

Abstract
The A1-adenosine receptor (A1-AR) is a G protein-coupled receptor that mediates many of the physiological effects of adenosine in the brain, heart, kidney, and adipocytes. Currently, ligand interactions with the A1-AR can be quantified on large cell populations only by using radioligand binding. To increase the resolution of these measurements, we have designed and characterized a previously undescribed fluorescent antagonist for the A1-AR, XAC-BY630, based on xanthine amine congener (XAC). This compound has been used to quantify ligand-receptor binding at a single cell level using fluorescence correlation spectroscopy (FCS). XAC-BY630 was a competitive antagonist of A1-AR-mediated inhibition of cAMP accumulation [log10 of the affinity constant (pKb) = 6.7)] and stimulation of inositol phosphate accumulation (pKb = 6.5). Specific binding of XAC-BY630 to cell surface A1-AR could also be visualized in living Chinese hamster ovary (CHO)-A1 cells by using confocal microscopy. FCS analysis of XAC-BY630 binding to the membrane of CHO-A1 cells revealed three components with diffusion times (τD) of 62 μs (τD1, free ligand), 17 ms (τD2, A1-AR-ligand), and 320 ms (τD3). Confirmation that τD2 resulted from diffusion of ligand-receptor complexes came from the similar diffusion time observed for the fluorescent A1-AR-Topaz fusion protein (15 ms). Quantification of τD2 showed that the number of receptor-ligand complexes increased with increasing free ligand concentration and was decreased by the selective A1-AR antagonist, 8-cyclopentyl-1,3-dipropylxanthine. The combination of FCS with XAC-BY630 will be a powerful tool for the characterization of ligand-A1-AR interactions in single living cells in health and disease.
Adenosine acts throughout the body as a hormone, autocrine factor, and neuromodulator (1). It exerts its actions at the cellular level by acting through a family of four transmembrane receptors belonging to the G protein-coupled receptor (GPCR) superfamily. These receptors, designated A1, A2A, A2B, and A3, all activate heterotrimeric G proteins to modulate the activity of adenylyl cyclase and phospholipase C (PLC) (1-3). More specifically, the A1-adenosine receptor (A1-AR) signals via Gαi/o proteins to inhibit adenylyl cyclase and stimulate PLC (2-4).
A major obstacle to the detailed investigation of ligand interactions with the A1-AR in health and disease has been the inability to study its pharmacology directly at the single cell level. This is crucial because changes in A1-AR localization, ligand-binding characteristics, and signaling in membrane compartments of individual cells may be important in a number of different pathologies (1). It is also clear that GPCRs, including the A1-AR, are not distributed uniformly over the cell surface but are localized in discrete membrane microdomains such as caveolae or cholesterol-rich lipid rafts (5). The exact nature and function of these domains are not yet clear, but they do appear to have an important role in the signaling, desensitization, and intracellular trafficking of a number of receptors (6, 7).
Comparison of ligand-binding characteristics in normal and diseased tissue samples is difficult using radioligand-binding techniques, which require a large (>104) cell population. This invariably means that primary human cells need to be expanded in cell culture, which may alter their phenotype. Fluorescent tagging of pharmacological agents is one way in which ligand-receptor interactions can be localized within individual cells. Such an approach has been used successfully for a number of GPCRs (8-11). However, obtaining quantitative ligand-binding data using fluorescence imaging techniques can be complex. In this study, we have chosen to use the noninvasive technique of fluorescence correlation spectroscopy (FCS) (12, 13) to perform quantitative analysis of ligand binding to the human A1-AR in single cells. FCS is based on confocal detection of fluctuations in signal intensity as fluorescent species diffuse through a small excitation volume (<0.5 fl). Autocorrelation analysis of the size and temporal nature of these fluctuations yields information about both their diffusion coefficient and concentration. Because the diffusion coefficient is related to molecular weight, this technique allows, for example, a small fast-diffusing fluorescent ligand to be distinguished from its slow-moving receptor-bound form (12). Positioning of the confocal volume on the cell membrane therefore allows simultaneous determination of the number of both free ligand molecules and receptor-ligand complexes within a small area (0.1-0.2 μm2) of cell membrane (13).
Previously, FCS has been used to measure ligand binding to cell surface receptors of both the tyrosine kinase and GPCR superfamilies (14-17). However, these studies have been performed with peptide ligands, which bind to the extracellular region of the receptor. No similar studies with antagonist ligands that bind to the intramembrane region of GPCRs have been performed.
This paper describes the design and pharmacological characterization of a fluorescent variant of xanthine amine congener (XAC; Fig. 1; refs. 18 and 19). We have used FCS to measure directly the interaction of this ligand with the A1-AR at subcellular resolution.
Fig. 1.

The chemical structure of XAC (a) and XAC-BY630 (b).
Materials and Methods
Synthesis and Fluorescence Spectroscopy of XAC-BY630. The primary amine of XAC (Sigma) was acylated by using BODIPY 630/650-X succinimydyl ester (Molecular Probes) by stirring for 2 h in N,N-dimethylformamide. The product was purified by RP-HPLC on a C8 column by using photodiode array detection. The structure of the ligand was confirmed by mass spectrometry (m/z time-of-flight ES+ found 974.3998; C50H55BF2N9O7S requires 974.4006) and 1H-NMR spectroscopy (see supporting information, which is published on the PNAS web site). Excitation and emission spectra and emission lifetime were obtained by using an Edinburgh Instruments (Livingston, U.K.) FLS920 fluorimeter.
Cell Lines. Chinese hamster ovary (CHO) cells expressing the wild-type A1-AR (CHO-A1 cells) (4) or an A1-AR with a C-terminal GFP tag (CHO-A1Tpz cells) were used. The fluorescent protein ORF of pGFPtpz-basic (Packard Bioscience) was subcloned into PCR3.1 (Invitrogen) after PCR (primers: CTCGAGCCTGGTGAGCAAGGGCGAG and CGACTTCTAGAAGCCCGGGTAACTTGTACAGCTCGTC) to produce a modified GFPtpz gene with the start codon replaced with CTG, a 5′-XhoI site, and a 3′-XbaI site. PCR (primers: GGGAGCTCTGCCAGCTTTGGTGAC and TCTAGACCCGGGAGCTTCTCGAGTCATCAGGCCTCTCTTC) was used to produce a modified A1-AR in PCR3.1 with the stop codon replaced with a XhoI site and an additional 3′ XbaI site. The tpz ORF was then excised from pCR3.1 by using XhoI and XbaI and ligated into the A1-AR pCR3.1 construct to generate the plasmid pCR3.1A1-TPZ. Cells were transfected by using Lipofectamine (GIBCO/BRL) according to the manufacturer's instructions. The cell line used here was produced after dilution cloning and selection with 600 μg/ml geneticin before functional characterization.
8-Cyclopentyl-1,3-[3H]dipropylxanthine (DPCPX) Binding to Membranes and Whole Cells. Radioligand binding to CHO-A1 cell membranes was carried out as described (4). For [3H]DPCPX binding in intact CHO-A1 or -A1Tpz cells, cells were grown to confluence in 96-well white viewplates (Corning). The media were removed and replaced with 100 μl of serum-free DMEM/F-12 media containing competing ligands, where appropriate. [3H]DPCPX was added in 100 μl of DMEM/F-12 media and the plates incubated at 37°C for 1 h. Nonspecific binding was determined by using 10 μM XAC. Cells were washed twice with 200 μl of PBS before addition of 200 μl of Microscint-20 (Packard Bioscience). The radioactive content of each well was determined by using a Topcount (Packard Bioscience), and the protein content of the wells was determined by the method of Lowry (20).
Assays for Second Messenger Generation. Measurement of [3H]inositol phosphate ([3H]IPx) and [3H]cAMP accumulation was performed as described (4), except that for cAMP measurements, [3H]cAMP was separated on acid alumina columns by using the method of Alvarez and Daniels (21).
Confocal Microscopy. For live cell confocal microscopy, cells were grown on 8-well coverglasses (Nunc, Fisher Scientific) in phenol red-free DMEM/F-12 media containing 5% (vol/vol) FCS and 2 mM glutamine. Cells were washed three times in Hepes-buffered saline solution (HBS; 147 mM NaCl/24 mM KCl/1.3 mM CaCl2/1 mM MgSO4/10 mM Hepes, pH 7.4) before equilibration in 360 μl of HBS for 15 min. Experiments were performed at 22 ± 2°C, with XAC-BY630 added for the times shown, after 30-min incubation with antagonist where indicated. Images were obtained by using a Zeiss LSM510 confocal microscope with a Plan-Neofluar 40 × 1.3 numerical aperture oil-immersion objective. XAC-BY630 images were taken by using 633-nm excitation, with emission collected through an LP650 filter. For simultaneous imaging of A1-adenosine receptor Topaz fusion protein (A1-AR-Tpz), additional excitation was with 488 nm and emission collected using a BP505-560 filter. Images within each experiment were collected by using identical laser-power, offset, and gain. Colocalization analysis was performed by using Zeiss aim software (Zeiss).
Fluorescence Correlation Spectroscopy (FCS). FCS is based on autocorrelation analysis of fluorescence intensity fluctuations within a small defined confocal volume element. The normalized intensity autocorrelation function [G(τ)] for fluctuations about a mean intensity, I, is described as follows, with the angular brackets representing an ensemble average:
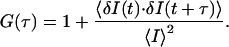 |
1 |
Here, the intensity fluctuation, δI(t), about the average intensity, I, at time, t, is correlated with the fluctuation at a given time later, δI(t + τ). The algebraic form of this equation relating to 3D diffusion of a number of fluorescent species through a Gaussian volume is:
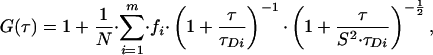 |
2 |
where fi is the fraction of species, i, from a total number of species, m, with a mean dwell time in the volume of τDi. N is the number of fluorescent particles in the volume, and S is a structure parameter, representing the ratio of the vertical and radial axes, ω1 and ω2, of the confocal volume. The confocal measurement volume was then estimated according to:
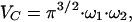 |
3 |
allowing the concentrations of fluorescent species to be calculated. For a species diffusing in two dimensions, such as a membrane receptor, S → ∞, and Eq. 2 simplifies to:
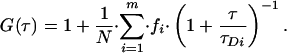 |
4 |
FCS measurements were performed on a Zeiss Confocor 2 fluorescence correlation spectrometer using a c-Apochromat 40 × 1.2 numerical aperture water-immersion objective. For each experiment, the diffusion time of either Cy5 (D = 3.16 × 10-10·m2/s) (Amersham Pharmacia Bioscience) or Rhodamine 6G (D = 2.8 × 10-10·m2/s) (Molecular Probes) was used to calculate the dimensions of the confocal volume (Eq. 3: ω1 = 0.25 μm, ω2 = 1.5 μm at 633 nm, and ω1 = 0.16 μm, ω2 = 0.84 μm at 514 nm) (13). Cells were prepared and exposed to drugs as described for confocal imaging. The confocal volume was positioned in cells in the x-y plane by using a live image from a Zeiss Axiocam HR camera. The center of the confocal volume was positioned either on the membrane (A1-AR-Tpz) or at the 50% peak intensity of the membrane (XAC-BY630 binding) using an intensity scan through the cell in the z axis. In the case of XAC-BY630, data were collected (10-s prebleach, 2 × 30 s) by using 633-nm excitation light (0.5-1 kW/cm2), with emission collected through an LP650 filter. For CHO-A1Tpz cells, 514-nm excitation (2.0-2.5 kW/cm2) was used with emission collected through a BP530-600 filter and readings taken for 2 × 20 s after a 15-s prebleach.
For XAC-BY630 binding experiments, autocorrelation data were fitted to a multicomponent diffusional model (Eq. 2) within Zeiss aim software, incorporating extra variables to account for triplet state formation. Values were minimized by using a Marquardt algorithm with values for structure parameter and free ligand diffusion time fixed to calibration values. Fit quality was assessed on residuals to the fit. The concentrations of free and bound components were then calculated directly from the autocorrelation amplitude. Autocorrelation data from CHO-A1Tpz cells were fitted in a similar way according to a 2D diffusion model (Eq. 4).
Data Analysis and Statistics. EC50 and IC50 values were obtained from concentration-response and radioligand-binding curves following nonlinear curve fitting to a multicomponent logistical equation by using prism (GraphPad, San Diego). pKi values were calculated from the Cheng-Prussoff equation, and apparent log10 of the dissociation constant (log Kb) values were determined by using the Gaddum Equation as described (8).
Data are presented as mean ± SEM, and n represents either the number of independent experiments performed or, in FCS experiments, the number of cells on which independent measurements were taken. Statistical analysis was by unpaired Student's t test, with a two-tailed P value <0.05 taken to indicate statistical significance.
Results
Synthesis and Characterization of XAC-BY630. To provide a fluorescent probe useful for studying both recombinant and endogenous A1-ARs, we designed and characterized a fluorescent antagonist for this receptor based on the moderate affinity ligand xanthine amine congener (XAC) (Fig. 1a). The primary alkyl amine of XAC was acylated, with the succinimydyl ester BODIPY 630/650-X-SE, incorporating a six-atom linker between fluorophore and ligand. The resulting product, XAC-BY630 (Fig. 1b), was purified to >99% by RP-HPLC and its chemical identity confirmed by 1H-NMR spectroscopy and mass spectrometry. Spectroscopic analysis of XAC-BY630 in Hepes-buffered saline solution showed that the excitation and emission spectra of the fluorophore remained virtually unchanged on coupling to XAC [excitation/emission maxima = 627/647 nm (BODIPY 630/650-X-SE) and 636/651 nm (XAC-BY630)]. Similarly conjugation had only a slight effect on the emission lifetime (6.0 ± 0.6 ns vs. 5.1 ± 0.5 ns for BODIPY 630/650-X-SE and XAC-BY630, respectively).
The pharmacological properties of XAC-BY630 were characterized in CHO cells transfected to express the human A1-AR (CHO-A1 cells). Whole-cell saturation-binding experiments using the radiolabeled antagonist [3H]DPCPX showed that CHO-A1 cells expressed the A1-AR at 2.8 ± 0.2 pmol/mg protein, with the radioligand showing an affinity consistent with its binding to the human A1-AR (log Kd = -8.64 ± 0.04, n = 9). Whole-cell competition-binding studies vs. [3H]DPCPX (1 nM) also showed typical affinity values for DPCPX and XAC at the A1-AR (log Kds = -8.05 ± 0.11 and -7.37 ± 0.06, n = 9 and 3, respectively). The 4-fold difference in affinity values for DPCPX between saturation and competition studies is significant (P < 0.01), although the reason for this difference is unclear.
The affinities of XAC and XAC-BY630 for the A1-AR were also compared in radioligand-binding and functional assays. Competition radioligand-binding studies using [3H]DPCPX-labeled CHO-A1 cell membranes showed XAC-BY630 to have an affinity ≈10-fold lower than that of XAC itself (log Kd = -6.82 ± 0.11 and -7.79 ± 0.13, respectively, n = 4). BODIPY 630/650-X-SE itself had no effect on [3H]DPCPX binding at concentrations of up to 10-5 M. In CHO-A1 cells, the A1-AR agonist 5′-N-ethylcarboxamido adenosine (NECA) inhibited forskolin-stimulated cAMP production in a dose-dependent manner, consistent with activation of Gi/o-family G proteins. This response was antagonized in a competitive manner by both 1 μM XAC and XAC-BY630 with apparent log Kb values of -7.94 ± 0.15 (n = 5) and -6.68 ± 0.05 (n = 7), respectively (see supporting information). Similar results were found for NECA-stimulated inositol phosphate production, which is mediated by Gβγ activation of PLC (2, 3) (apparent log Kb values = -7.46 ± 0.14 and -6.45 ± 0.14 for XAC and XAC-BY630, respectively, n = 3). Thus, both radioligand-binding and functional assays consistently indicate a 10-fold lower affinity of XAC-BY630 for the A1-AR compared to XAC itself (P < 0.01 in each case). For both XAC and XAC-BY630, there was no significant difference between the affinity values determined by radioligand binding and in functional assays (P > 0.05).
Visualizing Binding of XAC-BY630 to the A1-AR. Binding of XAC-BY630 to the A1-AR in living CHO-A1 cells was visualized by using confocal microscopy. A 15-min incubation with XAC-BY630 (10-100 nM) produced a concentration-dependent binding of ligand that was predominantly to the cell membrane (Fig. 2a). At higher concentrations (250 nM), intracellular uptake of the ligand was apparent, with no increase in membrane binding. After addition of 50 nM XAC-BY630, membrane binding was detected after only 5 min and then increased steadily for up to 20 min (Fig. 2b). There was little subsequent increase in membrane binding, although a small increase in intracellular ligand was seen up to 90 min after ligand addition (data not shown). No ligand was detected in the nucleus under any of the above conditions. Binding of XAC-BY630 to CHO-A1 cell membranes was inhibited in a concentration-dependent manner by DPCPX over a range (1-20 nM) consistent with its affinity at the A1-AR (Fig. 2c). In the presence of DPCPX, intracellular ligand was still detected, albeit at a decreased level. Specific binding of XAC-BY630 to the A1-AR in the membranes of CHO-A1 cells expressing lower levels of receptor (150 fmol/mg protein) was also detected (data not shown). Importantly, XAC-BY630 (100 nM, 30 min) showed no binding to the membranes of untransfected CHO-K1 cells, although a diffuse cytoplasmic signal was seen (Fig. 2c Right). Similarly, BODIPY 630/650-X-SE did not bind to the membranes of either CHO-A1 or native CHO-K1 cells (not shown) but did give a strong punctate cytoplasmic signal.
Fig. 2.

Visualization of the A1-AR in live CHO-A1 cells. CHO-A1 cells were incubated at 22°C with 10-250 nM XAC-BY630 for 15 min (a), 50 nM XAC-BY630 for 0-30 min (b), or DPCPX (1-20 nM, 30 min, 37°C) (c) followed by 50 nM XAC-BY630 for 15 min before capture of single confocal images. (c Right) Native CHO-K1 cells were incubated with XAC-BY630 (100 nM,15 min). Each experiment shown is representative of four performed.
To investigate further the nature of the membrane binding of XAC-BY630 to the A1-AR, we generated CHO cells expressing the A1-AR fused on its C terminus to the Topaz variant of GFP (CHO-A1Tpz cells). The receptor expression level in this clone was similar to that seen in CHO-A1 cells (2.7 ± 0.3 pmol/mg protein, [3H]DPCPX log Kd = -8.61 ± 0.06, n = 5), as were the affinities of A1-AR ligands for the receptor (log Kd for DPCPX and XAC = -8.00 ± 0.04 and -7.16 ± 0.06, respectively, n = 3, P > 0.05 vs. CHO-A1 cells). As with CHO-A1 cells, there was a significant difference between -log Kd values for DPCPX obtained from saturation and competition binding (P < 0.001). Confocal imaging showed that the fluorescent A1-AR was predominantly localized to the cell membrane (Fig. 3a), although areas of strong intracellular fluorescence were detected, particularly within the perinuclear region of some cells. Incubation of CHO-A1Tpz cells with XAC-BY630 (100 nM, 15 min) showed clear membrane binding of the ligand, with the most intense binding seen in areas showing the highest receptor expression, as demonstrated by colocalization analysis (Fig. 3 c and d). Incubation with XAC-BY630 (100 nM, 45 min) did not cause any significant change in receptor distribution. As with CHO-A1 cells, membrane binding of XAC-BY630 to CHO-A1Tpz cells was DPCPX-sensitive (data not shown).
Fig. 3.

Visualization of XAC-BY630 binding to CHO-A1Tpz cells. Cells were incubated with XAC-BY630 (100 nM, 30 min, 22°C), and simultaneous confocal images were captured with 488-nm excitation (a,A1-ARTpz) and 633-nm excitation (b, XAC-BY630). The overlay image (c) represents colocalized pixels as yellow/orange. Colocalization analysis showed those pixels with the highest degree of colocalization (d). The image shown is representative of five similar experiments performed.
FCS Analysis of the Membrane Localized A1-AR-Tpz Fusion. To estimate the diffusion characteristics of the ligand-bound A1-AR, FCS was used to analyze diffusion of the A1-AR-Tpz fusion protein in the membrane of CHO-A1Tpz cells. Autofluorescence was assessed in CHO-A1 cells by using 514-nm excitation at powers of <1.5 kW/cm2. This produced a count rate of 0.5-1 kHz, which corresponded to a single species with a diffusion time of ≈2-3 ms (data not shown). Measurements in CHO-A1 Tpz cells were therefore limited to those where the average count rate was >15 kHz to minimize interference from this autofluorescence. For measurements of A1-AR-Tpz, the confocal volume was positioned in the x-y plane over the cell nucleus, where the subsequent z-intensity profile allowed clear distinction of the lower (LM) and upper membranes (UM) (Fig. 4a). FCS measurements taken on the UM gave an autocorrelation function that was best fit by a model containing two diffusion components (Fig. 4b). These components were present in equal quantities and had diffusion times of τD1 = 107 ± 10 μs (104/104 cells) and τD2 = 15.0 ± 0.7ms (100/104 cells, range 3.2-43.6 ms). The fast-diffusing component, τD1, is too fast to represent diffusion of a membrane protein and most likely represents an intramolecular photophysical process, such as flickering of the Topaz fluorophore (22, 23). The slower-diffusing component, τD2, (diffusion coefficient, D = 4.27 × 10-9·cm2/s), therefore represents diffusion of membrane-localized A1-AR-Tpz.
Fig. 4.

FCS analysis of A1-AR-Tpz expressed in CHO cells. FCS measurements were performed in CHO-A1Tpz cells at 22°C as described. (a) The confocal volume was positioned over the cell nucleus in x-y, and a z-intensity scan was performed. Peaks in intensity corresponded to the lower membrane (LM) and upper cell membranes (UM). The vertical line represents the measurement position. (b) Intensity fluctuations (Upper) were collected for 30 s, producing the normalized autocorrelation curve shown (Lower). Data were best fit by a two-component 2D model, which, in this instance, gave τD1 = 45 μs [fraction (f1) = 0.46], τD2 = 12.4 ms (f2 = 0.54).
FCS Measurements of XAC-BY630 Binding to the A1-AR. Having established the diffusion characteristics of the A1-AR itself, the binding of XAC-BY630 to CHO-A1 cells was investigated. Both BODIPY 630/650-X-SE and XAC-BY630 showed fluorescent properties suitable for use with FCS, having low triplet fractions (<10%) and being resistant to photobleaching. Fluctuation analysis of XAC-BY630 in Hepes-buffered saline solution showed a simple monophasic autocorrelation curve with τD = 62.3 ± 1.6 μs (D = 2.51 × 10-6·cm2/s) (n = 21).
For XAC-BY630 binding, positioning of the measurement volume was carried out as described for CHO-A1Tpz cells (Fig. 5a). Autofluorescence at 633 nm was negligible relative to the subsequent signal (<1 kHz). In cells incubated with XAC-BY630 (2.5 nM added), distinct differences in the diffusion characteristics were seen with movement of the confocal volume through the cell (Fig. 5). Measurements 5 μm above the cell surface (position A) indicated a single diffusing species (τD = 63 μs), equivalent to diffusion of the ligand in solution. Measurements on the upper cell membrane (position B) showed two additional slow-diffusing components. Movement into the cell cytoplasm (position C) resulted in a monophasic autocorrelation curve in most cases, with an increased diffusion time compared to position A (τD1 = 5.0 ± 1.5 ms, n = 7). FCS measurements taken on cell membranes over a range of XAC-BY630 concentrations (1-40 nM added, 5-45 min) consistently produced autocorrelation functions containing two components in addition to that of free ligand (τD1) (Fig. 6a). These had mean diffusion times of 17.2 ± 1.4 ms (τD2) and 321 ± 25 ms (τD3) (D = 9.1 × 10-9·cm2/s and 4.7 × 10-10·cm2/s, respectively) (Table 1). Component τD2 had a diffusion time essentially the same as that seen for the membrane localized A1-AR-Tpz fusion and is therefore likely to represent the ligand-A1-AR complex. The slower component, τD3, was not found when measuring receptor diffusion directly in A1-AR-Tpz cells and possibly represents nonspecific binding of ligand or even movement of the cells themselves during the measurement. Species with similar diffusion characteristics were also seen when XAC-BY630 bound to CHO-A1Tpz cell membranes (τD2 = 14.6 ± 4.5 ms and τD3 = 168 ± 23 ms, n = 12). Further experiments were carried out to quantify τD2 and confirm its identity as ligand-A1-AR complex. The amount of τD2 seen in CHO-A1 cells incubated with XAC-BY630 (1-40 nM added, 30 min) was concentration-dependent (Fig. 6b). FCS measurements at free ligand concentrations of >60 nM were difficult, because the low amplitude of the autocorrelation curve precluded accurate determination of particle number. Fitting of a classic saturation binding isotherm (R2 = 0.92) to these data yields a log Kd value of -7.47 ± 0.34, and a Bmax of 73.9 ± 16.5 nM, which corresponds to 55 receptors/μm2. Preincubation of cells with 1 μM DPCPX substantially reduced the amount of τD2 seen. In 33% of DPCPX-treated cells, τD2 was absent entirely (Table 1), whereas in the remaining 67% it was significantly reduced (Fig. 6b). However, the diffusion times obtained for τD2 and τD3 were not significantly different in DPCPX-treated cells compared to control cells. In contrast, the amount of τD3 was not significantly decreased by preincubation with DPCPX, suggesting that this represents nonspecific ligand binding.
Fig. 5.

Diffusional analysis of XAC-BY630 binding to CHO-A1 cells by using FCS. CHO-A1 cells were incubated with XAC-BY630 (2.5 nM, 15 min). (a) After a z-intensity scan (Right). FCS readings were taken at three different z-positions within the same cell; A, extracellular; B, upper membrane; and C, intracellular. (b) Intensity fluctuations (Upper) were recorded for 30 s, after a 15-s prebleach, and subsequent normalized autocorrelation analyses are shown (Lower). Position A, τD1 = 65 μs(f1 = 1); position B, τD1 = 65 μs(f1 = 0.50), τD2 = 18.4 ms (f2 = 0.27), τD3 = 264 ms (f3 = 0.23); position C, τD1 = 7.3 ms (f1 = 1).
Fig. 6.

XAC-BY630 binding to the membrane of single CHO-A1 cells quantified by using FCS. (a) CHO-A1 cells were incubated with XAC-BY630 (2.5 nM, 30 min) and the confocal volume positioned either (i) in the extracellular buffer or (ii) on the upper membrane. Intensity fluctuations were recorded (Upper), and the subsequent normalized autocorrelation curves are shown (Lower). Diffusion components were assigned as follows: (i) τD1 = 64 μs(f1 = 1), (ii) τD1 = 64 μs(f1 = 0.24), τD2 = 12.5 ms (f2 = 0.65), τD3 = 300 ms (f3 = 0.11). (b) Measurements as described in a were performed on cells incubated with 1-40 nM XAC-BY630 (30 min, 22°C). For each cell, the free ligand (τD1) and corresponding amount of receptor-ligand complex (τD2) were calculated. The plot of [Free] vs. [Bound] is shown (closed circles). Each point is representative of measurements taken on 7-16 cells, in four to eight experiments. The data have been fitted to a single component binding isotherm (solid line), which yields Bmax = 75 nM, and Kd = 33 nM. A further set of experiments were performed in CHO-A1 cells exposed to DPCPX (1 μM, 30 min) before addition of XAC-BY630 (open circles). Each point is representative of measurements taken on 6-12 cells, within three to six independent experiments.
Table 1. Diffusion times of binding components revealed by FCS analysis of XAC-BY630 binding to CHO-A1 cell membranes.
| Control
|
+DPCPX
|
|||
|---|---|---|---|---|
| Component | Diffusion time, ms | Number of cells, % | Diffusion time, ms | Number of cells, % |
| τd2 | 17.2 ± 1.4 | 95/101 (94) | 18.0 ± 2.4 | 43/64 (67) |
| τd3 | 321 ± 25 | 84/101 (83) | 281 ± 30 | 53/64 (83) |
FCS measurements were performed on the upper membrane of CHO-A1 cells that had been incubated with buffer or 1μM DPCPX prior to addition of XAC-BY630 (1-40 nM added). Autocorrelation analysis was performed as described in Materials and Methods. The mean diffusion times of each component are shown, with the number of cells in each group in which that diffusion component was found.
Discussion
The ability to gain quantitative and spatially resolved ligand-binding information for the A1-AR in single cells provides an important step toward understanding the organization of the receptor and its signaling. We have developed a fluorescent A1-AR antagonist, XAC-BY630, which is functional as both a pharmacological agent and a fluorescent probe, and which allows such information to be obtained.
The design of a fluorescent ligand for the A1-AR was complicated by the intramembrane location of the ligand-binding site for this receptor, and we incorporated an appropriate linker to preserve the access of the pharmacophore to its site. The choice of XAC as the basis of this fluorescent probe was based on structure-activity relationships that show that substituents on the C8 position of the xanthine ring are tolerated for A1-AR binding (19, 24-26). The fluorophore BODIPY 630/650 was chosen because it is pharmacologically inactive at the A1-AR and has photophysical properties suitable for single-cell applications (27). The affinities of DPCPX and XAC for the A1-AR in both CHO-A1 and -A1Tpz cells were similar and consistent with previously reported values (1, 3, 18). Radioligand-binding and functional assays both showed that XAC-BY630 was only 10-fold less potent as a competitive antagonist at the A1-AR than XAC itself.
XAC-BY630 allowed the visualization of ligand-receptor complexes by confocal microscopy, with clear and specific membrane labeling seen in both CHO-A1 and -A1Tpz cells. It had a fast on-rate, allowing binding to be visualized after as little as 5 min, with intracellular ligand accumulating only at longer time points and higher concentrations. Interestingly, this accumulation was also blocked to some extent by DPCPX, suggesting that XAC-BY630 enters the cell through both receptor-dependent and -independent mechanisms. This is supported by the changes in localization of ligand over time seen in CHO-A1Tpz cells. At early time points, XAC-BY630/A1-AR-Tpz colocalization was at the cell membrane. However, at longer time points, colocalization was also seen in some intracellular areas rich in A1-AR-Tpz, suggesting that XAC-BY630 may also bind to receptor that is cycling to/from the membrane. This may account for the substantially slower diffusion time of ligand in the intracellular milieu.
FCS analysis initially allowed the characterization of the diffusion of A1-AR-Tpz in CHO cell membranes before performing ligand-binding analysis. Intensity scans in z allowed easy localization of the cell membrane, because the profiles were consistent with the images obtained with standard fluorescence and confocal microscopy (28). FCS measurements taken on the upper membrane consistently yielded autocorrelation data suggesting two components of 100 μs and 15 ms. The 15-ms component most likely represents translational diffusion of the receptor and is similar to that seen for other receptors, such as those for epidermal growth factor, insulin, and galanin, when measured by FCS analysis of fluorescent ligand binding (14, 15, 17, 29). The faster 100-μs component is most likely a result of a fast intramolecular chemical process within the Topaz protein, such as proton exchange, producing flickering. This process has been demonstrated in yellow-shifted GFP variants such as Topaz and depends on both excitation intensity and the local pH (22, 23).
FCS analysis also allowed simultaneous quantification of receptor-bound XAC-BY630 and free ligand in the vicinity of the receptor. Two slower-diffusing species were consistently detected in these experiments in addition to fast-diffusing free ligand. These components were localized to the membrane because the diffusion time was different from that of intracellular ligand, and they were not present when the measurement volume was moved upward by 1 μm. One of these species (τD2) had a diffusion time that was very similar to that seen for A1-AR-Tpz and most probably represents the ligand-receptor complex. This interpretation is supported by the significant reduction in the concentration of τD2 at all ligand concentrations in the presence of an A1-AR antagonist. Additionally, the concentration of τD2 depended on free ligand concentration and showed an affinity similar to that seen for XAC-BY630 in other assays. An equivalent species to τD2 was also seen when XAC-BY630 binding was measured in CHO-A1Tpz cells.
The third and slowest-diffusing component (τD3, 320 ms) possibly represents nonspecific binding of XAC-BY630 to the cell membrane, because this is a factor found in most radioligand-binding studies, particularly with hydrophobic ligands. This is consistent with the presence of this slow component in both CHO-A1 and -A1Tpz cells and its insensitivity to DPCPX. The lack of this component when measuring diffusion of A1-AR-Tpz itself also suggests it is unlikely to represent a ligand-receptor complex. This cannot be ruled out, however, because ligand-induced movement of the receptor to a more rigid area of membrane may produce a substantial slowing of translational diffusion. Such a situation has been described for the μ-opioid receptor, where fast diffusion of the receptor was superimposed on a slower diffusion of the domain within which the receptor resided (30). FCS analysis of ligand binding to cell surface receptors for insulin C-peptide, galanin, and GABA has also identified two distinct membrane species, although in these studies both components were sensitive to exposure to unlabeled ligand (17, 29, 31). Interestingly, Roettger et al. (32) demonstrated two subsets of cholecystokinin receptors whose diffusion characteristics were differentially affected by antagonist binding.
Conclusion
FCS analysis of the binding of a previously undescribed functional fluorescent antagonist, XAC-BY630, allows quantification of A1-AR ligand interaction in a small area (≈0.1-0.2 μm2) of cell membrane. At this resolution, the average number of receptor-ligand complexes within the confocal volume (0.5-15) may allow single ligand-receptor complexes to be studied. Because the technique is noninvasive, has a large dynamic range, and is sensitive, it should be possible to use XAC-BY630 to quantitatively study the pharmacology of the endogenous A1-AR expressed in human cells from healthy and diseased tissue. Furthermore, localizing the detection volume to specific areas of the cell membrane, such as caveolae, will give insight into differences in receptor-ligand interactions within different membrane domains of the same cell.
Supplementary Material
Acknowledgments
This work was supported by Wellcome Trust Grants 057199 and 066817 and the University of Nottingham.
Abbreviations: A1-AR, A1-adenosine receptor; A1-AR-Tpz, A1-AR Topaz fusion protein; DPCPX, 8-cyclopentyl-1,3-dipropylxanthine; FCS, fluorescence correlation spectroscopy; GPCR, G protein-coupled receptor; τD, diffusion time; XAC, xanthine amine congener; CHO, Chinese hamster ovary.
References
- 1.Ralevic, V. R. & Burnstock, G. (1998) Pharmacol. Rev. 50, 415-475. [PubMed] [Google Scholar]
- 2.Friedholm, B. B., Ijzerman, A. P., Jacobson, K. A., Klotz, K.-N. & Linden, J. (2001) Pharmacol. Rev. 53, 527-552. [PMC free article] [PubMed] [Google Scholar]
- 3.Klotz, K.-N. (2000) Naunyn-Schmiedeberg's Arch. Pharmacol. 362, 382-391. [DOI] [PubMed] [Google Scholar]
- 4.Cordeaux, Y., Briddon, S. J., Megson, A. E., McDonnell, J., Dickenson, J. M. & Hill, S. J. (2001) Mol. Pharmacol. 58, 1075-1084. [DOI] [PubMed] [Google Scholar]
- 5.Gines, S., Ciruela, F., Burgueno, J., Casado, V., Canela, E. I., Mallol, J., Lluis, C. & Franco, R. (2001) Mol. Pharmacol. 59, 1314-1323. [PubMed] [Google Scholar]
- 6.Zajchowski, L. D. & Robbins, S. M. (2002) Eur. J. Biochem. 269, 737-752. [DOI] [PubMed] [Google Scholar]
- 7.Helmreich, E. J. M. (2003) Biophys. Chem. 100, 519-534. [DOI] [PubMed] [Google Scholar]
- 8.Baker, J. G., Hall, I. P. & Hill, S. J. (2003) Br. J. Pharmacol. 139, 232-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harikumar, K. G., Pinon, D. I., Wessels, W. S., Prendergast, F. G. & Miller, L. J. (2002) J. Biol. Chem. 277, 18552-18560. [DOI] [PubMed] [Google Scholar]
- 10.Emmerson, P. J., Archer, S., El-Hamouly, W., Mansour, A., Akil, H. & Medzihradsky, F. (1997) Biochem. Pharmacol. 54, 1315-1322. [DOI] [PubMed] [Google Scholar]
- 11.Bennett, V. J. & Simmons, M. A. (2001) BMC Chem. Biol. 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwille, P. (2001) Cell Biochem. Biophys. 34, 383-408. [DOI] [PubMed] [Google Scholar]
- 13.Pramanik, A. & Rigler, R. (2001) in Fluorescence Correlation Spectroscopy: Theory and Applications, eds. Elson, E. & Rigler, R. (Springer, Heidelberg), pp. 101-129.
- 14.Pramanik, A. & Rigler, R. (2001) Biol. Chem. 382, 371-378. [DOI] [PubMed] [Google Scholar]
- 15.Zhong, Z.-H., Pramanik, A., Ekberg, K., Jansson, O. T., Jörnvall, H., Wahren, J. & Rigler, R. (2001) Daibetologia 44, 1184-1188. [DOI] [PubMed] [Google Scholar]
- 16.Patel, R. C., Kumar, U., Lamb, D. C., Eid, J. S., Rocheville, M., Grant, M., Rani, A., Hazlett, T., Patel, S. S., Gratton, E. & Patel, Y. C. (2002) Proc. Natl. Acad. Sci. USA 99, 3294-3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rigler, R., Pramanik, A., Jonasson, P., Kratz, G., Jansson, O. T., Nygren, P.-Å., Ståhl, S., Ekberg, K., Johansson, B.-L., Uhlén, S., et al. (1999) Proc. Nat. Acad. Sci. USA 96, 13318-13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobson, K. A., Kirk, K. L., Padget, W. L. & Daly, J. W. (1985) J. Med. Chem. 28, 1341-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobson, K. A., De la Cruz, R., Schulick, R., Kiriasis, L., Padgett, W., Pfeiderer, W., Kirk, K. L., Neumayer, J. L. & Daly, J. W. (1988) Biochem. Pharmacol. 37, 3653-3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowry, O. H., Rosenbrough, N. J., Farr, A. L. & Randell, R. J. (1951) J. Biol. Chem. 193, 265-275. [PubMed] [Google Scholar]
- 21.Alvarez, R. & Daniels, D. V. (1992) Anal. Biochem. 203, 76-82. [DOI] [PubMed] [Google Scholar]
- 22.Schwille, P., Kummer, S., Heikal, A. A., Moerner, W. E. & Webb, W. W. (2000) Proc. Natl. Acad. Sci USA 97, 151-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haupts, U., Maiti, S., Schwille, P. & Webb, W. W. (1998) Proc. Natl. Acad. Sci. USA 95, 13573-13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobson, K. A., Ukena, D., Padgett, W., Kirk, K. A. & Daly, J. W. (1987) Biochem. Pharmacol. 36, 1697-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacobson, K. A., Zimmet, J., Schulick, R., Barone, S., Daly, J. W. & Kirk, K. L. (1987) FEBS Lett. 225, 97-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boring, D. L., Ji, X.-D., Zimmet, J., Taylor, K. E., Stiles, G. L. & Jacobson, K. A. (1991) Bioconjugate Chem. 2, 77-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buschmann, V., Weston, K. D. & Sauer, M. (2003) Bioconjugate Chem. 14, 195-201. [DOI] [PubMed] [Google Scholar]
- 28.Brock, R. & Jovin, T. M. (1998) Cell Mol. Biol. 44, 847-856. [PubMed] [Google Scholar]
- 29.Pramanik, A., Olsson, M., Langel, U., Bartfai, T. & Rigler, R. (2001) Biochemistry 40, 10839-10845. [DOI] [PubMed] [Google Scholar]
- 30.Daumas, F., Destainville, N., Millot, C., Lopez, A., Dean, D. & Salome, L. (2003) Biophys. J. 84, 356-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meissner, O. & Häberlein, H. (2003) Biochemistry 42, 1667-1672. [DOI] [PubMed] [Google Scholar]
- 32.Roettger, B. F., Hellen, E. H., Burghardt, T. P. & Miller, L. J. (2001) J. Fluor. 11, 237-246. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.