

ABSTRACT
The Mycobacterium tuberculosis genome includes the large family of pe_pgrs genes, whose functions are unknown. Because of precedents in other pathogens in which gene families showing high sequence variation are involved in antigenic variation, a similar role has been proposed for the pe_pgrs genes. However, the impact of immune selection on pe_pgrs genes has not been examined. Here, we sequenced 27 pe_pgrs genes in 94 clinical strains from five phylogenetic lineages of the M. tuberculosis complex (MTBC). We found that pe_pgrs genes were overall more diverse than the remainder of the MTBC genome, but individual members of the family varied widely in their nucleotide diversity and insertion/deletion (indel) content: some were more, and others were much less, diverse than the genome average. Individual pe_pgrs genes also differed in the ratio of nonsynonymous to synonymous mutations, suggesting that different selection pressures act on individual pe_pgrs genes. Using bioinformatic methods, we tested whether sequence diversity in pe_pgrs genes might be selected by human T cell recognition, the major mechanism of adaptive immunity to MTBC. We found that the large majority of predicted human T cell epitopes were confined to the conserved PE domain and experimentally confirmed the antigenicity of this domain in tuberculosis patients. In contrast, despite being genetically diverse, the PGRS domains harbored few predicted T cell epitopes. These results indicate that human T cell recognition is not a significant force driving sequence diversity in pe_pgrs genes, which is consistent with the previously reported conservation of human T cell epitopes in the MTBC.
IMPORTANCE
Recognition of Mycobacterium tuberculosis antigens by T lymphocytes is known to be important for immune protection against tuberculosis, but it is unclear whether human T cell recognition drives antigenic variation in M. tuberculosis. We previously discovered that the known human T cell epitopes in the M. tuberculosis complex are highly conserved, but we hypothesized that undiscovered epitopes with naturally occurring sequence variants might exist. To test this hypothesis, we examined the pe_pgrs genes, a large family of genes that has been proposed to function in immune evasion by M. tuberculosis. We found that the pe_pgrs genes exhibit considerable sequence variation, but the regions containing T cell epitopes and the regions of variation are distinct. These findings confirm that the majority of human T cell epitopes of M. tuberculosis are highly conserved and indicate that selection forces other than T cell recognition drive sequence variation in the pe_pgrs genes.
INTRODUCTION
Pathogens have evolved numerous mechanisms to overcome, evade, or exploit host immune responses. One such mechanism, antigenic variation, consists of generating escape mutants that are not recognized by existing host molecules. Antigenic variation is used by a wide range of pathogens, including bacteria, fungi, parasites (1), and viruses (2). Well-documented examples of gene families involved in antigenic variation include the highly diverse Neisseria species opa gene family and the Plasmodium falciparum var. gene family. Antigenic variation in Neisseria opa genes occurs by slipped-strand mispairing and depends on CTCTT pentamer units that vary in number and lead to the translational shift of the protein reading frame (3). In P. falciparum, antigenic variation of the parasite is mediated by the differential expression of a surface molecule encoded by ~60 var gene paralogs. Each individual parasite expresses a single var gene at a time, and variation is determined by epigenetic modifications (4). In both cases, antigenic diversity is generated by conversion events among gene family members sharing intragenic tandem repeats. Thus, gene families with similar but not identical sequences are often involved in strategies used by pathogens to evade recognition by adaptive immune responses.
The mechanisms involved in antigenic variation may shape the diversity of both pathogen and host loci. Indeed, the diversity of the most polymorphic regions of the human genome, the human leukocyte antigen (HLA) loci, is thought to be a response to pathogen escape variants (5, 6). In turn, host immune pressure is likely to imprint a pathogen’s genome by selecting for escape variants (7, 8). Hence, analysis of genetic diversity in pathogen populations can reveal mechanisms involved in the interaction with the host immune system.
Tuberculosis (TB) is caused by a group of phylogenetically closely related bacteria, the Mycobacterium tuberculosis complex (MTBC) (9), which is characterized by a largely clonal population structure and low overall genetic diversity (10, 11, 12). MTBC has been classified into seven main phylogenetic lineages which are associated with different geographic regions and human populations (11–18). Despite the availability of drug treatment, TB remains a major public health problem, causing an estimated 1.4 million deaths in 2011 (19). This is due in part to the lack of an efficacious vaccine, whose design is complicated by insufficient understanding of the host-pathogen interaction.
T cell-mediated immunity is critical for resistance to M. tuberculosis, as T cell-deficient humans, nonhuman primates, and mice are susceptible to rapidly progressive disease (20–22). Among T lymphocytes, CD4+ T cells are essential for protective immunity to TB. In HIV-infected humans, loss of CD4+ T cells greatly increases susceptibility to TB (20). Humans (23), nonhuman primates (24), and mice (25, 26) also generate CD8+ T cell responses to M. tuberculosis antigens during infection. In addition to the role for T cells in protective immunity in TB, there is also evidence that human T cell responses are involved in inflammatory tissue damage and in transmission of TB (reviewed in reference 20). Antigen-specific T cells recognize short peptide epitopes generated by proteolysis of pathogen proteins, bound to HLA molecules on the surface of dendritic cells and macrophages. CD4+ T cells recognize peptide antigens bound to HLA class II molecules (HLA-DR, -DQ, -DP), while CD8+ T cells recognize peptides bound to HLA class Ia molecules (HLA-A, -B, -C).
M. tuberculosis possesses multiple strategies to subvert host immunity, including downregulation of antigen gene expression, sequestration in immature phagosomes, manipulation of cytokine production, blockade of class II antigen presentation, and inhibition of T cell effector functions (27–31). However, no evidence of antigenic variation has yet been found in M. tuberculosis. Indeed, contrary to what the model of immune escape by antigenic variation would predict, recent comparison of 491 human T cell epitopes in 21 genetically diverse MTBC strains revealed that the known human T cell epitopes in the MTBC are hyperconserved and under strong purifying selection (13). That study utilized next-generation DNA sequencing that generates short sequence reads and was thus unable to comprehensively analyze one of the most variable gene families in the MTBC: the PE superfamily (13).
The PE superfamily is restricted to Mycobacteriaceae (32). The name is derived from a Pro-Glu motif within the first 10 amino acids of an N-terminal domain of ~110 amino acids. PE proteins have been divided into three families, of which the polymorphic GC-rich-repetitive sequence (PE_PGRS) family is the largest, comprising 64 members (9). The other two families either contain a PE domain only (33) or a PE domain followed by a C-terminal unique sequence that can be as long as 400 amino acids (PE_unique domain). The PGRS domain varies in size from tens of to several hundred amino acids and is characterized by tandem repeats of Gly-Gly-Ala and Gly-Gly-Asn motifs that vary in number and can be intercalated by regions of diverse sequence up to 60 amino acids long. In addition, some PE_PGRS proteins contain a unique domain linked to the C terminus of the PGRS domain (34). Because of the unique repetitive sequence of the PGRS domain and the high redundancy within the family, pe_pgrs genes are thought to be hot spots of recombination and single nucleotide polymorphisms (SNPs). Indeed, analysis of three pe_pgrs genes showed that clinical isolates of the MTBC can harbor polymorphisms in these genes, with a predominance of mutations occurring within the PGRS domain (35, 36). These observations are consistent with the widely held view that immune recognition is the force driving sequence diversity in pe_pgrs genes (36, 37).
To better understand the genetic diversity of the pe_pgrs gene family, and to characterize the impact of immune recognition on sequence variation of these genes, we sequenced 25 pe_pgrs and 2 PE_unique domain genes in 94 phylogeographically diverse clinical isolates of human-associated MTBC. We then analyzed the 64 PE_PGRS proteins in the H37Rv reference genome for predicted CD4+ and CD8+ T cell epitopes. Finally, we combined these epitope predictions with our sequence diversity data. We found that individual pe_pgrs genes differed widely in their genetic diversity and in the direction and extent of selective pressures acting on them, suggesting these genes have distinct and nonredundant functions. We found that the large majority of predicted CD4+ and CD8+ T cell epitopes were confined to the conserved PE domain and that despite being genetically diverse, the PGRS domains harbored few predicted T cell epitopes. These results weigh against the view that PE_PGRS proteins vary as a consequence of T cell-mediated immune selection.
RESULTS
DNA sequence diversity and selection in PE_PGRS proteins.
To characterize the sequence diversity of PE_PGRS proteins and the impact of human T cell recognition, we selected 27 PE_PGRS family members for comparative DNA sequencing. These included 22 classical PE_PGRS proteins, 3 members with a unique domain linked to the C terminus of the PGRS domain (PE_PGRS 6, 35, and 39), and 2 PE_unique domain proteins (PE_PGRS 62 and LipY) (Fig. 1; see also Table S1 in the supplemental material). The 27 genes encoding these proteins were sequenced in 94 clinical isolates representative of five of the seven global MTBC lineages (see Table S2). Lineage 5 and lineage 7 were not represented in this study. All genes, except for wag22, were amplified and sequenced in at least 80% of the strains (Table 1). Due to deletion of wag22 in 47 strains, and a frameshift which disrupts the open reading frame (ORF) at the beginning of wag22 in other strains (confirmed using alternative primer sets and by extraction of data from whole genome sequencing [14]), only 15/94 (14%) strains had an intact wag22 ORF, and genetic diversity analyses were not computed for this gene. In addition, we found that pe_pgrs 2 was deleted in all strains of lineage 6.
FIG. 1 .

Individual pe_pgrs genes vary widely in frequency and type of variation in the M. tuberculosis complex (MTBC). (A) Nucleotide diversity (π) of individual pe_pgrs genes sequenced in this study. The median nucleotide diversity of the 24 selected pe_pgrs genes is indicated by the black horizontal line. The overall median nucleotide diversity of the MTBC genome is indicated by a dashed horizontal line (13). (B) Ratio of substitution rates at nonsynonymous and synonymous sites (dN/dS ratio). The median dN/dS ratio for all 24 pe_pgrs genes analyzed is indicated by a dashed line, which is the same as the dN/dS ratio for the whole MTBC genome (13). (C) Indel diversity of the individual pe_pgrs genes. The pe_unique domain genes (pe_pgrs 62 and lipY) are each designated by an asterisk.
TABLE 1 .
Strain and gene analysisa
| Gene | No. of strains |
||
|---|---|---|---|
| Analyzed | Excludedb | With no PCR product | |
| pe_pgrs 1 | 94 | 1 | 0 |
| pe_pgrs 2 | 81 | 6 | 8 |
| pe_pgrs 5 | 87 | 8 | 0 |
| pe_pgrs 6 | 62 | 27 | 6 |
| pe_pgrs 15 | 92 | 1 | 2 |
| pe_pgrs 23 | 81 | 9 | 5 |
| pe_pgrs 24 | 73 | 1 | 21 |
| pe_pgrs 25 | 91 | 1 | 3 |
| pe_pgrs 26 | 94 | 0 | 1 |
| pe_pgrs 29 | 83 | 0 | 12 |
| pe_pgrs 31 | 93 | 2 | 0 |
| pe_pgrs 33 | 95 | 0 | 0 |
| pe_pgrs 34 | 95 | 0 | 0 |
| pe_pgrs 35 | 91 | 3 | 1 |
| pe_pgrs 38 | 76 | 2 | 17 |
| pe_pgrs 39 | 92 | 1 | 2 |
| pe_pgrs 41 | 87 | 1 | 7 |
| pe_pgrs 44 | 93 | 0 | 2 |
| pe_pgrs 46 | 76 | 1 | 18 |
| pe_pgrs 47 | 86 | 1 | 8 |
| pe_pgrs 48 | 86 | 0 | 9 |
| pe_pgrs 51 | 91 | 3 | 1 |
| pe_pgrs 58 | 92 | 0 | 3 |
| pe_pgrs 59 | 91 | 0 | 4 |
| pe_pgrs 62 | 92 | 0 | 3 |
| lipY | 93 | 2 | 0 |
wag22 was omitted because of its high frequency of disruption or deletion.
Strains were excluded from analysis if a frameshift indel disrupted the open reading frame or if the gene contained gaps larger than 20% of the gene length.
We first used the DNA sequences to determine the nucleotide diversity (π) for each pe_pgrs gene. We found that pe_pgrs genes as a group were more diverse than the rest of the MTBC genome (π = 0.00042 compared to 0.0003) (Wilcoxon signed-rank test, P < 0.01) (Fig. 1A). However, not all pe_pgrs genes were equally diverse; some were very conserved (π < 0.0002) and others highly variable (π > 0.001) (Fig. 1A; see also Table S3 in the supplemental material). Similarly, the pe_pgrs genes varied widely in their rate of nonsynonymous/synonymous substitutions (dN/dS ratio) (Fig. 1B; see Table S3), suggesting that the selection pressures acting on the individual genes differ: 7 genes showed evidence of diversifying selection (dN/dS > 1.5), 10 were under purifying selection (dN/dS < 0.7), and 7 appeared to evolve neutrally (dN/dS = 0.7 to 1.5) (Fig. 1B). Of note, the two genes encoding PE_unique proteins, pe_pgrs 62 and lipY, were among the group exhibiting the highest dN/dS ratio. The lowest dN/dS values were found in pe_pgrs 44, 47, and 59, which were more conserved than the genome overall (Fig. 1B). These results indicate that the pe_pgrs genes vary greatly in their sequence diversity, suggesting that individual pe_pgrs genes are subjected to distinct selection pressures.
Insertions and deletions.
Insertions or deletions (indels) can also contribute to genetic diversity and be under selective pressure (38). We calculated the number of nonredundant indel events and the indel diversity per site, defined as the average number of indels per nucleotide site between any two DNA sequences chosen at random. As we found for SNPs, the number of indel events and the indel diversity per site were heterogeneous among the pe_pgrs genes (Fig. 1C; see also Table S4 in the supplemental material). Some contained only one indel event (pe_pgrs 29 and 39), while others (pe_pgrs 26 and 33) contained 10 to 12 indel events (see Fig. S1 and Table S4). The indel distribution along the gene sequence was not random: 138/145 (95%) of the indels were in the PGRS domains (χ2 with Yates correction for continuity = 13.72, P < 0.001). Indels often occur in repeated sequences (39); hence, the GC-rich-repetitive sequence and homopolymeric tracts in the PGRS domain likely make this region prone to indels.
To explore differences in selection pressure on indels, we classified indels according to whether or not they led to frameshifts (see Fig. S1). Frameshifts frequently lead to a premature stop codon in the corresponding ORF, and if no other indel restores the ORF, most frameshift mutations will be deleterious and removed by purifying selection. In the absence of selection, one-third of indels will contain multiples of three nucleotides, hence maintaining the ORF, and two-thirds of indels will not be multiples of three, causing a frameshift. We found that 21 out of 23 (87%) of the pe_pgrs genes that contain indels had a ratio of in-frame to frameshift indels greater than 0.33 (see Fig. S1 and Table S4), which suggests that natural selection is acting on these indels. However, based on these data alone, it is not possible to determine whether selection is acting positively or negatively. Together, the findings that pe_pgrs genes vary greatly in both sequence diversity and indel diversity indicate that the individual genes are under distinct selection pressures that operate within and/or between human hosts. The findings also imply that members of the pe_pgrs family serve distinct, and not redundant, functions.
PE_PGRS diversity across phylogenetic lineages.
Comparison of the diversity of individual pe_pgrs genes by lineage in the MTBC allows identification of homoplasies and comparison of intralineage and interlineage variation. Nonsynonymous SNPs (nSNPs) in individual pe_pgrs genes exhibited considerable variation in their intra- and interlineage frequency (see Fig. S2 in the supplemental material). Four genes, pe_pgrs 6, 25, 33, and 58, contained nSNPs in all 5 lineages examined, while variants in 17 genes were present in strains in 3 or fewer lineages (see Fig. S2A). With the exception of one possible gene conversion event in two strains, we found no evidence of homoplasy, or convergent evolution, in different lineages involving nSNPs. Individual pe_pgrs genes also exhibited considerable variation in the intra- and interlineage frequency of indels (see Fig. S2B).
To seek higher-order grouping of the lineages, we performed a three-dimensional principal component analysis (PCA) of the genetic variation of 45 of the strains used in this study, based on the non-pe_pgrs polymorphisms found in them as previously reported (12) (Fig. 2A). PCA distinguished three major groups: group 1 contains all strains belonging to lineages 2, 3, and 4 (considered evolutionarily “modern”), group 2 contains strains belonging to lineage 1, and group 3 consists of strains belonging to lineage 6 (Fig. 2B). Examination of the variation of individual pe_pgrs within the individual lineages using analysis of molecular variance (AMOVA) (69), among the “modern” lineages, and among the three groups identified by PCA revealed three distinct patterns (Fig. 2C). In one pattern (exemplified by pe_pgrs 58), nearly all of the variation (92%) was found within the individual lineages; in the second pattern (exemplified by pe_pgrs 15), most of the variation was found among the lineages in PCA group 1 (75%); and in the third (exemplified by pe_pgrs 5), the variation was distributed among the groups defined by PCA (93%). This result further indicates that individual pe_pgrs genes exhibit differences in genetic drift and/or are under distinct selection pressure.
FIG 2 .

Sequence variation in individual pe_pgrs genes is differentially distributed within and among lineage groups. (A) Neighbor-joining phylogeny adapted from reference 13. The tree is rooted with Mycobacterium canettii, the closest known outgroup, and node support after 1,000 bootstrap replication is indicated. For each lineage, branches are collapsed and colored according to the main MTBC lineages as defined previously (12, 16). The shaded boxes highlight the combinations used for the comparative analysis in panel C. Light gray, comparison within lineages; dark gray, comparison among lineages 2, 3, and 4; black, comparison among the three lineage groups identified by PCA. (B) Three-dimensional PCA plot of the strains used in this study based on non-pe_pgrs polymorphisms (12) highlights three major groups separating the three evolutionarily “modern” lineages from lineage 1 and lineage 6. (C) Percentage of variation of individual pe_pgrs genes within the 5 lineages, among the “modern” lineages, and among the three groups identified by PCA. The pe_unique domain genes (pe_pgrs 62 and lipY) are each designated by an asterisk.
In contrast to our findings involving SNPs, indels exhibited several instances of convergent evolution. Although the majority of these involved the appearance of a given indel in only one strain in each of two lineages, pe_pgrs 23, 26, and 33 contained indels shared by more than one strain in at least one of the affected lineages (see Fig. S2B). For example, indel 6 in pe_pgrs 33 was present in all strains of lineage 3 and one strain of lineage 2, and indel 7 of the same gene was present in all strains of lineage 1 and one strain of lineage 4.
Identification of human CD4+ and CD8+ T cell epitopes in PE_PGRS proteins.
T cell-mediated immunity is essential for preventing progression of TB and depends on binding of bacterial peptides to HLA molecules for recognition by T cell antigen receptors. Since the existence of multiple family members and the diversity in the PE_PGRS family have been interpreted as evidence for a role in immune evasion (9), we tested the hypothesis that the sequence diversity within PE_PGRS proteins alters HLA binding of putative human T cell epitopes and thereby allows escape from T cell recognition.
To test this hypothesis, we used the amino acid sequences of the 64 PE_PGRS proteins of strain H37Rv (NC_000962.2) for epitope prediction, using NetMHCpan and NetMHCIIpan (http://tools.immuneepitope.org/main/html/tcell_tools.html) (40, 41). For this analysis, we chose 15 HLA class I (7 HLA-A and 8 HLA-B) and 8 HLA class II alleles as representative of diverse human populations (see Table S5 in the supplemental material). HLA class II analysis predicted a mean of 52 high-affinity epitopes per PE_PGRS protein, ranging from 0 (PE_PGRS 40) to 170 epitopes (PE_PGRS 50), whereas HLA class I analysis predicted a mean of 18 high-affinity epitopes per protein. Overall, we identified 3,150 predicted CD4+ and 1,053 predicted CD8+ T cell epitopes in the 64 proteins. Analysis of the distribution of the predicted high-affinity HLA class II epitopes along the protein sequences revealed that 2,301 of the 3,150 (73%) predicted epitopes were located within the PE domains (Fig. 3A). Although the same pattern was observed for high-affinity CD8+ T cell epitopes, this result was less striking but was similar when class I epitopes with predicted intermediate affinity (50% inhibitory concentration [IC50] < 500 nM) were included (Fig. 3B). Alternative prediction algorithms, including the stabilized matrix method (SMM and SMM_align) (42–45), yielded similar results (see Fig. S3). Closer inspection of the PE domains revealed a cluster of epitopes between amino acids 50 to 85: this region harbored 70% of all predicted HLA class I and class II epitopes (Fig. 3). Notably, 32% of the predicted HLA class II epitopes localized to a region consisting of 9 amino acids at positions 76 to 84, with high conservation of phenylalanine, valine, and leucine at positions 76, 77, and 80.
FIG 3 .

MHC class I and class II epitopes are concentrated in the PE domain of PE_PGRS proteins. The amino acid sequences of all 64 annotated PE_PGRS were used for in silico epitope prediction. The graph represents the number of high binding affinity (IC50 < 50 nM; solid line) and intermediate binding affinity (IC50 < 500 nM; dashed line) epitopes for the selected MHC class II (A) and MHC class I (B) alleles, by amino acid position of the proteins. The lower part of each panel shows an expanded view of the PE domain containing a high density of predicted epitopes. The observed results deviate significantly from what would be predicted with a random distribution (P < 0.001; χ2 with Yates correction for continuity).
We verified the immunogenicity of the predicted epitopes in the PE domains (Fig. 4A), using cells from newly diagnosed sputum smear-positive, HIV-seronegative tuberculosis patients in the Gambia. Synthetic peptides representing consensus sequences from predicted epitopes in the PE domains of multiple PE_PGRS proteins were used to stimulate freshly obtained diluted whole blood samples followed by quantitation of secreted gamma interferon (IFN-γ). This revealed a close association between the density of the predicted epitopes at a given position and the amount of gamma interferon released (Fig. 4B; analysis of variance [ANOVA], P = 0.02).
FIG 4 .
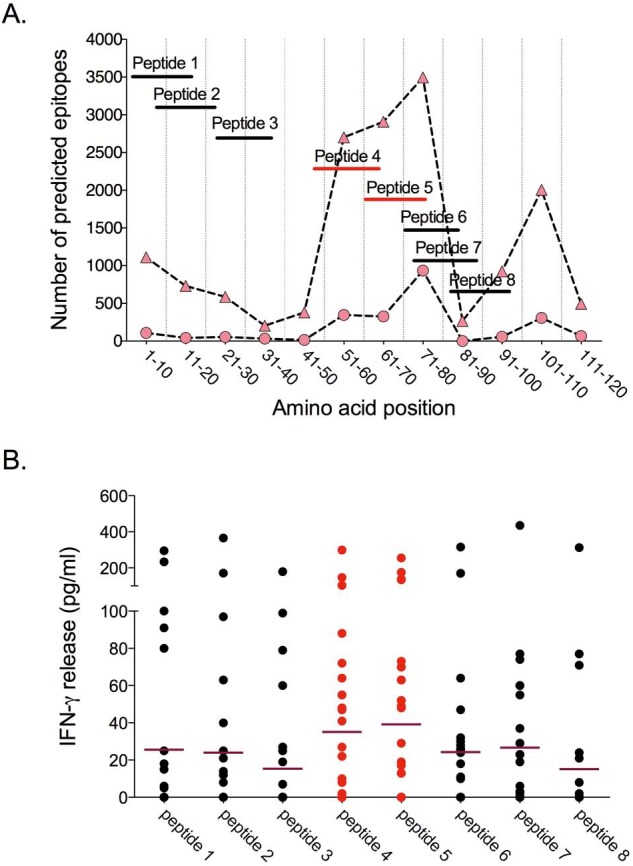
Antigenicity of peptides representing predicted CD4 T cell epitopes in the PE domain. Peptides were selected based on the frequency of the corresponding sequences in the PE domains of 64 PE_PGRS proteins and on the predicted binding affinity to common human HLA class II alleles. (A) Position of the candidate epitope peptides within the PE domain, compared with predicted T cell epitope frequencies in PE_PGRS proteins. The graph represents the number of predicted high binding affinity (IC50 < 50 nM; circles) and intermediate binding affinity (IC50 < 500 nM; triangles) HLA class II-restricted epitopes relative to the position within the PE domain of PE_PGRS proteins. (B) Responses (release of IFN-γ) to the peptides shown in panel A, in a diluted whole blood assay performed on fresh samples of newly diagnosed pulmonary tuberculosis patients in the Gambia. Each point represents the response of a single subject; the horizontal line represents the mean response for the group. Each value is the net concentration after subtraction of background determined with an unstimulated sample run in parallel. Responses induced by peptides 4 to 7 are the strongest compared to that of peptides 1, 2, 3, and 8 (peptides 6 and 7, P = 0.01; peptides 4 and 5, P < 0.0001 by nonparametric Friedman test). Peptides 4 and 5 (red lines) correspond to the region with the highest density of predicted epitopes.
Based on the distribution of predicted epitopes, we identified two groups of PE_PGRS proteins. One group (45 proteins) had no predicted class I or class II epitopes within the C-terminal PGRS domain. The second group (19 proteins) was characterized by one or more clusters of predicted class I and class II epitopes within the PGRS region or at the C terminus. The regions in which these epitopes were located did not contain the tandem repeats of Gly-Gly-Ala and Gly-Gly-Asn, which are the hallmarks of the PGRS domain (Fig. 5). The unique C-terminal domain of the two PE_unique domain proteins (PE_PGRS 62 and LipY) was similarly characterized by a large proportion of predicted epitopes. Thus, the classically defined Gly-Ala-rich PGRS domain was devoid of predicted epitopes (Fig. 5), suggesting that this domain is not recognized by T cells of individuals with the globally prevalent HLA class I and class II alleles considered here.
FIG 5 .
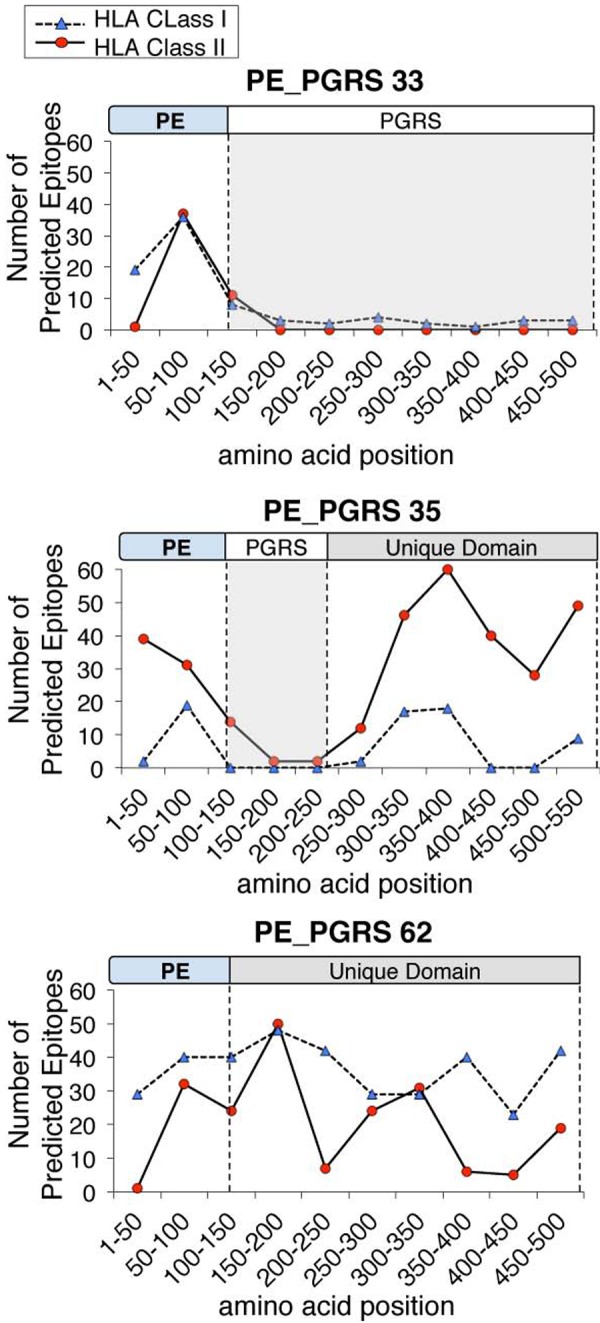
T cell epitopes are associated with specific domains of PE_PGRS proteins. Epitope profiles are shown for representatives of 3 variants of PE family members: a typical PE_PGRS protein (PE_PGRS 33), a PE_unique protein (PE_PGRS 62), and a PE_PGRS with a region of unique sequences located at the C terminus (PE_PGRS 35). Profiles for high affinity (IC50 < 50 nM) MHC class II epitopes and intermediate affinity (IC50 < 500 nM) MHC class I epitopes are shown.
Epitope prediction and genetic diversity.
To characterize the impact of sequence diversity of PE_PGRS proteins on HLA binding and T cell recognition, we analyzed the consequences of amino acid changes on epitope prediction in the three domains of the PE_PGRS proteins: the PE domain, the PGRS domain, and the C-terminal unique domain. The PE domain was highly conserved across all paralogs of the family, with an average identity and similarity of ~46% and ~65%, respectively. The PE domain was significantly more conserved than the rest of the PE_PGRS proteins: of 381 polymorphisms (SNPs and indels) in the 26 sequenced proteins (excluding Wag22), only 9.2% (12 synonymous SNPs [sSNPs], 19 nSNPs, 4 indels) occurred in the PE domain (χ2 with Yates correction for continuity = 13.9, P < 0.001). Consistent with this, nucleotide diversity (π) was significantly lower in the PE domain than in the PGRS domain (Wilcoxon signed-rank test, P < 0.05) (Fig. 6A), and the overall ratio of nonsynonymous to synonymous evolutionary changes (dN/dS ratio) of the PE domain was 0.6, indicating purifying selection acting on this domain (Fig. 6B). Finally, the nSNPs in the PE domain did not affect the predicted HLA class I and class II epitopes. The single exception was in PE_PGRS 26, where an nSNP affecting all strains from lineage 6 (West Africa) occurred within the conserved epitope hot spot in residues 76 to 84 and was associated with a marked change in the HLA class II prediction: conversion of Gln to His in position 78 changed the number of predicted epitopes from 27 to 42.
FIG 6 .
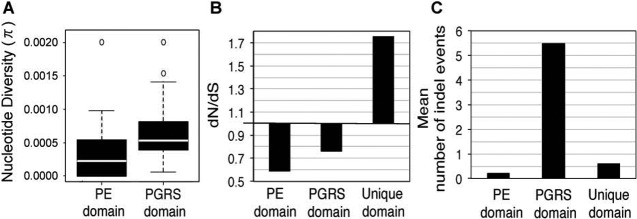
Sequence variations are unevenly distributed in the domains of pe_pgrs genes. (A) Nucleotide diversity by domain: median (horizontal line), interquartile range (box), minimum and maximum values (whiskers), and outliers (circles). pe_unique domains are not included, due to the small number analyzed. (B) dN/dS ratio by domain of the pe_pgrs genes and pe_unique genes. (C) Indel distribution by protein domain.
In contrast to the conserved PE domain, the PGRS domains, which contained few predicted T cell epitopes in the reference sequence, were highly polymorphic, with 70 sSNPs, 124 nSNPs, and 132 indels among the 24 proteins. However, none of the nSNPs or indels in these domains resulted in either loss or appearance of predicted CD4+ or CD8+ T cell epitopes. Therefore, the hypothesis that the high genetic diversity and large number of indels concentrated in the PGRS domains mediate evasion of T cell recognition is not supported by these results. The bias toward synonymous substitutions in the PGRS domains (dN/dS = 0.75; Fig. 6B) further argues against the notion that PGRS domains exhibit antigenic diversity. Although numerous in-frame indels were present in the PGRS domains (Fig. 6C), the amino acid sequences of variants harboring in-frame indels were similar to the reference sequence, suggesting these in-frame indels have a limited impact on gene function or antigenicity.
Analysis of the C-terminal unique domain present in PE_PGRS 6, 35, 39, and 62 and LipY revealed that this domain was under positive selection (dN/dS = 1.7) (Fig. 6B), and a significant proportion of the predicted epitopes were associated with this domain. In contrast to the PE domain, the frameshift indels and nSNPs occurring in this portion of the protein did affect the results of epitope predictions. For example, PE_PGRS 6, 35, and 39 were all characterized by a 1-bp indel that disrupted the entire C-terminal domain but maintained the integrity of the remainder of the protein. The insertion in PE_PGRS 6 occurred in 26 strains from lineages 1 and 6, implying that the C-terminal domain of this PE_PGRS is not essential in these lineages. PE_PGRS 62 and LipY, both PE_unique domain proteins, harbored 6 and 4 nSNPs, respectively, in their C-terminal unique domain. Epitope prediction analyses showed that each mutation altered either the predicted affinity or the number of HLA alleles potentially involved in the interaction, especially for predicted HLA class II-restricted epitopes. Overall, these results show that although human T cell recognition is clearly not the force driving the variation of the PE and PGRS domains, this may not be true for the C-terminal unique domain present in a subset of PE_PGRS proteins.
DISCUSSION
Our results extend previous findings that considerable sequence diversity exists within members of the pe_pgrs family (35, 36, 46, 47). We found that this was reflected by a significantly higher nucleotide diversity than that in the rest of the genome and by a high frequency of indels. In contrast to a previous study of five pe_pgrs genes (47), our analysis revealed that individual members of the pe_pgrs family differ widely in their nucleotide diversity and indel frequency. Intriguingly, we found that some genes with the lowest nucleotide diversity had the highest indel diversity (pe_pgrs 23 and 33 in Fig. 1A and C). Moreover, our data showed that the individual pe_pgrs genes had very different dN/dS values. A recent study used sequence data from five pe_pgrs genes and calculated an overall dN/dS ratio of 0.88, which was interpreted as evidence for limited selection pressure acting on these genes (47). However, our data suggest that the selection pressures acting on individual pe_pgrs genes differ, with some evolving neutrally, while others showed dN/dS consistent with purifying or diversifying selection.
In addition to the differences among the various pe_pgrs genes, we observed important differences between the PE and the PGRS domains. Specifically, the N-terminal PE domain in all of the 27 genes examined was conserved among strains, and most of the diversity localized to the PGRS and C-terminal unique domains. Indel diversity was also predominantly associated with the PGRS domains. However, most of these indels were in frame, suggesting that selection is acting to preserve the reading frame of most pe_pgrs genes. Taken together, our results suggest that pe_pgrs genes differ in function, that many of these functions are likely to be nonredundant, and that the overrepresentation of in-frame indels supports a role for natural selection.
One of the most widely studied pe_pgrs genes is pe_pgrs 33 (35, 48, 49). We found that this gene exhibited a low nucleotide diversity (π) and low dN/dS ratio but a large number of in-frame indels. Prior studies of pe_pgrs 33 sequence variants found that large deletions in this gene were associated with reduced induction of tumor necrosis factor alpha (TNF-α) (50), reduced patient clustering, and absence of lung cavitation (49). In our study, 30% of the strains contained large indels in pe_pgrs 33. Moreover, all strains from lineage 1 harbored a frameshift indel and a premature stop codon, resulting in a protein reduced in length at the C terminus by ~30%. These data support the notion that natural genetic variation in the MTBC, even in a single gene, may have a significant impact on clinical and epidemiological phenotypes (51).
While the findings noted above suggest a function for PE_PGRS 33 in modulating innate immune and inflammatory responses, our results indicate that the findings for one member of the pe_pgrs family are unlikely to be generalizable to the family as a whole. Despite a common molecular architecture, our analysis showed that pe_pgrs can be categorized into groups according to their genetic diversity or indel diversity and also revealed that while some members are evolutionarily conserved, others are clearly under diversifying selection. Identification of these groups suggests that further studies to understand the role of PE_PGRS proteins may benefit by considering PE_PGRS as a superfamily with multiple functions rather than as a unique group of proteins sharing a common role.
Since the proposed involvement of PE_PGRS proteins in antigenic variation is based on the highly polymorphic nature of their PGRS domains, and since prevailing evidence supports a dominant role for T cells in immunity to TB, we expected to find T cell epitopes concentrated in the PGRS domain. However, most predicted epitopes were located in the PE domain, and amino acid sequence variation did not affect the predicted T cell epitopes, which is consistent with our recent finding that the experimentally verified human T cell epitopes in MTBC are conserved (13). Analysis of the data available in the Immune Epitope Database (http://www.iedb.org; accessed 1 August 2013) reinforced this conclusion: of the currently known 1,649 M. tuberculosis epitopes that induce human T cell responses, 30 are encoded in pe_pgrs genes, and only three of these T cell epitopes are located in the PGRS domain. Finally, our experimental findings confirm that predicted T cell epitopes are targets for recognition by T cells of humans with pulmonary tuberculosis. Thus, the findings reported here indicate that T cell recognition is not the diversifying selection pressure acting on members of the pe_pgrs family and emphasize the need to identify the selection mechanisms that are responsible. Since we and others have found that the greatest variation is in the PGRS domains, and since the PGRS domains of at least some of the family members are targets for recognition by antibodies (52, 53), it is possible that the PGRS variants are selected by antibody recognition. However, the possibility of a role for antibody responses in protective immunity against TB has only recently been considered (54), and no data currently exist to indicate that antibody recognition of a PE_PGRS protein can contribute to control of tuberculosis or select for antigenic variants. In mice, Delogu and Brennan observed that DNA immunization with full-length pe_pgrs 33 promoted a nonprotective, B cell-skewed response, while vaccination with the PE domain alone elicited predominantly cell-mediated immunity and protection against challenge (55). In contrast, our finding that the predicted T cell epitopes in the PE_PGRS proteins are conserved, suggesting little evolutionary advantage to escaping recognition by human T cells, is consistent with two major possibilities that are not mutually exclusive. One is that M. tuberculosis uses mechanisms other than epitope sequence variation to evade elimination by T cell responses. The second is that epitope sequence conservation is driven by an evolutionary benefit to the bacteria by T cell recognition, such as by facilitating TB transmission. The former is supported by evidence for multiple mechanisms of M. tuberculosis evasion of cellular immune responses (56), while the latter is supported by the observation that CD4+ T cell-deficient HIV-infected individuals transmit TB less efficiently than do immunocompetent individuals (20).
In conclusion, although we show that pe_pgrs genes are overall more diverse than the remainder of the MTBC genome, we observed a wide range of genetic diversity across the paralogs of this unique family of genes. Our results strongly suggest that human T cell recognition is not the evolutionary force driving the sequence variation of these genes and compel the examination of alternative hypotheses regarding the selective forces acting on the pe_pgrs genes.
MATERIALS AND METHODS
Bacteria.
PE_PGRS sequences were determined in 94 strains chosen from a worldwide MTBC collection (see Table S2 in the supplemental material). Ethical approval for strains isolated in Nepal (prospectively collected) was obtained from the Nepal Health Research Council (NHRC), Kathmandu, Nepal, and the Ethics Committee of Basel, Switzerland (EKBB). Written informed consent was obtained for all Nepalese patients. All other MTBC strains were obtained from established reference collections (12, 16, 57). Bacterial strains were grown from single colonies, and genomic DNA was extracted using Qiagen kits.
We determined the main MTBC lineages by single nucleotide polymorphisms (SNPs) using multiplex real-time PCR with fluorescence-labeled probes (TaqMan, Applied Biosystems, USA) adopted from previous studies (58).
PE_PGRS sequencing.
Twenty-seven pe_pgrs genes were amplified in their entirety in 94 DNA samples by using primers anchoring conserved flanking regions (see Table S6); therefore, pe_pgrs flanked by other PE/PPE genes were excluded from analysis, to take advantage of conserved primer sites. As multiple sequencing reactions were necessary to obtain full-length gene sequences, additional sequencing primers were designed within each gene to achieve overlaps of at least 70 bp. Deletion of wag22 and pe_pgrs 2 in some strains was confirmed using independent sets of amplification primers and by analysis of whole genome sequences as generated in reference 14. Sequences were obtained at Macrogen Laboratories (Seoul, South Korea; Amsterdam, the Netherlands) and GENEWIZ, Inc. (New Jersey).
Sequences showing unusual patterns, such as convergent evolution mutations or out-of-frame indels, were verified by sequencing an independent PCR product from the same sample. A total of 128 samples for 26 loci were resequenced (402,127 bp). Only two mutations failed to be confirmed.
Sequence analysis.
Sequences were assembled using the Staden package (59) to obtain one sequence per gene and strain. For each pe_pgrs, sequences from all strains were aligned by using Clustal (60) and MAAFT (61) and verified manually.
Genetic variability and positive selection analyses were performed using DnaSP4 (62) and MEGA 4 (63). For the nucleotide diversity analysis, polymorphic sites were excluded from analysis if other strains contained sequences with large indels (larger than 25% of the gene) that affected the same region. Sequences with frameshift indels disrupting the ORF were also excluded. Genetic diversity parameters for all sequenced pe_pgrs (except wag22) are shown in Table S3 in the supplemental material. The Nei method was used to calculate the haplotype diversity and the nucleotide diversity (64). The dN/dS ratio was calculated by implementing the method of Nei and Gojobori (65), using the number of nonredundant sSNPs and nSNPs. Table S4 shows the parameters of the indel analysis for all sequenced pe_pgrs.
The average nucleotide diversity and indel diversity per site were calculated for each domain using the Nei method (64). In order to calculate the dN/dS ratio for each domain, we generated a concatenated alignment combining all individual domains from each gene. For each concatenate, we performed pairwise comparisons with the reference strain (H37Rv) to define synonymous and nonsynonymous substitutions implementing the method of Nei and Gojobori as described above. To group strains and lineages in our data set, a principal component analysis (PCA) was performed using BioNumerics software version 6.6 based on 370 polymorphisms in 89 non-pe_pgrs genes (12) for 45 strains included in our study (see Table S2). PCA distinguished three major groups: strains belonging to lineage 6 (Mycobacterium africanum), strains belonging to lineage 1, and strains belonging to the lineages 2, 3, or 4 (these three lineages have been referred to as evolutionarily “modern”) (12).
Epitope prediction.
Complete PE_PGRS amino acid sequences of MTBC strain H37Rv were retrieved from GenBank (http://www.ncbi.nlm.nih.gov/Genbank/). NetMHCpan 2.3 and NetMHCIIpan 2.2 were used for predicting CD8+ and CD4+ T cell epitopes in MTBC protein sequences, respectively (40, 41).
NetMHCpan and NetMHCIIpan servers are reported to be the most accurate prediction algorithms currently available based on the artificial neural network algorithm (66). The output of these predictions is in the form of a table, showing binding affinity of each possible putative epitope sequence with selected HLA alleles. Threshold cutoff values corresponding to IC50 values of <50 nM and <500 nM were used for both MHC class II and class I predictions. CD8+ T-cell peptide length was not included as a criterion in the analysis, and multiple allele/length pairs were submitted at a time.
HLA allele selection.
In order to represent the diverse human population, prediction analyses were performed using the most prevalent HLA-A, -B, and -DR alleles in representative populations as defined by the Allele Frequency Database (http://www.allelefrequencies.net/). The results of population coverage for the 10 geographical regions for which the allele frequencies are available are shown in Table S5 in the supplemental material.
Human T cell responses to predicted PE domain epitope peptides.
The subjects studied were newly diagnosed pulmonary tuberculosis patients enrolled in a prospective study that will be described in detail elsewhere. Briefly, after informed consent, HIV-seronegative adults with sputum smear-positive tuberculosis at the MRC Laboratories, the Gambia, donated blood for analysis in a diluted whole blood assay, as previously described (67, 68). Peptides were synthesized by EZ Biolabs and were added at a final concentration of 10 µg/ml to heparinized whole blood diluted with 9 volumes of RPMI 1640. Samples without stimulus and with phytohemagglutinin (PHA; 5 µg/ml) were used as negative and positive controls, respectively. After a 7-day incubation at 37°C in a humidified CO2 incubator, supernatants were removed and assayed for the concentration of gamma interferon (IFN-γ) by enzyme-linked immunosorbent assay (ELISA). Samples in which release of IFN-γ in response to PHA did not exceed the mean plus 2 standard deviations above the concentration in the unstimulated controls were excluded from analysis. The studies were reviewed and approved by the NY University Institutional Review Board and by the Gambia Government/Medical Research Council (MRC) Joint Ethics Committee.
Statistical analysis.
Wilcoxon signed-rank (Mann-Whitney) test was used to compare the genetic diversity of (i) pe_pgrs genes with the rest of the M. tuberculosis genes and (ii) the PE and PGRS domains of pe_pgrs, using Stata 10 (StataCorp LP, College Station, TX, USA). Epitope distribution analyses were performed by using the χ2 test with Yates correction for continuity to compare the observed and expected frequencies of T cell epitopes distributed in PE_PGRS proteins (df = 1). Partitioning of genetic variation among and within groups identified by PCA was tested by an analysis of molecular variance (AMOVA) using Arlequin version v3.1.5.3 (69). The numerical vectors provided from PCA were graphically represented, and the groupings were used to group haplotypes and compute the AMOVA. We used PE_PGRS sequence haplotypes as individuals, and we grouped haplotypes into groups according to results from PCA analysis. Significance tests were assessed using 1,023 permutations, and then Bonferroni adjustment was applied for multiple comparisons.
Results of interferon gamma release in response to each epitope peptide by all included subjects were compared to that of the unstimulated samples by nonparametric Friedman test, using Prism 6.0 (GraphPad, San Diego, CA).
Nucleotide sequence accession numbers.
Sequences have been deposited in GenBank and assigned accession numbers JX179300 through JX181656.
SUPPLEMENTAL MATERIAL
The frequency of in-frame and frameshift indels varies by pe_pgrs gene. Percentage of indels in each pe_pgrs gene that are in-frame (light gray) or frameshift (darker gray). The number of indel events in each category is indicated in the appropriate segment of the bar. pe_unique domain genes are indicated by an asterisk. The fraction of in-frame indels predicted if random is indicated by a dashed line. Download
Variation in intralineage and interlineage distribution of nSNPs and indels in individual pe_pgrs genes. For each of the 26 sequenced pe_pgrs, nSNPs (A) and indels (B) are shown for the lineage of the strain in which they were identified. For each nSNP (polymorphic site [PS]) or indel (ID), a color gradient shows the proportion of strains affected by each genetic change. Black, 0 strain; yellow, 1 strain; orange, >1 strain but less than the entire lineage; and red, the entire lineage. Variants found exclusively in the reference strain, H37Rv, are designated by Rv in the associated box. Indels 1 bp in length are designated by “1 bp” in the associated box. With the exception of one potential gene conversion event in two strains, we found no evidence of homoplasy, or convergent evolution, in different lineages involving SNPs. The only homoplasic SNPs were found in pe_pgrs 2, where two strains from lineage 4 (Euro-American) showed 3 nearly consecutive amino acid substitutions in the PGRS domain of PE_PGRS 2. The two affected strains show different regions of difference, indicating a gene conversion event of 12 bp rather than linked mutations occurring independently in the two different sublineages. Download
Domain distribution of intermediate affinity MHC class I and class II epitopes predicted by the alternative SMM and SMM_align methods. Graphs represent the number of predicted MHC class I and class II epitopes in all PE_PGRS, by amino acid position. Threshold value corresponding to an IC50 of <500 nM was used for the analysis. The following HLA alleles were used: SMM MHC class I predictions, HLA-A 01:01, 02:01, 11:01, 23:01, 24:02, 31:01 and HLA-B 07:02, 40:01, 40:02, 44:03, 51:01, 53:01; SMM_align MHC class II predictions, HLA-DRB1*03:01 and 15:01. The observed results deviate significantly (P < 0.001) from that expected by a random distribution. Download
Nucleotide and amino acid sequence characteristics of the pe_pgrs family.
Characteristics of bacterial isolates used in this study.
Genetic diversity parameters of the pe_pgrs genes sequenced in this study.
Indel diversity parameters of the pe_pgrs genes sequenced in this study.
The prevalent HLA genes representing human populations worldwide used for epitope prediction. From the dbMHC database (http://www.ncbi.nlm.nih.gov/projects/gv/mhc/).
Primer sequences.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01 AI090928 and HHSN266200700022C) and the Swiss National Science Foundation (PP0033-119205). R.C. was supported by the Potts Memorial Foundation and the Belgian American Educational Foundation. M.C. was supported by the Programa VALi+d.
We thank Thembela Huna, Stephanie Birrer, and Daphne Davis for technical assistance. We also thank Martin Ota for his contributions to the early stages of the project and the clinicians, subjects, and microbiology staff at the MRC Unit for their contributions to these studies.
Footnotes
Citation Copin R, Coscollá M, Seiffert SN, Bothamley G, Sutherland J, Mbayo G, Gagneux S, Ernst JD. 2014. Sequence diversity in the pe_pgrs genes of Mycobacterium tuberculosis is independent of human T cell recognition. mBio 5(1):e00960-13. doi:10.1128/mBio.00960-13.
REFERENCES
- 1. Deitsch KW, Lukehart SA, Stringer JR. 2009. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat. Rev. Microbiol. 7:493–503. 10.1038/nrmicro2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burton DR, Poignard P, Stanfield RL, Wilson IA. 2012. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 337:183–186. 10.1126/science.1225416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Virji M. 2009. Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat. Rev. Microbiol. 7:274–286. 10.1038/nrmicro2097 [DOI] [PubMed] [Google Scholar]
- 4. Freitas-Junior LH, Hernandez-Rivas R, Ralph SA, Montiel-Condado D, Ruvalcaba-Salazar OK, Rojas-Meza AP, Mâncio-Silva L, Leal-Silvestre RJ, Gontijo AM, Shorte S, Scherf A. 2005. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 121:25–36. 10.1016/j.cell.2005.01.037 [DOI] [PubMed] [Google Scholar]
- 5. Hill AV, Allsopp CE, Kwiatkowski D, Anstey NM, Twumasi P, Rowe PA, Bennett S, Brewster D, McMichael AJ, Greenwood BM. 1991. Common West African HLA antigens are associated with protection from severe malaria. Nature 352:595–1195. 10.1038/352595a0 [DOI] [PubMed] [Google Scholar]
- 6. Howard JC. 1991. Immunology. Disease and evolution. Nature 352:565–572. 10.1038/352565a0 [DOI] [PubMed] [Google Scholar]
- 7. Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, Bentley SD. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434. 10.1126/science.1198545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, Kuse N, Oka S, Duda A, Prendergast A, Crawford H, Leslie A, Brumme Z, Brumme C, Allen T, Brander C, Kaslow R, Tang J, Hunter E, Allen S, Mulenga J, Branch S, Roach T, John M, Mallal S, Ogwu A, Shapiro R, Prado JG, Fidler S, Weber J, Pybus OG, Klenerman P, Ndung'u T, Phillips R, Heckerman D, Harrigan PR, Walker BD, Takiguchi M, Goulder P. 2009. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458:641–645. 10.1038/nature07746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. 10.1038/31159 [DOI] [PubMed] [Google Scholar]
- 10. Achtman M. 2008. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu. Rev. Microbiol. 62:53–123. 10.1146/annurev.micro.62.081307.162832 [DOI] [PubMed] [Google Scholar]
- 11. Comas I, Homolka S, Niemann S, Gagneux S. 2009. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS One 4:e7815. 10.1371/journal.pone.0007815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, Roach JC, Kremer K, Petrov DA, Feldman MW, Gagneux S. 2008. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 6(12):e311. 10.1371/journal.pbio.0060311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S. 2010. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 42:498–503. 10.1038/ng.590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, Parkhill J, Malla B, Berg S, Thwaites G, Yeboah-Manu D, Bothamley G, Mei J, Wei L, Bentley S, Harris SR, Niemann S, Diel R, Aseffa A, Gao Q, Young D, Gagneux S. 2013. Out-of-Africa migration and neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45:1176–1182. 10.1038/ng.2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Firdessa R, Berg S, Hailu E, Schelling E, Gumi B, Erenso G, Gadisa E, Kiros T, Habtamu M, Hussein J, Zinsstag J, Robertson BD, Ameni G, Lohan AJ, Loftus B, Comas I, Gagneux S, Tschopp R, Yamuah L, Hewinson G, Gordon SV, Young DB, Aseffa A. 2013. Mycobacterial lineages causing pulmonary and extrapulmonary tuberculosis, Ethiopia. Emerg. Infect. Dis. 19:460–463. 10.3201/eid1903.120256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, Nicol M, Niemann S, Kremer K, Gutierrez MC, Hilty M, Hopewell PC, Small PM. 2006. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 103:2869–2942. 10.1073/pnas.0511240103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hirsh AE, Tsolaki AG, DeRiemer K, Feldman MW, Small PM. 2004. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc. Natl. Acad. Sci. U. S. A. 101:4871–4877. 10.1073/pnas.0305627101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reed MB, Pichler VK, McIntosh F, Mattia A, Fallow A, Masala S, Domenech P, Zwerling A, Thibert L, Menzies D, Schwartzman K, Behr MA. 2009. Major Mycobacterium tuberculosis lineages associate with patient country of origin. J. Clin. Microbiol. 47:1119–1147. 10.1128/JCM.02142-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. WHO 2011. Global tuberculosis control. World Health Organization, Geneva, Switzerland [Google Scholar]
- 20. Kwan CK, Ernst JD. 2011. HIV and tuberculosis: a deadly human syndemic. Clin. Microbiol. Rev. 24:351–427. 10.1128/CMR.00042-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin PL, Rodgers M, Smith L, Bigbee M, Myers A, Bigbee C, Chiosea I, Capuano SV, Fuhrman C, Klein E, Flynn JL. 2009. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect. Immun. 77:4631–4673. 10.1128/IAI.00592-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. North RJ, Jung Y-J. 2004. Immunity to tuberculosis. Annu. Rev. Immunol. 22:599–1222. 10.1146/annurev.immunol.22.012703.104635 [DOI] [PubMed] [Google Scholar]
- 23. Lancioni C, Nyendak M, Kiguli S, Zalwango S, Mori T, Mayanja-Kizza H, Balyejusa S, Null M, Baseke J, Mulindwa D, Byrd L, Swarbrick G, Scott C, Johnson D, Malone L, Mudido-Musoke P, Boom W, Lewinsohn D, Lewinsohn DA, Tuberculosis Research Unit 2012. CD8+ T cells provide an immunologic signature of tuberculosis in young children. Am. J. Respir. Crit. Care Med. 185:206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen C, Huang D, Wang R, Shen L, Zeng G, Yao S, Shen Y, Halliday L, Fortman J, McAllister M, Estep J, Hunt R, Vasconcelos D, Du G, Porcelli S, Larsen M, Jacobs W, Haynes B, Letvin N, Chen Z. 2009. A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PLoS Pathog. 5:e1000392. 10.1371/journal.ppat.1000392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. 2001. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 193:271–351. 10.1084/jem.193.3.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woodworth JS, Shin D, Volman M, Nunes-Alves C, Fortune SM, Behar SM. 2011. Mycobacterium tuberculosis directs immunofocusing of CD8+ T cell responses despite vaccination. J. Immunol. 186:1627–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bold TD, Banaei N, Wolf AJ, Ernst JD. 2011. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS Pathog. 7:e42716. 10.1371/journal.ppat.1002063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cooper AM. 2009. Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 27:393–815. 10.1146/annurev.immunol.021908.132703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tufariello JM, Chan J, Flynn JL. 2003. Latent tuberculosis: mechanisms of host and bacillus that contribute to persistent infection. Lancet Infect. Dis. 3:578–668. 10.1016/S1473-3099(03)00741-2 [DOI] [PubMed] [Google Scholar]
- 30. Winslow GM, Cooper A, Reiley W, Chatterjee M, Woodland DL. 2008. Early T-cell responses in tuberculosis immunity. Immunol. Rev. 225:284–383. 10.1111/j.1600-065X.2008.00693.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Young D, Hussell T, Dougan G. 2002. Chronic bacterial infections: living with unwanted guests. Nat. Immunol. 3:1026–1058. 10.1038/ni1102-1026 [DOI] [PubMed] [Google Scholar]
- 32. Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. 2006. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol. Biol. 6:95. 10.1186/1471-2148-6-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. 2006. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 103:8060–8065. 10.1073/pnas.0602606103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delogu G, Cole ST, Brosch R. 2008. The PE and PPE protein families of Mtb, p 131–150 In Kaufmann SH, Rubin E. (ed), Handbook of tuberculosis, vol 1 Wiley -VCH Verlag GmbH & Co., KGaA, Weinheim, Germany [Google Scholar]
- 35. Talarico S, Cave MD, Marrs CF, Foxman B, Zhang L, Yang Z. 2005. Variation of the Mycobacterium tuberculosis PE_PGRS 33 gene among clinical isolates. J. Clin. Microbiol. 43:4954–4960. 10.1128/JCM.43.10.4954-4960.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Talarico S, Zhang L, Marrs CF, Foxman B, Cave MD, Brennan MJ, Yang Z. 2008. Mycobacterium tuberculosis PE_PGRS16 and PE_PGRS26 genetic polymorphism among clinical isolates. Tuberculosis 88:283–294. 10.1016/j.tube.2008.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gagneux S, Brennan M. 2010. Strain and antigen variation in Mycobacterium tuberculosis: implications for the development of new tools for tuberculosis. In Acosta A, Sarmiento M. (ed), The art and science of tuberculosis vaccine development. Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 38. Podlaha O, Zhang J. 2003. Positive selection on protein-length in the evolution of a primate sperm ion channel. Proc. Natl. Acad. Sci. U. S. A. 100:12241–12247. 10.1073/pnas.2033555100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ripley LS. 1990. Frameshift mutation: determinants of specificity. Annu. Rev. Genet. 24:189–402. 10.1146/annurev.ge.24.120190.001201 [DOI] [PubMed] [Google Scholar]
- 40. Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. 2009. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61:1–14. 10.1007/s00251-008-0341-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nielsen M, Justesen S, Lund O, Lundegaard C, Buus S. 2010. NetMHCIIpan-2.0—improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res. 6:9. 10.1186/1745-7580-6-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moutaftsi M, Peters B, Pasquetto V, Tscharke DC, Sidney J, Bui H.H., Grey H, Sette A. 2006. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat. Biotechnol. 24:817–826. 10.1038/nbt1215 [DOI] [PubMed] [Google Scholar]
- 43. Nielsen M, Lundegaard C, Lund O. 2007. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics 8:238. 10.1186/1471-2105-8-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peters B, Sette A. 2005. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6:132. 10.1186/1471-2105-6-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. 2008. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 4:e1000048. 10.1371/journal.pcbi.1000048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karboul A, Gey van Pittius NC, Namouchi A, Vincent V, Sola C, Rastogi N, Suffys P, Fabre M, Cataldi A, Huard RC, Kurepina N, Kreiswirth B, Ho JL, Gutierrez MC, Mardassi H. 2006. Insights into the evolutionary history of tubercle bacilli as disclosed by genetic rearrangements within a PE_PGRS duplicated gene pair. BMC Evol. Biol. 6:107. 10.1186/1471-2148-6-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McEvoy CR, Cloete R, Müller B, Schürch AC, van Helden PD, Gagneux S, Warren RM, Gey van Pittius NC. 2012. Comparative analysis of mycobacterium tuberculosis pe and ppe genes reveals high sequence variation and an apparent absence of selective constraints. PLoS One 7:e30593. 10.1371/journal.pone.0030593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cadieux N, Parra M, Cohen H, Maric D, Morris SL, Brennan MJ. 2011. Induction of cell death after localization to the host cell mitochondria by the Mycobacterium tuberculosis PE_PGRS33 protein. Microbiology 157:793–1597. 10.1099/mic.0.041996-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Talarico S, Cave MD, Foxman B, Marrs CF, Zhang L, Bates JH, Yang Z. 2007. Association of Mycobacterium tuberculosis PE PGRS33 polymorphism with clinical and epidemiological characteristics. Tuberculosis 87:338–384. 10.1016/j.tube.2007.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Basu S, Pathak SK, Banerjee A, Pathak S, Bhattacharyya A, Yang Z, Talarico S, Kundu M, Basu J. 2007. Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-alpha. J. Biol. Chem. 282:1039–1089. 10.1074/jbc.M604379200 [DOI] [PubMed] [Google Scholar]
- 51. Coscolla M, Gagneux S. 2010. Does M. tuberculosis genomic diversity explain disease diversity? Drug Discov. Today Dis. Mech. 7:e43–e59. 10.1016/j.ddmod.2010.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Banu S, Honoré N, Saint-Joanis B, Philpott D, Prévost MC, Cole ST. 2002. Are the PE-PGRS proteins of Mycobacterium tuberculosis variable surface antigens? Mol. Microbiol. 44:9–28. 10.1046/j.1365-2958.2002.02813.x [DOI] [PubMed] [Google Scholar]
- 53. Singh H, Raghava GP. 2001. ProPred: prediction of HLA-DR binding sites. Bioinformatics 17:1236–1237. 10.1093/bioinformatics/17.12.1236 [DOI] [PubMed] [Google Scholar]
- 54. Achkar JM, Casadevall A. 2013. Antibody-mediated immunity against tuberculosis: implications for vaccine development. Cell Host Microbe 13:250–262. 10.1016/j.chom.2013.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Delogu G, Brennan MJ. 2001. Comparative immune response to PE and PE_PGRS antigens of Mycobacterium tuberculosis. Infect. Immun. 69:5606–5617. 10.1128/IAI.69.9.5606-5611.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ernst JD. 2012. The immunological life cycle of tuberculosis. Nat. Rev. Immunol. 12:581–591. 10.1038/nri3259 [DOI] [PubMed] [Google Scholar]
- 57. Wirth T, Hildebrand F, Allix-Béguec C, Wölbeling F, Kubica T, Kremer K, van Soolingen D, Rüsch-Gerdes S, Locht C, Brisse S, Meyer A, Supply P, Niemann S. 2008. Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 4(9):e1000160. 10.1371/journal.ppat.1000160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stucki D, Malla B, Hostettler S, Huna T, Feldmann J, Yeboah-Manu D, Borrell S, Fenner L, Comas I, Coscollà M, Gagneux S. 2012. Two new rapid SNP-typing methods for classifying Mycobacterium tuberculosis complex into the main phylogenetic lineages. PLoS One 7:e41253. 10.1371/journal.pone.0041253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Staden R. 1996. The Staden sequence analysis package. Mol. Biotechnol. 5:233–274. 10.1007/BF02900361 [DOI] [PubMed] [Google Scholar]
- 60. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4958. 10.1093/nar/25.24.4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Katoh K, Kuma K, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33:511–519. 10.1093/nar/gki198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rozas J, Sánchez-nchez-DelBarrio JC, Messeguer X, Rozas R. 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2503 [DOI] [PubMed] [Google Scholar]
- 63. Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1605. 10.1093/molbev/msm092 [DOI] [PubMed] [Google Scholar]
- 64. Nei M. 1987. Molecular evolutionary genetics. Columbia University Press, New York, NY. [Google Scholar]
- 65. Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3:418–426 [DOI] [PubMed] [Google Scholar]
- 66. Chaves FA, Lee AH, Nayak JL, Richards KA, Sant AJ. 2012. The utility and limitations of current Web-available algorithms to predict peptides recognized by CD4 T cells in response to pathogen infection. J. Immunol. 188:4235–4283. 10.4049/jimmunol.1103640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Black GF, Fine PEM, Warndorff DK, Floyd S, Weir RE, Blackwell JM, Bliss L, Sichali L, Mwaungulu L, Chaguluka S, Jarman E, Ngwira B, Dockrell HM. 2001. Relationship between IFN-gamma and skin test responsiveness to Mycobacterium tuberculosis PPD in healthy, non-BCG-vaccinated young adults in Northern Malawi. Int. J. Tuberc. Lung Dis. 5:664–672 [PubMed] [Google Scholar]
- 68. Black GF, Thiel BA, Ota MO, Parida SK, Adegbola R, Boom WH, Dockrell HM, Franken KL, Friggen AH, Hill PC, Klein MR, Lalor MK, Mayanja H, Schoolnik G, Stanley K, Weldingh K, Kaufmann SH, Walzl G, Ottenhoff TH, Biomarkers GCGH, for TB Consortium 2009. Immunogenicity of novel DosR regulon-encoded candidate antigens of Mycobacterium tuberculosis in three high-burden populations in Africa. Clin. Vaccine Immunol. 16:1203–1212. 10.1128/CVI.00111-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Excoffier L, Laval G, Schneider S. 2005. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol. Bioinform. Online 1:47–50 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The frequency of in-frame and frameshift indels varies by pe_pgrs gene. Percentage of indels in each pe_pgrs gene that are in-frame (light gray) or frameshift (darker gray). The number of indel events in each category is indicated in the appropriate segment of the bar. pe_unique domain genes are indicated by an asterisk. The fraction of in-frame indels predicted if random is indicated by a dashed line. Download
Variation in intralineage and interlineage distribution of nSNPs and indels in individual pe_pgrs genes. For each of the 26 sequenced pe_pgrs, nSNPs (A) and indels (B) are shown for the lineage of the strain in which they were identified. For each nSNP (polymorphic site [PS]) or indel (ID), a color gradient shows the proportion of strains affected by each genetic change. Black, 0 strain; yellow, 1 strain; orange, >1 strain but less than the entire lineage; and red, the entire lineage. Variants found exclusively in the reference strain, H37Rv, are designated by Rv in the associated box. Indels 1 bp in length are designated by “1 bp” in the associated box. With the exception of one potential gene conversion event in two strains, we found no evidence of homoplasy, or convergent evolution, in different lineages involving SNPs. The only homoplasic SNPs were found in pe_pgrs 2, where two strains from lineage 4 (Euro-American) showed 3 nearly consecutive amino acid substitutions in the PGRS domain of PE_PGRS 2. The two affected strains show different regions of difference, indicating a gene conversion event of 12 bp rather than linked mutations occurring independently in the two different sublineages. Download
Domain distribution of intermediate affinity MHC class I and class II epitopes predicted by the alternative SMM and SMM_align methods. Graphs represent the number of predicted MHC class I and class II epitopes in all PE_PGRS, by amino acid position. Threshold value corresponding to an IC50 of <500 nM was used for the analysis. The following HLA alleles were used: SMM MHC class I predictions, HLA-A 01:01, 02:01, 11:01, 23:01, 24:02, 31:01 and HLA-B 07:02, 40:01, 40:02, 44:03, 51:01, 53:01; SMM_align MHC class II predictions, HLA-DRB1*03:01 and 15:01. The observed results deviate significantly (P < 0.001) from that expected by a random distribution. Download
Nucleotide and amino acid sequence characteristics of the pe_pgrs family.
Characteristics of bacterial isolates used in this study.
Genetic diversity parameters of the pe_pgrs genes sequenced in this study.
Indel diversity parameters of the pe_pgrs genes sequenced in this study.
The prevalent HLA genes representing human populations worldwide used for epitope prediction. From the dbMHC database (http://www.ncbi.nlm.nih.gov/projects/gv/mhc/).
Primer sequences.