

ABSTRACT
The twin-arginine translocation (Tat) system, needed to transport folded proteins across biological membranes, has not been characterized in the gastric pathogen Helicobacter pylori. Analysis of all H. pylori genome sequences available thus far reveals the presence of single copies of tatA, tatB, and tatC needed for the synthesis of a fully functional Tat system. Based on the presence of the twin-arginine hallmark in their signal sequence, only four H. pylori proteins appear to be Tat dependent: hydrogenase (HydA), catalase-associated protein (KapA), biotin sulfoxide reductase (BisC), and the ubiquinol cytochrome oxidoreductase Rieske protein (FbcF). In the present study, targeted mutations were aimed at tatA, tatB, tatC, or queA (downstream gene control). While double homologous recombination mutations in tatB and queA were easily obtained, attempts at disrupting tatA proved unsuccessful, while deletion of tatC led to partial mutants following single homologous recombination, with cells retaining a chromosomal copy of tatC. Double homologous recombination tatC mutants were obtained only when a plasmid-borne, isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible copy of tatC was introduced prior to transformation. These conditional tatC mutants could grow only in the presence of IPTG, suggesting that tatC is essential in H. pylori. tatB and tatC mutants had lower hydrogenase and catalase activities than the wild-type strain did, and the ability of tatC mutants to colonize mouse stomachs was severely affected compared to the wild type. Chromosomal complementation of tatC mutants restored hydrogenase and catalase activities to wild-type levels, and additional expression of tatC in wild-type cells resulted in elevated Tat-dependent enzyme activities. Unexpectedly, the tat strains had cell envelope defects.
IMPORTANCE
This work reports the first characterization of the twin-arginine translocation (Tat) system in the gastric pathogen Helicobacter pylori. While tatB mutants were easily obtained, only single-crossover partial tatC mutants or conditional tatC mutants could be generated, indicating that tatC is essential in H. pylori, a surprising finding given the fact that only four proteins are predicted to be translocated by the Tat system in this bacterium. The levels of activity of hydrogenase and catalase, two of the predicted Tat-dependent enzymes, were affected in these mutants. In addition, all tat mutants displayed cell envelope defects, and tatC mutants were deficient in mouse colonization.
INTRODUCTION
The twin-arginine translocation (Tat) system is needed for protein export across the cytoplasmic membranes of bacteria and archaea, as well as for protein import into the thylakoids of chloroplasts. The key feature of the Tat pathway is its ability to transport folded proteins across biological membranes, while another translocation system, the Sec system, can transport only unfolded, nascent proteins that fold after they cross the membrane (1). The Tat system enables cofactors such as flavins or iron-sulfur clusters to be retained during transit across the membrane. The target proteins are often predicted to perform redox functions; therefore, such cofactor stability is crucial to translocated enzyme activity. In addition, some Tat-transported proteins are involved in metabolism, metal acquisition, or cell envelope maintenance (2, 3). Precursor proteins that are translocated through the Tat pathway contain a conserved, distinctive (S/T)-R-R-X-F-L-K motif, in which X can be any polar amino acid and the consecutive arginine residues are almost invariant (4). The minimal set of components required for Tat translocation in Escherichia coli, the most extensively studied organism, consists of three integral membrane proteins: TatA, TatB, and TatC (5). Two other genes designated tatD and tatE can also be found in bacteria such as E. coli. While tatD has no apparent function in Tat-dependent protein transport (6), tatE is a cryptic gene duplication of tatA, and the proteins encoded by these genes are functionally interchangeable (7). In other bacteria, the TatB component does not seem to be essential for export, as some genomes (Staphylococcus aureus for instance) carry genes that encode only a single TatA and TatC (8).
The importance of the Tat system varies among microorganisms. It has been shown to be required for virulence in several animal, human, or plant pathogens, including Salmonella enterica serovar Enteritidis (9), Yersinia pseudotuberculosis (10), Vibrio cholerae (11), Dickeya dadantii 3937 (12), or Campylobacter jejuni (13). The last pathogen is of significance for the present study, because C. jejuni and Helicobacter pylori are closely related microorganisms that belong to the same group of Epsilonproteobacteria. While the Tat system is dispensable in most microorganisms characterized so far (including C. jejuni), it has been shown to be essential in only a few bacterial or archaeal species, including Sinorhizobium meliloti (14), Bdellovibrio bacteriovorus (15), Mycobacterium tuberculosis (16), and the halophilic archaea Halobacterium salinarum and Haloferax volcanii (17, 18).
H. pylori, the causative agent of peptic ulcers, colonizes the gastric epithelium (19) in about 50% of the world’s population (20); therefore, it is probably the most successful pathogenic bacterium in the world. So far, very little is known about the Tat system in H. pylori. Analysis of all H. pylori genomes available thus far suggests that there is a single copy of tatA, tatB, and tatC genes. Both tatB and tatC (hp1060 and hp1061, respectively, in sequenced strain 26695 [21]) are part of the same six-gene operon, while tatA (hp0320) is located elsewhere on the chromosome and is part of a five-gene polycistronic unit (22). Based on a study by Sargent and coworkers who successfully complemented an E. coli tatA mutant with a plasmid expressing H. pylori tatA (23), H. pylori tatA appears to be functional, at least in E. coli. In the present study, we strived to determine the role of the Tat pathway in H. pylori and its importance for survival and pathogenesis by generating a set of mutations in tatB and tatC genes in various H. pylori parental strains. Our results reveal the essentiality of TatC, but not TatB, for viability of this gastric pathogen and an intriguing and unexpected role of TatC in cell envelope defects and host cell colonization.
RESULTS
Putative components and predicted targets of the Tat pathway in H. pylori.
Analysis of several H. pylori genome sequences, including those of H. pylori strains 26695 (21), J99 (24), HPAG-1 (25), G27 (26), 98-10 (27), and B128 (27), reveals the presence of one copy of tatA, tatB, and tatC, the minimum set of genes required for a functional Tat translocase in Gram-negative bacteria (5, 28). For the well-characterized E. coli model system, tatA, tatB, and tatC genes are part of one unique tatABC operon; however, in H. pylori, the tatA gene is located on another locus (hp0320 for strain 26695) unrelated to tatBC (hp1060-hp1061 for strain 26695) (see Fig. S1 in the supplemental material). Another feature of the H. pylori Tat pathway is the absence of a tatE ortholog, which is present in E. coli. Similarities and differences between E. coli and H. pylori Tat systems have been previously highlighted by the fact that H. pylori tatA could functionally complement an E. coli tatA mutant, whereas H. pylori tatB cannot complement an E. coli tatB mutant (complementation of E. coli tatC by H. pylori tatC was not tested) (23). These findings are supported by comparison analysis of H. pylori and E. coli Tat protein sequences: TatA homologs share 46% identity and 63% similarity, while TatB homologs share only 27% identity and 46% similarity; TatC homologs display 33% identity and 54% similarity.
Compared to most bacteria, H. pylori appears to have a surprisingly limited list of putative Tat targets (Table 1). Indeed, a search based on the presence of the conserved (S/T)-R-R-X-F-L-K motif (X is any polar amino acid) (the hallmark of the Tat system) in putative signal sequences using three different prediction programs—PRED-TAT (29), TATFIND (30), and TatP (31)—suggests that there are only four proteins believed to depend upon Tat for their translocation in the gastric pathogen: the catalase accessory protein KapA; the hydrogenase small-subunit protein HydA; a putative biotin sulfoxide reductase, BisC; and the cytochrome oxidase Rieske subunit protein FbcF. Only two of these (KapA and FbcF) are predicted to be Tat-dependent proteins by all Tat prediction programs.
TABLE 1 .
Predicted Tat-transported proteins in H. pylori
| Proteina | N-terminal sequenceb | Description | Protein predicted by the following Tat signal prediction programc: |
||
|---|---|---|---|---|---|
| PRED-TAT | TATFIND | TatP | |||
| HP0407 | MSISRRSILTKIPIALASANVLKA | Biotin sulfoxide reductase (BisC) | Yes | No | No |
| HP0631 | MFYDEKKTYQKIEERLDIVRSFNAHNEHKNLQDEFKGAGISRRDLLKWAGMMSTALALPASFAPLTLKA | Hydrogenase small subunit (HydA) | Yes | Yes | No |
| HP0874 | MKRRDFIKTTTLGATGAVLGAQILQA | Catalase-associated protein (KapA) | Yes | Yes | Yes |
| HP1540 | MADIQRRDFLGMSLASVTAIGAIAASLVAMKKTWDPLPSVVSA | Ubiquinol cytochrome c oxidase, Rieske (FbcF) | Yes | Yes | Yes |
HP numbers refer to H. pylori strain 26695 (21).
The signal sequence with the twin-arginine motif (the two arginines shown in boldface type) [consensus sequence (S/T)RRXFLK] is shown up to the cleavage site predicted by PRED-TAT (29). Conserved amino acids found in the Tat consensus sequence are underlined.
Prediction programs used in this study are PRED-TAT (29), TATFIND (30), and TatP (31). The proteins that were predicted by the programs are shown in boldface type.
Construction of tatC, ΔtatC, and ΔtatA mutants.
Our initial attempt to inactivate the Tat system in H. pylori consisted of transforming several wild-type (WT) strains (26695, 43504, and SS1) with a suicide plasmid (pSLB129) that contained the tatC gene (hp1061) disrupted with a kanamycin resistance cassette (aphA3). Multiple attempts were mostly unsuccessful and led to only one kanamycin-resistant mutant (SLB1066), generated from strain 26695. This mutant was analyzed further but with caution because of the possibility of compensatory mutations. PCR analysis of this unique clone revealed a single homologous recombination (SHR) with (i) the entire plasmid inserted at the tatC locus, as detected by PCR using primers specific for the bla plasmid marker (data not shown), and (ii) the presence of the disrupted tatC::aphA3 construct and an intact copy of tatC within the chromosome (see Fig. S2 in the supplemental material).
Given the possibility that a truncated TatC polypeptide (still synthesized from the tatC::aphA3 construct) would be detrimental to H. pylori, we constructed an alternative suicide plasmid in which tatC was almost completely deleted (pSLB137 [see Table S1 and Fig. S1 in the supplemental material]). Transformation of the three H. pylori strains described above either with pSLB137 or with a purified PCR product containing the same (ΔtatC::aphA3) sequence yielded dozens of Kanr transformants, but only in strain 43504. PCR analysis revealed that none of the mutants was the result of double homologous recombination (DHR); instead, they all retained an intact copy of tatC while integrating ΔtatC::aphA3 within their chromosome (Fig. S2). To increase our chances of obtaining DHR-tatC mutants, the aphA3 marker was replaced by a chloramphenicol (Cm) acetyltransferase (cat) marker, previously shown to be a better tool to generate mutations in H. pylori, according to Gorrell et al. (32). Transformation with plasmids or PCR products containing ΔtatC::cat yielded Cmr clones; however, once again, PCR analysis revealed that they resulted from SHR (data not shown). Even though all attempts at generating tatC and ΔtatC mutants led to SHR, we reasoned that recombination within the tatBC locus or in its vicinity might still yield useful strains (i.e., affecting the Tat system). These partial mutants (from here on referred to as “SHR-tat mutants”) were therefore analyzed further by (i) looking at Tat-dependent enzyme activities (hydrogenase and catalase) and (ii) complementing them with a chromosomal copy of tatC (see below). Finally, attempts at constructing ΔtatA mutants by introducing PCR products containing ΔtatA::cat in several parental strains were unsuccessful, suggesting that tatA is an essential gene in H. pylori.
Unlike tatA or tatC, the tatB gene can be inactivated by double homologous recombination.
Since disruption of tatA or tatC proved to be a challenge, we targeted another component of the Tat machinery, TatB. The tatB gene (hp1060) was cloned into two different vectors, and a cat cassette was inserted into two unique restriction sites naturally present within tatB (see Fig. S1 in the supplemental material). Transformation of wild-type H. pylori with both plasmids yielded dozens of Cmr clones, and PCR analysis of the mutants’ genomes revealed that recombination by double crossover had occurred in each case (Fig. S2), in contrast with what had been observed for SHR-tatC or SHR-ΔtatC mutants (Table S3). This result suggests that, unlike tatA or tatC, tatB can be successfully mutagenized. Therefore, tatB does not appear to be essential in H. pylori.
Hydrogenase and catalase activities are affected in tatB mutants, SHR-ΔtatC mutants, and merodiploid cells.
Hydrogenase and catalase are among the proteins whose location, and as a consequence, activity, is expected to be affected if the Tat machinery is impaired in H. pylori. Indeed, H. pylori possesses only one (hydrogen uptake) [Ni-Fe] hydrogenase, previously shown to be membrane-bound (33), and there is a twin-arginine consensus in the sequence of the small-subunit HydA (Table 1). Catalase (KatA, HP0875) does not have a signal sequence; however, the catalase-associated protein (KapA, HP0874) possesses one signal sequence that includes a Tat motif (Table 1), and it is therefore hypothesized that KatA relies on KapA to be translocated on the periplasmic side of the membrane through a “hitchhiking” mechanism (34). This hypothesis is strengthened by the fact that KatA and KapA have been shown to interact (35) and that catalase activity is reduced in periplasmic fractions of kapA mutants (34). Therefore, whole-cell hydrogenase and catalase activities were determined on wild-type and various mutant strains (Fig. 1). While hydrogenase and catalase activities were only slightly decreased in tatB (SLB1085) mutants (approximately 80% of the level in the wild type), they were significantly reduced in SHR-ΔtatC (SLB1049) mutants. The latter suggests that the Tat machinery is indeed impaired in single-crossover ΔtatC mutants. By inserting a copy of tatC in an unrelated region of the chromosome of SHR-ΔtatC mutants, we generated a strain (SLB1093) in which hydrogenase and catalase activities were partially and fully restored, respectively (Fig. 1). Expression of the same PureA-tatC construct in wild-type cells (merodiploid cells, strain SLB1087) led to higher hydrogenase and catalase activities than those measured in the wild type (Fig. 1).
FIG 1 .
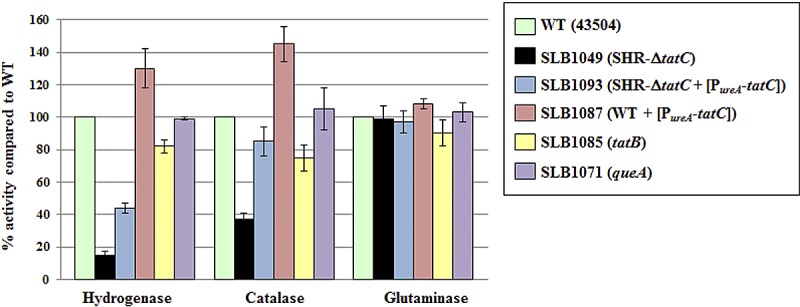
Hydrogenase, catalase, and glutaminase activities of wild-type and various mutant strains. Each enzyme activity was determined using live whole cells. The strains are shown in the symbol key to the right of the graph. Results shown are means ± standard deviations (error bars) of activities relative to that of the wild type and represent 2 to 4 independent growth experiments, with at least three replicate assays.
Hydrogenase and catalase activities in a queA strain (our control strain for assessing possible polar effects) were similar to the activities in the wild type (Fig. 1); this confirmed that the phenotype of tat mutant strains can be assigned to the absence or disruption of tatB or tatC. All mutant strains and the wild type were also assayed for glutaminase activity, our control enzyme for non-Tat-dependent periplasmic activity. Glutaminase (also known as γ-glutamyl transpeptidase) was previously shown to be detected in periplasmic and extracellular fractions (36, 37). Despite the presence of an almost perfect Tat-specific consensus (RRSFLK) in its N-terminal sequence, it is not predicted to be translocated by the Tat system by any of the programs; instead, the PRED-TAT program predicts that glutaminase is Sec dependent (29). Wild-type-like glutaminase activities were recorded for all mutants (Fig. 1), confirming that glutaminase is indeed not a Tat target and suggesting that cellular integrity of tat mutants is not compromised.
To confirm that the differences in catalase activities in these different strains were indeed due to cellular mislocalization rather than to differences in catalase activities per se, complementary assays were carried out using cell-free protein extracts. As shown in Fig. 1, whole-cell catalase activity of SHR-ΔtatC mutants was only 37% ± 4% of that of the WT; however, when cell-free extracts of the same SHR-ΔtatC mutant strain were used for the assay, there was no noticeable difference in catalase activity levels between SHR-ΔtatC and wild-type cells (data not shown). From this control experiment it is concluded that the observed decreased catalase activity in whole cells of tatC mutants is likely due to mislocalization of the catalase enzyme. Taken together, these results suggest that SHR-tatC mutants are indeed tat mutants and that hydrogenase and catalase, as previously hypothesized, rely on the Tat machinery to be transported to their final destination.
H. pylori tat mutants have cell division defects.
Phase-contrast microscopy analysis of various strains with mutations in genes encoding components of the Tat system revealed unusually long cells that appear to result from deficient cell division (Fig. 2). Indeed, cells from strain 26695 SHR-tatC (SLB1066 [Fig. 2B]) were longer than cells from the parental strain (26695 [Fig. 2A]), while cells from strains 43504 SHR-ΔtatC::aphA3 (SLB1049 [Fig. 2E]), 43504 SHR-ΔtatC::cat (SLB1075 [Fig. 2G]), and 43504 tatB::cat (SLB1085 [Fig. 2H]) were all longer than WT cells (43504 [Fig. 2D]). The other tatB mutant (strain SLB1086) also displayed the same phenotype (data not shown). Chromosomal complementation of SLB1066 and SLB1049 strains with a copy of tatC restored wild-type-like cellular morphology (SLB1091 and SLB1093 [Fig. 2C and F], respectively). The morphologies of mutant strains with mutations generated in the queA control gene in strain 43504 (SLB1097 [Fig. 2I]), 26695, or X47 (data not shown) were similar to that of the wild type, suggesting that the observed phenotype is not due to a polar effect on the queA gene; rather, it can be attributed to deficiencies of the Tat pathway in these different mutants.
FIG 2 .

Phase-contrast microscopy of various wild-type, tatB, tatC, and queA mutant cells.
SHR-ΔtatC mutants are deficient in mouse colonization.
Since hydrogenase and catalase activities are required for full colonization of the mouse stomach (38, 39), we hypothesized that the ability to colonize mice might be affected in tatC mutants. Therefore, the mouse-adapted X47 strain was transformed with either plasmid pSLB137, containing ΔtatC::aphA3, or plasmid pSLB130, containing queA::aphA3 (control). Kanr transformants were isolated in both cases; however, PCR analysis revealed that there was double crossover for X47 queA::aphA3 mutants (SLB1108 strain), but not for X47 ΔtatC::aphA3 mutants (SLB1107 strain) as previously observed with other H. pylori strains in the current study (data not shown). Hydrogenase and catalase activities of SLB1107 mutants were approximately 80% and 50% compared to that of the wild-type X47 strain, respectively, and these were statistically significant differences. Also, SLB1107 mutant cells (Fig. 2J) were longer than their parental strain (X47), albeit less morphologically altered than other tat mutants. X47 (wild-type), SLB1107, and SLB1108 strains were orally given to mice, and colonization levels in the stomach were assessed 3 weeks later (Fig. 3). While the wild-type strain X47 colonized all 9 inoculated mice and the SLB1108 queA control strain colonized all 4 inoculated animals with colonization levels similar to that of the wild type, SLB1107 was severely deficient in its ability to colonize mice: most mice had lower colonization levels, and two of them had no detectable recovered CFU from stomach homogenates (Fig. 3). Therefore, a fully functional Tat system is required to efficiently colonize mouse stomachs.
FIG 3 .
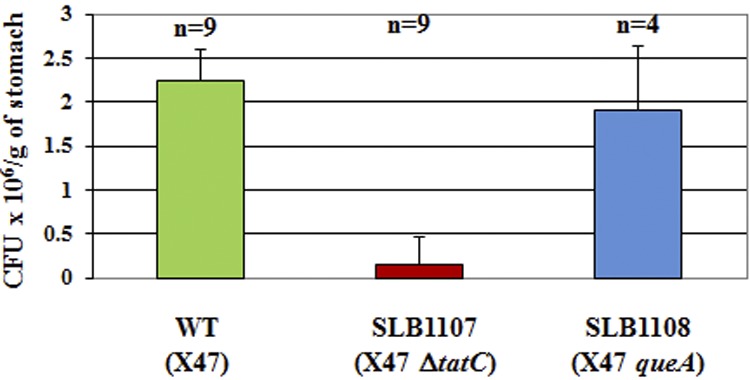
Mouse stomach colonization levels of wild-type, SHR-tatC, and queA mutant strains. Mice were orally given approximately 1.5 × 108 H. pylori cells of a strain. After 3 weeks, their stomachs were harvested, homogenized, diluted, and plated, and the number of CFU was determined 3 to 5 days after harvest. The number of mice used for each strain is shown above the columns.
Use of conditional tatC mutants reveals that tatC is an essential gene in H. pylori.
The fact that the activities of predicted Tat-dependent enzymes (hydrogenase and catalase) were decreased in SHR-ΔtatC mutants (or increased upon the addition of chromosomal tatC) strongly suggests that TatC is indeed affected in SHR-ΔtatC strains. Nevertheless, we sought to complement these data by creating strains that contained inducible tatC and chromosomal inactivated tatC. Therefore, we constructed tatC conditional mutants by (i) cloning tatC under the control of a Ptac promoter in a shuttle vector able to replicate in H. pylori (40), (ii) introducing this plasmid containing Ptac-tatC (pSLB495) into wild-type cells (strain SLB1308), and (iii) eventually targeting the chromosomal tatC locus in the presence of isopropyl-β-d-thiogalactopyranoside (IPTG). Using this approach, for the first time the chromosomal copy of tatC was inactivated by double homologous recombination (strain SLB1310 [see Fig. S2 in the supplemental material]). In the presence of Cm and IPTG, strain SLB1310 was able to grow as well as the wild-type strain (Table 2) and had similar cell morphology (Fig. 2L); however, when strain SLB1310 was diluted with brain heart infusion (BHI) broth and spread on blood agar (BA) medium without Cm or IPTG, it was unable to grow, indicating that tatC is indeed essential in H. pylori (Table 2).
TABLE 2 .
tatC is essential for H. pylori viability
| Strain | Genotype | Medium | Growtha | OD600b |
|---|---|---|---|---|
| 43504 | WT | BA | Yes | 27 ± 3 |
| SLB1308 | WT carrying pSLB495 (Ptac-tatC) | BA | Yes | 30 ± 2 |
| BA-Cm-IPTG | Yes | 23 ± 2 | ||
| SLB1310 | ΔtatC mutant carrying pSLB495 (Ptac-tatC) | BA | No | ND |
| BA-Cm-IPTG | Yes | 21 ± 5 |
Cells were grown on BA (for WT) or BA-Cm-IPTG (for SLB1308 and SLB1310 strains) for 24 h. The cells were harvested and washed in BHI broth, and 0.2-ml suspension (OD600 of 0.1) was plated on either BA or BA-Cm-IPTG medium. Cells from each plate were harvested after 48 h and resuspended in 1 ml of PBS, and OD600 was determined.
Results shown are the means ± standard deviations for 4 independent experiments. ND, not determined.
Directly restreaking SLB1310 cells from selective medium (Cm-IPTG) to nonselective medium resulted in a mixed population of live and dying cells that had lower hydrogenase and catalase activity than the wild type, while retaining the same glutaminase activity (Fig. 4). These conditional mutants grown on nonselective plates displayed the same abnormal cellular morphology as SHR-ΔtatC or tatB mutants, confirming the link between Tat and cell morphology in H. pylori (Fig. 2K). Interestingly, the SLB1308 strain (the intermediate strain used to construct conditional tatC mutants), which possess chromosomal and plasmid tatC copies, had higher hydrogenase and catalase activities than the wild type when grown in the presence of Cm and IPTG (Fig. 4). Taken together, these results confirm that a fully functional Tat pathway is required for hydrogenase and catalase activities, as previously hypothesized based on signal peptide analysis. In addition, they highlight the unexpected link between the Tat system and cell division in H. pylori. Most importantly, they strongly suggest that the Tat pathway, or more specifically TatC, is required for H. pylori.
FIG 4 .
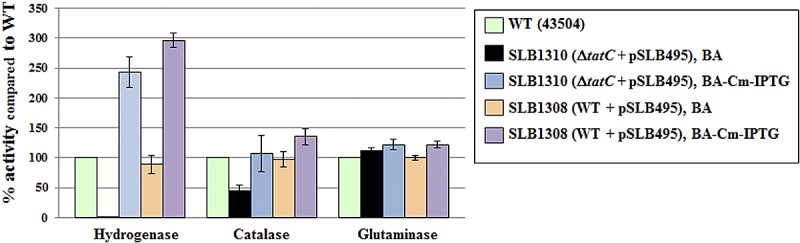
Hydrogenase, catalase, and glutaminase activities of WT and conditional tatC mutants. Each enzyme activity was determined using whole cells. Strains and growth conditions are indicated in the symbol key to the right of the graph. BA is blood agar, a noninducible medium. BA-Cm-IPTG is blood agar with Cm and IPTG, an inducible medium. Results shown are means ± standard deviations of activities relative to that of the wild type and represent 2 to 4 independent growth experiments, with at least three replicate assays.
The requirement for tatC could be linked to FbcF.
Among the four proteins expected to be translocated via the Tat pathway in H. pylori, two of these proteins, KapA (and its partner KatA) and hydrogenase have already been shown to be dispensable in H. pylori. Indeed, kapA can be successfully disrupted and even though kapA mutants cannot colonize mice anymore, they are viable under standard lab conditions (38). Likewise, a previous study revealed that strains with mutations in the large hydrogenase subunit HydB (encoded by hp0632 in strain 26695 [21]) are viable, despite attenuated ability to colonize mice (39). To rule out the possibility that deletion of hp0631, the gene encoding the twin-arginine-containing subunit HydA, would be lethal, we deleted the entire hyd operon (hp0631 through hp0635) in two H. pylori parental strains, 26695 and 43504 (data not shown). As expected, these ΔhydABCDE mutants were devoid of hydrogenase activity; however, their growth and morphology were similar to those of the wild type (data not shown). Therefore, HydA, or more broadly hydrogenase, is not an essential enzyme in H. pylori. Besides, mutations in the hp0407 (bisC) gene were also generated in three different parental strains in the present study. These mutants were viable and microscopically similar to the wild type (data not shown). Finally, the fourth putative Tat-dependent protein, FbcF, was targeted. This time, multiple attempts at constructing fbcF mutants were unsuccessful, suggesting that fbcF is essential in H. pylori. Albeit indirect, this result links the essentiality of the Tat system at least in part to an incorrect localization of the essential cytochrome oxidase Rieske subunit.
DISCUSSION
In the present study, we showed that H. pylori tatB (hp1060 in strain 26695 [21]) can be inactivated while tatA (hp0320) or tatC (hp1061) appear to be essential in H. pylori. Indeed, using two independent suicide plasmids containing part of the hp1060 DNA sequence disrupted by a chloramphenicol resistance cassette, we were able to obtain dozens of chloramphenicol-resistant clones in each case. Genetic analysis of the mutants unambiguously confirmed that in both cases tatB was disrupted by insertion of the marker, following double homologous recombination. Since it appears there is no tatB homolog (duplicated copy) in H. pylori, tatB does not seem to be essential in H. pylori. Mutations in tatB appear to have limited effect on hydrogenase and catalase activities in H. pylori, with cells retaining approximately 80% of activity in each case. Following the same mutant construction strategy, we were able to successfully disrupt hp1062 (queA), the gene located downstream of tatC (see Fig. S1 in the supplemental material). In contrast, multiple attempts to disrupt tatA were unsuccessful, and attempts at deleting tatC were also unsuccessful or led to partial mutants retaining an undisrupted copy of tatC in their chromosome (Table S3). These gene disruption results occurred regardless of the source of DNA (circular or linear suicide plasmid or PCR), the marker used (chloramphenicol or kanamycin resistance), the H. pylori recipient strain (SS1, 26695, 43504, or X47) or the DNA treatment (methylation with H. pylori cell extracts). The occurrence of single homologous recombination within the tatBC locus is expected to have a wide range of effects on Tat(B)C expression and stability and on the assembly of the final TatABC complex; this could include decreased tatC levels or the formation of truncated nonfunctional Tat chimeras that would compete with intact Tat machinery, etc., and these phenotypes have yet to be characterized.
Results from a global transposon mutagenesis study aimed at determining essential genes in H. pylori (41) showed that there was no transposon insertion in hp1061 (tatC), while there was one insertion in hp1062 (queA), suggesting that tatC is essential, while queA is not, in agreement with results from the present study. However, these results have to be taken with caution, as the same study reported no transposon insertion in hp1060 (tatB) and one transposon “hit” in hp0320 (tatA) (41); we now know from the current study that tatB mutants can be constructed, while tatA mutants cannot. The fact that tatB appears to be dispensable (while tatA and tatC are not) suggests that one or several Tat-dependent targets (yet to be determined) can be translocated in a TatB-independent manner. It also suggests that TatAC, rather than TatABC, constitutes the minimum translocase set in H. pylori. This would differentiate H. pylori from other Gram-negative bacteria, whose vast majority rely on an integral TatABC complex (5). However, this has to be stated with caution, given the fact that tatB mutants showed the same abnormal microscopic morphology as partial or conditional tatC mutants (Fig. 2). To determine whether the phenotypes observed for tatB mutants were due to a possible polar effect on tatC expression levels, we used quantitative reverse transcription-PCR (qRT-PCR) to compare tatC mRNA levels in tatB mutants (SLB1085 and SLB1086) to that of the wild-type strain. The levels of expression of tatC standardized to gyrA (see Text S1 in the supplemental material) were comparable in tatB mutant and wild-type strains (data not shown). Therefore, phenotypes observed for tatB mutants (decreased hydrogenase and catalase activities, abnormal morphology) are not the consequence of decreased tatC expression but rather suggest that TatB plays an important role, albeit one that is probably less critical than TatA or TatC, among the Tat machinery in H. pylori.
More generally, the finding that the Tat system—or more precisely TatAC—appears to be essential in H. pylori is surprising considering the limited number (four) of predicted target proteins in the gastric pathogen. The H. pylori Tat pathway is not the first bacterial pathway shown to be essential; however, the three other bacterial species requiring Tat for survival, M. tuberculosis (16), S. meliloti (14), and B. bacteriovorus (15), have significantly higher numbers of predicted Tat-dependent substrates with 31, 94, and 21 substrates, respectively (5, 42). In addition, the Tat system has been found to be also essential in halophilic archaea, such as H. salinarum and H. volcanii (17, 18). Most haloarchaea rely almost exclusively on Tat (instead of Sec) because the proteins need to be properly folded in a timely manner (prior to translocation) before being exposed to the high-salinity environment. For instance, H. salinarum and H. volcanii appear to have 60 and 68 Tat-dependent substrates, respectively (5).
The search for Tat-specific signal peptides in H. pylori was done on translated DNA sequences from the sequenced strain 26695 (21). In order to account for possible start codon misannotation that would hamper our quest for Tat signal peptides, alternate start codons (still resulting in valid reading frames) were identified upstream and downstream of existing annotated start codons (Govind Chandra and Dave Widdick, personal communication). This search found only three proteins believed to be Tat targets in H. pylori, HP0874 (KapA), HP1031 (HydA), and HP1540 (FbcF), in agreement with previously published genome mining data (42). The HP0407 (BisC) protein was not found in this search because only TatP (31) and TATFIND (30) programs were used and none of them predicts HP0407 to be a Tat-translocated protein (Table 1). However, the most recent prediction program, PRED-TAT (29), categorizes HP0407 as Tat dependent. HP0407 is annotated as biotin sulfoxide reductase (based on sequence homology), but its role has yet to be determined. When expressed in E. coli, recombinant HP0407 localized to the membrane, consistent with its expected final localization (S. Benoit, unpublished data). In addition, a selenocysteine codon and a molybdopterin-guanine dinucleotide (MGD) binding motif can be identified in the sequence of HP0407 (J. Craig Venter Institute [JCVI] Comprehensive Microbial Resource [CMR] at http://cmr.jcvi.org). These features are shared by various reductases, such as formate dehydrogenases, dimethyl sulfoxide (DMSO) or trimethylamine N-oxide (TMAO) reductases, all of which are known to be translocated by the Tat system (3); therefore, HP0407 appears to be a prime candidate for Tat dependency in H. pylori. In the current study, we report the construction of viable hp0407 mutants (by double homologous recombination) in three different H. pylori strains (43504, X47, and 26695), indicating that the hp0407 gene is not essential in H. pylori.
KapA (HP0874) is one of the other proteins predicted to be dependent on Tat (Table 1). A previous two-hybrid study revealed that KapA can interact with KatA (HP0875), the catalytic catalase subunit (35). Through a mechanism commonly described as “hitchhiking” (43), KatA would bind to KapA, and the heterologous protein complex would be translocated by the Tat system through the cytoplasmic membrane (using KapA signal peptide) to reach the periplasm. In agreement with this model, Harris and Hazell showed that there was 5.5-fold-less catalase activity in periplasmic fractions of kapA mutants compared to periplasmic fractions of wild-type cells (34). Results from the present study also support this model: we found less catalase activity in whole cells of tatB mutants, SHR-ΔtatC partial mutants, or tatC conditional mutants grown in the absence of IPTG compared to whole-cell activity in the parental strain, suggesting that less catalase was effectively transported toward the periplasm. The decrease in catalase activity observed in SHR-ΔtatC mutants was not due to decreased overall activity in these mutants, because catalase activity in cell-free extracts (total protein) of mutant and wild-type cells was shown to be almost identical. Besides, increasing the expression of tatC in mutant or wild-type cells, either through expression of a PureA-tatC chromosomal copy or a Ptac-tatC plasmid copy in the presence of IPTG, led to increased levels of whole-cell catalase activity, indicating a correlation between tatC levels and catalase distribution within the cell. The construction of kapA and katA mutants in H. pylori has been previously reported (34); therefore, none of these genes appear to be essential under lab conditions. Hence, it is unlikely that the lethality observed for conditional tatC mutants is linked to mislocalization of KapA and KatA.
Whole-cell enzyme assays were also used as an indirect way to assess hydrogenase distribution in H. pylori tat mutants. There is only one hydrogenase in H. pylori, previously shown to be the H2 uptake type and membrane bound (33). The heterotrimeric HydABC complex is expected to rely on HydA and its Tat signal peptide (Table 1) to be translocated to the cytoplasmic membrane. If the Tat machinery is absent or nonfunctional, the hydrogenase complex will still be synthesized but unable to reach the membrane and electron transport components, and H2 oxidation coupled to cytochrome oxidases will probably be decreased. Indeed, a decrease of hydrogenase activity was observed in whole cells of SHR-ΔtatC partial mutants or uninduced conditional tatC mutants and, to a lesser extent, in tatB mutants. In contrast, the addition of tatC (complemented SHR-ΔtatC partial mutants, merodiploid strain, or induced conditional tatC mutants) led to increased whole-cell hydrogenase activities. These results strongly suggest a correlation between tatC levels and hydrogenase distribution within the cell. HydB-deficient or hydrogenase-negative (ΔhydABCDE) mutants are viable (reference 39 and our current study); therefore, it is not expected that mislocalization of the hydrogenase complex (due to Tat deficiency) would lead to cellular death.
The fourth hypothesized target is FbcF (HP1540). Based on protein sequence analysis, it is a [2Fe-2S]-containing Rieske subunit of a cytochrome b oxidase. Rieske subunits from a variety of microorganisms, such as Synechocystis, Paracoccus denitrificans, or Legionella pneumophila, have been shown to be Tat dependent (44–46). Mislocalization of FbcF would lead to impaired (oxygen) respiration and aerobic growth defects, as recently shown with Rieske protein homologs in the obligate aerobe Streptomyces coelicolor or the facultative anaerobe Shewanella oneidensis (47, 48). H. pylori is microaerophilic and has very limited respiration capacity, with no formate dehydrogenase, nitrate reductase, or DMSO or TMAO reductase; as stated above, the role of the HP0407 protein is still unknown. In addition, the gastric pathogen appears to possess only one cbb3-type cytochrome c terminal oxidase (21, 49). The closely related epsilonproteobacterium C. jejuni also possesses a Tat-dependent ubiquinol cytochrome b oxidase (PetA or Cj1186c [50]); however, it has a branched respiratory chain with two different terminal oxidases, a cbb3-type cytochrome c oxidase and a bd-type quinol oxidase (50). Therefore, mislocalization of FbcF is expected to have a bigger impact on H. pylori respiration (and viability) than mislocalization of PetA on C. jejuni. This difference could explain why C. jejuni tatC (13) mutants are viable while H. pylori tatC mutants are not. To determine whether FbcF is important for H. pylori, we attempted to generate fbcF mutants; however, this approach was unsuccessful, suggesting that fbcF is essential in H. pylori. Therefore, mislocalization of FbcF, along with the mislocalization of the other Tat-dependent proteins and their associated complex, HydA-hydrogenase, KapA-catalase, and HP0407, probably accounts for the lethality of conditional tatC mutants, as well as the deficiency in mouse stomach colonization observed for X47 SHR-ΔtatC partial mutants.
Finally, another phenotype was observed for all the tat mutants: long cells that appeared to be unable to properly divide and had envelope defects were observed for tatB, SHR-tatC, SHR-ΔtatC, and conditional tatC mutants grown under uninduced conditions. Complementation with chromosomal tatC or plasmid-borne tatC in the presence of IPTG resulted in wild-type-like cell morphology. This phenotype was not completely unexpected because E. coli tat mutants have been reported to exhibit similar abnormal cellular morphology (51). The cause of this phenotype was attributed to mislocalization of two cell wall hydrolases, AmiA and AmiC, both of which possess a signal peptide with a twin-arginine motif (51). H. pylori possesses only one AmiA protein (HP0772 in strain 26695 [21]), which shares 28% identity with E. coli AmiA and 32% identity with E. coli AmiC. Analysis of the protein sequence of H. pylori AmiA (HpAmiA) reveals the presence of a signal sequence (see Fig. S3 in the supplemental material); however, there is no twin-arginine motif, and it is predicted to be Sec dependent by programs such as PRED-TAT (29) or SignalP-4.1 (52). Therefore, AmiA is not expected to be mislocalized in H. pylori tat mutants, and it is probably not the reason for the observed abnormal cell morphology. Since none of the (four) proteins predicted to be Tat dependent are supposed to play an active role in cell envelope synthesis or cell division in H. pylori, this phenotype suggests one of the following. (i) There are more Tat-dependent targets yet to be discovered than the four described in this article. (ii) The mislocalization of some of the Tat-dependent proteins, especially hydrogenase and cytochrome oxidase, might introduce global changes in the cell redox state that in turn will have effects such as the one observed. Further work is currently under way to elucidate which of these two hypothesis account for the observed phenotype in tatB or tatC mutants.
MATERIALS AND METHODS
Bacterial strains and plasmids.
E. coli and H. pylori strains and plasmids used in this study are listed in Table S1 in the supplemental material. Genomic DNA from H. pylori strain 26695 was used as the template for all PCR amplifications. All DNA plasmids or PCR products used to generate mutants were sequenced on both strands at the Georgia Genomics Facility, University of Georgia, Athens, GA.
Growth conditions.
E. coli cells were grown aerobically in Luria-Bertani (LB) medium or plates at 37°C. Ampicillin (100 µg/ml), chloramphenicol (25 µg/ml), or kanamycin (30 µg/ml) was added as needed. H. pylori was routinely grown on brucella agar (BA) plates supplemented with 10% defibrinated sheep blood at 37°C under microaerophilic conditions (5% CO2, 4% O2, and 91% N2). Brain heart infusion (BHI) broth was used to resuspend the cells for microscopy analysis. Chloramphenicol (8 or 25 µg/ml) or kanamycin (20 µg/ml) was added as needed. BA medium was supplemented with amphotericin B (10 µg/ml), vancomycin (10 µg/ml), and bacitracin (50 µg/ml) for mouse colonization experiments.
Construction of H. pylori ΔtatA mutants.
Attempts at generating ΔtatA mutants were done using a splicing by overlap extension PCR (SOE PCR) method. Briefly, primers TASOE1 and TASOE2 (see Table S2 in the supplemental material) were used to amplify a 460-bp-long DNA sequence located upstream of tatA (hp0320). Primers TASOE3 and TASOE4 were used to amplify a 410-bp-long sequence located downstream of tatA. The final amplification step included each purified PCR product, a cat cassette, and primers TASOE1 and TASOE4. The resulting 1,670-bp-long PCR product was introduced into H. pylori strains X47, 43504, and 26695. The same procedure was carried out two more times without success.
Construction of H. pylori tatB mutants.
Two different tatB mutants were generated using two restriction sites naturally present within the tatB (hp1060) gene, SspI and HindIII, located 164 bp and 324 bp downstream of the tatB start codon, respectively. Primers TB1 and TB2 (see Table S2 and Fig. S1 in the supplemental material) were used to PCR amplify a 720-bp-long DNA sequence containing the 482-bp-long hp1060 gene and flanking sequences from hp1059 (ruvB) and hp1061 (tatC) genes. The PCR product was either cloned directly into a pGEM-T vector to generate plasmid pSLB208 or digested with BamHI and ligated into BamHI-cut pSLB112 plasmid (SspI-free pUC19 derivative [Table S1]) to yield plasmid pSLB209. Plasmids pSLB208 and pSLB209 were digested with HindIII and SspI, respectively. Following digestion, each plasmid was subsequently blunt ended with T4 polymerase before being ligated with a blunt-ended 800-bp-long cat cassette (chloramphenicol resistance [Cmr]). Finally, each newly generated plasmid, pSLB210 or pSLB212 (Table S1), was introduced into various H. pylori strains by natural transformation or electroporation to generate tatB::cat-I or tatB::cat-II mutants, respectively. H. pylori cells were transferred after 16 h onto BA plates supplemented with 25 µg/ml chloramphenicol. When using strain 43504 as the recipient strain, hundreds of clones appeared after 3 to 5 days of incubation. The disruption of the tatB gene and the insertion of the cat cassette (by double crossover) were confirmed by PCR using genomic DNA from each mutant as a template and primers TB1 and TB2 (Fig. S1 and Fig. S2).
Construction of H. pylori tatC and ΔtatC mutants.
Two different approaches were followed to either disrupt or delete the tatC (hp1061) gene in H. pylori. First, a unique SspI site present within tatC was used to insert an ahpA3 (Kanr) cassette. Briefly, primers TC3 and TC4 (see Table S2 and Fig. S1 in the supplemental material) were used to amplify a 717-bp-long DNA fragment containing part of tatC. The PCR product was digested with HindIII and cloned into similarly digested plasmid pSLB112 to yield plasmid pSLB113. A 1.3-kb-long, blunt-ended aphA3 cassette was cloned into tatC (SspI; 415 bp downstream of the tatC start codon), generating plasmid pSLB129 (Table S1). This plasmid was introduced by natural transformation or electroporation into several H. pylori strains (26695, 43504, or X47). H. pylori cells were transferred after 16 to 24 h onto BA plates supplemented with either kanamycin or chloramphenicol. After numerous attempts, only one kanamycin-resistant clone (SLB1066) could be recovered using 26695 as the recipient strain. This mutant was analyzed by PCR using internal primers TC3 and TC4 and external primers TC1 and TC6 (Fig. S1 and Fig. S2).
In order to generate ΔtatC::aphA3 or ΔtatC::cat deletion mutants, the following sequential method was used. First, a 416-bp-long DNA sequence containing part of tatB and the first four codons of tatC was amplified by PCR using primers TC1 and TC2 (see Table S2 and Fig. S1 in the supplemental material). The PCR product was digested with BamHI and EcoRI and cloned into similarly digested vector pBS-KS to generate plasmid pSLB135. This plasmid was subsequently digested with EcoRI and ligated with an EcoRI-cut aphA3 cassette, yielding plasmid pSLB136. Next, a 410-bp-long PCR product that contained the last three codons of the tatC gene and the first 397 bp of the queA gene (obtained by using primers TC5 and TC6) was digested with SalI and XhoI and ligated with similarly cut pSLB136 plasmid to generate pSLB137 (Fig. S1). Alternatively, the aphA3 cassette from pSLB137 was excised using EcoRI and replaced by a cat cassette, yielding plasmid pSLB196 (Table S1). Plasmids pSLB137 and pSLB196 were used to generate kanamycin-resistant or chloramphenicol-resistant tatC deletion mutants in various H. pylori parental strains, respectively. No Kanr or Cmr clone could be obtained in H. pylori strain 26695 despite numerous attempts, including DNA methylation with H. pylori cell extracts prior to transformation. In contrast, dozens of Kanr or Cmr transformants were isolated when strain 43504 or X47 was used. Mutants were genetically analyzed by PCR using internal primers TC3 and TC4 or external primers TC1 and TC6 (Fig. S1 and Fig. S2).
Chromosomal complementation of SHR-tatC or SHR-ΔtatC mutants.
Primers TC7 and TC8 (see Table S2 and Fig. S1 in the supplemental material) were used to amplify a 763-bp-long DNA sequence containing the tatC coding sequence and to introduce an NdeI restriction site (at the ATG start codon) and an XhoI restriction site, respectively. The PCR product was digested with NdeI and XhoI and ligated into similarly digested plasmid pPA (53) to place the tatC gene under the control of the 200-bp-long H. pylori PureA promoter. The plasmid generated, pSLB217, was subsequently digested with BglII and XhoI (to release the 963-bp-long PureA-tatC construct) and blunt ended using T4 polymerase before being ligated into EcoRV-digested plasmid pEU39Cm (54), yielding plasmid pSLB218 (Table S1). This suicide plasmid was introduced into H. pylori wild-type strain 43504 and into 26695 tatC::aphA3 (SLB1066) or 43504 ΔtatC::aphA3 (SLB1049) mutant cells, and Cmr mutants were isolated following homologous recombination of the 1.76-kb-long PureA-tatC-cat construct within the hp0405 gene in the chromosome, as confirmed by PCR, using genomic DNA from each mutant as a template and primers HP405 and Cat2 (data not shown).
Construction of a conditional tatC mutant.
The NdeI- and BamHI-digested tatC PCR product obtained with primers TC7 and TC8 (see above and see Table S2 and Fig. S1 in the supplemental material) was ligated into similarly digested plasmid pILL2150 (40) to generate plasmid pSLB495. In this plasmid, the tatC gene is under the control of a (IPTG-inducible) Ptac promoter. Plasmid pSLB495 was methylated in the presence of S-adenosylmethionine (New England Biolabs, Ipswich, MA) and cell-free extracts from H. pylori parental strain 43504, as described by Boneca et al. (40). Methylated plasmid pSLB495 was then introduced into strain 43504, and chloramphenicol-resistant mutants were isolated. The resulting strain, SLB1308, was transformed with plasmid pSLB137 (suicide vector harboring ΔtatC::aphA3 [see above and Fig. S1]) in the presence of Cm (8 µg/ml) and IPTG (1 mM). Kanamycin-resistant transformants were isolated on plates supplemented with kanamycin (20 µg/ml) as well as chloramphenicol (8 µg/ml) and 1 mM IPTG. The disruption of tatC by double crossover was confirmed by PCR using external primers TC1 and TC6 (Fig. S1 and S2). These mutants were viable only in the presence of 1 mM IPTG.
Construction of H. pylori queA mutant.
Primers QA1 and QA2 (see Table S2 and Fig. S1 in the supplemental material) were used to PCR amplify a 937-bp-long DNA sequence containing part of the 1,038-bp-long queA gene (hp1062). The PCR product was digested with EcoRI and inserted within similarly cut pBS-KS vector to generate plasmid pSLB114. A unique AfeI restriction site located within queA was used to introduce a 1.3-kb-long aphA3 cassette. The resulting plasmid (pSLB130) was introduced into various H. pylori strains by natural transformation to generate queA::aphA3 chromosomal mutants. Kanamycin-resistant transformants were obtained from strain 26695, 43504, or X47, generating strain SLB1071, SLB1097, or SLB1108, respectively (Table S1). The concomitant chromosomal disruption of the queA gene and the insertion of aphA3 were confirmed by PCR using genomic DNA from each mutant as a template and primers QA1 and QA2 (Fig. S1 and Fig. S2).
Whole-cell enzyme assays.
Hydrogenase, catalase, or glutaminase activity was determined on whole cells. Cells were grown on BA plates (with Cm and IPTG as reported) for 24 to 48 h. While the mutant to wild-type enzyme activity ratio was similar between experiments, there was significant variation in the net values in independent experiments due to the use of whole cells and growth condition variables (blood batches, gas conditions, etc.). To minimize this natural variability while still reflecting the respective enzyme activity of each mutant compared to the wild type, all enzyme activities are given as means ± standard deviations of percentages of each activity relative to the wild-type value. One unit of optical density at 600 nm (OD600) corresponds to approximately 1 × 109 H. pylori cells per ml.
(i) Hydrogenase assays.
Cells were grown in presence of H2 (in sealed anaerobic jars with CampyPak Plus microaerophilic envelopes, Becton Dickinson, Sparks, MD), harvested and resuspended in phosphate-buffered saline (PBS), cell density (OD600) was measured and hydrogen uptake was followed using a previously described amperometric method (33). Hydrogenase activity of wild-type (43504) cells ranged from 0.5 to 1.6 nmol of H2 used per min per 108 cells. Results represent 2 to 4 independent growth experiments, each with at least 3 assay replicates.
(ii) Catalase assays.
Cells were washed and resuspended in phosphate-buffered saline (PBS) to a final OD600 of 1.0. Five microliters of whole cells was mixed with 495 μl of PBS containing 15 mM H2O2, and the initial H2O2 disappearance (decrease in OD240) was monitored for up to 1 min. Catalase activity of wild-type (43504) cells ranged from 20 to 110 µmol H2O2 per min per 108 cells depending on the experiment. Results are averages ± standard deviations from 2 to 4 independent growth experiments, each with 5 to 10 assay replicates. For a control, catalase activities were also determined on cell-free protein extracts. In this case, cells were broken by sonication and spun down, and total protein concentration was determined using the bicinchoninic acid (BCA) kit (Thermo, Fisher Pierce, Rockford, IL). Catalase assays were carried out as described above, using 0.5 µg total protein.
(iii) Glutaminase assays.
Glutaminase activity was monitored by measuring glutamine-dependent ammonium production by whole cells, a combination of two previously published methods (55, 56). Cells were harvested, washed, and resuspended to a final OD600 of 1.0 in 50 mM HEPES (pH 7.5) and 25 mM NaCl. The reaction was started by the addition of 10 mM glutamine, and ammonium production was determined (after 15 to 30 min) using the phenol-sodium hypochlorite method of Weatherburn (56). Glutaminase activity of wild-type (43504) cells ranged from 15 to 80 nmol NH3 produced per min per 109 cells between independent growth experiments. Results are averages ± standard deviations from 2 to 4 growth independent experiments, each with at least 3 assay replicates.
Microscopy analysis.
H. pylori cells grown for 24 to 48 h on BA plates were resuspended in a drop of BHI broth and examined with a phase-contrast microscope (Leica Microsystems, DM55008). Digital images were obtained at a magnification of ×1,000 using a QIQICAM Fast 1394 camera (Compix Inc.).
Mouse colonization experiments.
H. pylori X47 (mouse-adapted, parental strain), X47 SHR-ΔtatC, and X47 ΔqueA mutant strains were grown on BA plates, harvested in sterile PBS, and resuspended (in PBS) to a final OD600 of 1.7. Each mouse (n = 9 for the WT, n = 9 for the SHR-ΔtatC mutant, and n = 4 for the queA mutant) was orally given 0.15 ml of bacterial suspension (approximately 1.5 × 108 cells). Mice were sacrificed 3 weeks postinoculation. Their stomachs were quickly removed, weighed, and gently homogenized in 5-ml phosphate-buffered saline using a Dounce hand homogenizer. The homogenates were diluted in PBS and plated (0.1 ml) in duplicate on plates supplemented with bacitracin, amphotericin B, and vancomycin. The plates were incubated for 5 to 7 days at 37°C in a 4% O2 partial pressure atmosphere for colony counting. Data are expressed as CFU recovered per gram of stomach.
SUPPLEMENTAL MATERIAL
Supplemental Materials and Methods Download
(A) Genetic organization of the tatBC locus in H. pylori 26695 chromosome. Restriction sites used to generate tatB, tatC, or queA insertion mutants are indicated. The locations of primers used to construct ΔtatC deletion mutant or to verify mutants by PCR are indicated. (B) Linear map of suicide plasmid pKS-ΔtatC::aphA3 (pSLB137) and locations of primers used to verify mutants. This plasmid was used to generate SHR-ΔtatC mutants and DHR-ΔtatC conditional mutants. (C) Linear map of H. pylori-replicating plasmid pSLB495 used to express tatC in conditional mutants. Download
PCR analysis of various tatB, tatC, ΔtatC, and queA mutants Download
(A) Nucleotide sequence of amiA (hp0772) and its upstream sequence (http://www.ncbi.nlm.nih.gov/nuccore/AE000511). The region upstream of amiA (hp0772) is shown, with part of hp0773 (in italic). The amiA gene is shown in boldface type. Putative start and stop codons are underlined (J. F. Tomb et al., Nature 388:539–547, 1997). (B) Protein sequence of AmiA (http://www.cmr.jcvi.org). A putative Sec-dependent signal sequence is underlined. The cleavage site is predicted to occur at residue Ala19, according to PRED-TAT (P. G. Bagos, E. P. Nikolaou, T. D. Liakopoulos, and K. D. Tsirigos, Bioinformatics 26:2811–2817, 2010) or SignalP-4.1 (T. N. Petersen, S. Brunak, G. von Heijne, and H. Nielsen, Nat. Methods 8:785–786, 2011). Download
Strains and plasmids used in this study
Primers used in this study. All primers were purchased from Integrated DNA Technologies, Coralville, IA. The uppercase letters in the Sequence* column indicate H. pylori-derived sequences. Restriction sites are underlined.
Strategies used to generate tatA, tatB, tatC, and queA knockout mutants in H. pylori. In the Outcome* column, HR stands for homologous recombination.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grant number RO1 AI077569).
We thank Lawrence Shimkets and members of his lab (University of Georgia) for their help with phase-contrast microscopy. We are grateful to Dave Widdick and Govind Chandra (John Innes Centre, Norwich, United Kingdom) for their help with Tat motif search. We thank Hilde de Reuse (Pasteur Institute, Paris, France) for the kind gift of plasmid pILL2150.
Footnotes
Citation Benoit SL, Maier RJ. 2014. Twin-arginine translocation system in Helicobacter pylori: TatC, but not TatB, is essential for viability. mBio 5(1):e01016-13. doi:10.1128/mBio.01016-13.
REFERENCES
- 1. Wickner W, Schekman R. 2005. Protein translocation across biological membranes. Science 310:1452–1456. 10.1126/science.1113752 [DOI] [PubMed] [Google Scholar]
- 2. Berks BC, Palmer T, Sargent F. 2005. Protein targeting by the bacterial twin-arginine translocation (Tat) pathway. Curr. Opin. Microbiol. 8:174–181. 10.1016/j.mib.2005.02.010 [DOI] [PubMed] [Google Scholar]
- 3. Palmer T, Sargent F, Berks BC. 2005. Export of complex cofactor-containing proteins by the bacterial Tat pathway. Trends Microbiol. 13:175–180. 10.1016/j.tim.2005.02.002 [DOI] [PubMed] [Google Scholar]
- 4. Stanley NR, Palmer T, Berks BC. 2000. The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J. Biol. Chem. 275:11591–11596. 10.1074/jbc.275.16.11591 [DOI] [PubMed] [Google Scholar]
- 5. Palmer T, Berks BC. 2012. The twin-arginine translocation (Tat) protein export pathway. Nat. Rev. Microbiol. 10:483–496. 10.1038/nrmicro2814 [DOI] [PubMed] [Google Scholar]
- 6. Wexler M, Sargent F, Jack RL, Stanley NR, Bogsch EG, Robinson C, Berks BC, Palmer T. 2000. TatD is a cytoplasmic protein with DNase activity. No requirement for TatD family proteins in sec-independent protein export. J. Biol. Chem. 275:16717–16722. 10.1074/jbc.M000800200 [DOI] [PubMed] [Google Scholar]
- 7. Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, Palmer T. 1998. Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J. 17:3640–3650. 10.1093/emboj/17.13.3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee PA, Orriss GL, Buchanan G, Greene NP, Bond PJ, Punginelli C, Jack RL, Sansom MS, Berks BC, Palmer T. 2006. Cysteine-scanning mutagenesis and disulfide mapping studies of the conserved domain of the twin-arginine translocase TatB component. J. Biol. Chem. 281:34072–34085. 10.1074/jbc.M607295200 [DOI] [PubMed] [Google Scholar]
- 9. Mickael CS, Lam PK, Berberov EM, Allan B, Potter AA, Köster W. 2010. Salmonella enterica serovar Enteritidis tatB and tatC mutants are impaired in Caco-2 cell invasion in vitro and show reduced systemic spread in chickens. Infect. Immun. 78:3493–3505. 10.1128/IAI.00090-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lavander M, Ericsson SK, Bröms JE, Forsberg A. 2006. The twin arginine translocation system is essential for virulence of Yersinia pseudotuberculosis. Infect. Immun. 74:1768–1776. 10.1128/IAI.74.3.1768-1776.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang L, Zhu Z, Jing H, Zhang J, Xiong Y, Yan M, Gao S, Wu LF, Xu J, Kan B. 2009. Pleiotropic effects of the twin-arginine translocation system on biofilm formation, colonization, and virulence in Vibrio cholerae. BMC Microbiol. 9:114. 10.1186/1471-2180-9-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodríguez-Sanz M, Antúnez-Lamas M, Rojas C, López-Solanilla E, Palacios JM, Rodríguez-Palenzuela P, Rey L. 2010. The Tat pathway of plant pathogen Dickeya dadantii 3937 contributes to virulence and fitness. FEMS Microbiol. Lett. 302:151–158. 10.1111/j.1574-6968.2009.01844.x [DOI] [PubMed] [Google Scholar]
- 13. Rajashekara G, Drozd M, Gangaiah D, Jeon B, Liu Z, Zhang Q. 2009. Functional characterization of the twin-arginine translocation system in Campylobacter jejuni. Foodborne Pathog. Dis. 6:935–945. 10.1089/fpd.2009.0298 [DOI] [PubMed] [Google Scholar]
- 14. Pickering BS, Oresnik IJ. 2010. The twin arginine transport system appears to be essential for viability in Sinorhizobium meliloti. J. Bacteriol. 192:5173–5180. 10.1128/JB.00206-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang CY, Hobley L, Till R, Capeness M, Kanna M, Burtt W, Jagtap P, Aizawa S, Sockett RE. 2011. The Bdellovibrio bacteriovorus twin-arginine transport system has roles in predatory and prey-independent growth. Microbiology 157:3079–3093. 10.1099/mic.0.052449-0 [DOI] [PubMed] [Google Scholar]
- 16. Saint-Joanis B, Demangel C, Jackson M, Brodin P, Marsollier L, Boshoff H, Cole ST. 2006. Inactivation of Rv2525c, a substrate of the twin arginine translocation (Tat) system of Mycobacterium tuberculosis, increases beta-lactam susceptibility and virulence. J. Bacteriol. 188:6669–6679. 10.1128/JB.00631-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dilks K, Giménez MI, Pohlschröder M. 2005. Genetic and biochemical analysis of the twin-arginine translocation pathway in halophilic archaea. J. Bacteriol. 187:8104–8113. 10.1128/JB.187.23.8104-8113.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas JR, Bolhuis A. 2006. The tatC gene cluster is essential for viability in halophilic archaea. FEMS Microbiol. Lett. 256:44–49. 10.1111/j.1574-6968.2006.00107.x [DOI] [PubMed] [Google Scholar]
- 19. Marshall BJ, Warren JR. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1:1311–1315 [DOI] [PubMed] [Google Scholar]
- 20. Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R. 1999. Helicobacter pylori virulence and genetic geography. Science 284:1328–1333. 10.1126/science.284.5418.1328 [DOI] [PubMed] [Google Scholar]
- 21. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547. 10.1038/41483 [DOI] [PubMed] [Google Scholar]
- 22. Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, Stadler PF, Vogel J. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255. 10.1038/nature08756 [DOI] [PubMed] [Google Scholar]
- 23. Sargent F, Stanley NR, Berks BC, Palmer T. 1999. Sec-independent protein translocation in Escherichia coli. A distinct and pivotal role for the TatB protein. J. Biol. Chem. 274:36073–36082 [DOI] [PubMed] [Google Scholar]
- 24. Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176–180. 10.1038/16495 [DOI] [PubMed] [Google Scholar]
- 25. Oh JD, Kling-Bäckhed H, Giannakis M, Xu J, Fulton RS, Fulton LA, Cordum HS, Wang C, Elliott G, Edwards J, Mardis ER, Engstrand LG, Gordon JI. 2006. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: evolution during disease progression. Proc. Natl. Acad. Sci. U. S. A. 103:9999–10004. 10.1073/pnas.0603784103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baltrus DA, Amieva MR, Covacci A, Lowe TM, Merrell DS, Ottemann KM, Stein M, Salama NR, Guillemin K. 2009. The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol. 191:447–448. 10.1128/JB.01416-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McClain MS, Shaffer CL, Israel DA, Peek RM, Jr, Cover TL. 2009. Genome sequence analysis of Helicobacter pylori strains associated with gastric ulceration and gastric cancer. BMC Genomics 10:3. 10.1186/1471-2164-10-S1-S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Robinson C, Matos CF, Beck D, Ren C, Lawrence J, Vasisht N, Mendel S. 2011. Transport and proofreading of proteins by the twin-arginine translocation (Tat) system in bacteria. Biochim. Biophys. Acta 1808:876–884. 10.1016/j.bbamem.2010.11.023 [DOI] [PubMed] [Google Scholar]
- 29. Bagos PG, Nikolaou EP, Liakopoulos TD, Tsirigos KD. 2010. Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics 26:2811–2817. 10.1093/bioinformatics/btq530 [DOI] [PubMed] [Google Scholar]
- 30. Rose RW, Brüser T, Kissinger JC, Pohlschröder M. 2002. Adaptation of protein secretion to extremely high-salt conditions by extensive use of the twin-arginine translocation pathway. Mol. Microbiol. 45:943–950. 10.1046/j.1365-2958.2002.03090.x [DOI] [PubMed] [Google Scholar]
- 31. Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S. 2005. Prediction of twin-arginine signal peptides. BMC Bioinformatics 6:167. 10.1186/1471-2105-6-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gorrell RJ, Yang J, Kusters JG, van Vliet AH, Robins-Browne RM. 2005. Restriction of DNA encoding selectable markers decreases the transformation efficiency of Helicobacter pylori. FEMS Immunol. Med. Microbiol. 44:213–219. 10.1016/j.femsim.2004.10.019 [DOI] [PubMed] [Google Scholar]
- 33. Maier RJ, Fu C, Gilbert J, Moshiri F, Olson J, Plaut AG. 1996. Hydrogen uptake hydrogenase in Helicobacter pylori. FEMS Microbiol. Lett. 141:71–76. 10.1111/j.1574-6968.1996.tb08365.x [DOI] [PubMed] [Google Scholar]
- 34. Harris AG, Hazell SL. 2003. Localisation of Helicobacter pylori catalase in both the periplasm and cytoplasm, and its dependence on the twin-arginine target protein, KapA, for activity. FEMS Microbiol. Lett. 229:283–289. 10.1016/S0378-1097(03)00850-4 [DOI] [PubMed] [Google Scholar]
- 35. Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, Schächter V, Chemama Y, Labigne A, Legrain P. 2001. The protein-protein interaction map of Helicobacter pylori. Nature 409:211–215. 10.1038/35051615 [DOI] [PubMed] [Google Scholar]
- 36. Chevalier C, Thiberge JM, Ferrero RL, Labigne A. 1999. Essential role of Helicobacter pylori gamma-glutamyltranspeptidase for the colonization of the gastric mucosa of mice. Mol. Microbiol. 31:1359–1372. 10.1046/j.1365-2958.1999.01271.x [DOI] [PubMed] [Google Scholar]
- 37. Valenzuela M, Bravo D, Canales J, Sanhueza C, Díaz N, Almarza O, Toledo H, Quest AF. 2013. Helicobacter pylori-induced loss of survivin and gastric cell viability is attributable to secreted bacterial gamma-glutamyl transpeptidase activity. J. Infect. Dis. 208:1131–1141. 10.1093/infdis/jit286 [DOI] [PubMed] [Google Scholar]
- 38. Harris AG, Wilson JE, Danon SJ, Dixon MF, Donegan K, Hazell SL. 2003. Catalase (KatA) and KatA-associated protein (KapA) are essential to persistent colonization in the Helicobacter pylori SS1 mouse model. Microbiology 149:665–672. 10.1099/mic.0.26012-0 [DOI] [PubMed] [Google Scholar]
- 39. Olson JW, Maier RJ. 2002. Molecular hydrogen as an energy source for Helicobacter pylori. Science 298:1788–1790. 10.1126/science.1077123 [DOI] [PubMed] [Google Scholar]
- 40. Boneca IG, Ecobichon C, Chaput C, Mathieu A, Guadagnini S, Prévost MC, Colland F, Labigne A, de Reuse H. 2008. Development of inducible systems to engineer conditional mutants of essential genes of Helicobacter pylori. Appl. Environ. Microbiol. 74:2095–2102. 10.1128/AEM.01348-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Salama NR, Shepherd B, Falkow S. 2004. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J. Bacteriol. 186:7926–7935. 10.1128/JB.186.23.7926-7935.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dilks K, Rose RW, Hartmann E, Pohlschröder M. 2003. Prokaryotic utilization of the twin-arginine translocation pathway: a genomic survey. J. Bacteriol. 185:1478–1483. 10.1128/JB.185.4.1478-1483.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rodrigue A, Chanal A, Beck K, Müller M, Wu LF. 1999. Co-translocation of a periplasmic enzyme complex by a hitchhiker mechanism through the bacterial tat pathway. J. Biol. Chem. 274:13223–13228. 10.1074/jbc.274.19.13223 [DOI] [PubMed] [Google Scholar]
- 44. Aldridge C, Spence E, Kirkilionis MA, Frigerio L, Robinson C. 2008. Tat-dependent targeting of Rieske iron-sulphur proteins to both the plasma and thylakoid membranes in the cyanobacterium Synechocystis PCC6803. Mol. Microbiol. 70:140–150. 10.1111/j.1365-2958.2008.06401.x [DOI] [PubMed] [Google Scholar]
- 45. Bachmann J, Bauer B, Zwicker K, Ludwig B, Anderka O. 2006. The Rieske protein from Paracoccus denitrificans is inserted into the cytoplasmic membrane by the twin-arginine translocase. FEBS J 273:4817–4830. 10.1111/j.1742-4658.2006.05480.x [DOI] [PubMed] [Google Scholar]
- 46. De Buck E, Vranckx L, Meyen E, Maes L, Vandersmissen L, Anné J, Lammertyn E. 2007. The twin-arginine translocation pathway is necessary for correct membrane insertion of the Rieske Fe/S protein in Legionella pneumophila. FEBS Lett. 581:259–264. 10.1016/j.febslet.2006.12.022 [DOI] [PubMed] [Google Scholar]
- 47. Hopkins A, Buchanan G, Palmer T. 2013. The role of the twin arginine protein transport pathway in the assembly of the Streptomyces coelicolor A3(2) cytochrome bc1 complex. J. Bacteriol. 196:50–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luo Q, Dong Y, Chen H, Gao H. 2013. Mislocalization of Rieske protein PetA predominantly accounts for the aerobic growth defect of Tat mutants in Shewanella oneidensis. PLoS One 8:e62064. 10.1371/journal.pone.0062064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kelly DJ. 1998. The physiology and metabolism of the human gastric pathogen Helicobacter pylori. Adv. Microb. Physiol. 40:137–189. 10.1016/S0065-2911(08)60131-9 [DOI] [PubMed] [Google Scholar]
- 50. Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG. 2000. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403:665–668. 10.1038/35001088 [DOI] [PubMed] [Google Scholar]
- 51. Ize B, Stanley NR, Buchanan G, Palmer T. 2003. Role of the Escherichia coli Tat pathway in outer membrane integrity. Mol. Microbiol. 48:1183–1193. 10.1046/j.1365-2958.2003.03504.x [DOI] [PubMed] [Google Scholar]
- 52. Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 53. Benoit S, Maier RJ. 2003. Dependence of Helicobacter pylori urease activity on the nickel-sequestering ability of the UreE accessory protein. J. Bacteriol. 185:4787–4795. 10.1128/JB.185.16.4787-4795.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Olson JW, Agar JN, Johnson MK, Maier RJ. 2000. Characterization of the NifU and NifS Fe-S cluster formation proteins essential for viability in Helicobacter pylori. Biochemistry 39:16213–16219. 10.1021/bi001744s [DOI] [PubMed] [Google Scholar]
- 55. Stark RM, Suleiman MS, Hassan IJ, Greenman J, Millar MR. 1997. Amino acid utilisation and deamination of glutamine and asparagine by Helicobacter pylori. J. Med. Microbiol. 46:793–800. 10.1099/00222615-46-9-793 [DOI] [PubMed] [Google Scholar]
- 56. Weatherburn MW. 1967. Phenol-hypochlorite reaction for determination of ammonia. Anal. Chem. 39:971–974. 10.1021/ac60252a045 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials and Methods Download
(A) Genetic organization of the tatBC locus in H. pylori 26695 chromosome. Restriction sites used to generate tatB, tatC, or queA insertion mutants are indicated. The locations of primers used to construct ΔtatC deletion mutant or to verify mutants by PCR are indicated. (B) Linear map of suicide plasmid pKS-ΔtatC::aphA3 (pSLB137) and locations of primers used to verify mutants. This plasmid was used to generate SHR-ΔtatC mutants and DHR-ΔtatC conditional mutants. (C) Linear map of H. pylori-replicating plasmid pSLB495 used to express tatC in conditional mutants. Download
PCR analysis of various tatB, tatC, ΔtatC, and queA mutants Download
(A) Nucleotide sequence of amiA (hp0772) and its upstream sequence (http://www.ncbi.nlm.nih.gov/nuccore/AE000511). The region upstream of amiA (hp0772) is shown, with part of hp0773 (in italic). The amiA gene is shown in boldface type. Putative start and stop codons are underlined (J. F. Tomb et al., Nature 388:539–547, 1997). (B) Protein sequence of AmiA (http://www.cmr.jcvi.org). A putative Sec-dependent signal sequence is underlined. The cleavage site is predicted to occur at residue Ala19, according to PRED-TAT (P. G. Bagos, E. P. Nikolaou, T. D. Liakopoulos, and K. D. Tsirigos, Bioinformatics 26:2811–2817, 2010) or SignalP-4.1 (T. N. Petersen, S. Brunak, G. von Heijne, and H. Nielsen, Nat. Methods 8:785–786, 2011). Download
Strains and plasmids used in this study
Primers used in this study. All primers were purchased from Integrated DNA Technologies, Coralville, IA. The uppercase letters in the Sequence* column indicate H. pylori-derived sequences. Restriction sites are underlined.
Strategies used to generate tatA, tatB, tatC, and queA knockout mutants in H. pylori. In the Outcome* column, HR stands for homologous recombination.