

Graphical abstract
Abbreviations: BSF, bloodstream form; EbS, 2-phenyl-12-benzisothiazol-3(2H)-one; EbSe, ebselen (2-phenyl-12-benzisoselenazol-3(2H)-one); G6-P, glucose-6-phosphate; G6PDH, glucose-6-phosphate dehydrogenase; Gly3P, glycerol-3-phosphate; GK, glycerol kinase; HK, hexokinase; PF, procyclic form; rTbHK1, recombinant Trypanosoma brucei hexokinase 1; TbHK, T. brucei hexokinase
Keywords: Ebselen, Hexokinase, Inhibitors, Trypanosoma brucei
Highlights
-
•
Trypanosoma brucei hexokinase 1 is irreversibly inhibited by ebselen.
-
•
Mutation of Cys residues did not change hexamer abundance.
-
•
Active variants bearing Cys mutations were inhibited by ebselen.
-
•
ESI–MS/MS indicated that the essential Cys327 was oxidized by ebselen.
Abstract
Glycolysis is essential to Trypanosoma brucei, the causative agent of African sleeping sickness, suggesting enzymes in the pathway could be targets for drug development. Ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one, EbSe) was identified in a screen as a potent inhibitor of T. brucei hexokinase 1 (TbHK1), the first enzyme in the pathway. EbSe has a history of promiscuity as an enzyme inhibitor, inactivating proteins through seleno-sulfide conjugation with Cys residues. Indeed, dilution of TbHK1 and inhibitor following incubation did not temper inhibition suggesting conjugate formation. Using mass spectrometry to analyze EbSe-based modifications revealed that two Cys residues (C327 and C369) were oxidized after treatment. Site-directed mutagenesis of C327 led to enzyme inactivation indicating that C327 was essential for catalysis. C369 was not essential, suggesting that EbSe inhibition of TbHK1 was the consequence of modification of C327 via thiol oxidation. Additionally, neither EbSe treatment nor mutation of the nine TbHK1 Cys residues appreciably altered enzyme quaternary structure.
1. Introduction
Trypanosoma brucei is the causative agent of African sleeping sickness in humans and nagana in livestock, both of which have tremendous impact on the lives of people in sub-Saharan Africa (Brun et al., 2010). Bloodstream form (BSF) T. brucei, the lifecycle stage that grows rapidly in the blood of the mammalian host, depends solely on glycolysis for ATP production. As a consequence, glycolytic enzymes from the parasite have been considered as potential targets for therapeutic design.
The first enzyme activity in glycolysis, which transfers a phosphoryl group from ATP to glucose, is catalyzed by hexokinases (HK). T. brucei harbors two hexokinase genes, TbHK1 and TbHK2, and both gene products are essential to BSF parasite (Chambers et al., 2008a). The TbHKs, whether purified from parasites or heterologously expressed in Escherichia coli, oligomerize into hexamers (MIsset et al., 1986; Chambers et al., 2008b). Due to the 98% identity of TbHK1 and TbHK2, the contribution of both to hexamer formation in vivo has remained elusive. Nonetheless, recombinant heterohexamers generated in vitro with known ratios of TbHK1 and TbHK2 have kinetic properties more similar to those reported for T. brucei-derived TbHK than recombinant TbHK homohexamers (Chambers et al., 2008b), suggesting that in the parasite oligomers are most likely heterohexamers. Notably, the composition of the heterohexamers in vivo is regulated in response to the nutritional environment in which the cells are cultured. The mechanisms behind this dynamic hexamerization, including the enzyme protein domains that participate in oligomerization, remain to be elucidated.
TbHK1 has previously been genetically and chemically validated as a potential target for therapeutic design. Further, the enzyme has been the subject of both structure-based approaches and high throughput screening (HTS) campaigns to identify compounds with potential as leads in therapeutic development. The HTS campaign included the screening of 220,233 compounds for inhibitors of TbHK1 (Sharlow et al., 2010a,b). From this effort ten inhibitors, including six structurally related isobenzothiazolinone inhibitors have been identified. Ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one, EbSe, Fig. 1A), a selenium containing variant of isobenzothiazolinones, was the most potent TbHK1 inhibitor found in the HTS campaign, with an IC50 = 0.05 ± 0.03 μM. Notably, 2-phenyl-1,2-benzisothiazol-3(2H)-one (SID 17387000, EbS, Fig. 1A), which differs from EbSe by replacement of the selenium atom with sulfur, was also identified in the HTS as a potent TbHK1 inhibitor (IC50 = 2.0 ± 0.5 μM).
Fig. 1.
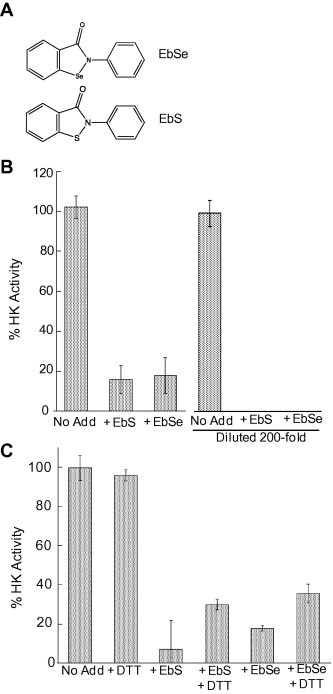
EbSe, a known Cys-reactive compound, inhibits TbHK1 activity. (A) Structures of ebselen (EbSe) and ebsulfur (EbS), SID 17387000. (B) EbS and EbSe inhibition are irreversible by dilution. TbHK1 (32 ng) was incubated with EbSe or EbS in the assay for 15 min. Alternatively, inhibitor was incubated with enzyme prior to addition of other assay components, which yielded a 200-fold dilution of enzyme and inhibitor. (C) DTT can block but not reverse TbHK1 inhibition by EbSe. TbHK1 (32 ng) was incubated with EbS or EbSe (hatched bars) followed by the addition of DTT (100 mM) prior to assay. Experiments were performed in triplicate and standard deviation is indicated.
EbSe is known to form seleno-sulfide adducts with target protein Cys residues. For example, EbSe inhibited human indoleamine 2,3-dioxygenase (IDO) through covalent modification of multiple IDO Cys residues, causing a change in enzyme conformation and inactivation (Terentis et al., 2010). This EbSe-based oxidization of critical Cys residues can also result in the generation of inappropriate disulfide linkages (Sakurai et al., 2006). Here we explore the role Cys residues have in EbSe-based TbHK1 inhibition. Through these efforts, we have found that EbSe oxidizes a single critical Cys residue, rather than promiscuously modifying Cys residues. Additionally, we have characterized the role of TbHK1 Cys residues in enzyme activity and the impact of their modification on oligomerization, finding that several of the Cys residues are essential for catalysis and can influence hexamerization.
2. Materials and methods
2.1. Reagents
Tris (2-carboxyethyl)phosphine (TCEP), glucose-6-phosphate dehydrogenase, β-nicotinamide adenine dinucleotide (NAD+), adenosine triphosphate (ATP), and glucose were purchased from Sigma (St. Louis, MO). Dimethyl sulfoxide (DMSO) was purchased from Fisher Scientific (Pittsburgh, PA), while phosphoenol pyruvate (PEP), 2-phenyl-1,2-benzisoselenazol-3(2H)-one (ebselen, EbSe, PubChem SID 856002) and glucosamine were obtained from VWR International (West Chester, PA). 3-(N-maleimidopropionyl)-biocytin was purchased from Cayman Chemical (Ann Arbor, MI).
2.2. Recombinant enzyme purification and assay conditions
Recombinant TbHK1 was purified as described from a culture of E. coli M15(pREP) harboring pQE30 (Qiagen, Valencia, CA) with TbHK1 cloned in frame of a 6-His tagging sequence (Morris et al., 2006). Briefly, a 10 mL bacterial culture was used to inoculate a 1 L culture which was grown to an OD of ∼1 and then induced for 24 h at room temperature with 250 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) and purified as described (Morris et al., 2006).
TbHK1 Cys variants were generated using the parental pQE30 TbHK1 construct and a QuikChange II Site-Directed Mutagenesis Kit (Stratagene, LaJolla, CA). Primers used for the mutagenesis are listed in Supplementary Table S1 and generation of all variants was confirmed by sequencing. Please note that despite repeated efforts, C103A was not successfully generated. Protein expression and purification were performed as described above, with fractions from the purification probed by western blotting using an anti-RGS-His6 antibody (Qiagen, Valencia, CA) to identify those harboring the recombinant protein. All variants were at least 90% pure based on coomassie blue staining of proteins resolved by SDS–PAGE.
HK assays were performed in triplicate as described using a coupled reaction to measure enzyme activity (Misset and Opperdoes, 1984; Morris et al., 2006). In short, the coupled assay uses glucose-6-phosphate dehydrogenase (G6PDH) to convert glucose-6-phosphate (G6-P) generated by HK to 6-phosphogluconate with coincident reduction of NADP to NADPH, which is monitored spectrophotometrically at 340 nm. Note that EbSe was found to be ineffective in a counterscreen for inhibition of G6PDH. Kinetic analyses were performed using KaleidaGraph 4.1 (Synergy Software, Reading, PA).
2.3. Mass spectrometry analysis of EbSe-treated TbHK1
Mass spectrometry was performed to map EbSe modification on TbHK1. First, solution samples were C4 zip tipped (Millipore) following the manufacturers protocol. The desalted protein was dried down and re-suspended in 100 ng of trypsin (Sigma, proteomics grade) and digested overnight at 37 °C. The digested sample was then placed in an auto sampler vial for LC–MS/MS analysis.
Enzymatically digested samples were analyzed via liquid chromatography (LC)–electrospray ionization (ESI)–tandem mass spectrometry (MS/MS) on an Orbitrap Elite mass spectrometer (Thermo) coupled to a Dionex 3000 nano LC system. A 20 cm 75 micron C-18 reversed phase LC column (packed in house, with Waters ODS C18) was utilized with a 200 nL/min flow rate and a 120 min gradient from 2% acetonitrile, 0.2% formic acid to 50% acetonitrile, 0.2% formic acid. MS data were acquired in a data-dependent strategy selecting the fragmentation events based on the precursor abundance in the survey scan (400–1700 Th). The resolution of the survey scan was 60,000 at m/z 400 Th with a target value of 1e6 ions and 1 microscan. Low resolution CID MS/MS spectra were acquired on the top 20 ions, with a target value of 1000 ions in normal CID scan mode. MS/MS acquisition in the linear ion trap was partially carried out in parallel to the survey scan in the Orbitrap analyzer by using the preview mode (first 192 ms of the MS transient). The maximum injection time for MS/MS was 100 ms. Dynamic exclusion was 120 s and early expiration was enabled. The isolation window for MS/MS fragmentation was set to 2 Th.
MS/MS data was searched against a custom database containing the users provided sequence using the searching algorithm node Sequest HT in Proteome Discoverer 1.4 (Thermo). Variable modifications of oxidation on methionine and Cys, di-oxidation and tri-oxidation of Cys, the addition of EbSe to cysteine, the substitution of S with Se, and the conversion of Cys to dehydro-Ala were all considered.
2.4. Native gel analysis and negative stain transmission electron microscopy
For native gels, protein samples were diluted in native gel loading buffer (10% (v/v) glycerol, 2.7 mM Tris–HCL, pH 6.8, 0.1% bromphenol blue) and resolved on a 4% polyacrylamide gel (4% bis acrylamide, 375 mM Tris–Cl, pH 8.8, 0.05% (v/v) TEMED, and 0.05% ammonium persulfate) using a Tris/glycine buffer (2.7 mM Tris–HCl and 192 mM glycine, pH 6.9) (Chambers et al., 2008b). Proteins were detected by silver staining.
For electron microscopy, samples were prepared by placing a drop of protein in solution on a formvar coated copper TEM grid held in anti-capillary forceps. The protein was allowed to settle to the grid surface for 60 s and excess liquid wicked away with a filter paper. The grids were then stained with a drop of aqueous 2% uranyl acetate for 30 s after which the excess stain was wicked away. Images of the negatively stained protein particles were collected using a JEOL JEM 1200 EX transmission electron microscopy equipped with a Gatan Orius 830 camera. The images were prepared for publication using Adobe Photoshop.
2.5. 3-(N-Maleimidopropionyl)-biocytin (MPB) assays
TbHK1 and variants (equal amounts as determined by coomassie staining of an SDS–PAGE gel) were incubated (30 min, RT) with MPB (50 μM) in buffer (20 mM Na2HPO4, 5 mM glucose, 0.4 M (NH4)SO4), resolved by 4% native gel electrophoresis, and monomers analyzed following transfer to nitrocellulose by western blotting using an anti-biotin antibody (1:10,000; Cell Signaling Technology Danvers, MA). Relative darkness was quantitated using ImageJ software. To score the consequences of inhibitor on MPB labeling, TbHK1 was incubated with increasing concentrations of inhibitor (15 min, RT) followed by addition of MPB (50 μM), and the mixture incubated for an additional 30 min at RT.
3. Results
In two independent screens for TbHK1 inhibitors, we identified EbSe (2-phenyl-1,2-benzisoselenazol-3(2H)-one) as a potent TbHK1 inhibitor. In the first, a LOPAC screen, EbSe inhibited TbHK1 88.1 ± 0.6% at 10 μM. Also, EbSe and five other structurally related isobenzothiazolinones were identified as TbHK1 inhibitors during a high throughput screen (HTS) of 220,233 small molecules (Sharlow et al., 2010b). While EbSe was the most potent inhibitor identified (IC50 = 0.05 ± 0.03 μM), the finding that it can form covalent seleno-sulifide conjugates with Cys residues or act as a Cys oxidant suggested that it was not an ideal lead for further development. Supporting this supposition, EbSe had been identified as an inhibitor of a number of different enzymes in multiple validated HTS campaigns, possibly because of its Cys-reactive nature (Sharlow et al., 2010b).
Other Cys reactive TbHK1 inhibitors have also been identified. For example, the thiol-reactive reagent 4-chloromercuribenzoic acid was identified in the LOPAC screen as a potent inhibitor of TbHK1 (84.7 ± 0.4% inhibition at 10 μM) (Sharlow et al., 2010b). Additionally, the irreversible disulfide-reactive reagent tris (2-carboxyethyl)phosphine (TCEP) inhibited TbHK1 (IC50 = 6.6 ± 0.4 μM), supporting the possibility that a Cys residue (or multiple Cys residues) is important for catalysis.
3.1. Neither dilution, DTT, nor excess Mg2+ can reverse EbSe inhibition of TbHK1
Six isobenzothiazolinones were identified in the HTS as potent inhibitors of TbHK1, including EbS (SID 17387000), which is a structural analog of EbSe that harbors a S atom in place of the Se found in EbSe (Fig. 1). Inhibition by this compound or by EbSe was not reversible by 200-fold dilution of the inhibitor after pre-incubation with enzyme, suggesting a covalent modification of TbHK1 (Fig. 1B). Because EbSe inhibition of human IDO had been reported to be reversible by inclusion of the reducing reagent dithiothreitol (DTT) (Terentis et al., 2010), we assessed the consequences of this reagent on both EbS and EbSe inhibition (Fig. 1C). Alone, DTT (100 mM) had little impact on enzyme activity. Addition of DTT after either EbS or EbSe incubation only modestly rescued enzyme activity. While pre-incubation of TbHK1 with DTT prevented EbSe inhibition (data not shown), it is likely this occurred as a result of DTT interacting directly with EbSe to block association of the small molecule with the enzyme. A report describing the formation of DTT/EbSe adducts supports this possibility (Borges et al., 2005). Last, EbSe could be inhibitory to TbHK1 as a result of causing inappropriate coordination of the essential Mg2+ cofactor. Addition of excess Mg2+ did not relieve EbSe inhibition, even at ∼15-fold higher concentrations than are used in the standard reaction (data not shown).
3.2. MPB as a probe for exposed Cys interactions with inhibitors
To further explore the role of surface-exposed Cys residues in inhibitor binding, the Cys modifying reagent 3-(N-maleimidopropionyl)-biocytin (MPB) was used to modify surface-exposed Cys residues (Bayer et al., 1985). At concentrations up to 50 μM, MPB did not impact enzyme activity; however, pre-incubation with MPB did not appreciably alter EbSe inhibition, suggesting that the residues accessible to MPB were not involved in the enzyme inhibition or that EbSe could compete with the MPB modification or interact with free MPB.
3.3. Mass spectrometry to identify covalent modifications of TbHK1
Because the MPB experiments did not clarify the role of Cys residues in EbSe inhibition, we pursued scoring the direct consequences of EbSe treatment by ESI–MS/MS. This was performed keeping in mind that Cys residues have been described in the literature as the target of EbSe modification, either by formation of a selano-Cys bond between the enzyme and inhibitor or with the EbSe serving as a thiol oxidant.
Modifications of TbHK1 were scored using digested peptides from EbSe treated and untreated enzyme that were subjected to ESI–MS/MS (Supplemental Data set 1). Using this approach, species were identified with dioxidized (sulfone) and trioxidized (sulfonic acid) modified C103 in both untreated and treated samples. In contrast, dioxidized and trioxidized C327 and C369 modifications were identified only in the EbSe treated samples (Fig. 2). Peptides bearing oxidized C327 and C369 were not observed in the untreated samples, suggesting that they were either not present in the untreated sample or that the modified peptides were present but simply not detected in the untreated samples. Lastly, EbSe-conjugated through a seleno-sulfide bond to TbHK1 was not detected in the treated samples.
Fig. 2.
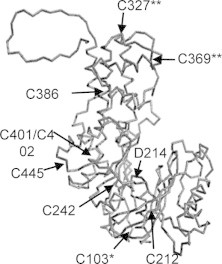
Two Cys residues on the large lobe of TbHK1 are modified in EbSe treated samples. The predicted distribution of TbHK1 Cys residues, based on modeling to the yeast structure, with the nine Cys residues and the catalytic base (D214) included to indicate the active site (Chambers et al., 2008c). The * indicates the Cys found by ESI–MS/MS to be oxidized in both untreated and EbSe-treated TbHK1, while the ** indicates the two oxidized Cys residues observed only in peptides from treated samples.
3.4. Mutagenesis of individual Cys residues in TbHK1
The observation that EbSe modified both C327 and C369 suggested that one or both were important for catalysis and/or EbSe inhibition. Further, it hinted at the possibility of generating EbSe-insensitive enzyme by alteration of the putative target residues. To explore the role of the nine TbHK1 Cys residues (Fig. 2) in catalysis and inhibition, variants harboring Cys to Ala changes of the residues were generated.
The consequences of these modifications on enzyme activity were varied. Three of the variants (C212A, C327A, and C386A) lacked detectable activity (Table 1), while three other variants (C369A, C401A, and C402A) had subtly reduced specific activities. The remaining two variants (C242A and C445A) had specific activities similar to the unaltered enzyme. EbSe and TCEP were then tested against the active variants. Both compounds were potent inhibitors of all of the active variants (including C369A), with no detectable difference in sensitivity compared to unmodified TbHK1 (Table 1).
Table 1.
Specific activity and sensitivity to of TbHK1 Cys variants.a
| Variant | Specific activity (mmol min−1 μg−1) | EbSe IC50 (μM) | TCEP IC50 (mM) |
|---|---|---|---|
| WT | 0.60 ± 0.03 | 0.35 ± 0.07 | 6.6 ± 0.4 |
| C103A | NAb | ||
| C103S | 0.17 ± 0.00 | <WTc | |
| C212A | NDd | ||
| C212S | 0.23 ± 0.01 | <WT | |
| C242A | 1.1 ± 0.12 | 0.55 ± 0.02 | 4.1 ± 0.3 |
| C327A | ND | ||
| C327S | ND | ||
| C369A | 0.24 ± 0.02 | 0.26 ± 0.02 | 7.5 ± 0.4 |
| C386A | ND | ||
| C386S | ND | ||
| C401A | 0.17 ± 0.03 | 0.29 ± 0.04 | 10 ± 0.7 |
| C402A | 0.23 ± 0.01 | 0.43 ± 0.11 | 7.2 ± 0.2 |
| C445A | 0.79 ± 0.01 | 0.52 ± 0.05 | 5.4 ± 0.1 |
Experiments were performed in triplicate and standard deviation is indicated.
NA, protein not expressed.
Inhibition was 100% using EbSe at a concentration that inhibited WT protein 50%.
Not determined, as activity was not detected.
In an effort to recover activity from the inactive Cys to Ala variants (C212A, C327A, and C386A), the three Cys residues were altered by mutation to a more conserved residue, Ser. The C212S variant had reduced yet detectable activity, while the other two yielded activity barely above the threshold of the variability in the assay. Notably, C212S remained sensitive to EbSe inhibition (Table 1).
3.5. Effects of Cys variants on oligomerization
To explore the impact of Cys residue alteration on the protein tertiary structure, native gels were used to resolve hexamer and monomer abundance, which were then characterized by silver staining and western blotting, respectively (Fig. 3A) (Chambers et al., 2008b). Hexamer abundance was not altered in most of the variants with C386A and C445A having ∼2-fold increases in oligomer abundance (Fig. 3A). Two TbHK1 variants (C327A and C369A) had slightly reduced levels of monomer (as determined by probing with the anti-six His antibody), while C445A had a ∼10-fold reduction in monomer abundance, suggesting this residue influences oligomerization.
Fig. 3.
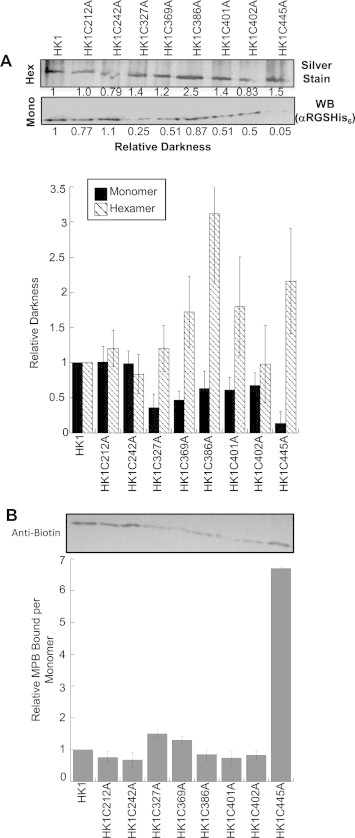
Relative hexamer, monomer, and free Cys abundance for TbHK1 and variants. (A.) Relative hexamer and monomer abundance of TbHK1 and Cys variants. Relative hexamer abundance (hatched bars) was determined by densitometry of appropriately sized bands visualized by silver staining of a 4% native gel followed by comparison to TbHK1 (lane 1). Relative monomer abundance (black bars) was determined by western blot analysis of the appropriate region of a 4% native gel using an anti-RGSH6 antibody and comparison to TbHK1. (B.) MPB modification of the monomer was determined by incubating variants with MPB (50 μM) for 30 min and monitoring the appropriate region of the gel by western blotting using an anti-biotin antibody. Experiments were performed in triplicate and standard deviation is indicated, with representative data included above each bar graph.
The proteins were also incubated with MPB and biotinylation of monomeric TbHK1 scored in order to further assess the consequences of Cys mutation on structure. Protein was resolved by native gel electrophoresis (4%), with monomer transferred to nitrocellulose followed by western blotting with antisera to the six His tag used for affinity purification to score relative protein concentration or an anti-biotin antibody to determine the relative level of biotinylation.
Using MPB to characterize surface exposed Cys residues in the TbHK1 variants, seven of the eight variants were labeled similarly to WT protein (Fig. 3B). However, C445A had an increased number of free Cys residues available for biotinylation by MPB per monomer as compared to TbHK1. This amino acid alteration may lead to an unstable monomer that, as a result of the modification, has many of its Cys residues exposed and available for MPB modification. Additionally, neither EbSe nor TCEP impact oligomerization of TbHK1 (data not shown).
3.6. Oligomers of C445A are indistinguishable from TbHK1 by electron microscopy
To date, the oligomers of TbHK1 have been refractory to structural studies due to an inability to generate useful crystals. However, EM analysis of recombinant protein suggests that enzyme oligomers are globular and roughly symmetrical, with an average diameter of 18.25 ± 0.19 nm (Fig. 4). The C445A variant, which has slightly greater abundance of hexamer in the native gel analysis (Fig. 3A), had a similar distribution of sizes (average diameter = 16.5 ± 2.8 nm), suggesting the mutation had relatively little impact on gross architecture. Notably, the overall structure of the hexamers suggests a ring-shaped tertiary structure.
Fig. 4.

Negative stain TEM analysis of globular TbHK1 hexamers. Samples of WT TbHK1 (upper) and C445A variant TbHK1 (lower) were analyzed using a JEOL JEM 1200 EX transmission electron microscope after negative staining. Inset contains enlargement of four representative protein particles. Scale bar = 100 nm.
4. Discussion
Currently there are a limited number of drugs available for the treatment of African trypanosomiasis. These agents have drawbacks – several are limited to treatment of infections before the parasite has crossed the blood–brain barrier, while others are active against one but not both human-infective subspecies of T. brucei. Target-based drug discovery offers the advantage of assessing drug/target interaction and allowing potential anticipation of possible resistance mechanisms.
TbHK1, which has been genetically and chemically validated as a suitable target for therapeutic development, has been the subject of several screening campaigns for small molecule inhibitors (Chambers et al., 2008a; Dodson et al., 2010; Sharlow et al., 2010b). The small molecule EbSe, the sulfur derivative EbS, and several structural analogs of EbS were identified in these campaigns, suggesting that the compounds could prove useful for probing the enzymology of the protein. Further analysis revealed that both EbSe and EbS were mixed inhibitors of TbHK1 (with respect to ATP) and both were toxic to parasites (Sharlow et al., 2010b). While the potency against recombinant enzyme and parasites was different (with IC50 values of 0.05 ± 0.03 and 2.0 ± 0.5 for EbSe and EbS compared to EC50 values of 2.9 ± 0.28 and 0.030 ± 0.067 for the two compounds, respectively), both were able to reduce parasite G6-P levels after acute exposure, suggesting that at least one possible in vivo targets was TbHK1 (Sharlow et al., 2010a,b). Recently, trypanothione reductase has been identified as a second potential target (Lu et al., in press). With both targets being essential to the parasite, it is difficult to resolve which (if either) are responsible for the toxic action of the molecules.
EbSe had previously been characterized as a mimic of glutathione peroxidase, utilizing reduced glutathione to reduce hydrogen peroxide and lipid hydroperoxides. Additionally, EbSe has also been identified as a potent electrophile, facilitating inter- and intramolecular disulfide linkages (Sakurai et al., 2006). For example, EbSe was found to have reacted with up to 8 Cys residues of human IDO, causing a change in conformation that led to IDO inactivation (Terentis et al., 2010). Here, we have pursued a further dissection of the mode of action of TbHK1 inhibition by this group of compounds with a particular focus on covalent Cys modification. The results of these efforts suggest that the inhibitory activity of EbSe against TbHK1 is likely the result of thiol oxidation of C327.
Initially, we found that TbHK1 inhibition by EbSe was not relieved by enzyme/inhibitor dilution, which supported the idea that the compound was indeed irreversibly modifying the enzyme. This was in stark contrast to inhibition of a HK from Plasmodium falciparum (PfHK), which we have recently characterized (Harris et al., 2013). While EbSe and EbS were ∼10-fold more potent inhibitors of PfHK, inhibition by both was completely relieved by dilution (Harris et al., 2013).
While the dilution experiments suggested that EbSe was irreversibly modifying TbHK1, the finding that EbSe inhibition was not reversible on treatment with DTT supported the notion that the inhibition was not due to the canonical promiscuous seleno-sulfide formation that has been observed previously with EbSe. This supposition was supported by the mass spectrometry analysis, which revealed oxidized Cys species including sulfone and sulfonic acid functionality at C103, C327, and C369, with the latter two sites of oxidations detected only in the EbSe treated samples. Mutational analysis revealed that C327 is essential while C369 could be altered without notable consequence on either catalysis or EbSe sensitivity, suggesting that inhibition of TbHK1 is a consequence of oxidation of the former Cys. Lacking a solved structure for either monomeric or heteromeric TbHK1, it is difficult to resolve the reasons for the observed inhibition as both C327 and C369 are predicted (based on modeling) to be on the large lobe of the enzyme at some distance from the active site.
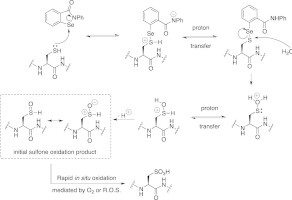
EbSe has been described as a thiol oxidant in other studies (Schewe, 1995; Sakurai et al., 2006) suggesting a model for TbHK1 thiol oxidation (Fig. 5). In the model, the Se atom of EbSe is attacked by the Cys SH followed by a proton transfer. The resultant S-Se conjugate is then hydrolyzed, with a subsequent proton transfer followed by deprotonation of the OH to yield the initial Cys sulfoxide. This functionality was not observed by MS/MS analysis, and is expected to undergo rapid in situ oxidation to the corresponding sulfone and sulfonic acids that were observed by MS. In principle, the initial conjugation with EbSe would be reversible prior to hydrolysis, accounting for the finding that promiscuous Cys oxidation was not observed. This mechanism would also potentially explain the reduced potency of EbS. EbS is structurally nearly-identical to EbSe but the Se-bearing molecule would likely be a more potent oxidizing agent.
Fig. 5.
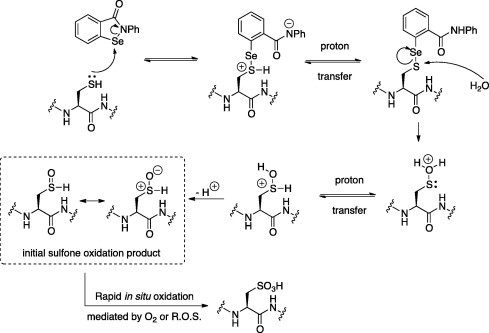
Potential scheme for EbSe-based oxidation of Cys residues in TbHK1.
Despite its potentially promiscuous nature, EbSe is non-toxic to humans and is currently deployed in phase III clinical trials for treatment of ischemic stroke (Stroke Trials Registry Home Page, as of 08/16/2013) and was found to improve the outcome of patients suffering stroke (Yamaguchi et al., 1998). These studies and others that indicate that EbSe has antibacterial properties as a consequence of inhibition of bacterial thioredoxin reductases (Lu et al., 2013) suggest that the benzisoselenazol derivatives could prove useful for therapeutic development.
Acknowledgements
This work was supported in part by the US National Institutes of Health 1R15AI075326 to JCM. We thank Jennifer Bethard (Department of Cell and Molecular Pharmacology, Mass Spectrometry Facility, Medical University of South Carolina) for the mass spectrometry analysis.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary data
LC-ESI-MS/MS analysis of untreated TbHK1. Desalted TbHK1 was digested with trypsin and subject to MS/MS. MS/MS data was searched against a custom database containing TbHK1 sequence using the searching algorithm node Sequest HT in Proteome Discoverer 1.4 (Thermo) to consider possible oxidation on Met and Cys, di-oxidation and tri-oxidation of Cys, the addition of EbSe to cysteine, the substitution of S with Se, and the conversion of Cys to dehydro-Ala.
LC-ESI-MS/MS analysis of EbSe TbHK1. Enzyme was treated with EbSe in solution, desalted, and analyzed as described in Fig. 1S.
Primers used for Cys variant production.
References
- Bayer E.A., Zalis M.G., Wilchek M. 3-(N-Maleimido-propionyl)biocytin: a versatile thiol-specific biotinylating reagent. Anal. Biochem. 1985;149:529–536. doi: 10.1016/0003-2697(85)90609-8. [DOI] [PubMed] [Google Scholar]
- Borges V.C., Rocha J.B., Nogueira C.W. Effect of diphenyl diselenide, diphenyl ditelluride and ebselen on cerebral Na(+), K(+)-ATPase activity in rats. Toxicology. 2005;215:191–197. doi: 10.1016/j.tox.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Brun R., Blum J., Chappuis F., Burri C. Human African trypanosomiasis. Lancet. 2010;375:148–159. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- Chambers J.W., Fowler M.L., Morris M.T., Morris J.C. The anti-trypanosomal agent lonidamine inhibits Trypanosoma brucei hexokinase 1. Mol. Biochem. Parasitol. 2008;158:202–207. doi: 10.1016/j.molbiopara.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Chambers J.W., Kearns M.T., Morris M.T., Morris J.C. Assembly of heterohexameric trypanosome hexokinases reveals that hexokinase 2 is a regulable enzyme. J. Biol. Chem. 2008;283:14963–14970. doi: 10.1074/jbc.M802124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J.W., Morris M.T., Smith K.S., Morris J.C. Residues in an ATP binding domain influence sugar binding in a trypanosome hexokinase. Biochem. Biophys. Res. Commun. 2008;365:420–425. doi: 10.1016/j.bbrc.2007.10.192. [DOI] [PubMed] [Google Scholar]
- Dodson H.C., Lyda T.A., Chambers J.W., Morris M.T., Christensen K.A., Morris J.C. Quercetin, a fluorescent bioflavanoid, inhibits Trypanosoma brucei hexokinase 1. Exp. Parasitol. 2010;127:423–428. doi: 10.1016/j.exppara.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M.T., Walker D.M., Drew M.E., Mitchell W.G., Dao K., Schroeder C.E., Flaherty D.P., Weiner W.S., Golden J.E., Morris J.C. Interrogating a hexokinase-selected small molecule library for inhibitors of Plasmodium falciparum hexokinase. Antimicrob. Agents Chemother. 2013;57:3717–3731. doi: 10.1128/AAC.00662-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Vlamis-Gardikas A., Kandasamy K., Zhao R., Gustafsson T.N., Engstrand L., Hoffner S., Engman L., Holmgren A. Inhibition of bacterial thioredoxin reductase: an antibiotic mechanism targeting bacteria lacking glutathione. FASEB J. 2013;27:1394–1403. doi: 10.1096/fj.12-223305. [DOI] [PubMed] [Google Scholar]
- Lu, J., Vodnala, S.K., Gustavsson, A.L., Gustafsson, T.N., Sjoberg, B., Johansson, H.A., Kumar, S., Tjernberg, A., Engman, L., Rottenberg, M.E., Holmgren, A., in press. Ebsulfur is a benzisothiazolone cytocidal inhibitor targeting the trypanothione reductase of Trypanosoma brucei. J. Biol. Chem. http://dx.doi.org/jbc.M113.495101 [DOI] [PMC free article] [PubMed]
- Misset O., Opperdoes F.R. Simultaneous purification of hexokinase, class-I fructose-bisphosphate aldolase, triosephosphate isomerase and phosphoglycerate kinase from Trypanosoma brucei. Eur. J. Biochem. 1984;144:475–483. doi: 10.1111/j.1432-1033.1984.tb08490.x. [DOI] [PubMed] [Google Scholar]
- Misset O., Bos O.J., Opperdoes F.R. Glycolytic enzymes of Trypanosoma brucei. Simultaneous purification, intraglycosomal concentrations and physical properties. Eur. J. Biochem. 1986;157:441–453. doi: 10.1111/j.1432-1033.1986.tb09687.x. [DOI] [PubMed] [Google Scholar]
- Morris M.T., DeBruin C., Yang Z., Chambers J.W., Smith K.S., Morris J.C. Activity of a second Trypanosoma brucei hexokinase is controlled by an 18-amino-acid C-terminal tail. Eukaryot. Cell. 2006;5:2014–2023. doi: 10.1128/EC.00146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T., Kanayama M., Shibata T., Itoh K., Kobayashi A., Yamamoto M., Uchida K. Ebselen, a seleno-organic antioxidant, as an electrophile. Chem. Res. Toxicol. 2006;19:1196–1204. doi: 10.1021/tx0601105. [DOI] [PubMed] [Google Scholar]
- Schewe T. Molecular actions of ebselen – an antiinflammatory antioxidant. Gen. Pharmacol. 1995;26:1153–1169. doi: 10.1016/0306-3623(95)00003-j. [DOI] [PubMed] [Google Scholar]
- Sharlow, E., Golden, J.E., Dodson, H., Morris, M., Hesser, M., Lyda, T., Leimgruber, S., Schroeder, C.E., Flaherty, D.P., Weiner, W.S., Simpson, D., Lazo, J.S., Aube, J., Morris, J.C., 2010a. Identification of Inhibitors of Trypanosoma brucei Hexokinases, Probe Reports from the NIH Molecular Libraries Program, Bethesda (MD). [PubMed]
- Sharlow E.R., Lyda T.A., Dodson H.C., Mustata G., Morris M.T., Leimgruber S.S., Lee K.H., Kashiwada Y., Close D., Lazo J.S., Morris J.C. A target-based high throughput screen yields Trypanosoma brucei hexokinase small molecule inhibitors with antiparasitic activity. PLoS Negl. Trop. Dis. 2010;4:e659. doi: 10.1371/journal.pntd.0000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentis A.C., Freewan M., Sempertegui Plaza T.S., Raftery M.J., Stocker R., Thomas S.R. The selenazal drug ebselen potently inhibits indoleamine 2,3-dioxygenase by targeting enzyme cysteine residues. Biochemistry. 2010;49:591–600. doi: 10.1021/bi901546e. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T., Sano K., Takakura K., Saito I., Shinohara Y., Asano T., Yasuhara H. Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Ebselen Study Group. Stroke. 1998;29:12–17. doi: 10.1161/01.str.29.1.12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
LC-ESI-MS/MS analysis of untreated TbHK1. Desalted TbHK1 was digested with trypsin and subject to MS/MS. MS/MS data was searched against a custom database containing TbHK1 sequence using the searching algorithm node Sequest HT in Proteome Discoverer 1.4 (Thermo) to consider possible oxidation on Met and Cys, di-oxidation and tri-oxidation of Cys, the addition of EbSe to cysteine, the substitution of S with Se, and the conversion of Cys to dehydro-Ala.
LC-ESI-MS/MS analysis of EbSe TbHK1. Enzyme was treated with EbSe in solution, desalted, and analyzed as described in Fig. 1S.
Primers used for Cys variant production.