimica et biophysica acta. 2003;1619:9–14. doi: 10.1016/s0304-4165(02)00422-1. [DOI] [PubMed] [Google Scholar]
- 25.Hu KQ, Liu C, Ernst H, Krinsky NI, Russell RM, Wang XD. The Journal of biological chemistry. 2006;281:19327–19338. doi: 10.1074/jbc.M512095200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiefer C, Hessel S, Lampert JM, Vogt K, Lederer MO, Breithaupt DE, von Lintig J. The Journal of biological chemistry. 2001;276:14110–14116. doi: 10.1074/jbc.M011510200. [DOI] [PubMed] [Google Scholar]
- 27.Amengual J, Gouranton E, van Helden YG, Hessel S, Ribot J, Kramer E, Kiec-Wilk B, Razny U, Lietz G, Wyss A, Dembinska-Kiec A, Palou A, Keijer J, Landrier JF, Bonet ML, von Lintig J. PloS one. 2011;6:e20644. doi: 10.1371/journal.pone.0020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lobo GP, Isken A, Hoff S, Babino D, von Lintig J. Development. 2012;139:2966–2977. doi: 10.1242/dev.079632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin M, Li S, Moghrabi WN, Sun H, Travis GH. Cell. 2005;122:449–459. doi: 10.1016/j.cell.2005.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:12413–12418. doi: 10.1073/pnas.0503460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Redmond TM, Gentleman S, Duncan T, Yu S, Wiggert B, Gantt E, Cunningham FX., Jr The Journal of biological chemistry. 2001;276:6560–6565. doi: 10.1074/jbc.M009030200. [DOI] [PubMed] [Google Scholar]
- 32.Lindqvist A, Andersson S. The Journal of biological chemistry. 2002;277:23942–23948. doi: 10.1074/jbc.M202756200. [DOI] [PubMed] [Google Scholar]
- 33.Moiseyev G, Takahashi Y, Chen Y, Gentleman S, Redmond TM, Crouch RK, Ma JX. The Journal of biological chemistry. 2006;281:2835–2840. doi: 10.1074/jbc.M508903200. [DOI] [PubMed] [Google Scholar]
- 34.Borowski T, Blomberg MR, Siegbahn PE. Chemistry. 2008;14:2264–2276. doi: 10.1002/chem.200701344. [DOI] [PubMed] [Google Scholar]
- 35.Kiser PD, Farquhar ER, Shi W, Sui X, Chance MR, Palczewski K. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2747–2756. doi: 10.1073/pnas.1212025109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi Y, Moiseyev G, Chen Y, Ma JX. FEBS letters. 2005;579:5414–5418. doi: 10.1016/j.febslet.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 37.Redmond TM, Poliakov E, Yu S, Tsai JY, Lu ZJ, Gentleman S. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13658–13663. doi: 10.1073/pnas.0504167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poliakov E, Gentleman S, Cunningham FX, Jr, Miller-Ihli NJ, Redmond TM. The Journal of biological chemistry. 2005;280:29217–29223. doi: 10.1074/jbc.M500409200. [DOI] [PubMed] [Google Scholar]
- 39.Que L, Jr, Ho RY. Chemical reviews. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 40.Costas M, Mehn MP, Jensen MP, Que L., Jr Chemical reviews. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 41.Pau MY, Lipscomb JD, Solomon EI. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:18355–18362. doi: 10.1073/pnas.0704191104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gillmor SA, Villasenor A, Fletterick R, Sigal E, Browner MF. Nature structural biology. 1997;4:1003–1009. doi: 10.1038/nsb1297-1003. [DOI] [PubMed] [Google Scholar]
- 43.Katona G, Andreasson U, Landau EM, Andreasson LE, Neutze R. Journal of molecular biology. 2003;331:681–692. doi: 10.1016/s0022-2836(03)00751-4. [DOI] [PubMed] [Google Scholar]
- 44.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 45.Ruch S, Beyer P, Ernst H, Al-Babili S. Molecular microbiology. 2005;55:1015–1024. doi: 10.1111/j.1365-2958.2004.04460.x. [DOI] [PubMed] [Google Scholar]
- 46.Markwell J, Bruce BD, Keegstra K. The Journal of biological chemistry. 1992;267:13933–13937. [PubMed] [Google Scholar]
- 47.Britton G. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1995;9:1551–1558. [PubMed] [Google Scholar]
- 48.Socaciu C, Jessel R, Diehl HA. Spectrochimica acta Part A, Molecular and biomolecular spectroscopy. 2000;56:2799–2809. doi: 10.1016/s1386-1425(00)00375-9. [DOI] [PubMed] [Google Scholar]
- 49.Socaciu C, Jessel R, Diehl HA. Chemistry and physics of lipids. 2000;106:79–88. doi: 10.1016/s0009-3084(00)00135-3. [DOI] [PubMed] [Google Scholar]
- 50.Leuenberger MG, Engeloch-Jarret C, Woggon WD. Angewandte Chemie. 2001;40:2613–2617. doi: 10.1002/1521-3773(20010716)40:14<2613::AID-ANIE2613>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 51.Golczak M, Kiser PD, Lodowski DT, Maeda A, Palczewski K. The Journal of biological chemistry. 2010;285:9667–9682. doi: 10.1074/jbc.M109.063941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scherzinger D, Ruch S, Kloer DP, Wilde A, Al-Babili S. The Biochemical journal. 2006;398:361–369. doi: 10.1042/BJ20060592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takahashi Y, Moiseyev G, Chen Y, Ma JX. Investigative ophthalmology & visual science. 2006;47:5191–5196. doi: 10.1167/iovs.06-0614. [DOI] [PubMed] [Google Scholar]
- 54.Xue L, Jahng WJ, Gollapalli D, Rando RR. Biochemistry. 2006;45:10710–10718. doi: 10.1021/bi060897y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xue L, Gollapalli DR, Maiti P, Jahng WJ, Rando RR. Cell. 2004;117:761–771. doi: 10.1016/j.cell.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 56.Jin M, Yuan Q, Li S, Travis GH. The Journal of biological chemistry. 2007;282:20915–20924. doi: 10.1074/jbc.M701432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hamel CP, Tsilou E, Pfeffer BA, Hooks JJ, Detrick B, Redmond TM. The Journal of biological chemistry. 1993;268:15751–15757. [PubMed] [Google Scholar]
- 58.Takahashi Y, Moiseyev G, Ablonczy Z, Chen Y, Crouch RK, Ma JX. The Journal of biological chemistry. 2009;284:3211–3218. doi: 10.1074/jbc.M807248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chander P, Gentleman S, Poliakov E, Redmond TM. The Journal of biological chemistry. 2012;287:30552–30559. doi: 10.1074/jbc.M112.364596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Redmond TM, Poliakov E, Kuo S, Chander P, Gentleman S. The Journal of biological chemistry. 2010;285:1919–1927. doi: 10.1074/jbc.M109.027458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poliakov E, Parikh T, Ayele M, Kuo S, Chander P, Gentleman S, Redmond TM. Biochemistry. 2011;50:6739–6741. doi: 10.1021/bi200532m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmidt H, Kurtzer R, Eisenreich W, Schwab W. The Journal of biological chemistry. 2006;281:9845–9851. doi: 10.1074/jbc.M511668200. [DOI] [PubMed] [Google Scholar]
- 63.Devery J, Milborrow BV. The British journal of nutrition. 1994;72:397–414. doi: 10.1079/bjn19940042. [DOI] [PubMed] [Google Scholar]
- 64.Lindqvist A, Sharvill J, Sharvill DE, Andersson S. The Journal of nutrition. 2007;137:2346–2350. doi: 10.1093/jn/137.11.2346. [DOI] [PubMed] [Google Scholar]
- 65.Leung WC, Hessel S, Meplan C, Flint J, Oberhauser V, Tourniaire F, Hesketh JE, von Lintig J, Lietz G. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2009;23:1041–1053. doi: 10.1096/fj.08-121962. [DOI] [PubMed] [Google Scholar]
- 66.von Lintig J. Annual review of nutrition. 2010;30:35–56. doi: 10.1146/annurev-nutr-080508-141027. [DOI] [PubMed] [Google Scholar]
- 67.Lobo GP, Amengual J, Palczewski G, Babino D, von Lintig J. Biochimica et biophysica acta. 2012;1821:78–87. doi: 10.1016/j.bbalip.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, Eliaou C, Liu SY, Harris E, Redmond TM, Arnaud B, Claustres M, Hamel CP. Nature genetics. 1997;17:139–141. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- 69.Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A. Nature genetics. 1997;17:194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 70.Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3088–3093. doi: 10.1073/pnas.95.6.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bereta G, Kiser PD, Golczak M, Sun W, Heon E, Saperstein DA, Palczewski K. Biochemistry. 2008;47:9856–9865. doi: 10.1021/bi800905v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walia S, Fishman GA, Jacobson SG, Aleman TS, Koenekoop RK, Traboulsi EI, Weleber RG, Pennesi ME, Heon E, Drack A, Lam BL, Allikmets R, Stone EM. Ophthalmology. 2010;117:1190–1198. doi: 10.1016/j.ophtha.2009.09.056. [DOI] [PubMed] [Google Scholar]
- 73.von Lintig J, Dreher A, Kiefer C, Wernet MF, Vogt K. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:1130–1135. doi: 10.1073/pnas.031576398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oberhauser V, Voolstra O, Bangert A, von Lintig J, Vogt K. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19000–19005. doi: 10.1073/pnas.0807805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Notredame C, Higgins DG, Heringa J. Journal of molecular biology. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 76.Gouet P, Courcelle E, Stuart DI, Metoz F. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]