

Abstract
Muscleblind-like proteins (MBNL) are RNA binding proteins that bind to the poly(CUG) and poly(CCUG) sequences that are the causative agents of myotonic dystrophy. It has been suggested that as a result of binding to the repeating RNA sequences, MBNL1 is abnormally expressed and translocated, which leads to many of the misregulated events in myotonic dystrophy. In this work, steady-state fluorescence quenching experiments suggest that MBNL1 alters the structure of helical RNA targets upon binding, which may explain the selectivity of MBNL1 for less structured RNA target sites. The removal of one pair of zinc fingers greatly impairs the binding affinity of MBNL1, which indicates that the two pairs of zinc fingers could possibly interact with RNA targets cooperatively. Alanine scanning mutagenesis suggested that the binding energy may be distributed across the protein. Overall, the results presented here suggest that small molecules that stabilize the helical structure of poly(CUG) and poly(CCUG) RNAs will inhibit the formation of complexes with MBNL1.
Keywords: protein-RNA complex, helix destabilization, myotonic dystrophy, RNA, Fluorescence quenching
Introduction
Muscleblind-like (MBNL) proteins have been shown to be involved in regulation of the alternative splicing of pre-mRNA.[1-3] A toxic RNA gain-of-function model has suggested that the down-regulation of muscleblind-like proteins by an expansion of repeated RNA sequences is one molecular cause of myotonic dystrophy (dystrophia myotonica, DM), which is a multisystem inherited disease characterized by progressive muscle wasting and weakness, cardiac conduction defects, and other neuromuscular problems. There are two types of DM, each associated with a repeated sequence of RNA. Patients with myotonic dystrophy type 1 (DM1) have a poly(CTG) repeat in the 3′-untranslated region of the dystrophia myotonic protein kinase gene (DMPK). Patients with myotonic dystrophy type 2 (DM2) bear a poly(CCTG) repeat in the CCHC-type zinc finger 9 gene (ZNF9).[1-5] There is considerable support for a toxic RNA gain-of-function of the poly(CUG) or poly(CCUG) transcripts, which tend to accumulate in nuclear foci, as the molecular basis of DM.[6-9]
MBNL1, a muscleblind-like protein that is involved in regulating pre-mRNA splicing, is strongly recruited into ribo-nuclear foci comprised of poly(CUG) or poly(CCUG) RNA in muscle tissues from patients.[6] It has been shown that loss of MBNL1 to the nuclear foci accounts for about 80%-90% of the total mis-splicing events observed in DM.[10] Among these mis-splicing events, at least four have been identified to be directly regulated by MBNL1 through binding with specific regions of pre-mRNA.[11-19] MBNL1 is overexpressed in heart and skeletal muscle, which are the two tissues prominently affected in myotonic dystrophy.[7, 20] MBNL1 deficient mice show similar symptoms as patients with myotonic dystrophy, and over-expression of MBNL1 reverses some symptoms of myotonic dystrophy in a mouse poly(CUG) model.[20-23]
MBNL1 contains four CCCH zinc finger domains, positioned as tandem pairs at the N-terminus (ZnF1 and ZnF2) and the middle (ZnF3 and ZnF4) of the protein. ZnF1 and ZnF3 share similar amino acid sequence, and ZnF2 and ZnF4 share similar sequences. In ZnF1 and ZnF3 domains, the Cys are found in CX7CX6CX3H sequences, while in ZnF2 and ZnF4 domains, the Cys are found in CX7CX4CX3H sequences. The RNA binding activity of MBNL1 resides in the two tandem zinc finger domains (Figure 1).[18, 19]
Figure 1.
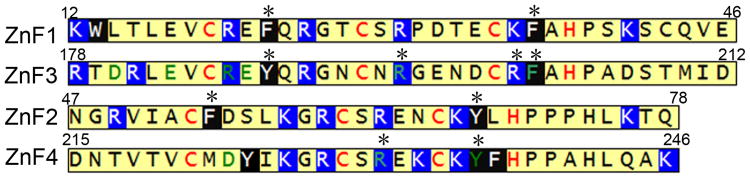
Sequences of the four zinc fingers of MBNL1. The sites of mutations are marked with asterisks on top of the amino acids. Cys and His residues that chelate with zinc ion are in red. Amino acids with side chains that interact with RNA in the structure of the complex[24] are in green. Positively charged amino acids are highlighted in blue background and aromatic amino acids are highlighted in black background.
The MBNL1 protein binds to RNA targets, including both helical and single-stranded sequences, containing 5′-YCGY-3′ sequences.[11, 25] Structured helical sequences include the poly-CUG repeat sequences of DM1, which form structures that resemble A-form RNA.[26, 27] Sequences with less stable secondary structures include cardiac troponin T (cTNT), which is targeted by MBNL1 to regulate alternative splicing. MBNL1 can bind to a 32mer segment of the cTNT pre-mRNA and promote the inclusion of exon 5 in cTNT. [16, 18, 24, 28] Here, we have used in vitro binding assays and steady-state fluorescence measure-ments with purified recombinant MBNL1 protein to probe the interaction of MBNL1 protein with RNAs with different structures. Our data suggest that that MBNL1 disrupts helical RNA structure upon binding and that the binding energy is distributed throughout the two pairs of the zinc fingers of MBNL1.
Results
Analysis of the requirements of RNA secondary structure for MBNL1 binding
To investigate the binding of MBNL1 to RNA targets with different structures, the oligonucleotide (CUG)12, which includes a stabilizing GC at the end of the stem, was selected as a mimic of pathogenic, structured RNA in DM1 (Figure 2). As a mimic of the natural target sites of MBNL1, a segment of cTNT comprised of 18 nucleotides (cTNT18) was selected. Although the most stable secondary structure of cTNT18 that is predicted by UNAfold is shown in Figure 2,[29, 30] the Tm was determined to be 26 ± 6 °C. Thus, the structure of this RNA is likely to be an equilibrating mixture of stem loop and single-stranded forms. In contrast, (CUG)12 forms a more stable structure with a Tm of 71 ± 2 °C. The homodimers formed by 5′-G(CUG)2C-3′ and 5′-(CUG)6-3′ were found to be A-form RNA helices in crystal structures.[26, 27] All experiments reported here were carried out with MBNL1, which is comprised of amino acids 1-260 of MBNL, and binds to RNA with similar affinity as the full-length protein.[18, 31] Apparent dissociation constants for MBNL1-RNA complexes were determined using gel electrophoresis mobility shift assays. The apparent dissociation constants for binding to (CUG)12 are much larger than those for binding to cTNT18, and are similar to previously reported values.[18, 25]
Figure 2.

Most stable secondary structures predicted by UNAfold[29, 30] of (CUG)12, cTNT32, and cTNT18. The box in the cTNT32 RNA indicates the sequence of cTNT18 RNA.
Because most RNA targets of MBNL1 contain more than one 5′-YGCY-3′ binding motif, we investigated the contribution of each of the 5′-YGCY-3′ motifs to complex stability. We compared the binding affinities of two cTNT18 variants in which either of the two 5′-GC-3′ sequences was replaced with a 5′-AA-3′ sequence (Figure 3). The mutations disrupt only one binding motif, while leaving the other motif intact. Adenines were chosen to replace GC because substitution with A is unlikely to introduce a new MBNL1 binding site. Modification at the 5′-GC binding motif (cTNT18_AA1) resulted in an approximately 6000-fold increase in Kd, while modification at the second binding motif (cTNT18_AA2) resulted in an approximately 20-fold increase in Kd (Table 1). These results suggest that both 5′-YGCY-3′ motifs contribute to the interaction with MBNL1. The different apparent dissociation constants for the two modified cTNT18 RNAs suggest that the presence of 5′-YGCY-3′ is not the only factor that influences the binding affinity of MBNL1. However, it is worth noting that these modifications not only disrupt the binding motif, but also change the folding of the RNA structure. It is possible that the AA sequence substituted for the more 3′-GC motif can pair with 5′-UU-3′ in the loop region and make the 5′-GC more accessible to binding MBNL1.
Figure 3.

Electrophoretic gel mobility shift assays of MBNL1 with cTNT18 and analogs. One representative gel is presented for each RNA target. The RNA structures shown in the figures are the most stable secondary structures predicted by UNAfold.[29, 30] Sequences highlighted in boxes in the structures are speculated binding sites. Positions of mutations in 5′-YGCY-3′ motifs in cTNT18_AA1 and cTNT18_AA2 are noted with arrows. In all gels the far right lane contains RNA only.
Table 1.
RNA melting temperatures, free energies of folding, and equilibrium dissociation constants of complexes formed with MBNL1.a
| RNA Name | Tm (°C) | ΔG (kcal·mol-1) | Kd (nM) |
|---|---|---|---|
| (CUG)12 | 71 ± 2 | -12.8 ± 0.4 | 150 ± 20 |
| cTNT18 | 26 ± 6 | NDb | 1.3 ± 0.2 |
| cTNT18_AA1 | ND | ND | >6000 |
| cTNT18_AA2 | ND | ND | 26 ± 4 |
| cTNT18_m1 | >89 | ND | 0.5 ± 0.3 |
| cTNT18_m2 | 69 ± 3 | -7.8 ± 0.3 | >1000 |
| cTNT21* | 52 ± 3 | -3.9 ± 0.3 | 14 ± 3 |
Errors are the standard deviation of at least three independent measurements.
ND indicates that the free energy of folding could not be unambiguously determined from the absorbance versus temperature curve.
To probe the ability of MBNL1 to bind 5′-YCGY-3′ target sites in different structural contexts, the cTNT18 RNA was modified so that the two 5′-YCGY-3′ motifs would be separated (cTNT18_m1) or closely associated (cTNT18_m2) in the most stable secondary structures predicted by UNAfold. Similar dissociation constants were obtained for complexes formed with cTNT18 and cTNT18_m1 RNA even though in cTNT18_m1 the two 5′-YCGY-3′ motifs are forced apart by a double-stranded region formed between the original loop region of cTNT18 and additional RNA sequence at the 3′ end of cTNT18 RNA. In contrast, when a stable stem region is located in the RNA structure to bring the two 5′YGCY3′ motifs into close proximity, as in cTNT18_m2 RNA, the complex is destabilized (Kd> 1000 nM). The high Tm's and low folding free energies of the cTNT18_m1 and cTNT18_m2 RNAs indicate that they fold into stable secondary structures at the temperatures used for the gel mobility shift assays.
Changes of RNA conformation upon binding MBNL1
To investigate conformational changes of the RNA upon binding, we designed an RNA target (cTNT21, Figure 4A) with a fluorophore (FAM) and a quencher (Dabcyl) appended to the 5′- and 3′- termini, respectively. Analysis of the structure with UNAfold[29, 30] suggests that this RNA forms a duplex structure in which the quencher and fluorophore are in close proximity. Consistent with this structure, the Tm of the labeled RNA (cTNT21*) was determined to be 52 ± 3 °C (Supporting Information Figure 3A). Because all the fluorescence measurements were performed at 20°C, the labeled RNA molecules should form stable stem loop structures in these experiments. The affinity of cTNT21 for MBNL1 was 14 ± 3 nm, which is approximately 10-fold weaker than that of cTNT18, presumably because of the two additional base pairs, which increase the similarity of this RNA target to (CUG)12.
Figure 4.
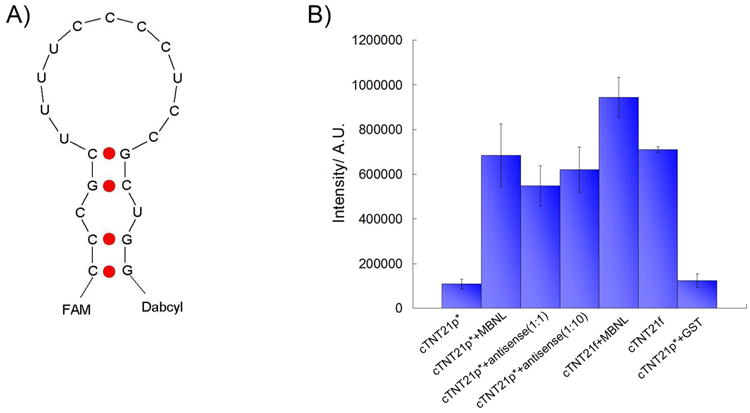
A) Schematic illustration of the structure of the cTNT21 RNA labelled with the fluorophore and quencher, FAM and Dabcyl. B) Quantification of fluorescence emission intensity at 520 nm under different conditions; cTNT21* is labelled with FAM and Dabcyl; cTNT21f is labelled with FAM only. The concentration of MBNL1 is 1.5 μM and RNA is 20 nM.
The fluorescence intensity of cTNT21* should increase if the two strands are separated because quenching efficiency is directly correlated to the distance between the quencher and fluorophore.[32] We observed a weak signal from the free RNA and a significant increase in fluorescence intensity upon addition of protein (Figure 4B). The increase in fluorescence intensity does not originate from the protein because a mixture of MBNL1 and unlabeled RNA gave no fluorescence signal. As a positive control, antisense DNA was added to the labelled RNA solution in 1:1 or 10:1 ratios, which resulted in a large increase in fluorescence intensity, presumably because the formation of the DNA-RNA duplex disrupts the RNA stem-loop structure. In addition, binding of MBNL1 to an RNA molecule labelled only with the fluorophore FAM resulted in only a small increase in fluorescence signal compared to the RNA labelled with both the fluorophore and quencher. Together, these results suggest that the conformation of the RNA molecule is changed upon protein binding such that the 3′- and 5′- ends of the stem loop are separated.
The conformational changes of the RNA target upon MBNL1 binding suggested by the fluorescence studies described above are supported by circular dichroism spectroscopy. The CD spectra of (CUG)12 cTNT21, and cTNT18 have maxima at 266, 273, and 275 nm, respectively. The CD spectrum of (CUG)12 is similar to those of (CUG)4 and (CUG)5 spectra that were reported previously.[18, 33] The (CUG)12 spectrum is consistent with A-form RNA, which has a maximum between 260 and 270 nm.[34, 35] The maxima of cTNT18 and cTNT21 are at longer wavelength than (CUG)12, which is consistent with these RNA sequences having reduced helical structure compared to (CUG)12.
Upon binding of the RNA targets to MBNL1, the peak intensity of (CUG)12 decreased by 23 %, that of cTNT21 decreased by 14 %, and that of cTNT18 remained nearly unchanged. The decrease in peak intensity implies reduced base stacking in the bound structures of (CUG)12 and cTNT21.[36-38] Upon binding MBNL1, the wavelength maximum was red-shifted by 3 nm for (CUG)12, 3 nm for cTNT18, and 5 nm for cTNT21, which is consistent with changes in RNA structure occurring upon MBNL1 binding. A decrease in peak intensity and shifting towards longer wavelengths upon MBNL1 binding was previously observed with (CUG)4 RNA.[18] This type of change is also typical when ssDNA binding protein binds DNA and RNA targets.[36, 39] These data support a model in which the structures of (CUG)12 and cTNT21 are altered towards single-stranded upon binding MBNL1, while that of cTNT18 undergoes structural changes. The CD spectra of the bound cTNT18p and cTNT18 RNAs are nearly identical, although those of the free RNA differ, suggesting that the structures of these RNA sequences become similar upon binding MBNL1.
Evaluation of the RNA binding affinities of truncated and mutated MBNL1 proteins
To investigate whether pairs of the zinc finger motifs found in MBNL1 are able to bind RNA, we created ZnF12, which is comprised of the two tandem zinc finger motifs at the N-terminus of the protein, and ZnF34, which is comprised of the more C-terminal tandem zinc finger motifs. ICP-MS analysis suggests that the truncated proteins have a similar zinc inclusion rate per zinc finger compared to MBNL1N (supporting information Figure 4). The complexes formed with both CUG12 and cTNT18 RNAs are destabilized by truncation of either pair of zinc fingers (Table 2). No binding of either construct was observed to (CUG)12 RNA, while binding to cTNT18 RNA of either ZnF12 or ZnF34 was observed only at the highest protein concentrations (Kd>3750 for GST-ZnF12; Kd>5000 for GST-ZnF34; see Supporting Information Figure 4).
Table 2.
Equilibrium dissociation constants of wild type and mutant MBNL1 proteins with (CUG)12 and cTNT18 RNA sequences.a
| Protein | (CUG)12 (nM) | cTNT18 (nM) |
|---|---|---|
| Wild type | 150 ± 20 | 1.3 ± 0.2 |
| Phe22Ala | 270 ± 50 | 0.6 ± 0.4 |
| Phe36Ala | 390 ± 20 | 1.4 ± 0.1 |
| Phe54Ala | 510 ± 50 | 2 ± 1 |
| Tyr68Ala | 540 ± 50 | 1.6 ± 0.2 |
| Tyr188Ala | 160 ±10 | 1.3 ± 0.4 |
| Arg195Ala | 180 ± 50 | 1.0 ± 0.4 |
| Arg201Ala | 500 ± 100 | 0.6 ± 0.2 |
| Phe202Ala | 220 ± 50 | 1.3 ± 0.3 |
| Arg231Ala | 190 ± 30 | 1.3 ± 0.2 |
| Tyr236Ala | 70 ± 20 | 0.9 ± 0.3 |
| Wild type – GST | 160 ± 30 | 4 ± 2 |
| ZnF12-GST | NBb | >3750 |
| ZnF34-GST | NB | >5000 |
Errors are the standard deviation of at least three independent measurements.
NB indicates that no binding was observed in the gel mobility shift assay.
To probe the contribution of individual amino acids to the recognition of RNA by MBNL1 we carried out site-directed mutagenesis involving 10 amino acids (Figure 1). Conserved aromatic amino acid residues in all four motifs were selected. Phe202 and Tyr236 were shown in the crystal structure of the complex formed between zinc fingers 3 and 4 and CGCUGU to interact with G and C bases in the RNA target.[24] Phe36 and Tyr68 are placed at similar positions in zinc fingers 1 and 2. In addition, similarly placed aromatic amino acids in the CCCH motifs of the TIS11d protein intercalate between UU and AU dinucleotides.[40] Two arginine residues that interact with RNA in the crystal structure were mutated, Arg195 and Arg201.[24]
Each selected amino acid was replaced with alanine to avoid introducing other functional groups or a bias for a particular peptide structure or solvent exposure. The dissociation constants of mutant proteins with (CUG)12 and cTNT18 RNAs are given in Table 2 (for representative gel pictures see supporting information Figure 5). The binding affinities of the mutated proteins for cTNT18 were nearly identical. The mutations resulted in up to a 4-fold destabilization of the complexes formed with (CUG)12 with the largest destabilization observed for Phe54Ala, Tyr68Ala, and Arg201Ala mutations. These modest destabilizations show that none of these individual amino acids are essential to binding.
Discussion
Truncation of MBNL1 destabilizes complexes with RNA
The weak binding of ZnF12 and ZnF34 to RNA suggests that one tandem pair of zinc fingers can bind RNA, but with much lower affinity than MBL1N. There is conflicting data in the literature about whether tandem pairs of zinc fingers can bind RNA. These inconsistencies may result from different buffers being used during binding measurements.[24, 31] Because our goal is to compare the binding affinities of truncated and full length MBNL1N, binding measurements were performed using the same conditions for the truncated and full-length proteins. The data imply that the two pairs of zinc fingers may cooperatively interact with RNA substrates. However, more experiments need to be carried out in the future to confirm the cooperativity between the zinc fingers. The small complex destabilization caused by substitution of individual amino acids with alanine may indicate that the binding energy is distributed through large portions of the zinc finger regions. The similarity of the effects of MBNL1 truncation and mutation on the stability of complexes formed with (CUG)12 and cTNT18 RNAs supports similar recognition of structured and unstructured RNA targets by MBNL1.
MBNL1 alters the structure of helical RNA targets
The data presented here suggest that RNA targets with high helicity, such as poly(CUG), are shifted towards single-stranded structures upon binding MBNL1. Thus, when MBNL1 binds to RNA that has a significant secondary structure, the association of the RNA with MBNL1 is in competition with the formation of the RNA secondary structure. As a result the binding affinity of structured RNA targets for MBNL1 is reduced compared to less structured RNA targets.
These observations may explain the affinities of MBNL1 for RNA targets with different secondary structures. For example, in poly(CUG) repeats, although the presence of the U-U mismatch lowers the folding energy of RNAs, the secondary structure is stable. As a result the dissociation constant of the complex formed with (CUG)12 RNA is two orders of magnitude greater than that of the complex formed with cTNT18 RNA, a nearly single-stranded target site. The difference in binding free energy of MBNL1 for (CUG)12 and cTNT18 RNA is similar to the calculated energetic penalty for disrupting the secondary structure of (CUG)12 at an MBNL1 binding site.[41-43] Consistent with this hypothesis, the dissociation constant of the complex formed with cTNT21 RNA is in between those of the complexes formed with (CUG)12 and cTNT18 RNA. When the stability of the secondary structure of the RNA substrate is increased, for example cTNT18_m2 in Figure 3, the protein no longer binds to the RNA because the protein-RNA interaction cannot compete with the formation of the RNA secondary structure. Thus, there is an inverse relationship between the stability of the RNA secondary structure and that of the complex.
RNA secondary structure may play a role in regulation of alternative splicing by MBNL1
The preference of MBNL1 for RNA targets with little secondary structure may be important for MBNL1 function. MBNL1 recognizes 5′-YGCY-3′ (Y=C/U) motifs in pre-mRNA,[11] but such motifs are expected to have a high occurrence in pre-mRNA sequences. If each of the four bases were to have a 25% chance of occupying a certain position on the pre-mRNA sequence, the 5′-YGCY-3′ motif would be found every 64 nucleotides on average. Indeed, a sequence search by Berglund and coworkers revealed hundreds of potential MBNL1 binding motifs in pre-mRNAs that are misregulated in myotonic dystrophy.[11] However, it may be possible to rule out some of these potential binding sites by considering the preference of MBNL1 for RNA substrates with low folding energy. We carried out a sliding window analysis of pre-mRNA folding on the two best studied MBNL1 binding sites in the human genome, intron 4 of cTNT pre-mRNA and intron 11 of insulin receptor pre-mRNA.[16, 18, 44] As expected, there is a relatively low mRNA free folding energy at previously identified binding sites, which are highlighted in black in Figure 6.[16, 18, 44]
Figure 6.

Sliding window analysis of the free energy of folding pre-mRNA. A) human cTNT; B) human insulin receptor. Identified binding sites are in black.
The data presented here suggest that MBNL1 may be a new example of an RNA chaperone.[45, 46] It could possibly regulate alternative splicing by helping the pre-mRNAs fold into conformations favorable for splicing without consumption of ATP. One of the best studied RNA and DNA chaperones is the HIV-1 nucleocapsid protein, which uses CCHC zinc fingers to bind RNA.[47-49] Like MBNL1, the NC protein binds with higher affinity to single-stranded than double-stranded nucleic acids. Binding of the NC protein to hairpin DNA sequences shifts the equilibrium towards a partial unwinding of the helix.[50] Although it has been proposed that MBNL1 could block the spliceosome assembly to promote the exclusion of exons,[18] it is still unknown how MBNL1 promotes the inclusion of certain exons. The conformational change of RNA induced by MBNL1 may play an important role in these activities.
New perspectives for drug development for myotonic dystrophy
Our results provide new insights for destabilization of the interactions of MBNL1 with poly(CUG) or poly(CCUG) RNA sequences.[51-56] First, our data suggest that binding of MBNL1 to cTNT21* results in at least a partial dissociation of the duplex structure. Therefore, to bind to an RNA target, MBNL1 may need to overcome an energy barrier for melting the RNA, and any molecule that stabilizes poly(CUG) or poly(CCUG) in a duplex structure could inhibit binding of MBNL1. Second, the results of the experiments performed with truncated MBNL1 proteins suggest that for an inhibitor that targets the protein, blocking the RNA binding of one pair of zinc fingers may be sufficient to destabilize the complex because the protein with only one pair of zinc fingers binds RNA with low binding affinity. These findings suggest approaches for drug development and may contribute to an understanding of molecules identified from screening approaches.
Experimental Section
Expression vectors
The expression vector pGEX-6p-1/MBNL1N was obtained from Maurice S. Swanson (University of Florida, College of Medicine, Gainesville, FL, USA).[19] The expressed protein is comprised of amino acids 1-260 from human MBNL1 and a His6-tag at the C-terminus. The expression vectors for ZnF12 and ZnF34 truncated proteins were constructed by amplifying the appropriate sequences of pGEX-6p-1/MBNL1N using the primers:
ZnF12
5′-CGGCGGGATCCATGGCTGTTAGTGTCA-3′
5′-CGGCCGCTCGAGGATTACTCGTCCATT-3′
ZnF34
5′-CGGGATCCATGTTAATGCGAACAGAC-3′
5′-CCGCTCGAGCTGGTATTGGG-3′
The amplified sequences were subcloned into pGEX-6p-1 using the BamHI and XhoI (Invitrogen) restriction sites. The correct sequences were confirmed by DNA sequencing.
For site directed mutagenesis, the pGEX-6p-1/MBNL1N vector was amplified using the following primers:
Phe22Ala
5′-GTATGTAGAGAGGCCCAGAGGG-3′
5′-GTCCCCCTCTGGGCCTCTCTAC-3′
Phe36Ala
5′-GGAATGTAAAGCTGCACATCCTTCG-3′
5′-CGAAGGATGTGCAGCTTTACATTCC-3′
Phe54Ala
5′-ATCGCCTGCGCTGATTCATT-3′
5′-AATGAATCAGCGCAGGCGAT-3′
Tyr68Ala
5′-GAACTGCAAAGCTCTTCATCCA-3′
5′-TGGATGAAGAGCTTTGCAGTTC-3′
Tyr188Ala
5′-TATGTCGAGAGGCCCAACGT-3′
5′-ACGTTGGGCCTCTCGACATA-3′
Arg195Ala
5′-AATTGCAACGCAGGAGAAAATG-3′
5′-CATTTTCTCCTGCGTTGCAATT-3′
Arg201Ala
5′-GAAAATGATTGTGCGTTTGCTC-3′
5′-GAGCAAACGCACAATCATTTTC-3′
Phe202Ala
5′-TGATTGTCGGGCTGCTCATCC-3′
5′-AGGATGAGCAGCCCGACAATCA-3′
Arg231Ala
5′-GGAGATGCTCTGCGGAAAAGTG-3′
5′-CACTTTTCCGCAGAGCATCTCC-3′
Tyr236Ala
5′-AAGTGCAAAGCCTTTCATCCCC-3′
5′-GGGGATGAAAGGCTTTGCACTT-3′
The PCR products were treated with DpnI (Invitrogen), transformed into Xl1-blue competent cells (Stratagene), and isolated using QIAquick Spin Miniprep Kit (QIAGEN). The correct sequences were confirmed by DNA sequencing.
Recombinant protein preparation and purification
Proteins were expressed in BL21-CodonPlus(DE3)-RP cells (Stratagene) in LB media and induced with 1 mM IPTG at OD600 0.6. The cultures were grown for an additional 2 h at 37 °C after induction. The cells were collected by centrifugation, resuspended in lysis buffer (25 mM Tris•HCl (pH 8), 0.5 M NaCl, 10 mM imidazole, 2 mM BME, 5% glycerol, 0.1% Triton X-100, 2 mg/ml lysozyme, 0.1 mM PMSF, 1 μM pepstatin, 1 μM leupeptin), and lysed by ultrasonication. The cell lysate was centrifuged for 10 min at 10000 rpm, and the supernatant was collected and filtered through a 45 μm Millex Filter (Millipore). The sample was incubated with Ni-NTA agarose (QIAGEN) for 1 h at 4 °C, washed with a buffer comprised of 25 mM Tris•HCl (pH 8), 0.5 M NaCl, 20 mM imidazole, and 0.1% Triton X-100, and eluted with a buffer comprised of 25 mM Tris•HCl (pH 8), 0.5 M NaCl, 250 mM imidazole, and 0.1%Triton X-100. The eluent was collected and incubated with Glutathione Sepharose 4B (GE Healthcare) for 1 h at 4 °C. After washing with a buffer containing 25 mM Tris•HCl (pH 8), 300 mM NaCl, 5 mM BME and 0.1% TritonX-100, the beads were collected and incubated with PreScission Protease (GE Healthcare) in the washing buffer overnight at 4 °C. The protein was collected in the flow-through of the column and was concentrated with a Microcon Centrifugal Filter 3000 MWCO (Millipore).
For fluorescence measurements, ICP-MS analysis and electrophoretic mobility shift assays with truncated proteins, the eluent containing the GST fusion protein was dialyzed against a storage buffer (20 mM Tris•HCl (pH 7.5), 100 mM NaCl, 5 mM MgCl2, 1 mM BME). The GST fusion proteins were directly used in these assays. The Kd of the complex formed with the GST fusion of MBNL1 with RNA is within error of that of MBNL1N (supporting information, Figure 3).
For all proteins, the molecular weights were confirmed by MALDI mass spectrometry, purity was determined by SDS-PAGE, and the concentrations of protein samples were determined from Bradford assays (Bio-Rad). ICP-MS analyses of the proteins were performed by Microanalytical Lab, University of Illinois at Urbana-Champaign.
Electrophoretic mobility shift assays
All RNA oligonucleotides were purchased from IDT DNA HPLC purified. RNA was labeled with [γ-32P]-ATP using T4 polynucleotide kinase (Invitrogen). Radiolabeled RNAs were heated to 95 °C for 2 min and then placed on ice for 20 min. RNA samples were diluted to 0.2 nM with buffer containing 27 mM Tris•HCl (pH 7.5), 66 mM NaCl, and 6.7 mM MgCl2. Proteins were serially diluted in buffer containing 20 mM Tris•HCl (pH 7.5), 175 mM NaCl, 5 mM MgCl2, 1.25 mM BME, 12.5% glycerol, 2 mg/mL BSA, and 0.1 mg/mL heparin. Non GST tagged protein was used in all the assays except the assays with truncated proteins, in which GST tag was not removed to improve the yield of proteins. Protein and RNA solutions were mixed in a 1:1 ratio by volume and incubated at room temperature for 20 min. The samples were loaded on a 6% polyacrylamide gel (37:1, acryl:bisacrylamide). The gel was run at 360 V in 0.5 X Tris-Borate buffer (pH 8.3) for 30 min and visualized on a Molecular Dynamics Storm PhosphorImager. The apparent Kd values were obtained by fitting fraction RNA bound versus protein concentration using the following equation: fraction RNA bound = 1/(1+Kd/[protein]total). All measurements were performed with greater than 10-fold excess of protein over RNA so that [protein] would be approximately equal to [protein]total. For representative gel pictures, see supporting information Figure 1.
Thermal denaturation studies
The melting curves (absorbance versus temperature) of the RNAs were measured on a Shimadzu UV2450 spectrophotometer equipped with a temperature-controller. The absorbance of each RNA sample (∼3 μM RNA in 27 mM Tris•HCl (pH 7.5), 66 mM NaCl, and 6.7 mM MgCl2) was monitored at 260 nm from 10 °C to 90 °C at a ramp rate of 0.5 °C/min. The melting temperature (Tm) of each sample was determined from the maximum point of the first derivative of the melting curve determined using LabSolutions – Tm Analysis version 1.0. Free energy values were determined from the melting curve using Meltwin 3.5. (http://www.meltwin.com/)
Circular dichroism experiments
CD spectra were recorded on a Jasco J-700 spectropolarimeter. Protein or RNA samples were dissolved in 250 μl buffer containing 27 mM Tris•HCl (pH 7.5), 66 mM NaCl, and 6.7 mM MgCl2. The final concentration of protein was 12 μM and of RNA was 2.5 μM. Before taking measurements, RNA samples were heated at 95°C for 5 min and then cooled on ice for 20 min. The CD spectra were monitored from 300 to 250 nm at a rate of 200nm/min. Data points were acquired every 0.5 nm. The spectra were not evaluated at wavelengths below 250 because of the strong CD spectrum of the protein in this region. The spectra obtained were averages of 50 accumulations.
Steady-state fluorescence emission measurements
Fluorescein-labeled RNA oligonucleotides were purchased from IDT DNA HPLC purified. All the fluorescence measurements were obtained using GST fusion MBNL1N, which was shown to have similar binding affinity as non-fusion MBNL1N (supporting information, Figure 3). Steady-state fluorescence spectra were recorded on a FluoroMax-2 spectrometer equipped with a 150W xenon lamp and modified Czerny-Turner spectrometers in both the excitation and emission position utilizing Datamax Spectroscopy Software. The instrument was interfaced with a Neslab RTE-111 temperature controller with a remote sensor. The excitation and emission slits were 4nm and the excitation wavelength was 485nm. All fluorescence scans were recorded at 20 °C using 1 nm wavelength increments. Samples prepared for fluorescence measurements were dissolved in a buffer containing 20 mM Tris•HCl (pH 7.5), 100 mM NaCl, 5 mM MgCl2, and1 mM BME.
Sliding window analysis of mRNA folding energy
The RNA folding energy was calculated using the software RNAstructure 4.6, using the default setting: 37 °C, 1 μM RNA, and 29nt window length.
Supplementary Material
Figure 5.

Circular dichroism spectrum of A) (CUG)12 RNA free (red) and bound to MBNL1 (blue) and B) Free cTNT18 (green) and cTNT21 (red) free and cTNT18 (blue) and cTNT21 (black) bound to MBNL1N. The RNA concentration is 2.5 μM and the protein concentration is 12 μM.
Acknowledgments
We thank Dr. Madhavaiah Chandra for assistance with PCR and Dr. Maurice S. Swanson for the MBNL1N construct. This work was supported by the National Institutes of Health (RO1AR058361).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Amack JD, Mahadevan MS. Dev Biol. 2004;265:294. doi: 10.1016/j.ydbio.2003.07.021. [DOI] [PubMed] [Google Scholar]
- 2.Mankodi A, Thornton CA. Curr Opin Neurol. 2002;15:545. doi: 10.1097/00019052-200210000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Ranum LP, Cooper TA. Annu Rev Neurosci. 2006;29:259. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 4.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Science. 2001;293:864. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 5.Mahadevan MS, Yadava RS, Yu Q, Balijepalli S, Frenzel-McCardell CD, Bourne TD, Phillips LH. Nat Genet. 2006;38:1066. doi: 10.1038/ng1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fardaei M, Larkin K, Brook JD, Hamshere MG. Nucleic Acids Res. 2001;29:2766. doi: 10.1093/nar/29.13.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, Brook JD. Hum Mol Genet. 2002;11:805. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 8.Mankodi A, Lin X, Blaxall BC, Swanson MS, Thornton CA. Circ Res. 2005;97:1152. doi: 10.1161/01.RES.0000193598.89753.e3. [DOI] [PubMed] [Google Scholar]
- 9.Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. J Cell Biol. 1995;128:995. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, Ares M., Jr Nat Struct Mol Biol. 2010;17:187. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goers ES, Purcell J, Voelker RB, Gates DP, Berglund JA. Nucleic Acids Res. 2010;38:2467. doi: 10.1093/nar/gkp1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hino S, Kondo S, Sekiya H, Saito A, Kanemoto S, Murakami T, Chihara K, Aoki Y, Nakamori M, Takahashi MP, Imaizumi K. Hum Mol Genet. 2007;16:2834. doi: 10.1093/hmg/ddm239. [DOI] [PubMed] [Google Scholar]
- 13.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Hum Mol Genet. 2004;13:3079. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 14.Kino Y, Washizu C, Oma Y, Onishi H, Nezu Y, Sasagawa N, Nukina N, Ishiura S. Nucleic Acids Res. 2009;37:6477. doi: 10.1093/nar/gkp681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dansithong W, Paul S, Comai L, Reddy S. J Biol Chem. 2005;280:5773. doi: 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- 16.Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA. EMBO J. 2004;23:3103. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pascual M, Vicente M, Monferrer L, Artero R. Differentiation. 2006;74:65. doi: 10.1111/j.1432-0436.2006.00060.x. [DOI] [PubMed] [Google Scholar]
- 18.Warf MB, Berglund JA. RNA. 2007;13:2238. doi: 10.1261/rna.610607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, Swanson MS. Nucleic Acids Res. 2007;35:5474. doi: 10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanadia RN, Urbinati CR, Crusselle VJ, Luo D, Lee YJ, Harrison JK, Oh SP, Swanson MS. Gene Expr Patterns. 2003;3:459. doi: 10.1016/s1567-133x(03)00064-4. [DOI] [PubMed] [Google Scholar]
- 21.Hao M, Akrami K, Wei K, De Diego C, Che N, Ku JH, Tidball J, Graves MC, Shieh PB, Chen F. Dev Dyn. 2008;237:403. doi: 10.1002/dvdy.21428. [DOI] [PubMed] [Google Scholar]
- 22.Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, Swanson MS. Proc Natl Acad Sci U S A. 2006;103:11748. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matynia A, Ng CH, Dansithong W, Chiang A, Silva AJ, Reddy S. PLoS One. 2010;5:e9857. doi: 10.1371/journal.pone.0009857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teplova M, Patel DJ. Nat Struct Mol Biol. 2008;15:1343. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cass DM, Hotchko R, Barber P, Jones K, Gates DP, Berglund JA. BMC Mol Biol. 2011;12 doi: 10.1186/1471-2199-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mooers BH, Logue JS, Berglund JA. Proc Natl Acad Sci U S A. 2005;102:16626. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiliszek A, Kierzek R, Krzyzosiak WJ, Rypniewski W. Nucleic Acids Res. 2009;37:4149. doi: 10.1093/nar/gkp350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warf MB, Diegel JV, von Hippel PH, Berglund JA. Proc Natl Acad Sci U S A. 2009;106:9203. doi: 10.1073/pnas.0900342106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markham NR, Zuker M. Nucleic Acids Res. 2005;33:W577. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markham NR, Zuker M. Methods Mol Biol. 2008;453:3. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- 31.Kino Y, Mori D, Oma Y, Takeshita Y, Sasagawa N, Ishiura S. Hum Mol Genet. 2004;13:495. doi: 10.1093/hmg/ddh056. [DOI] [PubMed] [Google Scholar]
- 32.Lakowicz JR. Principles of Fluorescence Spectroscopy: Quenching of Fluorescence. Springer Science+Business Media; New York: 2006. p. 278. [Google Scholar]
- 33.Pinheiro P, Scarlett G, Rodgers A, Rodger PM, Murray A, Brown T, Newbury SF, McClellan JA. J Biol Chem. 2002;277:35183. doi: 10.1074/jbc.M202235200. [DOI] [PubMed] [Google Scholar]
- 34.Gray DM, Liu JJ, Ratliff RL, Allen FS. Biopolymers. 1981;20:1337. [Google Scholar]
- 35.Gray DM, Hung SH, Johnson KH. Methods Enzymol. 1995;246:19. doi: 10.1016/0076-6879(95)46005-5. [DOI] [PubMed] [Google Scholar]
- 36.Mark BL, Gray D. Biopolymers. 1997;42:337. doi: 10.1002/(SICI)1097-0282(199709)42:3<337::AID-BIP6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 37.Tan R, Frankel AD. Biochemistry. 1992;31:10288. doi: 10.1021/bi00157a016. [DOI] [PubMed] [Google Scholar]
- 38.Daly TJ, Rusche JR, Maione TE, Frankel AD. Biochemistry. 1990;29:9791. doi: 10.1021/bi00494a005. [DOI] [PubMed] [Google Scholar]
- 39.Berova N, Nakanishi K, Woody RW. Circular dichroism: principles and applications. Wiley-VCH, Inc; New York: 2000. [Google Scholar]
- 40.Hudson BP, Martinez-Yamout MA, Dyson HJ, Wright PE. Nat Struct Mol Biol. 2004;11:257. doi: 10.1038/nsmb738. [DOI] [PubMed] [Google Scholar]
- 41.Davis AR, Znosko BM. Biochemistry. 2007;46:13425. doi: 10.1021/bi701311c. [DOI] [PubMed] [Google Scholar]
- 42.Mathews DH, Savina J, Zuker M, Turner DH. J Mol Biol. 1999;288:911. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 43.Xia T, SantaLucia J, Burkard ME, Kierzek R, Schroeder SJ, Jiao X, Cox C, Turner DH. Biochemistry. 1998;37:14719. doi: 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]
- 44.Sen S, Talukdar I, Liu Y, Tam J, Reddy S, Webster NJ. J Biol Chem. 2010;285:25426. doi: 10.1074/jbc.M109.095224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rajkowitsch L, Chen D, Stampfl S, Semrad K, Waldsich C, Mayer O, Jantsch MF, Konrat R, Blasi U, Schroeder R. RNA Biol. 2007;4:118. doi: 10.4161/rna.4.3.5445. [DOI] [PubMed] [Google Scholar]
- 46.Semrad K. Biochem Res Internat. 2011;2011:1. doi: 10.1155/2011/532908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cosa G, Zeng Y, Liu HW, Landes CF, Makarove DE, Musier-Forsyth K, Barbara PF. J Phys Chem B. 2006;110:2419. doi: 10.1021/jp054189i. [DOI] [PubMed] [Google Scholar]
- 48.Azoulay J, Clamme JP, Darlix JL, Roques BP, Mély Y. J Mol Biol. 2003;326:691. doi: 10.1016/s0022-2836(02)01430-4. [DOI] [PubMed] [Google Scholar]
- 49.Hong MK, Harbron EJ, O'Connor DB, Guo J, Barbara PF, Levin JG, Musier-Forsyth K. J Mol Biol. 2003;325:1. doi: 10.1016/s0022-2836(02)01177-4. [DOI] [PubMed] [Google Scholar]
- 50.Cosa G, Harbron EJ, Zeng Y, Liu HW, O'Connor DB, Eta-Hosokawa C, Musier-Forsyth K, Barbara PF. Biophys J. 2004;87:2759. doi: 10.1529/biophysj.104.043083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gareiss PC, Sobczak K, McNaughton BR, Palde PB, Thornton CA, Miller BL. J Am Chem Soc. 2008;130:16254. doi: 10.1021/ja804398y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arambula JF, Ramisetty SR, Baranger AM, Zimmerman SC. Proc Natl Acad Sci U S A. 2009;106:16068. doi: 10.1073/pnas.0901824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Disney MD, Lee MM, Pushechnikov A, Childs-Disney JL. Chembiochem. 2010;11:375. doi: 10.1002/cbic.200900716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee MM, Pushechnikov A, Disney MD. ACS Chem Biol. 2009;4:345. doi: 10.1021/cb900025w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, Thornton CA, Disney MD. J Am Chem Soc. 2009;131:9767. doi: 10.1021/ja9020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Proc Natl Acad Sci U S A. 2009;106:18551. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.