

Abstract
The complete sequence of retroperitoneal fibromatosis-associated herpesvirus Macaca nemestrina (RFHVMn), the pig-tailed macaque homolog of Kaposi's sarcoma-associated herpesvirus (KSHV), was determined by next-generation sequence analysis of a Kaposi's sarcoma (KS)-like macaque tumor. Colinearity of genes was observed with the KSHV genome, and the core herpesvirus genes had strong sequence homology to the corresponding KSHV genes. RFHVMn lacked homologs of open reading frame 11 (ORF11) and KSHV ORFs K5 and K6, which appear to have been generated by duplication of ORFs K3 and K4 after the divergence of KSHV and RFHV. RFHVMn contained positional homologs of all other unique KSHV genes, although some showed limited sequence similarity. RFHVMn contained a number of candidate microRNA genes. Although there was little sequence similarity with KSHV microRNAs, one candidate contained the same seed sequence as the positional homolog, kshv-miR-K12-10a, suggesting functional overlap. RNA transcript splicing was highly conserved between RFHVMn and KSHV, and strong sequence conservation was noted in specific promoters and putative origins of replication, predicting important functional similarities. Sequence comparisons indicated that RFHVMn and KSHV developed in long-term synchrony with the evolution of their hosts, and both viruses phylogenetically group within the RV1 lineage of Old World primate rhadinoviruses. RFHVMn is the closest homolog of KSHV to be completely sequenced and the first sequenced RV1 rhadinovirus homolog of KSHV from a nonhuman Old World primate. The strong genetic and sequence similarity between RFHVMn and KSHV, coupled with similarities in biology and pathology, demonstrate that RFHVMn infection in macaques offers an important and relevant model for the study of KSHV in humans.
INTRODUCTION
Kaposi's sarcoma (KS) continues to be the world's most common AIDS-associated malignancy despite the strong research effort over the past 30 years to study its cause and to develop effective therapies (1). KS presents as a highly vascularized neoplasm containing characteristic elongated, spindle-shaped tumor cells expressing endothelial cell markers (2). Kaposi's sarcoma-associated herpesvirus (KSHV)/human herpesvirus 8 (HHV8) was discovered in 1994 (3) and is now known to be the causative agent of all forms of KS, including both classical (non-HIV) and HIV-associated KS (4). KS tumors are uniformly infected with KSHV (5), and tumor cells express a number of KSHV genes associated with viral latency, including the open reading frame 73 (ORF73) latency-associated nuclear antigen (LANA) (6). Subsequent studies have shown that KSHV also plays an etiological role in AIDS-associated pleural effusion B-cell lymphoma and multicentric Castleman's disease, two lymphoproliferative disorders (7).
Sequence analysis of the KSHV genome revealed strong similarities with the New World primate rhadinovirus herpesvirus saimiri (HVS), which infects the squirrel monkey and causes T-cell lymphomas in heterologous hosts (8). The linear arrangement of genes in the KSHV genome was so similar to that in the HVS genome that the gene nomenclature for KSHV was patterned after that of HVS, with KSHV ORF designations ranging from 2 to 75 and 15 novel KSHV genes given a KSHV-specific “K” designation (K1 to K15). Many of these KSHV-specific genes are captured cellular genes that endow the virus with important immune evasion and pathogenic properties that are important to the biology and life cycle of the virus and play roles in tumorigenesis (9). KSHV is more distantly related to Epstein-Barr virus (EBV), which also causes lymphoproliferative disorders, placing KSHV, along with EBV, in the gammaherpesvirus subfamily of tumorigenic herpesviruses. KSHV is formally grouped with HVS within the genus Rhadinovirus, while EBV and related nonhuman primate viruses group together in the genus Lymphocryptovirus of gammaherpesviruses.
An epidemic of KS-like tumors occurred in several different macaque species at the Washington National Primate Research Center (WaNPRC) in the early 1980s (10, 11). This syndrome, called retroperitoneal fibromatosis (RF), was characterized by multifocal subcutaneous and systemic lesions involving retroperitoneal tissues that progressively expanded, forming large masses in the abdominal cavity, resulting in 10% mortality in colony-born juveniles and 1% in all colony macaques at the WaNPRC. The epidemic of RF was associated with a form of simian AIDS (SAIDS) caused by infection with a simian D-type retrovirus (simian retrovirus type 2 [SRV-2]) (11). Morphological and histochemical analysis of the characteristic RF tumors revealed many similarities with KS, including the presence of proliferating spindle-shaped mesenchymal tumor cells and neoangiogenesis (12). The multifocal nature of the tumor and the association with a form of AIDS caused by retroviral infection suggested a common etiology with KS.
Using the consensus-degenerate hybrid oligonucleotide primer (CODEHOP) approach (13), we detected DNA sequences of two closely related novel viruses in RF tumors from different macaque species and designated these viruses retroperitoneal fibromatosis-associated herpesvirus Macaca mulatta (RFHVMm) from rhesus macaques and retroperitoneal fibromatosis-associated herpesvirus Macaca nemestrina (RFHVMn) from pig-tailed macaques (14). Using additional PCR-based approaches, 10-kb flanking regions of the RFHVMm and RFHVMn genomes were sequenced, which showed a high level of sequence conservation with KSHV and a strong conservation of genomic structure, including homologs of several KSHV-specific ORFs (15). Based on these sequence and genetic similarities, RFHV was grouped with KSHV within the genus Rhadinovirus of gammaherpesviruses. Using quantitative-PCR (qPCR) techniques, high levels of RFHV genomic DNA were detected in RF tumors from different macaque species, with viral loads reaching several viral genomes per cell (16, 17), corresponding to the viral load of latent KSHV in KS tumors (3). Furthermore, immunohistochemical studies revealed high levels of the RFHV ORF73 LANA homolog in the nuclei of the RF spindloid tumor cells, detected with a monoclonal antibody to KSHV LANA (16, 18). These findings suggested that RFHV had an etiological association with RF tumors in macaques.
Subsequent to the discovery of RFHV, another macaque rhadinovirus was identified at the New England National Primate Research Center (NENPRC). This virus, called rhesus rhadinovirus (RRV), was cultured from blood cells of an infected macaque, viral DNA was isolated, and a 10-kb fragment was cloned and sequenced (19). A closely related rhadinovirus, MneRV2/PRV, was also identified in pig-tailed macaques (20, 21). The full genome sequence of RRV was obtained from purified viral DNA from the original New England strain (H26-95) (22) and another RRV strain (17577) from a rhesus macaque at the Oregon National Primate Research Center (ONPRC) (23). Sequence analysis revealed strong conservation of genomic structure between RRV, KSHV, and HVS, and RRV was also grouped within the genus Rhadinovirus of gammaherpesviruses. However, both RRV strains lacked homologs of most of the KSHV-specific ORFs, suggesting important biological differences between RRV and KSHV.
Phylogenetic analysis of available sequences revealed that the members of the genus Rhadinovirus in primates segregated into two phylogenetic groups, the New World and Old World primate rhadinoviruses, and that the Old World rhadinoviruses further segregated into two distinct rhadinovirus lineages. The Rhadinovirus-1 (RV1) lineage consisted of KSHV and close homologs from other nonhuman Old World primates, including RFHV strains from different macaque species, while the Rhadinovirus-2 (RV2) lineage consisted of RRV, MneRV2, and homologs from other nonhuman Old World primate species (21, 24). Whereas members of both RV1 and RV2 rhadinovirus lineages have been identified in numerous Old World primate species, including chimpanzees, gorillas, macaques, and different monkey species (14, 19, 21, 24–30), only the RV1 rhadinovirus KSHV has been identified in humans.
While the full genome of RRV was sequenced from highly purified viral DNA obtained from primary lytic infection of cultured cells, it has not been possible to purify RFHV DNA for genomic sequence analysis due to the lack of a permissive infection system. Previously, fragments of the RFHV genome were laboriously sequenced from PCR products or cloned genome fragments (15, 18, 21, 31). With the reduced cost of high-throughput next-generation (Next-Gen) sequencing techniques, we have now obtained the complete genome sequence of RFHVMn from an archived frozen RF tumor carrying ∼3 RFHV genomes per cell by Illumina-based sequencing of the complete complement of DNA present in the tumor, reducing the DNA reads from the macaque genome bioinformatically, and performing a de novo assembly of the RFHV genome sequence. This is the first sequence from a clinical isolate of an RV1 rhadinovirus genome determined by Next-Gen approaches. Comparison of the RFHVMn genome with the KSHV genome revealed close genetic and genomic similarities between the macaque and human RV1 rhadinoviruses, suggesting a common biology, life cycle, and pathology. These similarities demonstrate that RFHV infection in macaques is a close animal model of KSHV infection in humans.
MATERIALS AND METHODS
Tissue and DNA preparation.
A subcutaneous fibromatosis tumor from a female pig-tailed macaque, M78114, was removed at necropsy (age, 5.8 years) and stored frozen at −70°C (animal 2) (12). DNA was isolated from a portion of the tumor by digestion with proteinase K, followed by phenol-chloroform extraction and ethanol precipitation. This DNA was found to contain 3.4 copies of the RFHVMn genome per cellular genome by real-time PCR (17). A library was constructed from the tumor DNA using the Illumina Genomic Sample DNA Preparation Kit according to the manufacturer's instructions.
Next-Gen sequence analysis.
The library was clonally amplified on a cluster generation station using Illumina version 4 cluster generation reagents to achieve a target density of approximately 700,000/mm2 in a single channel of a flow cell. The resulting library was then sequenced on a Genome Analyzer IIx using Illumina version 5.0 sequencing reagents, which generated non-paired-end reads of 50 nucleotides. Image analysis, base calling, and error estimation were performed using Illumina Analysis Pipeline (version 2.8). A similar analysis was performed on the Illumina HiSeq instrument with a single lane of 100-bp reads. The sequence reads were first mapped onto the human and rhesus macaque genomes using Bowtie (32). The unmapped sequences were assembled into contigs using the de novo assembler Trinity (33). The contigs were analyzed using BLASTX to identify matches in a library of all KSHV proteins. The contigs with homology to KSHV proteins and the known RFHV sequences were assembled onto a KSHV genome sequence template using Sequencher (Gene Codes), and the gaps were filled in by PCR and Sanger sequencing. The sequence reads were mapped onto the resulting RFHVMn genome and visualized using Tablet (34). Any discrepancies in the mapped sequences were resolved by PCR and Sanger sequencing.
Gene annotations.
The annotation of the RFHVMn genome was initially performed using the Gene Annotation Transfer Utility (GATU) provided at the Viral Bioinformatics Resource Center (35) based on the reference sequence for KSHV, NC_009333. Additional open reading frames were identified using BLASTX (NCBI).
Dot plot alignments.
Alignments of the complete genome sequences of RFHVMn and KSHV were performed using the Java Dot Plot Alignment program (JDotter) provided at the Viral Bioinformatics Resource Center (36). Dot plot alignments of smaller DNA regions were performed using GenePro software (Riverside Scientific).
Comparative and phylogenetic analyses.
Sequence alignments were performed using GenePro software (Riverside Scientific) or Muscle (37). Phylogenetic analyses were performed using the protein distance (ProtDist), neighbor-joining (Neighbor), or protein maximum-likelihood with molecular clock (Promlk) algorithms implemented in Phylip (Seattle, WA) 3.62.
MicroRNA prediction.
Ab initio prediction of pre-microRNA (miRNA) hairpin structures was carried out as described previously (38–40) using the VMir software package with default settings.
Nucleotide sequence accession number.
The RFHVMn genome was submitted to GenBank under accession number KF703446.
RESULTS AND DISCUSSION
Next-Gen sequence analysis of the RFHVMn genome.
We previously reported that macaque RF tumors are latently infected with RFHV, with several viral genomes per cell (17). This level of virus infection is similar to that seen in latent KSHV infections in KS tumor biopsy specimens and constitutes the highest level of RFHV that has been identified in infected macaque tissue. Total DNA was extracted from a subcutaneous RF tumor, obtained from pig-tailed macaque M78114 at necropsy, that had been stored frozen at −70°C. This animal had multiple nodular cutaneous lesions that were aggressively enlarging (12). At necropsy, tumors were found in the lower abdomen wall, groins, thighs, lung, kidney, adrenal, and liver. There was moderate enlargement of the spleen and lymph nodes. The small subcutaneous nodules were composed of fibrous tissue with abundant spindle-shaped fibroblasts, compressed capillary structures, and focal hemosiderin deposits. Occasionally, two or three endothelial cells formed slit-like lumen structures. Electron microscopy showed evidence of type D retrovirus particles within the tumor nodules, and subsequent PCR sequence analysis identified this as SRV-2 strain 2B (41). Real-time qPCR analysis of the tumor DNA sample revealed 3.4 RFHVMn genomes per cell and 0.02 MneRV2 genome per cell (macaque RF2 in reference 17). This RF tumor DNA had previously been used to generate a lambda library from which several RFHVMn genomic fragments had been isolated (18).
To attempt Next-Gen sequence analysis of the RFHVMn genome, total DNA from this tumor was used to prepare an Illumina non-paired-end library and was sequenced on an Illumina Genome Analyzer II with two lanes of 50-bp reads. This resulted in 66 million reads, or approximately 3.3 × 109 bases, which was not sufficient to allow de novo assembly in the presence of the remaining nonviral genomic sequences.
The same Illumina library was used for a single-lane 100-bp analysis on the Illumina HiSeq, which gave 9.5 × 107 reads, or approximately 9.5 × 109 bases. These reads were mapped onto the human and rhesus macaque genomes using Bowtie. The unmapped sequence reads were assembled into contigs with the de novo assembler Trinity. The contigs were analyzed with BLASTX to determine which ones had homology to KSHV proteins in a local database. One hundred twelve contigs with homology to KSHV proteins were identified and aligned based on the genomic structure of KSHV. There were 46 contigs longer than 1 kb, with the largest contig having 6.7 kb of sequence. A BLASTN search of the subtracted read library using the 3′ 25 nucleotides of each contig as the query identified sequence reads that bridged several of the gaps between the contigs. Twenty gaps remained in the sequence, which were targeted for PCR amplification using positional information derived from the KSHV genome. PCR primers derived from adjacent contigs were used to amplify the DNA region spanning the contig gaps in the RFHVMn sequence. Sanger sequencing provided sequences bridging these 20 gaps with gap sizes ranging from 4 to 1,325 bp, with an average of 206 bp. The largest gaps occurred within direct- and inverted-repeat regions of the genome. To verify the final sequence, the reads from both the 50-bp runs and the 100-bp run were combined into a single read library and mapped using Bowtie onto the final sequence derived from the Illumina-generated contigs and the Sanger-generated gap sequences. Visualization of the mapped Illumina reads with Tablet revealed coverage of 99%, with an average depth of 7.4 reads per nucleotide. With the additional Sanger-sequencing reads, the sequence coverage reached 100%.
The RFHVMn genome is colinear with the KSHV genome.
The final sequence of the RFHVMn genome contained 127,320 bp and included all of the unique sequence and a portion of the terminal-repeat region. The G+C content was 53.2%, which is similar to that of the KSHV genome at 53.5%. The RFHVMn genome sequence was annotated based on the KSHV genome, as shown in Fig. 1, using the KSHV ORF nomenclature with positional/structural homologs of the KSHV-specific “K” ORFs designated “RF” ORFs. The sequence starts at the left end of the RFHV genome, 405 bp upstream of ORF RF1, a structural and positional homolog of the KSHV ORF K1, and ends 438 bp upstream of the initiation codon of ORF RF15, a structural and positional homolog of the KSHV ORF K15. Initially, the protein-coding ORFs were identified, and BLASTX was used to determine homology to other proteins in the nonredundant protein database. In cases of more than one possible initiating ATG codon, homology with KSHV and Kozak's consensus rules for codon initiation (42) were used to predict a protein initiation sequence. The ORFs predicted from the RFHVMn genome sequence are shown in Table 1, and the size and sequence homology to the corresponding ORFs from the KSHV and RRV genomes are indicated.
Fig 1.

Comparative map of the rhadinovirus and lymphocryptovirus genomes. The positions and transcription directions of the ORFs identified in the RFHVMn genome (KF703446; this study) are compared to the corresponding ORFs in KSHV (NC_009333), RRV (AF210726 and NC_003401), and EBV (NC_007605), as identified in the NCBI accession records (the basic design of this map was patterned after Fig. 1 of reference 22). The numbering of the KSHV, RFHVMn, and RRV ORFs is patterned after the genome structure of the prototype rhadinovirus, HVS, with KSHV–specific ORFs designated K1 to K15 and their homologs in RFHVMn and RRV designated RF1 to RF15 and R1 to R15, respectively. The EBV ORFs are indicated, using the typical EBV nomenclature above the ORF and the corresponding rhadinovirus designation below. The approximate positions of the ORFs are shown with regard to their positions within the KSHV genome (i.e., 1 to 140 kb). The sizes of the ORF markers are approximately consistent with the sizes of the actual encoded proteins. Vertical black lines within an ORF indicate splicing events or internal initiations, while longer–range splices of protein–coding exons are indicated with bars. The ORFs are color coded with regard to their conservation in other herpesvirus genomes, showing core HV genes conserved in most herpesvirus families, genes conserved in beta– and gammaherpesviruses, gammherpesviruses only, gamma–2–herpesviruses, RV1 and RV2 rhadinoviruses, RV1 rhadinoviruses only, KSHV only, and EBV only, as indicated. The RRV nomenclature from AF210726 was used for consistency.
Table 1.
ORFs predicted from the RFHVMn genome sequence, with size and sequence homology to the corresponding ORFs from the KSHV and RRV genomes
| ORF | Function or homolog | RFHVa size (aa) | KSHVb |
RRVc |
||
|---|---|---|---|---|---|---|
| Size (aa) | % Identityd | Size (aa) | % Identity | |||
| RF1 | ITAM-containing signal transducing membrane protein; K1 | 528 | 289 | 22.6 | 423 | 16.1 |
| 4 | Complement control protein; membrane protein; KCP | 515 | 558 | 33.6 | 395 | 30.4 |
| 6 | Single-stranded DNA binding protein (ssDBP); UL29 | 1,132 | 1,133 | 70.9 | 1,132 | 60.5 |
| 7 | Subunit of terminase; UL28 | 688 | 695 | 56.9 | 686 | 51.3 |
| 8 | Glycoprotein B; UL27 | 847 | 845 | 70.6 | 829 | 62.8 |
| 9 | DNA polymerase; UL30 | 1,013 | 1,012 | 75.7 | 1,014 | 67.5 |
| 10 | Derived from dUTPase; CMV UL82/83/84 | 411 | 418 | 44.0 | 416 | 30.1 |
| - | (ORF 11; derived from dUTPase; CMV UL82/83/84)f | - | 407 | 409 | ||
| RF2 | vIL-6; K2 | 209 | 204 | 32.4 | 207 | 15.1 |
| 2 | Dihydrofolate reductase (vDHFR) | 208 | 210 | 54.1 | 188 | 40.7 |
| RF3 | E3 ubiquitin ligase membrane protein; K3; MIR1; ORF12 | 232 | 333 | 38.7 | - | |
| 70 | Thymidylate synthase (vTS) | 344 | 337 | 64.6 | 333 | 61.6 |
| RF4 | Interleukin 8-like CC chemokine; K4; vMIP-II; vCCL-2 | 94 | 94 | 59.6 | 118 | 32.3 |
| RF4.1 | Interleukin 8-like CC chemokine; K4.1; vMIP-Ibeta; vCCL-3 | 102 | 114 | 48.2 | - | |
| RF4.2 | Contains hydrophobic domains; K4.2 | 125 | 182 | 14.5 | - | |
| - | (E3 ubiquitin ligase membrane protein; K5; MIR2)f | - | 256 | - | ||
| - | (Interleukin 8-like CC chemokine; vMIP-I; vCCL-1)f | - | 95 | - | ||
| RF7 | IAP-like inhibitor of apoptosis; K7; vIAP | 171 | 126 | 17.3 | - | |
| 16 | Bcl-2-like inhibitor of apoptosis; vBcl-2 | 178 | 175 | 42.3 | 187 | 39.4 |
| 17 | Minor capsid scaffold protein; UL26 | 546 | 553 | 47.7 | 536 | 41.1 |
| 17.5 | Major capsid scaffold protein; UL26.5 | 300 | 288 | 35.0 | 291 | 32.2 |
| 18 | Related to CMV UL79 | 255 | 257 | 66.1 | 257 | 54.9 |
| 19 | Putative portal-capping protein; UL25 | 547 | 549 | 59.0 | 547 | 47.3 |
| 20 | Nuclear protein; UL24 | 287 | 320 | 51.0 | 350 | 39.5 |
| 21 | Thymidine kinase; UL23 | 564 | 580 | 55.5 | 557 | 38.2 |
| 22 | Glycoprotein H; UL22 | 713 | 730 | 47.9 | 726 | 35.6 |
| 23 | Tegument protein;UL21 | 411 | 404 | 53.1 | 402 | 46.1 |
| 24 | Related to CMV UL87 | 752 | 752 | 64.1 | 732 | 54.4 |
| 25 | Major capsid protein; UL19 | 1,378 | 1,376 | 82.1 | 1,378 | 74.5 |
| 26 | Capsid triplex protein; UL18 | 305 | 305 | 78.0 | 305 | 63.3 |
| 27 | Related to EBV BDLF2 | 293 | 290 | 41.7 | 269 | 28.0 |
| 28 | Membrane protein | 92 | 102 | 50.5 | 91 | 32.1 |
| 29 | Putative ATPase subunit of terminase; UL15 | 685 | 687 | 72.1 | 690 | 52.7 |
| 30 | Related to CMV UL91 | 96 | 77 | 62.3 | 76 | 31.7 |
| 31 | Related to CMV UL92 | 202 | 224 | 64.9 | 217 | 46.3 |
| 32 | Tegument protein; DNA packaging; UL17 | 451 | 454 | 44.0 | 464 | 36.3 |
| 33 | Tegument protein; UL16 | 335 | 312 | 57.8 | 336 | 42.3 |
| 34 | Related to CMV UL95 | 329 | 327 | 60.3 | 327 | 48.5 |
| 35 | Tegument protein; UL14 | 150 | 151 | 56.0 | 149 | 32.0 |
| 36 | Serine-threonine protein kinase; UL13 | 444 | 444 | 67.1 | 435 | 44.3 |
| 37 | SOX; host shutoff; DNase; UL12 | 487 | 486 | 71.7 | 472 | 65.9 |
| 38 | alkaline exonuclease; UL11 | 63 | 61 | 52.4 | 69 | 36.4 |
| 39 | Glycoprotein M;UL10 | 393 | 400 | 64.3 | 378 | 60.3 |
| 40 | Subunit of helicase-primase complex; UL8 | 642 | 669 | 38.2 | 654 | 31.2 |
| 42 | Tegument protein; UL7 | 277 | 278 | 59.9 | 272 | 45.7 |
| 43 | Portal protein; UL6 | 575 | 605 | 75.1 | 576 | 60.5 |
| 44 | Helicase subunit of helicase-primase complex; UL5 | 782 | 788 | 74.4 | 790 | 66.0 |
| 45 | Inhibition of IRF-7; virion phosphoprotein; | 362 | 407 | 27.5 | 353 | 24.0 |
| 46 | Uracil-DNA glycosylase; UL2 | 254 | 255 | 68.5 | 230 | 54.2 |
| 47 | Glycoprotein gL; UL1 | 166 | 167 | 38.0 | 163 | 28.3 |
| 48 | Unknown function; related to BRRF2 EBV | 433 | 402 | 30.4 | 389 | 28.4 |
| 49 | Unknown function; related to BRRF1 EBV | 302 | 302 | 63.9 | 301 | 52.8 |
| 50 | RTA; transactivator; related to BRLF1 EBV | 665 | 631 | 48.3 | 577 | 36.7 |
| RF8α | bZIP transcription factor; K8α | 238 | 237 | 34.3 | 234 | 22.7 |
| RF8.1 | ORF51; K8.1; related to gp42 EBV | 300 | 228 | 18.5 | 275 | 12.0 |
| 52 | Related to BLRF2 EBV | 133 | 131 | 51.9 | 139 | 44.6 |
| 53 | Glycoprotein N; UL49A | 104 | 108 | 43.5 | 104 | 42.3 |
| 54 | Deoxyuridine triphosphatase; UL50 | 309 | 318 | 50.3 | 290 | 38.5 |
| 55 | Tegument protein; UL51 | 222 | 227 | 66.7 | 210 | 51.1 |
| 56 | Primase subunit of helicase-primase complex; UL52 | 838 | 843 | 57.8 | 828 | 50.5 |
| 57 | Posttranscriptional regulator; UL54 | 462 | 455 | 53.5 | 442 | 42.0 |
| RF9 | vIRF-1; K9 | 466 | 449 | 35.6 | 415e | 16.9e |
| RF10 | vIRF-4; K10 | 773 | 911 | 20.3 | - | - |
| RF10.5 | vIRF-3; K10.5 | 601 | 566 | 22.0 | - | - |
| RF11 | vIRF-2; K11 | 424 | 467 | 27.5 | - | - |
| 58 | Multiple transmembrane protein; UL43 | 358 | 357 | 51.8 | 360 | 38.2 |
| 59 | DNA polymerase processivity factor; UL42 | 400 | 396 | 57.8 | 394 | 50.3 |
| 60 | Ribonucleotide reductase small subunit; UL40 | 305 | 305 | 79.7 | 304 | 69.5 |
| 61 | Ribonucleotide reductase large subunit; UL39 | 790 | 792 | 44.3 | 788 | 61.5 |
| 62 | Capsid triplex protein; UL38 | 328 | 331 | 66.5 | 331 | 51.8 |
| 63 | Tegument protein; UL37 | 926 | 928 | 44.5 | 939 | 36.8 |
| 64 | Large tegument protein; UL36 | 2,604 | 2,635 | 45.8 | 2,548 | 16.2 |
| 65 | Small capsid protein on hexon tips; UL35 | 187 | 170 | 42.8 | 169 | 38.3 |
| 66 | BFRF2 EBV homolog; related to CMV UL49 | 427 | 429 | 57.3 | 448 | 43.4 |
| 67 | Capsid docking protein on nuclear lamina; UL34 | 269 | 271 | 63.8 | 222 | 61.7 |
| 67A | DNA packaging; UL33 | 84 | 80 | 56.8 | 86 | 51.2 |
| 68 | Nuclear localization of capsids and DNA packaging; UL32 | 462 | 467 | 60.2 | 457 | 47.6 |
| 69 | Egress of capsids from nucleus; UL31 | 296 | 302 | 65.9 | 296 | 59.7 |
| RF12 | Related to Kaposin A; K12 | 212 | 60 | 19.0 | - | |
| RF13 | FLIP-like inhibitor of apoptosis; vFLIP; K13 | 267 | 188 | 48.4 | 174 | 35.2 |
| 72 | Cyclin D-like protein; vCyc | 265 | 257 | 46.3 | 254 | 34.7 |
| 73 | Latency-associated nuclear antigen; LANA | 1,071 | 1,162 | 24.3 | 448 | 20.4 |
| RF14 | vOX2; contains two Ig domains; K14 | 300 | 314 | 33.3 | 253 | 32.0 |
| 74 | vGPCR; G protein coupled receptor; similar to interleukin 8 receptor | 336 | 342 | 56.4 | 342 | 43.1 |
| 75 | Tegument protein; related to FGARAT | 1,303 | 1,296 | 62.2 | 1,298 | 42.4 |
| RF15 | Signal transducing membrane protein; K15; LAMP | 487 | 489 | 21.5 | 531 | 20.0 |
RFHV, RFHVMn genome (KF703446).
KSHV, KSHV genome (NC_009333).
RRV, RRV-26-95 (AF10726); KSHV-specific ORFs missing in RRV are indicated with hyphens.
Percent identity to the corresponding ORF from the RFHVMn genome.
Eightfold amplification of the RF9/K9 gene homolog—RRV ORF9-1 to ORF9-8.
KSHV-specific ORFs missing in RFHVMn are indicated in parentheses, and their relative positions within the RFHVMn genome are indicated by hyphens.
Homologs of all of the major KSHV ORFs, except ORF11, K5/MIR2, and K6/vCCL-1, were identified in the RFHVMn genome. The sequence homology with KSHV ranged from 14% (RF4.2/K4.2) to 82% (ORF25 major capsid protein), while the homology with RRV ranged from 12% (RF8.1/R8.1) to 75% (ORF25 major capsid protein). In all cases, the RFHV ORFs were more similar to the corresponding KSHV ORFs than they were to the RRV ORFs. Most of the core herpesvirus genes were highly conserved between RFHVMn and KSHV (Table 1 and Fig. 1). Ten core genes encoded proteins nearly identical in size to the KSHV homolog, with sequence identities ranging from 70 to 82%. For example, the ORF25 major capsid protein homolog of RFHVMn was 1,376 amino acids (aa) in length compared to 1,378 aa for the KSHV homolog, with 82% identity. An additional 37 herpesvirus core genes in RFHVMn encoded proteins that had 50 to 70% identity to the corresponding KSHV-encoded proteins.
The complete RFHVMn genomic DNA sequence was aligned with the complete KSHV genomic DNA sequence using the Java Dot Plot alignment program (JDotter), which generates dot plot alignments of large DNA and protein sequences (36). The RFHVMn and KSHV genomes were colinear and showed a high degree of nucleotide sequence conservation that was evident throughout the genomes (Fig. 2). Five divergent loci with regions of lower DNA sequence similarity were identified.
Fig 2.
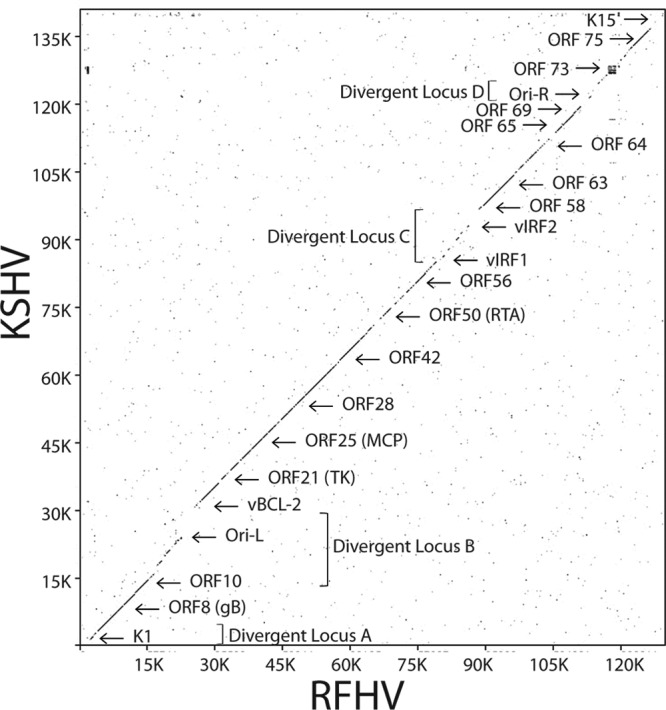
Nucleotide dot plot alignment of the complete RFHVMn and KSHV genomes. The nucleotide sequence of the RFHVMn genome (KF703446) was compared to the sequence of the KSHV genome (NC_009333) using the Java Dot Plot alignment program (JDotter). The positions of specific ORFs and the divergent loci are indicated.
Divergent locus A.
Divergent locus A corresponds to the left end of the genomic region of RFHVMn and KSHV, containing the junction of the terminal-repeat region with the beginning of the unique genomic sequence (Fig. 1 and 2). The first 206 bp of the RFHVMn genomic sequence contained a repeating pattern of the hexanucleotide sequence AACCTT and closely related motifs. At bp 405, the ORF RF1, which encoded a structural and positional homolog of KSHV ORF K1, was identified (43) (Fig. 1 and 2). Like ORF K1, the ORF RF1 protein contained a strongly conserved N-terminal domain related to the immunoglobulin receptor family (44), with a C-terminal hydrophobic transmembrane domain and a conserved immunoglobulin receptor tyrosine-based activation motif (ITAM) (Fig. 3). Unlike K1, the RF1 transmembrane domain was flanked by two protein repeat regions. The upstream region (repeat 1) encoded 9 repeating units of the peptide sequence QAPTTRETTTTSGHTT, whereas the downstream region (repeat 2) encoded large numbers of glutamine residues interspersed with either histidines or prolines (Fig. 3).
Fig 3.

Comparison of the KSHV ORF K1 and the RFHVMn ORF RF1 protein sequences. The sequences of KSHV ORF K1 (YP_001129350) and the RFHVMn ORF RF1 (AGY30683) positional homolog were aligned. Identical residues are highlighted in black. The regions conserved with the immunoglobulin (Ig) superfamily and the immunoglobulin receptor tyrosine–based activation motif (ITAM) are indicated, and the variable regions (VR1 and VR2) identified in the KSHV subtypes (45) are shown. The residues predicted to form a hydrophobic transmembrane (TM) domain and the positions of two repeat domains present in the RF1 sequence are indicated. Potential N-linked glycosylation sites are underlined, and sites conserved between K1 and RF1 are indicated with asterisks. Dashed lines represent gaps in the sequence, while solid lines represent continuations of the left and right brackets.
The KSHV K1 gene exhibits high sequence variability among virus subtypes and variants, with changes concentrated within two 40-amino-acid extracellular-domain variable regions (45). The first variable region (VR1) of K1 contains two N-linked glycosylation sites (Asn53 and Asn73) and a highly conserved cysteine residue (Cys76) in common with RF1 (Fig. 3). Surprisingly, RF1 lacks two cysteine residues (Cys52/Cys72) (Fig. 3) that flank a hypervariable region in K1, which was postulated to form a 23-amino-acid hypervariable loop with a disulfide bridge between Cys52 and Cys76 (45). The absence of homologs of Cys52 and Cys72 in RF1 suggests that the hypervariable loop in K1 is actually 19 amino acids, with a disulfide bridge between Cys52 and Cys72. The unique repeat region (repeat 1) present in RF1 is positioned in the middle of the second variable region (VR2) of K1. The amino acid blocks conserved between RF1 and K1 flank these variable regions. It is unknown whether the RF1 gene displays the same high sequence variability seen in K1 in different subtypes or RFHVMn variants.
Divergent locus B.
Divergent locus B (Fig. 1) (15 to 30 kb) corresponds to the KSHV genomic region containing a number of viral homologs of cellular genes, including ORF K2, the viral interleukin 6 (vIL-6) homolog; ORF2, the viral dihydrofolate reductase (vDHFR) homolog; ORF K3, the viral modulator of immune response (vMIR1) homolog; ORF70, the viral thymidylate synthase (vTS) homolog; ORF K4, the viral CC chemokine homolog (vCCL2); ORF K4.1, the viral CC chemokine homolog (vCCL3); and ORF16, the viral BCL-2 homolog (vBCL-2). The viral homologs present in divergent locus B of KSHV are lacking in the more distantly related EBV genome (8) (Fig. 1), suggesting that they were acquired from an ancestral primate host genome after the divergence of the Rhadinovirus and Lymphocryptovirus gammaherpesvirus lineages.
We have previously shown, using PCR-based approaches, that the divergent locus B of the RFHV genome contained conserved homologs of ORF K2 vIL-6, ORF2 vDHFR, ORF K3 vMIR1, and ORF70 vTS (15). The localizations of the vDHFR genes in RFHV and KSHV (Fig. 1; position. ∼18 kb,) are identical and different from those seen in RRV and HVS (Fig. 1; position, ∼2 kb), suggesting a different capture event for the ancestral RV1 vDHFR gene (15). As we have reported previously, the RFHV genome lacks a homolog of ORF11, a gene of unknown function that is present in all other known gammaherpesviruses (15). The ORF11 homologs of KSHV, RRV, and EBV (LF2) are distantly related to the ORF10 homologs in KSHV, RFHVMn, RRV, and EBV (LF1) (15), and both appear to have evolved through duplication of a dUTPase captured from an ancestral host genome (46). This suggests that the lack of an ORF11 homolog in RFHV may be compensated for by ORF10.
The Next-Gen sequence analysis revealed that the RFHVMn genome also contained homologs of vCCL2 (ORF RF4), vCCL3 (ORF RF4.1), and vBCL2 (ORF16), which were highly conserved with the corresponding KSHV genes (Fig. 4A, B, and D, respectively). However, the RFHVMn genome was missing homologs of vMIR2 (K5) and vCCL-1 (K6) (Fig. 1; positions, ∼25 to 28 kb). The KSHV ORF K5 is a close homolog of ORF K3, while ORF K6 is a close homolog of ORF K4. The pairing of ORFs K3 and K4 and the pairing of ORFs K5 and K6, coupled with the absence of the ORF K5 and K6 pair in RFHVMn, suggests that ORF K5 and K6 resulted from a duplication of the ancestral K3 and K4 genes after the divergence of RFHVMn and KSHV, supporting previous conclusions (47). An ori-Lyt-associated transcript has been identified in KSHV immediately adjacent to K5 (48). No evidence for a homologous sequence was identified in the RFHVMn genome, suggesting that this gene evolved after the divergence of RFHVMn and KSHV, possibly associated with the duplication event that generated the K5 and K6 genes.
Fig 4.

Amino acid alignments of the KSHV ORFs K4, K4.1, K4.2, and vBCL2 with the corresponding RFHVMn homologs. The amino acid alignments of ORFs K4 (YP_001129362) and RF4 (AGY30694) (A), K4.1 (YP_001129363) and RF4.1 (AGY30695), (B), K4.2 (YP_001129364) and RF4.2 (AGY30696) (C), and KvBCL2 (YP_001129368) and RFvBCL2 (AGY30698) (D) are shown. Putative hydrophobic TM domains and N-terminal signal sequences with predicted cleavage sites are underlined, and cysteine residues are highlighted. BLAST analysis revealed that the K4/RF4 and K4.1/RF4.1 ORFs are conserved members of the family of interleukin 8-like CC cytokines. The K4.2 and RF4.2 ORFs both contained putative transmembrane domains (two in K4.2 and one in RF4.2). *, identical residue; :, conservative substitution; ., semiconservative substitution.
The RFHVMn genome included ORF RF4.2 and ORF RF7, positional homologs of KSHV ORFs K4.2 and K7. While K4.2 contains two potential membrane-spanning domains at the C terminus, RF4.2 contains only a single membrane-spanning domain in the middle of the protein (Fig. 4C). Only minimal sequence similarity was evident between K4.2 and RF4.2, and the functions are unknown. ORF K7 has been identified as an inhibitor of apoptosis with an unknown mechanism (49). Previously, sequence similarity was detected between K7 and a splice variant of human survivin (50). Analysis of ORF RF7 revealed the presence of an N-terminal hydrophobic domain similar to that seen in ORF K7, which is predicted to function as a membrane-spanning domain (Fig. 5, underlined). Alignment of the RF7 and K7 sequences revealed a low level of sequence similarity (Fig. 5, highlighted in green). However, alignment of RF7, K7, and human survivin revealed clear sequence homology between RF7 and survivin (Fig. 5, highlighted in red), which was even more pronounced than the homology between K7 and survivin (Fig. 5, highlighted in blue). While some sequence motifs were conserved across the three sequences (Fig. 5, highlighted in black), most of the amino acids conserved between RF7 and survivin were not those conserved between K7 and survivin. However, the overall similarity between RF7, K7, and survivin adds support to the hypothesis that both RF7 and K7 have evolved from an ancestral survivin-like gene that had been captured from an ancestral host genome. The similarities detected between the antiapoptotic functions of K7 and survivin (50, 51) suggest that RF7 would also function as an inhibitor of apoptosis.
Fig 5.
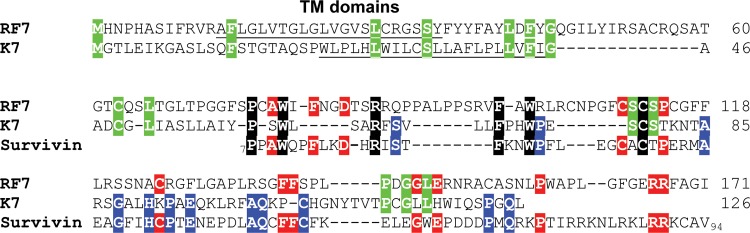
Comparison of the KSHV ORF K7 and RFHV ORF RF7 protein sequences. The sequences of KSHV ORF K7 (YP_001129367) and the RFHVMn ORF RF7 (AGY30697) positional homolog were aligned. Since the K7 sequence was previously shown to have sequence similarity to human survivin, the sequence of survivin/baculoviral IAP repeat containing protein 5 isoform 2 (NP_001012270) from aa 7 to aa 94 was aligned to the RF7 and K7 sequences. Amino acid residues conserved among all three sequences are highlighted in black, those conserved between RF7 and K7 are highlighted in green, those conserved between RF7 and survivin are highlighted in red, and those conserved between K7 and survivin are highlighted in blue. Putative N-terminal hydrophobic transmembrane domains are underlined.
Divergent locus C.
The third divergent locus identified in the DNA dot plot (Fig. 2) contained a region of duplicated viral homologs of the interferon regulatory factor (vIRF) (Fig. 1; positions, ∼84 to 94 kb). In the KSHV genome, the vIRF homologs include ORF K9, a nonspliced gene, and ORFs K10, K10.5, and 11, which all encode gene products with a spliced mRNA transcript. The analogous region within the related RRV genome contains eight duplicated, unspliced vIRF homologs (Fig. 1). The RFHV genome contained four vIRF homologs that correspond directly to the KSHV vIRF genes (Fig. 1): RF9, a nonspliced homolog of K9; RF10, a spliced homolog of K10; RF10.5, a spliced homolog of K10.5; and RF11, a spliced homolog of K11. Alignment of the RFHV vIRF sequences revealed strong sequence conservation with the corresponding KSHV vIRF sequences, and the phasing and positions of the splice sites were conserved (data not shown). Phylogenetic analysis revealed that the K9-RF9, K10-RF10, K10.5-RF10.5, and K11-RF11 gene pairs clustered, indicating that the four genes evolved from an ancestral RV1 rhadinovirus before the divergence of RFHV and KSHV (Fig. 6). The nonspliced K9-RF9 proteins clustered more closely with the vIRF homologs in RRV and the macaque and human homologs of the cellular interferon regulatory factor 8 (IRF8) (Fig. 6), suggesting functional homology. The spliced genes K10/RF10, K10.5/RF10.5, and K11/RF11 all clustered together, suggesting that they had evolved from a common spliced ancestral gene, and the long branch lengths in the maximum-likelihood tree were evidence of significant divergence of these vIRF proteins.
Fig 6.
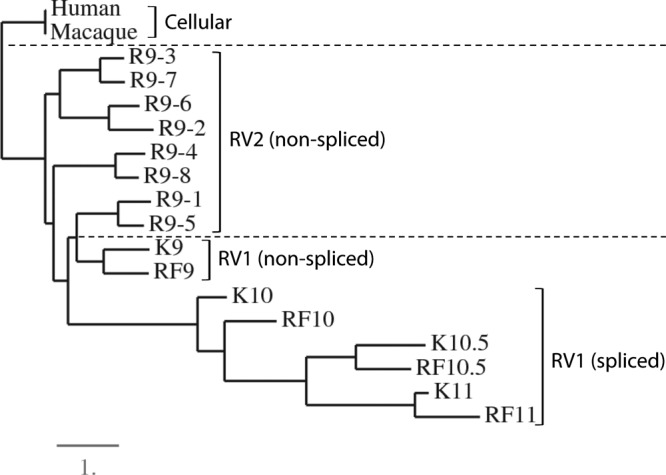
Phylogenetic analysis of the vIRF homologs. The encoded amino acid sequences of the vIRF homologs of KSHV (K9 [YP_001129411], K10 [YP_001129414], K10.5 [YP_001129413], and K11 [YP_001129412]), RFHVMn (RF9-11 [AGY30740-43; this study]), and RRV (R9.1 [AAF60036], R9.2 [AAF60037], R9.3 [AAF60038], R9.4 [AAF60039], R9.5 [AAF60040], R9.6 [AAF60041], R9.7 [AAF60042], and R9.8 [AAF60043]) were aligned using MUSCLE, and phylogeny was determined by protein maximum likelihood. The human (NP_002154) and rhesus macaque (NP_001252887) cellular interferon regulatory factor 8 sequences were used as an outgroup. The lineage divisions of the RV1 and RV2 rhadinovirus proteins and the cellular proteins are indicated with dashed lines, and the presence of spliced transcripts is noted. The numbers of substitutions per site are indicated.
Divergent locus D.
Divergent locus D is located between ORF69 and ORF71 within the latency locus of KSHV. The K12 Kaposin A ORF (60 aa) is located between ORF69 and the long inverted repeat 1 (LIR1) in the KSHV genome and initiates with an ATG codon just downstream of the DR5 and DR6 direct repeats flanking LIR1 (Fig. 7A). No repeat regions homologous to DR5 or DR6 were identified in the RFHVMn genome in this region; however, a large RF12 ORF (212 aa) was identified as a positional homolog of K12 Kaposin A (Fig. 7A). Alignment of the K12 Kaposin A protein sequence and RF12 revealed N-terminal sequence homology with an interesting conserved motif CX(6)VPPSX(1,2)RGP [where X(6) means any 6 amino acids and X(1,2) means either 1 or 2 amino acids] (Fig. 7B), suggesting that the two ORFs were evolutionarily related and that this motif was functionally important. While Kaposin A terminated 12 aa after this motif, the RF12 ORF continued with a large proline-rich sequence with an unusual domain of 12 basic arginine and lysine residues near the C terminus (Fig. 7B). Additional ORFs initiating with CUG/GUG within the DR5/6 repeat region have been identified in the KSHV genome, including Kaposin B and C (52). Homologous ORFs were not detected in the RFHVMn genome due to the lack of similar direct repeats.
Fig 7.

Comparison of the divergent D loci of RFHVMn and KSHV. (A) Graphical representation of the structure of the Ori-Lyt-R regions of KSHV (NC_009333) and RFHVMn (KF703446) between ORF69 and ORF71; the presence of encoded proteins and microRNAs is shown. The position of the 18-bp TATA repeat (red box) and C/EBP palindromic motifs (stars) are indicated. The known KSHV microRNA genes (purple) and the predicted RFHVMn candidate microRNAs with bona fide pre-microRNA hairpins (blue) are shown. (B) Alignment of ORF K12 (YP_001129428) and the positional ORF RF12 (AGY30757) homolog. Identical residues and the basic domain of RF12 are highlighted. (C) Alignment of the kshv-miR-K12-10a-3p and rfhvmn-miRc-RF9-3p pre-miRNAs. The predicted hairpin structures are indicated by parentheses. Boldface and underlined letters denote sequences of the mature kshv-miR-K12-10a-3p RNA (74) and the mature rfhvmn-miRc-RF9-3p, as predicted (pred) according to the guidelines of Zeng et al. (75). The conserved seed sequences are highlighted.
Further analysis of the divergent locus D of RFHVMn using combined structure/sequence alignment methods, as described in Materials and Methods, revealed a cluster of nine high-scoring hairpins located in the region between ORF69 and ORF71 (Fig. 7A), a genomic position that is analogous to those of miRNA clusters in other rhadinovirus genomes (53). Although most of the miRNA candidates did not exhibit sequence homology to KSHV pre-miRNAs, the predicted mature 3p miRNA of rfhvmn-miRc-RF9 shared its seed sequence with kshv-miR-K12-10a (Fig. 7C), suggesting that these two miRNAs may target overlapping sets of host transcripts. Interestingly, kshv-miR-K12-10a and rfhvmn-miRc-RF9 are both analogously positioned at the end of the homologous Kaposin A-like ORFs, K12 and RF12 (Fig. 7A). kshv-miR-K12-10a has been found to repress expression of the tumor necrosis factor-like weak inducer of apoptosis receptor (TWEAK-R) via seed-dependent targeting of two sites in the 3′-untranslated region (UTR) of the TWEAK-R transcript (54). Both sites are perfectly conserved in the rhesus macaque genome, suggesting that rfhvmn-miRc-RF9-3p may share the ability to suppress TWEAK-induced apoptosis and inflammatory cytokine expression.
Divergent loci B and D contain conserved sequence motifs in the left- and right-end long inverted repeats.
The left origin of lytic replication (Ori-L) of KSHV (Fig. 1; position, ∼23 to 24 kb,) is positioned within a long (∼1-kb) inverted repeat (LIR1) of a region at the right end of the KSHV genome containing the right origin of lytic replication (Ori-R) (Fig. 1; position, ∼ 119 to 120 kb) (55). A dot plot alignment of the KSHV Ori-L and Ori-R (inverted) revealed strong nucleotide conservation, interrupted only by the position of the kshv-miR-K12-9 microRNA gene in the Ori-R region (Fig. 8A). The sequence similarity between the left and right regions averaged 90% identity across the 1-kb repeat, with 500 bp of identical nucleotides (55, 56). A dot plot alignment of the analogous regions within the RFHVMn genome revealed little nucleotide conservation between the left and right regions of the genome, except for a small sequence located between bp 22,271 and 22,319 on the left and bp 111,405 and 111,517 on the right (Fig. 8B, TATATA repeat). Alignment of the two nucleotide sequences from the left and right (inverted) regions of RFHVMn revealed the presence of a highly conserved TATATA repeat element (Fig. 8C, 18-bp AT, highlighted in blue) flanked by additional sequences (CR4 and CR5, highlighted in yellow) conserved between the two regions. Alignment of the RFHVMn left and right sequences with the nucleotide sequences of the KSHV Ori-L and Ori-R, which were identical in this region, revealed that the TATATA repeat element and flanking elements were also conserved in the KSHV Ori-L and Ori-R regions (Fig. 8C). The KSHV TATATA element was previously identified as an 18-bp AT palindrome critical for lytic DNA replication (48, 56), suggesting that the conserved RFHV TATATA elements correspond to the left (Ori-L) and right (Ori-R) origins of lytic replication of RFHV. An additional 16-bp AT-rich region identified as critical for KSHV lytic DNA replication (57) was only partially conserved in the RFHV Ori-L region (Fig. 8C, 16-bp AT, highlighted in blue).
Fig 8.

Comparison of the long-inverted-repeat regions of Ori-Lyt-R and Ori-Lyt-L of KSHV and RFHVMn. (A and B) Dot plot of the nucleotide sequence alignment of the inverted-repeat regions of Ori-Lyt-L and Ori-Lyt-R for KSHV (A) and RFHVMn (B), with the approximate positions of the C/EBP palindromes (M1/2, M5/6, M7/8, and M9/10), the 18-bp TATATA repeat, and adjacent microRNA genes, illustrated in panel C. (C) Alignment of the nucleotide sequences of KSHV (U75698) containing the Ori-Lyt-L (bp 23223 to 23685) (KSL), the inverted sequence of KSHV containing the Ori-Lyt-R region bp (119245 to 119782) (KSRi), the nucleotide sequences of RFHVMn (KF703446; this study) containing the putative Ori-Lyt-L (bp 21937 to 22462) (RFL), and the inverted sequence of RFHVMn containing the putative Ori-Lyt-R (bp 111254 to 111800) (RFRi). The conserved AT-rich motifs (blue) and C/EBP motifs (red) are indicated (see the text). Other conserved regions (CR1 to CR6) are highlighted in yellow or gray. The kshv-miR-K12-K9 microRNA with the mature microRNA (purple) and stem-loops (green) and the predicted rfhvmn-miRc-RF6 are shown.
Further examination of the aligned nucleotide sequences of the RFHVMn and KSHV Ori-L and Ori-R regions revealed complete conservation of the four CCAAT/enhancer binding protein (C/EBP) motifs that have been identified in the KSHV Ori-L and Ori-R regions as critical for binding the ORF K8/bZIP homolog of KSHV and lytic DNA replication (48, 58) (Fig. 8C, M1/2, M5/6, M7/8, and M9/10, highlighted in red). An additional C/EBP binding motif (Fig. 8C, M3/4, highlighted in red) that was partially conserved in the RFHV Ori-R and the KSHV Ori-R and Ori-L was identified in the RFHVMn Ori-L. Furthermore, an additional distal C/EBP binding motif was identified in the RFHVMn Ori-R (Fig. 8C, M11/12, highlighted in red). While the KSHV Ori-R and Ori-L inverted repeats had identical DNA sequences across the C/EBP binding domain and the AT palindrome, extending for more than 1 kb, the RFHV Ori-R and Ori-L nucleotide sequences were conserved only at the C/EBP binding sites and the AT palindrome and flanking region. This limited sequence homology between the Ori-R and Ori-L regions is unusual, as other gammaherpesviruses, including KSHV and EBV, contain identical or nearly identical inverted repeats in the left- and right-hand ends of their genomes. The conservation of the C/EBP motifs and the TATATATA functional elements in the RFHV Ori-L and Ori-R regions strongly indicates that the regulation of lytic DNA replication in RFHV and KSHV is analogous. It is also interesting that the nucleotide alignment of the RFHVMn and KSHV Ori-R and Ori-L sequences revealed the presence of conserved nucleotide motifs that have not been functionally assessed (Fig. 8C, CR1 to -6, highlighted in yellow). The conservation of these sequences suggests that they play additional roles in the regulation of lytic DNA replication.
As shown in the dot plot in Fig. 8A, the KSHV Ori-L and Ori-R inverted repeats were interrupted by the presence of the kshv-miR-K12-9 microRNA gene in Ori-R (Fig. 8C, highlighted in green and purple). The sequence of the 5′ end of the primary kshv-miR-K12-9 transcript (Fig. 8C, highlighted in green) was highly conserved in the Ori-L of KSHV (twice) and RFHV (once) (Fig. 8C, highlighted in gray). A candidate microRNA (rfhvmn-miRc-RF6) was identified in the RFHVMn Ori-R sequence (Fig. 8C) as a near-positional homolog of kshv-miR-K12-9; however, no seed sequence similarity was obvious. An additional TATATA motif was detected within the rfhvmn-miRc-RF6 sequence in RFHVMn Ori-R that was conserved in RFHVMn Ori-L (Fig. 8C, highlighted in blue).
The RFHVMn and KSHV LANA promoters are conserved.
We previously cloned and sequenced the ORF73 major LANA from RFHVMn and identified functional and structural domains conserved with KSHV LANA (18). Previous analysis of the KSHV LANA transcripts and promoter region revealed the presence of two alternate promoters. The constitutive promoter, LANApc, is located in the upstream K14-coding sequence and generates a latency-associated spliced LANA transcript containing a 5′ noncoding exon and a 3′ exon encoding the complete LANA protein (59) (Fig. 9A and B). In addition, an alternate promoter, LANApi, located within the intron region that is removed from the spliced transcript, generates a nonspliced transcript encoding the complete LANA protein (Fig. 9A and B). This alternate promoter is induced by ORF50, the viral replication and transactivator protein (Rta), to produce LANA transcripts during the early stages of KSHV infection (60). An RBP-Jκ binding site adjacent to a TATA promoter element was found to be critical for LANApi function (61) (Fig. 9A and B).
Fig 9.

Conservation of the bidirectional ORF73 LANA and K14/RF14 promoter structure of KSHV and RFHVMn. (A and B) Diagrammatic rendering (A) and sequence alignment (B) of the promoter region of the KSHV LANA/K14 genes (NC_009333) and the RFHV LANA/RF14 genes, showing the initiating codons and the positions of the conserved splice donor (SD) and splice acceptor (SA) sites generating the latent spliced LANA transcript from the constitutive promoter (LANApc). The Rta-inducible LANApi promoter of KSHV LANA is indicated, with its associated RBP-Jκ binding sites and Rta response element (RRE) located within the intron region spliced out of the LANApc transcript (see the text). The position of a putative LANApi promoter for RFHV LANA is shown. (C) Alignment of the RBP-Jκ binding motifs of KSHV and RFHV indicated in panels A and B. Nucleotide numbers are based on the LANA translational initiation codon.
Using a series of primers derived from the region upstream of the RFHVMn LANA coding sequence, we detected a spliced LANA transcript in RNA obtained from an RFHV-infected RF tumor (unpublished observations). Sequence analysis of the spliced RFHVMn LANA transcript revealed splice donor and acceptor sites that were conserved with the donor and acceptor sites determined for the major spliced transcript of KSHV LANA (59, 62) (Fig. 9A, SA and SD, and B). Furthermore, comparison of the sequences upstream of the LANA-coding region revealed strong conservation of the KSHV K14p, LANApc, and LANApi promoter elements within the RFHVMn sequence (Fig. 9A and B), including the presence of an RBP-Jκ element immediately upstream of the putative LANApi promoter (Fig. 9A, B, and C). This suggests that RFHVMn contains a bidirectional promoter for the RF14 and LANA genes, as shown for KSHV (63), although the RFHVMn sequence lacked an obvious homolog of the KSHV Rta response element (RRE) and the second RBP-Jκ binding site proximal to ORF K14.
RF8.1, K8.1, and EBV gp42 are positional homologs with a conserved promoter element.
Alignment of the RFHVMn genome sequence with the genome sequences of KSHV and EBV revealed positional homology between ORF RF8.1 (RFHVMn), ORF K8.1 (KSHV), and gp42 (EBV) (Fig. 1) that had previously been unrecognized. All three coding sequences initiated at an ATG codon 94 to 124 bp downstream of the termination of the bZIP homologs (RF8, K8, and bZIP, respectively) (Fig. 10). A comparison of the corresponding sequences of the three viruses revealed complete conservation of AATATTAA, a TATA-like element previously identified in the promoter of the EBV gp42 gene. Immediately downstream (29 bp) of the TATA-like element in KSHV is a transcriptional start site for K8.1 (64) (Fig. 10), suggesting that the AATATTAA sequence also functions as a promoter for both KSHV K8.1 and RFHVMn RF8.1. Interestingly, integral to the TATA-like motif in EBV gp42 is a polyadenylation site, AATATTAATAAA (underlined), for the upstream bicistronic EBV Rta/bZIP transcript. The KSHV Rta/K8 transcripts do not terminate in this region and instead terminate after the K8.1 C-terminal spliced exon with a polyadenylation signal (AATAAA) that is conserved in the KSHV and RFHVMn sequences (bp 76,813 to 76,818 and bp 71,222 to 71,227, respectively).
Fig 10.

Conservation of the junctional regions of the K8 and K8.1 genes and their homologs in RFHVMn and EBV. The nucleotide sequences of KSHV (NC_009333), RFHV (this study), and EBV (NC_007605) from the junctional regions of K8/K8.1, RF8/RF8.1, and bZIP/GP42 were aligned, and the translational stop and initiation codons are highlighted. The transcriptional start site of the K8.1 mRNA is indicated, and the polyadenylation signal for EBV bZIP is marked with asterisks (see the text). The conserved AATATTAA motif within the promoters for K8.1, RF8.1, and GP42 is highlighted.
Conservation of spliced gene transcripts.
A number of genes with spliced protein-coding transcripts have been described for KSHV, including ORFs 29, 40, 50, K8, K8.1, 57, K10, K10.5, K11, and K15 (Fig. 1, vertical black bars in coding regions). Analogs of the splice junctions generating these spliced transcripts were detected in the RFHVMn sequence for all of these genes except ORF RF8.1, the homolog of ORF K8.1. Alternate spliced transcripts of ORF K8.1 generate three different proteins, K8.1α (228 aa), K8.1β (167 aa), and K8.1γ (197 aa) (65) (Fig. 11A). K8.1α is generated by read-through of exons 1 and 2A with a splice to exon 3, encoding the hydrophobic transmembrane domain, which is in an alternate reading frame (Fig. 11A). K8.1β is generated by a cryptic splice site that joins exon 1 directly with exon 3, while K8.1γ is generated by read-through of exons 1, 2A, and 2B with no splices (Fig. 11A). In contrast, the RF8.1 gene has a continuous open reading frame that encodes a sequence homologous to that of K8.1α with a C-terminal hydrophobic transmembrane domain in a single nonspliced open reading frame (Fig. 11B). An additional domain in RF8.1 (Fig. 11B, orange) is located between the domains conserved with exons 2A and 3 of KSHV. It is unknown whether the RF8.1 gene has a spliced transcript generating a homolog of K8.1β.
Fig 11.

Comparison of the transcripts of KSHV ORF K8.1 and its homolog RFHVMn ORF RF8.1. (A) Gene structure of KSHV ORF K8.1 with the exons color coded and amino acids at the junctions indicated. Exons 2 and 3 are in different reading frames, as indicated, and exon 2B indicates a read-through of a cryptic splice site. Alternate splice variants that have been detected are labeled with red stars (see the text). NCBI accession numbers: K8.1α, AAC63270; K8.1β, AAC63271; K8.1γ, AAB62630. The N-terminal signal peptide (SP) and C-terminal hydrophobic TM are shown. (B) Gene structure of RFHV ORF RF8.1, with regions homologous to the K8.1 exons color coded and labeled. The entire RF8.1 ORF (AGY30733) is in the same reading frame, unlike K8.1.
RNA transcripts for the K8 gene that encode three different alternately spliced K8-related proteins (K8α, 237 aa; K8β, 190 aa; and K8γ, 239 aa) have been previously identified (66, 67) (Fig. 12A). While the K8α transcript is generated by two splice events and contains three separate exons, K8β and K8γ are derived by differential read-through of the splice sites utilized for generating K8α. RFHV encodes RF8α (242 aa), a homolog of K8α, which would also be generated by two splice events (Fig. 12B). In addition, RFHV could generate RF8β (205 aa), homologous to K8β, via a similar splicing strategy (Fig. 12B). KSHV also generates a nonspliced K8 transcript, K8γ (239 aa), containing both exons 1 and 2 with a read-through of the intron that is spliced out in K8α and K8β and a read-through of the second splice junction terminating immediately after a valine codon (Fig. 12A). In RF8, however, exons 1 and 2 are not in the same reading frame, as is found in K8, so that a nonspliced transcript would yield a truncated protein, RF8γ (208 aa), lacking exon 2 (Fig. 12B). Interestingly, the EBV bZIP homolog of K8 has a gene structure similar to that of RF8, in which exons 1 and 2 are not in the same reading frame (Fig. 12C). A doubly spliced bZIP mRNA encoding bZIP (246 aa) that corresponds to K8α/RF8α, containing all three exons, has been identified (68). In addition, a truncated form of bZIP, bZIPΔ (210 aa), containing only exons 1 and 3, has been detected (69) (Fig. 12C). Whether RFHVMn or EBV generates read-through transcripts encoding RF8β/bZIPβ or RF8γ/bZIPγ spliced variants, similar to what is seen in KSHV K8, has not been determined.
Fig 12.

Comparison of the spliced transcripts of KSHV ORF K8, RFHVMn ORF RF8, and EBV bZIP. (A) Gene structure of KSHV ORF K8, with the exons color coded and amino acids at the junctions indicated. Exons 1 and 2 are in the same reading frame, and exons 2 and 3 are separated by an intron sequence. Transcript K8γ is generated by a read-through of cryptic splice junctions between exons 1A and 1B and between exons 2A and 2B (accession no. AAD25322). Transcript K8α is generated by splicing exons 1A, 2A, and 3 (accession no. AAD25316). Transcript K8β is generated by splicing exons 1A and 2A with read-through of the cryptic splice junction between exons 2A and 2B (accession no. AAD25319). (B) Gene structure of RFHVMn ORF RF8. Exons 1 and 2 are in different reading frames, while exons 2 and 3 are separated by an intron sequence. Possible transcripts corresponding to the K8 transcripts are shown (RF8α; accession no. AGY30732). (C) Gene structure of EBV bZIP, revealing close similarity to the gene structure of RF8. Possible transcripts corresponding to the K8 transcripts are shown, and detected transcripts are indicated with red stars. The bZIP accession no. is NC_007605; for more information regarding bZIPΔ, see reference 69.
In all other cases of putative spliced transcripts in RFHVMn, the splice donor and acceptor sites were conserved with those found in KSHV (Fig. 1; splice junctions are indicated with black vertical bars in the ORFs), and the splice phasing and coding sequences across the splice junctions were highly conserved. Like KSHV ORF K15, ORF RF15 is generated from a transcript with seven introns, and the locations and phasing are conserved (data not shown). The splice junctions and phasing for the viral IRF homologs, RF10, RF10.5, and RF11, are conserved, as discussed above. Current studies using transcriptome-sequencing (RNA-seq) approaches in macaque tumor and normal tissue samples are ongoing to identify and confirm the presence of these and additional spliced transcripts in RFHVMn.
RFHV phylogenetically clusters with KSHV within an RV1 rhadinovirus lineage distinct from RRV.
To examine the evolutionary relationship between RFHV, RRV, and KSHV, multiple alignments and phylogenetic analyses were performed on a subset of conserved core herpesvirus genes, including ORF8 (glycoprotein B; UL27 family), ORF9 (DNA polymerase; UL30 family), ORF37 (SOX shutoff exonuclease; UL12 family), and ORF64 (large tegument protein; UL36 family). In all cases, the RFHVMn homolog of these core genes clustered with the encoded KSHV homolog (Fig. 13A). The core gene homologs of RRV (rhesus macaque) were more distantly related and clustered separately, as shown earlier with a partial ORF9 DNA polymerase sequence (21) and the full-length ORF59 DNA polymerase processivity factor (31). These data confirm the separation of the macaque and human rhadinoviruses into two distinct RV1 and RV2 lineages (21, 24). Additional phylogenetic analyses performed on the rhadinovirus-specific genes, including ORFK2 (vIL-6), ORF2 (DHFR), ORFK3 (MIR1), ORF70 (TS), and ORFK4 (vCCL-2), mirrored the phylogenetic relationship determined with the core herpesvirus genes for these viruses (reference 15 and data not shown).
Fig 13.
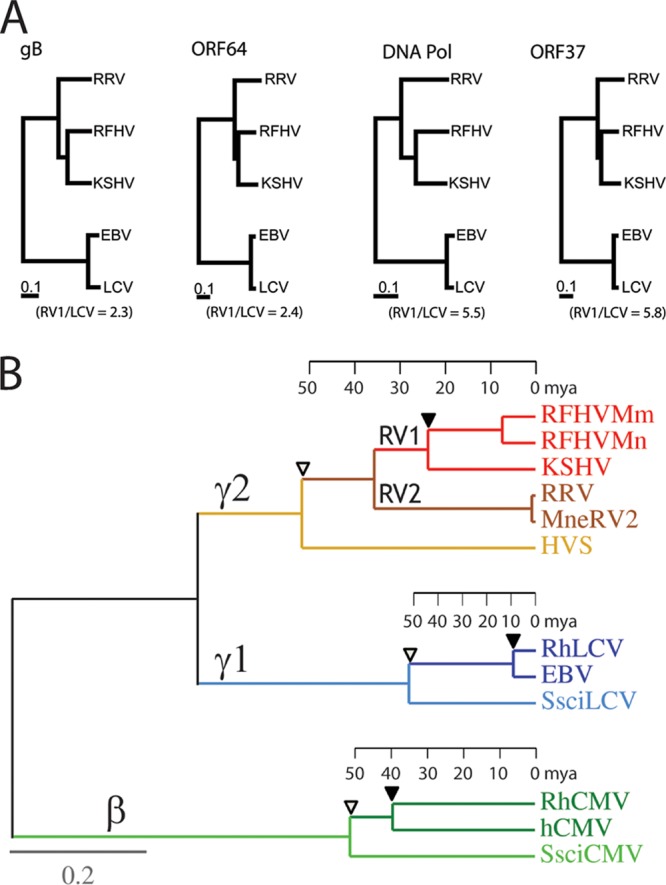
Phylogenetic comparison of KSHV and EBV and their macaque homologs. (A) The amino acid sequences for the homologs of glycoprotein B (gB), ORF64/BPLF1, DNA polymerase (DNA Pol), and ORF37/BGLF5 from KSHV, RFHVMn (this study), RRV, EBV, and rhesus macaque RhLCV were aligned using ClustalW, and a distance tree was generated for each gene using neighbor joining as implemented in Phylip 3.2. The numbers of substitutions per site are indicated, and the ratios of branch lengths between the human and macaque RV1 rhadinoviruses (KSHV and RFHVMn) and human and macaque lymphocryptoviruses (LCV) are shown. (B) Protein maximum-likelihood analysis with a molecular clock of a 1,313-aa concatenated sequence from the DNA polymerase and glycoprotein B of the Old World primate rhadinoviruses, lymphocryptoviruses, and cytomegaloviruses from humans (KSHV, EBV, and hCMV) and rhesus macaques (RFHVMm, RRV, RhLCV, and RhCMV) and New World primate rhadinovirus, lymphocryptovirus, and cytomegalovirus from the squirrel monkey (HVS, SsciLCV, and SsciCMV). Rhadinovirus sequences from the pig-tailed macaque (RFHVMn and MneRV2) were also analyzed. The betaherpesvirus (β), gamma-1-herpesvirus (γ1)/lymphocryptovirus, and gamma-2-herpesvirus (γ2)/rhadinovirus branches are indicated. The tentative evolutionary time scale in millions of years before the present) is positioned on each branch based on the same date for separation of the Old and New World herpesvirus lineages as for separation of the Old and New World primate lineages (76) (open triangles). The separation of the macaque and human herpesvirus lineages is indicated with solid triangles. Protein sequences were obtained from the complete genome sequences of KSHV (NC_009333), RFHVMn (KF703446; this study), RRV (NC_003401), HVS (NC_001350), EBV (NC_007605), RhLCV (AY037858), hCMV (X17403), RhCMV (NC_006150), and SsciCMV (FJ483967) and partial sequences of SsciLCV (AY139024), RFHVMm (AF005479), and MneRV2 (unpublished data).
Divergence of the macaque and human lymphocryptovirus and rhadinovirus species.
In the phylogenetic analysis of the rhadinovirus core herpesvirus genes above, the homologous lymphocryptovirus genes from EBV and its macaque homolog (RhLCV) were included as an outgroup. Examination of the trees revealed that the branch lengths between the human (EBV) and macaque (RhLCV) lymphocryptovirus proteins were 2- to 6-fold shorter than those between the human (KSHV) and macaque (RFHV) rhadinovirus proteins (Fig. 13A). This low rate of change in the lymphocryptovirus lineage has been detected previously (70). In the previous study, the phylogenetic trees for the New World and Old World lymphocryptovirus lineages were compared to those of their primate hosts, using available protein sequences encoded by the conserved DNA polymerase and glycoprotein B genes (946 aa concatenated) with imposition of a global molecular clock. A tentative evolutionary time scale was determined based on the divergence of the New and Old World primate hosts. Two lineages of hominoid lymphocryptovirus lineages were identified, with different divergence characteristics. One hominoid lineage showed divergence levels between macaque RhLCV1 and other Old World monkey lymphocryptoviruses that were characteristic of long-term synchronous evolution with their host species. In contrast, the other hominoid lineage, containing human EBV and chimpanzee PtroLCV-1, showed much smaller divergence with the monkey lymphocryptoviruses, suggesting that the hominoid lineage containing EBV and PtroLCV-1 arose by interspecies transfer from a lineage of Old World monkey viruses around 12 million years ago (70).
To compare the evolution of the human and macaque rhadinoviruses, we performed a protein maximum-likelihood analysis of the available macaque and human lymphocryptovirus (gamma-1-herpesvirus) and rhadinovirus (gamma-2-herpesvirus) species using a 1,313-aa concatenated DNA polymerase and a glycoprotein B sequence similar to that employed in the Ehlers study (70), with imposition of a molecular clock. Sequences of the macaque and human cytomegaloviruses (betaherpesvirus) were included for comparison. In addition, the sequences from the lymphocryptovirus, rhadinovirus, and cytomegalovirus homologs of the New World squirrel monkey (Saimiri sciureus) virus were included to position the time scale for the New World-Old World divergence. As observed previously, the sequence divergence of the rhadinovirus lineage after the New World-Old World split was much greater than the divergence seen in the lymphocryptovirus lineage during the same time frame (Fig. 13B, open triangles). Similarly, the divergence of the macaque RFHV and human KSHV RV1 rhadinoviruses was significantly greater than the divergence of the macaque RhLCV and human EBV (Fig. 13B, solid triangles). This indicates that RFHV and KSHV have developed in long-term synchrony with the evolution of their host species, unlike the macaque RhLCV and human EBV. The divergence of the CMV lineage after the New World-Old World split was intermediate to that seen with the RV and LCV lineages.
DNA polymerase and glycoprotein B sequences were available for two macaque RV1 species (RFHVMn and RFHVMm) and two macaque RV2 species (MneRV2 and RRV) from pig-tailed and rhesus macaques, respectively. Phylogenetic analysis revealed that the macaque RV2 sequences clustered distinct from the macaque and human RV1 lineage. The divergence of the RV2 and RV1 lineages mapped to a time point after the New World-Old World split (approximately 50 million years ago [mya]) but before the divergence of the human and macaque lineages (approximately 35 mya). Interestingly, the branch lengths between the RV1 rhadinoviruses from rhesus and pig-tailed macaques were much greater than the branch lengths between the RV2 rhadinoviruses from the same macaque species, indicating substantially different evolutionary rates between these two Old World primate rhadinovirus lineages. Phylogenetic analysis of other viral genes from these rhadinoviruses supports this conclusion (15, 31).
RF tumor infectome.
As indicated above, the Illumina reads from the combined 50-bp and 100-bp runs from the RF tumor sample were mapped onto the de novo-assembled RFHVMn genome sequence using Tablet, yielding an average depth of 7.4 reads per nucleotide. The Illumina reads were also mapped onto the pig-tailed macaque oncostatin M gene as a marker for the macaque genome. This yielded an average depth of 3.3 reads per nucleotide. Since the oncostatin M gene and the macaque genome are diploid in each cell, the Illumina read data indicated that RFHV was present at 4.5 copies per cell, strongly correlating with our previous qPCR quantitation of 3.4 viral genomes per cell (17). We also mapped the Illumina reads onto the complete genome sequence of SRV-2, which we had detected previously in the RF tumor sample by PCR (41). Our analysis revealed coverage of 6.3 reads per nucleotide, indicating that the SRV2 DNA levels were very similar to the RFHV levels in the tumor sample.
We had previously shown by qPCR that this RF tumor contained 0.021 genome per cell of MneRV2, the pig-tailed macaque RV2 rhadinovirus homolog of RRV (17). This indicated that the RV2 virus was not present uniformly in the tumor cells and was probably present in infected lymphocytes infiltrating the tumor. Mapping the Illumina reads onto the complete genome sequence of MneRV2, which we had recently determined (unpublished observations), revealed coverage of 0.04 read per nucleotide, yielding a prevalence of 0.02 MneRV2 genome per cell in the RF tumor sample, confirming our previous qPCR data. The level of MneRV2 in the pig-tailed macaque RF tumors correlates closely with the level of RRV in the rhesus macaque RF tumors (17). To determine whether the pig-tailed macaque lymphocryptovirus (MnLCV) was present in the RF tumor sample, the Illumina reads were mapped onto a segment of the DNA polymerase gene of MnLCV that we had previously determined (unpublished results), since the complete MnLCV genome sequence has not been determined. No Illumina reads mapped to the MnLCV sequence, indicating the absence of LCV in this RF tumor sample.
Summary.
Our analysis revealed that the RFHMn genome is the closest homolog of the human tumor virus KSHV to be completely sequenced. In addition, the RFHVMn genome is the first rhadinovirus genome to be sequenced directly from a clinical tumor sample using next-generation sequencing techniques. The RFHVMn genome contained close homologs of all of the core rhadinovirus genes, as well as the majority of the unique genes present in the KSHV genome that play critical roles in the biology of KSHV and its induced pathology. Of the KSHV-specific genes, RFHVMn notably lacked homologs of the KSHV ORF K5 (MIR2) and K6 (vCCL-1) genes, which appear to have been generated by a duplication event of the ancestral gene homologs of the ORF K3/RF3 (MIR1) and ORF K4/RF4 (vCCL-2) genes after the divergence of KSHV and RFHVMn. Members of both the K3/RF3 and K4/RF4 gene families have been shown to play important roles in the modulation of the immune response during virus infection. The related ORF K3 and K5 proteins of KSHV mediate the downregulation of a number of immunomodulatory proteins from the surfaces of virus-infected cells (71). Although the RF3 homolog in RFHVMn can modulate immune proteins targeted by both K3 and K5 (72), the presence of the duplicated K5 gene in KSHV presumably provides additional functionality for immune modulation in the hominoid RV1 rhadinovirus lineage. KSHV encodes three chemokine homologs of macrophage inflammatory protein 1α (MIP-1α) that can function to polarize the adaptive immune response, potentially altering the efficacy of the antiviral response (73). While RFHVMn lacks ORF K6 (vCCL-1), it contains homologs of both ORF K4 (vCCL-2) and ORF K4.1 (vCCL-3). As K4 and K6 are closely related, it appears that K6 arose by a duplication of the ancestral K4 gene after the divergence of KSHV and RFHVMn.
The exact functional outcome of the addition of the K5 and K6 genes to the hominoid RV1 rhadinovirus genome is unclear. However, it appears that there was strong selective advantage to increasing the immunomodulatory capabilities of KSHV compared to RFHVMn during virus evolution. Few differences were noted within the core herpesvirus genes between RFHVMn and KSHV, suggesting strong similarities in virus biology. While the RNA transcript splicing was predicted to be very similar between RFHVMn and KSHV based on conservation of splice junctions, it is not clear how well alternate splicing of specific genes, such as RF8 and RF8.1, generates similar viral protein species. High levels of sequence conservation were noted in specific promoter sequences and within the putative origins of replication, suggesting similar transcriptional and replication processes in KSHV and RFHV. Sequence comparisons indicated that RFHV and KSHV cluster within the RV1 lineage of Old World primate rhadinoviruses and have developed in long-term synchrony with the evolution of their hosts. The overall strong genetic and sequence similarity between RFHVMn and KSHV, coupled with the apparent similarities in biology and pathology of the viruses, suggest that RFHVMn infections in macaques offer important and relevant models for the study of KSHV infections in humans.
ACKNOWLEDGMENTS
We thank Michael G. Katze and the Center for Systems and Translational Research in Infectious Diseases (STRIDE), University of Washington, for encouraging and supporting the use of Next-Gen sequencing techniques to obtain the RFHV genome sequence; Jim Casey and his laboratory at Cornell University for their help in preparing the initial RF tumor DNA lambda library, which provided the first large regions of RFHV genome sequence; Emily Schultz, Kurt Strand, and Marnix Bosch, who were instrumental in the initial discovery and characterization of RFHV and the RF tumors in macaques; Mitra Barahimi, who determined the RFHV LANA splice junctions; Emilia Gan, who provided the whole-genome dot plot comparison of RFHV and KSHV; and the Washington National Primate Research Center (WaNPRC) for access to their archive of macaque tissues and biopsy specimens.
This work was supported by the National Center for Research Resources (NCRR) and the Office of Research Infrastructure Programs (ORIP) of the National Institutes of Health (NIH) through grant RR023343 (to T. M. Rose). Additional funding for sequence generation and analysis was provided by Public Health Service grants R24OD011157 and P51OD010425 (WaNPRC Division of Nonhuman Primate Systems Biology) to Michael G. Katze, STRIDE.
Footnotes
Published ahead of print 9 October 2013
REFERENCES
- 1.Antman K, Chang Y. 2000. Kaposi's sarcoma. N. Engl. J. Med. 342:1027–1038 [DOI] [PubMed] [Google Scholar]
- 2.Russell Jones R, Orchard G, Zelger B, Wilson Jones E. 1995. Immunostaining for CD31 and CD34 in Kaposi sarcoma. J. Clin. Pathol. 48:1011–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 4.Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 120:939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dittmer DP. 2003. Transcription profile of Kaposi's sarcoma-associated herpesvirus in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 63:2010–2015 [PubMed] [Google Scholar]
- 7.Cesarman E. 2011. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. 305:163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholas J, Zong JC, Alcendor DJ, Ciufo DM, Poole LJ, Sarisky RT, Chiou CJ, Zhang X, Wan X, Guo HG, Reitz MS, Hayward GS. 1998. Novel organizational features, captured cellular genes, and strain variability within the genome of KSHV/HHV8. J. Natl. Cancer Inst. Monogr. 23:79–88 [DOI] [PubMed] [Google Scholar]
- 10.Giddens WE, Jr, Tsai CC, Morton WR, Ochs HD, Knitter GH, Blakley GA. 1985. Retroperitoneal fibromatosis and acquired immunodeficiency syndrome in macaques. Pathologic observations and transmission studies. Am. J. Pathol. 119:253–263 [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai C-C. 1993. Fibromatosis in macaques infected with type D retroviruses, p 48–57 In Jones TC, Mohr U, Hunt RD. (ed), Nonhuman primates I. Spinger-Verlag, Berlin, Germany [Google Scholar]
- 12.Tsai CC, Warner TF, Uno H, Giddens WE, Jr, Ochs HD. 1985. Subcutaneous fibromatosis associated with an acquired immune deficiency syndrome in pig-tailed macaques. Am. J. Pathol. 120:30–37 [PMC free article] [PubMed] [Google Scholar]
- 13.Rose TM, Schultz ER, Henikoff JG, Pietrokovski S, McCallum CM, Henikoff S. 1998. Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res. 26:1628–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rose TM, Strand KB, Schultz ER, Schaefer G, Rankin GW, Jr, Thouless ME, Tsai CC, Bosch ML. 1997. Identification of two homologs of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in retroperitoneal fibromatosis of different macaque species. J. Virol. 71:4138–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rose TM, Ryan JT, Schultz ER, Raden BW, Tsai CC. 2003. Analysis of 4.3 kilobases of divergent locus B of macaque retroperitoneal fibromatosis-associated herpesvirus reveals a close similarity in gene sequence and genome organization to Kaposi's sarcoma-associated herpesvirus. J. Virol. 77:5084–5097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bielefeldt-Ohmann H, Barouch DH, Bakke AM, Bruce AG, Durning M, Grant R, Letvin NL, Ryan JT, Schmidt A, Thouless ME, Rose TM. 2005. Intestinal stromal tumors in a simian immunodeficiency virus-infected, simian retrovirus-2 negative rhesus macaque (Macaca mulatta). Vet. Pathol. 42:391–396 [DOI] [PubMed] [Google Scholar]
- 17.Bruce AG, Bakke AM, Bielefeldt-Ohmann H, Ryan JT, Thouless ME, Tsai CC, Rose TM. 2006. High levels of retroperitoneal fibromatosis (RF)-associated herpesvirus in RF lesions in macaques are associated with ORF73 LANA expression in spindleoid tumour cells. J. Gen. Virol. 87:3529–3538 [DOI] [PubMed] [Google Scholar]
- 18.Burnside KL, Ryan JT, Bielefeldt-Ohmann H, Gregory Bruce A, Thouless ME, Tsai CC, Rose TM. 2006. RFHVMn ORF73 is structurally related to the KSHV ORF73 latency-associated nuclear antigen (LANA) and is expressed in retroperitoneal fibromatosis (RF) tumor cells. Virology 354:103–115 [DOI] [PubMed] [Google Scholar]
- 19.Desrosiers RC, Sasseville VG, Czajak SC, Zhang X, Mansfield KG, Kaur A, Johnson RP, Lackner AA, Jung JU. 1997. A herpesvirus of rhesus monkeys related to the human Kaposi's sarcoma-associated herpesvirus. J. Virol. 71:9764–9769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mansfield KG, Westmoreland SV, DeBakker CD, Czajak S, Lackner AA, Desrosiers RC. 1999. Experimental infection of rhesus and pig-tailed macaques with macaque rhadinoviruses. J. Virol. 73:10320–10328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schultz ER, Rankin GW, Jr, Blanc MP, Raden BW, Tsai CC, Rose TM. 2000. Characterization of two divergent lineages of macaque rhadinoviruses related to Kaposi's sarcoma-associated herpesvirus. J. Virol. 74:4919–4928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexander L, Denekamp L, Knapp A, Auerbach MR, Damania B, Desrosiers RC. 2000. The primary sequence of rhesus monkey rhadinovirus isolate 26-95: sequence similarities to Kaposi's sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J. Virol. 74:3388–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Searles RP, Bergquam EP, Axthelm MK, Wong SW. 1999. Sequence and genomic analysis of a Rhesus macaque rhadinovirus with similarity to Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 73:3040–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greensill J, Sheldon JA, Renwick NM, Beer BE, Norley S, Goudsmit J, Schulz TF. 2000. Two distinct gamma-2 herpesviruses in African green monkeys: a second gamma-2 herpesvirus lineage among old world primates? J. Virol. 74:1572–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duprez R, Boulanger E, Roman Y, Gessain A. 2004. Novel gamma-2-herpesvirus of the Rhadinovirus 2 lineage in gibbons. Emerg. Infect. Dis. 10:899–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greensill J, Sheldon JA, Murthy KK, Bessonette JS, Beer BE, Schulz TF. 2000. A chimpanzee rhadinovirus sequence related to Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8: increased detection after HIV-1 infection in the absence of disease. AIDS 14:F129–F135 [DOI] [PubMed] [Google Scholar]
- 27.Lacoste V, Mauclere P, Dubreuil G, Lewis J, Georges-Courbot MC, Gessain A. 2000. KSHV-like herpesviruses in chimps and gorillas. Nature 407:151–152 [DOI] [PubMed] [Google Scholar]
- 28.Lacoste V, Mauclere P, Dubreuil G, Lewis J, Georges-Courbot MC, Gessain A. 2001. A novel gamma 2-herpesvirus of the Rhadinovirus 2 lineage in chimpanzees. Genome Res. 11:1511–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lacoste V, Mauclere P, Dubreuil G, Lewis J, Georges-Courbot MC, Rigoulet J, Petit T, Gessain A. 2000. Simian homologues of human gamma-2 and betaherpesviruses in mandrill and drill monkeys. J. Virol. 74:11993–11999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitby D, Stossel A, Gamache C, Papin J, Bosch M, Smith A, Kedes DH, White G, Kennedy R, Dittmer DP. 2003. Novel Kaposi's sarcoma-associated herpesvirus homolog in baboons. J. Virol. 77:8159–8165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruce AG, Bakke AM, Gravett CA, DeMaster LK, Bielefeldt-Ohmann H, Burnside KL, Rose TM. 2009. The ORF59 DNA polymerase processivity factor homologs of Old World primate RV2 rhadinoviruses are highly conserved nuclear antigens expressed in differentiated epithelium in infected macaques. Virol. J. 6:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29:644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. 2010. Tablet—next generation sequence assembly visualization. Bioinformatics 26:401–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tcherepanov V, Ehlers A, Upton C. 2006. Genome Annotation Transfer Utility (GATU): rapid annotation of viral genomes using a closely related reference genome. BMC Genomics 7:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brodie R, Roper RL, Upton C. 2004. JDotter: a Java interface to multiple dotplots generated by dotter. Bioinformatics 20:279–281 [DOI] [PubMed] [Google Scholar]
- 37.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grundhoff A. 2011. Computational prediction of viral miRNAs. Methods Mol. Biol. 721:143–152 [DOI] [PubMed] [Google Scholar]
- 39.Grundhoff A, Sullivan CS, Ganem D. 2006. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 12:733–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. 2005. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 435:682–686 [DOI] [PubMed] [Google Scholar]
- 41.Philipp-Staheli J, Marquardt T, Thouless ME, Bruce AG, Grant RF, Tsai CC, Rose TM. 2006. Genetic variability of the envelope gene of Type D simian retrovirus-2 (SRV-2) subtypes associated with SAIDS-related retroperitoneal fibromatosis in different macaque species. Virol. J. 3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozak M. 1987. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15:8125–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lagunoff M, Ganem D. 1997. The structure and coding organization of the genomic termini of Kaposi's sarcoma-associated herpesvirus. Virology 236:147–154 [DOI] [PubMed] [Google Scholar]
- 44.Lee H, Guo J, Li M, Choi JK, DeMaria M, Rosenzweig M, Jung JU. 1998. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi's sarcoma-associated herpesvirus. Mol. Cell. Biol. 18:5219–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zong JC, Ciufo DM, Alcendor DJ, Wan X, Nicholas J, Browning PJ, Rady PL, Tyring SK, Orenstein JM, Rabkin CS, Su IJ, Powell KF, Croxson M, Foreman KE, Nickoloff BJ, Alkan S, Hayward GS. 1999. High-level variability in the ORF-K1 membrane protein gene at the left end of the Kaposi's sarcoma-associated herpesvirus genome defines four major virus subtypes and multiple variants or clades in different human populations. J. Virol. 73:4156–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davison AJ, Stow ND. 2005. New genes from old: redeployment of dUTPase by herpesviruses. J. Virol. 79:12880–12892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGeoch DJ. 2001. Molecular evolution of the gamma-Herpesvirinae. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:421–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Li H, Chan MY, Zhu FX, Lukac DM, Yuan Y. 2004. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 78:8615–8629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng P, Park J, Lee BS, Lee SH, Bram RJ, Jung JU. 2002. Kaposi's sarcoma-associated herpesvirus mitochondrial K7 protein targets a cellular calcium-modulating cyclophilin ligand to modulate intracellular calcium concentration and inhibit apoptosis. J. Virol. 76:11491–11504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang HW, Sharp TV, Koumi A, Koentges G, Boshoff C. 2002. Characterization of an anti-apoptotic glycoprotein encoded by Kaposi's sarcoma-associated herpesvirus which resembles a spliced variant of human survivin. EMBO J. 21:2602–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng P, Scott CW, Cho NH, Nakamura H, Chung YH, Monteiro MJ, Jung JU. 2004. Kaposi's sarcoma-associated herpesvirus K7 protein targets a ubiquitin-like/ubiquitin-associated domain-containing protein to promote protein degradation. Mol. Cell. Biol. 24:3938–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sadler R, Wu L, Forghani B, Renne R, Zhong W, Herndier B, Ganem D. 1999. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5722–5730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walz N, Christalla T, Tessmer U, Grundhoff A. 2010. A global analysis of evolutionary conservation among known and predicted gammaherpesvirus microRNAs. J. Virol. 84:716–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abend JR, Uldrick T, Ziegelbauer JM. 2010. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi's sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J. Virol. 84:12139–12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.AuCoin DP, Colletti KS, Xu Y, Cei SA, Pari GS. 2002. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) contains two functional lytic origins of DNA replication. J. Virol. 76:7890–7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin CL, Li H, Wang Y, Zhu FX, Kudchodkar S, Yuan Y. 2003. Kaposi's sarcoma-associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication: identification of the ori-Lyt and association of K8 bZip protein with the origin. J. Virol. 77:5578–5588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.AuCoin DP, Colletti KS, Cei SA, Papouskova I, Tarrant M, Pari GS. 2004. Amplification of the Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 318:542–555 [DOI] [PubMed] [Google Scholar]
- 58.Wu FY, Wang SE, Tang QQ, Fujimuro M, Chiou CJ, Zheng Q, Chen H, Hayward SD, Lane MD, Hayward GS. 2003. Cell cycle arrest by Kaposi's sarcoma-associated herpesvirus replication-associated protein is mediated at both the transcriptional and posttranslational levels by binding to CCAAT/enhancer-binding protein alpha and p21(CIP-1). J. Virol. 77:8893–8914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Talbot SJ, Weiss RA, Kellam P, Boshoff C. 1999. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 257:84–94 [DOI] [PubMed] [Google Scholar]
- 60.Lan K, Kuppers DA, Verma SC, Robertson ES. 2004. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J. Virol. 78:6585–6594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu J, Verma SC, Cai Q, Robertson ES. 2011. The single RBP-Jkappa site within the LANA promoter is crucial for establishing Kaposi's sarcoma-associated herpesvirus latency during primary infection. J. Virol. 85:6148–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sarid R, Wiezorek JS, Moore PS, Chang Y. 1999. Characterization and cell cycle regulation of the major Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J. Virol. 73:1438–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hilton IB, Dittmer DP. 2012. Quantitative analysis of the bidirectional viral G-protein-coupled receptor and lytic latency-associated nuclear antigen promoter of Kaposi's sarcoma-associated herpesvirus. J. Virol. 86:9683–9695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang S, Zheng ZM. 2002. Kaposi's sarcoma-associated herpesvirus K8 exon 3 contains three 5′-splice sites and harbors a K8.1 transcription start site. J. Biol. Chem. 277:14547–14556 [DOI] [PubMed] [Google Scholar]
- 65.Zhu FX, Cusano T, Yuan Y. 1999. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin SF, Robinson DR, Miller G, Kung HJ. 1999. Kaposi's sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein-Barr virus. J. Virol. 73:1909–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seaman WT, Ye D, Wang RX, Hale EE, Weisse M, Quinlivan EB. 1999. Gene expression from the ORF50/K8 region of Kaposi's sarcoma-associated herpesvirus. Virology 263:436–449 [DOI] [PubMed] [Google Scholar]
- 68.Speck SH, Chatila T, Flemington E. 1997. Reactivation of Epstein-Barr virus: regulation and function of the BZLF1 gene. Trends Microbiol. 5:399–405 [DOI] [PubMed] [Google Scholar]
- 69.Lau R, Packham G, Farrell PJ. 1992. Differential splicing of Epstein-Barr virus immediate-early RNA. J. Virol. 66:6233–6236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ehlers B, Spiess K, Leendertz F, Peeters M, Boesch C, Gatherer D, McGeoch DJ. 2010. Lymphocryptovirus phylogeny and the origins of Epstein-Barr virus. J. Gen. Virol. 91:630–642 [DOI] [PubMed] [Google Scholar]
- 71.Areste C, Blackbourn DJ. 2009. Modulation of the immune system by Kaposi's sarcoma-associated herpesvirus. Trends Microbiol. 17:119–129 [DOI] [PubMed] [Google Scholar]
- 72.Harris S, Lang SM, Means RE. 2010. Characterization of the rhesus fibromatosis herpesvirus MARCH family member rfK3. Virology 398:214–223 [DOI] [PubMed] [Google Scholar]
- 73.Rezaee SA, Cunningham C, Davison AJ, Blackbourn DJ. 2006. Kaposi's sarcoma-associated herpesvirus immune modulation: an overview. J. Gen. Virol. 87:1781–1804 [DOI] [PubMed] [Google Scholar]
- 74.Samols MA, Hu J, Skalsky RL, Renne R. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:9301–9305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeng Y, Yi R, Cullen BR. 2005. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J. 24:138–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bininda-Emonds OR, Cardillo M, Jones KE, MacPhee RD, Beck RM, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A. 2007. The delayed rise of present-day mammals. Nature 446:507–512 [DOI] [PubMed] [Google Scholar]