

Abstract
The history of mankind remains one of the most challenging fields of study. However, the emergence of anatomically modern humans has been so recent that only a few genetically informative polymorphisms have accumulated. Here, we show that DNA sequences from Helicobacter pylori, a bacterium that colonizes the stomachs of most humans and is usually transmitted within families, can distinguish between closely related human populations and are superior in this respect to classical human genetic markers. H. pylori from Buddhists and Muslims, the two major ethnic communities in Ladakh (India), differ in their population-genetic structure. Moreover, the prokaryotic diversity is consistent with the Buddhists having arisen from an introgression of Tibetan speakers into an ancient Ladakhi population. H. pylori from Muslims contain a much stronger ancestral Ladakhi component, except for several isolates with an Indo-European signature, probably reflecting genetic flux from the Near East. These signatures in H. pylori sequences are congruent with the recent history of population movements in Ladakh, whereas similar signatures in human microsatellites or mtDNA were only marginally significant. H. pylori sequence analysis has the potential to become an important tool for unraveling short-term genetic changes in human populations.
Keywords: mtDNA, microsatellites, admixture, human migrations, population genetics
Polymorphic genetic markers have been used repeatedly in attempted reconstructions of human evolutionary history and population migrations (1–3). However, the well known “out of Africa” bottleneck ≈200,000 years ago (4) reduced the genetic diversity of modern humans in Asia and Europe dramatically, such that the differentiation of modern populations requires genetic markers with rapid evolutionary rates. Beginning in the 1970s, sequence diversity within the mitochondrial D-loop hypervariable segments (HVS1 and HVS2) was introduced as a tool for this purpose (5). However, these hypermutable mtDNA fragments are only a few hundred base pairs long and, therefore, provide only a limited number of informative single-nucleotide polymorphisms for recognizing haplotypes and haplogroups. Furthermore, certain polymorphic nucleotides in mtDNA have arisen on independent occasions (homoplasies) because of mutational hot-spots (6). Many mitochondrial haplotypes are present in many human populations and can only be used to differentiate populations on the basis of their frequencies of occurrence.
Because of these limitations, alternative genetic markers have been used increasingly since the mid-1990s, including microsatellites (7–9) and single-nucleotide polymorphisms on the Y chromosome (10–14) and in autosomal genes (15–18). Each of these newer genetic markers also has limitations. For example, the exchange of microsatellite allelic profiles and human genotypes between scientific groups by means of a central, global microsatellite database is just beginning (19). Furthermore, it is not obvious whether hundreds of microsatellite markers can be scored identically in many laboratories. Sequence polymorphisms in autosomal and Y chromosome genes are easier to standardize, but they are much less polymorphic than mtDNA hypervariable segments, thus requiring the use of numerous independent single-nucleotide polymorphisms, which is technologically demanding.
In the absence of a universal “gold standard” marker panel for anthropological genetics, it seems advisable to investigate alternative markers for human populations. Alternative approaches have focused on commensals and parasites that accompany human migrations, including viruses such as JC virus (20, 21) and human T-lymphotrophic virus (22), fungi (23), rats (24), and the bacterium Helicobacter pylori (25–27). In the present study, we investigated the usefulness of H. pylori, a Gram-negative bacterium that persistently colonizes the stomach of >50% of humans, as a potential genetic marker for deciphering recent human migrations. We also genotyped their human hosts with microsatellites and mtDNA HVS1 sequences to compare the resolution and power of the host and bacterial markers.
H. pylori are highly genetically diverse, providing ≈1,400 informative sites within 4.5 kb of sequence, and their global genetic structure parallels that of humans (28). Moreover, epidemiological studies have shown that transmission occurs predominantly within families (29–32). This point is critical because vertical transmission is more likely to reflect human migrations than is the horizontal transmission that is typical of many microbes.
We hypothesized that H. pylori might provide more information on recent human admixture than classical human markers, and we tested this concept in a remote area with diverse ethnic groups, well known recent history, and a limited contemporary gene flow. Ladakh, in northern India, is an isolated trans-Himalayan region with low population density. The Ladhakhi languages have both Mongolian and Indo-Iranian components, and various religions are represented in Ladakh (Buddhists, 48%; Muslims, 47%; and Hindus, 5%). The original population may have been Dards who colonized the western Himalayas via the Indus valley (33). However, the Dard culture was overwhelmed by the immigration of shepherds and nomads from Tibet ≈1,000 years ago (34). The present-day Buddhists are primarily descendants of Mongolians, and they bear a close physiognomic affinity with Tibetans. In contrast, Arghuns, the majority of the Muslims, are the descendants of Sufi masters from Pakistan who settled in Ladakh after the 14th century (34, 35).
We compare the genetic relationships between Buddhists and Muslims in Ladakh that are revealed by human genetic markers (HVS1 and microsatellites) and by sequences of eight gene fragments of the H. pylori that they carry. The sequence information from H. pylori was found to have the highest resolution and discriminatory power for unraveling human population structure and evolutionary history.
Methods
Sampling and DNA Extraction. Biopsies were taken for diagnostic purposes during gastroendoscopy of 50 Ladakhi patients with dyspeptic symptoms (36). H. pylori from the biopsies was cultivated after single-colony isolation and used to prepare bacterial DNA (36). Human DNA was also isolated from the biopsies by using the DNeasy tissue kit (Qiagen, Valencia, CA), according to the manufacturer's instructions. The experiments described here received the permission of Prince Jigmet Namgyal (Leh, Ladakh), and they were approved by the Institutional Board of Research Associates (New York University School of Medicine).
DNA Sequencing and Microsatellite Genotyping. Details on the PCR technology are available in Supporting Materials and Methods, which is published as supporting information on the PNAS web site. The multilocus haplotypes are available at http://pubmlst.org/helicobacter/projects/ladakh, and the individual alleles can be downloaded at http://pubmlst.org/helicobacter.
Statistical Analysis. Phylogenetic analyses, population-genetic statistics and details on how structure (available at http://pritch.bsd.uchicago.edu/software.html) was used are also available in Supporting Text.
Results
HVS1 Diversity and Genetic Structure. mtDNA HVS1 was sequenced (positions 16,001–16,388) from 39 subjects, and the sequences were aligned relative to the Cambridge Reference Sequence (37). We identified 32 unique sequences, which varied at 52 polymorphic sites (see Table 2, which is published as supporting information on the PNAS web site). The ratio of transitions to transversions was 10-fold, similar to prior analyses (38). The 32 sequences clustered into two groups that were weakly supported by phylogenetic analyses (Fig. 1a): one containing the B and F haplogroups that are found preferentially in Southeast Asia, as well as typically European haplogroups (U, H, J, and R), and the other one containing haplogroups D and M that are common in East and Northern Asia. However, the phylogenetic tree did not distinguish between Muslims and Buddhists, whose mtDNA sequences are distributed through all of the haplogroups. Therefore, we could not differentiate ethnic groups on the basis of this maternally inherited marker.
Fig. 1.

Phylogenetic analysis of mtDNA HVS1 sequences. The designations for individual probands consist of L plus an arbitrary number (followed by the haplogroup in a) and are color-coded as indicated. (a) Maximum-likelihood tree of the HVS1 sequences (388 bp) using a Tamura–Nei model of evolution (I = 0.4806; G = 0.7551; starting tree = neighbor-joining; nearest-neighbor interchange branch swapping limited to 20,000 rearrangements). Data were constructed by using paup* and bootstrapped with 1,000 replicates. Only bootstrap values >70% are shown. The tree includes the Cambridge Reference Sequence and was rooted with a pygmy sequence. (b) Phylogenetic network relating HVS1 sequences. Colors of the filled circles correspond to the six main radiation groups described in Oota et al. (38), and small open circles correspond to missing haplotypes. The network obtained from Ladakhi HVS1 sequences is completely congruent with the network of Oota et al. (38), except that proband L133 shares typical Group II variant sites (C at np362 and G at np319) but is placed at a terminal node of Group VI.
In an attempt to infer the evolutionary status of the Ladakhi population within Eurasia, we used a subset of 185 nucleotides for which a large-scale analysis has been described (38). This region (16,194–16,378) contained 28 polymorphisms in 26 distinct haplotypes that were used to construct a reticulated network (Fig. 1b). The reticulated network was reduced to a minimum-spanning tree as described (38). Interestingly, the network from the 39 Ladhakis contained all six radiation groups (I–VI) described by Oota et al. (38) on the basis of 414 haplotypes. Each of the six radiation groups is characterized by specific single-nucleotide polymorphisms and occurs in nearly all Asiatic populations. However, the radiation groups show clear geographical differences in their frequency distribution; e.g., Group IV is particularly prevalent (67%) in West Asia. We, therefore, compared the frequencies of the radiation groups between Buddhists and Muslims in Ladakh. There is no significant difference between the frequencies of the radiation groups in Buddhists and Muslims because the null hypothesis of no difference between them could not be rejected, even after correction for small sample sizes (P = 0.72). We then compared the frequencies of the radiation groups within the entire sample from Ladakh with data from West Asia, Far-East Asia, continental East Asia, and Southeast Asia. The Ladakhi sample was not significantly different (P = 0.533) from populations in continental East Asia (Taiwan Han Chinese, Cantonese, Chinese, Mongols, and Altai from central Siberia) but did differ significantly (P < 0.001) from all other major geographical populations that had been described (38).
Microsatellite Markers. Similar to the results with mtDNA, we could not distinguish Buddhists from Muslims by using size variation of 17 autosomal microsatellites. By using the population-differentiation test, the two ethnic groups were indistinguishable (P = 0.8), indicating that they share nearly identical allelic frequencies. A common measure of population genetic differences between the two ethnic groups was both low and insignificant (FST = 0.007, P < 0.14). We also attempted to detect a weak genetic structure with an alternative Bayesian statistics tool, structure (39), which measures the posterior probability of data based on K, the number of populations. Analyses with K = 1 yielded a higher likelihood than K = 2 or K = 3, again indicating that a single population was sampled. However, a three-dimensional correspondence analysis revealed slight ethnically related differences in the distributions of individual genotypes (Fig. 2). Microsatellite genotypes from Buddhists and Muslims overlap, but genotypes from the former are relatively homogeneous, whereas those from Muslims are more diverse and scatter throughout the three dimensions. The Muslim sample is marked by a significant deficit of heterozygotes (FIS = 0.084, P = 0.02), which is unlike the Buddhist population (FIS = 0.019, P = 0.24), possibly reflecting a Wahlund effect, which occurs when a sample contains more than one discrete population.
Fig. 2.

Three-dimensional correspondence analysis (FCA) of microsatellite genotypes from Ladakhis. Yellow, Buddhists; blue, Muslims.
H. pylori. A total of 3,850 bp from eight unlinked neutral genes (40) was sequenced for 50 isolates from Ladakh and compared with sequences from 69 H. pylori strains from Indo-Europeans and Asians (28). Although phylogenetic methods are inappropriate for sequences resulting from frequent recombination, we present a haplotype tree (Fig. 3a) based on the 974 polymorphic sites within the multilocus sequences from H. pylori to permit direct comparison with the HVS-1 results. European and East-Asian sequences formed two distinct clusters, whereas all but three H. pylori sequences from Ladakh fell into a separate bush, most closely related to the East-Asian sequences. Most of the isolates from Ladakhi Muslims and a few of the isolates from Buddhists clustered together, whereas most Buddhist isolates fell into an intermediate paraphyletic “bridge” to the East-Asian cluster (Fig. 3a).
Fig. 3.

A multilocus haplotype tree and quantitative sources of nucleotides from three ancestral populations for 119 H. pylori isolates from Ladakh, East Asia, and Indo-Europe. (a) Neighbor-joining tree of individual haplotypes (Kimura two-parameter model). Fig. 6, which is published as supporting information on the PNAS web site, includes the designation of each individual isolate. (b) Sources of nucleotides. The diameters of the three circles indicate the genetic diversity of the three ancestral populations and the filled arcs indicate the proportion of the nucleotides in the 119 haplotypes that are derived from each of these populations. The black lines joining the ancestral populations represent a neighbor-joining population tree as measured by 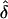 , the net nucleotide distance between populations.
, the net nucleotide distance between populations.
The same data were analyzed with structure to determine the number of ancestral populations and the sources of nucleotides (Fig. 3b). The analysis indicated three ancestral populations because that value yielded a higher posterior probability than did runs assuming one, two, four, or five populations. We have retained the same designations for these three ancestral sources as were used in ref. 28, in which it was shown that modern isolates from East Asians have a strong ancestral East-Asian component whereas European isolates are admixtures from ancestral Europe1 and Europe2. In the current analysis, approximately equal proportions of the polymorphic nucleotides in the 119 sequences were derived from each of the three ancestral populations.
We calculated the proportion of ancestry from each of these three ancestral sources for each of the 119 isolates. For Ladakhi isolates, 37 of the 50 have inherited at least 50% of their polymorphic nucleotides from ancestral Europe1, whereas only 11 have the majority of their nucleotides from ancestral East Asia. The large proportion of ancestry from ancestral Europe1 is highly significant (χ2 = 25.6; P < 10–5; 2 df). Isolates from Ladakh differ from East-Asian isolates, which are predominantly derived from ancestral East Asia, or European isolates (Turks, Germans, and Spaniards; predominantly from ancestral Europe2) (Figs. 4a and 7, which is published as supporting information on the PNAS web site). Bangladeshi isolates split into two groups (Fig. 7) with predominant ancestry from ancestral Europe2 (seven isolates) or ancestral Europe1 (three isolates).
Fig. 4.

Ancestry of nucleotides in H. pylori isolates from Ladakh as inferred by structure.(a) Ternary plots of proportion of ancestry from three sources of nucleotides designated ancestral East Asia, ancestral Europe1, and ancestral Europe2. Each data point corresponds to a single isolate whose proportion of ancestry from each of the three sources is represented by its proximity to the corresponding corner of the triangle. Note that the proportion of ancestry ranges between 0 and 1.0, regardless of the true genetic distances between the ancestral sources. (b) Proportion of ancestry from ancestral East Asia for H. pylori isolates by ethnic source.
Furthermore, and unlike the data from either mtDNA or microsatellites, the H. pylori sequences show clear patterns of genetic variability that distinguish Buddhists and Muslims (Fig. 4). H. pylori from Buddhists show a gradient of introgression from pure EastAsian ancestry to almost-pure Europe1 ancestry, including all hybrid intermediates (Fig. 4a). Isolates from Muslims have a more uniform Europe1 ancestry, except for three isolates that possess considerable ancestry from Europe2. Furthermore, the contribution of nucleotides from ancestral East Asia is rather modest among isolates from Muslims, unlike the Buddhist isolates (Fig. 4b).
Selection might blur genetic ancestry or generate spurious signals. Therefore, we tested all sequence data for selection by using Fu and Li's test and Tajima's D test. Both test statistics did not deviate significantly from a neutral distribution (see Table 3, which is published as supporting information on the PNAS web site), showing that the results do not reflect differential selection.
Pairwise Mismatch Analysis. Pairwise mismatch distributions were unimodal for both human mtDNA and H. pylori sequences within the Buddhist community (Fig. 5), as expected after a sudden expansion of population size. The distribution of the mtDNA data did not differ significantly from the expectations of an expansion model (sum of squared deviations under the least-squares approach, P = 0.570; Harpending's raggedness index, P = 0.500) but did differ significantly from the expectations for a stable, nonexpanding population (Kolmogorov–Smirnov test, D = 0.454, P = 0.014). The mean time since expansion was estimated as 32,000–125,000 years, assuming a substitution rate of 9 × 10–8 per site per year (41) (expansion parameter τ = 5.70; 95% confidence interval; 2.23–8.70). Similar calculations are not possible for the H. pylori sequences because the substitution rate has not yet been estimated.
Fig. 5.

Mismatch distributions of differences between all possible pairs of HVS1 and H. pylori sequences from Buddhists (Left) and Muslims (Right). The assignment of data in the lower right to within- and between-population components is based on the observation that the between-population component disappeared when the three isolates with considerable ancestry from Europe2 were excluded.
The mismatch distributions for mtDNA and H. pylori sequences from Muslims are clearly not unimodal. The mtDNA mismatch distribution is ragged, as expected for a constant population size, and it might correspond to a bimodal distribution. The H. pylori mismatch distribution is unambiguously bimodal, and it displayed a very high variance. These observations suggest that two distinct populations have mixed in Ladakh: one corresponding to the ancient Ladakhi population, and another introduced by a more recent migration from Europe or the Near East. The bimodal bacterial mismatch distribution probably represents population subdivision (42, 43) because the distribution became largely unimodal after excluding the three H. pylori isolates with extensive ancestry from Europe2.
Discussion
Signatures Left by Migrations. Buddhists and Muslims in Ladakh have been socially distinct for 500–1,000 years because of cultural and religious differences. Such separation might be expected to leave signatures on population structure but mtDNA sequences and microsatellite genotypes did not detect significant genetic differentiation between the two ethnic groups, by using either F statistics or the analysis of molecular variance (AMOVA) approach (Table 1). A lack of differentiation might reflect a lack of resolution by these markers and/or small sample sizes. Nevertheless, under the same conditions, significant genetic differentiation (FST = 0.031, P < 0.007) was observed with the H. pylori sequences from Ladakh and 3% of the genetic variance observed by analysis of molecular variance (AMOVA) is attributable to the variance between ethnic groups (Table 1). A second and very striking feature is the different ancestry of H. pylori from Buddhists and Muslims (Fig. 4a); such a clear differentiation was not achieved with the human genetic markers. H. pylori from Buddhists are the product of an asymmetric gene flow between two ancestral populations, ancestral East Asia and Europe1.
Table 1. Comparison of genetic divergence between Buddhists and Muslims on the basis of both human and bacterial genetic markers.
| Marker | Source | FST | AMOVA |
|---|---|---|---|
| mtDNA | Human | 0.021 (P < 0.16) | 0.020 (P < 0.11) |
| Microsatellites | Human | 0.007 (P < 0.14) | 0.008 (P < 0.13) |
| H. pylori | Bacterial | 0.031 (P < 0.007) | 0.031 (P < 0.001) |
AMOVA, analysis of molecular variance.
“Ancestral Europe1” and “ancestral Europe2” were invented as designations for sources of ancestry in a sample of numerous H. pylori from Europe plus 17 isolates from Ladakhi Muslims (28). These eurocentric designations were invented because of the predominance of isolates from Europe and are possibly misleading in the present context. Nucleotides from “ancestral Europe1” were interpreted as having been imported from Central Asia because they were particularly frequent in Finland and Estonia, which have imported Uralic languages from central Asia, and they were most frequent among the Ladakhi isolates (28). Nucleotides from “ancestral Europe2” were interpreted as having been imported with the Neolithic migrations from the Near East. Based on historical records, we now infer that the gene flow within H. pylori from Buddhists in Ladakh reflects the introgression of Mongoloid Tibetan migrants carrying hpEastAsia bacteria into a preexisting Central Asian population carrying ancestral Europe1. As a result of this migration, most of the strains from Buddhists have mixed ancestry and each genome is a mosaic of many small chromosomal fragments originating from either ancestral East Asia or Europe1. In contrast, H. pylori from Muslims are more homogeneous, and 62–82% of the nucleotides in 15 of 18 such isolates are derived from the ancestral Europe1 population. Only three strains from Muslims show a clear signature of Europe2 introgression and might correspond to an admixture of Muslims migrants from the Near East with local Ladakhis.
Bacteria from clinical disease might possibly differentiate between human ethnic groups because of differential selection for particular virulence factors and yield population structures that are different than samples from humans without clinical disease. However, the bacteria tested here were isolated from patients with dyspepsia, which does not correlate with the carriage of H. pylori, and almost none of them showed any clinical symptoms that are associated with these bacteria. Furthermore, selective differences related to the ethnic grouping of their host should have left signatures at the nucleotide level, which were not detected (see Table 3).
Religion Versus Genetic Impact. The H. pylori data also reveal differences in how religions spread: Buddhism was imported together with numerous East-Asian nucleotides, whereas the Muslims who settled in Ladakh left only few novel genetic signatures, although a significant proportion of Ladakhis converted to the Muslim religion. It remains unclear why the Muslim community does not contain more signatures of the earlier Mongoloid introgression. Possibly, the two main waves of genetic flux from East Asia and the Near East reached their optimum ≈500 years ago, splitting off a strongly introgressing Buddhist entity and a Muslim entity that preserved its ancestral Ladakhi population structure because of cultural and religious isolation. Alternatively, the late Muslim missionary wave appealed primarily to animists and other nonconverted Ladakhi, whose bacterial genomes had acquired fewer foreign nucleotides than their Buddhist counterparts. The latter scenario agrees well with the geographic restriction of Muslims to the Kargil and Suru Valleys as well as the capital city (Leh).
H. pylori Can Help Decipher Short-Term Human History. The data presented here illustrate shortcomings of classical human genetic markers for deciphering recent human history. For example, the ragged shape of the pairwise mismatch distribution in mtDNA sequences from Muslims (Fig. 5) suggests a constant population size with a single ancestral source. In contrast, the H. pylori sequences from Muslims possess a bimodal distribution, reflecting mixed ancestry from two populations. Although neither our microsatellite genotypes nor mtDNA sequences were able to differentiate between the population structures from Buddhists and Muslims, the H. pylori sequences showed different population structures that correlate well with recent history. The genetic diversity of H. pylori sequences is nearly twice as high as within the mtDNA hypervariable region HVS1 (see Table 3). Moreover, the bacterial sequence diversity is probably uniform over its entire 1.6-Mb genome, unlike mtDNA, in which the high sequence diversity is restricted to a region of only a few hundred base pairs. A global database (http://pubmlst.org/helicobacter) now exists that provides hundreds of H. pylori genotypes from numerous sources.
Analysis based on H. pylori sequences also possesses a number of drawbacks. Because of the possibility of mixed colonization, it is preferable to use DNA isolated from cultivated bacteria after single-colony purification as a PCR template rather than direct amplification from gastric fluid. Obtaining human DNA is simpler than microbiological manipulations. Experiments with ancient DNA also would pose more stringent requirements than for human DNA or, perhaps, be impossible. Finally, only half of the current global population is colonized with H. pylori and its prevalence is declining in industrialized countries. Despite these disadvantages, our comparative approach showed that analysis of H. pylori sequences provides more information about recent Ladakh history than do classical human genetic markers. We showed (28) a close correlation between H. pylori populations and ancient patterns of human migration. The results presented here concern a much shorter time scale, centering on events in the last 1,000 years. We conclude that H. pylori genetic diversity may be as informative as most human genetic markers, or even more informative, for deducing population migrations in the last 5,000 years.
Another important aspect of H. pylori biology concerns its high recombination rate (44): it is an “unusually sexual bacterium” (45). High recombination rates facilitate the exchange of genes or gene fragments between genetically distant isolates that cocolonize single humans in interethnic marriages. When such genetic influx is largely restricted to families, it results in a reproductive pattern resembling that of humans, except for being haploid. Thus, H. pylori is more than a proxy measure of human ancestry and might even be considered to represent one of the most promising alternatives to classical human genetic markers for analyzing introgression between human populations.
Supplementary Material
Acknowledgments
We thank Dr. Tsering Norboo (SNM Hospital, Leh, Ladakh) without whose help the study would not have been possible; Ines Diehl, Ulrike Reichard, Gabriele Schönian, and Judith Romero-Gallo for technical assistance; Philippe Roumagnac for numerous discussions; and two anonymous reviewers for helpful comments. This work was supported by Deutsche Forschungsgemeinschaft Grants Ac36/9-3 and Ac36/10-3 and National Institutes of Health Grant R0 IGM 63270.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Cann, R. L. (2001) Science 291, 1742–1748. [DOI] [PubMed] [Google Scholar]
- 2.Cavalli-Sforza, L. L. (1998) TIG 14, 60–65. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein, D. B. & Chikhi, L. (2002) Annu. Rev. Genomics Hum. Genet. 3, 129–152. [DOI] [PubMed] [Google Scholar]
- 4.Cann, R. L., Stoneking, M. & Wilson, A. C. (1987) Nature 325, 31–36. [DOI] [PubMed] [Google Scholar]
- 5.Cavalli-Sforza, L. L. & Feldman, M. W. (2003) Nat. Genet. 33, Suppl., 266–275. [DOI] [PubMed] [Google Scholar]
- 6.Wakeley, J. (1993) J. Mol. Evol. 37, 613–623. [DOI] [PubMed] [Google Scholar]
- 7.Ruiz-Linares, A., Ortiz-Barrientos, D., Figueroa, M., Mesa, N., Munera, J. G., Bedoya, G., Velez, I. D., Garcia, L. F., Perez-Lezaun, A., Bertranpetit, J., et al. (1999) Proc. Natl. Acad. Sci. USA 96, 6312–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldstein, D. B., Ruiz, L. A., Cavalli-Sforza, L. L. & Feldman, M. W. (1995) Proc. Natl. Acad. Sci. USA 92, 6723–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jorde, L. B., Rogers, A. R., Bamshad, M., Watkins, W. S., Krakowiak, P., Sung, S., Kere, J. & Harpending, H. C. (1997) Proc. Natl. Acad. Sci. USA 94, 3100–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ke, Y., Su, B., Song, X., Lu, D., Chen, L., Li, H., Qi, C., Marzuki, S., Deka, R., Underhill, P., et al. (2001) Science 292, 1151–1153. [DOI] [PubMed] [Google Scholar]
- 11.Poloni, E. S., Semino, O., Passarino, G., Santachiara-Benerecetti, A. S., Dupanloup, I., Langaney, A. & Excoffier, L. (1997) Am. J. Hum. Genet. 61, 1015–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semino, O., Passarino, G., Oefner, P. J., Lin, A. A., Arbuzova, S., Beckman, L. E., De Benedictis, G., Francalacci, P., Kouvatsi, A., Limborska, S., et al. (2000) Science 290, 1155–1159. [DOI] [PubMed] [Google Scholar]
- 13.Su, B., Jin, L., Underhill, P., Martinson, J., Saha, N., McGarvey, S. T., Shriver, M. D., Chu, J., Oefner, P., Chakraborty, R., et al. (2000) Proc. Natl. Acad. Sci. USA 97, 8225–8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Underhill, P. A., Shen, P., Lin, A. A., Jin, L., Passarino, G., Yang, W. H., Kauffman, E., Bonne-Tamir, B., Bertranpetit, J., Francalacci, P., et al. (2000) Nat. Genet. 26, 358–361. [DOI] [PubMed] [Google Scholar]
- 15.Collins, F. S., Guyer, M. S. & Charkravarti, A. (1997) Science 278, 1580–1581. [DOI] [PubMed] [Google Scholar]
- 16.Wang, D. G., Fan, J. B., Siao, C. J., Berno, A., Young, P., Sapolsky, R., Ghandour, G., Perkins, N., Winchester, E., Spencer, J., et al. (1998) Science 280, 1077–1082. [DOI] [PubMed] [Google Scholar]
- 17.Zhao, Z., Jin, L., Fu, Y. X., Ramsay, M., Jenkins, T., Leskinen, E., Pamilo, P., Trexler, M., Patthy, L., Jorde, L. B., et al. (2000) Proc. Natl. Acad. Sci. USA 97, 11354–11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reich, D. E., Cargill, M., Bolk, S., Ireland, J., Sabeti, P. C., Richter, D. J., Lavery, T., Kouyoumjian, R., Farhadian, S. F., Ward, R., et al. (2001) Nature 411, 199–204. [DOI] [PubMed] [Google Scholar]
- 19.Rosenberg, N. A., Pritchard, J. K., Weber, J. L., Cann, H. M., Kidd, K. K., Zhivotovsky, L. A. & Feldman, M. W. (2002) Science 298, 2381–2385. [DOI] [PubMed] [Google Scholar]
- 20.Sugimoto, C., Kitamura, T., Guo, J., Al Ahdal, M. N., Shchelkunov, S. N., Otova, B., Ondrejka, P., Chollet, J. Y., El Safi, S., Ettayebi, M., et al. (1997) Proc. Natl. Acad. Sci. USA 94, 9191–9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo, J., Sugimoto, C., Kitamura, T., Ebihara, H., Kato, A., Guo, Z., Liu, J., Zheng, S. P., Wang, Y. L., Na, Y. Q., et al. (1998) J. Gen. Virol. 79, 2499–2505. [DOI] [PubMed] [Google Scholar]
- 22.Neel, J. V., Biggar, R. J. & Sukernik, R. I. (1994) Proc. Natl. Acad. Sci. USA 91, 10737–10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher, M. C., Koenig, G. L., White, T. J., San Blas, G., Negroni, R., Alvarez, I. G., Wanke, B. & Taylor, J. W. (2001) Proc. Natl. Acad. Sci. USA 98, 4558–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matisoo-Smith, E., Roberts, R. M., Irwin, G. J., Allen, J. S., Penny, D. & Lambert, D. M. (1998) Proc. Natl. Acad. Sci. USA 95, 15145–15150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kersulyte, D., Mukhopadhyay, A. K., Velapatino, B., Su, W., Pan, Z., Garcia, C., Hernandez, V., Valdez, Y., Mistry, R. S., Gilman, R. H., et al. (2000) J. Bacteriol. 182, 3210–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamaoka, Y., Orito, E., Mizokami, M., Gutierrez, O., Saitou, N., Kodama, T., Osato, M. S., Kim, J. G., Ramirez, F. C., Mahachai, V., et al. (2002) FEBS Lett. 517, 180–184. [DOI] [PubMed] [Google Scholar]
- 27.Ghose, C., Perez-Perez, G. I., Dominguez-Bello, M. G., Pride, D. T., Bravi, C. M. & Blaser, M. J. (2002) Proc. Natl. Acad. Sci. USA 99, 15107–15111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falush, D., Wirth, T., Linz, B., Pritchard, J. K., Stephens, M., Kidd, M., Blaser, M. J., Graham, D. Y., Vacher, S., Perez-Perez, G. I., et al. (2003) Science 299, 1582–1585. [DOI] [PubMed] [Google Scholar]
- 29.Drumm, B., Perez-Perez, G. I., Blaser, M. J. & Sherman, P. M. (1990) N. Engl. J. Med. 322, 359–363. [DOI] [PubMed] [Google Scholar]
- 30.Rothenbacher, D., Bode, G., Berg, G., Knayer, U., Gonser, T., Adler, G. & Brenner, H. (1999) J. Infect. Dis. 179, 398–402. [DOI] [PubMed] [Google Scholar]
- 31.Tindberg, Y., Bengtsson, C., Granath, F., Blennow, M., Nyren, O. & Granstrom, M. (2001) Gastroenterology 121, 310–316. [DOI] [PubMed] [Google Scholar]
- 32.Kivi, M., Tindberg, Y., Sorberg, M., Casswall, T. H., Befrits, R., Hellstrom, P. M., Bengtsson, C., Engstrand, L. & Granstrom, M. (2003) J. Clin. Microbiol. 41, 5604–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jina, P. S. (2001) in Religious History of Ladakh, ed. Jina, P. S. (Sri Satguru, Delhi, India), pp. 20–23.
- 34.Kaul, S. & Kaul, H. N. (1992) in Ladakh Through the Ages, Towards a New Identity (Nataraj Books, Springfield, VA), pp. 118–141.
- 35.Srinivas, S. (1998) The Mouths of People, the Voice of god (Oxford Univ. Press, New York).
- 36.Romero-Gallo, J., Perez-Perez, G. I., Novick, R. P., Kamath, P., Norbu, T. & Blaser, M. J. (2002) Clin. Diagn. Lab Immunol. 9, 1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson, S., Bankier, A. T., Barrell, B. G., de Bruijn, M. H., Coulson, A. R., Drouin, J., Eperon, I. C., Nierlich, D. P., Roe, B. A., Sanger, F., et al. (1981) Nature 290, 457–465. [DOI] [PubMed] [Google Scholar]
- 38.Oota, H., Saitou, N. & Ueda, S. (2002) Anthropol. Sci. 110, 293–312. [Google Scholar]
- 39.Pritchard, J. K., Stephens, M. & Donnelly, P. (2000) Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Achtman, M., Azuma, T., Berg, D. E., Ito, Y., Morelli, G., Pan, Z.-J., Suerbaum, S., Thompson, S., van der Ende, A. & van Doorn, L. J. (1999) Mol. Microbiol. 32, 459–470. [DOI] [PubMed] [Google Scholar]
- 41.Bonatto, S. L. & Salzano, F. M. (1997) Am. J. Hum. Genet. 61, 1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marjoram, P. & Donnelly, P. (1994) Genetics 136, 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wakeley, J. (1996) Theor. Popul. Biol. 49, 369–386. [DOI] [PubMed] [Google Scholar]
- 44.Falush, D., Kraft, C., Taylor, N. S., Correa, P., Fox, J. G., Achtman, M. & Suerbaum, S. (2001) Proc. Natl. Acad. Sci. USA 98, 15056–15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spratt, B. G. (2003) Science 299, 1528–1529. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.