

Abstract
Apurinic/apyrimidinic endonuclease 1 (APE1) is an Mg2+-dependent enzyme responsible for incising the DNA backbone 5′ to an apurinic/apyrimidinic (AP) site. Here, we use rapid quench flow (RQF) techniques to provide a comprehensive kinetic analysis of the strand incision activity (kchemistry) of APE1 acting on an authentic AP site along with two widely-used analogs, a reduced AP site and a tetrahydrofuran (THF) site. In the presence of biologically-relevant Mg2+, APE1 incises all three substrates at a rate faster than the resolution of the RQF, ≥700 s−1. To obtain quantitative values of kchemistry and facilitate comparison of the authentic substrate versus the substrate analogs, we replaced Mg2+ with Mn2+ or Ni2+, or introduced a mismatch 5′ to the lesion site. Both strategies were sufficient to slow kchemistry and resulted in rates within the resolution of the RQF. In all cases where quantitative rates were obtained, kchemistry for the reduced AP site is indistinguishable from the authentic AP site. Notably, there is a small decrease, ~1.5-fold, in kchemistry for the THF site relative to the authentic AP site. These results highlight a role in strand incision for the C1′ oxygen of the AP site, and warrant consideration when designing experiments using substrate analogs.
Apurinic/apyrimidinic (AP) sites are generated by the repair activity of DNA glycosylases, damaging chemical agents, and by spontaneous hydrolysis of the purine N-glycosidic bond (1, 2). If left unrepaired, AP sites are both cytotoxic and mutagenic to a cell, and therefore repair of these sites is essential to maintaining genomic integrity (3, 4).
Apurinic/apyrimidinic endonuclease 1 (APE1), a Mg2+-dependent base excision repair (BER) enzyme, is the major human AP endonuclease responsible for incising the DNA phosphodiester backbone 5′ to AP sites, generating a nick with 3′-hydroxyl and 5′-deoxyribose phosphate (dRP) termini (Figure 1) (5). Repair of the resulting nick is completed by DNA polymerase and DNA ligase. In addition to its endonuclease activity, APE1 is known to have 3′-phosphodiesterase and 3′-phosphatase activity, 3′ to 5′ exonuclease activity, as well as a role in regulating the redox state of several transcription factors (6–8). APE1 also stimulates the rate of product release for many DNA glycosylases, where the action of a glycosylase precedes APE1 in the BER cascade (9–13). Finally, APE1 has been shown to stimulate the dRPase activity as well as the strand displacement synthesis activity of DNA polymerase β (14, 15).
FIGURE 1.

Structures of AP, AP-Red, and THF. The authentic AP site exists as an equilibrium between the ring-closed hemiacetal (A) and ring-opened aldehyde (B). The ring-opened aldehyde is subject to β-elimination, and can further be converted to the β,δ-elimination product under basic conditions. The ring-opened aldehyde can be reduced by NaBH4 to generate the AP-Red substrate. APE1 incises the AP site, to create a nick with a 3′-OH and 5′-dRP group.
An important consideration for DNA containing an AP site is that the lesion exists as an equilibrium between the ring-closed hemiacetal (Figure 1A) and ring-opened aldehyde (Figure 1B) forms. Furthermore, the ring-opened aldehyde is known to undergo β-elimination, resulting in a strand break with 3′-α,β-unsaturated aldehyde and 5′-phosphate termini (16). Because of the instability of the authentic AP site, analogs that do not undergo β-elimination are often used. Two commonly-used analogs are the reduced AP site, which is generated by reduction of an authentic AP site with NaBH4, and the tetrahydrofuran (THF) site. To date, all x-ray crystal structures of APE1 bound to DNA utilize the THF AP site analog (17, 18).
Several previous studies examined the kinetics of APE1 endonuclease activity. Steady-state rates for APE1 acting on an authentic AP site (19–21), a THF site (20–24), and a reduced AP site (16, 25) are the same within error, ~2–10 s−1. More recently, the first transient-state characterization of APE1 strand incision was reported (24); using rapid quench flow (RQF) techniques and a THF-containing substrate, it was reported that the steady-state rate of APE1 is limited by a slow step that follows strand incision chemistry, likely product release, and that the rate of strand incision (kchemistry) is at least 850 s−1. With respect to the authentic AP site, to date all transient-state kinetics have been performed using stop-flow fluorescence (SFF) techniques, which monitor conformational changes within APE1 (20, 21). One notable SFF study compared APE1 processing the authentic AP and THF sites; interestingly, for the THF substrate an additional conformational change in the APE1/DNA complex was observed prior to the chemistry step (21).
In this work we use RQF techniques and report the first experiments to monitor directly the strand incision activity of APE1 on the biologically-relevant authentic AP site. The rate of APE1 strand incision for authentic, reduced and THF AP sites was determined in the presence of Mg2+, Mn2+, or Ni2+. Furthermore, we also investigated APE1 strand incision chemistry on substrates with mismatches 5′ to the lesion. These varied experimental conditions allow for a comprehensive comparison of the strand incision activity of APE1 acting on an authentic AP site and the substrate analogs. The results highlight the destabilizing effects of different metal ions, and mismatches located 5′ to the AP site. Furthermore, these results merit consideration when designing experiments using substrate analogs.
EXPERIMENTAL PROCEDURES
Oligonucleotide Synthesis and Purification
DNA oligonucleotides used in this work are listed in Table 1, and were synthesized using standard phosphoramidite chemistry on a BioAutomation DNA/RNA synthesizer. The modified phosphoramidites deoxyuridine (5′-dimethoxytrityl-5-O-acetyl-2′-deoxyuridine,3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite) and tetrahydrofuran (5′-O-dimethoxytrityl-1′, 2′-dideoxyribose-3′-[(2-cyanothyl)-(N,N-diisopropyl)]-phosphoramidite) were used in synthesis of lesion-containing stands (denoted as LS in Table 1) and were obtained from Glen Research. During synthesis the 5′-DMT was retained to aid in HPLC purification. Two rounds of HPLC purification were performed on each oligonucleotide as previously described (26). The 5′-DMT group was removed and quantification of each oligonucleotide was performed using the ε260 values estimated for single-stranded DNA (27) and a Beckman Coulter DU800 UV-VIS Spectrophotometer.
Table 1.
DNA Oligonucleotides Used in this Study
| Name | Sequence |
|---|---|
| LSa | 5′-CGTTCAACGTGCACTXACAGCACGTCCCAT-3′ |
|
| |
| WM | 3′-GCAAGTTGCACGTGACTGTCGTGCAGGGTA-5′ |
| MM1b | 3′-GCAAGTTGCACGTGTCTGTCGTGCAGGGTA-5′ |
| MM2b | 3′-GCAAGTTGCACGTCACTGTCGTGCAGGGTA-5′ |
| MM3b | 3′-GCAAGTTGCACGAGACTGTCGTGCAGGGTA-5′ |
| MM4b | 3′-GCAAGTTGCACCTGACTGTCGTGCAGGGTA-5′ |
LS indicates the lesion-containing strand where X denotes location of AP, AP-Red, THF, or Uracil site.
Mismatched nucleotide is underlined.
DNA Duplex Assembly and Characterization
Oligonucleotides containing THF or Uracil were 5′-32P end-labeled using T4 Polynucleotide Kinase (New England Biolabs) following the manufacturer’s protocol. Assembly of the 30-mer duplex substrate was achieved by annealing 30 pmol (for transient-state experiments) or 300 pmol (for steady-state experiments) of the 5′-radiolabeled lesion-containing strand in the presence of a 1.5-fold excess of the desired complement [either well matched (WM) or containing a mismatch (MM1, MM2, MM3, or MM4)] in 300 μL of 50 mM HEPES-KOH, 100 mM KCl, pH 7.5.
To generate a duplex containing the reduced AP site, the uracil-containing duplex was incubated with 1.5 units of uracil DNA glycosylase (UDG; New England Biolabs) and freshly prepared NaBH4 (final concentration of NaBH4 was 0.1 M) overnight at 37 °C. The DNA was desalted using a 0.5 mL, 3,000 MW Amicon centrifugal filter. To generate a duplex containing the authentic AP site, the uracil-containing duplex was incubated with 1.5 units of UDG for 30 min at 37 °C. Due to the lability of the authentic AP substrate, UDG was not removed from the authentic AP site samples, therefore THF DNA was also incubated with 1.5 units of UDG for 30 min at 37 °C.
The stability of the duplex substrates formed using the WM complement was assessed immediately following the incubation with UDG. For each substrate, three 15 μL aliquots of the lesion-containing duplexes were prepared and 15 μL of Buffer A (50 mM HEPES-KOH, 100 mM KCl, 10 mM MgCl2 and 0.24 mg/mL BSA, pH 7.5) was added. The first aliquot was then quenched with 50 μL 100 mM EDTA, followed by 40 μL of denaturing dye (80% formamide, 10 mM EDTA, 1 mg/mL xylene cyanol). The second aliquot was quenched with 50 μL of 0.1 M NaOH followed by 40 μL of denaturing dye. The first and second aliquots were placed on dry ice until gel electrophoresis. A third aliquot was quenched with 50 μL of 100 mM EDTA and immediately dried in vacuo, resuspended in 105 μL denaturing dye, and heated to 90 °C for 3 min prior to gel loading. A separate 15 μL aliquot of each duplex was incubated with 15 μL of APE1 (final concentration of 500 nM APE1), diluted in Buffer A, for 5 min at 37 °C, and quenched with 50 μL of 100 mM EDTA followed by 40 μL denaturing dye and placed on dry ice. All reactions were also carried out on uracil-containing duplex not treated with UDG, as a control. All samples were loaded onto an 18% denaturing PAGE gel (33 × 42 cm, 0.4 mm thick) and were electrophoresed for ~3 h at 80 W and the results were visualized using phosphorimagery.
Expression and Purification of human APE1
Escherichia coli BL21(DE3) pLysS cells were transformed via heat shock with the pXC53 plasmid carrying the APE1 gene (16). The transformed cells were grown at 37 °C in 2 L of LB media containing 100 μg/mL ampicillin to an OD600 of 0.6–0.7, at which time cells were induced with isopropyl β-D-1-thiogalactopyranoside to a final concentration of 1 mM. After 2 h of growth at 37 °C, the cells were pelleted by centrifugation (3,000 × g, 30 min, 4 °C). The supernatant was discarded, and the cells were frozen with liquid nitrogen and stored at −80 °C until purification of APE1. Upon thawing, cells were resuspended in lysozyme buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM PMSF, 1 μg/mL pepstatin, 1 μg/mL leupeptin) at room temperature for 30 min. The incubation continued for another 20 min after the addition of NaCl to a final concentration of 1 M. Cells were then lysed using a French press. Lysates were clarified by centrifugation (24,336 × g, 20 min, 4 °C). Purification of the protein was completed using both HiTrap SP HP and Heparin HP columns (GE Healthcare) as previously described (21). The purity of APE1 was >90% as assessed by SDS-PAGE. The total concentration of APE1 was determined by the Bradford method using bovine γ-globulin as a standard. The APE1 preparation was >80% active as determined by steady-state kinetic experiments and all APE1 concentrations given below or in figure captions are active enzyme concentrations.
It is noteworthy that the purified APE1 contained trace amounts of Mg2+. For transient-state kinetics experiments using different metal ions, it was important to ensure that this trace Mg2+ is removed. Therefore, we performed an EDTA chelation experiment (Figure S1). The authentic AP site-WM duplex was prepared as described above. Two 40 μL APE1 aliquots (1000 nM) were prepared in either Buffer A or Buffer B (50 mM HEPES-KOH, 100 mM KCl and 0.24 mg/mL BSA, pH 7.5). A 5 μL aliquot of AP-WM DNA was mixed with 5 μL Buffer A. A second 5 μL DNA aliquot was mixed with 5 μL of APE1 in Buffer A. Seven 5 μL aliquots of APE1 in Buffer B were initially mixed with EDTA to a concentration of 0–40 mM, followed by addition of 5 μL of AP-WM DNA, which created samples with a final concentration of 0–20 mM EDTA. All samples were incubated at 37 °C for 3 min, followed by addition of 10 μL denaturing dye and placed on dry ice. All samples were loaded onto an 18% denaturing PAGE gel (33 × 42 cm, 0.4 mm thick) and were electrophoresed for ~3 h at 80 W and the results were visualized using phosphorimagery. We found that a final reaction concentration of 0.25 mM EDTA was sufficient to chelate the trace amount of Mg2+ present following expression and purification of APE1 (Figure S1).
APE1 Transient-State Kinetic Assays
APE1 transient-state kinetic assays were performed using a Rapid Quench Flow instrument (RQF-3, KinTek Corp.). DNA duplex and enzyme aliquots were prepared freshly for each time course. To limit β-elimination in the authentic AP site DNA, a separate UDG reaction was performed prior to each time course.
For metal ion dependence kinetics, the lesion-containing strand was annealed to the WM complement. For each time course, a 300 μL aliquot of 1000 nM APE1 in Buffer B containing EDTA to chelate Mg2+ (50 mM HEPES-KOH, 100 mM KCl, 500 nM EDTA, and 0.24 mg/mL BSA, pH 7.5), was prepared. For each time course, two controls were performed. In the first control, used to demonstrate inactivity of the APE1 aliquot in the absence of additional metal ions, a 15 μL aliquot of DNA was manually mixed with 15 μL of the prepared APE1 aliquot and incubated for 3 min at 37 °C, followed by addition of 50 μL of 100 mM EDTA, to quench the reaction, and 40 μL of denaturing dye, then placed on dry ice. The second control, performed at the end of the RQF time course, was used to demonstrate the stability of the DNA throughout the time course; DNA and Buffer B, in the absence of enzyme, were mixed in the RQF instrument, followed by quenching with 100 mM EDTA by the RQF and manual addition of 40 μL denaturing dye and placed on dry ice until electrophoresis. Following initial chelation reaction and immediately prior to RQF reactions, MgCl2, MnCl2 or NiCl2, to a concentration of 10 mM, was added to the APE1 aliquot. It is important to note that by mixing an equal volume of enzyme and DNA by the RQF, the final concentration of DNA and enzyme was 50 nM and 500 nM, respectively, and final concentration of EDTA and divalent metal ions was 250 nM and 5 mM, respectively. RQF reactions were allowed to proceed for 2–15,000 msec at 37 °C prior to quenching by the addition of 50 μL of 100 mM EDTA by the RQF. Once the sample was expelled from the RQF, 40 μL of denaturing dye was added and samples were placed on dry ice until separation on a 18% denaturing PAGE gel (33 × 42 cm, 0.4 mm thick). The samples were run for ~2 hrs at 80 W. The products were visualized by phosphorimagery. Sample autoradiograms can be found in Supporting Information (Figure S2). Additionally, a transient-state kinetic time course was run, as described above, for each lesion in the presence of MgCl2 but lacking EDTA, to ensure addition of EDTA did not affect strand incision chemistry rates. Kinetic experiments were also performed using authentic AP DNA annealed to the WM complement as described above, in the presence of 0.5, 1, 2.5, and 5 mM MgCl2; such experiments allowed us to determine the Mg2+ concentration that provided maximal strand incision, 5 mM, which was used in subsequent experiments (Figure S3).
For 5′ mismatch kinetics, the lesion-containing strand was annealed to either MM1, MM2, MM3, or MM4. For each time course, a 300 μL aliquot of 1000 nM APE1 in Buffer A was prepared. RQF reactions and gel electrophoresis proceeded as described above.
For all kinetic time courses, 100 mM EDTA was used to quench each time point. A control was performed to ensure a concentration of 100 mM EDTA was sufficient to quench the reaction (data not shown). A 15 μL aliquot of prepared APE1, after addition of either Mg2+, Mn2+, or Ni2+, was mixed with 50 μL of 100 mM EDTA, followed by 15 μL of DNA and incubated for 3 min at 37 °C followed by the addition of 40 μL of denaturing dye and placed on dry ice until gel electrophoresis. The results were visualized by phosphorimagery.
For all transient-state kinetic experiments, Kaleidagraph was used to fit data from the full time course as previously described to obtain the rate of strand incision chemistry (kchemistry) (28). Importantly, the fastest reaction time that can be performed on the RQF instrument is 2 msec, which provides an upper limit to the rate that can be determined of 700 s−1. This rate is calculated by assuming two half-lives have passed, or at least 75% of substrate has been converted to product at the 2 msec time point. This 75% substrate turnover was chosen as the cut-off point as rates obtained from burst kinetic plots are determined by the slope of the burst; 75% product formation is a point reliably on the burst portion and not the plateau. It is of note that this methodology provides a conservative estimate of the capability of the RQF instrument. Two-tailed, student’s T-test was performed to obtain the p-values for transient-state rates for THF-containing DNA compared to authentic AP DNA.
APE1 Steady-State Kinetic Assays
DNA substrates, annealed to WM complement, were created as described above. For each time course, a 300 μL aliquot of 100 nM APE1 in Buffer A was prepared. RQF instrumentation was used to rapidly mix and quench DNA and APE1 to a final concentration of 500 nM DNA and 50 nM APE1. RQF reactions were allowed to proceed for 2–100 msec and quenched and gel electrophoresed as described above. The products were visualized by phosphorimagery. Both a burst phase and a linear phase are observed with the slope of the linear phase equal to kss × [active enzyme] (28). It is important to note that we have defined the steady-state rate as kss while this reference defines the steady-state rate as k3. The active enzyme concentration of >80% was determined by extrapolating the linear phase line through the y-axis. The reported steady-state rates are from a single data set and reported error is the error associated with the fit. Two additional steady-state kinetic assays were performed with varying substrate concentrations (250 nM and 1000 nM) and rates comparable to those reported here were obtained.
RESULTS
Characterization of DNA Substrates
DNA duplexes containing an authentic AP site (AP DNA), reduced AP site (AP-Red DNA), or THF site (THF DNA) were used to obtain the kinetic parameters of human APE1. Prior to performing kinetic experiments, we confirmed the lability of the authentic AP site, and observed formation of the β-elimination product during gel electrophoresis as seen in Figure 2 as a smear in the gel (AP Lane 1). This smear, which was also observed in our kinetic time course experiments with AP DNA, was included as substrate during quantitation, and is likely due to the increased temperature incurred during electrophoresis. The β-elimination product is formed prior to electrophoresis when the AP DNA is dried in vacuo and heated to 90 °C prior to loading (AP Lane 2). Furthermore, we can convert the AP site to the β,δ-elimination product in the presence of 0.1 M NaOH (AP Lane 3). Finally, reaction of AP DNA with APE1 leads to complete conversion to the APE1 product (AP Lane 4). Important for the experiments conducted here, β-elimination of the AP site can be avoided by not heating the samples above 37 °C, and not drying the samples.
FIGURE 2.
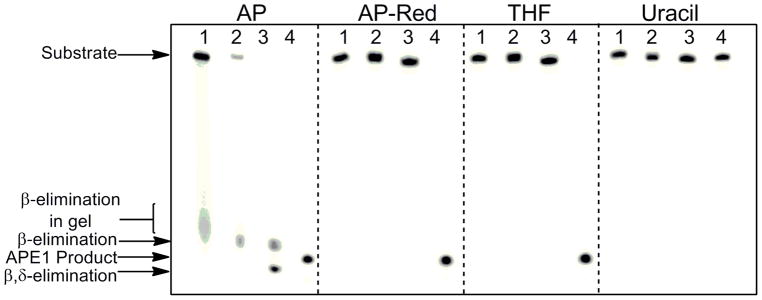
Susceptibility of DNA substrates to β and β,δ-elimination. Autoradiogram revealing β-elimination of authentic AP site before and during gel electrophoresis. Samples contained 30 pmol uracil or THF-containing duplex (annealed to WM complement) in 300 μL of 50 mM HEPES-KOH, 100 mM KCl, pH 7.5 and were incubated with 1.5 units of UDG for 30 min at 37 °C. For AP-Red, uracil-containing DNA and NaBH4 (final concentration of 0.1M) was incubated overnight with 1.5 units of UDG at 37 °C. Following incubation, samples were mixed with an equal volume of Buffer A and subjected to the following: quenching by addition of EDTA and denaturing dye (lanes 1); addition of EDTA followed by in vacuo drying, resuspension in denaturing dye, and heating to 90 °C for 3 min (lanes 2); quenching with 0.1 M NaOH and denaturing dye (lanes 3). All substrates were also allowed to react with APE1, to a final concentration of 500 nM, for 3 min at 37 °C (lane 4). Uracil-containing duplex in the absence of UDG was also used as a control.
As anticipated, the AP-Red and THF DNA are not susceptible to β-elimination and no strand cleavage is observed when the DNA is dried, heated, treated with NaOH, or electrophoresed (Figure 2; AP-Red and THF Lanes 1–3). Treatment of both the AP-Red and THF DNA with APE1 leads to conversion to the APE1 product (Figure 2; AP-Red and THF Lanes 4).
Transient-State Kinetics of APE1: Dependence on Metal Ion
In order to determine kchemistry for APE1 incising the AP, AP-Red, and THF DNA, transient-state kinetic time courses were performed in which the concentration of APE1 was 10-fold greater than the concentration of DNA. Mixing of the enzyme with each DNA substrate and rapid quenching of the reaction was achieved using an RQF instrument. Experiments were performed in the presence of 5 mM Mg2+, Mn2+, or Ni2+. This concentration of Mg2+ is biologically relevant, and we also observed maximal strand incision at this concentration (Figure S3). For all three DNA substrates, in the presence of Mg2+, Mn2+, or Ni2+, we observe a rapid burst in product formation followed by a product plateau (Figure 3A–C). In the presence of the biologically-relevant ion Mg2+, APE1 incises all three DNA substrates very rapidly, and is faster than the resolution of the RQF with kchemistry ≥ 700 s−1 (Table 2). Initial attempts to slow strand incision by performing experiments at 4 °C, instead of 37 °C, were unsuccessful as kchemistry was still faster than the resolution of the RQF (data not shown). However, in the presence of Mn2+ or Ni2+, kchemistry is slowed for all three substrates and is within the resolution of the instrument; this decrease in kchemistry can be observed qualitatively in Figure 3 (inset) as more points in the burst region of the graph for Mn2+ or Ni2+, relative to Mg2+. Furthermore, in the presence of Mn2+ or Ni2+, kchemistry for the THF DNA is ~1.5 times slower than AP DNA and AP-Red DNA.
FIGURE 3.
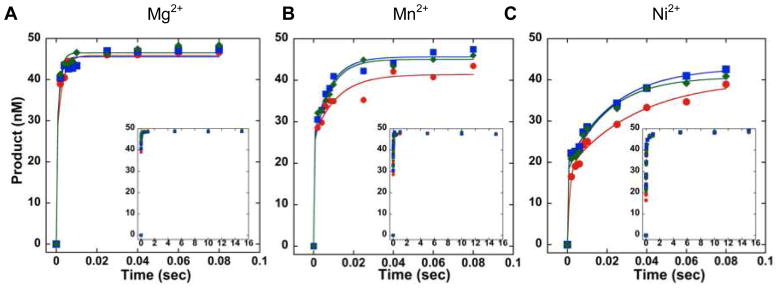
Transient-state kinetic time courses of strand incision activity of APE1 acting on AP (blue), AP-Red (green) or THF (red) DNA in the presence of 5 mM (A) Mg2+, (B) Mn2+, (C) or Ni2+. Inset depicts the full time course. Experimental conditions were 50 nM DNA, 500 nM APE1, 50 mM HEPES-KOH, 100 mM KCl, 0.12 mg/mL BSA, 0.25 mM EDTA, and 5 mM MgCl2, MnCl2, or NiCl2, pH 7.5. Reactions were performed at 37 °C using a RQF instrument and quenched with 100 mM EDTA.
Table 2.
| kchemistry (s−1) | |||
|---|---|---|---|
|
| |||
| Substrate | Mg2+ | Mn2+ | Ni2+ |
| AP | ≥700 | 329 ± 16 | 155 ± 17 |
| AP-Red | ≥700 | 325 ± 30 | 167 ± 11 |
| THF | ≥700 | 234 ± 46c | 111 ± 18d |
Measured at 37 °C under transient-state conditions using RQF instrumentation.
Error represents the standard deviation from at least four experiments.
p=0.008 when compared to AP DNA using student’s T test.
p=0.01 when compared to AP DNA using student’s T test.
Transient-State Kinetics of APE1: Dependence on 5′ Mismatch
We also determined the influence on kchemistry of mismatches placed 1, 2, 3, or 4 base pairs (bp) from the 5′ side of the AP, AP-Red, or THF site (Table 1). These experiments were performed in the presence of 5 mM Mg2+. In all cases we observe a rapid burst in product formation followed by a product plateau (Figure 4A-D). We observed the slowest value of kchemistry when the mismatch is adjacent to the lesion, and observe a recovery in kchemistry as the mismatch is moved away from the lesion site (Table 3). Indeed, for AP DNA and AP-Red DNA, when the mismatch is 3 bp away, kchemistry is ≥ 700 s−1 and is again faster than the resolution of the RQF. For THF DNA, the mismatch must be 4 bp away before the resolution of the RQF is exceeded. Furthermore, in the presence of a 5′ mismatch 1, or 2 bp away from the lesion, kchemistry for the THF DNA is ~1.5 times slower than AP DNA and AP-Red DNA. Experiments were also performed in which an A•A mismatch was placed immediately to the 3′ side of each lesion; kchemistry was faster than the resolution of the RQF for all three lesions (Figure S4).
FIGURE 4.
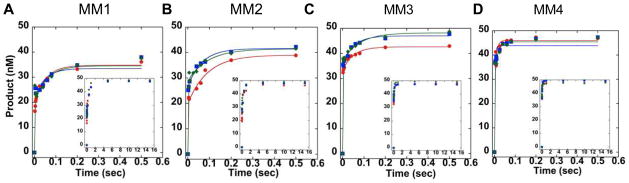
Transient-state kinetic time courses of strand incision activity of APE1 acting on AP (blue), AP-Red (green), or THF (red) DNA containing a mismatch (A) 1, (B) 2, (C) 3, or (D) 4 bp on the 5′ side of the lesion. Inset depicts the full time course. Experimental conditions were 50 nM DNA, 500 nM APE1, 50 mM HEPES-KOH, 100 mM KCl, 0.12 mg/mL BSA, 0.25 mM EDTA, and 5 mM MgCl2, pH 7.5. Reactions were performed at 37 °C using a RQF instrument and quenched with 100 mM EDTA.
Table 3.
| kchemistry (s−1)a,b | ||||
|---|---|---|---|---|
|
| ||||
| Substrate | MM-1 | MM-2 | MM-3 | MM-4 |
| AP | 156 ± 29 | 312 ± 29 | ≥700 | ≥700 |
| AP-Red | 163 ± 21 | 307 ± 24 | ≥700 | ≥700 |
| THF | 116 ± 29c | 194 ± 32d | 575 ± 65 | ≥700 |
Measured at 37 °C under transient-state conditions using RQF instrumentation.
Error represents the standard deviation from at least four experiments.
p=0.04 when compared to AP DNA using student’s T test.
p=0.008 when compared to AP DNA using student’s T test.
Steady-State Kinetics of APE1
In order to compare the steady-state rate (kss) of APE1 acting on authentic AP, AP-Red, and THF DNA, we performed steady-state kinetic time courses in which the concentration of DNA was 10 times greater than the concentration of APE1. As with the transient-state experiments, we used the RQF instrument to rapidly mix and quench each reaction. As seen in Figure 5, for each time course we observe an initial burst of product formation followed by a linear accumulation of product. The initial burst represents the fast chemistry step that leads to the initial turnover of product. The linear phase signifies a slow step that occurs after chemistry, likely product release, and defines the steady-state rate. The steady-state rates for each lesion, 2.2 ± 0.3 s−1, 2.4 ± 0.3 s−1, and 2.4 ± 0.2 s−1, for AP, AP-Red and THF, respectively, are the same within error.
FIGURE 5.

Steady-state kinetic time courses of APE1 acting on an AP (blue), AP-Red (green) or THF (red) site in the presence of 5 mM MgCl2. Experimental conditions were 500 nM DNA and 50 nM APE1 in 50 mM HEPES-KOH, 100 mM KCl, 0.24 mg/mL BSA, 0.25 mM EDTA 5 mM MgCl2, pH 7.5. Reactions were performed at 37 °C using a RQF instrument and quenched with 100 mM EDTA.
DISCUSSION
Steady-State and Transient-State Kinetics of APE1 in the Presence of Mg2+
We report the first RQF-derived value of kchemistry for APE1 incising an authentic AP site, ≥ 700 s−1, making APE1 one of the fastest BER enzymes known to date. Indeed, in the presence of Mg2+, strand incision by APE1 is very rapid and kchemistry is faster than the resolution of the RQF for AP, AP-Red, and THF DNA. Our results are consistent with the only other APE1 transient-state experiments performed using RQF instrumentation in which kchemistry for THF DNA is limited by the resolution of the instrument (24). Also consistent with literature reports, our steady-state rates for APE1 acting on AP-Red and THF analogs are indistinguishable from the authentic AP DNA (16, 19–25). It has previously been suggested that the high copy number of APE1 (350,000–7,000,000 per cell) may serve to counteract the slow steady-state rate (7).
In other experiments performed using SFF, kchemistry of 68 s−1 and 97 s−1 have been reported for APE1 processing a THF and authentic AP site, respectively (20). Notably at ≥700 s−1, the rates we obtain are significantly faster; however, SFF experiments do not directly measure strand incision, but rather monitor conformational changes of the enzyme during processing of the DNA substrate, and may underestimate the rate.
Transient-State Kinetics of APE1 in the Presence of Mn2+ or Ni2+
A divalent metal ion is required for APE1 strand incision (29–32). Examination of x-ray crystal structures of APE1 bound to the incised product of THF-containing DNA, and the results of several site-directed mutagenesis studies have led to the proposal that the divalent metal ion coordinates to an oxygen of the electrophilic phosphate, as well as residues in the APE1 active site pocket, such as E96 (29–32). Via these interactions the metal ion is thought to stabilize and correctly orient the DNA backbone for strand incision, as well as polarize the P-O bond to facilitate nucleophilic attack of an activated water molecule. A previous study reported a qualitative comparison of APE1 activity in the presence of several metal ions, including Mg2+, Mn2+, and Ni2+(29); activity was greatest with Mg2+, and reduced with Mn2+ or Ni2+. Indeed, depending on the lesion, kchemistry was reduced at least 2–3 fold in the presence of Mn2+, and at least 4–6.5 fold in the presence of Ni2+. This reduction of strand incision chemistry is likely due to slightly altered positioning of the larger Mn2+ or Ni2+ ions in the active site, and an altered orientation of the DNA backbone. However, we cannot rule out the possibility that different metal ions may change the mechanism of strand incision all together. Nevertheless, by slowing strand incision and obtaining quantitative values, we reveal that in the presence of Mn2+ or Ni2+, kchemistry for AP DNA and AP-Red DNA are indistinguishable. Furthermore, albeit a small difference, the rate of strand incision is statistically different for THF DNA where kchemistry is ~1.5 times slower than the authentic AP DNA.
Transient-State Kinetics of APE1 in the Presence of 5′ Mismatches
While varying the metal ion allowed us to obtain quantitative values for kchemistry, and to compare directly APE1 strand incision on the authentic AP site versus the substrate analogs, we considered the possibility that changing the metal ion also changed the mechanism of APE1; if this were the case, the observed difference between THF and the authentic AP site may not be biologically relevant. Thus, we sought an alternate method to slow strand incision. Previous work has qualitatively shown that activity of APE1 on THF-containing DNA is reduced when a mismatch is directly 5′ or 3′ to the lesion site, with the 5′ mismatch having a more dramatic effect (22). Similar results were also observed for the removal of an 8-oxo-7,8-dihydroguanine lesion by oxoguanine glycosylase 1, another BER enzyme; the presence of a 5′ mismatch was more detrimental to activity than a 3′ mismatch (33). Not surprisingly, when we positioned a mismatch 1, 2, 3, or 4 bp from the lesion, we saw the greatest reduction in kchemistry, at least ~5-fold, when the mismatch was immediately adjacent to the lesion. As the mismatch moved away from the lesion, kchemistry recovered to values comparable to those obtained for the well-matched substrates. The decreased rate of strand incision in the presence of the 5′ mismatches is likely due to distortion of the DNA backbone within the APE1 active site, in particular, distortion of the electrophilic phosphate 5′ to the lesion site; as the mismatch is moved further from the lesion, the effect is diminished. Interestingly, similar results have been established for the repair of bistranded authentic AP sites by APE1. Duplex substrates contained two AP sites on opposite strands, positioned 1, 3, or 5 bp apart, and strand incision was monitored at one of the AP sites. A reduction in incision by APE1 was observed when the second AP site was 1 or 3 bp away; conversely, when the second AP site was 5 bp away, no reduction in incision was observed (34). Although the identity of the mismatch varies in different locations (i.e., MM-1 contains a T•T mismatch whereas MM-2 has a C•C mismatch), the purpose of these experiments is not to compare the effect of mismatches at different locations, but rather to compare kchemistry obtained for AP, AP-Red, and THF when the mismatch is the same distance from the lesion. Indeed, consistent with the results obtained in the presence of Mn2+ or Ni2+, a ~1.5-fold reduction in kchemistry is observed for THF DNA relative to AP and AP-Red DNA when the mismatch is 1 or 2 bp away.
Importance and Function of C1′ Oxygen
Transient-state kinetic experiments revealed a small yet significant decrease in kchemistry for APE1 incising THF DNA compared to substrates containing an authentic AP or reduced AP site. This decrease was apparent when experiments were conducted in the presence of Mn2+ or Ni2+, or when a 5′ mismatch was 1 or 2 bp from the lesion. It is important to note, for experiments performed in the presence of Mg2+, differences in the way that APE1 may process the THF lesion are likely masked by the limitation of the RQF instrument. A feature distinguishing the THF site from the authentic and reduced AP site is the lack of a C1′ oxygen. Since strand incision is observed for THF DNA, this oxygen is not required for catalytic activity; however, its presence (or the presence of a C1′-OH) may play a role in stabilizing and optimally orienting the authentic AP and AP-Red substrates in the APE1 active site pocket. An extensive hydrogen bonding network is present in active site, and contributes to formation of the activated water molecule which serves as the nucleophile for the strand incision chemistry (35, 36). Interestingly, examination of several x-ray crystal structures of APE1 bound to THF DNA reveal that the primary amine of N212 would be within hydrogen-bonding distance of a C1′ oxygen (17). However, we cannot be certain that the APE1 binding pocket would be the same when an authentic AP substrate is bound. While the role of N212 remains unclear, site-directed mutagenesis studies using authentic AP DNA suggest this residue is important for substrate recognition; the role of N212 on THF containing DNA has not been explored (37). A THF lesion would lack the ability to form a hydrogen bond at the C1′ position, and may be oriented slightly differently within the active site, explaining the small reduction in kchemistry. It is noteworthy that previous APE1 kinetic studies also observed differences in the processing of THF DNA compared to authentic AP DNA; such studies used SFF techniques which detected conformational changes in APE1 (21). When observing directly strand incision we obtain similar results indicating that the C1′ oxygen directly influences strand incision.
Due to the wide spectrum of APE1 activity, as well as its cellular importance, it is not surprising that APE1 is a widely-studied enzyme. Here within, by examining the kinetics of APE1 on an authentic AP site and commonly-used analogs, we highlight the destabilizing effects of different metal ions and 5′ mismatches on APE1 stand incision chemistry. These kinetic-based experiments suggest a role for the C1′ oxygen of the AP site for optimal strand incision, and reveal that with respect to strand incision, the AP-Red analog cannot be distinguished from the authentic substrates, whereas the THF analog is processed slightly differently. This work calls to attention the importance of choosing substrate analogs, and understanding the implications such analogs can have on experimental results.
Supplementary Material
Acknowledgments
We are grateful to Dr. Samuel H. Wilson (NIH/NIEHS) for providing the expression plasmid for APE1, Dr. Wayne Chou and Prof. David Cane (Brown University) for assistance with protein expression and purification, and Katharina Bilotti for helpful discussions.
Abbreviations
- AP
apurinic/apyrimidinic
- APE1
apurinic/apyrimidinic endonuclease 1
- bp
base pair
- dRP
5′-deoxyribose phosphate
- RQF
rapid quench flow
- SFF
stop-flow fluorescence
- THF
tetrahydrofuran
- UDG
uracil DNA glycosylase
Footnotes
This work was supported by the National Institute of Environmental Health Sciences (R01ES019296).
SUPPORTING INFORMATION AVAILABLE
Autoradiogram revealing chelation of Mg2+ ions from APE1 preparation, autoradiogram revealing incision activity of APE1 on THF site with varying metal ions, transient-state kinetic time courses of strand incision activity of APE1 on authentic AP DNA in the presence of varying concentrations of Mg2+, and transient-state kinetic time courses of strand incision activity of APE1 acting on AP, AP-Red, or THF DNA containing a 3′ mismatch.
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 2.Wilson DM, III, Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mut Res/DNA Repair. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 3.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Ann Rev Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 4.Schaaper RM, Kunkel TA, Loeb LA. Infidelity of DNA synthesis associated with bypass of apurinic sites. Proc Natl Acad Sci USA. 1983;80:487–491. doi: 10.1073/pnas.80.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demple B, Harrison L. Repair of Oxidative Damage to DNA: Enzymology and Biology. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 6.Seki S, Hatsushika M, Watanabe S, Akiyama K, Nagao K, Tsutsui K. cDNA cloning, sequencing, expression and possible domain structure of human APEX nuclease homologous to Escherichia coli exonuclease III. Biochim Biophys Acta. 1992;1131:287–299. doi: 10.1016/0167-4781(92)90027-w. [DOI] [PubMed] [Google Scholar]
- 7.Chen DS, Herman T, Demple B. Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res. 1991;19:5907–5914. doi: 10.1093/nar/19.21.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vidal AE, Hickson ID, Boiteux S, Radicella JP. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: bypass of the AP lyase activity step. Nucleic Acids Res. 2001;29:1285–1292. doi: 10.1093/nar/29.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitzgerald ME, Drohat AC. Coordinating the Initial Steps of Base Excision Repair: Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J Biol Chem. 2008;283:32680–32690. doi: 10.1074/jbc.M805504200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldwin MR, O’Brien PJ. Human AP endonuclease 1 stimulates multiple-turnover base excision by alkyladenine DNA glycosylase. Biochemistry. 2009;48:6022–6033. doi: 10.1021/bi900517y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldwin MR, O’Brien PJ. Nonspecific DNA binding and coordination of the first two steps of base excision repair. Biochemistry. 2010;49:7879–7891. doi: 10.1021/bi100889r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett RA, Wilson DM, Wong D, Demple B. Interaction of human apurinic endonuclease and DNA polymerase beta in the base excision repair pathway. Proc Natl Acad Sci USA. 1997;94:7166–7169. doi: 10.1073/pnas.94.14.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sukhanova MV, Khodyreva SN, Lebedeva NA, Prasad R, Wilson SH, Lavrik OI. Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase beta and poly(ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 2005;33:1222–1229. doi: 10.1093/nar/gki266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strauss PR, Beard WA, Patterson TA, Wilson SH. Substrate Binding by Human Apurinic/Apyrimidinic Endonuclease Indicates a Briggs-Haldane Mechanism. J Biol Chem. 1997;272:1302–1307. doi: 10.1074/jbc.272.2.1302. [DOI] [PubMed] [Google Scholar]
- 17.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 18.Tsutakawa SE, Shin DS, Mol CD, Izumi T. Conserved Structural Chemistry for Incision Activity in Structurally Non-homologous Apurinic/Apyrimidinic Endonuclease APE1 and Endonuclease IV DNA repair. J Biol Chem. 2013;288:8445–8455. doi: 10.1074/jbc.M112.422774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu YJ, DeMott MS, Hwang JT, Greenberg MM, Demple B. Action of human apurinic endonuclease (Ape1) on C1′-oxidized deoxyribose damage in DNA. DNA Repair (Amst ) 2003;2:175–185. doi: 10.1016/s1568-7864(02)00194-5. [DOI] [PubMed] [Google Scholar]
- 20.Timofeyeva NA, Koval VV, Knorre DG, Zharkov DO, Saparbaev MK, Ishchenko AA, Fedorova OS. Conformational dynamics of human AP endonuclease in base excision and nucleotide incision repair pathways. J Biomol Struct Dyn. 2009;26:637–652. doi: 10.1080/07391102.2009.10507278. [DOI] [PubMed] [Google Scholar]
- 21.Kanazhevskaya LY, Koval VV, Zharkov DO, Strauss PR, Fedorova OS. Conformational transitions in human AP endonuclease 1 and its active site mutant during abasic site repair. Biochemistry. 2010;49:6451–6461. doi: 10.1021/bi100769k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson DM, Takeshita M, Grollman AP, Demple B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J Biol Chem. 1995;270:16002–16007. doi: 10.1074/jbc.270.27.16002. [DOI] [PubMed] [Google Scholar]
- 23.Mundle ST, Delaney JC, Essigmann JM, Strauss PR. Enzymatic mechanism of human apurinic/apyrimidinic endonuclease against a THF AP site model substrate. Biochemistry. 2009;48:19–26. doi: 10.1021/bi8016137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maher RL, Bloom LB. Pre-steady-state kinetic characterization of the AP endonuclease activity of human AP endonuclease 1. J Biol Chem. 2007;282:30577–30585. doi: 10.1074/jbc.M704341200. [DOI] [PubMed] [Google Scholar]
- 25.Lucas JA, Masuda Y, Bennett RAO, Strauss NS, Strauss PR. Single-Turnover Analysis of Mutant Human Apurinic/Apyrimidinic Endonuclease. Biochemistry. 1999;38:4958–4964. doi: 10.1021/bi982052v. [DOI] [PubMed] [Google Scholar]
- 26.Jarem DA, Wilson NR, Delaney S. Structure-dependent DNA damage and repair in a trinucleotide repeat sequence. Biochemistry. 2009;48:6655–6663. doi: 10.1021/bi9007403. [DOI] [PubMed] [Google Scholar]
- 27.Warshaw MM, Tinoco I. Optical properties of sixteen dinucleoside phosphates. J Mol Biol. 1966;20:29–38. doi: 10.1016/0022-2836(66)90115-x. [DOI] [PubMed] [Google Scholar]
- 28.Leipold MD, Workman H, Muller JG, Burrows CJ, David SS. Recognition and removal of oxidized guanines in duplex DNA by the base excision repair enzymes hOGG1, yOGG1, and yOGG2. Biochemistry. 2003;42:11373–11381. doi: 10.1021/bi034951b. [DOI] [PubMed] [Google Scholar]
- 29.Barzilay G, Mol CD, Robson CN, Walker LJ, Cunningham RP, Tainer JA, Hickson ID. Identification of critical active-site residues in the multifunctional human DNA repair enzyme HAP1. Nat Struct Biol. 1995;2:561–568. doi: 10.1038/nsb0795-561. [DOI] [PubMed] [Google Scholar]
- 30.Kane CM, Linn S. Purification and characterization of an apurinic/apyrimidinic endonuclease from HeLa cells. J Biol Chem. 1981;256:3405–3414. [PubMed] [Google Scholar]
- 31.Masuda Y, Bennett RA, Demple B. Rapid dissociation of human apurinic endonuclease (Ape1) from incised DNA induced by magnesium. J Biol Chem. 1998;273:30360–30365. doi: 10.1074/jbc.273.46.30360. [DOI] [PubMed] [Google Scholar]
- 32.Erzberger JP, Wilson DM. The role of Mg2+ and specific amino acid residues in the catalytic reaction of the major human abasic endonuclease: new insights from EDTA-resistant incision of acyclic abasic site analogs and site-directed mutagenesis. J Mol Biol. 1999;290:447–457. doi: 10.1006/jmbi.1999.2888. [DOI] [PubMed] [Google Scholar]
- 33.Sassa A, Beard WA, Prasad R, Wilson SH. DNA sequence context effects on the glycosylase activity of human 8-oxoguanine DNA glycosylase. J Biol Chem. 2012;287:36702–36710. doi: 10.1074/jbc.M112.397786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaudhry MA, Weinfeld M. Reactivity of human apurinic/apyrimidinic endonuclease and Escherichia coli exonuclease III with bistranded abasic sites in DNA. J Biol Chem. 1997;272:15650–15655. doi: 10.1074/jbc.272.25.15650. [DOI] [PubMed] [Google Scholar]
- 35.Gorman MA. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997;16:6548–6558. doi: 10.1093/emboj/16.21.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothwell DG, Hang B, Gorman MA. Substitution of Asp-210 in HAP1 (APE/Ref-1) eliminates endonuclease activity but stabilizes substrate binding. Nucleic Acids Res. 2000;28:2202–2213. doi: 10.1093/nar/28.11.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothwell DG, Hickson ID. Asparagine 212 is essential for abasic site recognition by the human DNA repair endonuclease HAP1. Nucleic Acids Res. 1996;24:4217–4221. doi: 10.1093/nar/24.21.4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.