

Abstract
Mesenchymal stem cells (MSC) are multipotent cells, functioning as precursors to a variety of cell types including adipocytes, osteoblasts, and chondrocytes. Between osteogenic and adipogenic lineage commitment and differentiation, a theoretical inverse relationship exists, such that differentiation towards an osteoblast phenotype occurs at the expense of an adipocytic phenotype. This balance is regulated by numerous, intersecting signaling pathways that converge on the regulation of two main transcription factors: peroxisome proliferator-activated receptor-γ (PPARγ) and Runt-related transcription factor 2 (Runx2). These two transcription factors, PPARγ and Runx2, are generally regarded as the master regulators of adipogenesis and osteogenesis. This review will summarize signaling pathways that govern MSC fate towards osteogenic or adipocytic differentiation. A number of signaling pathways follow the inverse balance between osteogenic and adipogenic differentiation and are generally proosteogenic/antiadipogenic stimuli. These include β-catenin dependent Wnt signaling, Hedgehog signaling, and NELL-1 signaling. However, other signaling pathways exhibit more context-dependent effects on adipogenic and osteogenic differentiation. These include bone morphogenic protein (BMP) signaling and insulin growth factor (IGF) signaling, which display both proosteogenic and proadipogenic effects. In summary, understanding those factors that govern osteogenic versus adipogenic MSC differentiation has significant implications in diverse areas of human health, from obesity to osteoporosis to regenerative medicine.
1. Introduction
Mesenchymal stem cells (MSC) are multipotent stromal cells capable of self-renewal and capable of multilineage mesenchymal differentiation [1]. These nonhematopoietic cells can differentiate down multiple mesenchymal lineages, including osteogenic, chondrogenic, adipogenic, myogenic, and neurogenic lineages [2] (Figure 1). Originally identified in the bone marrow, MSC are readily obtained from numerous mesenchymal tissue types, including skeletal muscle and adipose depots. In particular, adipose tissue is an attractive source for MSC isolation, as it is readily accessible with minimal morbidity by routine liposuction procedures [3–5]. Indeed, human adipose-derived stromal cells (or hASC) have been demonstrated to have significant potential for use in tissue engineering applications, as shown in preclinical animal models [6]. However, the uncultured stromal vascular fraction of adipose tissue represents a heterogeneous cell population that is not immediately suitable for bone formation, prompting investigators to search for alternative methods for MSC purification other than culture propagation [5, 7]. Alternative sources for MSC derivation include nearly any vascularized tissue, from umbilical cord to oral gingiva [8, 9]. Indeed, the perivascular origin of MSC has become an increasingly accepted theory [10–13].
Figure 1.
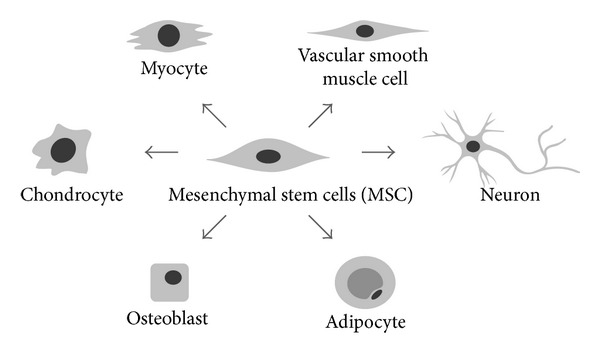
Multilineage differentiation of mesenchymal stem/stromal cells (MSC). Multipotent mesenchymal stem cells (MSC) have been derived from numerous vascularized tissue sources, including bone marrow, adipose, and skeletal muscle tissue, among others. Multilineage differentiation includes osteoblastic, chondrogenic, myogenic, smooth muscle, and neurogenic differentiation. With progressive differentiation toward a mature cell phenotype, often the capacity for differentiation down a competing lineage is lost.
MSC derived from bone marrow (BMSC) are relatively scarce in number but like all MSC have a capacity for repeated culture expansion while retaining their growth potential and multipotency [2]. BMSC typically express cell markers such as CD29, CD44, CD73, CD105, and CD166 and are negative for hematopoietic markers [2, 14]. However, it is worth noting that, with the diversity in sources and protocols for derivation, MSC cell identity remains relatively poorly defined across species, tissue type, and culture strain [15]. Upon induction and differentiation towards a specific mesenchymal lineage, the gene expression of MSC shifts until the phenotype is characteristic of the target cell. While MSC differentiation can be directed by multiple microenvironmental factors (such as mechanical forces [16], electrical currents [17–19], and magnetic fields [20]), this review will specifically focus on cytokine signaling that govern MSC lineage differentiation.
As mentioned, MSC function as precursors to a variety of mature mesenchymal cell types, including adipocytes. Various theoretical definitions of the process of adipocyte differentiation, or adipogenesis, have been put forth. Sinal and colleagues characterize adipogenesis in two phases: the determination phase and the terminal differentiation phase [21]. During the determination phase, multipotent MSC commit to the adipocyte lineage. Morphologically, preadipocytes have a fibroblastic phenotype and are not readily distinguishable from their MSC precursors. During the terminal differentiation phase, preadipocytes become adipocytes and acquire new functions, including lipid synthesis and storage, as well as adipocyte-specific protein production [22]. Rosen and colleagues define adipogenesis as a shift in gene expression from MSC to a phenotype that defines mature adipocytes [23], including expression of CD24, CD29, CD34, and CD36, among others [24–26]. Overall, adipogenesis is a sequentially and temporally ordered process involving multiple signaling cascades that converge at the level of peroxisome proliferator-activated receptor-γ (PPARγ) transcriptional activity [21, 23].
Of course, MSC also give rise to osteoblasts to form bone [2]. The process starts with commitment of osteoprogenitor cells and differentiation into pre-osteoblasts, which eventually develop into mature osteoblasts [27]. In turn, mature osteoblasts will become entombed in osteoid to become osteocytes. At its most basic level, osteoblast differentiation requires expression of the key transcription factor, Runt-related transcription factor 2 (Runx2) [27], which will be reviewed in the coming sections. However, Runx2 expression is not sufficient for osteoblast maturation, as other transcriptions factors and extracellular signals reviewed in this chapter are also involved [28]. The development of an immature osteoblast into a mature one can be categorized into phases of proliferation, maturation, matrix synthesis, and matrix mineralization (reviewed in [27]). Osteoblasts synthesize bone matrix to initially form bone and later function in bone remodeling and mineral metabolism [28].
The commitment and differentiation of MSC towards an adipogenic or osteogenic cell fate depend on a variety of signaling and transcription factors. A large body of experimental evidence suggests that an inverse correlation exists between adipogenesis and osteogenesis (Figure 2) [29, 30]. The evidence for an inverse relationship is primarily based on in vitro studies in which culture supplements upregulate osteogenic differentiation with associated downregulation of adipogenic differentiation, or vice versa [31–34]. Several bipotent or multipotent cell lines are commonly used. These include the pluripotent C3H10T1/2 cell line and the murine BMSC line M2-10B4 [35, 36]. Several cell signaling cascades exemplify proosteogenic/antiadipocytic stimuli and will be discussed below. These include β-catenin dependent Wnt signaling (as well as β-catenin independent signaling) [37, 38], Hedgehog signaling [39, 40], and NELL-1 (NEL-like protein 1) signaling [41, 42]. Dissimilarly, various signaling cascades demonstrate positive regulation of both osteogenesis and adipogenesis. Perhaps the most clinically relevant examples are bone morphogenetic proteins (BMPs), of which BMP-2 and BMP-7 are available for orthopaedic application [43, 44]. While the majority of BMPs promotes osteogenic commitment and differentiation of MSC [45, 46], BMPs also demonstrate proadipogenic effects [47, 48]. Insulin-like growth factor (IGF) signaling likewise demonstrates dual proosteogenic/proadipogenic effects. This review will sequentially discuss the effects of these diverse signaling cascades that coordinately govern MSC osteogenesis and adipogenesis.
Figure 2.
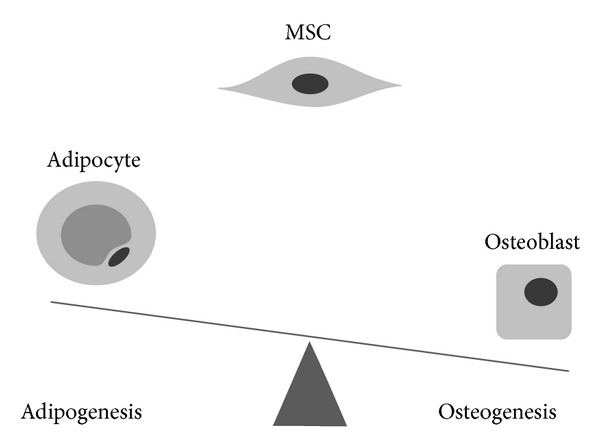
Theoretical inverse relationship between osteogenic and adipogenic programming. Multiple signaling pathways have been demonstrated to preferentially induce osteogenic programming at the expense of adipogenesis, or vice versa. In this regard, the differentiation of an MSC into either an adipocytic or osteoblastic phenotype can be theorized as a seesaw, where induction of one lineage comes at the expense of the other. However, numerous exceptions exist to this simplification.
2. Control of Adipogenesis and Osteogenesis by Transcription Factor Activity: Runx2 and PPARγ
Signaling cascades which promote MSC osteogenic and/or adipogenic lineage differentiation generally converge on two key transcription factors: PPARγ and Runx2. PPARγ is generally considered the master regulator of adipogenesis and also has well-described anti-osteoblastogenic effects. Likewise, Runx2 is regarded as the master regulator of osteogenesis. Together, they are in large part responsible for mediating the effects of various cytokines in determination of adipogenic versus osteogenic MSC differentiation. Typically, increased expression of one transcription factor is associated with downregulation of the other [49–52]. Of course, a number of other key transcriptional factors exert effects independent and in association with Runx2 and PPARγ. For example, Osterix and CCAAT/enhancer-binding family of proteins (C/EBP) play important adjunctive roles (see [53, 54] for a comprehensive review of the osteogenic and adipogenic functions of Osterix and C/EBP).
3. The Master Osteogenic Transcription Factor, Runx2
Originally identified as the binding site for polyomavirus enhancer binding protein (PEBP), Runx was later identified as the Moloney murine leukemia virus enhancer core binding protein [55]. The Runx family consists of three distinct proteins: Runx1-3, all of which are comprised of a varying α subunit with the same β subunit [56, 57]. In order to bind to DNA, Runx proteins must form a heterodimer with transcriptional coactivator core binding factor β (Cbfβ), a cotranscription factor [56]. The DNA binding domain of the Runx family, known as Runt, is homologous to the Runt sequence in Drosophila. Members of the Runx family have various roles in determining stem cell commitment; Runx1 determines hematopoietic stem cell differentiation [58], Runx2 determines osteoblastic and chondrogenic cell differentiation [59], and Runx3 has roles in epithelial differentiation, neurogenesis, and chondrocyte differentiation [60, 61]. Runx has also been postulated as both an oncogene and tumor suppressor: Runx family loss of function seems to be a key event in certain myeloid, lymphoid, and epithelial cancers [62, 63]. Retroviral overexpression of Runx2 has demonstrated oncogenic functions [64]. However, data does suggest that Runx3 acts as a tumor suppressor, as it is methylated and downregulated in cancer derived cell lines [65–68]. As the Runx family is structurally similar, it is possible that tissue-specific Runx activity allows for its complex role in carcinogenesis. In regard to osteogenic differentiation, Runx2 activates and regulates osteogenesis as the targeted gene of many signaling pathways, including but not limited to transforming growth factor-beta 1 (TGF-β1), BMP, Wingless type (Wnt), Hedgehog (HH), and (Nel)-like protein type 1 (NELL-1) [69–71]. Mice with a homozygous mutation for Cbfa-1 deficiency (Runx2 −/−) have an absence of differentiated osteoblasts and bone and die shortly after birth [72]. Such Runx2 null phenotypes cannot be rescued by the overexpression of other osteogenic factors, although the cleidocranial dysplasia-like phenotype of Runx2 +/− mice can be partially rescued [73, 74]. While Runx2 is not a key regulator of adipocyte differentiation, its function in promoting osteogenesis may subvert potential adipocyte lineage differentiation in MSC.
4. The Master Adipogenic Transcription Factor, PPARγ
Peroxisome proliferator-activated receptors are members of the steroid/thyroid hormone receptor gene superfamily [75]. Initially named for PPARα [76], subsequent structural analogs PPARδ and PPARγ were since discovered. All three PPARs are found in mammals and are activated by polyunsaturated fatty acids [77], interacting with binding sites on targeted genes by forming heterodimers with the retinoid X receptor (RXR) in order to recruit transcriptional coactivator proteins [78]. While both PPARα and PPARδ are expressed during adipogenesis, PPARγ is adipocyte restricted and more rapidly increases in expression during early adipogenesis [79, 80]. PPARγ is expressed during adipogenesis as two isoforms, PPARγ1 and PPARγ2, the latter being predominant in adipose tissue [21]. PPARγ1 is expressed at lower levels in adipose tissue among other tissues, including breast and prostatic tissue [81–83]. PPARγ is principally regarded as the master regulator of adipogenesis, for no other factor can rescue adipocyte formation in the event of PPARγ knockout, and generally all proadipogenic cell signaling pathways converge with PPARγ [84].
It is currently believed that a ligand-dependent activation of PPARγ must occur for any proadipogenic effects. Even then, the ligand is only necessary in the commitment phase for the adipocyte lineage, whereas PPARγ expression is necessary for both commitment and differentiation phases [84, 85]. One study demonstrated that differentiation of non-adipogenic fibroblasts required PPARγ activation through exposure to an exogenous ligand. By contrast, preadipocytes were able to continue with adipogenic differentiation without exposure to ligand [84]. One such set of ligands for PPARγ is thiazolidinediones (TZDs), which are potent PPARγ agonist among several other derivatives of polyunsaturated acids [86]. Recently, there have been several endogenous molecules derived from fatty acids found to bind and activate PPARγ, although induced adipogenesis [84, 85]. Moreover, recent studies show that ectopic expression of a mutant form of PPARγ without functional ligand-binding domains was able to support adipocyte differentiation [87], which inserts some doubt into the absolute requirement for PPARγ ligand activation.
Studies from genetic manipulation of PPARγ in mice have confirmed its central role in adipogenic differentiation. Cells derived from PPARγ +/− mice demonstrate a reduced ability to differentiate into adipocytes [84]. PPARγ-deficient embryonic stem cells fail to differentiate into adipocytes and instead differentiate into osteoblasts. Additionally, PPARγ +/− mice have demonstrated increased bone mass with increased osteoblastogenesis, while having a marked decrease in fat stores [84]. Similarly, mice with mutation in PPARγ2 have decreased expression of both PPARγ1 and PPARγ2 in white adipose tissue, while exhibiting increased bone formation [47]. In another approach, selective deletion of PPARγ in murine adipose tissue led to a loss of both brown and white adipocytes [22].
There is much evidence supporting the anti-osteoblastogenic and proadipogenic properties of PPARγ. Several PPARγ agonists/ligands, namely, TZD rosiglitazone and 15-deoxy-delta (12,14)-PGJ2, promote BMSC adipogenesis while inhibiting osteogenesis [88, 89]. However, not all agonists obtain this effect, as it depends on affinity of the ligand. For example, the partial agonist GW0072 inhibits MSC osteogenesis without necessarily affecting adipogenesis. In contrast, 9-hydroxyoctadecadienoic acid stimulates adipogenesis while not affecting osteoblastogenesis [88]. A similar pattern is seen in vivo, where chronic treatment of mice with low-affinity TZD troglitazone induces increased bone marrow adipocytes, without affecting bone mass [90]. Conversely, treatment with high-affinity TZD rosiglitazone decreases bone mineral density, rate of bone formation, and trabecular bone volume in addition to upregulating bone marrow adiposity [90, 91]. This inhibition of osteogenesis by high-affinity rosiglitazone was also associated with suppression of osteogenic transcription factors, including Runx2 [89]. Low-affinity agonist, netoglitazone, weakly inhibited osteoblastogenesis while inducing adipogenesis in vitro in a PPARγ2-dependent manner [89]. In vivo, neglitazone did not demonstrate an effect on bone, with unaffected expression levels of Runx2 [89].
5. Control of Adipogenesis and Osteogenesis by Wnt Signaling
Over the course of the past several decades, wingless-type MMTV integration site (Wnt) signaling has been identified to play an essential role in cell fate determination, proliferation, and differentiation [92, 93]. Dysregulation/hyperactivation of Wnt signaling is associated with numerous diseases such as neurodegeneration [94], gastrointestinal cancers [95], and osteoporosis [92]. To date, over nineteen Wnt receptors and coreceptors have been identified throughout seven families of proteins [93]. Collectively, Wnt signaling has demonstrated both proosteogenic and antiadipogenic activities, through both canonical (β-catenin dependent) and noncanonical (β-catenin independent) pathways (Figure 3).
Figure 3.

Schematic of β-catenin dependent and independent Wnt signaling pathways. Wnt signaling transduction occurs via β-catenin dependent or β-catenin independent signaling pathways. In β-catenin dependent signaling, extracellular Wnt ligands bind to the LRP5-Frizzled (Frz) complex to activate intracellular disheveled (DSH). This subsequently inhibits the intracellular complex comprised of axin, glycogen synthase kinase 3 (GSK3), and adenomatosis polyposis coli (APC) protein. This inhibits the cytosolic degradation of β-catenin, which accumulates and is free to enter the nucleus to heterodimerize with lymphoid enhancer-binding factor/T cell factor (LEF/TCF1) and mediate effects of gene transcription. Under the β-catenin independent signaling pathway, a similar transmembrane complex forms between Wnt, Frz, DSH, and Ror2 and activates secondary messengers.
The β-catenin dependent pathway initiates with the binding of extracellular Wnt ligands to the seven-pass transmembrane frizzled receptors (Frz) expressed at the cell surface [96]. This induces complex formation with transmembrane low-density lipoprotein receptor (LRP5/6) coreceptor, as well as intracellular proteins of the disheveled (DSH) family [97]. The resulting activation of DSH then functions to inhibit a second, intracellular complex comprised of axin, glycogen synthase kinase 3 (GSK3), and adenomatosis polyposis coli (APC) protein (Figure 3). GSK3 normally phosphorylates β-catenin, promoting its degradation. Wnt stimulation inhibits the Axin/GSK3/APC complex, and β-catenin accumulates rather than being degraded, and levels of nuclear β-catenin increase. Once inside the nucleus, β-catenin can heterodimerize with lymphoid enhancer-binding factor/T cell factor [97]. Ultimately, β-catenin dependent Wnt signaling elicits gene transcriptional activity to influence MSC lineage determination [98] (see [92] for a more comprehensive review). While the noncanonical Wnt pathway is similar in that it involves extracellular Wnt binding to frizzled receptors (Frz) and DSH downstream, it otherwise diverges to mediate its effects through a β-catenin independent manner [99–101]. Please see [102] for a more detailed review of noncanonical Wnt signaling.
Canonical Wnt signaling has well-established effects on bone mass in both animal models and human patients. LRP5 mutational studies first identified a critical role for Wnt signaling in bone maintenance [103]. LRP5 loss-of-function mutations cause pseudo-glioma syndrome, characterized by a low bone mass phenotype. Conversely, LRP5 gain-of-function mutations result in a high bone mass phenotype [104–106]. A direct role for β-catenin in regulating osteoblast and osteoclast activity has been repeatedly observed [107]. For example, in mesenchymal osteoblastic precursors, β-catenin deficiency leads to arrest of osteoblast development at an early stage and consequent embryonic skeletal defects [107–110]. Similarly, in committed osteoblasts, β-catenin deficiency results in impaired maturation and mineralization [111, 112]. As well, Wnt/β-catenin signaling activity in both mature and osteoblastic precursors leads to altered OPG/RANKL elaboration and secondary reductions in osteoclast activity and bone resorption [113, 114]. Accordingly, current clinical applications for osteoporosis target Wnt inhibitors to stimulate formation of new bone and inhibit bone resorption, or so-called “inhibitors to Wnt inhibitors.” Currently targeted Wnt signaling antagonists include Sclerostin (SOST) and Dickkopf-1 (DKK1) [115]. Expectedly, inhibition of these antagonists, via anti-SOST and anti-DKK1, respectively, has been shown to stimulate bone formation and increase bone mineral density, with phase II clinical trials (for anti-SOST) and preclinical trials (for anti-DKK1) underway [116–118]
Various members of the Wnt signaling family have been identified to inhibit the early stages of adipogenesis [119]. For example, WNT10B has been shown to maintain 3T3-L1 preadipocytes in an undifferentiated state via inhibition of PPARγ and C/EBP-α [120–122]. Similarly, activation of β-catenin via ectopic expression of Wnt1 also leads to direct suppression of PPARγ and prevention of 3T3-L1 cell adipogenic differentiation [120, 121]. Interestingly, this negative inhibition is reciprocal, in that upregulation of PPARγ functions to inhibit β-catenin signaling [120, 121, 123]. Conversely, inhibition of Wnt/β-catenin signaling via treatment with DKK family proteins positively regulates adipogenesis [119, 120, 124]. Further studies suggest that the canonical ligand Wnt3a, among several others, inhibits activation of both PPARγ and C/EBPα in order to elicit its antiadipogenic effects [125]. However, while PPARγ upregulation may negatively regulate Wnt/β-catenin signaling, overexpression of PPARγ and/or C/EBPα is not sufficient in rescuing Wnt/β-catenin-mediated inhibition of adipogenesis [21, 125].
In general, Wnt/β-catenin signaling pathway activation follows the inverse pattern between the induction of MSC osteogenic and adipogenic differentiation. The activation of Wnt/β-catenin, via lithium chloride, for instance, inhibits GSK3b, which results in general in both the promotion osteogenesis and the suppression of adipogenesis [126, 127]. Similarly, Wnt10b stimulates osteogenesis in vivo to increase bone mass while blocking adipogenesis in preadipocytes in vitro via stabilization of free cystolic β-catenin [120, 124, 128]. Other canonical Wnt ligands, such as Wnt6 and Wnt10a, exhibit similar effects in stimulating osteogenesis while also inhibiting adipogenesis [129]. Not surprisingly, disruption of Wnt/β-catenin impairs osteogenesis in vitro [111, 112] while increasing adipogenesis both in vitro and in vivo [120, 124, 130]. Moreover, inhibitors of the Wnt/β-catenin pathway also demonstrate consistency with this inverse relationship between osteo- and adipogenic differentiation. DKK1, for instance, which is secreted by preadipocyte cells, inhibits osteogenesis while promoting adipogenesis in vitro [131]. The inverse relationship carries over to the noncanonical branch of Wnt signaling as well. Wnt5a, for instance, has been shown to suppress proadipogenic PPARγ transactivation when coinduced with proosteogenic Runx2 in MSC [21, 132]. Thus, seen across multiple ligands and inhibitors, Wnt signaling generally exerts proosteogenic and antiadipogenic effects in both canonical or noncanonical signal transduction pathways.
6. Control of Adipogenesis and Osteogenesis by Hedgehog Signaling
Since its original discovery in Drosophila, the Hedgehog (HH) protein family has been identified in all vertebrates and classified into three structural homologues: Sonic Hedgehog (SHH), Indian Hedgehog (IHH), and Desert Hedgehog (DHH). DHH expression is typically limited to male reproductive tract [133] and will not be further discussed. SHH and IHH are critical during embryological development. In particular, SHH plays a key role during skeletogenesis, involved in patterning of the axial, appendicular, and facial skeleton [134, 135]. Closely related to SHH through gene duplication, IHH regulates both chondrogenesis and endochondral bone formation [136]. In fact, disruption of HH signaling results in severe skeletal abnormalities, the most common of which is holoprosencephaly [137]. In regulation of stem cells, SHH is a critical moderator of cell differentiation, as it demonstrates proosteogenic and antiadipogenic properties in multiple MSC types [39].
All three HH morphogens follow the same, highly conserved HH signaling pathway (Figure 4). First, the insoluble HH polypeptide precursor undergoes conversion into a soluble, multimeric form capable of diffusing across the cell membrane. This is then autocatalytically processed from a 45 kD to a 19 kD protein, with modifications for a cholesterol moiety at the C-terminal and palmitate at the N-terminal [138]. Subsequently, the modified HH morphogen is secreted from the cell via Dispatched, a large transmembrane protein, after which it binds to the receptor Patched (PTCH), a 12-pass transmembrane protein, on the receiving cell. This binding to PTCH relinquishes Smoothened (SMO), a 7-pass transmembrane protein, from PTCH suppression, thereby enabling activation of the glioblastoma gene products (Gli) family of transcription factors (Gli1-3). Since Gli1 is a target gene of the HH pathway, it is used as a reliable marker for HH signaling activity [84]. It is important to note that HH signal transduction occurs at the primary cilia and that intraflagellar transport (IFT) proteins are required to preserve cilia during HH signaling [135]. Accordingly, these IFT proteins are essential in transferring transmembrane proteins PTCH and SMO, as movement through the cilium is required to upregulate genes targeted by HH signaling [84]. While being not fully understood, it is currently believed that HH signal transduction is mainly mediated though the Gli transcription factors, and that they are responsible for HH-induced lineage commitment during MSC differentiation.
Figure 4.

Schematic of Hedgehog signaling pathway. The initially insoluble Hedgehog (HH) ligand precursor undergoes a series of intracellular modifications before reaching an active, multimeric form. Following release from the membrane by Dispatched (DISP), the morphogen binds to Patched (PTCH), which releases Smoothened (SMO) from constitutive inhibition by PTCH. This activates the Gli2/3 complex, which goes on to promote gene expression of Gli1, while repressing the transcriptional repressor Gli3.
The antiadipogenic potential of HH signaling in MSC has been observed across a variety of adipocyte and multipotent cell lineages. Generally, adipogenesis in MSC, as it relates to HH signaling, occurs as a result of decreased Gli1, Gli2, Gli3, and PTCH expression [40]. Conversely, when the HH pathway is upregulated via SMO-activated inducer of HH signaling, such as purmorphamine [139], there is a significant decrease in adipocyte-specific markers: adipocyte fatty acid binding protein, adipsin, CD36, adiponectin, and leptin. Through the inhibition of adipogenic genes, HH signaling ultimately decreases sensitivity to insulin, which in turn reduces the expression of adipogenic transcription factors, C/EBPα and PPARγ [40]. Moreover, in vitro studies evaluating RNAi scans on Drosophila genome have confirmed the antiadipogenic function of HH signaling. Specifically, HH signaling blocked differentiation of white adipocytes. Likewise, transgenic activation of HH signaling in both Drosophila and mammalian models impaired fat formation [140, 141]. Using multipotent C3H10T1/2 cells, treatment with SHH resulted in the suppression of the proadipogenic effects of bone morphogenetic protein (BMP)2 [142].
In addition to its antiadipogenic properties, HH signaling is well known to stimulate MSC osteogenic differentiation. While the exact mechanism and stage at which HH acts during osteoblastogenesis are not completely understood, both in vivo and in vitro data suggest that bone formation occurs via a positive feedback loop. That is, HH-induced osteoblastogenesis requires BMP signaling, and together they elicit a synergistic expression of alkaline phosphatase activity [143]. This positive feedback loop is further mediated by Gli2 transcription, which serves to upregulate BMP-2 expression, which in turn activates Gli transcription [144]. In the murine MSC line C3H10T1/2, HH simultaneously induced osteoblastic differentiation while inhibiting adipogenesis [145–147]. In KS483 cells, a similar induction of osteogenesis via SHH was observed alongside inhibited adipogenesis, despite adipogenic culture conditions [148]. It is important to note that SHH induced differentiation was only observed in immature mesenchymal cell lines 3H10T1/2 and not pre-osteoblastic MC3T3-E1 or osteoblastic cell lines OS 17/2.8 and ROB-C26 [143, 147]. These data imply that SHH activity may be key in stimulating osteoblastogenesis only during early stages of cell differentiation. In summary, current data suggest that HH signaling promotes MSC osteogenic differentiation over adipogenic differentiation, primarily via Gli transcriptional factor activity.
7. Control of Adipogenesis and Osteogenesis by NELL-1 Signaling
The secreted molecule NELL-1 (NEL-like protein 1) was first discovered to have osteoinductive properties by its overexpression during premature bone formation in human sporadic coronal craniosynostosis [149, 150]. NELL-1 is expressed during both intramembranous and endochondral bone formation. Overexpression increases both differentiation and mineralization selectively in osteoblasts and is highly specific to the osteochondral lineage [151]. Transgenic mice overexpressing NELL-1 show premature cranial suture fusion and bone overgrowth, thus replicating the human observed phenotype [152]. Interestingly, the nontissue specific overexpression of NELL-1 in mice only manifested phenotypes in the calvarial bone. This finding suggests a relative osteo-specific effect of NELL-1 signaling. Conversely, downregulation of NELL-1 resulted in inhibited osteoblastogenesis in vitro in primary cultures of fetal rat calvarial cells and MC3T3 cell line cultures [152]. Moreover, complete loss of NELL-1 in mice results in significant reduction in the mineralization of calvarial bones and attenuated osteoblastogenesis [153]. Thus, NELL-1 has been shown to have a critical role in craniofacial osteogenic differentiation and bone formation [152].
The osteoblastogenic effects of NELL-1 have been studied in the context of bone tissue engineering. For example, in vivo NELL-1 administration induces significant calvarial defect healing in rats [154]. When NELL-1 was applied to a PLGA scaffold in a rat calvarial defect, decreased Osterix-producing cells were observed, concomitantly with increased bone sialoprotein, osteocalcin, and BMP-7 [149]. In vivo, several studies have demonstrated that NELL-1 has comparable bone regeneration capacity as BMP-2, in both calvarial defect and spinal fusion models, among others [149, 155]. NELL-1 has also been applied to critical-sized femoral segment defect models in rats, observing to enhanced bone regeneration/osseous union [156]. A variety of spinal fusion models have also been investigated across several animal models. For example, NELL-1 demonstrated osteoinductive properties in rat spinal fusions [154, 157], using apatite coated alginate/chitosan microparticles and β-TCP scaffolds [158]. In a sheep spinal fusion model using demineralized bone graft, NELL-1 increased both bone volume and mineral density at three months, with a similar bone-forming efficacy to BMP-2 [155]. Overall, NELL-1 demonstrates robust induction of bone throughout many in vivo models, ranging from rodents to large preclinical animals [151].
Mechanistically, NELL-1 is directly regulated by the transcription factor Runx2 [74, 151, 154]. NELL-1 is preferentially expressed in osteoblasts in levels similar to Runx2 and is most highly expressed during skeletogenesis [74, 151]. In Runx2 deficient mice, overexpression of NELL-1 was not sufficient to rescue mineralization, whereas absence of NELL-1 significantly decreased Runx2 activity in vitro [74]. Integrinβ1 was recently identified as the first cell surface receptor of NELL-1 [159]. Cell surface binding in a pre-osteoblast cell line required Integrinβ1 expression [159]. Moreover, siRNA for Integrinβ1 blocked at least some of the cellular effects of NELL-1, including induction of pre-osteoblast attachment [159]. NELL-1 is known to promote osteogenesis accompanied by activation of MAPK, canonical Wnt and HH signaling [41, 42, 160, 161] (Figure 5). NELL-1 activates both ERK1/2 and JNK1 MAPK pathways in Saos-2 osteosarcoma cell type [160]. This activation of MAPK signaling is associated with Runx2 protein phosphorylation (activation) [160]. In addition, NELL-1 induced MAPK activity is accompanied by activation of phosphate transporters Pit1 and Pit2 to increase pre-osteoblast mineralization [162]. NELL-1 induction of Wnt signaling has been observed in both osteoblastic and osteoclastic cell types and is associated with its proosteogenic and antiosteoclastic effects [161]. The activation of HH signaling by NELL-1 has thus far been observed in preadipocytes only [42].
Figure 5.
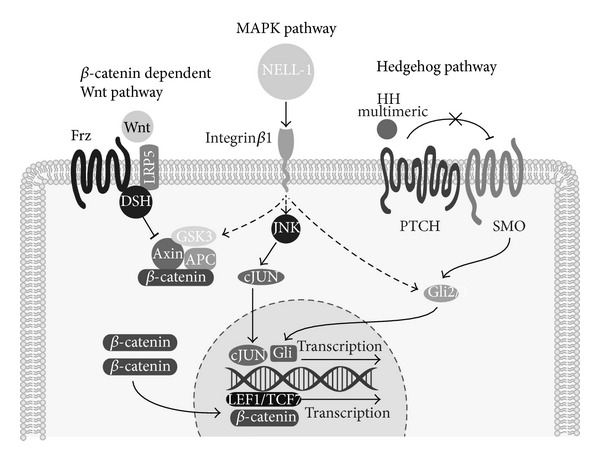
Schematic of NELL-1 signaling pathway. NELL-1 is a secreted osteoinductive protein that binds to the cell surface receptor Integrinβ1. Binding to Integrinα3 has also been reported. Multiple intracellular signaling pathways have been shown to increase after NELL-1 stimulation, including MAPK, Hedgehog, and β-catenin dependent Wnt signaling. Although the relative importance of these pathways is still undefined, NELL-1 treatment results in increased Runx2 transcription, Runx2 phosphorylation, and induction of osteogenic programming.
Recent data has shown that NELL-1 also exerts antiadipogenic effects [41]. These effects were found both in the preadipocyte cell line 3T3-L1 cells, as well as primary adipose-derived MSC (ASC) [41]. This was observed both in adipocyte specific gene expression and intracellular lipid accumulation. Recent in vivo studies have confirmed the antiadipogenic effects of NELL-1, in which direct intramedullary injection of NELL-1 reduced intramarrow adipocytes in a senile rat model [163]. This antiadipogenic effects of NELL-1 in preadipocytes is associated with activation of HH signaling, including HH signaling markers Ihh, Gli1, and Ptc1. Further studies found that coapplication of NELL-1 with cyclopamine, an antagonist for Smoothened, completely reversed or blunted the proosteogenic effects of NELL-1 [42]. Thus, NELL-1 is an osteoinductive cytokine with concomitant antiadipogenic properties. These effects may be through activation/intersection with MAPK, Wnt, and HH signaling.
8. Control of Adipogenesis and Osteogenesis by BMP Signaling
Bone morphogenetic proteins (BMPs), members of transforming growth factor-β (TGF-β) superfamily, are extracellular cytokines originally isolated from bone extract and found to induce of ectopic chondrogenesis and osteogenesis [164]. BMPs are responsible for numerous cell regulatory processes, including the differentiation and patterning of bone and cartilage [165]. Over 20 different BMPs have been identified, of which BMP-2, -4, -7, -9, and -13 are most commonly studied in the context of MSC differentiation [45, 166]. Both recombinant BMP-2 and -7 are approved by the FDA for the regeneration of bone in spinal fusion surgery and commonly used off-label for other orthopaedic applications [167, 168].
BMPs produce their effects through interaction with two serine-threonine kinase cell surface BMP receptors (BMPRs). Type II BMPRs initiate signaling upon binding to a BMP ligand, following which recruitment, phosphorylation, and activation of type I BMPRs occurs [165, 169, 170]. While there are several different type I BMPRs, only a few are involved in MSC differentiation, including BMPR-IA and BMPR-IB [47]. Several downstream BMP signaling elements exist, including Smad1/5/8, MAP Kinase, and c-Jun N-terminal kinase (JNK) signaling pathways, which are phosphorylated and thereby activated [47, 84, 171]. Of these, Smad1/5/8 signaling transduction is the most pertinent to MSC differentiation, as it is principally through the Smad-protein complexes that transcriptional regulation of adipogenic and osteogenic programming is regulated [165, 169, 170] (see [172] for a more detailed review of BMP signaling transduction).
BMP induced adipogenesis involves both Smad1/5/8 and MAPK activation [173]. BMP induced Smad1/5/8 signaling activates PPARγ via zinc finger transcription factor Schnurri-2 and C/EBPα, which exhibit synergistic, adipogenic effects [33, 174]. Accordingly, a Smad antagonist such as Smad6 reduces both PPARγ signaling and BMP-associated adipogenesis [173]. Similar to Smad1/5/8 signaling, BMP induced activation of MAPK signaling is associated with PPARγ activation and adipogenic differentiation [173]. Conversely, disruption of MAPK signaling also inhibits both PPARγ expression and BMP-associated adipogenesis [173]. Investigators have identified BMP signaling activity at the earliest stages of MSC adipogenesis [175, 176]. When MSC are forced into a preadipocyte cell lineage via exposure to 5-azacytidine, a potent inhibitor of DNA methylation, BMP-4 expression increases [175, 176]. BMP-4 has also been shown to have significance in brown adipose tissue, which prioritizes heat production over energy storage [177, 178]. Forced expression of BMP-4 in white adipocytes induces a brown adipocyte phenotype, including increased energy expenditure and insulin sensitivity [179]. Moreover, once MSC have been forced into preadipocyte cells, BMP-4 overexpression is sufficient to induce commitment to adipocyte lineage differentiation [45, 175, 180].
BMP signaling is one of the central signaling pathways involved in the induction of osteogenic differentiation and regulation of bone formation. Multiple murine studies involving genetically modified BMP ligands, BMP receptors, and BMP inhibitors demonstrate a critical role for BMP signaling in bone formation [181–184]. For example, transgenic mice with modified BMPR-IA receptors exhibit low bone mass and irregular calcification [181]. Inhibitors of BMP signaling, such as Noggin and Gremlin, impair bone formation when overexpressed [179, 185, 186]. In general, BMP induced osteogenesis utilizes both autocrine and paracrine pathways [187, 188] and works in conjunction with Osterix via both Runx2 dependent and independent pathways. BMP receptor activation in osteogenesis, as in adipogenesis, involves both Smad1/5/8 and MAPK downstream signaling activation. While 31 different BMP ligands are identified to date, only several actually promote MSC osteogenic differentiation [189]. Specifically, BMP-2, -4, -6, -7, and -9 have been shown to promote osteogenic commitment, as well as terminal osteogenic differentiation in MSC [45, 46]. BMP-2, the most commonly studied BMP ligand, induces MSC osteogenesis both in vitro and in vivo [190–197]. Furthermore, investigators have found that short-term BMP-2 treatment is both necessary and sufficient for osteogenic commitment in the C3H10T1/2 cell line [198]. It is important to note that murine-derived MSC in general show a robust osteogenic response to BMP signaling, whereas human MSC show a more variable response. For example, several studies evaluating BMP-2, -4, or -7 in human MSC did not observe reliably increased osteogenic differentiation [199]. Further investigation has suggested that higher expression of the BMP antagonist Noggin may underlie the variable response of human MSC to BMP-induced osteogenesis [200, 201].
The precise determinants that govern BMP signaling induced adipogenesis versus osteogenesis in MSC are not well understood. Two variables that may determine the effects of BMP on MSC differentiation have been observed: dosage and receptor type. In terms of dosage, lower concentrations of BMP-2 have been shown to directs towards adipocyte formation, while higher concentrations favor osteogenic differentiation in C3H10T1/2 [48]. However, these effects of dosage may be ligand- and cell-type dependent. In terms of receptor type, signaling through BMPR-IA in general induces adipogenic effects, while signaling via BMPR-1B induces osteogenic effects. For example, expression of constitutively active BMPR-IA induces adipogenic differentiation, while overexpression of inactive BMPR-IA inhibits adipogenic differentiation [47]. The converse effects were obtained by manipulation of BMPR-IB expression. Namely, constitutive BMPR-IB activation induces osteogenic differentiation while inactive BMPR-IB inhibited osteogenic differentiation [47]. However, conflicting data does exist regarding the specificity of BMPRs for lineage differentiation. For example, osteoblast-selective interference of BMPR-IA demonstrated anti-osteogenic effects including irregular calcification and decreased bone mass [181]. Thus, BMP receptor type and dosage are two known variables that have effect on MSC lineage determination, although no global rule applies [202].
9. Control of Adipogenesis and Osteogenesis by IGF Signaling
Discovered over fifty years ago, insulin-like growth factor-I (IGF-I) was originally identified as a soluble factor with insulin-like properties and induced by a growth hormone. Since then, we have developed a better understanding of this cytokine, especially in regard to its contribution towards bone formation and remodeling [203, 204] and adipogenesis. As a peptide hormone that acts in an endocrine, paracrine, and autocrine manner [205], IGF-1 primarily elicits effects via the IGF-I receptor (IGF1R) and IGF-binding proteins (IGFBPs) 1–6 [206]. While IGF-1 is primarily concentrated in the liver, it can be found systemically and is present in most peripheral tissues, including bone [204, 206, 207]. The functions of IGF-1 in bone have been well documented.
IGF-1 produces its effect by inducing several intracellular signaling pathways. IGF-1 first binds to the IGF-1 receptor, which autophosphorylates the receptor intracellularly at the kinase domain. With the receptor now activated, various protein substrates are consequently activated, including insulin receptor substrate-1 (IRS-1) and Src homology and collagen protein (SHC) [206]. IRS-1 goes on to activate the phosphoinositol 3-kinase (PI3-K), 3-PI-dependent kinase- (PDK-1), and Akt pathways, while SHC is responsible for activating the Ras/Raf/mitogen-activated protein (MAP) kinase pathways [207]. IRS-1 elicits its effect through interaction with and activation of PI3K, thereby catalyzing the phosphorylation of PIP2 to PIP3. The elevated levels of PIP3 consequently activate PDK-1 and Akt [208]. Activation of PI3K, PDK-1, and Akt has been shown to be important in skeletal growth [208, 209]. In fact, knockout Akt1/Akt2 mice demonstrate significantly impaired bone development and skeletal growth [208]. Meanwhile, SHC, which forms a complex with Grb2 and SOC, is responsible for increasing cell proliferation through activation of the Ras/Raf-1/MAPK pathway [206].
During bone remodeling, IGF-1 is released from the bone matrix to stimulate MSC osteoblastogenesis via activation of mammalian target of rapamycin (mTOR). This allows for the maintenance of both bone structure and mass, both of which were downregulated in mice with knockout of IGF-1 receptors in pre-osteoblastic cells [210]. Similarly, mice with deleted IGF-1 receptors in osteoclasts exhibit increased bone formation from decreased osteoclast formation [211]. Interestingly, IGF binding protein 3 is also a corequisite for IGF-1 in the bone matrix to stimulate new bone formation in rats [210]. Interestingly, while IGF binding protein 5 has exhibited proosteogenic properties in several studies, it also demonstrates inhibition of bone formation through impairing IGF-induced osteoblastogenesis [212]. Additionally, in serum-deprived conditions, MSC were shown to proliferate in response to IGF-1 [213]. Upstream, serum response factor (SRF) is found to regulate both IGF-1 and Runx2 signaling to control bone formation. In mice with conditional deletion of SRF in osteoblasts, Runx2 transactivity was restored via overexpression of SRF. SRF then plays an important role for IGF-1-induced osteoblastogenesis and mineralization through regulation of IGF-1 expression and Runx2 transactivity [214]. Collectively, these studies confirm the importance of IGF-1, its receptor, and respective binding protein for osteogenic differentiation and bone remodeling.
Combination of IGF-1 with various other growth factors provides additional insight on the mechanism of bone formation by IGF-1. For example, the addition of PDGF with IGF has been demonstrated to be more efficacious than either alone in terms of osteogenic induction in ASC [215]. Likewise, the combination of IGF-1 with AMD3100, an antagonist of chemokine receptor of CxCR4, showed significant augmentation of bone growth in segmental fracture murine models, associated with facilitation by the Akt/PI3K, MEK1/2-Erk1/2, and Smad2/3 signaling pathways [216]. In a distraction osteogenesis sheep model, application of both IGF-1 and TGF-β1 led to accelerated bone healing [217]. Another study found that growth hormone (GH) could increase to compensate for IGF-1 deficiency in mice to protect against inhibition of bone modeling during growth [218]. PTH is also known to stimulate both osteoblast and osteoclast function [211], with a role in modulating IGF-1 signaling through mechanisms involving IHH and ephrins [219]. Furthermore, there is a potential crosstalk between IGF-1 signaling and the integrin mechanosensing pathways, as evidenced by the failure of skeletal unloading to aid in bone growth despite IGF-1 infusion [219].
Interestingly, IGF-1 has been found to promote both adipogenic and osteogenic differentiation. For example, IGF-1 induces cell division of adipocyte precursor cells [220]. In addition, IGF receptors are involved in promoting adipogenesis through induction of advanced glycation end products (AGEs). AGEs activate both NAD(P)H oxidase and Src, which ultimately leads to the phosphorylation/activation of both IGF-1 receptor and Akt downstream in 3T3-L1 preadipocyte cells [221]. Further, Akt1/Akt2 knockout mice demonstrate impaired adipogenesis [208]. In fact, it has been shown that both Akt1 and Akt2 are necessary to induce PPARγ, the key regulator for adipogenesis. Thus, a critical threshold of Akt activity, as regulated by IGF-1, contributes to the maintenance of cell proliferation, growth, and adipogenic differentiation [208].
10. Discussion
Numerous signaling pathways induce the adipogenic and/or osteogenic differentiation of MSC, not all of which were covered in this review. The majority of signaling pathways ultimately converge downstream affecting PPARγ or Runx2 expression, transcriptional activity, or both. Although the mechanisms have not been fully discerned, many of these growth factors tend to elicit an “inverse relationship” between adipogenic and osteogenic differentiation. As discussed, Wnt, HH, and NELL-1 signaling follow this pattern, exhibiting proosteogenic/antiadipogenic effects [222]. Other well-studied signaling pathways further support this inverse relationship, including fibroblast growth factor-2 (FGF-2) [223], TGF-β1 [69, 224], and Notch signaling pathway [225], to name a few. Likewise, other transcription factors besides Runx2 demonstrate a proosteogenic, antiadipocytic relationship, one example being the recently described transcriptional activator TAZ (transcriptional activator with PDZ binding motif) [226]. However, there are a few exceptions to this pattern. For example, both IGF and BMP signaling have pleotropic, proosteogenic and proadipocytic properties [198, 227–229]. In summary, an inverse relationship exists between adipogenic and osteogenic lineage differentiation in MSC governed by diverse signaling pathways. The understanding of this relationship has far-reaching implications for the understanding of human health and treatment of human disease.
Conflict of Interests
The author declares no conflict of interests.
Acknowledgment
The author would like to thank A. Nguyen for his excellent technical assistance.
References
- 1.Jackson WM, Nesti LJ, Tuan RS. Concise review: clinical translation of wound healing therapies based on mesenchymal stem cells. Stem Cells Translational Medicine. 2012;1(1):44–50. doi: 10.5966/sctm.2011-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chamberlain G, Fox J, Ashton B, Middleton J. Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells. 2007;25(11):2739–2749. doi: 10.1634/stemcells.2007-0197. [DOI] [PubMed] [Google Scholar]
- 3.Levi B, Longaker MT. Concise review: adipose-derived stromal cells for skeletal regenerative medicine. Stem Cells. 2011;29(4):576–582. doi: 10.1002/stem.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizuno H, Tobita M, Uysal AC. Concise review: adipose-derived stem cells as a novel tool for future regenerative medicine. Stem Cells. 2012;30(5):804–810. doi: 10.1002/stem.1076. [DOI] [PubMed] [Google Scholar]
- 5.James AW, Zara JN, Zhang X, et al. Perivascular stem cells: a prospectively purified mesenchymal stem cell population for bone tissue engineering. Stem Cells Translational Medicine. 2012;1(6):510–519. doi: 10.5966/sctm.2012-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levi B, James AW, Nelson ER, et al. Human adipose derived stromal cells heal critical size mouse calvarial defects. PLoS ONE. 2010;5(6) doi: 10.1371/journal.pone.0011177.e11177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levi B, Wan DC, Glotzbach JP, et al. CD105 protein depletion enhances human adipose-derived stromal cell osteogenesis through reduction of transforming growth factor β1 (TGF-β1) signaling. The Journal of Biological Chemistry. 2011;286(45):39497–39509. doi: 10.1074/jbc.M111.256529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim DW, Staples M, Shinozuka K, et al. Wharton's Jelly-derived mesenchymal stem cells: phenotypic characterization and optimizing their therapeutic potential for clinical applications. International Journal of Molecular Sciences. 2013;14(6):11692–11712. doi: 10.3390/ijms140611692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corrao S, la Rocca G, Lo Iacono M, Corsello T, Farina F, Anzalone R. Umbilical cord revisited: from Wharton's jelly myofibroblasts to mesenchymal stem cells. Histology and Histopathology. 2013;28(10):1235–1244. doi: 10.14670/HH-28.1235. [DOI] [PubMed] [Google Scholar]
- 10.Corselli M, Crisan M, Murray IR, et al. Identification of perivascular mesenchymal stromal/stem cells by flow cytometry. Cytometry A. 2013;83(8):714–720. doi: 10.1002/cyto.a.22313. [DOI] [PubMed] [Google Scholar]
- 11.James AW, Zarab JN, Corselli M, et al. An abundant perivascular source of stem cells for bone tissue engineering. Stem Cells Translational Medicine. 2012;1(9):673–684. doi: 10.5966/sctm.2012-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corselli M, Chen CW, Sun B, et al. The tunica adventitia of human arteries and veins as a source of mesenchymal stem cells. Stem Cells and Development. 2012;21(8):1299–1308. doi: 10.1089/scd.2011.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crisan M, Yap S, Casteilla L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3(3):301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Sammons J, Ahmed N, El-Sheemy M, Hassan HT. The role of BMP-6, IL-6, and BMP-4 in mesenchymal stem cell-dependent bone development: effects on osteoblastic differentiation induced by parathyroid hormone and vitamin D3. Stem Cells and Development. 2004;13(3):273–280. doi: 10.1089/154732804323099208. [DOI] [PubMed] [Google Scholar]
- 15.Augello A, De Bari C. The regulation of differentiation in mesenchymal stem cells. Human Gene Therapy. 2010;21(10):1226–1238. doi: 10.1089/hum.2010.173. [DOI] [PubMed] [Google Scholar]
- 16.Kelly DJ, Jacobs CR. The role of mechanical signals in regulating chondrogenesis and osteogenesis of mesenchymal stem cells. Birth Defects Research C. 2010;90(1):75–85. doi: 10.1002/bdrc.20173. [DOI] [PubMed] [Google Scholar]
- 17.Hronik-Tupaj M, Rice WL, Cronin-Golomb M, Kaplan DL, Georgakoudi I. Osteoblastic differentiation and stress response of human mesenchymal stem cells exposed to alternating current electric fields. BioMedical Engineering Online. 2011;10, article 9 doi: 10.1186/1475-925X-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Creecy CM, O'Neill CF, Arulanandam BP, Sylvia VL, Navara CS, Bizios R. Mesenchymal stem cell osteodifferentiation in response to alternating electric current. Tissue Engineering A. 19(3-4):467–474. doi: 10.1089/ten.tea.2012.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammerick KE, James AW, Huang Z, Prinz FB, Longaker MT. Pulsed direct current electric fields enhance osteogenesis in adipose-derived stromal cells. Tissue Engineering A. 2010;16(3):917–931. doi: 10.1089/ten.tea.2009.0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan J, Dong L, Zhang B, Qi N. Effects of extremely low-frequency magnetic field on growth and differentiation of human mesenchymal stem cells. Electromagnetic Biology and Medicine. 2010;29(4):165–176. doi: 10.3109/01676830.2010.505490. [DOI] [PubMed] [Google Scholar]
- 21.Muruganandan S, Roman AA, Sinal CJ. Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: cross talk with the osteoblastogenic program. Cellular and Molecular Life Sciences. 2009;66(2):236–253. doi: 10.1007/s00018-008-8429-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nature Reviews Molecular Cell Biology. 2006;7(12):885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 23.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes and Development. 2000;14(11):1293–1307. [PubMed] [Google Scholar]
- 24.Berry R, Rodeheffer MS. Characterization of the adipocyte cellular lineage in vivo. Nature Cell Biology. 15(3):302–308. doi: 10.1038/ncb2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jumabay M, Abdmaulen R, Urs S, et al. Endothelial differentiation in multipotent cells derived from mouse and human white mature adipocytes. Journal of Molecular and Cellular Cardiology. 2012;53(6):790–800. doi: 10.1016/j.yjmcc.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vroegrijk IO, van Klinken JB, van Diepen JA, et al. Cd36 is important for adipocyte recruitment and affects lipolysis. Obesity. 2013 doi: 10.1002/oby.20354. [DOI] [PubMed] [Google Scholar]
- 27.Neve A, Corrado A, Cantatore FP. Osteoblast physiology in normal and pathological conditions. Cell and Tissue Research. 2011;343(2):289–302. doi: 10.1007/s00441-010-1086-1. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe K, Ikeda K. Osteoblast differentiation and bone formation. Nippon Rinsho. 2009;67(5):879–886. [PubMed] [Google Scholar]
- 29.James AW, Pang S, Askarinam A, Corselli M, et al. Additive effects of sonic hedgehog and Nell-1 signaling in osteogenic versus adipogenic differentiation of human adipose-derived stromal cells. Stem Cells and Development. 21(12):2170–2178. doi: 10.1089/scd.2011.0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pei L, Tontonoz P. Fat’s loss is bone’s gain. Journal of Clinical Investigation. 2004;113(6):805–806. doi: 10.1172/JCI21311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beresford JN, Bennett JH, Devlin C, Leboy PS, Owen ME. Evidence for an inverse relationship between the differentiation of adipocytic and osteogenic cells in rat marrow stromal cell cultures. Journal of Cell Science. 1992;102(2):341–351. doi: 10.1242/jcs.102.2.341. [DOI] [PubMed] [Google Scholar]
- 32.Dorheim M-A, Sullivan M, Dandapani V, et al. Osteoblastic gene expression during adipogenesis in hematopoietic supporting murine bone marrow stromal cells. Journal of Cellular Physiology. 1993;154(2):317–328. doi: 10.1002/jcp.1041540215. [DOI] [PubMed] [Google Scholar]
- 33.Krishnan V, Bryant HU, MacDougald OA. Regulation of bone mass by Wnt signaling. Journal of Clinical Investigation. 2006;116(5):1202–1209. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bennett CN, Longo KA, Wright WS, et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(9):3324–3329. doi: 10.1073/pnas.0408742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang Q-Q, Otto TC, Lane MD. Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(26):9607–9611. doi: 10.1073/pnas.0403100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burroughs J, Gupta P, Blazar BR, Verfaillie CM. Diffusible factors from the murine cell line M2-10B4 support human in vitro hematopoiesis. Experimental Hematology. 1994;22(11):1095–1101. [PubMed] [Google Scholar]
- 37.D'Alimonte I, Lannutti A, Pipino C, et al. Wnt signaling behaves as a “master regulator” in the osteogenic and adipogenic commitment of human amniotic fluid mesenchymal stem cells. Stem Cell Reviews and Reports. 2013;9(5):642–654. doi: 10.1007/s12015-013-9436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taipaleenmäki H, Abdallah BM, AlDahmash A, Säämänen A-M, Kassem M. Wnt signalling mediates the cross-talk between bone marrow derived pre-adipocytic and pre-osteoblastic cell populations. Experimental Cell Research. 2011;317(6):745–756. doi: 10.1016/j.yexcr.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 39.James AW, Leucht P, Levi B, et al. Sonic hedgehog influences the balance of osteogenesis and adipogenesis in mouse adipose-derived stromal cells. Tissue Engineering A. 2010;16(8):2605–2616. doi: 10.1089/ten.tea.2010.0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fontaine C, Cousin W, Plaisant M, Dani C, Peraldi P. Hedgehog signaling alters adipocyte maturation of human mesenchymal stem cells. Stem Cells. 2008;26(4):1037–1046. doi: 10.1634/stemcells.2007-0974. [DOI] [PubMed] [Google Scholar]
- 41.James AW, Pan A, Chiang M, et al. A new function of Nell-1 protein in repressing adipogenic differentiation. Biochemical and Biophysical Research Communications. 2011;411(1):126–131. doi: 10.1016/j.bbrc.2011.06.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.James AW, Pang S, Askarinam A, et al. Additive effects of sonic hedgehog and Nell-1 signaling in osteogenic versus adipogenic differentiation of human adipose-derived stromal cells. Stem Cells and Development. 2012;21(12):2170–2178. doi: 10.1089/scd.2011.0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jung RE, Windisch SI, Eggenschwiler AM, Thoma DS, Weber FE, Hämmerle CHF. A randomized-controlled clinical trial evaluating clinical and radiological outcomes after 3 and 5 years of dental implants placed in bone regenerated by means of GBR techniques with or without the addition of BMP-2. Clinical Oral Implants Research. 2009;20(7):660–666. doi: 10.1111/j.1600-0501.2008.01648.x. [DOI] [PubMed] [Google Scholar]
- 44.Bessa PC, Casal M, Reis RL. Bone morphogenetic proteins in tissue engineering: the road from laboratory to clinic, part II (BMP delivery) Journal of Tissue Engineering and Regenerative Medicine. 2008;2(2-3):81–96. doi: 10.1002/term.74. [DOI] [PubMed] [Google Scholar]
- 45.Kang Q, Song W-X, Luo Q, et al. A Comprehensive analysis of the dual roles of BMPs in regulating adipogenic and osteogenic differentiation of mesenchymal progenitor cells. Stem Cells and Development. 2009;18(4):545–558. doi: 10.1089/scd.2008.0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dorman LJ, Tucci M, Benghuzzi H. In vitro effects of bmp-2, bmp-, and bmp-13 on proliferation and differentation of mouse mesenchymal stem cells. Biomedical Sciences Instrumentation. 2012;48:81–87. [PubMed] [Google Scholar]
- 47.Chen D, Ji X, Harris MA, et al. Differential roles for bone morphogenetic protein (BMP) receptor type IB and IA in differentiation and specification of mesenchymal precursor cells to osteoblast and adipocyte lineages. Journal of Cell Biology. 1998;142(1):295–305. doi: 10.1083/jcb.142.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang EA, Israel DI, Kelly S, Luxenberg DP. Bone morphogenetic protein-2 causes commitment and differentiation in C3H10T1/2 and 3T3 cells. Growth Factors. 1993;9(1):57–71. doi: 10.3109/08977199308991582. [DOI] [PubMed] [Google Scholar]
- 49.Valenti MT, Garbin U, Pasini A, et al. Role of Ox-PAPCs in the differentiation of mesenchymal stem cells (MSCs) and Runx2 and PPARγ2 expression in MSCs-like of osteoporotic patients. PLoS ONE. 2011;6(6) doi: 10.1371/journal.pone.0020363.e20363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Su P, Xu C, et al. Melatonin inhibits adipogenesis and enhances osteogenesis of human mesenchymal stem cells by suppressing PPARγ expression and enhancing Runx2 expression. Journal of Pineal Research. 2010;49(4):364–372. doi: 10.1111/j.1600-079X.2010.00803.x. [DOI] [PubMed] [Google Scholar]
- 51.Li X, Cui Q, Kao C, Wang G-J, Balian G. Lovastatin inhibits adipogenic and stimulates osteogenic differentiation by suppressing PPARγ2 and increasing Cbfa1/Runx2 expression in bone marrow mesenchymal cell cultures. Bone. 2003;33(4):652–659. doi: 10.1016/s8756-3282(03)00239-4. [DOI] [PubMed] [Google Scholar]
- 52.Zhang X, Yang M, Lin L, et al. Runx2 overexpression enhances osteoblastic differentiation and mineralization in adipose—derived stem cells in vitro and in vivo. Calcified Tissue International. 2006;79(3):169–178. doi: 10.1007/s00223-006-0083-6. [DOI] [PubMed] [Google Scholar]
- 53.Zhou X, Zhang Z, Feng JQ, et al. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(29):12919–12924. doi: 10.1073/pnas.0912855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Darlington GJ, Ross SE, MacDougald OA. The role of C/EBP genes in adipocyte differentiation. The Journal of Biological Chemistry. 1998;273(46):30057–30060. doi: 10.1074/jbc.273.46.30057. [DOI] [PubMed] [Google Scholar]
- 55.Wang S, Wang Q, Crute BE, Melnikova IN, Keller SR, Speck NA. Cloning and characterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor. Molecular and Cellular Biology. 1993;13(6):3324–3339. doi: 10.1128/mcb.13.6.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bae S-C. Regulation mechanisms for the heterodimeric transcription factor, PEBP2/CBF. Histology and Histopathology. 1999;14(4):1213–1221. doi: 10.14670/HH-14.1213. [DOI] [PubMed] [Google Scholar]
- 57.Ogawa E, Inuzuka M, Maruyama M, et al. Molecular cloning and characterization of PEBP2β, the heterodimeric partner of a novel Drosophila runt-related DNA binding protein PEBP2α . Virology. 1993;194(1):314–331. doi: 10.1006/viro.1993.1262. [DOI] [PubMed] [Google Scholar]
- 58.North TE, Stacy T, Matheny CJ, Speck NA, de Bruijn MFTR. Runx1 Is expressed in adult mouse hematopoietic stem cells and differentiating myeloid and lymphoid cells, but not in maturing erythroid cells. Stem Cells. 2004;22(2):158–168. doi: 10.1634/stemcells.22-2-158. [DOI] [PubMed] [Google Scholar]
- 59.Yoshida CA, Yamamoto H, Fujita T, et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes and Development. 2004;18(8):952–963. doi: 10.1101/gad.1174704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Levanon D, Bettoun D, Harris-Cerruti C, et al. The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. EMBO Journal. 2002;21(13):3454–3463. doi: 10.1093/emboj/cdf370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brenner O, Levanon D, Negreanu V, et al. Loss of Runx3 function in leukocytes is associated with spontaneously developed colitis and gastric mucosal hyperplasia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(45):16016–16021. doi: 10.1073/pnas.0407180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ito Y. RUNX genes in development and cancer: regulation of viral gene expression and the discovery of RUNX family genes. Advances in Cancer Research. 2008;99:33–76. doi: 10.1016/S0065-230X(07)99002-8. [DOI] [PubMed] [Google Scholar]
- 63.Blyth K, Cameron ER, Neil JC. The RUNX genes: gain or loss of function in cancer. Nature Reviews Cancer. 2005;5(5):376–387. doi: 10.1038/nrc1607. [DOI] [PubMed] [Google Scholar]
- 64.Stewart M, Terry A, Hu M, et al. Proviral insertions induce the expression of bone-specific isoforms of PEBP2αA (CBFA1): evidence for a new myc collaborating oncogene. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(16):8646–8651. doi: 10.1073/pnas.94.16.8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goel A, Arnold CN, Tassone P, et al. Epigenetic inactivation of RUNX3 in microsatellite unstable sporadic colon cancers. International Journal of Cancer. 2004;112(5):754–759. doi: 10.1002/ijc.20472. [DOI] [PubMed] [Google Scholar]
- 66.Li Q-L, Kim H-R, Kim W-J, et al. Transcriptional silencing of the RUNX3 gene by CpG hypermethylation is associated with lung cancer. Biochemical and Biophysical Research Communications. 2004;314(1):223–228. doi: 10.1016/j.bbrc.2003.12.079. [DOI] [PubMed] [Google Scholar]
- 67.Kim TY, Lee HJ, Hwang KS, et al. Methylation of RUNX3 in various types of human cancers and premalignant stages of gastric carcinoma. Laboratory Investigation. 2004;84(4):479–484. doi: 10.1038/labinvest.3700060. [DOI] [PubMed] [Google Scholar]
- 68.Guo W-H, Weng L-Q, Ito K, et al. Inhibition of growth of mouse gastric cancer cells by Runx3, a novel tumor suppressor. Oncogene. 2002;21(54):8351–8355. doi: 10.1038/sj.onc.1206037. [DOI] [PubMed] [Google Scholar]
- 69.Lee K-S, Kim H-J, Li Q-L, et al. Runx2 is a common target of transforming growth factor β1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Molecular and Cellular Biology. 2000;20(23):8783–8792. doi: 10.1128/mcb.20.23.8783-8792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Komori T. Regulation of osteoblast differentiation by runx2. Advances in Experimental Medicine and Biology. 2010;658:43–49. doi: 10.1007/978-1-4419-1050-9_5. [DOI] [PubMed] [Google Scholar]
- 71.Pratap J, Wixted JJ, Gaur T, et al. Runx2 transcriptional activation of Indian Hedgehog and a downstream bone metastatic pathway in breast cancer cells. Cancer Research. 2008;68(19):7795–7802. doi: 10.1158/0008-5472.CAN-08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 73.Hesse E, Saito H, Kiviranta R, et al. Zfp521 controls bone mass by HDAC3-dependent attenuation of Runx2 activity. Journal of Cell Biology. 2010;191(7):1271–1283. doi: 10.1083/jcb.201009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang X, Ting K, Bessette CM, et al. Nell-1, a key functional mediator of Runx2, partially rescues calvarial defects in Runx2+/- mice. Journal of Bone and Mineral Research. 2011;26(4):777–791. doi: 10.1002/jbmr.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARγ . Annual Review of Biochemistry. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 76.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 77.Gottlicher M, Widmark E, Li Q, Gustafsson J-A. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(10):4653–4657. doi: 10.1073/pnas.89.10.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Current Opinion in Cell Biology. 1997;9(2):222–232. doi: 10.1016/s0955-0674(97)80066-x. [DOI] [PubMed] [Google Scholar]
- 79.Chawla A, Lazar MA. Peroxisome proliferator and retinoid signaling pathways co-regulate preadipocyte phenotype and survival. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(5):1786–1790. doi: 10.1073/pnas.91.5.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Adams M, Montague CT, Prins JB, et al. Activators of peroxisome proliferator-activated receptor γ have depot- specific effects on human preadipocyte differentiation. Journal of Clinical Investigation. 1997;100(12):3149–3153. doi: 10.1172/JCI119870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPARγ2: tissue-specific regulator of an adipocyte enhancer. Genes and Development. 1994;8(10):1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- 82.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 83.Zhu Y, Qi C, Korenberg JR, et al. Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(17):7921–7925. doi: 10.1073/pnas.92.17.7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tzameli I, Fang H, Ollero M, et al. Regulated production of a peroxisome proliferator-activated receptor-γ ligand during an early phase of adipocyte differentiation in 3T3-L1 adipocytes. The Journal of Biological Chemistry. 2004;279(34):36093–36102. doi: 10.1074/jbc.M405346200. [DOI] [PubMed] [Google Scholar]
- 85.Schopfer FJ, Lin Y, Baker PRS, et al. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor γ ligand. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ) The Journal of Biological Chemistry. 1995;270(22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 87.Walkey CJ, Spiegelman BM. A functional peroxisome proliferator-activated receptor-γ ligand-binding domain is not required for adipogenesis. The Journal of Biological Chemistry. 2008;283(36):24290–24294. doi: 10.1074/jbc.C800139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lecka-Czernik B, Moerman EJ, Grant DF, Lehmann JM, Manolagas SC, Jilka RL. Divergent effects of selective peroxisome proliferator-activated receptor-γ2 ligands on adipocyte versus osteoblast differentiation. Endocrinology. 2002;143(6):2376–2384. doi: 10.1210/endo.143.6.8834. [DOI] [PubMed] [Google Scholar]
- 89.Lazarenko OP, Rzonca SO, Suva LJ, Lecka-Czernik B. Netoglitazone is a PPAR-gamma ligand with selective effects on bone and fat. Bone. 2006;38(1):74–84. doi: 10.1016/j.bone.2005.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146(3):1226–1235. doi: 10.1210/en.2004-0735. [DOI] [PubMed] [Google Scholar]
- 91.Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B. Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology. 2004;145(1):401–406. doi: 10.1210/en.2003-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim W, Kim M, Jho EH. Wnt/beta-catenin signalling: from plasma membrane to nucleus. Biochemical Journal. 2013;450(1):9–21. doi: 10.1042/BJ20121284. [DOI] [PubMed] [Google Scholar]
- 93.Niehrs C. The complex world of WNT receptor signalling. Nature Reviews Molecular Cell Biology. 13(12):767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- 94.Berwick DC, Harvey K. The importance of Wnt signalling for neurodegeneration in Parkinson's disease. Biochemical Society Transactions. 40(5):1123–1128. doi: 10.1042/BST20120122. [DOI] [PubMed] [Google Scholar]
- 95.White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012;142(2):219–232. doi: 10.1053/j.gastro.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xavier CP, Melikova M, Chuman Y, et al. Secreted Frizzled-related protein potentiation versus inhibition of Wnt3a/beta-catenin signaling. Cell Signal. 2013;26(1):94–101. doi: 10.1016/j.cellsig.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pandur P, Maurus D, Kühl M. Increasingly complex: new players enter the Wnt signaling network. BioEssays. 2002;24(10):881–884. doi: 10.1002/bies.10164. [DOI] [PubMed] [Google Scholar]
- 98.Etheridge SL, Spencer GJ, Heath DJ, Genever PG. Expression profiling and functional analysis of Wnt signaling mechanisms in mesenchymal stem cells. Stem Cells. 2004;22(5):849–860. doi: 10.1634/stemcells.22-5-849. [DOI] [PubMed] [Google Scholar]
- 99.Wang H-Y, Malbon CC. Wnt-frizzled signaling to G-protein-coupled effectors. Cellular and Molecular Life Sciences. 2004;61(1):69–75. doi: 10.1007/s00018-003-3165-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li F, Chong ZZ, Maiese K. Winding through the WNT pathway during cellular development and demise. Histology and Histopathology. 2006;21(1–3):103–124. doi: 10.14670/hh-21.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Davis LA, Zur Nieden NI. Mesodermal fate decisions of a stem cell: the Wnt switch. Cellular and Molecular Life Sciences. 2008;65(17):2658–2674. doi: 10.1007/s00018-008-8042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van Amerongen R. Alternative Wnt pathways and receptors. Cold Spring Harbor Perspectives in Biology. 2012;4(10) doi: 10.1101/cshperspect.a007914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Case N, Rubin J. β-catenin—a supporting role in the skeleton. Journal of Cellular Biochemistry. 2010;110(3):545–553. doi: 10.1002/jcb.22574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Little RD, Carulli JP, Del Mastro RG, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. American Journal of Human Genetics. 2002;70(1):11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gong Y, Roger B, Slee B, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 106.Boyden LM, Mao J, Belsky J, et al. High bone density due to a mutation in LDL-receptor-related protein 5. The New England Journal of Medicine. 2002;346(20):1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 107.Chen J, Long F. beta-catenin promotes bone formation and suppresses bone resorption in postnatal growing mice. Journal of Bone and Mineral Research. 2013;28(5):1160–1169. doi: 10.1002/jbmr.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132(1):49–60. doi: 10.1242/dev.01564. [DOI] [PubMed] [Google Scholar]
- 109.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Developmental Cell. 2005;8(5):739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 110.Hill TP, Später D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Developmental Cell. 2005;8(5):727–738. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 111.Holmen SL, Zylstra CR, Mukherjee A, et al. Essential role of β-catenin in postnatal bone acquisition. The Journal of Biological Chemistry. 2005;280(22):21162–21168. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- 112.Glass DA, II, Bialek P, Ahn JD, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Developmental Cell. 2005;8(5):751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 113.Takahashi N, Maeda K, Ishihara A, Uehara S, Kobayashi Y. Regulatory mechanism of osteoclastogenesis by RANKL and Wnt signals. Frontiers in Bioscience. 2011;16(1):21–30. doi: 10.2741/3673. [DOI] [PubMed] [Google Scholar]
- 114.Nie B, Zhou S, Fang X, et al. Implication of receptor activator of NF-kappaB ligand in Wnt/beta-catenin pathway promoting osteoblast-like cell differentiation. Journal of Huazhong University of Science and Technology. 2012;32(6):818–822. doi: 10.1007/s11596-012-1040-4. [DOI] [PubMed] [Google Scholar]
- 115.Gatti D, Viapiana O, Fracassi E, et al. Sclerostin and DKK1 in postmenopausal osteoporosis treated with denosumab. Journal of Bone and Mineral Research. 2012;27(11):2259–2263. doi: 10.1002/jbmr.1681. [DOI] [PubMed] [Google Scholar]
- 116.Lim V, Clarke BL. New therapeutic targets for osteoporosis: beyond denosumab. Maturitas. 2012;73(3):269–272. doi: 10.1016/j.maturitas.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 117.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. Journal of Bone and Mineral Research. 2011;26(1):19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 118.Papapoulos SE. Targeting sclerostin as potential treatment of osteoporosis. Annals of the Rheumatic Diseases. 2011;70(1):i119–i122. doi: 10.1136/ard.2010.141150. [DOI] [PubMed] [Google Scholar]
- 119.Laudes M. Role of WNT signalling in the determination of human mesenchymal stem cells into preadipocytes. Journal of Molecular Endocrinology. 2011;46(2):65–72. doi: 10.1530/JME-10-0169. [DOI] [PubMed] [Google Scholar]
- 120.Ross SE, Hemati N, Longo KA, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289(5481):950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- 121.Liu J, Farmer SR. Regulating the balance between peroxisome proliferator-activated receptor γ and β-catenin signaling during adipogenesis: a glycogen synthase kinase 3β phosphorylation-defective mutant of β-catenin inhibits expression of a subset of adipogenic genes. The Journal of Biological Chemistry. 2004;279(43):45020–45027. doi: 10.1074/jbc.M407050200. [DOI] [PubMed] [Google Scholar]
- 122.Laudes M. Role of WNT signalling in the determination of human mesenchymal stem cells into preadipocytes. Journal of Molecular Endocrinology. 2011;46(2):65–72. doi: 10.1530/JME-10-0169. [DOI] [PubMed] [Google Scholar]
- 123.Moldes M, Zuo Y, Morrison RF, et al. Peroxisome-proliferator-activated receptor γ suppresses Wnt/β-catenin signalling during adipogenesis. Biochemical Journal. 2003;376(3):607–613. doi: 10.1042/BJ20030426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bennett CN, Ross SE, Longo KA, et al. Regulation of Wnt signaling during adipogenesis. The Journal of Biological Chemistry. 2002;277(34):30998–31004. doi: 10.1074/jbc.M204527200. [DOI] [PubMed] [Google Scholar]
- 125.Kawai M, Mushiakea S, Bessho K, et al. Wnt/Lrp/beta-catenin signaling suppresses adipogenesis by inhibiting mutual activation of PPARgamma and C/EBPalpha. Biochemical and Biophysical Research Communications. 2007;363(2):276–282. doi: 10.1016/j.bbrc.2007.08.088. [DOI] [PubMed] [Google Scholar]
- 126.Li H-X, Luo X, Liu R-X, Yang Y-J, Yang G-S. Roles of Wnt/β-catenin signaling in adipogenic differentiation potential of adipose-derived mesenchymal stem cells. Molecular and Cellular Endocrinology. 2008;291(1-2):116–124. doi: 10.1016/j.mce.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 127.Galli C, Piemontese M, Lumetti S, et al. GSK3b-inhibitor lithium chloride enhances activation of Wnt canonical signaling and osteoblast differentiation on hydrophilic titanium surfaces. Clinical Oral Implants Research. 2013;24(8):921–927. doi: 10.1111/j.1600-0501.2012.02488.x. [DOI] [PubMed] [Google Scholar]
- 128.Bennett CN, Ouyang H, Ma YL, et al. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. Journal of Bone and Mineral Research. 2007;22(12):1924–1932. doi: 10.1359/jbmr.070810. [DOI] [PubMed] [Google Scholar]
- 129.Cawthorn WP, Bree AJ, Yao Y, et al. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone. 2012;50(2):477–489. doi: 10.1016/j.bone.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Castro CHM, Shin CS, Stains JP, et al. Targeted expression of a dominant-negative N-cadherin in vivo delays peak bone mass and increases adipogenesis. Journal of Cell Science. 2004;117(13):2853–2864. doi: 10.1242/jcs.01133. [DOI] [PubMed] [Google Scholar]
- 131.Gustafson B, Eliasson B, Smith U. Thiazolidinediones increase the wingless-type MMTV integration site family (WNT) inhibitor Dickkopf-1 in adipocytes: a link with osteogenesis. Diabetologia. 2010;53(3):536–540. doi: 10.1007/s00125-009-1615-1. [DOI] [PubMed] [Google Scholar]
- 132.Takada I, Mihara M, Suzawa M, et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-γ transactivation. Nature Cell Biology. 2007;9(11):1273–1285. doi: 10.1038/ncb1647. [DOI] [PubMed] [Google Scholar]
- 133.Yao HH-C, Whoriskey W, Capel B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes and Development. 2002;16(11):1433–1440. doi: 10.1101/gad.981202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75(7):1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- 135.Ruat M, Roudaut H, Ferent J, Traiffort E. Hedgehog trafficking, cilia and brain functions. Differentiation. 2012;83(2):S97–S104. doi: 10.1016/j.diff.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 136.Bitgood MJ, McMahon AP. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Developmental Biology. 1995;172(1):126–138. doi: 10.1006/dbio.1995.0010. [DOI] [PubMed] [Google Scholar]
- 137.Nanni L, Ming JE, Bocian M, et al. The mutational spectrum of the Sonic Hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Human Molecular Genetics. 1999;8(13):2479–2488. doi: 10.1093/hmg/8.13.2479. [DOI] [PubMed] [Google Scholar]
- 138.Simpson F, Kerr MC, Wicking C. Trafficking, development and hedgehog. Mechanisms of Development. 2009;126(5-6):279–288. doi: 10.1016/j.mod.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 139.Sinha S, Chen JK. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nature Chemical Biology. 2006;2(1):29–30. doi: 10.1038/nchembio753. [DOI] [PubMed] [Google Scholar]
- 140.Pospisilik JA, Schramek D, Schnidar H, et al. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell. 2010;140(1):148–160. doi: 10.1016/j.cell.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 141.Suh JM, Gao X, McKay J, McKay R, Salo Z, Graff JM. Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metabolism. 2006;3(1):25–34. doi: 10.1016/j.cmet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 142.Zehentner BK, Leser U, Burtscher H. BMP-2 and sonic hedgehog have contrary effects on adipocyte-like differentiation of C3H10T1/2 cells. DNA and Cell Biology. 2000;19(5):275–281. doi: 10.1089/10445490050021186. [DOI] [PubMed] [Google Scholar]
- 143.Yuasa T, Kataoka H, Kinto N, et al. Sonic hedgehog is involved in osteoblast differentiation by cooperating with BMP-2. Journal of Cellular Physiology. 2002;193(2):225–232. doi: 10.1002/jcp.10166. [DOI] [PubMed] [Google Scholar]
- 144.Zhao M, Qiao M, Harris SE, Chen D, Oyajobi BO, Mundy GR. The zinc finger transcription factor Gli2 mediates bone morphogenetic protein 2 expression in osteoblasts in response to hedgehog signaling. Molecular and Cellular Biology. 2006;26(16):6197–6208. doi: 10.1128/MCB.02214-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nakamura T, Aikawaa T, Iwamoto-Enomoto M, et al. Induction of osteogenic differentiation by hedgehog proteins. Biochemical and Biophysical Research Communications. 1997;237(2):465–469. doi: 10.1006/bbrc.1997.7156. [DOI] [PubMed] [Google Scholar]
- 146.Kinto N, Iwamoto M, Enomoto-Iwamoto M, et al. Fibroblasts expressing Sonic hedgehog induce osteoblast differentiation and ectopic bone formation. FEBS Letters. 1997;404(2-3):319–323. doi: 10.1016/s0014-5793(97)00014-8. [DOI] [PubMed] [Google Scholar]
- 147.Spinella-Jaegle S, Rawadi G, Kawai S, et al. Sonic hedgehog increases the commitment of pluripotent mesenchymal cells into the osteoblastic lineage and abolishes adipocytic differentiation. Journal of Cell Science. 2001;114(11):2085–2094. doi: 10.1242/jcs.114.11.2085. [DOI] [PubMed] [Google Scholar]
- 148.van der Horst G, Farih-Sips H, Löwik CWGM, Karperien M. Hedgehog stimulates only osteoblastic differentiation of undifferentiated KS483 cells. Bone. 2003;33(6):899–910. doi: 10.1016/j.bone.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 149.Aghaloo T, Cowan CM, Chou Y-F, et al. Nell-1-induced bone regeneration in calvarial defects. American Journal of Pathology. 2006;169(3):903–915. doi: 10.2353/ajpath.2006.051210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ting K, Vastardis H, Mulliken JB, et al. Human NELL-1 expressed in unilateral coronal synostosis. Journal of Bone and Mineral Research. 1999;14(1):80–89. doi: 10.1359/jbmr.1999.14.1.80. [DOI] [PubMed] [Google Scholar]
- 151.Zhang X, Zara J, Siu RK, Ting K, Soo C. The role of NELL-1, a growth factor associated with craniosynostosis, in promoting bone regeneration. Journal of Dental Research. 2010;89(9):865–878. doi: 10.1177/0022034510376401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang X, Kuroda S, Carpenter D, et al. Craniosynostosis in transgenic mice overexpressing Nell-1. Journal of Clinical Investigation. 2002;110(6):861–870. doi: 10.1172/JCI15375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang X, Ting K, Pathmanathan D, et al. Calvarial cleidocraniodysplasia-like defects with ENU-induced Nell-1 deficiency. Journal of Craniofacial Surgery. 2012;23(1):61–66. doi: 10.1097/SCS.0b013e318240c8c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lu SS, Zhang X, Soo C, et al. The osteoinductive properties of Nell-1 in a rat spinal fusion model. Spine Journal. 2007;7(1):50–60. doi: 10.1016/j.spinee.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 155.Siu RK, Lu SS, Li W, et al. Nell-1 protein promotes bone formation in a sheep spinal fusion model. Tissue Engineering A. 2011;17(7-8):1123–1135. doi: 10.1089/ten.tea.2010.0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Li W, Zara JN, Siu RK, et al. Nell-1 enhances bone regeneration in a rat critical-sized femoral segmental defect model. Plastic and Reconstructive Surgery. 2011;127(2):580–587. doi: 10.1097/PRS.0b013e3181fed5ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lee M, Li W, Siu RK, et al. Biomimetic apatite-coated alginate/chitosan microparticles as osteogenic protein carriers. Biomaterials. 2009;30(30):6094–6101. doi: 10.1016/j.biomaterials.2009.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li W, Lee M, Whang J, et al. Delivery of lyophilized nell-1 in a rat spinal fusion model. Tissue Engineering A. 2010;16(9):2861–2870. doi: 10.1089/ten.tea.2009.0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shen J, Aaron W, Chung J, et al. NELL-1 promotes cell adhesion and differentiation via Integrinbeta1. Journal of Cellular Biochemistry. 2012;113(12):3620–3628. doi: 10.1002/jcb.24253. [DOI] [PubMed] [Google Scholar]
- 160.Chen F, Walder B, James AW, et al. NELL-1-dependent mineralisation of Saos-2 human osteosarcoma cells is mediated via c-Jun N-terminal kinase pathway activation. International Orthopaedics. 2012;36(10):2181–2187. doi: 10.1007/s00264-012-1590-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.James AW, et al. A new mode of action of Nell-1 protein via potentiation of Canonical Wnt signaling; 2011; Los Angeles, Ca, USA. Conferenece (Wnt '11); [Google Scholar]
- 162.Cowan CM, Zhangb X, James AW, et al. NELL-1 increases pre-osteoblast mineralization using both phosphate transporter Pit1 and Pit2. Biochemical and Biophysical Research Communications. 2012;422(3):351–357. doi: 10.1016/j.bbrc.2012.04.077. [DOI] [PubMed] [Google Scholar]
- 163.Kwak J, Zara JN, Chiang M, et al. NELL-1 injection maintains long-bone quantity and quality in an ovariectomy-induced osteoporotic senile rat model. Tissue Engineering A. 2013;19(3-4):426–436. doi: 10.1089/ten.tea.2012.0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wozney JM, Rosen V, Celeste AJ, et al. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242(4885):1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 165.Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22(4):233–241. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- 166.Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A. Bone morphogenetic proteins: a critical review. Cellular Signalling. 2011;23(4):609–620. doi: 10.1016/j.cellsig.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 167.Freire MO, You H-K, Kook J-K, Choi J-H, Zadeh HH. Antibody-mediated osseous regeneration: a novel strategy for bioengineering bone by immobilized anti-bone morphogenetic protein-2 antibodies. Tissue Engineering A. 2011;17(23-24):2911–2918. doi: 10.1089/ten.tea.2010.0584. [DOI] [PubMed] [Google Scholar]
- 168.Friedlaender GE, Perry CR, Cole JD, et al. Osteogenic protein-1 (bone morphogenetic protein-7) in the treatment of tibial nonunions. The Journal of Bone and Joint Surgery. 2001;83(2):S151–158. [PMC free article] [PubMed] [Google Scholar]
- 169.Miyazono K, Maeda S, Imamura T. BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine and Growth Factor Reviews. 2005;16(3):251–263. doi: 10.1016/j.cytogfr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 170.Nohe A, Keating E, Knaus P, Petersen NO. Signal transduction of bone morphogenetic protein receptors. Cellular Signalling. 2004;16(3):291–299. doi: 10.1016/j.cellsig.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 171.Nishimura R, Hata K, Matsubara T, Wakabayashi M, Yoneda T. Regulation of bone and cartilage development by network between BMP signalling and transcription factors. Journal of Biochemistry. 2012;151(3):247–254. doi: 10.1093/jb/mvs004. [DOI] [PubMed] [Google Scholar]
- 172.Li X, Cao X. BMP signaling and skeletogenesis. Annals of the New York Academy of Sciences. 2006;1068(1):26–40. doi: 10.1196/annals.1346.006. [DOI] [PubMed] [Google Scholar]
- 173.Hata K, Nishimura R, Ikeda F, et al. Differential roles of Smad1 and p38 kinase in regulation of peroxisome proliferator-activating receptor γ during bone morphogenetic protein 2-induced adipogenesis. Molecular Biology of the Cell. 2003;14(2):545–555. doi: 10.1091/mbc.E02-06-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jin W, Takagi T, Kanesashi S-N, et al. Schnurri-2 controls BMP-dependent adipogenesis via interaction with Smad proteins. Developmental Cell. 2006;10(4):461–471. doi: 10.1016/j.devcel.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 175.Bowers RR, Lane MD. A role for bone morphogenetic protein-4 in adipocyte development. Cell Cycle. 2007;6(4):385–389. doi: 10.4161/cc.6.4.3804. [DOI] [PubMed] [Google Scholar]
- 176.Bowers RR, Kim JW, Otto TC, Lane MD. Stable stem cell commitment to the adipocyte lineage by inhibition of DNA methylation: role of the BMP-4 gene. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(35):13022–13027. doi: 10.1073/pnas.0605789103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Herzig S, Wolfrum C. Brown and white fat: from signaling to disease. Biochim Biophys Acta. 2013;1831(5):p. 895. doi: 10.1016/j.bbalip.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 178.Hao R, Yuan L, Zhang N, et al. Brown adipose tissue: distribution and influencing factors on FDG PET/CT scan. Journal of Pediatric Endocrinology and Metabolism. 2012;25(3-4):233–237. doi: 10.1515/jpem-2012-0029. [DOI] [PubMed] [Google Scholar]
- 179.Qian SW, Tang Y, Li X, et al. BMP4-mediated brown fat-like changes in white adipose tissue alter glucose and energy homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(9):E798–E807. doi: 10.1073/pnas.1215236110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ahrens M, Ankenbauer T, Schroder D, Hollnagel A, Mayer H, Gross G. Expression of human bone morphogenetic proteins-2 or -4 in murine mesenchymal progenitor C3H10T 1/2 cells induces differentiation into distinct mesenchymal cell lineages. DNA and Cell Biology. 1993;12(10):871–880. doi: 10.1089/dna.1993.12.871. [DOI] [PubMed] [Google Scholar]
- 181.Mishina Y, Starbuck MW, Gentile MA, et al. Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. The Journal of Biological Chemistry. 2004;279(26):27560–27566. doi: 10.1074/jbc.M404222200. [DOI] [PubMed] [Google Scholar]
- 182.Okamoto M, Murai J, Yoshikawa H, Tsumaki N. Bone morphogenetic proteins in bone stimulate osteoclasts and osteoblasts during bone development. Journal of Bone and Mineral Research. 2006;21(7):1022–1033. doi: 10.1359/jbmr.060411. [DOI] [PubMed] [Google Scholar]
- 183.Gazzerro E, Smerdel-Ramoya A, Zanotti S, et al. Conditional deletion of gremlin causes a transient increase in bone formation and bone mass. The Journal of Biological Chemistry. 2007;282(43):31549–31557. doi: 10.1074/jbc.M701317200. [DOI] [PubMed] [Google Scholar]
- 184.Gazzerro E, Pereira RC, Jorgetti V, Olson S, Economides AN, Canalis E. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology. 2005;146(2):655–665. doi: 10.1210/en.2004-0766. [DOI] [PubMed] [Google Scholar]
- 185.Davis SW, Camper SA. Noggin regulates Bmp4 activity during pituitary induction. Developmental Biology. 2007;305(1):145–160. doi: 10.1016/j.ydbio.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhu W, Kim J, Cheng C, et al. Noggin regulation of bone morphogenetic protein (BMP) 2/7 heterodimer activity in vitro. Bone. 2006;39(1):61–71. doi: 10.1016/j.bone.2005.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cheng C. Osteogenic activity of the fourteen types of human bone morphogenetic proteins (BMPs) Journal of Bone and Joint Surgery A. 2004;85(8):1544–1552. doi: 10.2106/00004623-200308000-00017. [DOI] [PubMed] [Google Scholar]
- 188.Suzawa M, Takeuchi Y, Fukumoto S, et al. Extracellular matrix-associated bone morphogenetic proteins are essential for differentiation of murine osteoblastic cells in vitro. Endocrinology. 1999;140(5):2125–2133. doi: 10.1210/endo.140.5.6704. [DOI] [PubMed] [Google Scholar]
- 189.Ducy P, Karsenty G. The family of bone morphogenetic proteins. Kidney International. 2000;57(6):2207–2214. doi: 10.1046/j.1523-1755.2000.00081.x. [DOI] [PubMed] [Google Scholar]
- 190.Reid J, Gilmour HM, Holt S. Primary non-specific ulcer of the small intestine. Journal of the Royal College of Surgeons of Edinburgh. 1982;27(4):228–232. [PubMed] [Google Scholar]
- 191.Varkey M, Kucharski C, Haque T, Sebald W, Uludağ H. In vitro osteogenic response of rat bone marrow cells to bFGF and BMP-2 treatments. Clinical Orthopaedics and Related Research. 2006;(443):113–123. doi: 10.1097/01.blo.0000200236.84189.87. [DOI] [PubMed] [Google Scholar]
- 192.Partridge K, Yang X, Clarke NMP, et al. Adenoviral BMP-2 gene transfer in mesenchymal stem cells: In vitro and in vivo bone formation on biodegradable polymer scaffolds. Biochemical and Biophysical Research Communications. 2002;292(1):144–152. doi: 10.1006/bbrc.2002.6623. [DOI] [PubMed] [Google Scholar]
- 193.Wegman F, Bijenhof A, Schuijff L, Oner FC, Dhert WJ, Alblas J. Osteogenic differentiation as a result of BMP-2 plasmid DNA based gene therapy in vitro and in vivo. European Cells & Materials. 2011;21:230–242. doi: 10.22203/ecm.v021a18. [DOI] [PubMed] [Google Scholar]
- 194.Park K-H, Kim H, Moon S, Na K. Bone morphogenic protein-2 (BMP-2) loaded nanoparticles mixed with human mesenchymal stem cell in fibrin hydrogel for bone tissue engineering. Journal of Bioscience and Bioengineering. 2009;108(6):530–537. doi: 10.1016/j.jbiosc.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 195.Tang Y, Tang W, Lin Y, et al. Combination of bone tissue engineering and BMP-2 gene transfection promotes bone healing in osteoporotic rats. Cell Biology International. 2008;32(9):1150–1157. doi: 10.1016/j.cellbi.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 196.Kempen DHR, Lu L, Hefferan TE, et al. Retention of in vitro and in vivo BMP-2 bioactivities in sustained delivery vehicles for bone tissue engineering. Biomaterials. 2008;29(22):3245–3252. doi: 10.1016/j.biomaterials.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Cheng S-L, Lou J, Wright NM, Lai C-F, Avioli LV, Riew KD. In vitro and in vivo induction of bone formation using a recombinant adenoviral vector carrying the human BMP-2 gene. Calcified Tissue International. 2001;68(2):87–94. [PubMed] [Google Scholar]
- 198.Noël D, Gazit D, Bouquet C, et al. Short-term BMP-2 expression is sufficient for in vivo osteochondral differentiation of mesenchymal stem cells. Stem Cells. 2004;22(1):74–85. doi: 10.1634/stemcells.22-1-74. [DOI] [PubMed] [Google Scholar]
- 199.Osyczka AM, Diefenderfer DL, Bhargave G, Leboy PS. Different effects of BMP-2 on marrow stromal cells from human and rat bone. Cells Tissues Organs. 2004;176(1–3):109–119. doi: 10.1159/000075032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Deifenderfer DL, Osyczka AM, Reilly GC, Leboy PS. BMP responsiveness in human mesenchymal stem cells. Connective Tissue Research. 2003;44(1):305–311. [PubMed] [Google Scholar]
- 201.Chen C, Uludağ H, Wang Z, Jiang H. Noggin suppression decreases BMP-2-induced osteogenesis of human bone marrow-derived mesenchymal stem cells in vitro. Journal of Cellular Biochemistry. 2012;113(12):3672–3680. doi: 10.1002/jcb.24240. [DOI] [PubMed] [Google Scholar]
- 202.Rahman S, Czernik PJ, Lu Y, Lecka-Czernik B. β-catenin directly sequesters adipocytic and insulin sensitizing activities but not osteoblastic activity of PPARγ2 in marrow mesenchymal stem cells. PLoS ONE. 7(12) doi: 10.1371/journal.pone.0051746.e51746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yakar S, Kim H, Zhao H, et al. The growth hormone-insulin like growth factor axis revisited: lessons from IGF-1 and IGF-1 receptor gene targeting. Pediatric Nephrology. 2005;20(3):251–254. doi: 10.1007/s00467-004-1613-y. [DOI] [PubMed] [Google Scholar]
- 204.Giustina A, Mazziotti G, Canalis E. Growth hormone, insulin-like growth factors, and the skeleton. Endocrine Reviews. 2008;29(5):535–559. doi: 10.1210/er.2007-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Livingstone C. Insulin-like growth factor-I (IGF-I) and clinical nutrition. Clinical Science. 2013;125(6):265–280. doi: 10.1042/CS20120663. [DOI] [PubMed] [Google Scholar]
- 206.Kawai M, Rosen CJ. Insulin-like growth factor-I and bone: lessons from mice and men. Pediatric Nephrology. 2009;24(7):1277–1285. doi: 10.1007/s00467-008-1040-6. [DOI] [PubMed] [Google Scholar]
- 207.Govoni KE. Insulin-like growth factor-I molecular pathways in osteoblasts: potential targets for pharmacological manipulation. Current Molecular Pharmacology. 2012;5(2):143–152. [PubMed] [Google Scholar]
- 208.Peng X-D, Xu P-Z, Chen M-L, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes and Development. 2003;17(11):1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ghosh-Choudhury N, Abboud SL, Nishimura R, Celeste A, Mahimainathan L, Choudhury GG. Requirement of BMP-2-induced phosphatidylinositol 3-kinase and Akt serine/threonine kinase in osteoblast differentiation and Smad-dependent BMP-2 gene transcription. The Journal of Biological Chemistry. 2002;277(36):33361–33368. doi: 10.1074/jbc.M205053200. [DOI] [PubMed] [Google Scholar]
- 210.Xian L, Wu X, Pang L, et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nature Medicine. 2012;18(7):1095–1101. doi: 10.1038/nm.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bikle DD, Wang Y. Insulin like growth factor-I: a critical mediator of the skeletal response to parathyroid hormone. Current Molecular Pharmacology. 2012;5(2):135–142. [PMC free article] [PubMed] [Google Scholar]
- 212.Mukherjee A, Rotwein P. Insulin-like growth factor binding protein-5 in osteogenesis: facilitator or inhibitor? Growth Hormone and IGF Research. 2007;17(3):179–185. doi: 10.1016/j.ghir.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mccarty RC, Gronthos S, Zannettino AC, Foster BK, Xian CJ. Characterisation and developmental potential of ovine bone marrow derived mesenchymal stem cells. Journal of Cellular Physiology. 2009;219(2):324–333. doi: 10.1002/jcp.21670. [DOI] [PubMed] [Google Scholar]
- 214.Chen J, Yuan K, Mao X, et al. Serum response factor regulates bone formation via IGF-1 and Runx2 signals. Journal of Bone and Mineral Research. 2012;27(8):1659–1668. doi: 10.1002/jbmr.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Levi B, James AW, Wan DC, Glotzbach JP, Commons GW, Longaker MT. Regulation of human adipose-derived stromal cell osteogenic differentiation by insulin-like growth factor-1 and platelet-derived growth factor-α . Plastic and Reconstructive Surgery. 2010;126(1):41–52. doi: 10.1097/PRS.0b013e3181da8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kumar S, Ponnazhagan S. Mobilization of bone marrow mesenchymal stem cells in vivo augments bone healing in a mouse model of segmental bone defect. Bone. 2012;50(4):1012–1018. doi: 10.1016/j.bone.2012.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Bernstein A, Mayr HO, Hube R. Can bone healing in distraction osteogenesis be accelerated by local application of IGF-1 and TGF-β1? Journal of Biomedical Materials Research B. 2010;92(1):215–225. doi: 10.1002/jbm.b.31508. [DOI] [PubMed] [Google Scholar]
- 218.Courtland H-W, Sun H, Beth-On M, et al. Growth hormone mediates pubertal skeletal development independent of hepatic IGF-1 production. Journal of Bone and Mineral Research. 2011;26(4):761–768. doi: 10.1002/jbmr.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Tahimic CG, Wang Y, Bikle DD. Anabolic effects of IGF-1 signaling on the skeleton. Frontiers in Endocrinology. 2013;4, article 6 doi: 10.3389/fendo.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wabitsch M, Hauner H, Heinze E, Teller WM. The role of growth hormone/insulin-like growth factors in adipocyte differentiation. Metabolism. 1995;44(10):45–49. doi: 10.1016/0026-0495(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 221.Yang SJ, Chen CY, Chang GD, et al. Activation of Akt by advanced glycation end products (AGEs): involvement of IGF-1 receptor and caveolin-1. PLoS ONE. 2013;8(3) doi: 10.1371/journal.pone.0058100.e58100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.James AW, Pan A, Chiang M, et al. A new function of Nell-1 protein in repressing adipogenic differentiation. Biochemical and Biophysical Research Communications. 2011;411(1):126–131. doi: 10.1016/j.bbrc.2011.06.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Xiao L, Sobue T, Esliger A, et al. Disruption of the Fgf2 gene activates the adipogenic and suppresses the osteogenic program in mesenchymal marrow stromal stem cells. Bone. 2010;47(2):360–370. doi: 10.1016/j.bone.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Choy L, Skillington J, Derynck R. Roles of autocrine TGF-β receptor and Smad signaling in adipocyte differentiation. Journal of Cell Biology. 2000;149(3):667–681. doi: 10.1083/jcb.149.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Ugarte F, Ryser M, Thieme S, et al. Notch signaling enhances osteogenic differentiation while inhibiting adipogenesis in primary human bone marrow stromal cells. Experimental Hematology. 2009;37(7):867.e1–875.e1. doi: 10.1016/j.exphem.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 226.Byun MR, Jeong H, Bae SJ, Kim AR, Hwang ES, Hong J-H. TAZ is required for the osteogenic and anti-adipogenic activities of kaempferol. Bone. 2012;50(1):364–372. doi: 10.1016/j.bone.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 227.Wang S, Mu J, Fan Z, et al. Insulin-like growth factor 1 can promote the osteogenic differentiation and osteogenesis of stem cells from apical papilla. Stem Cell Research. 2012;8(3):346–356. doi: 10.1016/j.scr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 228.Teruel T, Valverde AM, Benito M, Lorenzo M. Insulin-like growth factor I and insulin induce adipogenic-related gene expression in fetal brown adipocyte primary cultures. Biochemical Journal. 1996;319(2):627–632. doi: 10.1042/bj3190627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Huang Y-L, Qiu R-F, Mai W-Y, et al. Effects of insulin-like growth factor-1 on the properties of mesenchymal stem cells in vitro. Journal of Zhejiang University B. 2012;13(1):20–28. doi: 10.1631/jzus.B1100117. [DOI] [PMC free article] [PubMed] [Google Scholar]