

Abstract
Augmenter of liver regeneration is a member of the ERV family of small flavin-dependent sulfhydryl oxidases that contain a redox-active CxxC disulfide bond in redox communication with the isoalloxazine ring of bound FAD. These enzymes catalyze the oxidation of thiol substrates with the reduction of molecular oxygen to hydrogen peroxide. This work studies the catalytic mechanism of the short, cytokine, form of augmenter of liver regeneration (sfALR) using model thiol substrates of the enzyme. The redox potential of the proximal disulfide in sfALR was found to be approximately 57 mV more reducing than the flavin chromophore, in agreement with titration experiments. Rapid reaction studies show that dithiothreitol (DTT) generates a transient mixed disulfide intermediate with sfALR signaled by a weak charge-transfer interaction between the thiolate of C145 and the oxidized flavin. The subsequent transfer of reducing equivalents to the flavin ring is relatively slow, with a limiting apparent rate constant of 12.4 s−1. However, reoxidation of the reduced flavin by molecular oxygen is even slower (2.3 s−1 at air saturation), and thus largely limits turnover at 5 mM DTT. The nature of the charge-transfer complexes observed with DTT was explored using a range of simple monothiols to mimic the initial nucleophilic attack on the proximal disulfide. While β–mercaptoethanol is a very poor substrate of sfALR (~ 0.3 min−1 at 100 mM thiol), it rapidly generates a mixed disulfide intermediate allowing the thiolate of C145 to form a strong charge-transfer complex with the flavin. Unlike the other monothiols tested, glutathione is unable to form charge-transfer complexes and is an undetectable substrate of the oxidase. These data are rationalized on the basis of the stringent steric requirements for thiol-disulfide exchange reactions. The inability of the relatively bulky glutathione to attain the in-line geometry required for efficient disulfide exchange in sfALR may be physiologically important in preventing the oxidase from catalyzing the potentially harmful oxidation of intracellular glutathione.
Augmenter of liver regeneration (ALR1; also abbreviated GFER, growth factor ERV1-like) was first described as a circulating growth factor that was released into mammalian blood following liver injury (1, 2). The observation that mammalian ALR and its yeast ortholog ERV1p share a domain found in the flavoprotein Quiescin-sulfhydryl oxidase (QSOX (3)) led to the realization that these smaller enzymes are also flavin-dependent sulfhydryl oxidases (4, 5) catalyzing the general reaction:
ALR exists in two splice variants. The short-form, sfALR, is an extracellular cytokine binding avidly to a receptor on the surface of hepatocytes and inducing the phosphorylation of the mitogen-activated kinases, MEK and MAPK (6, 7). sfALR is also found in the cytosol and nucleus (1, 2, 8–11). Nuclear sfALR interacts with the Jun activation-domain binding protein (JAB1) mediating the interaction between ALR and activator protein-1 (AP-1) via the phosphorylation of c-Jun (8, 12). sfALR has also been shown to protect against hydrogen peroxide and radiation-induced apoptosis (13, 14).
Crystal structures of rat (PDB 1OQC) (15) and human sfALR (PDB 3MBG and 3O55 (16, 17)) reveal a homodimer of helix-rich subunits (Figure 1A). The isoalloxazine ring of the flavin prosthetic group is bound at the mouth of a 4-helix bundle in each subunit with a 5th small helix adjacent to the adenine moiety of FAD. In common with many flavin-dependent disulfide oxidoreductases (18–20), a proximal redox-active disulfide is located adjacent to the C4a position of the isoalloxazine ring (Figure 1B). Each monomer contains an additional intra-chain structural disulfide (C171-C188) and participates in head-to-tail disulfides (C95-C204′ and C204-C95′; shown dotted in Figure 1C). The short-form of ALR shows very low sulfhydryl oxidase activity towards reduced unfolded lysozyme (5) and DTT has proved a useful model substrate of the enzyme (15, 21). ALR also exists in a longer splice variant with an 80-residue N-terminal extension containing an additional CxxC redox-active motif that interacts with MIA40 during oxidative protein folding in the mitochondrial intermembrane space (17, 22–24). In addition, the lfALR has cytosolic roles in modulation of mitochondrial morphology and in the regulation of hematopoietic stem cell proliferation (25, 26).
Figure 1.

Crystal structure and domain diagram of human ALR. Panel A shows the structure of human ALR (PDB 3MBG) with the two subunits in grey and green colors. The FAD cofactor is depicted in yellow and disulfides are shown as orange sticks. Panel B highlights the proximal disulfide (orange spheres) and its orientation with the isoalloxazine ring. The intervening two glutamic acid residues are depicted as sticks. Panel C shows bar diagrams of short and long splice variants of ALR applying a common numbering of amino acid residues (sfALR starts at M81). Cysteine residues that form disulfide bonds in ALR are shown in red. The proximal redox-active disulfide comprises C142-C145, the disulfide C171-C188 is structural, as are two interchain disulfides C95-C204′ and C204-C95′. lfALR contains an additional distal redox active disulfide C71-C74.
Both short and long forms of ALR share the helix-rich domain carrying the flavin and proximal disulfide moieties and the focus of this paper is to better understand how these redox centers communicate. A range of earlier anaerobic experiments showed that full reduction of the FAD prosthetic group required two electrons to be added per sfALR subunit (21) implying that the flavin was considerably more oxidizing than the proximal disulfide partner. Such behavior is not observed with flavin-dependent disulfide oxidoreductases of the pyridine nucleotide-disulfide oxidoreductase family (18–20). In those examples, the redox potential of the disulfide is usually comparable, or more oxidizing, than the FAD and hence a total of 4-electrons must be delivered before complete reduction of the flavin is achieved. We measured the redox potential of the proximal disulfide in sfALR to confirm that it is significantly more reducing than the flavin as a preliminary to the first examination of the pre-steady state kinetics of sfALR using DTT. Finally, results with this model substrate provide unexpected insights into the stabilization of mixed disulfide intermediates in catalysis by sfALR. In sum, these data provide for continuing studies of short and long isoforms of ALR and, more generally, for a greater appreciation of the catalytic potential of the ERV/ALR fold in the growing list of enzymes containing this diminutive FAD binding domain (27–29).
EXPERIMENTAL PROCEDURES
Materials and Reagents
Ampicillin, IPTG, DTT and PMSF were obtained from Gold Biotechnology Inc. Oxidized DTT, GnHCl and leupeptin were obtained from Sigma. Lysozyme and monobasic potassium phosphate were from Amresco. EDTA, LB broth and NaCl were from Fisher, and imidazole from Alfa Aesar.
General Methods
The enzymatic turnover of sfALR was determined in duplicate or triplicate using a Clark-type oxygen electrode as previously described (21). UV-VIS spectra were obtained as described previously (16, 21, 22). Where necessary, turbidity correction software supplied with the HP8453 diode-array spectrophotometer was used to correct for slight light scattering of the spectra of wild type and mutant sfALR.
Mutagenesis
Primers were purchased from Integrated DNA Technologies and mutagenesis was conducted as described previously (21, 22). DNA sequencing was performed by Genewiz Inc. The primers used in this work were: 5′-CTA AGT TTT ACC CCT GTG GGC CGT GTG CTG AAG ACC TAA G -3′ and 5′-CCT AGG TCT TCA GCA CAC GGC CCA CAG GGG TAA AAC TTA G -3′ for E143G/E144P; 5′-5′-CTA AGT TTT ACC CCT GTC ATC GTT GTG CTG AAG ACC TAA G -3′ and 5′-CCT AGG TCT TCA GCA CAG TGC GGA CAG GGG TAA AAC TTA G -3′ for E143P/E144H; and 5′-CTA AGT TTT ACC CCT GTA AGA AGT GTG CTG AAG ACC TAA G -3′ and 5′-CCT AGG TCT TCA GCA CAC TTC TTA CAG GGG TAA AAC TTA G -3′ for the E143K/E144K mutant.
Expression and Purification of sfALR
The pTrcHisA (Life Technologies, NY) expression vector (6xHis N-terminal affinity tag) encoded human short-form ALR (amino acids 81-205), in which two non-conserved and non-essential cysteine residues at positions 154 and 165 were mutated to alanine residues to minimize protein aggregation (21, 22). Expression was performed as previously described (16) utilizing 100 μg/mL ampicillin in the culture media. Cells were harvested at 5000 g for 8 min at 4 °C and immediately resuspended in 50 mM potassium phosphate, pH 7.5, containing 500 mM NaCl (binding buffer), flash frozen with liquid nitrogen and stored at −80 °C until purification.
Cells were thawed on ice and brought to 1 mM PMSF, 1 μM leupeptin and 0.1 mg/mL lysozyme immediately prior to two passes through the a French pressure cell at 10000 psi. The lysed cells were sonicated briefly to shear DNA and the suspension centrifuged at 15000 g for 1 h. The lysate was applied to a 5 mL HisTrap FF column (GE Healthcare), pre-equilibrated with binding buffer, and the column washed with 15 column volumes of this buffer. ALR was eluted using binding buffer supplemented with 0.02, 0.05, 0.2, 0.5 and 1 M imidazole. Yellow fractions were combined and analyzed using 12% Tris-Glycine SDS PAGE under reducing and non-reducing conditions. ALR fractions were combined and dialyzed into 50 mM potassium phosphate, pH 7.5, containing 1 mM EDTA. Unless otherwise stated this buffer was used for subsequent characterization of sfALR.
Preparation of the Apoprotein of sfALR and Reconstitution with 5-deaza-FAD
Apoprotein was prepared as described by Daithankar et al. (16), exchanged with 50 mM potassium phosphate buffer, pH 7.0, containing 1 mM EDTA, and reconstituted with 1.2 equivalents of 5-deaza-FAD. After 15 min incubation on ice, excess flavin was removed with 4 washes of buffer (pH 7.0) in an Amicon Ultra 0.5 mL centrifugal filter device (Millipore). The extinction coefficient of 5-deaza-FAD substituted sfALR is 11.5 mM−1 cm−1 at 408 nm and was determined as described previously (22).
Redox Potential Experiments
Solutions of 30 μM 5-deaza-FAD sfALR (0.13 mL in 50 mM potassium phosphate, 1 mM EDTA, pH 7.0, containing 20 mM oxidized DTT and 5 mM glucose) in a self-masking semimicro cuvette were incubated with 20 nM glucose oxidase and 5 nM catalase for 10 min to minimize subsequent aerobic oxidation of reduced DTT by residual traces of native ALR. Increments of 5 stock solutions of DTT (standardized with DTNB immediately prior to use and adjusted so that they contained 0.1, 1, 10, 20 and 100 mM DTT) were added to yield final cuvette concentrations of up to 5.1 mM DTT. Spectral changes were complete 1 min after addition of DTT at 25 °C. Data were corrected for light scattering, reagent depletion and dilution and the fraction reduced (the absorbance change at 439 nm divided by the maximal change at full reduction) was plotted as a function of the ratio of reduced to oxidized DTT. The same procedure was followed for the mutant enzymes with the exception that 100 mM oxidized DTT was used for the CPHC mutant. Redox curves were fit using the Hill Fit equation in GraphPad Prism version 6.0c yielding Kox and converted to redox potentials using the Nernst Equation using a value of −327 mV for the E∘′ of DTT (30).
Stopped-Flow Spectrophotometry
Stopped flow experiments were carried out at 25 °C in a Hi-Tech Scientific SF-61 SX2 double mixing stopped-flow system using KinetAsyst software from TgK Scientific. Prior to anaerobic experiments, the flow cell and driving syringes were soaked overnight in anaerobic buffer containing 5 mM glucose and 50 nM glucose oxidase. For studies of the reductive half-reaction with DTT, sfALR (60 μM in 50 mM phosphate buffer, pH 7.5 containing 1 mM EDTA, 5 mM glucose and 1 nM catalase) was deoxygenated in a tonometer prior to the anaerobic addition of 5 nM glucose oxidase. After further nitrogen/vacuum cycles, the tonometer was transferred to the stopped-flow instrument and mixed with various anaerobic solutions of DTT that had been deoxygenated in the same manner. DTT solutions were standardized using DTNB immediately prior to deoxygenation. The reduction of sfALR in the stopped-flow was followed in both diode-array and monochromator modes.
For the oxidative half-reaction, the 2-electron reduced protein (60 μM ALR in 50 mM potassium phosphate, pH 7.5, containing 1 mM EDTA, 5 mM glucose and 1 nM glucose oxidase) was prepared anaerobically in a tonometer equipped with an ancillary quartz cuvette (31). A standardized solution of sodium dithionite was added from a gas-tight syringe and the progress of the titration was followed spectrophotometrically to ensure generation of the 2-electron reduced enzyme without accumulation of excess reductant (21). Syringes containing various oxygen concentrations in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA were prepared as in DuPlessis et al. (32).
Rapid reaction data sets for the reductive and oxidative half-reactions were fit to the kinetic model presented in this paper using KinTek Explorer (33, 34). Observed rate constants are the average of at least 3 determinations. Analyses of the model were also run using Copasi Biochemical Network Simulator (35). Both programs utilized three consecutive equations (a–c) with rate constants as specified in Figure 3:
Figure 3.

Schematic representation of intermediates observed during catalysis of the aerobic oxidation of DTT by sfALR and the involvement of mixed disulfide intermediates in the reductive half-reaction. Panel A shows the rate of interconversion of observable intermediates deduced from stopped flow measurements (see Experimental Procedures). Panel B shows the involvement of two charge-transfer intermediates during the reductive half-reaction.
The reaction between βME and sfALR was followed in the stopped-flow spectrophotometer under pseudo-first order conditions (Figure 6). The observed rate constant for approach to equilibrium for Figure 7 is:
Figure 6.

βME forms mixed disulfide intermediates with sfALR. The spectrum of sfALR (30 μM) in air-saturated 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA was recorded before and 5 sec after the addition of 200 mM βME in a diode-array spectrophotometer (solid and dashed lines respectively). After 7 min the progressive depletion of oxygen from solution leads to the appearance of the blue neutral semiquinone (see the text). The inset plots the pseudo first-order rate constants obtained from the increase in absorbance at 540 nm following mixing sfALR in the stopped flow spectrophotometer with increasing concentration of βME (1 – 20 mM final). The slope of 3295 ± 59 M−1s−1 reflects the bimolecular association rate constant k1 and the intercept of 31.6 ± 0.6 s−1 corresponds to k−1. The corresponding equilibrium constant KD = 9.6 ± 0.25 mM.
Figure 7.
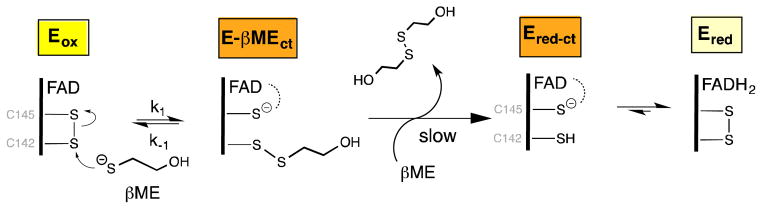
The reductive half-reaction between sfALR and β-mercaptoethanol. Rapid nucleophilic attack of the thiolate of βME reversibly yields the mixed disulfide charge-transfer species E-βMEct which is weakly reactive towards a second molecule of βME.
RESULTS AND DISCUSSION
Redox potential of the proximal disulfide
Anaerobic titrations of oxidized sfALR by sodium dithionite or with DTT both show full reduction of the bound FAD cofactor upon the addition of 2 electrons (21). Similarly, back titration of reduced sfALR (prepared by photoreducing an anaerobic solution of the protein until the FAD chromophore was bleached) gave a stoichiometry of 2 molecules of the 1-electron oxidant ferricyanide for complete return of the flavin absorbance (21). Collectively, these data show that the redox potential of the FAD is considerably more oxidizing than that of the proximal disulfide with which it communicates. Farrell and Thorpe measured the redox potential of the bound FAD moiety in sfALR, using equilibration with dyes, and found it to be −178 ± 2 mV (21). In the context of understanding the redox communication between proximal disulfides and the flavin prosthetic groups of ERV/ALR family members, we then wanted to determine the redox potential of the redox active disulfide in sfALR. Since we wished to measure this potential in the context of oxidized flavin, we explored the preparation of sfALR substituted with the much more reducing 5-deaza-FAD analog (36). sfALR apoprotein was prepared by washing sfALR bound to Ni-IDA beads with 6 M GuHCl until the FAD content of eluates was negligible ((16) see Experimental Procedures). The apoprotein was incubated with a slight molar excess of 5-deaza-FAD before excess free flavin was removed (see Experimental Procedures). The spectrum of 5-deaza-FAD sfALR is shown in Figure 2. The oxidized flavin envelope shows a typical spectrum for a 5-deaza-flavin substituted protein with an absorbance maximum at 408 nm. While reduction with 5 mM DTT leads to a small decrease in the extinction coefficient of the oxidized 5-deaza-FAD, scission of the proximal disulfide leads to a 3 nm blue shift of the leading edge of the oxidized 5-deaza-flavin absorbance envelope (and to the difference spectrum Figure 2B). Such blue shifts upon removal of proximal disulfides, by mutagenesis or chemical modification, have been noted in a number of flavin-dependent disulfide oxidoreductases (for example, in lipoamide dehydrogenase (37), QSOX (38, 39), Erv2p (40) and ALR itself (22)). Panel C of Figure 2 follows this spectral change (at 439 nm) as a function of the composition of a redox buffer formed by mixing DTT with its oxidized counterpart (DTTox; see Experimental Procedures). The midpoint potential is −235 ± 1.6 mV at pH 7; a value some 57 mV more reducing than that of the normal flavin cofactor in sfALR (21). Hence the equilibrium constant between 2-electron reduced forms of the enzyme in which either the FAD or the proximal disulfide undergoes 2-electron reduction is approximately 90-fold in favor of flavin reduction. This wide separation in redox potential provides confirmation of the results of reductive and oxidative titrations described earlier in this section. Our attempts to repeat the redox potential measurements using GSH/GSSG buffers were thwarted by the extremely slow reduction of even the normal FAD-substituted ALR by GSH (see later).
Figure 2.

Determination of the redox potential of the proximal disulfide in sfALR substituted with 5-deaza-FAD. Panel A shows the spectrum of 20 μM 5-deaza-FAD enzyme (see Experimental Procedures) in 50 mM phosphate buffer, pH 7.0, containining 1 mM EDTA recorded before and immediately after the addition of 5 mM DTT (solid and dashed lines respectively). Panel B shows the difference spectrum dominated at 439 nm by the blue shift in the leading edge of the oxidized 5-deazaflavin absorbance. Panel C follows the extent of these changes at 439 nm as a function of the redox poise of mixtures of reduced and oxidized DTT (see Experimental Procedures).
While not the major focus of this paper, we also explored the consequences of making three active site mutations involving the intervening E143 and E144 residues in the CxxC motif (Figure 1B). Surprisingly, substitution of the intervening EE dipeptide by the charge-reversed KK dipeptide showed minimal effect on the redox potential (Table 1). Two other mutations were chosen because they represent strongly reducing and oxidizing sequences in another family of redox protein containing CxxC motifs. Thus in thioredoxin family members, CGPC is prototypical of the strongly reducing bacterial thioredoxins (E∘′ = ~ −270 mV; (41–43) and, by contrast, the CPHC sequence is found in the highly oxidizing DsbA protein ((E∘′ = ~ −122 mV; (44, 45)) located in the periplasm of gram negative bacteria. Mutation of the GP dipeptide in E. coli thioredoxin to PH leads to a sizable 66 mV more positive redox potential (46). Reciprocally, replacement of the PH sequence in the oxidizing DsbA protein by the thioredoxin GP dipeptide generates a double mutant protein that is some 92 mV more reducing than the wild type protein (47, 48). A comparable insertion of these two dipeptides in the context of the ALR fold produced more limited modulation in redox potential (Table 1).
Table 1.
Redox potential of the proximal disulfide in wild type and mutant sfALR proteinsa
| Proximal CxxC motif | Redox potential (mV) |
|---|---|
| CEEC | −235 ± 1.6 |
| CGPC | −256 ± 1.2 |
| CPHC | −211 ± 2.9 |
| CKKC | −239 ± 1.4 |
Redox measurements were determined in 50 mM phosphate buffer, pH 7.0, containing 1 mM EDTA as described in Experimental Procedures.
Having confirmed the sizable difference in redox potentials between the two redox centers in wild type sfALR, we wished to explore the pre-steady state behavior of the enzyme in reductive and oxidative half-reactions and to reconcile these data with the steady-state kinetic parameters for the aerobic oxidation of DTT. This work provides new insights into the reactivity of the proximal CxxC motif in the context of the ALR fold.
Overall Model for Catalysis in sfALR
Figure 3 presents a scheme that incorporates all of the species that we have observed during the pre-steady state kinetic experiments to be described below. The covalent interaction of reduced DTT with oxidized sfALR yields, in principle, two charge-transfer species that are likely to have very similar spectra; both forms are combined within the orange box (Ect) in panel A of Figure 3. The first of these is the mixed disulfide shown more explicitly in panel B (E-DTTct), involving attack of a DTT thiolate on the C142 interchange surface accessible sulfur of the proximal disulfide (Figure 1B). After loss of DTTox the second charge-transfer species (Ered-ct) rapidly yields Ered which then enters the oxidative half-reaction (Panel A) leading to the net reduction of oxygen and to the generation of hydrogen peroxide.
Reductive Half-reaction of sfALR using DTT
We first investigated the interaction between sfALR and DTT under anaerobic conditions (see Experimental Procedures). The progression of spectra upon mixing sfALR with 5.4 mM DTT is shown in Figure 4. The low intensity wedge-shaped charge-transfer absorbance band extending to about 650 nm is formed maximally at about 10 ms in the stopped flow under these conditions (Figure S1). Thereafter the feature decays with accumulation of the reduced enzyme (Ered; see later). The Ered-ct form would be expected to show a long-wavelength charge-transfer species involving interaction between C145 (the charge-transfer thiol) and the oxidized flavin moiety (Figure 3). Precedent for this charge-transfer band at ~ 530 nm in ALR comes from the static spectrum of C142A and C142S mutants ((22, 49) and SSR unpublished observations) in which C145 thiolate would be within 3.1 – 3.2 Å of the C4a locus of the isoalloxazine ring ((49) PDB 3U2L; Schaefer-Ramadan, Dong, Bahnson and Thorpe, unpublished). However the Ered-ct species shown in Figure 3B is not observed upon 2-electron titration of wild type sfALR (21) because the reduced flavin species Ered is overwhelmingly favored (as shown by the redox measurements described above). The other possible contributor to the charge-transfer absorbance feature seen in Figure 4 is the mixed disulfide species with DTT (Figure 3B; E-DTTct). Such species have precedents in the mixed disulfides formed between two CxxC motifs in both Erv1p (40, 50, 51) and QSOX (38, 52, 53). In addition, Williams and coworkers have shown that oxidized yeast glutathione reductase treated with GSH exists largely in the mixed disulfide form (54). Later in the manuscript we will describe the surprising propensity of sfALR to form mixed disulfide species, but here we are concerned with identifying the main features of catalytic turnover; hence we aggregate all the observable charge-transfer species into the Ect form shown in Figure 3.
Figure 4.

Reduction of sfALR by DTT under anaerobic conditions. The main panel shows diode-array traces recorded 0.005, 0.145, 0.295, 0.445, 0.625, 0.845, and 1.405 s after mixing oxidized sfALR with DTT to give final concentrations of 30 μM and 5.4 mM respectively in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA (spectra 1–7 respectively; see Experimental Procedures). The inset shows limiting apparent pseudo first-order rate constants of 12.4 ± 0.2 s−1 and 11.2 ± 0.8 s−1 for the reduction of FAD at 454 nm (solid line) and the disappearance of the charge-transfer intermediate at 550 nm (dashed line), respectively. Data are the average of triplicate runs.
The DTT dependence of the apparent rate constants obtained from absorbance changes at 454 and 550 nm are comparable (Figure 4 with limiting kapp values of 12.4 ± 0.2 s−1 and 11.2 ± 0.8 s−1 respectively). Experiments conducted up to 18 mM DTT showed no detectable accumulation of the C4a-thiol-flavin adducts believed to be transitory intermediates in the reduction of flavins by thiols (55–57). The bimolecular reverse reaction k−2 (Figure 3) is negligible under the conditions of our experiments where, at most, stoichiometric levels of DTTox are formed in the presence of a large concentration of competing DTT. This reverse reaction, k−2, is set arbitrarily to 1 M−1s−1 in Figure 3; simulations reveal that it could be increased up to 10 M−1s−1 without significantly impacting the overall fits (Experimental Procedures).
Oxidative Half-reaction of 2-Electron Reduced sfALR with Oxygen
ALR has been shown to be an atypical flavoprotein oxidase (21, 22, 58). Firstly, ALR shows a high Km for oxygen (~240 μM) and a distinct preference for cytochrome c as an electron acceptor of the enzyme (21, 22). While lfALR could utilize this cytochrome as a means to transfer reducing equivalents to the respiratory chain, sfALR functions extramitochondrially (see above) where cytochrome c is not normally resident. Hence oxygen is a logical physiological electron acceptor for sfALR. A second unusual feature of sfALR is that it releases more superoxide ion than would be expected for a simple flavoprotein oxidase (58). For these reasons we investigated the reaction of the reduced sfALR with molecular oxygen.
The 2-electron reduced enzyme Ered (Figure 3) was generated by dithionite titration in an anaerobic tonometer and subsequently mixed with various concentrations of dissolved oxygen (Experimental Procedures). The course of the reaction, followed in the diode-array stopped-flow spectrophotometer, showed a monotonic reappearance of the spectrum of oxidized ALR over a range of wavelengths (Figure 5). Prior to mixing with oxygen very small levels of the blue semiquinone had accumulated during the anaerobic dithionite titration (21) and proved impractical to eliminate without over-reducing the enzyme. This species disappears during the subsequent reoxidation of the dihydroflavin form of sfALR monitored at 454 nm. The reappearance of Eox yields a bimolecular rate constant of 9650 ± 150 M−1s−1 with an essentially linear dependence on oxygen concentration over the range studied (Figure 5, inset). This irreversible reaction is significantly slower than the 105 – 106 M−1s−1 typically observed for flavoprotein oxidases (59–61); additionally the rate constant is 50- to 90-fold slower that reoxidation of the ERV-domain in avian (62) and Trypanosoma brucei QSOX (52).
Figure 5.

Oxidative half-reaction of sfALR. The 2-electron reduced enzyme (Experimental Procedures) was mixed with aerobic buffer to give final concentration of 30 μM enzyme and 120 μM oxygen. Spectra were recorded in the diode-array stopped-flow instrument 0.005, 0.055, 0.105, 0.155, 0.215, 0.285, 0.365 and 0.505 s after mixing (spectra 1–8 respectively). The inset plots the dependence on the apparent first-order rate constants for the return of the oxidized flavin at 454 nm as a function of dissolved oxygen concentration. Data points represent the average of 4 experiments at each oxygen concentration.
Summary of Reductive and Oxidative Reaction
The rate constants shown for the reductive half-reaction in Figure 3 predict steady-state kinetic parameters (see Experimental Procedures) which are in good agreement with those determined experimentally. In terms of kcat for DTT using 240 μM oxygen the observed and predicted values were 108 min−1 and 99 min−1 respectively. The corresponding Km values were 1.6 mM and 1.6 mM respectively. Farrell and Thorpe determined a Km of 240 μM for oxygen using 2 mM DTT (21); compared to a predicted value of 212 μM. Here the apparent Km value for oxygen reflects the emergence of the reductive half-reaction as the rate-limiting step with increasing magnitude of the k3[O2] term (Figure 3). While the very rapid formation of the small levels of the charge-transfer species (Figure 4 and Figure S1) made k1 impractical to determine experimentally, the values k1 and k−1 in Figure 3A provide good fits to the data (Experimental Procedures).
The Stabilization of Mixed Disulfide Intermediates in sfALR
We next consider whether a mixed disulfide intermediate with DTT could make a significant contribution to the aggregate Ect species shown in Figure 3A. To explore the propensity of DTT to form mixed disulfide intermediates in sfALR we used the monothiol βME which is structurally analogous to one half of DTT. We demonstrated that βME is a very poor substrate showing a turnover number of ~ 0.3 molecules of oxygen reduced/min at 100 mM thiol (Figure S2). Nevertheless βME forms facile and surprisingly intense charge-transfer intermediates when mixed with oxidized sfALR (Figure 6). Upon prolonged incubation there is a very slow subsequent appearance of the blue flavin semiquinone which always appears when oxygen is depleted from solution of ALR reduced with thiol substrates (15, 21, 22, 63). The pseudo first-order appearance of the mixed disulfide charge-transfer intermediate was followed at 540 nm in the stopped flow spectrophotometer (inset Figure 6) as a function of βME concentration giving rate constants of 3295 ± 59 M−1s−1 for k1 and 31.6 ± 0.6 s−1 for k−1, yielding an overall KD of 9.6 ± 0.25 mM; see Experimental Procedures). These data explain why plots of turnover versus βME concentration exhibit upward curvature followed by a linear increase (Figure S2). Thus, following a reversible mixed disulfide intermediate involving one molecule of βME (Figure 7), a second βME resolves this species in a kinetically sluggish bimolecular encounter.
Since βME is a monothiol structural mimic of half of DTT (Figure 3), there seems no reason to expect that these two reagents would not be comparable nucleophiles in the formation of the mixed disulfide intermediates shown in Figure 3B (64). For example, their thiol pK values are comparable (9.5 for βME and 9.3 for the first pK of DTT (64)). As a further validation of the similarity between the reactivity of these two reagents we explored their reactivity towards the chromophoric disulfide DTNB in the stopped-flow instrument by following the release of TNB under pseudo first-order conditions (Figure S3). Here the encounter between thiols and DTNB is second-order overall with the expected zero intercept for a reaction that is essentially irreversible given the strongly oxidizing redox potential of the DTNB disulfide. As expected, these bimolecular rate constants are similar, when compared on a per thiol basis at pH 7.5, 25 °C (1.90 ± 0.01 × 103 M−1s−1 for βME; and 2.72 ± 0.007 × 103 M−1s−1 for DTT). In sum, these data suggest that a significant fraction of the charge-transfer band seen in Figure 4 represents a mixed disulfide with one thiol of DTT and that the relatively slow reduction of the flavin of 12.4 s−1 may reflect the resolution of the mixed disulfide by the second thiol moiety of DTT. Typically the subsequent transfer of reducing equivalents from the reduced proximal disulfide in charge transfer with an oxidized flavin is rapid (800 s−1 for lipoamide dehydrogenase (18, 65, 66) and 110 s−1 for glutathione reductase (67)).
These data with βME suggest that ALR has an unexpected propensity to form mixed disulfides with a monothiol with which it would not have been expected to exhibit a particular affinity. Accordingly we surveyed several other monothiols (at 20 mM) for their abilities to generate these charge-transfer complexes at both pH 7.5 and pH 9 (Figure 8). While βME, N-acetylcysteamine and cysteamine (not shown) are rather comparable in their behavior, glutathione is conspicuously unable to form significant levels of mixed disulfide. This behavior is also seen at pH 9, where the other monothiols generate strong charge-transfer bands. All these monothiols, including GSH, have rather comparable pK values (from 8.2 – 9.5) and therefore would be expected to have correspondingly comparable intrinsic nucleophilicities (30, 68, 69). However GSH is notably bulkier than the other thiols, suggesting that steric factors are likely to explain why this abundant cellular monothiol is an undetectable substrate of sfALR (22). Mixed disulfide bond formation requires in-line approach of the attacking nucleophilic thiolate along the disulfide axis (70–73). While this first step is already disfavored for GSH, the completion of catalysis would require a second glutathione to resolve this sterically congested mixed disulfide. Thus although reduction of the proximal disulfide (E∘′ −235 mV) by GSH (E∘′ of − 240 mV (18)) is certainly feasible from a thermodynamic perspective, it is strongly disfavored kinetically. Hence intracellular sfALR can avoid the potentially damaging generation of hydrogen peroxide driven by millimolar levels of GSH (21, 58).
Figure 8.

Spectral changes on the addition of monothiols to ALR at pH 7.5 and 9.0. The spectrum of an aerobic solution of 30 μM ALR (black line) was recorded 10 s before the addition of 20 mM concentrations of glutathione (grey line), βME (red line) and N-acetylcysteamine (blue line). Panel A was conducted in phosphate buffer, pH 7.5, and panel B was recorded using Tris chloride buffer, pH 9.0.
Finally, we consider why monothiols can form significant complexes with sfALR when their bimolecular formation could be reversed intramolecularly by the locally enormous concentration of the neighboring C145 thiol shown in Figure 7. At least part of the answer may be the difference in pKs between the attacking monothiol and that of the C145 thiol that would be released upon mixed disulfide bond formation; as in all disulfide exchange reactions, the pK of attacking and leaving thiolates will profoundly modulate the position of the equilibrium (30, 68, 69, 74). Thus the pK of βME is 9.5 whereas the charge-transfer thiol has been reported to have a pK of 5.95 (4949) biasing the mixed disulfide equilibrium in favor of the charge-transfer species in Figure 7. The surprising stability of these initial mixed disulfide species then would allow sufficient time for attainment of a conformation in which in-line approach of the resolving substrate thiolate leads to the transient generation of Ered-ct and to the subsequent rapid formation of the free reduced enzyme Ered (Figure 3).
Supplementary Material
Acknowledgments
We thank Drs. Bruce Palfey and Kenneth Johnson for helpful discussion. Mr. Benjamin Israel generously provided the 5-deaza-FAD used in this work.
Footnotes
Funding
This work was supported in part by National Institutes of Health Grant GM26643 (CT) and USPHS Training Grant 1-T32-GM008550 (SSR). The content of this work is solely the responsibility of the authors, and does not necessarily reflect the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Abbreviations: ALR, augmenter of liver regeneration; sfALR, short-form augmenter of liver regeneration; lfALR, long-form augmenter of liver regeneration; ERV, essential for respiration and viability; EDTA, ethylenediaminetetraacetic acid; DTNB, 5,5′-dithiobis(2-nitrobenzoate); DTT, dithiothreitol; DTTox, oxidized dithiothreitol; GSH, reduced glutathione; GSSG, oxidized glutathione; GuHCl; guanidine hydrochloride; IPTG, isopropyl β-D-thiogalactopyranoside; βME, β-mercaptoethanol.
Figures S1–S3 providing rapid reaction kinetics of the formation of the charge-transfer intermediate, steady-state turnover of ALR using βME as a substrate, and a comparison of the nucleophilicity of DTT and βME using DTNB. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.LaBrecque DR, Pesch LA. Preparation and partial characterization of hepatic regenerative stimulator substance (SS) from rat liver. J Physiol. 1975;248:273–284. doi: 10.1113/jphysiol.1975.sp010973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hagiya M, Francavilla A, Polimeno L, Ihara I, Sakai H, Seki T, Shimonishi M, Porter KA, Starzl TE. Cloning and sequence analysis of the rat augmenter of liver regeneration (ALR) gene: expression of biologically active recombinant ALR and demonstration of tissue distribution. Proc Natl Acad Sci USA. 1994;91:8142–8146. doi: 10.1073/pnas.91.17.8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoober KL, Glynn NM, Burnside J, Coppock DL, Thorpe C. Homology between egg white sulfhydryl oxidase and quiescin Q6 defines a new class of flavin-linked sulfhydryl oxidases. J Biol Chem. 1999;274:31759–31762. doi: 10.1074/jbc.274.45.31759. [DOI] [PubMed] [Google Scholar]
- 4.Lee J, Hofhaus G, Lisowsky T. Erv1p from Saccharomyces cerevisiae is a FAD-linked sulfhydryl oxidase. FEBS Lett. 2000;477(1–2):62–66. doi: 10.1016/s0014-5793(00)01767-1. [DOI] [PubMed] [Google Scholar]
- 5.Lisowsky T, Lee JE, Polimeno L, Francavilla A, Hofhaus G. Mammalian augmenter of liver regeneration protein is a sulfhydryl oxidase. Dig Liver Dis. 2001;33:173–180. doi: 10.1016/s1590-8658(01)80074-8. [DOI] [PubMed] [Google Scholar]
- 6.Wang G, Yang X, Zhang Y, Wang Q, Chen H, Wei H, Xing G, Xie L, Hu Z, Zhang C, Fang D, Wu C, He F. Identification and characterization of receptor for mammalian hepatopoietin that is homologous to yeast ERV1. J Biol Chem. 1999;274:11469–11472. doi: 10.1074/jbc.274.17.11469. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Li M, Xing G, Hu Z, Wang Q, Dong C, Wei H, Fan G, Chen J, Yang X, Zhao S, Chen H, Guan K, Wu C, Zhang C, He F. Stimulation of the mitogen-activated protein kinase cascade and tyrosine phosphorylation of the epidermal growth factor receptor by hepatopoietin. J Biol Chem. 2000;275:37443–37447. doi: 10.1074/jbc.M004373200. [DOI] [PubMed] [Google Scholar]
- 8.Chen X, Li Y, Wei K, Li L, Liu W, Zhu Y, Qiu Z, He F. The potentiation role of hepatopoietin on activator protein-1 is dependent on its sulfhydryl oxidase activity. J Biol Chem. 2003;278:49022–49030. doi: 10.1074/jbc.M304057200. [DOI] [PubMed] [Google Scholar]
- 9.Zhang LM, Liu DW, Liu JB, Zhang XL, Wang XB, Tang LM, Wang LQ. Effect of naked eukaryotic expression plasmid encoding rat augmenter of liver regeneration on acute hepatic injury and hepatic failure in rats. World J Gastroenterol. 2005;11:3680–3685. doi: 10.3748/wjg.v11.i24.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pawlowski R, Jura J. ALR and Liver Regeneration. Mol Cell Biochem. 2006;288:159–169. doi: 10.1007/s11010-006-9133-7. [DOI] [PubMed] [Google Scholar]
- 11.Gatzidou E, Kouraklis G, Theocharis S. Insights on augmenter of liver regeneration cloning and function. World J Gastroenterol. 2006;12:4951–4958. doi: 10.3748/wjg.v12.i31.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu C, Li Y, Zhao Y, Xing G, Tang F, Wang Q, Sun Y, Wei H, Yang X, Wu C, Chen J, Guan KL, Zhang C, Chen H, He F. Intracrine hepatopoietin potentiates AP-1 activity through JAB1 independent of MAPK pathway. FASEB J. 2002;16:90–92. doi: 10.1096/fj.01-0506fje. [DOI] [PubMed] [Google Scholar]
- 13.Polimeno L, Pesetti B, Lisowsky T, Iannone F, Resta L, Giorgio F, Mallamaci R, Buttiglione M, Santovito D, Vitiello F, Mancini ME, Francavilla A. Protective effect of augmenter of liver regeneration on hydrogen peroxide-induced apoptosis in SH-SY5Y human neuroblastoma cells. Free Radic Res. 2009;43:865–875. doi: 10.1080/10715760903100125. [DOI] [PubMed] [Google Scholar]
- 14.Cao Y, Fu YL, Yu M, Yue PB, Ge CH, Xu WX, Zhan YQ, Li CY, Li W, Wang XH, Wang ZD, Li YH, Yang XM. Human augmenter of liver regeneration is important for hepatoma cell viability and resistance to radiation-induced oxidative stress. Free Radic Biol Med. 2009;47:1057–1066. doi: 10.1016/j.freeradbiomed.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 15.Wu CK, Dailey TA, Dailey HA, Wang BC, Rose JP. The crystal structure of augmenter of liver regeneration: A mammalian FAD-dependent sulfhydryl oxidase. Protein Sci. 2003;12:1109–1118. doi: 10.1110/ps.0238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daithankar VN, Schaefer SA, Dong M, Bahnson BJ, Thorpe C. Structure of the human sulfhydryl oxidase augmenter of liver regeneration and characterization of a human mutation causing an autosomal recessive myopathy. Biochemistry. 2010;49:6737–6745. doi: 10.1021/bi100912m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banci L, Bertini I, Calderone V, Cefaro C, Ciofi-Baffoni S, Gallo A, Kallergi E, Lionaki E, Pozidis C, Tokatlidis K. Molecular recognition and substrate mimicry drive the electron-transfer process between MIA40 and ALR. Proc Natl Acad Sci USA. 2011;108:4811–4816. doi: 10.1073/pnas.1014542108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams CH., Jr . Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase-A family of flavoenzyme transhydrogenases. In: Muller F, editor. Chemistry and Biochemistry of Flavoenzymes. CRC Press; 1992. pp. 121–211. Chemistry and Biochemistry of Flavoenzymes. [Google Scholar]
- 19.Argyrou A, Blanchard JS. Flavoprotein disulfide reductases: advances in chemistry and function. Prog Nucleic Acid Res Mol Biol. 2004;78:89–142. doi: 10.1016/S0079-6603(04)78003-4. [DOI] [PubMed] [Google Scholar]
- 20.Miller SM. Flavoprotein disulfide reductases and structurally related flvoprotein thiol/disulfide-linked oxidoreductases. In: Hille R, Miller SM, Palfey BA, editors. Handbook of Flavoproteins. De Gruyter; Berlin: 2013. pp. 165–201. [Google Scholar]
- 21.Farrell SR, Thorpe C. Augmenter of liver regeneration: a flavin dependent sulfhydryl oxidase with cytochrome C reductase activity. Biochemistry. 2005;44:1532–1541. doi: 10.1021/bi0479555. [DOI] [PubMed] [Google Scholar]
- 22.Daithankar VN, Farrell SR, Thorpe C. Augmenter of liver regeneration: substrate specificity of a flavin-dependent oxidoreductase from the mitochondrial intermembrane space. Biochemistry. 2009;48:4828–4837. doi: 10.1021/bi900347v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chacinska A, Pfannschmidt S, Wiedemann N, Kozjak V, Sanjuan Szklarz LK, Schulze-Specking A, Truscott KN, Guiard B, Meisinger C, Pfanner N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 2004;23:3735–3746. doi: 10.1038/sj.emboj.7600389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 25.Sankar U, Means AR. Gfer is a critical regulator of HSC proliferation. Cell Cycle. 2011;10:2263–2268. doi: 10.4161/cc.10.14.15919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Todd LR, Damin MN, Gomathinayagam R, Horn SR, Means AR, Sankar U. Growth Factor erv1-like Modulates Drp1 to Preserve Mitochondrial Dynamics and Function in Mouse Embryonic Stem Cells. Mol Biol Cell. 2010;21:1226–1236. doi: 10.1091/mbc.E09-11-0937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thorpe C. Flavoproteins in oxidative protein folding. In: Hill R, Miller SM, Palfey BA, editors. Handbook of Flavoproteins. de Gruyter; Berlin: 2013. pp. 248–269. [Google Scholar]
- 28.Fass D. The Erv family of sulfhydryl oxidases. Biochim Biophys Acta. 2008;1783:557–566. doi: 10.1016/j.bbamcr.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 29.Sevier C. Erv2 and Quiescin Sulfhydryl Oxidases: Erv-Domain Enzymes Associated with the Secretory Pathway. Antioxid Redox Sign. 2012;16:800–808. doi: 10.1089/ars.2011.4450. [DOI] [PubMed] [Google Scholar]
- 30.Lees WJ, Whitesides GM. Equilibrium-Constants for Thiol Disulfide Interchange Reactions - a Coherent, Corrected Set. J Org Chem. 1993;58:642–647. [Google Scholar]
- 31.Williams CH, Arscott LD, Matthews RG, Thorpe C, Wilkinson KD. Methodology employed for anaerobic spectrophotometric titrations and for computer-assisted data analysis. Methods Enzymol. 1980;62D:185–198. doi: 10.1016/0076-6879(79)62217-6. [DOI] [PubMed] [Google Scholar]
- 32.DuPlessis ER, Pellet J, Stankovich MT, Thorpe C. Oxidase activity of the acyl-CoA dehydrogenases. Biochemistry. 1998;37:10469–10477. doi: 10.1021/bi980767s. [DOI] [PubMed] [Google Scholar]
- 33.Johnson KA, Simpson ZB, Blom T. FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal Biochem. 2009;387:30–41. doi: 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 34.Johnson KA, Simpson ZB, Blom T. Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal Biochem. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 35.Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U. COPASI--a COmplex PAthway SImulator. Bioinformatics. 2006;22:3067–3074. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- 36.Walsh CT, Fisher J, Spencer R, Graham DW, Ashton WT, Brown JE, Brown RD, Rogers EF. Chemical and enzymatic properties of riboflavin analogues. Biochemistry. 1978;17:1942–1951. doi: 10.1021/bi00603a022. [DOI] [PubMed] [Google Scholar]
- 37.Thorpe C, Williams CHJ. Differential reactivity of the two active site cysteine residues generated on reduction of pig heart lipoamide dehydrogenase. J Biol Chem. 1976;251:3553–3557. [PubMed] [Google Scholar]
- 38.Heckler EJ, Alon A, Fass D, Thorpe C. Human quiescin-sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis. Biochemistry. 2008;47:4955–4963. doi: 10.1021/bi702522q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brohawn SG, Rudik I, Thorpe C. Avian sulfhydryl oxidase is not a metalloenzyme: adventitious binding of divalent metal ions to the enzyme. Biochemistry. 2003;42:11074–11082. doi: 10.1021/bi0301385. [DOI] [PubMed] [Google Scholar]
- 40.Wang W, Winther JR, Thorpe C. Erv2p: characterization of the redox behavior of a yeast sulfhydryl oxidase. Biochemistry. 2007;46:3246–3254. doi: 10.1021/bi602499t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore EC, Reichard P, Thelander L. Enzymatic Synthesis of Deoxyribonucleotides. V Purification and Properties of Thioredoxin Reductase from Escherichia Coli B. J Biol Chem. 1964;239:3445–3452. [PubMed] [Google Scholar]
- 42.Krause G, Lundstrom J, Barea JL, Pueyo de la Cuesta C, Holmgren A. Mimicking the active site of protein disulfide-isomerase by substitution of proline 34 in Escherichia coli thioredoxin. J Biol Chem. 1991;266:9494–9500. [PubMed] [Google Scholar]
- 43.Mossner E, Huber-Wunderlich M, Glockshuber R. Characterization of Escherichia coli thioredoxin variants mimicking the active-sites of other thiol/disulfide oxidoreductases. Prot Sci. 1998;7:1233–1244. doi: 10.1002/pro.5560070519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zapun A, Bardwell JCA, Creighton TE. The Reactive and Destabilizing Disulfide Bond of Dsba, a Protein Required for Protein Disulfide Bond Formation In Vivo. Biochemistry. 1993;32:5083–5092. doi: 10.1021/bi00070a016. [DOI] [PubMed] [Google Scholar]
- 45.Huber-Wunderlich M, Glockshuber R. A single dipeptide sequence modulates the redox properties of a whole enzyme family. Fold Des. 1998;3:161–171. doi: 10.1016/S1359-0278(98)00024-8. [DOI] [PubMed] [Google Scholar]
- 46.Jonda S, Huber-Wunderlich M, Glockshuber R, Mossner E. Complementation of DsbA deficiency with secreted thioredoxin variants reveals the crucial role of an efficient dithiol oxidant for catalyzed protein folding in the bacterial periplasm. EMBO J. 1999;18:3271–3281. doi: 10.1093/emboj/18.12.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aslund F, Berndt KD, Holmgren A. Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J Biol Chem. 1997;272:30780–30786. doi: 10.1074/jbc.272.49.30780. [DOI] [PubMed] [Google Scholar]
- 48.Inaba K, Ito K. Paradoxical redox properties of DsbB and DsbA in the protein disulfide-introducing reaction cascade. EMBO J. 2002;21:2646–2654. doi: 10.1093/emboj/21.11.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banci L, Bertini I, Calderone V, Cefaro C, Ciofi-Baffoni S, Gallo A, Tokatlidis K. An Electron-Transfer Path through an Extended Disulfide Relay System: The Case of the Redox Protein ALR. J Am Chem Soc. 2012:1442–1445. doi: 10.1021/ja209881f. [DOI] [PubMed] [Google Scholar]
- 50.Hofhaus G, Lee JE, Tews I, Rosenberg B, Lisowsky T. The N-terminal cysteine pair of yeast sulfhydryl oxidase Erv1p is essential for in vivo activity and interacts with the primary redox centre. Eur J Biochem. 2003;270:1528–1535. doi: 10.1046/j.1432-1033.2003.03519.x. [DOI] [PubMed] [Google Scholar]
- 51.Coppock DL, Thorpe C. Multidomain flavin-dependent sulfhydryl oxidases. Antioxid Redox Signal. 2006;8:300–311. doi: 10.1089/ars.2006.8.300. [DOI] [PubMed] [Google Scholar]
- 52.Kodali VK, Thorpe C. Quiescin sulfhydryl oxidase from Trypanosoma brucei: catalytic activity and mechanism of a QSOX family member with a single thioredoxin domain. Biochemistry. 2010;49:2075–2085. doi: 10.1021/bi902222s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alon A, Grossman I, Gat Y, Kodali VK, DiMaio F, Mehlman T, Haran G, Baker D, Thorpe C, Fass D. The dynamic disulphide relay of quiescin sulphydryl oxidase. Nature. 2012;488:414–418. doi: 10.1038/nature11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arscott LD, Veine DM, Williams CH., Jr Mixed disulfide with glutathione as an intermediate in the reaction catalyzed by glutathione reductase from yeast and as a major form of the enzyme in the cell. Biochemistry. 2000;39:4711–4721. doi: 10.1021/bi9926431. [DOI] [PubMed] [Google Scholar]
- 55.Thorpe C, Williams CH. Spectral evidence for a flavin adduct in a monoalkylated derivative of pig heart lipoamide dehydrogenase. J Biol Chem. 1976;251:7726–7728. [PubMed] [Google Scholar]
- 56.O’Donnell ME, Williams CH., Jr Reconstitution of Escherichia coli thioredoxin reductase with 1-deazaFAD. Evidence for 1-deazaFAD C-4a adduct formation linked to the ionization of an active site base. J Biol Chem. 1984;259:2243–2251. [PubMed] [Google Scholar]
- 57.Miller SM, Massey V, Ballou D, Williams CH, Jr, Distefano MD, Moore MJ, Walsh CT. Use of a site-directed triple mutant to trap intermediates: demonstration that the flavin C(4a)-thiol adduct and reduced flavin are kinetically competent intermediates in mercuric ion reductase. Biochemistry. 1990;29:2831–2841. doi: 10.1021/bi00463a028. [DOI] [PubMed] [Google Scholar]
- 58.Daithankar VN, Wang W, Trujillo JR, Thorpe C. Flavin-linked Erv-family sulfhydryl oxidases release superoxide anion during catalytic turnover. Biochemistry. 2012;51:265–272. doi: 10.1021/bi201672h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chaiyen P, Fraaije MW, Mattevi A. The enigmatic reaction of flavins with oxygen. Trends Biochem Sci. 2012;37:373–380. doi: 10.1016/j.tibs.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 60.Massey V. The reactivity of oxygen with flavoproteins. In: Chapman S, Perham R, Scrutton NS, editors. Flavins and Flavoproteins, International Congress Series. Walter De Gruyter; 2002. pp. 3–11. [Google Scholar]
- 61.Mattevi A. To be or not to be an oxidase: challenging the oxygen reactivity of flavoenzymes. Trends Biochem Sci. 2006;31:276–283. doi: 10.1016/j.tibs.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 62.Hoober KL, Thorpe C. Egg white sulfhydryl oxidase: Kinetic mechanism of the catalysis of disulfide bond formation. Biochemistry. 1999;38:3211–3217. doi: 10.1021/bi9820816. [DOI] [PubMed] [Google Scholar]
- 63.Kay CWM, Elsasser C, Bittl R, Farrell SR, Thorpe C. Determination of the distance between the two neutral flavin radicals in augmenter of liver regeneration by pulsed ELDOR. J Am Chem Soc. 2006;128:76–77. doi: 10.1021/ja057308g. [DOI] [PubMed] [Google Scholar]
- 64.Houk J, Singh R, Whitesides GM. Measurement of Thiol Disulfide Interchange Reactions and Thiol Pka Values. Meth Enzymol. 1987;143:129–140. doi: 10.1016/0076-6879(87)43023-1. [DOI] [PubMed] [Google Scholar]
- 65.Benen J, van Berkel W, Dieteren N, Arscott D, Williams C, Jr, Veeger C, de Kok A. Lipoamide dehydrogenase from Azotobacter vinelandii: site-directed mutagenesis of the His450-Glu455 diad. Kinetics of wild-type and mutated enzymes. Eur J Biochem. 1992;207:487–497. doi: 10.1111/j.1432-1033.1992.tb17075.x. [DOI] [PubMed] [Google Scholar]
- 66.Massey V, Gibson QH, Veeger C. Intermediates in the catalytic action of lipoyl dehydrogenase (diaphorase) Biochem J. 1960;77:341–351. doi: 10.1042/bj0770341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rietveld P, Arscott LD, Berry A, Scrutton NS, Deonarian MP, Perham RN, Williams CHJ. Reductive and oxidative half-reactions of glutathione reductase from Escherichia coli. Biochemistry. 1994;33:13888–13895. doi: 10.1021/bi00250a043. [DOI] [PubMed] [Google Scholar]
- 68.Houk J, Whitesides GM. Structure Reactivity Relations for Thiol Disulfide Interchange. J Am Chem Soc. 1987;109:6825–6836. [Google Scholar]
- 69.Singh R, Whitesides G. Thiol-disulfide Interchange. In: Patai S, Rappoport Z, editors. The chemistry of sulphur-containing functional groups. John Wiley; 1993. pp. 633–658. [Google Scholar]
- 70.Rosenfield RE, Parthasarathy R, Dunitz JD. Directional Preferences of Nonbonded Atomic Contacts with Divalent Sulfur 1. Electrophiles and Nucleophiles. J Am Chem Soc. 1977;99:4860–4862. [Google Scholar]
- 71.Fernandes PA, Ramos MJ. Theoretical insights into the mechanism for thiol/disulfide exchange. Chemistry. 2004;10:257–266. doi: 10.1002/chem.200305343. [DOI] [PubMed] [Google Scholar]
- 72.Bach RD, Dmitrenko O, Thorpe C. Mechanism of thiolate-disulfide interchange reactions in biochemistry. J Org Chem. 2008;12–21, 73:12–21. doi: 10.1021/jo702051f. [DOI] [PubMed] [Google Scholar]
- 73.Heckler EJ, Rancy PC, Kodali VK, Thorpe C. Generating disulfides with the Quiescin-sulfhydryl oxidases. Biochim Biophys Acta. 2008;1783:567–577. doi: 10.1016/j.bbamcr.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Winther JR, Thorpe C. Quantification of thiols and disulfides. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbagen.2013.03.031. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.