

Abstract
Lysine specific demethylase 1 (LSD1), the first identified histone demethylase, plays an important role in epigenetic regulation of gene activation and repression. The up-regulated LSD1's expression has been reported in several malignant tumors. In the current study, we designed and synthesized five series of 1, 2, 3-triazole-dithiocarbamate hybrids and screened their inhibitory activity toward LSD1. We found that some of these compounds, especially compound 26, exhibited the most specific and robust inhibition of LSD1. Interestingly, compound 26 also showed potent and selective cytotoxicity against LSD1 overexpressing gastric cancer cell lines MGC-803 and HGC-27, as well as marked inhibition of cell migration and invasion, compared to 2-PCPA. Furthermore, compound 26 effectively reduced the tumor growth bared by human gastric cancer cells in vivo with no signs of adverse side effects. These findings suggested that compound 26 deserves further investigation as a lead compound in the treatment of LSD1 overexpressing gastric cancer.
INTRODUCTION
Epigenetic post-transcriptional modifications on histone, including acetylation, methylation, and phosphorylation, modulate gene activation and repression. Among these modifications, methylation and demethylation of lysine are dynamically regulated by a number of histone lysine methyltransferases (HKMTs) and histone lysine demethylases (HKDMs) including lysine specific demethylase 1 (LSD1). LSD1, the first characterized demethylase in 2004,1 is a highly conserved flavin adenine dinucleotide (FAD)-dependent oxidative enzyme, containing amine oxidase domain. It demethylates mono-, di-methylated K4 and K9 of histone 3, as well as p53, E2F transcription factor 1 (E2F1) and DNA methyltransferases (DNMTs) and further regulates their downstream cellular function.2–7 LSD1 has been reported to be overexpressed in many malignant tumors, including breast, colon, prostate, lung, gastric cancers and others.8–16 Down regulation of LSD1 by RNAi or various kinds of inhibitors has been shown to effectively treat those cancers by inducing re-expression of aberrantly silenced genes.11, 12, 17–23 Therefore, LSD1 has been considered an important and promising anticancer target.
As a member of monoamine oxidase (MAO) family, LSD1 utilizes a noncovalently bound FAD as its cofactor to oxidatively remove the methyl groups of its substrates.1 MAO-A and MAO-B, another two members of MAO family, share the same mechanism and cofactor of LSD1 in the cleavage of the inactivated carbon-nitrogen bonds from their substrates.24 MAO inactivators (Figure 1), such as pargyline (1), phenelzine (2) and tranylcypromine (3, 2-PCPA), have been reported to function as non-selective LSD1 inhibitors.25 In addition, 2-PCPA derivatives, peptides and polyamine analogs have been synthesized as LSD1 inhibitors.12, 17, 18, 26–33 However, only three new classes of LSD1 chemical inhibitors, including amidoxime based compounds,22 amidino-guanidinium compounds,34 and phenyl oxazoles,19 have been reported during the past two years. Highly selective LSD1 inhibitors with strong toxicity toward cancer cells and less or no side effects on normal cells remain to be identified.
Figure 1.
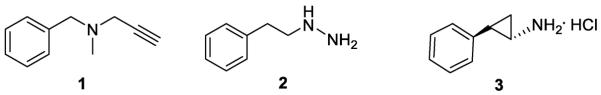
MAO inhibitors that inhibit LSD1
The azole heterocylces-pyrazole, including thiazole, imidazole, pyrrzole, oxadiazole and triazole-have attracted more attention by medicinal chemists for many years due to their numerous biological activities,35, 36 especially their MAO inhibitory effect.37–43 Among these azole heterocylces, 1, 2, 3-triazole was mainly used based on its synthetic accessibility by click chemistry as well as its capacity for binding of biomolecular targets. Recently, several inhibitors toward MAO-A were developed accordingly.44, 45 Furthermore, click chemistry has been widely used for synthesizing other inhibitors against epigenetic enzymes, such as HDAC.46, 47 Hence, in this study, 1, 2, 3-triazole was chosen as a part of our target compound skeleton.
Dithiocarbamates was selected due to their inhibitory activities against fungal, bacteria and malignant cancer.48–50 Disulfiram (DSF), as a supporting treatment of chronic alcoholism, is commercially available, and recently it has been reported as P-glycoprotein efflux pump modulator with antifugal potential.51 Furthermore, when disulfiram creates complexes with metals, it becomes a proteasome inhibitor and acts as a promising approach for anticancer therapy.51 BO-3482, a novel dithiocarbamate-containing carbapenem with activity against MRSA (Methicillin-Resistant Staphylococcus Aureus), has been tested in preclinical trail.52, 53 Meanwhile, we have previously reported a pool of novel butenolides-containing dithiocarbamates with good anticancer activity.54 Therefore, based on an existing pool of triazole-containing dithiocarbamates,55 a library with eighty-four 1, 2, 3-triazole-dithiocarbamate hybrids was synthesized by the click chemistry approach. Their anti-LSD1 activity and cytotoxicities were then determined. We found that compared to 2-PCPA, a non-selective LSD1 inhibitor, triazole-dithiocarbamate-based LSD1 inhibitors, especially compound 26, are more potent and exhibits selective inhibition of the growth of LSD1 over-expressing gastric cancer cell lines. Compound 26 also impaired cell migration and invasion and significantly inhibited tumor growth in vivo. These findings indicate that triazole-dithiocarbamate based LSD1 inhibitors represent a novel class of LSD1 inhibitors against LSD1 overexpressing gastric cancer.
CHEMISTRY
Alkyne intermediates 4a–4j were synthesized as shown in Scheme 1. Commercially available amines were treated with CS2 and propargyl bromide in the presence of Na3PO4.12H2O in one pot to provide 4a–4j (Figure 2).
Scheme 1.
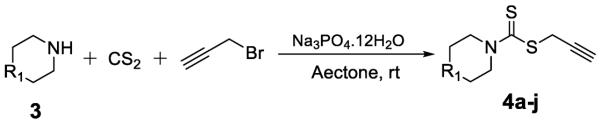
Synthesis of alkynes intermediates.
Figure 2.
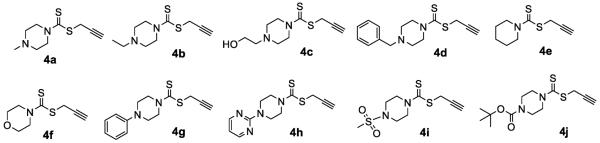
Alkynes intermediates 4a–4j used in the present study.
Azides 5a and 5e–5g (Figure 3) were obtained from commercially available bromides by direct azide displacement. Azides 5b were prepared using classical diazotation-azidation protocols.56 The synthesis of azide intermediates 5c, 5d, 5h and 5i (Figure 3) is shown in Scheme 2. Substituted phenethyl alcohol was esterified to tosylate 6, which was converted to azide derivative 5c by reacting with sodium azide in acetonitrile. Hydroxy coumarin was treated with 1, 2-dibromoethane or tosyl chloride and then with sodium azide to aquire 5d and 5i, respectively. Azide derivative 5h was prepared by a sodium azide substitution of 10, obtained from staring material 8 from the reported procedures.57, 58
Figure 3.
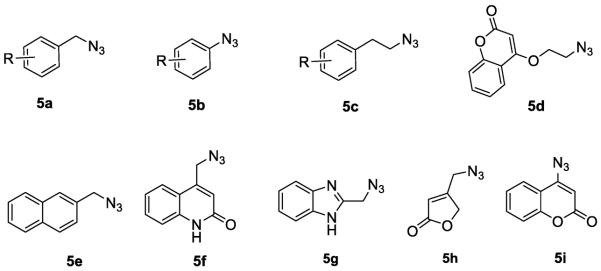
Azides 5a–5i used in the present study.
Scheme 2.

Synthesis of the azides 5c, 5d, 5h and 5ia
The 1, 2, 3-triazole-dithiocarbamate hybrids 12–72 were synthesized through a huisgen 1, 3-dipolar cycloaddition between alkynes 4a-j and azides 5a-i using copper sulfate and sodium ascorbate in aqueous THF (Scheme 3). The synthesis of 1, 2, 3-triazole-dithiocarbamate hybrids 73–95 is shown in Scheme 4. 22–28 and 38–40 were deprotected using trifluoroacetic acid in dichloromethane to afford compounds 73–82. Compounds 83–92 were obtained by reaction of 73–78 and 81–82 with corresponding chloroformic ester under basic condition. Compounds 73–75 and 77 were treatment with acetyl chloride and Et3N to obtain 93–95.
Scheme 3.
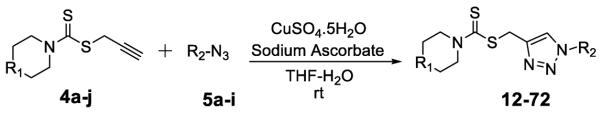
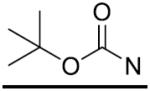
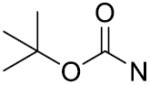
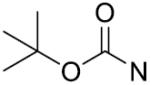
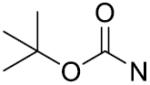
Synthesis of 1, 2, 3-triazole-dithiocarbamate hybrids 12–72 through click chemistry.
Scheme 4.

Synthesis of 1, 2, 3-triazole-dithiocarbamate hybrids 73–96a.
RESULTS AND DISCUSSION
Identification of LSD1 Inhibitors and structure-activity relationship (SAR) Studies
To determine the inhibitory activity of this compound library against LSD1, we generated a LSD1 recombinant expressing vector containing human LSD1 cDNA. The expression of recombinant LSD1 was then induced in Escherichia Coli (E. Coli) and purified as published.18 The demethylation activity of the recombinant LSD1 was further determined by a fluorescence-based method, using synthesized H3K4me2 as a substrate.32 All the compounds synthesized in this study were examined in vitro for their inhibitory effect on LSD1 activity, 2-PCPA was chosen as a positive control. The results were summarized as Table 1–5.
Table 1.
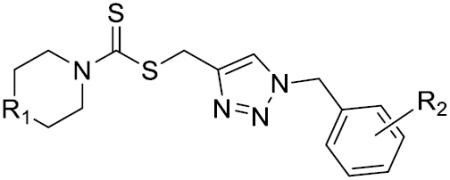
Structures of compounds 12–40 and their inhibition rates (IC50) to the purified LSD1 recombinant in vitro.
| |||||||
|---|---|---|---|---|---|---|---|
| Comp. | R1 | R2 | IC50(μM) | Comp. | R1 | R2 | IC50(μM) |
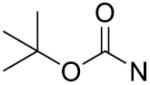
| 12 | CH2 | o-F | >125 | 27 |
|
p-OCH3 | 18.4±2.5 |
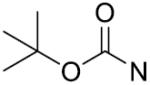
| 13 | CH2 | p-F | >125 | 28 |
|
m-CH3 | 43.3±5.9 |
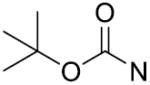
| 14 | CH2 | p-CH3 | >125 | 29 |
|
o-CH3 | >125 |
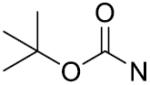
| 15 | CH2 | H | >125 | 30 |
|
p-CF3 | >125 |
| 16 | CH2 | p-Cl | >125 | 31 |
|
o-CF3 | >125 |
| 17 | 0 | H | >125 | 32 |
|
o-Br | 27.5±3.7 |
| 18 | 0 | o-F | >125 | 33 |
|
70.7±1.2 | |
| 19 | 0 | p-F | >125 | 34 |
|
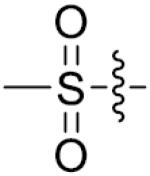
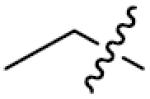
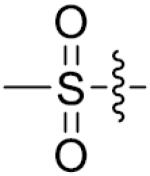
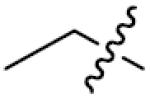
o-OH | 69.2±3.6 |
| 20 | 0 | p-CH3 | >125 | 35 |
|
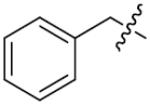
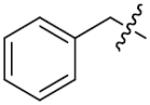
p-NO2 | >125 |
| 21 | 0 | p-Cl | >125 | 36 |
|
m-OH | 77.1±3.1 |
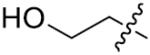
| 22 |
|
o-F | 8.0±1.0 | 37 |
|
o, p-diOH | 81.3±2.0 |
| 23 |
|
p-F | 21.6±2.3 | 38 |
|
m, p-diC1 | 88.9±3.2 |
| 24 |
|
o-Cl | 11.5±2.1 | 39 |
|
o, o-diF | 51.2±4.3 |
| 25 |
|
p-Cl | 56.5±2.8 | 40 |
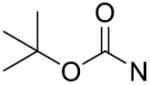
|
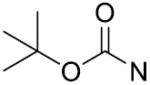
m,p,m-triOCH3 | 19.0±2.5 |
| 26 |
|
p-CH3 | 2.1±0.7 | 2-PCPA | - | - | 27.8±2.6 |
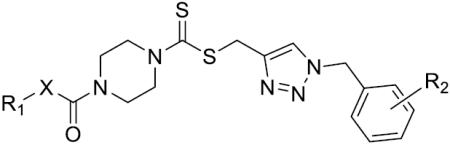
Table 5.
The Effect of the bulk of carbamate moiety on LSD1 inhibition.
| ||||
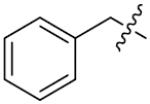
|---|---|---|---|---|
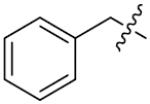
|
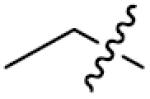
| ||||
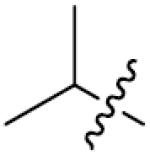
| Comp. | R1 | R2 | X | IC50(μM) |
| 83 |
|
o-F | 0 | 32.8±2.0 |
| 84 |
|
p-F | 0 | 47.6±4.3 |
| 85 |
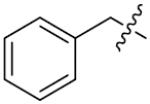
|
p-Cl | 0 | 53.2±2.9 |
| 86 |
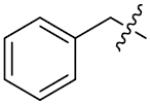
|
p-Cl | 0 | >125 |
| 87 |
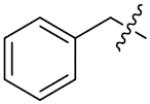
|
p-CH3 | 0 | 22.4±1.0 |
| 88 |
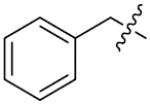
|
p-OCH3 | 0 | 35.1±2.1 |
| 89 |
|
m, p-diCl | 0 | 29.5±1.3 |
| 90 |
|
m,p,m-triOCH3 | 0 | 43.5±4.1 |
| 91 |
|
p-CH3 | 0 | 17.0±1.2 |
| 92 |
|
p-CH3 | 0 | 15.0±0.7 |
| 93 | H | o-F | CH2 | 30.2±1.9 |
| 94 | H | p-F | CH2 | 47.1±2.0 |
| 95 | H | p-CH3 | CH2 | 25.3±2.2 |
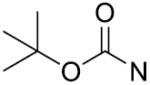
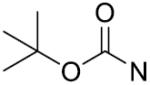
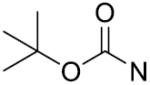
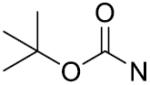
The in vitro inhibitory activity results of compounds 12–40 against LSD1 were determined initially and shown in Table 1. With the exception of compounds 29–31 and 36, all the compounds bearing a carbamate moiety exhibit moderate to good potency with IC50 values ranging from 2.1 to 88.9 μM. Among them, compound 26 shows the most potent activity to LSD1, with an IC50 of 2.1 μM, which is 14 times higher than that of 2-PCPA. Moreover, compounds 22–24, 27 and 40 are more potent than 2-PCPA, with IC50 values in the single- or double-digit micromolar range. During the SAR studies, we found that the substitution on the phenyl ring was important for the in vitro LSD1 inhibitory activity showing over 35-fold activity loss, when the methyl group was replaced with the hydroxyl group (compound 26 vs 33). We also observed that the para substitution was preferential: the p-CH3 derivative compound 26 was more potent against LSD1 than the m-CH3 derivative 28 (IC50 = 43.3 μM) and the o-CH3 derivative 29 without any detectable activity. The nitrogen of the piperidine was essential. When the nitrogen was replaced with a methylene or an oxygen atom (12–16, 17–21), their inhibitory activities to LSD1 were completely lost.
To determine whether the length of carbon tether between the triazole ring and the benzene ring might affect the compounds' binding of LSD1, compounds 41–46 were synthesized and their LSD1 inhibitory activities were shown in Table 2. Either extension or reduction of the carbon tether length by one carbon, a complete loss of potency was observed. Those results suggested that the linker between the benzene ring and the triazole plays an important role in their activities.
Table 2.
The effect of linker length on LSD1 inhibition.
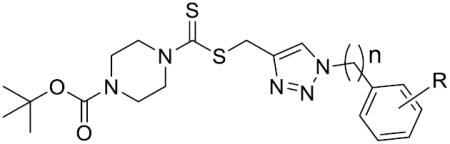
| |||
|---|---|---|---|
| Comp. | n | R | IC50(μM) |
| 41 | 0 | p-CH3 | >125 |
| 42 | 0 | p-OCH3 | >125 |
| 43 | 0 | o-F | >125 |
| 44 | 0 | m-CF3 | >125 |
| 45 | 2 | p-CH3 | >125 |
| 46 | 2 | p-F | >125 |
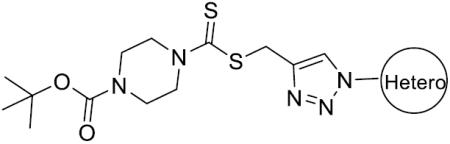
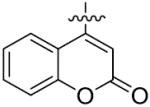
The importance of benzene ring in the LSD1 inhibitory activity was also explored (Table 3). Replacing the phenyl scaffold of compound 26 with coumarin ring (47), naphthalene ring (48), quinolinone ring (49), benzoxazole ring (50) or butenolide ring (51) led to a complete loss of activity or very weak binding affinity, indicating the significance of the benzene ring in retaining their activity.
Table 3.
The Effect of different aromatic rings on LSD1 inhibition.
| ||
|---|---|---|
| Comp. | Hetero | IC50(μM) |
| 47 |
|
>125 |
| 48 |
|
>125 |
| 49 |
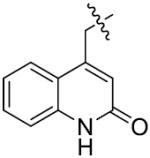
|
>125 |
| 50 |
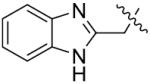
|
>125 |
| 51 |
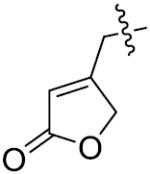
|
>125 |
| 52 |
|
>125 |
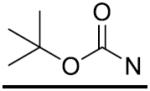
Furthermore, the importance of substituents on the N-atom (Table 4) was investigated. Changing the t-butyloxycarboryl group to alkyl chain methyl, ethyl, benzyl or hydroxyethyl resulted in inactive compounds 53–63. Replacing the t-butyloxycarboryl group by phenyl (as in 64–66), 2-pyrimidinyl (as in 67–68) or mesyl (as in 69–71) caused dramatic loss of activity. Removing the t-butyloxycarbonyl group was clearly detrimental for the inhibition of LSD1, such as, compound 26 (2.1 μM) compared to 77 (28.9 μM) and 73 (17.3 μM). These modifications and SAR studies revealed that the t-butyloxycarbonyl group is critical for their inhibitory activity.
Table 4.
The effect of different substituents at the N-atom on LSD1 inhibition.
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Comp. | R1 | R2 | IC50(μM) | Comp. | R1 | R2 | IC50(μM) |
| 53 | CH3 | o-F | >125 | 68 |
|
o-F | 39.8±1.1 |
| 54 | CH3 | p-CH3 | >125 | 69 |
|
p-F | 43.9±2.5 |
| 55 | CH3 | p-F | >125 | 70 |
|
p-CH3 | 35.7±2.0 |
| 56 | CH3 | o-Cl | >125 | 71 |
|
o-F | 45.7±1.9 |
| 57 |
|
p-CH3 | >125 | 72 |
|
p-F | 55.1±3.3 |
| 58 |
|
o-F | >125 | 73 | H | o-F | 17.3±1.7 |
| 59 |
|
p-CH3 | >125 | 74 | H | p-F | 69.3±1.8 |
| 60 |
|
o-F | >125 | 75 | H | o-Cl | >125 |
| 61 |
|
p-CH3 | 83.4±2.1 | 76 | H | p-Cl | >125 |
| 62 |
|
o-F | 100.1±9.3 | 77 | H | p-CH3 | 28.9±2.0 |
| 63 |
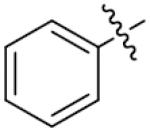
|
p-F | >125 | 78 | H | p-OCH3 | 19.5±1.9 |
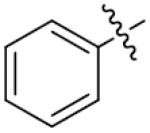
| 64 |
|
p-CH3 | 40.3±2.1 | 79 | H | p-CF3 | 87.4±2.3 |
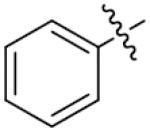
| 65 |
|
o-F | >125 | 80 | H | o, o-diF | >125 |
| 66 |
|
p-F | >125 | 81 | H | m, p-diCl | >125 |
| 67 |
|
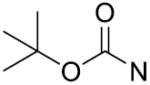
p-CH3 | 27.6±1.2 | 82 | H | m,p,m-triOCH3 | >125 |
We next synthesized compounds 83–95 to explore the bulk of carbamate moiety and the effect of the oxygen atom. The results were shown in Table 5. Changing the tert-butyl carbamates to benzyl carbamates (83–90) dramatically decreased the inhibitory activity, by comparing 87–88 with 26–27. Compared to compound 26, the ethyl carbamate 91 and isopropyl carbamate 92 showed 8.5- to 7.5-fold loss of activity in their IC50 values, respectively. This finding indicated that steric hindrance of the carbamoyl moiety was of pivotal importance in their activity. The role of the oxygen atom was investigated by preparing compounds 93–95, in which the oxygen atom was replaced with a methylene group. These compounds showed moderate activity with IC50 ranging from 25.3–47.1 μM, which is consistent with the results described above that the t-butyloxycarbonyl group is critical for the inhibitory activity.
In vitro inhibition properties of compound 26 to the recombinant LSD1 and its homologies: LSD2, MAO-A and MAO-B
Having identified compound 26 as a highly potent LSD1 inhibitor, we further determined the dissociation constant (Kd) of compound 26 by microscale thermophoresis (MST) experiment. The Kd values for compound 26 and 2-PCPA were 0.35 and 46.3 μM (Figure 4A, B), respectively, indicating the robust binding affinity of compound 26 to LSD1, compared to 2-PCPA (Figure 4A, B). Then, the inhibitory properties of compound 26 were characterized. We found that, during 60 min, the inhibition of compound 26 to LSD1 activity with three different concentrations (0.5, 2.5 and 12.5μM) exhibit time independent manner (Figure 4D). To test the reversibility of compound 26 for LSD1, a dilution assay was used. Our analysis suggested that dilution of the LSD1/compound 26 mixture by 80 folds resulted in the recovery of LSD1 activity, which means the molecule may interact noncovalently with the enzyme. However, in the presence of the covalently binding inhibitor 2-PCPA, LSD1 activity cannot be recovered after dilution (Figure 4E). These results indicate the reversibility of compound 26, compared to 2-PCPA. Dialysis experiment was also performed against hepes buffer after LSD1 was inactivated by high concentration of compound 26. After the dialysis, the activity can be restored either (Figure 4F), which further support the reversible inhibitory of compound 26. Next, the competitive analysis was performed to test the hypothesized binding sites of compound 26 on LSD1. With classic Lineweaver-Burk plots,59 we characterized that compound 26 was a non-competitive inhibitor over LSD1 substrate, H3K4me2 (Figure 4G), but a competitive inhibitor over LSD1 cofactor FAD (Figure 4H). These results indicate that compound 26 may penetrate into the cavity in LSD1 where FAD stands and reduce the recombinant activity. Another dialysis experiment was done to verify the FAD ejection with compound 26 treatment on LSD1. The inhibitor, FAD and LSD1 were incubated and dialyzed against the hepes buffer containing the same concentration of inhibitor. Then, the amount of FAD in the dialysis tubes was monitored at 468nm with UV/VIS spectrophotometer. After dialysis, the amount of FAD in the compound 26 dialysis tube was much less than that in control group (Data not shown), indicating that the FAD may be ejected with the treatment of compound 26. The Kd value of FAD is 0.182 μM (Figure 4C), smaller than that of compound 26, indicating that FAD, as the cofactor of LSD1, binds to this enzyme more tightly than compound 26. However, at a relative higher concentration, compound 26 may still exhibit competitive inhibition against FAD. It is not so common to create a FAD competitor. But, there are some published examples on FAD competitors. Quinine and mepacrine (atabrine) were stated to inhibit D-aminoacid oxidase by competing with FAD.60 Chlorpromazine (CPZ), a dopamine D2 receptor antagonist introduced in the treatment of schizophrenia in the early 1950s, acts as a FAD-competitive inhibitor of human D-amino acid oxidase (hDAAO) with similar structure.61K001 that competes for FAD binding, inhibits cryptochrome (CRY) ubiquitylation and lengthens the circadian cycle.62 There are some other papers published on FAD ejectors.63, 64 EN460, an inhibitor of endoplasmic reticulum oxidation 1(ERO1), was reported in 2010.64 This compound exhibits selectivity for ERO1 and displaces of bound FAD from the active site of the enzyme. Nevertheless, the structure of EN460 is different from that of FAD. Furthermore, several other compounds were obtained with virtual screening for a lead, which would compete with FAD.63 Hence, although the structure of compound 26 is not similar with FAD, it is still possibly for it to acts as FAD competitor or FAD ejector. On the other hand, compound 26 may also bind to an allosteric site of LSD1,65, 66, 67 resulting in an apparent change in binding affinity of another ligand at a distinctly different site. Furthermore docking analysis also illustrated the interaction between compound 26 and LSD1.
Figure 4.

Properties of compound 26's inhibitory effect to LSD1 activity in vitro. (A, B, C) Kd values for 2-PCPA (A), compounds 26 (B) and FAD (C) were obtained by microscale thermophoresis (MST) experiment; (D) Time dependent assay of compound 26 against LSD1. The stable progress curves for the inactivation of LSD1 were obtained by indicated concentrations of compound 26 treatment; (E, F) The reversibility of compound 26 to LSD1 activity was determined by dilution assay (E) and dialysis experiment (F), 2-PCPA was used a control; (G, H) Lineweaver–Burk plots demonstrate that compound 26 is non-competitive with the histone H3 substrate (G) and competitive with the LSD1 cofactor FAD (H). Data are mean±SD. P<0.01 was considered statistically highly significant. All experiments were carried out at least three times.
Activity of LSD1 and LSD2 recombinant was evaluated by quantifying the demethylase byproduct H2O2 with Amplex Red method coupled with HRP. Activity of MAO-A and MAO-B was evaluated by the commercialized kit. Data are mean±SD. All experiments were repeated at least three times.
As LSD1 belongs to the monoamine oxidase (MAO) family, the inhibitory effects of compound 22 and 26 to its homologies: MAO-A and MAO-B were also examined using commercially available kits. Compared to 2-PCPA, compounds 22 and 26 had no significant effects on MAO-A and MAO-B activities (Table 6). Recently, another FAD dependent lysine demethylase, named LSD2, was identified with 45% sequence identity with LSD1.68, 69 Therefore, the LSD2 recombinant encoding 22-822AA was also generated from E.Coli and the purified recombinant was then applied for its activity assay. As shown in Table 2, compound 22 and 26 showed poor inhibitory effect against LSD2 recombinant. These findings indicated the high selectivity of this class of inhibitors on LSD1 in vitro.
Table 6.
In vitro inhibitory activities of compound 22, 26 to LSD1 and its homologies: LSD2, MAO-A, and MAO-B. 2-PCPA was used as control.
| IC50(μM) |
||||
|---|---|---|---|---|
| Compd. | LSD1 | LSD2 | MAO-A | MAO-B |
| 22 | 11.5±2.1 | 49.1±7.3 | >1250 | 790.7±10.4 |
| 26 | 2.1±1.1 | 36.6±4.5 | >1250 | >1250 |
| 2-PCPA | 28.7±3.5 | 34.9±5.3 | 10.6±1.0 | 5.9±0.8 |
Docking and Molecular Dynamics (MD) Simulations
Predictions of the binding modes involve a comprehensive evaluation of the center structures of the clusters generated from a Perron-Cluster Cluster Analysis (PCCA) using the following three criteria: (1) the protein-ligand interaction energy obtained from the PCCA analysis; (2) the number of hydrogen bonds between the protein and the ligand calculated using a distance (donor-acceptor) and angle cutoff of 3Å and 20°, respectively; (3) the stability of the ligand in the binding site during MD simulations. Using the above procedure, two types of binding modes were identified for the ligand (Figure 5).
Figure 5.
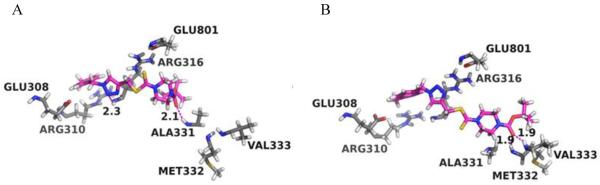
Stereo views of the putative binding modes of the ligands 26 in the FAD-binding site of LSD1. (A) 26-Pose1; (B) 26-Pose2. FAD-binding site residues are colored in grey while the ligand 26 is colored in magenta and green, respectively. Hydrogen bonds between the protein and ligands are shown in magenta dash lines with the distances (Å) between the donor hydrogen atom and the acceptor heavy atom labeled.
The binding modes predicted that the carbonyl oxygen (next to the tri-methyl group) is able to form hydrogen bonds to the backbone nitrogen of either ALA331 (Figure 5A) or both MET332 and VAL333 (Figure 5B). The triazole ring is also favorably positioned to form hydrogen bonds to the backbone nitrogen of ARG316 (Figure 5A). Furthermore, ALA331, MET332, VAL333 and ARG316 are part of the pocket that surround the isoalloxazine moiety of FAD.70 Occupation of these amino acids may result in the ejection of FAD. To further assess the dynamic stabilities of the predicted binding poses, short (1 ns) MD simulations were also performed on the binding poses of the two ligands. Overall, we found that both binding modes were well maintained during the short MD simulations for the ligands, which is in line with the experimental data showing that compound 26 stand in the cavity of LSD1 belong to the FAD-binding subdomain, while Arg 316 is one of the binding sites of FAD.71 These results further support our findings about the competitive inhibition of compound 26 over FAD, but specific mechanism on how does compound 26 act on LSD1 still need to be clarified in the further structure analysis.
Effects of compounds 22 and 26 on LSD1 activity in cultured gastric cancer cells and their cytotoxicities
To determine whether compounds 22 and 26 are cell active LSD1 inhibitors, the effect of these compounds on the methylation levels of LSD1 substrates H3K4 and H3K9 were analyzed in MGC-803 cells. We found that the amounts of H3K4me/me2 and H3K9me2 were dose dependently elevated after treatment of MGC-803 cells with these two compounds (Figure 6A, B). These results validated that compounds 22 and 26 can decrease LSD1 activity at the cellular level. To further determine the specificity of compounds 22 and 26 at the cellular level, the expression level of H3K4me3 that is the substrate of histone demethylase JARID1, was also evaluated. Figures 6A and B show that compounds 22 and 26 did not affect the expression of H3K4me3. These results suggested that compounds 22 and 26 are specific and cell active LSD1 inhibitors.
Figure 6.

Histone methylation in MGC-803 cells after treatment by compound 22(A) and 26(B) for 48h. The expressions of H3K4me1, me2 and H3K9me2, as well as H3K4me3 were then determined by western blot. The total levels of histone 3 (H3) were used as loading control. Data are mean±SD. *P<0.05 was considered significant, **P<0.01 was considered statistically highly significant. All experiments were carried out at least three times.
To further determine whether compounds 22 and 26 are potential candidates for chemotherapeutic agents against gastric cancer, the in vitro cytotoxicities of compounds 22 and 26 were evaluated by MTT method. Both compounds exhibited strong cytotoxicity against human gastric cancer cell lines MGC-803 and HGC-27 with higher LSD1 expression (IC50 of compounds 22 and 26 for MGC-803 line are 1.26μM and 0.89μM respectively; IC50 of compounds 22 and 26 for HGC-27 cell line are 1.56μM and 1.13μM, respectively) (Figures 7A, B, C, D). However, both of them did not show any marked effects on normal gastric epithelial cell line GES-1 and gastric cancer cell line SGC-7901 and both with lower LSD1 expression (IC50 of compounds 22 and 26 for SGC-7901 cell line are 69.5μM and 89.5μM respectively; IC50 of compounds 22 and 26 for GES-1 cell line are 50.26μM and 45.26 respectively) (Figure 7A, B, C, D, E). siRNA mediated LSD1 knock down (Figure 7F) in MGC-803 cells also resulted in a loss of cell viability (Figure 7G). These findings implicated that the cytotoxicities of compounds 22 and 26 on gastric cancer cells may be associated with their specific inhibition to the enhanced LSD1 expression in those cells.
Figure 7.

IC50 and cell viability of compounds 22 (A and B), and compound 26 (C and D) treatment on three gastric cancer cell lines: MGC-803, HGC-27, SGC-7901 and normal gastric epithelial cell line: GES-1; (E) The expression levels of LSD1 on the four gastric cell lines indicated above; (F) 3 days after LSD1 siRNA transfection in MGC-803 cells, LSD1 protein levels were determined by western blot, GAPDH was used as the loading control. (G) Cell viability upon LSD1 siRNA treatment. Data are mean±SD. **P <0.01 was considered statistically highly significant. All experiments were carried out at least three times.
Effect of Compound 26 on Apoptosis, Cell migration, Invasion
To explore cytotoxicity of 26 in MGC-803 cells, cell apoptosis was investigated with Hoechst 33258 staining. After 48h incubation with 26 at indicated concentrations, characteristic apoptotic morphological changes were observed by fluorescence microscope, including cell rounding, chromatin shrinkage and formation of apoptotic bodies (Figure 8A). The apoptotic analysis was also performed with Annexin V-FITC/PI double staining and quantitated by flow cytometry, compound 26 treatment of MGC-803 cells dose dependently increased the percentage of the apoptotic population up to 12.6%, 31.7% and 44.7%, respectively, compared to control (7.9%) (Figure 8B, C). As LSD1 shows a strong preference to reverse K370me2 of p53, which inhibits its association with the coactivator 53BP1 (p53-binding protein 1) through tandem Tudor domains in 53BP1 and represses p53 function,5 we suppose that 26 may prevent the demethylation at K370me2 of p53 by LSD1 and promote p53-mediated apoptosis.
Figure 8.
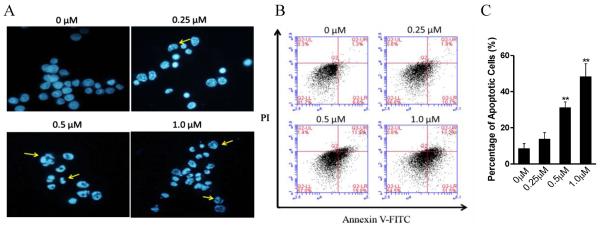
Compound 26 induced apoptosis in MGC-803 cells. (A) Apoptosis analysis with Hoechst-33258 staining after 48h treatment of 26 in MGC-803 cells; (B and C) Quantitative analysis of apoptotic cells using AnnexinV-FITC/PI double staining and flow-cytometry calculation. **P <0.01 was considered statistically highly significant. Data are mean±SD. All experiments were carried out at least three times.
In addition, MGC-803 cell migration was evaluated by wound healing and transwell experiments. Cell invasion capacity was determined by matrigel coated transwell experiment following a reported procedure.72 Microphotographs showed that untreated gastric cancer MGC-803 cells filled most of the wounded area two days after scratching the cell monolayer, whereas treatment with indicated doses of compound 26 markedly suppressed repairment of the wound (Figure 9A). The inhibitory effect of compound 26 on repopulation of the wounded area was not due to decreased proliferation, because the highest concentration (0.25 μM) of compound 26 we used in this assay cannot inhibit the cell proliferation as we discussed in Figure 8. Compound 26 also significantly inhibited the cell migration and invasion even at low concentration (0.02 μM) (Figure 9B and C), so did the siRNA mediated LSD1 knock down, when compared to the positive control 100μM 2-PCPA (Figure 9B and C). These results indicate that the effect of compound 26 on cell migration and invasion is much more powerful than 2-PCPA. LSD1 can form a complex with snai1 to act at the E-Box domain of E-Cadherin and restrain the expression of E-cadherin for promoting cell migration and invasion.53, 54 After incubation with compound 26, the protein levels of E-Cadherin were markedly elevated, without significant alteration of the total levels of LSD1 and snai1, compared to those on control (Figure 9D, E). To further determine the effects of compound 26 on the complex formation between LSD1 and snail, co-immunoprecipitation was performed, using LSD1 antibody as a bait. After compound 26 treatment for 48h, the binding of LSD1 to snai1 were significantly decreased, which is the same as that by the positive control 2-PCPA (Figure 9F, G). These results indicated that the enhanced expression of E-Cadherin by compound 26 may be associated with the decreased interaction of LSD1 and snail, and further clarified the inhibitory mechanism on the cell migration and invasion by this compound.
Figure 9.

Effects of compound 26 on cell migration, invasion in MGC-803 cells. (A) Wound healing assay; (B and C) Migration and invasion ability assay. Treatment of both compound 26 and LSD1 siRNA resulted in the depressed migration (B) and invasion (C) ability in MGC-803 cells, 2-PCPA was used as the positive control; (D and E) The expressions of E-Cadherin, snai1 and LSD1 after 48h treatment of compound 26; (F and G) Immunoprecipitation and densitomery quantitation of LSD1 and snai1 without (control) or with compound 26 and 100, 250μM 2-PCPA treatment in MGC-803 cells; input represents cell lysate controls. GAPDH was used as loading control. *P< 0.05 was considered statistically significant, **P<0.01 was considered statistically highly significant. Data are mean±SD. All experiments were carried out at least three times.
In vivo anti-tumor study
We next examined the effect of compound 26 on tumor growth in vivo in a xenograft model. Xenograft tumors were generated by subcutaneous implantation of MGC-803 cells into nude mice. After the treatment, body weights of the mice were monitored and their tumor sizes were measured and recorded every 3 days (Figure 10A). Compound 26 treatment at a dose of 20mg/kg significantly inhibited the growth of tumor over time and reduced tumor weight by 68.5% (Figures 10A, B and C). There were no apparent body weight loss during the treatment (Figure 10D). These data indicate that compound 26 was efficacious in inhibiting the growth of LSD1 overexpressing gastric tumor in vivo, but no obvious global toxicity.
Figure 10.

In vivo antitumor effects of compound 26 in MGC-803 bearing nude mice. MGC-803 cells were transplanted subcutaneously to the BALB-C nude mice and subjected to saline, hydroxypropyl-β-cyclodextrin (HP-β-CD) and compound 26 (5, 10, 20 mg/kg) for 21 days, respectively, and were then analyzed for tumor volume (A); (B) Represented tumors with the indicated treatment; (C) Tumor weight with the indicated treatment; (D) Body weight with the indicated treatment. *P < 0.05 was considered statistically significant, **P <0.01 was considered statistically highly significant. Data are mean±SD.
CONCLUSION
In summary, we have synthesized and identified a novel class of triazole-dithiocarbamate based, selective LSD1 inhibitors. Some of these triazole-dithiocarbamates, especially compound 26, exhibit reversible and FAD competitive LSD1 inhibition. To our best knowledge, compound 26 is the first identified highly selective LSD1 inhibitor with the ability to inhibit cell migration and invasion at lower concentrations. Our findings indicate that the lead triazole-dithiocarbamate based LSD1 inhibitor may be developed as a novel anti-cancer epigenetic drug to target gastric cancer with overexpression of LSD1.
METHODS
General Methods for Chemistry
Reagents and solvents were purchased from commercial sources and were used without further purification. Melting points were determined on a X-5 micromelting apparatus and. 1H NMR and 13C NMR spectra were recorded on a Bruker 400 MHz and 100 MHz spectrometer respectively. IR spectra were recorded on a Nicolet iS10 infrared spectrometer. High resolution mass spectra (HRMS) were recorded on a Waters Micromass Q-T of Micromass spectrometer. The purity of all biologically evaluated compounds was determined to be >95% by reversephase high performance liquid chromatography (HPLC) analysis. HPLC measurement was performed with a Phenomenex column (C18, 5.0 μm, 4.6 mm × 150 mm) on Dionex UltiMate 3000 UHPLC instrument from ThermoFisher. The signal was monitored at 254nm with a UV dector. A flow rate of 0.5 mL/min was used with mobile phase of MeOH in H2O (80:20, v/v).
General procedure for the synthesis of alkynes 4a-j
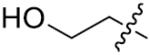
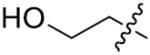
CS2 (2.28 g, 30 mmol) was added drop wise to the solution of tert-butyl piperazine-1-carboxylate (1.86g, 10 mmol) and Na3PO˙412H2O (2.28 g, 6 mmol) in acetone (40 mL). The reaction mixture was stirred at room temperature for 0.5 h. Then propargyl bromide (1.31 g, 11 mmol) was added to the mixture drop wise, the reaction mixture was stirred at room temperature for another 0.5 h. Then, the reaction mixture was filtered and the filtrate was concentrated under reduced pressure, the residue was dissolved in EtOAc (50 mL), washed with water (50 mL), brine (50 mL), dried over anhydrous Na2SO4 and concentrated under vacuum to afford alkyne 4j. Prop-2-ynyl 4-(2-hydroxyethyl)piperazine-1-carbodithioate (4c): Yield 82.2%, white solid. Mp: 96–97°C; 1H NMR (400 MHz, CDCl3) δ 4.36 (brs, 2H), 4.12 (d, J = 2.7 Hz, 2H), 3.96 (brs, 2H), 3.69 (t, J = 5.3 Hz, 2H), 2.64 (m, 6H), 2.27 (t, J = 2.7 Hz, 2H); HRMS (ESI) calcd for C10H17N2OS2 [M + H]+: 245.0782, found: 245.0781.
tert Butyl 4-((prop-2-ynylthio)carbonothioyl)piperazine-1-carboxylate (4j): Yield 92%, white solid. Mp: 87–88°C. 1H NMR (400 MHz, Acetone-d6, ppm): δ 4.28 (brs, 2H), 4.14 (d, J = 2.7 Hz, 2H), 4.00 (brs, 2H), 3.58 (brs, 4H), 2.78 (t, J = 2.7 Hz, 1H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3, ppm): δ 195.16, 154.39, 80.68, 78.19, 71.82, 28.36, 26.00; HRMS (ESI) calcd for C13H21N2O2S2 [M + H]+: 301.1044, found: 301.1046.
General procedure for the synthesis of compounds 12–72
In a round-bottom flask equipped with a magnetic stirred bar, alkyne derivatives 4 (5 mmol), azide derivatives 5 (5.5 mmol), CuSO4·5H2O (62mg, 0.25 mmol), sodium ascorbate (100 mg, 0.5 mmol), THF (20 mL) and H2O (20 mL) were added. The resulting mixture was stirred at room temperature for about 2 h. The disappearance of compounds 4 was monitored by TLC (silica gel, PE:EtOAc = 3:1). After the reaction, water (40 mL) was added and the reaction mixture was extracted with EtOAc (3×40 mL). The combined organic layer was washed with brine (100 mL), dried over anhydrous Na2SO4 and concentrated under vacuum to afford the crude products. The crude products were recrystallized from acetone to yield the pure products 12–72.
tert Butyl 4-(((1-(2-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methylthio)carbonothioyl)-piperazine-1-carboxylate (22): Yield 79.0%, white solid; Mp: 109–110°C; IR (KBr, cm−1) ν: 3447, 2979, 1693, 1494, 1478, 1424, 1279, 1167, 1012, 986, 932, 791, 757, 695; 1H NMR (400 MHz, CDCl3): δ 7.66 (s, 1H), 7.09–7.38 (m, 4H), 5.55 (s, 2H), 4.69 (s, 2H), 4.29 (brs, 2H), 3.91 (brs, 2H), 3.54 (t, J = 5.1 Hz, 4H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 196.42, 161.72, 159.26, 154.41, 144.00, 130.89, 130.81, 130.51, 130.48, 124.82, 124.78, 122.96, 121.98, 121.84, 115.92, 115.71, 80.63, 47.66, 47.61, 31.84, 28.34; 19F NMR (376 MHz, CDCl3) δ −118.03; HRMS (ESI) calcd for C20H27FN5O2S2 [M + H]+: 452.1590, found: 452.1598.
tert Butyl 4-(((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methylthio)carbonothioyl)-piperazine-1-carboxylate (23): Yield 81.5%, white solid. Mp: 171–172°C; IR (KBr, cm−1) ν: 3482, 3139, 2974, 1690, 1470, 1420, 1281, 1167, 1051, 994, 824, 781, 541; 1H NMR (400 MHz, CDCl3, ppm): δ 7.59 (s, 1H), 7.04–7.27 (m, 4H), 5.46 (s, 2H), 4.68 (s, 2H), 4.31 (brs, 2H), 3.90 (brs, 3H), 3.53 (s, 4H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, ppm): δ 196.40, 164.07, 161.60, 154.40, 130.49, 130.45, 129.96, 129.87, 116.20, 115.98, 80.66, 53.42, 31.78, 28.34; 19F NMR (376 MHz, CDCl3) δ −112.68; HRMS (ESI) calcd for C20H27FN5O2S2 [M + H]+: 452.1590, found: 452.1588.
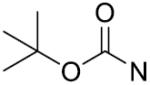
tert Butyl 4-(((1-(2-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methylthio)carbonothioyl)-piperazine-1-carboxylate (24): Yield 78.7%, white solid. Mp: 105–106°C; 1H NMR (400 MHz, CDCl3, ppm): δ 7.70 (s, 1H), 7.46 (dd, J1 = 1.4 Hz, J2 = 7.8 Hz, 1H), 7.23–7.33 (m, 2H), 7.18 (dd, J1 = 1.6 Hz, J2 = 7.4 Hz, 1H), 5.62 (s, 2H), 4.70 (s, 2H), 4.28 (brs, 2H), 3.93 (brs, 2H), 3.54 (t, J = 5.2 Hz, 4H), 1.47 (s, 9H); 13C NMR (100 MHz, Acetone-d6, ppm): δ 195.68, 154.33, 143.01, 133.30, 133.16, 130.60, 130.31, 129.74, 127.71, 123.79, 79.78, 50.99, 31.63, 27.63; HRMS (ESI) calcd for C20H27ClN5O2S2 [M + H]+: 468.1295, found: 468.1292.
tert Butyl 4-(((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methylthio)carbonothioyl)-piperazine-1-carboxylate (26): Yield 81.9%, white solid. Mp: 182–183°C; IR (KBr, cm−1) ν: 3454, 2975, 1682, 1457, 1422, 1224, 1078, 994, 933, 867, 772, 524; 1H NMR (400 MHz, CDCl3, ppm): δ 7.55 (s, 1H), 7.16–7.21 (m, 4H), 5.43 (s, 2H), 4.67 (s, 2H), 4.29 (brs, 2H), 3.91 (brs, 2H), 3.54 (t, J = 5.2 Hz, 4H), 2.35 (s, 3H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, ppm): δ 196.49, 154.41, 138.67, 131.57, 129.75, 128.09, 80.64, 54.01, 31.91, 28.35, 21.15; HRMS (ESI) calcd for C21H30N5O2S2 [M + H]+:448.1841, found: 448.1840.
tert Butyl 4-(((1-(4-methoxybenzyl)-1H-1,2,3-triazol-4yl)methylthio)carbonothioyl)-piperazine-1-carboxylate (27): Yield 85.8%, white solid. Mp: 129–130°C; 1H NMR (400 MHz, CDCl3, ppm): δ 7.54 (s, 1H), 7.21 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 5.41 (s, 2H), 4.67 (s, 2H), 4.29 (brs, 2H), 3.90 (brs, 2H), 3.80 (s, 3H), 3.51 (t, J = 5.2 Hz, 4H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, ppm): δ 196.47, 159.89, 154.40, 143.86, 129.62, 126.59, 122.52, 114.44, 80.61, 55.34, 53.70, 31.90, 28.35; HRMS (ESI) calcd for C21H30N5O3S2 [M + H]+: 464.1790, found: 464.1794.
tert Butyl 4-(((1-(2-bromobenzyl)-1H-1,2,3-triazol-4-yl)methylthio)carbonothioyl) piperazine-1-carboxylate (32): Yield 79.0%, white solid. Mp: 129–130°C; 1H NMR (400 MHz, CDCl3, ppm): δ 7.71 (s, 1H), 7.62 (dd, J1 = 1.1 Hz, J2 = 7.9 Hz, 1H), 7.32 (td, J1 = 1.1 Hz, J2 = 7.5 Hz, 1H), 7.24 (td, J1 = 1.5 Hz, J2 = 7.8 Hz, 1H), 7.12 (dd, J1 = 1.5 Hz, J2 = 7.6 Hz, 1H), 5.62 (s, 2H), 4.69 (s, 2H), 4.30 (brs, 2H), 3.92 (brs, 2H), 3.54 (t, J = 4.8 Hz, 4H,), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3, δ, ppm): 196.4, 154.4, 134.2, 133.2, 130.3, 130.2, 128.2, 123.4, 80.6, 53.8, 31.9, 28.4; HRMS(ESI) calcd for C20H27BrN5O2S2 [M + H]+: 512.0790, found: 512.0793.
General procedure for the synthesis of compounds 73–82
CF3COOH (4.56g, 40mmol) was added to a solution of 22 (0.90g, 2mmol) in CH2Cl2 (20mL) at 0°C. The reaction mixture was warmed to room temperature and stirred at the same temperature for about 8 h. The disappearance of compounds 22 was monitored by TLC (silica gel, CH2Cl2:CH3OH = 8:1). The reaction mixture was concentrated under vacuum, the residue was dissolved in CH2Cl2 (20 mL), washed with saturated NaHCO3 (50 mL), brine (50mL), dried over anhydrous Na2SO4 and concentrated under vacuum to afford compounds 73, which were used in the next reaction without further purification.
(1-(2-Fluorobenzyl)-1H-1,2,3-triazol-4-yl)methyl piperazine-1-carbodithioate (73): Yield 96.7%, white solid. Mp: 93–94°C; 1H NMR (400 MHz, CDCl3, ppm): δ 7.67 (s, 1H), 7.34–7.40 (m, 1H), 7.24–7.26 (m, 1H), 7.11–7.18 (m, 2H), 5.55 (s, 2H), 4.69 (s, 2H), 4.30 (brs, 2H), 3.93 (brs, 2H), 2.96 (t, J = 4.8 Hz, 4H); 13C NMR (100 MHz, CDCl3, ppm): δ 195.81, 161.72, 159.26,144.27, 130.88, 130.80, 130.50, 130.47, 124.82, 124.79, 122.98, 122.00, 121.86, 115.93, 115.72, 47.66, 47.61, 45.58, 31.76; HRMS (ESI) calcd for C15H19FN5S2 [M + H]+:352.1066, found: 352.1064. 1-(4-Methylbenzyl)-1H-1,2,3-triazol-4-yl)methyl piperazine-1-carbodithioate (78): yield 94.0%. white solid. Mp: 74–75°C; 1H NMR (400 MHz, CDCl3, ppm): δ 7.56 (s, 1H), 7.18 (s, 4H), 5.43 (s, 2H), 4.67 (s, 2H), 4.31 (brs, 2H), 3.92 (brs, 2H), 2.96 (brs, 4H), 2.35 (s, 3H); 13C NMR (100 MHz, CDCl3, ppm): δ 195.82, 138.61, 131.59, 129.74, 128.08, 122.72, 53.96, 31.80, 29.68, 21.17; HRMS (ESI) calcd for C16H22N5S2 [M + H]+:348.1317, found: 348.1319.
(1-(4-Methoxybenzyl)-1H-1,2,3-triazol-4-yl)methyl piperazine-1-carbodithioate (79): Yield 92.7%, white solid. Mp: 95–96°C; 1H NMR (400 MHz, CDCl3, ppm): δ 7.55 (s, 1H), 7.24 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 5.42 (s, 2H), 4.68 (s, 2H), 4.31 (brs, 2H), 3.95 (brs, 2H), 3.80 (s, 3H), 2.96 (t, J = 4.6 Hz, 4H); 13C NMR (100 MHz, CDCl3, ppm): δ 195.8, 159.9, 144.1, 129.6, 126.6, 122.5, 114.5, 55.3, 53.7, 45.6, 31.8; HRMS (ESI) calcd for C16H22N5OS2 [M + H]+:364.1266, found: 364.1263.
General procedure for the synthesis of compounds 83–92
A mixture of 73 (0.70 g, 2 mmol), K2CO3 (0.28 g, 2 mmol) and CbzCl (0.38 g, 2.2 mol) in CH2Cl2 (20mL) was stirred at room temperature for 7 h. The disappearance of compounds 73 was monitored by TLC (silica gel, PE:Actone = 3:1). After the reaction, K2CO3 was removed by filtration and the solvent was diluted with CH2Cl2 (10 mL), washed with water (40 mL), brine (40 mL), dried over anhydrous Na2SO4 and concentrated under vacuum to give the crude product, which was recrystallized from acetone to provide compound 83 as white solid.
Benzyl 4-(((1-(2-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methylthio)carbonothioyl) piperazine-1-carboxylate (83): Yield 74.6%, white solid. Mp: 138–139°C; 1H NMR (400 MHz, CDCl3, ppm): δ 7.66 (s, 1H), 7.08–7.37 (m, 9H), 5.54 (s, 2H), 5.15 (s, 2H), 4.69 (s, 2H), 4.30 (brs, 2H), 3.95 (brs, 2H), 3.61 (t, J = 5.2 Hz, 4H); 13C NMR (100MHz, CDCl3, ppm): δ 196.61, 161.73, 159.27, 155.04, 143.97, 136.21, 130.94, 130.85, 130.54, 130.51, 128.61, 128.30, 128.09, 124.84, 124.80, 123.04, 121.93, 121.79, 115.94, 115.73, 67.64, 47.72, 47.68, 43.02, 31.85; HRMS (ESI) calcd for C23H25FN5O2S2 [M + H]+: 486.1434, found: 486.1432.
Benzyl 4-(((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methylthio) carbonothioyl) piperazine-1-carboxylate (87): Yield 81.8%, white solid. Mp: 99–100°C; 1H NMR (400 MHz, CDCl3, ppm): δ 7.55 (s, 1H), 7.14–7.37 (m, 9H), 5.44 (s, 2H), 5.16 (s, 2H), 4.67 (s, 2H), 4.29 (brs, 2H), 3.96 (brs, 2H), 3.59 (t, J = 5.1 Hz, 4H), 2.35 (s, 3H); 13C NMR (100 MHz, CDCl3, ppm): δ 196.67, 155.04, 138.68, 136.21, 131.56, 129.76, 128.61, 128.31, 128.10, 122.73, 67.64, 54.02, 43.03, 31.95, 21.17; HRMS (ESI) calcd for C24H28N5O2S2 [M + H]+: 482.1684, found: 482.1683.
Benzyl 4-(((1-(3,4,5-trimethoxybenzyl)-1H-1,2,3-triazol-4-yl)methylthio)carbonthioyl) piperazine-1-carboxylate (90): Yield 82.0%, white solid. Mp: 134–135°C. 1H NMR (400 MHz, CDCl3, ppm): δ 7.63 (s, 1H), 7.35–7.37 (m, 5H), 6.47 (s, 2H), 5.40 (s, 2H), 5.15 (s, 2H), 4.68 (s, 2H), 4.28 (brs, 2H), 3.96 (brs, 2H), 3.83 (s, 3H), 3.82 (s, 6H), 3.62 (t, J = 5.0 Hz, 4H); 13C NMR (100 MHz, CDCl3, ppm): δ 196.60, 155.02, 153.69, 144.02, 138.30, 136.19, 130.08, 128.60, 128.30, 128.09, 122.82, 105.23, 67.64, 60.85, 56.25, 54.38, 43.01, 31.86; HRMS (ESI) calcd for C26H32N5O5S2 [M + H]+: 558.1845, found: 558.1841.
Ethyl 4-(((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methylthio)carbonothioyl) piperazine-1-carboxylate (91): Compound 91 was prepared from 77 and ethyl chloroformate using method above. Yield 77.4%, white solid. Mp: 100–101°C. 1H NMR (400 MHz, CDCl3, ppm): δ 7.57 (s, 1H), 7.17 (s, 4H), 5.44 (s, 2H), 4.70 (s, 2H), 4.31 (brs, 2H), 4.20 (q, J = 7.1 Hz, 2H), 3.93 (brs, 2H), 3.58 (s, 4H), 2.35 (s, 3H), 1.29 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3, ppm): δ 196.60, 155.25, 143.98, 138.70, 131.47, 129.77, 128.13, 122.82, 61.91, 54.08, 42.90, 31.85, 21.18, 14.65; HRMS (ESI) calcd for C19H26N5O2S2 [M + H]+: 420.1528, found: 420.1527.
Isopropyl 4-(((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methylthio)carbonothioyl)piperazine-1-carboxylate (92): Compound 92 was prepared from 77 and isopropyl chloroformate using method above. Yield 84.5%, Mp: 149–150°C. 1H NMR (400 MHz, CDCl3, ppm): δ 7.55 (s, 1H), 7.17 (s, 4H), 5.44 (s, 2H), 4.97 (m, 1H), 4.67 (s, 2H), 4.31 (brs, 2H), 3.93 (brs, 2H), 3.56 (s, 4H), 2.35 (s, 3H), 1.26 (d, J = 6.2 Hz, 6H); 13C NMR (100 MHz, CDCl3, ppm): δ 196.56, 154.91, 143.88, 138.66, 131.57, 129.75, 128.10, 122.65, 69.41, 53.97, 42.84, 31.90, 22.21, 21.18; HRMS (ESI) calcd for C20H27N5NaO2S2 [M + Na]+: 456.1504, found: 456.1503.
General procedure for the synthesis of compounds 93–95
A mixture of 73 (0.70g, 2mmol), Et3N (0.76g, 2.2mmol) and CH3COCl (0.17g, 2.2mol) in CH2Cl2 (10mL) was stirred at room temperature for 4.5 h. The disappearance of compounds 73 was monitored by TLC (silica gel, PE:Actone = 3:1). After the reaction, the solvent was washed with diluted hydrochloric acid, saturated NaHCO3 (40 mL) and brine (40 mL), dried over anhydrous Na2SO4 and concentrated under vacuum to give the crude product, which were recrystallized from acetone to provide compound 93 as white solid.
(1-(2-Fluorobenzyl)-1H-1,2,3-triazol-4-yl)methyl 4-acetylpiperazine-1-carbodithioate (93): Yield 71.3%,white solid, mp. 1H NMR (400 MHz, CDCl3) δ 7.67 (s, 1H), 7.09–7.38 (m, 4H), 5.55 (s, 2H), 4.68 (s, 2H), 4.30 (brs, 2H), 4.00 (brs, 2H), 3.73 (t, J = 5.1 Hz, 2H), 3.60 (t, J = 5.1 Hz, 2H), 2.13 (s, 3H); HRMS (ESI) calcd for C17H20FN5NaOS2 [M + Na]+: 416.0991, found: 416.0988.
Cells and Cell Viability Assay
The human gastric carcinoma cell lines MGC-803 and HGC-27 were supplied by the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences. Human gastric carcinoma cell line SGC-7901 was purchased from Shanghai Bogoo Biotechnology Company. Human gastric epithelial mucosa cell line GES-1 was preserved in our institute. Cells were cultured in DMEM medium or RPMI-1640 medium. All media were supplemented with 10% FBS. Cells were cultured in an incubator with 5% CO2 at 37 °C with medium changes every 2 days.
The cell viability was determined by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) assay according to the manufacturer's instructions. This assay measures dehydrogenase enzyme activity in metabolically active tumor cells, as reflected by the conversion of MTT to formazan, which is soluble in tissue culture medium and is detected by absorbance (A) at 570 nm. The production of formazan is proportional to the number of living cells, with the intensity of the produced color serving as an indicator of cell viability. The data were analyzed with SPSS 20.
Inhibitory Evaluation of the Synthesized Compounds Against LSD1, LSD2, MAO-A, MAO-B and Mechanism of Action Studies
cDNA encoding LSD1(157-852AA) was obtained by RT-PCR and cloned into pET28b to have the constructed plasmid pET28b-LSD1, then the plasmid was transfected into BL21(DE). The recombinant was induced with 0.25mM IPTG at 20°C and purified as published, as well as pET28a-LSD2, which is a kindly gift from Dr. Chen.18, 69 Then incubate the compounds with the recombinant and H3K4me2, after that, the fluoresce was measured at excitation wavelength 530 nm and emission wavelength 590 nm as reported in order to evaluate the inhibition rate of the candidate compound.73 Inhibitory effect of the candidate compounds against MAO-A and MAO-B were evaluated with the commercialized kit.
The dilution assay was done as published.21 Briefly, 2.5 μg of LSD1 recombinant were incubated with either 312.5 μM compound 26, 600 μM tranylcypromine, or DMSO. 1 h later, 1.25 μl aliquots were removed from all samples and diluted into HRP-assay solution containing substrate and coupling reagents to a final volume of 100 μl. This represents an 80-fold dilution of the inhibitor concentration, which is expected to yield the same inhibition rate for an irreversible inhibitor or significant difference for a reversible inhibitor.
For the dialysis experiment, compound 26 and LSD1 were incubate for 1 h at 37°C and then dialyzed against the hepes buffer at 4°C for 24 h. The buffer was changed every 12 h. Another two groups without inhibitor or with 2-PCPA were served as negative or positive controls. Then, the activity of LSD1 in the dialyzed tube was measured to evaluate the reversibility of compound 26 inhibitory. Another dialysis experiment was performed, using FAD and LSD1 incubated with or without the inhibitor against hepes buffer at 4°C for 24 h. In the inhibitor treatment group, the concentrations of inhibitors on both sides of dialysis tube were the same. Then, the amount of FAD in the dialysis tubes was measured at 468nm with UV/VIS spectrophotometer.
For the competitive analysis of compound 26, demethylase activity of LSD1 was assessed in the presence of different concentrations of the compound (0, 0.5, 2.5 and 12.5 μM) at a fixed concentration of FAD (2.5 μM) and peptide concentrations from 10 to 80 μM or fixed concentration of peptide (25 μM) and FAD concentrations from 0.6 to 9.0 μM. Assays were performed triplicate, and kinetic values were obtained using Lineweaver-Burk plots.59
MST Experiment
The LSD1 recombinant from Millipore was labeled with a red fluorescent dye using commercialized kit. The thermophoretic movement of the fluorescently labeled protein in complex with selected inhibitors was measured by monitoring the fluorescence distribution inside the capillary. The concentration of the labeled protein was kept constant at ~200 nM, while the concentration of the compound was varied. The samples were loaded into MST-grade glass capillaries. After a short incubation period, the MST analysis was performed using the Monolith NT.115.
Docking and Molecular Dynamics Simulations
The initial coordinates of LSD1 were obtained from Protein Data Bank (PDB) (PDB code: 2H9471). Two disordered loops with FASTA sequence EVKPPRDI and PSIPGAPQP were unresolved and thus built using MODELLER 9.10.74 The protein was then protonated at pH 7.0 using MCCE 2.2.75, 76 The coordinates of the ligands 26 were obtained from their SMILES using OEChem TK (OpenEye Scientific Software). AM1-BCC charges were assigned as the partial charges of the ligands using Antechamber.77, 78
Multiple conformers of ligand were generated using OMEGA (OpenEye Scientific Software) and docked into the FAD-binding site of LSD1 using OEDocking (OpenEye Scientific Software), which uses an exhaustive search algorithm and Chemgauss3 scoring function.79 One thousand binding poses were generated and clustered using a Root Mean Square Deviation (RMSD) metric to obtain ten most dissimilar clusters. Molecular dynamics (MD) simulations were subsequently performed on the centroid structures for each of the ten clusters using GROMACS 4.6.2.80 The amber99sb-ildn and GAFF force fields were used for the protein and ligands, respectively.81, 82 The protein-ligand complex was solvated in TIP3P water molecules. A 1500-step energy minimization and a 50 ps NVT equilibration simulation were performed at 300K before a 1ns production run was performed in NPT ensemble using a time step of 2 fs. Snapshots were saved every 1 ps for analysis. The ten 1 ns production trajectories were concatenated for a Perron-Cluster Cluster Analysis (PCCA),83 which generated ~20 distinct structure clusters.
SiRNA Transfection
6×105 cells were seeded in a 6 well plate and then incubated for 3 days in standard medium in the presence of 20–100 nM siRNA directed against LSD1 or control siRNA (Scrambled) complexed with Lipofectamine™ RNAiMAX Reagent according to the manufacturer's instructions.
Migration and Invasion Assay
For the wound healing assay, cells were plated to in a 24-well plate and the cell surface was scratched using a 10 μL pipette tip. Then, cells were treated with compound 26 with different concentrations followed by 48 h incubation and photographed on an inverted microscope.
For invasion assays, cells were plated in transwell 24 Well plates coated with diluted Matrigel. In the upper chamber, medium was supplemented with 1% heat-inactivated FBS. In the lower chamber, 20% FBS was used as chemoattractant. Different concentrations of compound 26 were added in the chamber. After 48 h, medium was removed and the chambers were washed twice with PBS. Non-invading cells were removed from the upper surface of the membrane by scrubbing with cotton tipped swab, and the invading cells were fixed with methanol for 15 min. Then the chambers were stained with 0.1% crystal violet for 30 min. 6 fields for each chamber were photographed on an inverted microscope and invading cells were counted in each field.
For migration assays, cells were plated in transwell 24 Well plates without Matrigel. The medium, attractant, staining and cell counting method were the same as invasion assay.
Immunoprecipitation and Western Blot
Cross linked immunoprecipitation was performed with the commercialized kit from Thermo Fisher. Western Blot was was perform with the total lysates by RIPA or histone purified with kit from Epigentek. Equal amounts of cell lysates were denatured, separated by SDS-PAGE and transferred to 0.2 μm nitrocellulose membranes. After blocking with PBS containing 5% non-fat dry milk, the membranes were incubated overnight at 4°C with primary antibody, followed by incubation with a horseradish peroxidase-conjugated secondary antibody. The immunoblots were visualized by enhanced chemiluminescence kit from Thermo Fisher.
Antibodies used were against histone H3K4me (Selleck Chemicals #A1237), H3K4me2 (Epitomics #1347-1), H3K9me2 (Epitomics #1349-1), H3K4me3 (Bioss, bs-4715R), total H3 (Sino Biological Inc #100005-MM01), LSD1 (Epitomics #5890-1), E-Cadherin (Epitomics #1702-1), Snai1 (Cell Signaling #3879), GAPDH (GoodHere #AB-M-M 001).
Apoptotic Analysis
Changes in cell morphology were analyzed by Hoechst-33258 staining. Cells were treated with compound 26 at the indicated concentrations for 48 h. Then the cells were fixed and stained with 5μg/mL Hoechst-33258. After staining, the cells were washed. Apoptotic cells were examined and identified according to the condensation and fragmentation of their nuclei by fluorescence microscopy.
The apoptosis was quantified by FACS with AnnexinV-FITC/PI Staining kit from Biovision. Ten thousand events for each sample were counted and analyzed by Accuri C6 flow cytometer. The early and late apoptotic cells were identified by the localization of Annexin V and PI.
Xenograft Studies
Xenografts models using human gastric cancer cell line (MGC-803) was established in BALB/C mice. Once the tumors reached 100 mm3, mice were divided into control groups (saline and hydroxypropyl-β-Cyclodextrin) and treatment groups. The treatment groups received compound 26 (5, 10, 20 mg/kg prepared with hydroxypropyl-β-Cyclodextrin) i.v. per day for a period of 21 days. Tumor volumes were measured at 3 days interval. After the 21st day, the mice were euthanized, and the tumors were isolated and weighed. Tumor size was determined by calliper measurements and the body weight was measured at 3 days interval to monitor drug toxicity.
Statistical analyses
Data were expressed as mean±SD. The significance of the difference between different groups was determined with analysis of variance (ANOVA) and Student t test. Results were considered statistically significant at P<0.05, P<0.01 was considered highly significant.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by National Natural Science Foundation of China (Project No. 81172937 by Dr. Hong-Min Liu, Project No. 81270270 by Dr. Wen Zhao). We are grateful to Dr. Shaomin Wang, Analysis and Testing Center, Zhengzhou University, for performing the HRESI-MS data; Mr. Bing Zhao, School of Pharmaceutical Sciences, Zhengzhou University, for the NMR analysis. We also thank Mr. Liang Zhou (Quantum Design China & NanoTemper Technologies GmbH) for providing the Microscale Thermophoresis (MST) assay.
ABBREVIATIONS USED
- LSD1
histone lysine specific demethylase 1
- LSD2
histone lysine specific demethylase 2
- HKMT
histone lysine methytransferase
- HKDM
histone demethylase
- DNMT
DNA methyltransferase
- MAO
monoamine oxidase
- HDAC
histone deacetylase
- DSF
disulfiram
- MRSA
methicillin-resistant staphylococcus aureus
- FAD
flavin adenine dinucleotide
- 2-PCPA
tranylcypromine
- HPLC
high performance liquid chromatography
- MST
microscale thermophoresis
- HP-β-CD
hydroxypropyl-β-cyclodextrin
- FBS
fetal bovine serum
- SD
standard deviation
Footnotes
Publisher's Disclaimer: “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a free service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are accessible to all readers and citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
Supporting Information. Characterization data for selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E, Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nature genetics. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 3.Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E, Chen T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature. 2009;461:415–418. doi: 10.1038/nature08315. [DOI] [PubMed] [Google Scholar]
- 5.Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, Berger SL. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- 6.Scoumanne A, Chen X. The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners. The Journal of biological chemistry. 2007;282:15471–15475. doi: 10.1074/jbc.M701023200. [DOI] [PubMed] [Google Scholar]
- 7.Kontaki H, Talianidis I. Lysine methylation regulates E2F1-induced cell death. Molecular cell. 2010;39:152–160. doi: 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 9.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, Nelson PS, Liu XS, Brown M, Balk SP. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer cell. 2011;20:457–471. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, Kirfel J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010;31:512–520. doi: 10.1093/carcin/bgp324. [DOI] [PubMed] [Google Scholar]
- 11.Pollock JA, Larrea MD, Jasper JS, McDonnell DP, McCafferty DG. Lysine-specific histone demethylase 1 inhibitors control breast cancer proliferation in ERalpha-dependent and -independent manners. ACS chemical biology. 2012;7:1221–1231. doi: 10.1021/cb300108c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Q, Huang Y, Marton LJ, Woster PM, Davidson NE, Casero RA., Jr Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1 (LSD1) and altering chromatin structure in human breast cancer cells. Amino acids. 2012;42:887–898. doi: 10.1007/s00726-011-1004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magerl C, Ellinger J, Braunschweig T, Kremmer E, Koch LK, Holler T, Buttner R, Luscher B, Gutgemann I. H3K4 dimethylation in hepatocellular carcinoma is rare compared with other hepatobiliary and gastrointestinal carcinomas and correlates with expression of the methylase Ash2 and the demethylase LSD1. Human pathology. 2010;41:181–189. doi: 10.1016/j.humpath.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Bennani-Baiti IM, Machado I, Llombart-Bosch A, Kovar H. Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing's sarcoma, osteosarcoma, and rhabdomyosarcoma. Human pathology. 2012;43:1300–1307. doi: 10.1016/j.humpath.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 15.Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. International Journal of Cancer. 2011;128:574–586. doi: 10.1002/ijc.25349. [DOI] [PubMed] [Google Scholar]
- 16.Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, Song Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PloS one. 2012;7:e35065. doi: 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki T, Miyata N. Lysine demethylases inhibitors. Journal of medicinal chemistry. 2011;54:8236–8250. doi: 10.1021/jm201048w. [DOI] [PubMed] [Google Scholar]
- 18.Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM, Casero RA., Jr. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dulla B, Kirla KT, Rathore V, Deora GS, Kavela S, Maddika S, Chatti K, Reiser O, Iqbal J, Pal M. Synthesis and evaluation of 3-amino/guanidine substituted phenyl oxazoles as a novel class of LSD1 inhibitors with anti-proliferative properties. Organic & Biomolecular Chemistry. 2013;11:3103–3107. doi: 10.1039/c3ob40217g. [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, Steinbergs N, Murray-Stewart T, Marton LJ, Casero RA. Oligoamine analogues in combination with 2-difluoromethylornithine synergistically induce re-expression of aberrantly silenced tumour-suppressor genes. The Biochemical journal. 2012;442:693–701. doi: 10.1042/BJ20111271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willmann D, Lim S, Wetzel S, Metzger E, Jandausch A, Wilk W, Jung M, Forne I, Imhof A, Janzer A, Kirfel J, Waldmann H, Schule R, Buettner R. Impairment of prostate cancer cell growth by a selective and reversible LSD1 inhibitor. International journal of cancer. Journal international du cancer. 2012;131:2704–2709. doi: 10.1002/ijc.27555. [DOI] [PubMed] [Google Scholar]
- 22.Hazeldine S, Pachaiyappan B, Steinbergs N, Nowotarski S, Hanson AS, Casero RA, Jr., Woster PM. Low Molecular Weight Amidoximes that Act as Potent Inhibitors of Lysine-Specific Demethylase 1. Journal of medicinal chemistry. 2012;55:7378–7391. doi: 10.1021/jm3002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohse B, Kristensen JL, Kristensen LH, Agger K, Helin K, Gajhede M, Clausen RP. Inhibitors of histone demethylases. Bioorganic & medicinal chemistry. 2011;19:3625–2636. doi: 10.1016/j.bmc.2011.01.046. [DOI] [PubMed] [Google Scholar]
- 24.Edmondson DE, Mattevi A, Binda C, Li M, Hubalek F. Structure and mechanism of monoamine oxidase. Current medicinal chemistry. 2004;11:1983–1993. doi: 10.2174/0929867043364784. [DOI] [PubMed] [Google Scholar]
- 25.Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chemistry & biology. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Culhane JC, Szewczuk LM, Liu X, Da G, Marmorstein R, Cole PA. A Mechanism-Based Inactivator for Histone Demethylase LSD1. Journal of the American Chemical Society. 2006;128:4536–4537. doi: 10.1021/ja0602748. [DOI] [PubMed] [Google Scholar]
- 27.Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, Jones RJ, Woster PM, Casero RA., Jr. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:7217–7128. doi: 10.1158/1078-0432.CCR-09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ueda R, Suzuki T, Mino K, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, Miyata N. Identification of Cell-Active Lysine Specific Demethylase 1-Selective Inhibitors. Journal of the American Chemical Society. 2009;131:17536–17537. doi: 10.1021/ja907055q. [DOI] [PubMed] [Google Scholar]
- 29.Sharma SK, Wu Y, Steinbergs N, Crowley ML, Hanson AS, Casero RA, Woster PM. (Bis)urea and (bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators. Journal of medicinal chemistry. 2010;53:5197–5212. doi: 10.1021/jm100217a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mimasu S, Umezawa N, Sato S, Higuchi T, Umehara T, Yokoyama S. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry. 2010;49:6494–6503. doi: 10.1021/bi100299r. [DOI] [PubMed] [Google Scholar]
- 31.Binda C, Valente S, Romanenghi M, Pilotto S, Cirilli R, Karytinos A, Ciossani G, Botrugno OA, Forneris F, Tardugno M, Edmondson DE, Minucci S, Mattevi A, Mai A. Biochemical, Structural, and Biological Evaluation of Tranylcypromine Derivatives as Inhibitors of Histone Demethylases LSD1 and LSD2. Journal of the American Chemical Society. 2010;132:6827–6833. doi: 10.1021/ja101557k. [DOI] [PubMed] [Google Scholar]
- 32.Culhane JC, Wang D, Yen PM, Cole PA. Comparative Analysis of Small Molecules and Histone Substrate Analogues as LSD1 Lysine Demethylase Inhibitors. Journal of the American Chemical Society. 2010;132:3164–3176. doi: 10.1021/ja909996p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tortorici M, Borrello MT, Tardugno M, Chiarelli LR, Pilotto S, Ciossani G, Vellore NA, Bailey SG, Cowan J, O'Connell M, Crabb SJ, Packham G, Mai A, Baron R, Ganesan A, Mattevi A. Protein Recognition by Short Peptide Reversible Inhibitors of the Chromatin-Modifying LSD1/CoREST Lysine Demethylase. ACS chemical biology. 2013;8:1677–1682. doi: 10.1021/cb4001926. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, Liu Y, Ward D, Quan J, Ye T, Zhang H. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer research. 2011;71:7238–7249. doi: 10.1158/0008-5472.CAN-11-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zificsak CA, Hlasta DJ. Current methods for the synthesis of 2-substituted azoles. Tetrahedron. 2004;60:8991–9016. [Google Scholar]
- 36.Davies JR, Kane PD, Moody CJ. N-H Insertion reactions of rhodium carbenoids. Part 5: A convenient route to 1,3-azoles. Tetrahedron. 2004;60:3967–3977. [Google Scholar]
- 37.Chimenti F, Maccioni E, Secci D, Bolasco A, Chimenti P, Granese A, Carradori S, Alcaro S, Ortuso F, Yanez M, Orallo F, Cirilli R, Ferretti R, La Torre F. Synthesis, stereochemical identification, and selective inhibitory activity against human monoamine oxidase-B of 2-methylcyclohexylidene-(4-arylthiazol-2-yl)hydrazones. Journal of medicinal chemistry. 2008;51:4874–4880. doi: 10.1021/jm800132g. [DOI] [PubMed] [Google Scholar]
- 38.Chimenti F, Fioravanti R, Bolasco A, Manna F, Chimenti P, Secci D, Befani O, Turini P, Ortuso F, Alcaro S. Monoamine oxidase isoform-dependent tautomeric influence in the recognition of 3,5-diaryl pyrazole inhibitors. Journal of medicinal chemistry. 2007;50:425–428. doi: 10.1021/jm060868l. [DOI] [PubMed] [Google Scholar]
- 39.La Regina G, Silvestri R, Artico M, Lavecchia A, Novellino E, Befani O, Turini P, Agostinelli E. New pyrrole inhibitors of monoamine oxidase: synthesis, biological evaluation, and structural determinants of MAO-A and MAO-B selectivity. Journal of medicinal chemistry. 2007;50:922–931. doi: 10.1021/jm060882y. [DOI] [PubMed] [Google Scholar]
- 40.Valente S, Tomassi S, Tempera G, Saccoccio S, Agostinelli E, Mai A. Novel reversible monoamine oxidase A inhibitors: highly potent and selective 3-(1H-pyrrol-3-yl)-2-oxazolidinones. Journal of medicinal chemistry. 2011;54:8228–8232. doi: 10.1021/jm201011x. [DOI] [PubMed] [Google Scholar]
- 41.Maccioni E, Alcaro S, Cirilli R, Vigo S, Cardia MC, Sanna ML, Meleddu R, Yanez M, Costa G, Casu L, Matyus P, Distinto S. 3-Acetyl-2,5-diaryl-2,3-dihydro-1,3,4-oxadiazoles: A New Scaffold for the Selective Inhibition of Monoamine Oxidase B. Journal of medicinal chemistry. 2011;54:6394–6398. doi: 10.1021/jm2002876. [DOI] [PubMed] [Google Scholar]
- 42.Gentili F, Pizzinat N, Ordener C, Marchal-Victorion S, Maurel A, Hofmann R, Renard P, Delagrange P, Pigini M, Parini A, Giannella M. 3-[5-(4,5-dihydro-1H-imidazol-2-yl)-furan-2-yl]phenylamine (Amifuraline), a promising reversible and selective peripheral MAO-A inhibitor. Journal of medicinal chemistry. 2006;49:5578–5586. doi: 10.1021/jm060605r. [DOI] [PubMed] [Google Scholar]
- 43.Chimenti F, Maccioni E, Secci D, Bolasco A, Chimenti P, Granese A, Befani O, Turini P, Alcaro S, Ortuso F, Cirilli R, La Torre F, Cardia MC, Distinto S. Synthesis, molecular modeling studies, and selective inhibitory activity against monoamine oxidase of 1-thiocarbamoyl-3,5-diaryl-4,5-dihydro-(1H)- pyrazole derivatives. Journal of medicinal chemistry. 2005;48:7113–7122. doi: 10.1021/jm040903t. [DOI] [PubMed] [Google Scholar]
- 44.Jia Z, Zhu Q. 'Click' assembly of selective inhibitors for MAO-A. Bioorganic & medicinal chemistry letters. 2010;20:6222–6225. doi: 10.1016/j.bmcl.2010.08.104. [DOI] [PubMed] [Google Scholar]
- 45.Reck F, Zhou F, Girardot M, Kern G, Eyermann CJ, Hales NJ, Ramsay RR, Gravestock MB. Identification of 4-substituted 1,2,3-triazoles as novel oxazolidinone antibacterial agents with reduced activity against monoamine oxidase A. Journal of medicinal chemistry. 2005;48:499–506. doi: 10.1021/jm0400810. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki T, Ota Y, Ri M, Bando M, Gotoh A, Ito Y, Tsumoto H, Tatum PR, Mizukami T, Nakagawa H, Iida S, Ueda R, Shirahige K, Miyata N. Rapid discovery of highly potent and selective inhibitors of histone deacetylase 8 using click chemistry to generate candidate libraries. Journal of medicinal chemistry. 2012;55:9562–9575. doi: 10.1021/jm300837y. [DOI] [PubMed] [Google Scholar]
- 47.Hou J, Li Z, Fang Q, Feng C, Zhang H, Guo W, Wang H, Gu G, Tian Y, Liu P, Liu R, Lin J, Shi Y.-k., Yin Z, Shen J, Wang PG. Discovery and Extensive in Vitro Evaluations of NK-HDAC-1: A Chiral Histone Deacetylase Inhibitor as a Promising Lead. Journal of medicinal chemistry. 2012;55:3066–3075. doi: 10.1021/jm201496g. [DOI] [PubMed] [Google Scholar]
- 48.Imamura H, Ohtake N, Jona H, Shimizu A, Moriya M, Sato H, Sugimoto Y, Ikeura C, Kiyonaga H, Nakano M, Nagano R, Abe S, Yamada K, Hashizume T, Morishima H. Dicationic dithiocarbamate carbapenems with anti-MRSA activity. Bioorganic & medicinal chemistry. 2001;9:1571–1578. doi: 10.1016/s0968-0896(01)00044-x. [DOI] [PubMed] [Google Scholar]
- 49.Ronconi L, Marzano C, Zanello P, Corsini M, Miolo G, Maccà C, Trevisan A, Fregona D. Gold(III) Dithiocarbamate Derivatives for the Treatment of Cancer: Solution Chemistry, DNA Binding, and Hemolytic Properties. Journal of medicinal chemistry. 2006;49:1648–1657. doi: 10.1021/jm0509288. [DOI] [PubMed] [Google Scholar]
- 50.Len C, Boulogne-Merlot A-S, Postel D, Ronco G, Villa P, Goubert C, Jeufrault E, Mathon B, Simon H. Synthesis and Antifungal Activity of Novel Bis(dithiocarbamate) Derivatives of Glycerol. Journal of Agricultural and Food Chemistry. 1996;44:2856–2858. [Google Scholar]
- 51.Cvek B, Dvorak Z. Targeting of nuclear factor-kappaB and proteasome by dithiocarbamate complexes with metals. Current pharmaceutical design. 2007;13:3155–3167. doi: 10.2174/138161207782110390. [DOI] [PubMed] [Google Scholar]
- 52.Adachi Y, Nakamura K, Kato Y, Hazumi N, Hashizume T, Nakagawa S. In vitro evaluation of BO-3482, a novel dithiocarbamate carbapenem with activity against methicillin-resistant staphylococci. Antimicrobial agents and chemotherapy. 1997;41:2282–2285. doi: 10.1128/aac.41.10.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagano R, Shibata K, Naito T, Fuse A, Asano K, Hashizume T, Nakagawa S. Therapeutic efficacy of BO-3482, a novel dithiocarbamate carbapenem, in mice infected with methicillin-resistant Staphylococcus aureus. Antimicrobial agents and chemotherapy. 1997;41:2278–2281. doi: 10.1128/aac.41.10.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X-J, Xu H-W, Guo L-L, Zheng J-X, Xu B, Guo X, Zheng C-X, Liu H-M. Synthesis and in vitro antitumor activity of new butenolide-containing dithiocarbamates. Bioorganic & medicinal chemistry letters. 2011;21:3074–3077. doi: 10.1016/j.bmcl.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 55.Duan Y-C, Ma Y-C, Zhang E, Shi X-J, Wang M-M, Ye X-W, Liu H-M. Design and synthesis of novel 1,2,3-triazole-dithiocarbamate hybrids as potential anticancer agents. European Journal of Medicinal Chemistry. 2013;62:11–19. doi: 10.1016/j.ejmech.2012.12.046. [DOI] [PubMed] [Google Scholar]
- 56.Hu M, Li J, Q. Yao S. In Situ “Click” Assembly of Small Molecule Matrix Metalloprotease Inhibitors Containing Zinc-Chelating Groups. Organic Letters. 2008;10:5529–5531. doi: 10.1021/ol802286g. [DOI] [PubMed] [Google Scholar]
- 57.Boeckman RK, Ko SS. Stereochemical control in the intramolecular Diels-Alder reaction. 2. Structural and electronic effects on reactivity and selectivity. Journal of the American Chemical Society. 1982;104:1033–1041. [Google Scholar]
- 58.Wang ES, Choy YM, Wong HNC. Synthetic studies on prehispanolone and 14,15-dihydroprehispanolone. Tetrahedron. 1996;52:12137–12158. [Google Scholar]
- 59.Lineweaver H, Burk D. The Determination of Enzyme Dissociation Constants. Journal of the American Chemical Society. 1934;56:658–666. [Google Scholar]
- 60.Burton K. The stabilization of D-amino acid oxidase by flavin-adenine dinucleotide, substrates and competitive inhibitors. The Biochemical journal. 1951;48:458–467. doi: 10.1042/bj0480458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caldinelli L, Molla G, Bracci L, Lelli B, Pileri S, Cappelletti P, Sacchi S, Pollegioni L. Effect of ligand binding on human D-amino acid oxidase: Implications for the development of new drugs for schizophrenia treatment. Protein Science. 2010;19:1500–1512. doi: 10.1002/pro.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirota T, Lee JW, St John PC, Sawa M, Iwaisako K, Noguchi T, Pongsawakul PY, Sonntag T, Welsh DK, Brenner DA, Doyle FJ, 3rd, Schultz PG, Kay SA. Identification of small molecule activators of cryptochrome. Science (New York, N.Y.) 2012;337:1094–1097. doi: 10.1126/science.1223710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chu Y, Chen X, Yang Y, Tang Y. Identification of small molecular inhibitors for Ero1p by structure-based virtual screening. Bioorganic & medicinal chemistry letters. 2011;21:1118–1121. doi: 10.1016/j.bmcl.2010.12.129. [DOI] [PubMed] [Google Scholar]
- 64.Blais JD, Chin KT, Zito E, Zhang Y, Heldman N, Harding HP, Fass D, Thorpe C, Ron D. A small molecule inhibitor of endoplasmic reticulum oxidation 1 (ERO1) with selectively reversible thiol reactivity. The Journal of biological chemistry. 2010;285:20993–21003. doi: 10.1074/jbc.M110.126599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stavropoulos P, Blobel G, Hoelz A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nature structural & molecular biology. 2006;13:626–632. doi: 10.1038/nsmb1113. [DOI] [PubMed] [Google Scholar]
- 66.Baron R, Vellore NA. LSD1/CoREST is an allosteric nanoscale clamp regulated by H3-histone-tail molecular recognition. Proceedings of the National Academy of Sciences. 2012;109:12509–12514. doi: 10.1073/pnas.1207892109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robertson JC, Hurley NC, Tortorici M, Ciossani G, Borrello MT, Vellore NA, Ganesan A, Mattevi A, Baron R. Expanding the Druggable Space of the LSD1/CoREST Epigenetic Target: New Potential Binding Regions for Drug-Like Molecules, Peptides, Protein Partners, and Chromatin. PLoS Comput Biol. 2013;9:e1003158. doi: 10.1371/journal.pcbi.1003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, Mattevi A. A novel mammalian flavin-dependent histone demethylase. The Journal of biological chemistry. 2009;284:17775–17782. doi: 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Q, Qi S, Xu M, Yu L, Tao Y, Deng Z, Wu W, Li J, Chen Z, Wong J. Structure-function analysis reveals a novel mechanism for regulation of histone demethylase LSD2/AOF1/KDM1b. Cell Res. 2013;23:225–241. doi: 10.1038/cr.2012.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karasulu B, Patil M, Thiel W. Amine Oxidation Mediated by Lysine-Specific Demethylase 1: Quantum Mechanics/Molecular Mechanics Insights into Mechanism and Role of Lysine 661. Journal of the American Chemical Society. doi: 10.1021/ja403582u. Online early access. [DOI] [PubMed] [Google Scholar]
- 71.Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferrari-Amorotti G, Fragliasso V, Esteki R, Prudente Z, Soliera AR, Cattelani S, Manzotti G, Grisendi G, Dominici M, Pieraccioli M, Raschella G, Chiodoni C, Colombo MP, Calabretta B. Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion. Cancer research. 2013;73:235–245. doi: 10.1158/0008-5472.CAN-12-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A Stable Nonfluorescent Derivative of Resorufin for the Fluorometric Determination of Trace Hydrogen Peroxide: Applications in Detecting the Activity of Phagocyte NADPH Oxidase and Other Oxidases. Analytical biochemistry. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 74.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annual review of biophysics and biomolecular structure. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 75.Georgescu RE, Alexov EG, Gunner MR. Combining conformational flexibility and continuum electrostatics for calculating pK(a)s in proteins. Biophysical journal. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alexov EG, Gunner MR. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophysical journal. 1997;72:2075–2093. doi: 10.1016/S0006-3495(97)78851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. Journal of computational chemistry. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 78.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. Journal of molecular graphics & modelling. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 79.McGann M. FRED Pose Prediction and Virtual Screening Accuracy. Journal of Chemical Information and Modeling. 2011;51:578–596. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 80.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. Journal of Chemical Theory and Computation. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 81.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Structure, Function, and Bioinformatics. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. Journal of computational chemistry. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 83.Deuflhard P, Huisinga W, Fischer A, Schütte C. Identification of almost invariant aggregates in reversible nearly uncoupled Markov chains. Linear Algebra and its Applications. 2000;315:39–59. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.