

Abstract
Protein aggregation is believed to occur through the formation of misfolded conformations. It is expected that, in order to minimize aggregation, an effective small molecule chaperone would destabilize these intermediates. To study the mechanism of a chemical chaperone, we have designed a series of mutant proteins in which a tryptophan residue experiences different local environments and solvent exposures. We show that these mutants correspond to a series of conformationally altered proteins with varying degree of misfolding stress and aggregation propensities. Using arginine as a model small molecule, we show that a combination of unfolded state contraction and denaturant like properties results in selective targeting and destabilization of the partially folded proteins. In comparison, the effect of arginine towards the folded like control mutant, which is not aggregation prone, is significantly less. Other small molecules, lacking either of the above two properties, do not offer any specificity towards the misfolded proteins.
Protein misfolding has been implicated in different neurodegenerative diseases1. Nature is equipped with complex biological machines to counter the misfolding threats in the form of molecular chaperones, which repair and rescue conformationally aberrant proteins2,3. Recent progress in chemical biology and biotechnology has necessitated the search for possible small molecule alternatives of the molecular chaperones, which would inhibit or reverse protein aggregation4,5. In addition, the use of small molecule chaperones has attracted enormous interests for their potential pharmaceutical roles and applications in the manufacturing and formulation development of protein based drugs6. Since protein aggregation often occurs through the formation of partially folded intermediate states with exposed hydrophobic surface, an ideal small molecule chemical chaperone is expected to target them and minimize their populations.
In this paper, we hypothesize that; an efficient small molecule chemical chaperone may contain two properties: first, it should contract the unfolded state and second, it should also behave like a denaturant. The rational for the unfolded state contraction comes from the fact that the contracted unfolded state would favor the folded state. The requirement for the denaturant like properties comes from the necessity to unfold the misfolded intermediates providing them with more opportunities to fold correctly. Although it is controversial, it is often believed that a denaturant like guanidinium hydrochloride (Gdn.HCl) or urea binds to the native state affecting the side chains. To put it differently, this hypothesis assumes that the ideal small molecule should affect not only the backbone (the unfolded state), but also the side chain (the folded state).
The first property (unfolded state contraction) may be conveniently satisfied by a group of small molecules popularly known as protecting osmolytes7,8. These are typically used as protein stabilizers and include free amino acids (like glycine or proline), carbohydrates (like sucrose, sorbitol or trehalose) and methylamines (such as betaine and trimethyl amine N oxide or TMAO). It has been established that the protecting osmolytes preferentially destabilize the unfolded states (affecting protein backbone), and have significantly less effect on the native folded state (or the side chains)8,9,10. While these osmolytes are expected to contract the unfolded state11 favoring indirectly the folded state, they may not necessarily offer any specificity towards the aggregation prone partially folded intermediates. The second property (denaturant like) can be satisfied using any common denaturant, like Gdn.HCl or urea.
In a small screening experiment involving twenty small molecules (mostly protecting osmolytes), we have found an additive, arginine, to be the most successful in its ability to minimize aggregation of two different recombinant proteins. Arginine can be used as a convenient model small molecule to test the above hypothesis because it contains a glycine backbone (colored green in Figure 1a) and hence it may behave like a protecting osmolyte. In addition, the denaturant like property of arginine would arise from its guanidine side chain (colored red in Figure 1a). Interestingly, arginine has found tremendous applications in biotechnology because of its roles in increasing protein solubility and refolding yields12,13,14,15. The mechanism of arginine's action remains controversial. While it has been shown that arginine can follow protecting osmolyte like behavior16, it has also been shown to unfold proteins, albeit partially17. The dual roles of arginine has been suggested by previous reports18.
Figure 1.

(a) The molecular structure of arginine: The backbone resembles glycine, which is a known protecting osmolyte. The side chain of arginine is identical to gdn.HCl, which is a common denaturant. (b) The predicted structure of KMP-11: The positions of the inserted tryptophan residues are shown using black arrows. The predicted structure was obtained using a combination of threading, ab-initio modeling and atomic-level structure refinement methods of I-TASSER28,29 from Zhanglab server. The modeled structure was equilibrated in aqueous environment using a molecular dynamic simulation run of 100 nsec (see materials and methods, Supporting Information); (c) SDS page electrophores of the purified mutants (d) Acrylamide quenching experiments of the tryptophan mutants of KMP-11: The change in F0/F with acrylamide concentration is the minimum for the Y48W mutant (red line with filled circles). The change in F0/F with the acrylamide concentration is the maximum for the F77W mutant (black line with filled squares). The change in F0/F with acrylamide concentrations for other mutants lies between these two extremes (black lines with hollow symbols); (e) The linear correlation observed between the experimentally determined KSV (M−1) values and the calculated solvent accessible surface area (SASA) (%). Three independent measurements were used to calculate the error bars for Ksv.
In this paper, we aim to show that the efficiency of arginine as an aggregation inhibitor arises due to its ability to combine unfolded state contraction and its property to unfold aggregation prone partially folded proteins. To achieve that, we have prepared different mutant proteins (Table S1, Supporting Information) of varying aggregation propensities using a combination of computational biology, bioinformatics and tryptophan scanning mutagenesis. Tryptophan is used as the probe since its conformation and surface accessibility can be determined easily and conveniently using steady state fluorescence.
A small (92 amino acids) protein, Kinetoplastid Membrane Protein-11 or KMP-11, found in Leishmania parasites is used as the template for designing these mutants. The protein KMP-11 is being developed as a potential vaccine candidate against visceral leishmaniasis19. Another reason why this protein has been chosen is because it does not have any tryptophan residue in its native folded state and hence the tryptophan probe can be inserted at different regions conveniently. Moreover, KMP11 does not have any proline or cysteine residue ruling out complications of proline related misfolding and cysteine-cysteine bond formation. In addition, a key prediction has been verified using an additional protein system, namely, the intestinal fatty acid binding proteins (IFABP).
Contraction of the unfolded states of the mutant proteins in the presence of different additives including arginine has been studied by fluorescence correlation spectroscopy (FCS) at single molecule resolution. In contrast, the conformation and stability of these mutant proteins have been studied by a variety of ensemble biophysical techniques. Our results show that arginine while contracting the unfolded states also destabilizes the partially folded mutants selectively. This results in more efficient refolding kinetics and a large decrease in the aggregation propensity. For arginine, the extent of destabilization and the increase in refolding efficiency correlate well. Two small molecule controls, a protecting osmolyte without any denaturant like behavior (sucrose) and a denaturant lacking any protecting osmolyte like behavior (Gdn.HCl), do not offer any specificity towards the aggregation prone partially folded mutants. Sucrose, for example, offers a generic stabilization and the extent of sucrose-induced stabilization is similar for all the mutants irrespective of their tertiary structures. We show further that the relative amplitude of these properties (unfolded state contraction and denaturant like) present in arginine may vary depending on the nature of target proteins, solution conditions and other factors.
Results
The KMP-11 mutants offer similar secondary structures but different extent of tryptophan surface accessibilities, tertiary structures and exposed hydrophobic surfaces
Different single site mutants have been designed by computationally calculating the solvent accessible surface area (SASA) of a tryptophan residue placed at different positions of KMP-11 (Table S1, Supporting Information). More information about the structural modeling and detailed validation of the modeled structure has been provided (Materials and Methods Section and also as Figures S1–S3, Supporting Information).These mutations span almost the entire length of KMP-11 (Figure 1b). The assumptions behind the design of the mutations are that, first, the inserted tryptophan residue would experience different extents of surface accessibility; and second, these slight changes in the local environment would not lead to any significant change in the overall secondary structure. Instead, these variations would result in observable change in the tertiary interactions in the protein. Alternatively, the overall goal is to design a number of partially folded proteins of identical secondary structure, but with varying degrees of tertiary interaction, exposed hydrophobic surface and aggregation propensity.
To experimentally test the first assumption, the computationally designed mutants have been cloned, expressed and purified (Figure 1c and Table S2, Supporting Information). The values of solvent accessibility have been measured experimentally by determining the Stern-Volmer constants (KSV, Figure 1d). Since the acrylamide quenching results obtained using steady state fluorescence methods may have complications arising from the static components of fluorescence quenching, key quenching experiments are repeated using time resolved fluorescence spectroscopy (Figure S4, Supporting Information). The values of computationally determined solvent accessible surface area (SASA) of the inserted tryptophan residue correlate well with the experimentally observed KSV values (Figure 1e and Table S1, Supporting Information).
We have used a combination of CD, tryptophan fluorescence and ANS binding experiments to test the second assumption (Figure 2). Indeed, the overall secondary structures of KMP-11 mutants have been found similar (Figure 2a and Figure S5a, Supporting Information) while their tertiary structures differ. One of these mutants, Y48W, is found to have strong tertiary structure as judged by near-UV CD (Figure 2b and Figure S5b, Supporting Information). Steady state fluorescence spectroscopy suggests that the tryptophan residue in Y48W mutant is buried well inside the protein hydrophobic core (Figure 2c and Figure S5c, Supporting Information). Furthermore, Y48W does not efficiently bind ANS (Figure 2d). In a nutshell, therefore, Y48W represents a well folded protein with native like tertiary structure.
Figure 2.

(a) Far-UV CD (b) Near-UV CD and (c) Tryptophan fluorescence spectra of Y48W (black), F73W (red) and F77W (blue) mutants of KMP-11(d) Steady state fluorescence emission spectra of ANS in the presence of Y48W (black), F73W (red), and F77W (blue). Also see Figure S5, Supporting Information for these data for all the mutants. For all ANS experiments, an ANS solution of the same concentration in 20 mM sodium phosphate buffer at pH 7.4 has been used as a background and subtracted from the experimentally observed data. An excitation wavelength of 360 nm has been used for the ANS experiments. For the steady state tryptophan fluorescence experiments, a 20 mM sodium phosphate buffer at pH 7.4 has been used as the background.
In sharp contrast, F77W shows diminished near-UV CD (Figure 2b and Figure S5b, Supporting Information). This mutant is characterized by the weakest fluorescence intensity and its emission maximum is red shifted to 350 nm (Figure 2c and Figure S5c, Supporting Information). The steady state fluorescence behavior of n-acetyl tryptophanamide (NATA, a free tryptophan analog) in aqueous buffer is found identical to that of F77W (Figure S6a, Supporting Information). Thus, the mutant Y77W represents a partially folded protein completely lacking any tertiary structure. Near UV CD, steady state fluorescence, acrylamide induced quenching behavior, and ANS binding properties of rest of the mutants remain between the above two extremes (Figure S5, Supporting Information).
Arginine affects the conformation of the folded state of the tryptophan mutants while sucrose does not
Figure 3 shows far-UV CD (Figure 3a), near-UV CD (Figure 3b) and fluorescence spectra (Figure 3c) of Y48W (left), F73W (middle), and F77W (right panel) mutants in the absence (black) and presence of 500 mM arginine (red), 500 mM gdn.HCl (blue) and 1 M sucrose (dark green) respectively. Sucrose and gdn.HCl have been used as controls representing a common protecting osmolyte and a popular protein denaturant respectively. These three mutants (Y48W, F73W, and F77W) are chosen because in these three mutants, the inserted tryptophan residue experiences large differences in the solvent accessibilities. While Y48W and F77W represent completely buried and exposed tryptophan residue respectively, the F73W mutant shows intermediate solvent exposure.
Figure 3.

(a) Far-UVCD (b) Near-UVCD and (c) Steady state tryptophan fluorescence measurements of Y48W (left), F73W (middle) and F77W (right column) KMP-11 mutants in the absence (black) and presence of 500 mM arginine (red), 500 mM gdn.HCl (blue) and 1 M sucrose (dark green). Necessary background corrections were made in each measurement. All these experiments were carried out in 20 mM sodium phosphate buffer at pH 7.4 at 25°C.
The addition of sucrose does not have any significant effect on the secondary (Figure 3a) or tertiary (Figure 3b) structures of any of the mutants studied. The local environment of the tryptophan residue has been found unperturbed by sucrose (Figure 3c). The addition of gdn.HCl, on the other hand, results in large decrease in the secondary and tertiary structures of the mutant proteins with significant alterations in the tryptophan environments.
It can be concluded that the effect of arginine is closer to that of gdn.HCl and is very different from that of sucrose. However, arginine and gdn.HCl results differ in two important aspects. First, the extent of conformational perturbation is significantly higher in gdn.HCl compared to that in arginine at identical concentration (500 mM). Second, the effect of arginine shows greater dependence on the nature of the mutants compared to gdn.HCl. As a matter of fact, the near-UV CD spectra of some of the partially folded mutants (for example F73W) are significantly different in their spectral characteristics in the presence of arginine (please refer to the red trace in the middle column of Figure 3b).
The above results have been complemented by ANS binding (Figure 4a) and acrylamide quenching measurements (Figure 4b, also shown in Table S3, Supporting Information). As shown before, the extent of ANS binding varies between different KMP-11 mutants. In the absence of additives, the mutants with partial or no tertiary structures bind ANS more efficiently than Y48W (buried tryptophan) (Figure 2d and 4a). The addition of arginine leads to complete loss of ANS binding (Figure 4a) and a large increase in the KSV values for the partially folded mutants (Figure 4b). In contrast, the addition of sucrose does not have any systematic effect on the ANS binding (Figure 4a) or KSV values (Figure 4b) of any of the mutants. The presence of gdn.HCl shows significantly higher effect on KSV compared to that of arginine (Figure 4b).
Figure 4.

(a) The extent of ANS binding in the absence (black) and presence of 500 mM arginine (red) and 1 M sucrose (blue). The extent of ANS binding has been determined by measuring the steady state fluorescence emission intensity at 475 nm. An ANS solution of the same buffer (in the absence and presence of sucrose or arginine of the same concentration) has been used as a background whose contribution has been subtracted at each data point. (b) The variation of KSV values for different KMP-11 mutants in the absence (black) and presence of 500 mM arginine (red), 1 M sucrose (blue), and 500 mM gdn.HCl (green). (c–d) FCS data: The representative correlation functions (c) obtained with the FCS experiments using the urea unfolded F77W/F9C mutant in the absence (black) and presence of 500 mM arginine (red). The fits of the data assuming a simple diffusion of the protein with the diffusion time of τD are shown by the residual distribution analyses (black and red in the absence and presence of arginine respectively, inset); (d) The variations of hydrodynamic radii (rH) of different unfolded mutants induced by increasing concentrations of arginine and sucrose; (e–f) Correlations between the thermal stability, ANS binding and Refolding efficiency: The variation in ΔRES,C (black) and ΔABS,C (red) with ΔTS,C in presence of 500 mM of (e) arginine and (f) sucrose. Both figures are plotted with the same scale for the ease of comparison. (g–i) Refolding kinetics data of the KMP11 mutants: (g) The refolding kinetics trace in the absence of any additive (black trace) could be fit to a sum of two exponentials. The nature of kinetics did not change in the presence of 500 mM sucrose (blue trace). The addition of 500 mM arginine simplifies the refolding kinetics removing the second slow component (red trace); (h) For the partially folded mutants (shown are F73W, F51W, and F30W), the values of k2 increased with the increase in the concentration of arginine, while the native like mutant (Y48W) showed no significant increase; (i) The addition of sucrose did not show any significant change in the values of k2. The error bars were calculated using three independent measurements.
Both arginine and sucrose contract the unfolded state of the tryptophan mutants
In the previous section, we have shown that both arginine and gdn.HCl have large effects on the conformations of the folded states of the KMP-11 mutants, and sucrose does not. In this section, we have investigated the effects of these small molecules on the unfolded states of KMP-11 mutants. Unfortunately, it is difficult, if not impossible, to study the conformation of the unfolded state using a traditional ensemble technique like CD or steady state fluorescence. Therefore, we have used fluorescence correlation spectroscopy (FCS) for this purpose. FCS offers information about the hydrodynamic radius and conformational dynamics at single molecule resolution. A brief discussion on FCS and the data analysis procedures are discussed in the material and methods section, Supporting Information.
The data presented in Figure 4c show representative correlation functions obtained with Alexa488Maleimide labeled Y48W/S9C mutant in the absence and presence of 500 mM arginine. Insertion of a cysteine residue (S9C) is needed for the labeling of the mutant proteins with the fluorescent dye, Alexa488Maleimide. Far UV CD spectroscopy shows that the attachment of the fluorescence dye does not result in any significant change in the secondary structure any of the mutants (Figure S6b, Supporting Information). The presence of arginine decreases the diffusion time (τD) of the protein and identical result has been obtained with sucrose. The values of τD have been used to determine the hydrodynamic radii (rH) of the protein in the presence of different concentrations of sucrose and arginine (Figure 4d). The results presented in Figure 4d show the presence of at least two conformers in the unfolded state of KMP-11 mutants. One of them (U) is extended while the other one (U′) is relatively more compact. The relative population of these two states depends on the presence of solution additives. For example, in the presence of 6 M urea and no additive, the extended conformer (U) is solely present and the population of U′ is negligible. With the increase in the concentration of sucrose or arginine keeping the urea concentration at 6 M, the population of U′ increases at the expense of U. The compact conformation (U′) is predominantly present in the presence of 500 mM sucrose or arginine (in 6 M urea). It is interesting to note that the behaviors of arginine and sucrose are almost identical in this regard. It may be pointed out that these two conformers (U and U′) are completely unfolded and indistinguishable by steady state fluorescence and CD.
Both sucrose and arginine increase the refolding efficiencies of the tryptophan mutants. However, arginine destabilizes the tryptophan mutants while sucrose stabilizes them
For these experiments, we have studied the effects of 500 mM arginine, sucrose and gdn.HCl on the thermal stability and refolding efficiency (RE) of KMP-11 mutants. The mid-points of the thermal unfolding transitions are used as the measure of thermal stability (Equation S4). RE has been used as the measure of aggregation propensity, which is measured by determining the reversibility of the thermal transitions (Equation S5). We have also assumed that the mutant proteins with partial or no tertiary structure and large exposed surface area would show less RE. This assumption effectively means that the ANS binding affinity of the mutants should have an inverse correlation with the RE and the data shown in Figure S7 (Supporting Information) establish that correlation.
Figure S8 (Supporting Information) shows thermal unfolding transitions of Y48W (strong native like tertiary structure), F73W (partially folded with loose tertiary structure) and F77W (no tertiary structure) mutants in the absence and presence of 500 mM arginine, 1 M sucrose and 500 mM gdn.HCl. The line drawn through data in each transition represents the fit using the two-state unfolding transition model (see Equation S4, Supporting Information).
In the case of Y48W mutant, the two-state fit of its thermal unfolding transition in the absence of any additive (Figure S8a, Supporting Information) yields a mid-point (Tm) value of 322 ± 0.2 K. The addition of arginine leads to a decrease in the thermal stability (Tm of 316 ± 0.5 K with 500 mM arginine). However, the extent of the decrease in the stability is significantly larger in the presence of same concentration of gdn.HCl (Tm of 304 ± 0.8 K with 500 mM gdn.HCl). The presence of sucrose, on the other hand, increases the thermal stability of Y48W mutant (Tm of 327 ± 0.2 K with 1 M sucrose).
Similar trend was also observed for other mutants. However, in the presence of 500 mM gdn.HCl, the thermal unfolding transitions are not straight forward. In some cases, for example with F77W, unfolding occurs very early and the baseline regions are not well defined (Figure S8b, Supporting Information). It is difficult to analyze this behavior using the two-state model we employed. The fitting parameters obtained from the thermal unfolding experiments of the tryptophan mutants are shown in Table S4, Supporting Information.
The values of RE are shown in Table S4. It is evident from the table that arginine increases the refolding of the KMP-11 mutants significantly. Addition of sucrose also increases the extent of refolding of KMP-11 mutants but its effect is less than that of arginine. The addition of 500 mM Gdn.HCl has a general decreasing effect on the refolding efficiency. This is not surprising and the presence of low concentration of Gdn.HCl has been shown to initiate aggregation in other proteins also20.
Sucrose influences the thermal stability and RE of the tryptophan mutants identically. The effect of arginine depends on the nature of the mutants
Assuming the mid-points of a particular mutant in the absence and presence of an additive (sucrose, arginine or gdn.HCl) of concentration [C] to be T and TS.C respectively, the change in stability of the mutant in the presence of that additive is
 |
Similarly, the change in the refolding efficiency (ΔRES,C) and the change in ANS binding efficiency (ΔABS,C) of a particular mutant induced by the additive of concentration [C] could be defined as
 |
and
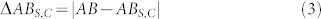 |
Figure 4e plots the variations of ΔRES,C and ΔABS,C with ΔTS,C for all the KMP-11 mutants in the presence of 500 mM arginine. The corresponding plots in the presence of sucrose are shown in Figure 4f. The x and y axes for both Figures 4e and 4f have been kept identical for easy comparison. A comparison between Figures 4e and 4f clearly shows that sucrose affects the thermal mid-points of all the mutants equally and the values of ΔTSuc,C occupy a very narrow region in Figure 4f. The presence of arginine, in contrast, affects the thermal stability of the mutants differently and the values of ΔTArg,C cover a wide range. Additionally, in the presence of arginine, both ΔREArg,C and ΔABArg,C correlate linearly with ΔTArg,C. In the presence of sucrose, no correlation exists between ΔRESuc,C, ΔABSuc,C and ΔTSuc,C.
The following points may be inferred from the above results. First, the addition of sucrose increases the stability and RE of all tryptophan mutants. The values of ΔTsuc,C and ΔREsuc,C depend solely on the concentration [C]suc and not the nature of the mutants. In contrast, the addition of arginine decreases the stability of all the mutants, but increases RE. The values of ΔTArg,C and ΔREArg,C depend both on arginine concentration [C]arg and on the nature of the mutants. Mutant proteins with stronger affinity towards ANS and lowest RE are the most destabilized by arginine, and there are direct correlations between ΔREArg,C, ΔABArg,C and ΔTArg,C. Second, while arginine competes with ANS for the exposed hydrophobic surface, sucrose does not recognize exposed hydrophobic surface. Third, the effect of gdn. HCl differ in two important aspects: a) at identical concentration, ΔTarg,C is much less compared to ΔTgdn,C and large instability induced by gdn.HCl makes data analyses difficult using a two-state model; b) at low concentration (e.g. 200–500 mM used in the present study), arginine increases while gdn.HCl typically decreases the RE.
Arginine simplifies the refolding kinetics of the partially folded mutants while sucrose does not have any significant effect
It has been shown that GroEL, one of the most extensively studied molecular chaperones, decreases protein aggregation by minimizing the number of possible routes a protein explores to reach its native folded state21. In order to get further insight into the effect of arginine, refolding kinetics experiments have been carried out using representative tryptophan mutants in the absence and presence of arginine and sucrose. Refolding has been initiated by diluting 11 times the protein unfolded at 6 M urea. Independent urea induced unfolding experiments with the tryptophan mutants helped us choose the initial (6 M urea) and final urea concentrations needed for these measurements (the representative data for the Y48W mutant are shown in Figure S9a–b, Supporting Information). Refolding kinetics measurements of the mutant proteins show the presence of two rate constants in the absence of any additives (Figure S10a–b, Supporting Information). A very rapid increase (rate constant, k1, of approx. 100 sec−1) followed by a relatively slow increase (rate constants, k2, varying between 2 sec−1 and 10 sec−1 depending on the mutants) in tryptophan fluorescence emission are observed by stopped flow fluorescence experiments. None of the rate constants and their relative amplitudes change at different excitation slit widths ruling out any contribution from photo-bleaching in any of the kinetic phase. The values of k1 and k2 do not have any dependence on protein concentrations, at least in the concentration range used in this study (Figure S10c–d, Supporting Information). The presence of 500 mM sucrose does not change the rate constants of any of the mutants (Figure 4g). The presence of 500 mM arginine, however, simplifies the refolding kinetics of most of the mutants (the kinetic trace for the F73W is shown in Figure 4g). In the presence of 500 mM arginine, the overall refolding kinetics of these proteins (Figure 4g shows the data for F73W) could be fit only to a single component. Interestingly, the addition of arginine to Y48W (strong tertiary structure with buried tryptophan), does not have any significant effect (see below).
The addition of sucrose or arginine does not have any significant effect on the rate constants of the fast phase (k1) of any of the KMP-11 mutants. The rates of the slow phase (k2) increase with the arginine concentration for the KMP-11 mutants (Figure 4h), except for Y48W (the folded mimic, Figure 4h), in which k2 remains unchanged. The amplitude of the slow phase (a2) decreases with arginine concentration and becomes negligible at high concentration (Figure S10h, Supporting Information). The negligible value of a2 at high arginine concentration explains the absence of the second component as described in the previous paragraph (Figure 4g). The presence of sucrose does not show any effect on either the amplitudes or the rate constants (see Figure 4i and Figure S10e–f, Supporting Information).
It is to be noted that the refolding kinetics could not be measured for F77W. This is because, F77W does not offer any change in tryptophan fluorescence intensity in the absence or presence of urea. Although the viscosity of sucrose solution has been shown to decrease the rate constant of refolding of CspB22, this effect is not observed in the present study. This is because; the use of 500 mM sucrose does not lead to a large enough change in viscosity to be able to influence the refolding kinetics.
Discussion
The mechanism of the action of sucrose may be described by a simple pathway as described in Figure 5a. In this pathway, at least two conformers of the unfolded state (U′ and U) have been assumed to be in equilibrium. U′ is relatively more compact with higher potential energy compared to the extended conformer (U). The presence of a compact and an extended conformer in the unfolded state has been established by the FCS results. In the presence of a protecting osmolyte, like sucrose, the higher energy compact state (U′) is preferred over the extended state (U). This in turn leads to an increase in the free energy of unfolding in the presence of sucrose (ΔGSuc,C > ΔG in Figure 5a), which is observed in the thermal unfolding results. Since, this pathway deals entirely with the free energy change between two unfolded conformers (ΔGU), and does not consider any aspect of folded (N) or folded like (N′) conformations, we can conclude
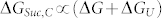 |
Figure 5. The potential energy diagrams for (a) sucrose and (b) arginine.
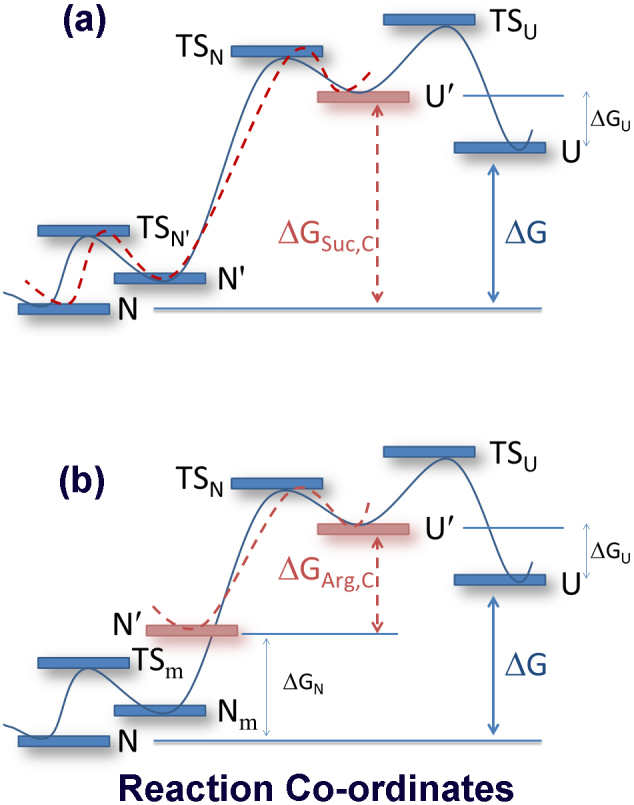
(a) For sucrose, two conformations of the unfolded state (U and U′) were found present in equilibrium. U is extended and U′ is relatively more compact and energetically unfavorable. The addition of sucrose enhanced the population of U′ resulting in higher free energy of unfolding (ΔGSuc,C ≫ ΔG). ΔG and ΔGSuc,C are the free energy of unfolding in the absence and presence of sucrose at concentration C, respectively. The folding pathways in the absence and presence of sucrose were differentiated using blue solid line (in the absence) and red dotted line (in the presence of sucrose), respectively. (b) The addition of arginine increases the free energy of the native states (and form N′). In the absence of arginine, refolding is complex and characterized by several pathways (stopped flow data, Figure 4g). The addition of arginine simplifies refolding and makes it relatively simpler (stopped flow data, Figure 4g) and more efficient (RE data, Figure 4e). The free energy diagrams in the absence and presence of the additives have been differentiated using blue solid line (in the absence) and red dotted line (in the presence of additives).
Since all KMP-11 mutants approximately contain the same back-bone surface area, the contribution of ΔGU (and hence ΔGSuc,C) would be similar for different mutants.
The mechanism of arginine's action is more complex (Figure 5b). Similar to sucrose and other protecting osmolytes, arginine increases the population of U′. Like a denaturant, it also increases the free energy of the folded states (increases the population of N′). The change in folding free energy in the presence of arginine (ΔGArg,C), hence, contains two components; one arising from the unfolded state destabilization/contraction (ΔGU) and the second one from the destabilization of the native state (ΔGN) as
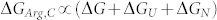 |
For KMP-11 mutants, the addition of arginine leads to an overall destabilization (i.e ΔGArg,C < ΔG), which implies that ΔGN has greater contribution compared to ΔGU. The present results also indicate that the value of ΔGN is higher for mutants with greater aggregation propensities. Alternatively, arginine interacts with the folded state of an aggregation prone mutant more efficiently compared to one which is less aggregation prone. Extrapolation of this to a protein with 100% refolding efficiency (and no aggregation propensity) even in the absence of any additive, effectively means, that arginine would behave like a protecting osmolyte (because ΔGArg,C = ΔG + ΔGU as ΔGN = 0). The intestinal fatty acid binding protein (IFABP) unfolds thermally with 100% refolding efficiency and in cooperative fashion. Like KMP-11, IFABP is a convenient model protein with no cysteine or proline residue and different aspects of its folding thermodynamics and kinetics have been extensively studied23,24,25. The melting temperature of its thermal unfolding and the extent of refolding are 344°K and 100% respectively. As predicted, the addition of arginine stabilizes the protein with mid-points of unfolding in the presence of 200 mM arginine being 357°K.
The data shown in Figure 6, directly show the presence of these two components in arginine. In contrast, sucrose has only one linear protecting osmolyte like component (Figure 6). More interestingly, the relative proportions of these two components (protecting osmolyte like and denaturant like) are not constant and they vary significantly for different mutants. Although several factors would affect the proportions of these two components, some of key ones may be the nature of proteins, the extent of misfolding stress and the concentration of arginine. A thorough understanding of these components and their fine-tuning could be useful to design a chemical chaperone, which is more potent and more specific in rescuing a mis-folded protein.
Figure 6. The components of protecting osmolyte like and denaturant like properties of arginine in the case of (a) Y48W (b) and F77W.

The arginine data for Y48W clearly showed the presence of two components, while the sucrose data for all the mutants have one linear stabilizing component.The transition points between these two components for the arginine data are shown by arrows. Three independent measurements were used to determine the error bars shown in this figure.
Finally, it is important to discuss the potential caveats of the present study. Although we have validated some of the data with a protein of known structure (IFABP), majority of the present results have been obtained with a protein with predicted structure, which may not be correct. While the present results establish strongly the superiority of arginine over sucrose (and gdn.HCl) towards the inhibition of protein aggregation, any generalized understanding of the mechanism of its action would require detailed studies involving other proteins of solved structures. Our laboratory has already presented results using two other proteins of predominately α-helical structure26,27. Several other proteins (with β-sheet and natively unfolded character) are also being investigated to find out if the differences in secondary structure (α-helix, β-sheet or the natively unfolded) play any roles in the mechanism of the small molecule chaperones. Nevertheless, we would argue that the results and the strategy presented in this paper would be beneficial for the stabilization of biologics pharmaceuticals and otherwise important proteins, whose structures are not available.
Methods
A short summary of the materials and methods section is provided in the following paragraph and the detailed description of every experiment has been supplied as Supporting Information.
The predicted structure of KMP-11 was obtained using a composite approach which combines threading, ab-initio modeling and atomic-level structure refinement methods. Molecular dynamics simulations were carried out with the tryptophan mutants of KMP-11 to computationally determine the solvent accessible surface area (SASA) of the tryptophan side chains. Far-UV and near-UV CD experiments were carried out in the absence and presence of different additives to study the effects of the additives on the secondary and tertiary structures. Steady state tryptophan fluorescence (excitation at 295 nm) was used to determine the local environment of the inserted tryptophan residues. ANS binding was measured using steady state fluorescence experiments using an excitation wavelength of 360 nm. Thermal unfolding transitions were measured using the temperature dependence of far UV CD and the data was fit using a two-state assumption. All the experiments were repeated at least three times to establish the trend. A free cysteine residue was inserted in every tryptophan mutant to label with Alexa488Maleimide, which is needed for the FCS measurements.
Author Contributions
K.C. and S.S. wrote the paper. S.R. provided the initial KMP-11 samples. S.P. carried out the computational studies. S.S. and S.S. carried out the experiments. K.C., S.S., S.P. and S.S. analyzed the data. All authors reviewed the manuscript.
Supplementary Material
Supporting Information
Acknowledgments
The authors would like to thank Dr. Sujoy Mukherjee of CSIR-IICB for his help in revising the original manuscript. The authors thank Prof. Carl Frieden of the Washington University School of Medicine for his critical evaluations of the initial manuscript and also for the IFABP plasmid constructs. This study has been funded by CSIR network project BSC 0114.
References
- Chiti F. & Dobson C. M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–66 (2006). [DOI] [PubMed] [Google Scholar]
- Horwich A. L., Farr G. W. & Fenton W. A. GroEL-GroES-mediated protein folding. Chem. Rev. 106, 1917–30 (2006). [DOI] [PubMed] [Google Scholar]
- Hartl F. U., Bracher A. & Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature 475, 324–32 (2011). [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A. et al. Chemical chaperones assist intracellular folding to buffer mutational variations. Nat. Chem. Biol. 8, 238–45 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon A. C., Moosa M. M., Gambin Y. & Deniz A. A. Counteracting chemical chaperone effects on the single-molecule alpha-synuclein structural landscape. Proc. Natl. Acad. Sci. U.S.A. 109, 17826–31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa T., Ejima D., Kita Y. & Tsumoto K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim. Biophys. Acta 1764, 1677–87 (2006). [DOI] [PubMed] [Google Scholar]
- Arakawa T. & Timasheff S. N. The stabilization of proteins by osmolytes. Biophys. J. 47, 411–4 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolen D. W. & Baskakov I. V. The osmophobic effect: natural selection of a thermodynamic force in protein folding. J. Mol. Biol. 310, 955–63 (2001). [DOI] [PubMed] [Google Scholar]
- Macchi F., Eisenkolb M., Kiefer H. & Otzen D. E. The effect of osmolytes on protein fibrillation. Int. J. Mol. Sci. 13, 3801–19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street T. O., Bolen D. W. & Rose G. D. A molecular mechanism for osmolyte-induced protein stability. Proc. Natl. Acad. Sci. U.S.A. 103, 13997–4002 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y., Bolen C. L. & Bolen D. W. Osmolyte-driven contraction of a random coil protein. Proc. Natl. Acad. Sci. U.S.A. 95, 9268–73 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umetsu M. et al. Nondenaturing solubilization of beta2 microglobulin from inclusion bodies by L-arginine. Biochem. Biophys. Res. Commun. 328, 189–97 (2005). [DOI] [PubMed] [Google Scholar]
- Ejima D. et al. High yield refolding and purification process for recombinant human interleukin-6 expressed in Escherichia coli. Biotechnol. Bioeng. 62, 301–10 (1999). [PubMed] [Google Scholar]
- Reddy K. R., Lilie H., Rudolph R. & Lange C. L-Arginine increases the solubility of unfolded species of hen egg white lysozyme. Protein Sci. 14, 929–35 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa T. & Tsumoto K. The effects of arginine on refolding of aggregated proteins: not facilitate refolding, but suppress aggregation. Biochem. Biophys. Res. Commun. 304, 148–52 (2003). [DOI] [PubMed] [Google Scholar]
- Baynes B. M., Wang D. I. & Trout B. L. Role of arginine in the stabilization of proteins against aggregation. Biochemistry 44, 4919–25 (2005). [DOI] [PubMed] [Google Scholar]
- Xie Q., Guo T., Lu J. & Zhou H. M. The guanidine like effects of arginine on aminoacylase and salt-induced molten globule state. Int. J. Biochem. Cell Biol. 36, 296–306 (2004). [DOI] [PubMed] [Google Scholar]
- Arakawa T. et al. Suppression of protein interactions by arginine: a proposed mechanism of the arginine effects. Biophys. Chem. 127, 1–8 (2007). [DOI] [PubMed] [Google Scholar]
- Basu R. et al. Kinetoplastid membrane protein-11 DNA vaccination induces complete protection against both pentavalent antimonial-sensitive and -resistant strains of Leishmania donovani that correlates with inducible nitric oxide synthase activity and IL-4 generation: evidence for mixed Th1- and Th2-like responses in visceral leishmaniasis. J. Immunol. 174, 7160–71 (2005). [DOI] [PubMed] [Google Scholar]
- Kendrick B. S., Carpenter J. F., Cleland J. L. & Randolph T. W. A transient expansion of the native state precedes aggregation of recombinant human interferon-gamma. Proc. Natl. Acad. Sci. U.S.A. 95, 14142–6 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutani N. & Udgaonkar J. B. GroEL channels the folding of thioredoxin along one kinetic route. J. Mol. Biol. 314, 1167–79 (2001). [DOI] [PubMed] [Google Scholar]
- Jacob M., Schindler T., Balbach J. & Schmid F. X. Diffusion control in an elementary protein folding reaction. Proc. Natl. Acad. Sci. U.S.A. 94, 5622–7 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay K., Elson E. L. & Frieden C. The kinetics of conformational fluctuations in an unfolded protein measured by fluorescence methods. Proc. Natl. Acad. Sci. U.S.A. 102, 2385–9 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay K., Zhong S., Yeh S. R., Rousseau D. L. & Frieden C. The intestinal fatty acid binding protein: the role of turns in fast and slow folding processes. Biochemistry 41, 4040–7 (2002). [DOI] [PubMed] [Google Scholar]
- Ropson I. J., Boyer J. A. & Dalessio P. M. A residual structure in unfolded intestinal fatty acid binding protein consists of amino acids that are neighbors in the native state. Biochemistry 45, 2608–17 (2006). [DOI] [PubMed] [Google Scholar]
- Ghosh R., Sharma S. & Chattopadhyay K. Effect of arginine on protein aggregation studied by fluorescence correlation spectroscopy and other biophysical methods. Biochemistry 48, 1135–43 (2009). [DOI] [PubMed] [Google Scholar]
- Haldar S., Mitra S. & Chattopadhyay K. Role of protein stabilizers on the conformation of the unfolded state of cytochrome c and its early folding kinetics: investigation at single molecular resolution. J. Biol. Chem. 285, 25314–23 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Kucukural A. & Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC bioinformatics 9, 40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information