

Abstract
Pertussis toxin (PTX) is an AB5-type exotoxin produced by the bacterium Bordetella pertussis, the causative agent of whooping cough. In vivo intoxication with PTX elicits a variety of immunologic and inflammatory responses, including vasoactive amine sensitization (VAAS) to histamine (HA), serotonin (5-HT), and bradykinin (BDK). Previously, by using a forward genetic approach, we identified the HA H1 receptor (Hrh1/H1R) as the gene in mice that controls differential susceptibility to B. pertussis PTX-induced HA sensitization (Bphs). Here we show, by using inbred strains of mice, F1 hybrids, and segregating populations, that, unlike Bphs, PTX-induced 5-HT sensitivity (Bpss) and BDK sensitivity (Bpbs) are recessive traits and are separately controlled by multiple loci unlinked to 5-HT and BDK receptors, respectively. Furthermore, we found that PTX sensitizes mice to HA independently of Toll-like receptor 4, a purported receptor for PTX, and that the VAAS properties of PTX are not dependent upon endothelial caveolae or endothelial nitric oxide synthase. Finally, by using mice deficient in individual Gαi/o G-protein subunits, we demonstrate that Gαi1 and Gαi3 are the critical in vivo targets of ADP-ribosylation underlying VAAS elicited by PTX exposure.
INTRODUCTION
Infection with the bacterium Bordetella pertussis can lead to whooping cough, usually typified by paroxysomal coughing and, in severe cases, oropharyngeal tissue swelling, hypertension, pneumothorax, and long-lasting aftereffects (1). The principal active agent in B. pertussis, pertussis toxin (PTX), is an AB5-type secreted exotoxin (2–4). The A (active) subunit catalyzes the ADP-ribosylation and thereby impairment of α-subunit signaling in Gαi/o-linked heterotrimeric guanine nucleotide regulatory protein (G-protein) complexes (5). This covalent modification prevents the Gαβγ complex from associating with G protein-coupled receptors (GPCRs) on the cell membrane (6). The B (binding) pentamer is thought to bind cell surface receptors on a variety of mammalian cells and facilitate the cellular entry of the A subunit (7). PTX elicits diverse physiological responses in vivo, including leukocytosis and altered glucose regulation (8), increased blood-brain barrier (BBB) permeability, and systemic vasoactive sensitization (VAAS) to biogenic amines (9). PTX is also used as an ancillary adjuvant to approximate an infectious environmental influence in animal models of tissue-specific autoimmune disease (10). Genetically, one of PTX's physiological effects, VAAS to histamine (HA), is controlled by a single autosomal dominant locus (Bphs, for B. pertussis-induced HA sensitization) and has been used as an intermediate phenotype for genetic studies of organ-specific autoimmune diseases (11, 12).
Although several cell types have the capacity to produce HA, mast cells are thought to be a main source during tissue inflammation (13). Mast cells also produce large amounts of serotonin (5-HT) and bradykinin (BDK), which, together with HA and other lipid and glycosylated mediators, are stored in granules. During inflammation, soluble and cell surface molecules can act as secretagogues to stimulate degranulation and release of factors from mast cells, which can affect vascular permeability (14), leading to antigen leakage from or inappropriate leukocyte access to tissues (13).
We previously studied VAAS in response to HA by using a forward genetic approach to identify the HA H1 receptor (Hrh1/H1R) as the gene underlying Bphs (11, 12), a subphenotype for tissue-specific autoimmune diseases elicited with the aid of PTX. A T cell-specific role for H1R in disease susceptibility was found (15), but a bone marrow chimeric approach revealed that expression of H1R in the nonhematopoietic compartment was responsible for Bphs (16), prompting the closer look at the mechanism of Bphs that we took in this study.
During the positional cloning of Bphs, we observed that SJL/J mice were also susceptible to PTX-induced 5-HT sensitization (Bpss, for B. pertussis-induced 5-HT sensitization) while C3H.BphsSJL/J mice were not, thereby excluding Hrh1 as a candidate for Bpss (17). These results indicate that the phenotypic variation in responsiveness to Bphs and Bpss reflects genetic control of distinct intermediate phenotypes rather than allelic variation in genes controlling overall susceptibility to VAAS. To address this possibility and to gain insight into the genetic control of VAAS to other vasoactive amines, we challenged a panel of PTX-sensitized inbred, F1, and N2 backcross mice with 5-HT and BDK and studied them for Bpss and Bpbs (B. pertussis-induced BDK sensitization), respectively. Unlike Bphs/Hrh1, which is dominant, we show that Bpss and Bpbs are recessive traits and are controlled by separate complex genetic mechanisms unlinked to 5-HT or BDK receptors.
It is known that PTX-induced VAAS to HA (VAASH) requires intoxication with active holotoxin (18) in a mechanism that presumably involves ADP-ribosylation of the Gαi/o class of G proteins (19). However, there are several Gαi/o proteins and splice variants but the specific in vivo target(s) associated with PTX-induced VAAS has remained unclear for nearly 30 years. We show here by using knockout (KO) mice that Gαi1 and Gαi3 are the specific targets of ADP-ribosylation required for PTX-induced VAASH.
MATERIALS AND METHODS
Ethics statement.
All experimental procedures were approved by the University of Vermont Institutional Animal Care and Use Committee (approval number 10-020) in accordance with relevant institutional guidelines and the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Mice.
Most of the inbred strains in Table 1 were purchased from The Jackson Laboratory, except for RFM/UnNCr, FVB/NCr, CBA/JCr, and C3H/HeN mice, which were purchased from the National Cancer Institute/Charles River Laboratories. F1 hybrid strains and N2 backcross mice, as in Tables 2 and 3, respectively, were generated in-house by using parental strains purchased from Jackson. Hrh1-deficient mice (B6.129P-Hrh1tm1Wat) (20), C3H.SJL-Bphss congenic mice (11), were maintained in-house at the University of Vermont. Bdkrb2-deficient mice (B6.129S7-Bdkrb2tm1Jfh/J) (21), Nos3-deficient mice (B6.129P2-Nos3tmiUnc/J) (22), caveolin-1-deficient mice (B6.Cg-Cav1tm1Mls/J) (23), Tlr4 mutant C57BL/10ScNJ mice (24), and control C57BL/10J mice were purchased from Jackson. PKCα-deficient (B6.129-Prkcatm1Jmk) (25), PKCβ-deficient (129/Sv-Prkcbtm1Tara) (26), PKCγ-deficient (B6.129P2-Prkcgtm1Stl) (27), PKCδ-deficient (129/SV×Ola.Prkcdtm1Qxu) (5), PKCε-deficient (129/Sv × C57BL/6.Prkcetm1Ltg) (28), and PKCζ-deficient (129/Sv.Prkcztm1Jmos) mice (29) were a kind gift from Michael Leitges, Max Planck Institute for Experimental Endocrinology, Hannover, Germany.
TABLE 1.
Bphs, Bpss, and Bpbs of inbred micea
| Strain | Bphs |
Bpss |
LD50 | Bpbs |
LD50 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Carrier + 5-HT (50 mg/kg) | PTX + 5-HT at: |
Carrier + Bdk (80 mg/kg) | PTX + Bdk at: |
||||||||||||
| Carrier + HA (25 mg/kg) | PTX + HA (25 mg/kg) | 50 mg/kg | 12.5 mg/kg | 3.1 mg/kg | 0.78 mg/kg | 0.39 mg/kg | 80 mg/kg | 40 mg/kg | 20 mg/kg | 10 mg/kg | |||||
| SM/J | 0/4 | 4/4b | 0/4 | 4/4b | 5/5 | 5/6 | 3/6 | 0/4 | 1.0 | 0/4 | 4/4b | 2/4 | 0/4 | 0/4 | 40.0 |
| SJL/J | 0/4 | 4/4b | 0/4 | 12/12c | 12/12 | 6/11 | 0/8 | 2.6 | 0/4 | 4/4b | 4/4 | 1/4 | 0/4 | 22.5 | |
| AKR/J | 0/4 | 0/4 | 0/4 | 12/12c | 10/10 | 8/17 | 0/8 | 3.2 | 0/4 | 4/4b | 3/4 | 0/4 | 0/4 | 35.6 | |
| MRL/MpJ | 0/4 | 0/4 | 0/4 | 4/4b | 6/6 | 4/8 | 0/4 | 2.9 | 0/4 | 2/4 | 1/4 | 0/4 | 0/4 | ||
| LG/J | 0/4 | 4/4b | 0/4 | 4/4b | 7/8 | 0/4 | 5.4 | 0/4 | 2/4 | 0/4 | 0/4 | 0/4 | |||
| RFM/UnNCr | 0/4 | 4/4b | 0/4 | 6/6d | 0/6 | 0/4 | 26.2 | ||||||||
| PL/J | 0/4 | 4/4b | 0/4 | 6/6d | 0/8 | 0/4 | 29.8 | ||||||||
| DBA/1J | 0/4 | 4/4b | 0/4 | 5/5d | 0/6 | 0/4 | 28.6 | ||||||||
| SWR/J | 0/4 | 4/4b | 0/4 | 6/6d | 0/4 | 0/4 | 22.0 | ||||||||
| C57BL/6J | 0/4 | 4/4b | 0/4 | 9/15 | 0/14 | 0/8 | 0/4 | 4/4b | 4/4 | 0/4 | 0/4 | 28.3 | |||
| DBA/2J | 0/4 | 4/4b | 0/4 | 4/15 | 0/14 | 0/4 | 0/4 | 4/4b | 2/4 | 0/4 | 0/4 | 40.0 | |||
| A/J | 0/4 | 4/4b | 0/4 | 2/15 | 0/12 | 0/4 | 0/4 | 4/4b | 2/4 | 0/4 | 0/4 | 40.0 | |||
| A/HeJ | 0/4 | 4/4b | 0/4 | 3/6 | 0/6 | 0/4 | 0/4 | 4/4b | 4/4 | 2/4 | 0/4 | 20.0 | |||
| NOD/LtJ | 0/4 | 4/4b | 0/4 | 2/6 | 0/8 | 0/4 | |||||||||
| FVB/NCr | 0/4 | 4/4b | 0/4 | 2/6 | 0/8 | 0/4 | |||||||||
| C57BL/10J | 0/4 | 4/4b | 0/4 | 1/6 | 0/4 | 0/4 | 0/4 | 2/4 | 0/4 | 0/4 | 0/4 | ||||
| NZB/BlnJ | 0/4 | 4/4b | 0/4 | 3/8 | 0/6 | 0/4 | 0/4 | 4/4b | 3/4 | 2/4 | 0/4 | 22.1 | |||
| NZW/LacJ | 0/4 | 4/4b | 0/4 | 2/9 | 0/6 | 0/4 | 0/4 | 4/4b | 4/4 | 2/4 | 0/4 | 20.0 | |||
| 129X1/SvJ | 0/4 | 4/4b | 0/4 | 6/10 | 5/14 | 1/5 | 0/4 | 0/4 | 4/4b | 3/4 | 2/4 | 0/4 | 22.1 | ||
| BALB/cJ | 0/4 | 4/4b | 1/4 | 3/10 | 6/17 | 0/5 | 0/4 | 0/4 | 4/4b | 4/4 | 2/4 | 0/4 | 20.0 | ||
| BALB/cByJ | 0/4 | 4/4b | 0/4 | 0/9 | 0/6 | 0/4 | 0/4 | 4/4b | 2/4 | 0/4 | 0/4 | 40.0 | |||
| CBA/JCr | 0/4 | 0/4 | 0/4 | 0/8 | 0/8 | 0/4 | |||||||||
| B10.D2n | 0/4 | 4/4b | 0/4 | 0/9 | 0/6 | 0/4 | 0/4 | 2/4 | 0/4 | 0/4 | 0/4 | ||||
| B10.S/SgSnJ | 0/4 | 4/4b | 0/4 | 1/10 | 0/6 | 0/4 | 0/4 | 1/4 | 0/5 | 0/5 | 0/5 | ||||
| C3H/HeJ | 0/4 | 0/4 | 0/4 | 4/14 | 4/15 | 0/4 | 0/4 | 4/4b | 4/4 | 1/4 | 0/4 | 22.5 | |||
| C3H/HeN | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 | 4/4b | 4/4 | 2/4 | 0/4 | 20.0 | |||
| C3H.BphsSJL | 0/4 | 4/4b | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 | 4/4b | 4/4 | 2/4 | 0/4 | 20.0 | |||
Animals were sensitized with 200 ng PTX by i.v. injection and challenged 3 days later by i.v. injection of HA (25 mg/kg), 5-HT (0.39 to 50 mg/kg), or BDK (10 to 80 mg/kg). Three or four mice were examined at a given dose. Dosages are based on mg/kg (dry weight) free base or peptide. Deaths were recorded at 30 min, and the results are expressed as the number dead/number studied. LD50s of 5-HT and HA were calculated with GraphPad Prism. Contrasts for combined phenotypes: SM = SJL = AKR = MRL; SM < LG = SJL = AKR = MRL; LG < RFM = PL = DBA/1 = SWR. The significance of differences between carrier- and PTX-treated groups was determined by Fisher's exact test. The significance of differences in LD50s among and between the strains was determined by the extra sum of squares F test. Bpss overall F = 15.9, P < 0.0001; Bpbs overall F = 1.3, P = 0.2.
P = 0.05.
P < 0.0001.
P < 0.01.
TABLE 2.
Inheritance of Bphs, Bpss, and Bpbs in F1 hybrid strainsa
| Phenotype and strain | Classification | No. of mice with phenotype/total (P value) |
Contrasts | ||
|---|---|---|---|---|---|
| Group 1 | Group 2 | Group 3 | |||
| Bphs | |||||
| SJL/J | Bphs+ Bpss+ Bpbs+ | 6/6b | 6/6c | 5/6d | |
| C3H/HeJ | Bphs− | 0/6b | 0/6c | 0/6d | |
| (C3H × SJL)F1 | 6/6b (0.0001) | 6/6c (0.0001) | 4/6d (0.0094) | SJL/J = (C3H × SJL)F1 > C3H/HeJ | |
| C57BL/6J | Bphs+ Bpbs+ | 6/6b | 6/6c | 1/6d | |
| CBA/J | Bphs− | 0/6b | 0/6c | 0/6d | |
| (B6 × CBA)F1 | 6/6b (0.0001) | 6/6c (0.0001) | 2/6d (0.3) | C57BL/6J = (B6 × CBA)F1 > CBA/J | |
| Bpss | |||||
| SJL/J | Bphs+ Bpss+ Bpbs+ | 12/12e | 12/12f | 6/11g | |
| PL/J (control) | Bpss+ | 6/6e | 0/8f | 0/4g | |
| (PL × SJL)F1 | 8/9e (0.35) | 1/9f (<0.0001) | 0/5g (0.03) | SJL/J > PL/J = (PL × SJL)F1 | |
| SJL/J | 12/12e | 12/12f | 6/11g | ||
| BALB/cJ | Bpss− | 3/10e | 6/17f | 0/5g | |
| (BALB × SJL)F1 | 6/9e (0.001) | 4/8f (0.002) | 0/5g (0.02) | SJL/J > BALB/cJ = (BALB × SJL)F1 | |
| AKR/J | Bphs+ Bpss+ Bpbs+ | 12/12e | 10/10f | 8/17g | |
| DBA/2J | Bpss− | 4/15e | 0/14f | 0/4g | |
| (AK × DBA)F1 | 4/8e (0.0003) | 0/8f (<0.0001) | 0/5g (0.05) | AKR/J > DBA/2J = (AK × DBA)F1 | |
| SJL | 10/10e | 10/10f | 6/10g | ||
| B10.S | Bpss− | 1/10e | 1/10f | 0/10g | |
| (B10 × SJL)F1 | 1/10e (<0.0001) | 0/10f (<0.0001) | 0/10g (0.0006) | SJL/J > B10.S = (B10 × SJL)F1 | |
| Bpbs | |||||
| SJL/J | Bphs+ Bpss+ Bpbs+ | 6/6h | 6/6i | 4/6j | |
| B10.S | Bpbs− | 1/6h | 0/6i | 0/6j | |
| (B10 × SJL)F1 | 2/6h (0.009) | 1/6i (0.0007) | 0/5j (0.008) | SJL/J > B10.S = (B10 × SJL)F1 | |
F1 hybrids were generated by using the indicated parental strains. Mice were then challenged for Bphs, Bpss, or Bpbs at the indicated doses. Significance of difference in susceptibility between parental strains and F1 hybrids was determined using Fisher's exact test.
HA challenge dose, 25 mg/kg free base.
HA challenge dose, 12.5 mg/kg free base.
HA challenge dose, 3.1 mg/kg free base.
5-HT challenge dose, 50 mg/kg free base.
5-HT challenge dose, 12.5 mg/kg free base.
5-HT challenge dose, 3.1 mg/kg free base.
BDK challenge dose, 80 mg/kg peptide.
BDK challenge dose, 40 mg/kg peptide.
BDK challenge dose, 20 mg/kg peptide.
TABLE 3.
Bphs, Bpss, and Bpbs in N2 backcross micea
| Phenotype and strain | No. of mice |
P value |
||
|---|---|---|---|---|
| Affected | Unaffected | 1 locus | 2 loci | |
| Bpss | ||||
| SJL | 10 | 0 | ||
| (B10.S × SJL)F1 | 0 | 10 | ||
| (B10.S × SJL) × B10.S N2 | 20 | 80 | 0.50 | 0.005 |
| Bpbs | ||||
| SJL | 10 | 0 | ||
| (B10.S × SJL)F1 | 0 | 10 | ||
| (B10.S × SJL) × B10.S N2 | 28 | 72 | 0.75 | <0.0001 |
SJL, (B10.S × SJL)F1, and (B10.S × SJL) × B10.S N2 mice were challenged for Bpss (25 mg/kg 5-HT) and Bpbs (40 mg/kg BDK) as in Table 1. Results were tested by chi-square goodness of fit for both one- and two-locus recessive models of inheritance.
Gαo-deficient mice (129SvEv.129SvEv-Gnao1tm1LBi) (30), Gαo1-deficient mice (129SvOla.129SvEv-Gnao1.1tm1LBi) (31), Gαo2-deficient mice (129SvOla.129SvEv-Gnao1.2tm1LBi) (31), Gαi1-deficient mice (129Sv.129SvEv-Gnai1tm1Drs) (32), Gαi2-deficient mice (B6.129SvEv-Gnai2tm1LBi) (33), Gαi3-deficient mice (129Sv.129SvEv-Gnai3tm1LBi) (31), and Gαi1/3 doubly deficient mice (129SvEv-Gnao1tm1LBi × 129SvEv-Gnai3tm1LBi) (31) were obtained from Lutz Birnbaumer (National Institute of Environmental Health Sciences, Research Triangle Park, NC) and maintained as wild-type (WT, +/+) mice, heterozygotes (+/−), or homozygotes (−/−) on 129SvEv (Gαo, Gαi1, Gαi2, Gαi3, Gαi1 × Gαi3), CD1 outbred (Gαo), FVB (Gαi2, 10th backcross), C57BL/6 (Gαi2, 8th backcross), and crossbred C57BL/6:129SvEv 50:50 (Gαo1, Gαo2, Gαi2) backgrounds. All animals were provided normal mouse chow and water ad libitum.
Vasoactive amine sensitization and challenge.
Bphs was carried out as described previously (11, 16). Bpss and Bpbs followed a similar protocol. Briefly, 200 ng of PTX (List Biological Laboratories, Campbell, CA) in phosphate-buffered saline was administered intravenously (i.v.) by tail vein injection in a volume of 0.1 ml on day 0. On day 3, mice were challenged i.v. with HA dihydrochloride, 5-HT hydrochloride, or BDK acetate (all from Sigma, dosed at milligrams per kilogram of body weight by using the free amine molecular weight). Deaths were recorded at 30 min postchallenge and are shown as the number of animals affected over the total.
Microsatellite genotyping.
We performed microsatellite typing as previously described, with liver DNA using informative microsatellite markers that distinguished B10.S and SJL/J mice and are linked to all known 5-HT receptors and the Bdkrb1/2 locus (34, 35).
RESULTS
Distinct genetic control of Bphs, Bpss, and Bpbs.
To further extend our hypothesis that distinct genetic mechanisms control susceptibility to PTX-induced sensitization to individual biogenic amines, we examined 27 different inbred mouse strains for Bphs, Bpss, or Bpbs. We identified two strains that were sensitive to a challenge with all three amines (SJL/J and SM/J), which is consistent with previous reports showing the SJL/J strain is sensitive to both HA and 5-HT challenges (17). Several strains were sensitive to only two amines, but we also observed strains that were monosensitive. For example, C57BL10/J mice were susceptible only to Bphs, C3H/HeJ or C3H/HeN mice were susceptible only to Bpbs, and MRL/MpJ mice were susceptible only to Bpss (see Table S1 in the supplemental material). Furthermore, even within a given phenotype, we observed variability in the 50% lethal doses (LD50s). The LD50 of Bpss for SM/J mice was 1 mg/kg 5-HT, whereas that for PL/J mice was 30 mg/kg (Table 1). Bpbs exhibited LD50s of 20 to 40 mg/kg. The dose of HA used as a control for all strains (25 mg/kg) is approximately 2 LD50s for susceptible strains and readily discriminates Bphs from Bphs-resistant strains (17). The observance of all possible amine sensitivity combinations across an inexhaustive survey of inbred mouse strains indicates that the phenotypic variation in susceptibility to VAAS reflects the genetic control of distinct intermediate phenotypes rather than allelic variation in genes controlling overall susceptibility to intoxication or VAAS.
Susceptibility to Bphs is controlled by Hrh1, and this phenotype is inherited in an autosomal dominant fashion (11). We expanded these studies to investigate the inheritance of Bpbs and Bpss by phenotyping (susceptible × resistant)F1 hybrid mice. (C3H/HeJ × SJL/J)F1 mice were Bphs, consistent with previous studies (12). Sudweeks et al. observed that (SJL/J × CBA/J)F1 mice were also Bphs (12). Here we extended this finding by phenotyping (C57BL/6J × CBA/J)F1 mice for Bphs and observed the same fully penetrant dominant inheritance (Table 2). These data are also consistent with genotype analysis of Hrh1, since both SJL/J and C57BL/6J mice harbor the Bphs (Hrh1s) allele while C3H/HeJ and CBA/J mice harbor the Bphs resistance Hrh1r allele (11).
Bpbs and Bpss are two-gene recessive traits.
To assess Bpbs and Bpss inheritance, we generated (susceptible × resistant)F1 hybrid mice on the basis of the phenotype data in Table 1. For both Bpss and Bpbs, we used SJL/J mice as the prototypical susceptible strain (SJL/J mice were susceptible to all three vasoactive amines, Bphs+ Bpss+ Bpbs+; Table 1). To rule out strain-specific contributions to the Bpss phenotype, we also employed an F1 hybrid generated by using AKR/J mice as the susceptible strain (like SJL/J mice, AKR/J mice are Bpss+ Bpbs+). As a positive control for interstrain crosses in Bpss, we assessed (SJL/J × PL/J)F1 mice (PL/J is Bpss+), and these mice indeed remained Bpss+ and were statistically indistinguishable from either parental strain (Table 2). By analyzing the Bpss phenotype of (SJL/J × BALB/cJ), (SJL/J × B10.S), and (AKR/J × DBA/2J)F1 hybrids, we found that Bpss was recessive since the F1 mice displayed the phenotype of the Bpss-resistant parental strain (Table 2). We observed a similar recessive inheritance pattern for Bpbs, where (SJL/J × B10.S)F1 mice were Bpbs resistant, like the B10.S parental strain (Table 2). Collectively, these data again confirm that the phenotypic variation in susceptibility to VAAS reflects the genetic control of responsiveness to individual VAAs rather than susceptibility and resistance to intoxication in general.
The fact that Bphs was reported to be controlled by a single autosomal dominant gene (36) was confirmed when we identified Hrh1 as the gene controlling Bphs (11). In order to determine the number of loci controlling Bpss and Bpbs, we generated (B10.S × SJL/J) × B10.S N2 backcross mice in sufficient numbers to be able to statistically evaluate by chi-square test both one- and two-locus models of genetic control. The data for both Bpss and Bpbs are consistent with a two-gene model of recessive inheritance (Table 3).
Lack of linkage of Bpss and Bpbs to 5-HT or BDK receptors, respectively.
Given that Bphs was identified as the product of Hrh1 (11), we wondered whether Bpss and Bpbs could be similarly controlled by 5-HT and BDK receptor polymorphism. To test this hypothesis, we determined whether or not Bpss or Bpbs was linked to any of the known 5-HT or BDK receptors, respectively. To this end, (B10.S × SJL/J) × B10.S N2 backcross mice were phenotyped for Bpss or Bpbs and genotyped by using informative microsatellite markers linked to the 14 5-HT genes encoding functional receptors on chromosomes 1, 4, 5, 9, 13, 14, 16, 18, 19, and X and to Bdkrb1/2 on chromosome 12. Four additional putative 5-HT receptor sequences (Htr1da, Htr1db, Htr1ea, and Htr1eb) have been deposited in PubMed Gene. Htr1da and Htr1db were based on partial Htr1d sequence data by Weydert et al. suggesting the possible existence of two Htr1d genes on mouse chromosome 4 (37). However, Htr1da was shown not to encode a unique functional receptor, and the record for Htr1db has been replaced with that for Htr1d, which we analyzed. With respect to Htr1ea and Htr1eb, the situation is similar; Htr1ea is actually Htr1e and Htr1eb is Htr1f, both of which we analyzed. Therefore, our analysis covered all known functional 5-HT receptor genes. By chi-square analysis, we did not observe significant linkage of Bpss to any of the known 5-HT receptors, nor did we observe linkage of Bpbs to Bdkrb1/2 (Table 4). However, we did observe that Bdkrb2-deficient mice (21) were completely protected from Bpbs (Table 5), indicating that while not genetically linked to Bpbs, Bdkrb2 signaling is required to elicit PTX-induced sensitization to BDK.
TABLE 4.
Bpss and Bpbs are not linked to 5-HT and BDK receptors, respectivelya
| Receptor | Chrb | Distance (cMc) | Size (Mbp) | Marker | Distance (cM) | Size (Mbp) | No. of mice |
χ2 value | P value | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Affected |
Unaffected |
|||||||||||||
| S | H | B | S | H | B | |||||||||
| Bpss | ||||||||||||||
| Htr2b | 1 | 52 | 88 | D1Mit10 | 56.6 | 93 | 3 | 12 | 2 | 22 | 14 | 39 | 4.8 | 0.4 |
| Htr5b | 63 | 123 | ||||||||||||
| Htr6 | 4 | 65 | 139 | D4Mit203 | 60 | 129 | 2 | 12 | 6 | 25 | 34 | 17 | 4.9 | 0.4 |
| Htr1d | 66 | 136 | ||||||||||||
| Htr5a | 5 | 15 | 28 | D5Mit148 | 18 | 32 | 6 | 9 | 3 | 16 | 46 | 14 | 4.5 | 0.5 |
| Htr3a | 9 | 29 | 49 | D9Mit105 | 35 | 63 | 9 | 6 | 3 | 21 | 34 | 19 | 6.6 | 0.3 |
| Htr3b | 29 | 49 | ||||||||||||
| Htr1b | 46 | 82 | ||||||||||||
| Htr1a | 13 | 58 | 106 | D13Mit75 | 59 | 107 | 3 | 11 | 6 | 20 | 37 | 18 | 1.2 | 0.9 |
| Htr2a | 14 | 42 | 75 | D14Mit71 | 44 | 97 | 6 | 11 | 3 | 17 | 36 | 23 | 2.3 | 0.8 |
| Htr1f | 16 | 39 | 65 | D16Mit14 | 34 | 51 | 4 | 9 | 7 | 19 | 32 | 25 | 3.9 | 0.6 |
| Htr4 | 18 | 37 | 62 | D18Mit81 | 41 | 67 | 4 | 11 | 5 | 19 | 39 | 18 | 0.4 | 1 |
| Htr7 | 19 | 33 | 36 | D19Mit88 | 34 | 37 | 6 | 8 | 6 | 20 | 34 | 22 | 1.8 | 0.9 |
| Htr2c | X | 66 | 143 | DXMit36 | 63 | 143 | 2/4d | 5 | 3/6d | 5/22d | 16 | 12/27d | 3.2/0.1d | 0.7/0.8d |
| Bpbs, Bdkrb1/2 | 12 | 52 | 107 | D12Mit7 | 50 | 104 | 7 | 11 | 10 | 14 | 36 | 15 | 3.4 | 0.6 |
(B10.S × SJL/J) × B10.S N2 backcross mice were phenotyped for Bpss or Bpbs as described for Table 3 (25 mg/kg 5-HT and 40 mg/kg BDK, respectively). B, B10.S; H, heterozygous; S, SJL/J (at indicated position with chromosome location for the gene and closest marker indicated). Genotype frequency differences among affected and unaffected BC1 mice were evaluated by chi-square analysis in two-by-two contingency tables. Significance was tested by Fisher's exact test.
Chr, chromosome.
cM, centimorgans.
Female/male values.
TABLE 5.
Bpbs in WT and Bdkrb2-deficient micea
| Strain | No. of mice dead/total after challenge with BDK at: |
χ2 value, df | P value | |||
|---|---|---|---|---|---|---|
| 80 mg/kg | 40 mg/kg | 20 mg/kg | 10 mg/kg | |||
| C57BL/6J | 3/3 | 3/3 | 1/3 | 0/3 | ||
| B6.129S7-Bdkrb2tm1Jfh/J | 0/4 | 0/4 | 0/4 | 0/4 | 26.0, 7 | 0.0005 |
WT or Bdkrb2-deficient mice were sensitized with 200 ng PTX by i.v. injection and challenged 3 days later by i.v. injection of BDK. Significance was determined by Fisher's exact test.
eNOS and caveolin-1 in Bphs.
To further investigate the molecular mechanism by which PTX intoxication elicits HA sensitization, we employed genetically modified mice lacking key individual components thought to be involved in PTX activity or the signal propagation involved in HA-mediated vasoregulation. Vascular endothelium expresses the enzyme nitric oxide synthase (eNOS, the protein encoded by Nos3), which catalyzes the formation of nitric oxide from l-arginine. This pathway plays a major role in the regulation of vascular tone and blood pressure through the generation of cyclic GMP (cGMP) (38–40). eNOS activation occurs in response to elevation of Ca2+ concentrations in the cell (17, 41). Activation of H1R increases intracellular Ca2+ levels as one of the proximal signaling events (42, 43). In addition, H1R activation has been shown to elevate cGMP levels in vivo (44). To address whether Nos3 is required for Bphs, we sensitized WT C57BL/6J mice and Nos3-deficient mice (B6.129 background) (22) with PTX and challenged them with HA. Nos3-deficient mice were just as Bphs as WT mice (Table 6), suggesting that VAAS following PTX intoxication is not dependent on Nos3. In endothelial cells (ECs), eNOS is highly expressed in caveolae, which are specialized membrane-associated organelles important for the barrier and vasoregulatory functions of these cells. To test whether other features of caveolae distinct from eNOS are important in the regulation of VAASH, we tested the Bphs response of caveolin-1-deficient mice. Since cross regulation of caveolin-1 and eNOS function has been shown (45), we also investigated Bphs in caveolin-1 (Cav1) and Nos3 doubly deficient mice. In contrast to control Hrh1-deficient mice, both Cav1−/− and Cav1−/− Nos3−/− mice were Bphs (Table 7). Together, these results indicate that neither the formation of caveolae nor activation of eNOS in ECs is a critical regulator of VAASH.
TABLE 6.
Bphs is not mediated by eNOSa
| Strain | No. of mice dead/total after challenge with HA at: |
||
|---|---|---|---|
| 50 mg/kg | 12.5 mg/kg | 3.13 mg/kg | |
| C57BL/6J | 4/4 | 4/4 | 2/4 |
| B6.129.P2-Nos3t,m,i/Unc/J | 4/4 | 4/4 | 1/4 |
WT and Nos3-deficient mice were analyzed for Bphs 3 days after the i.v. injection of 200 ng PTX. HA dosages are based on mg/kg (dry weight) free base. Deaths were recorded at 30 min, and the results expressed as the number dead/number studied.
TABLE 7.
Disruption of endothelial caveolae does not affect Bphsa
| Strain | No. of mice dead/total (% of total) after challenge with HA at: |
|||
|---|---|---|---|---|
| 25 mg/kg | 12.5 mg/kg | 6.25 mg/kg | 3.125 mg/kg | |
| WT | 2/2 (100) | 2/2 (100) | 2/2 (100) | 0/2 (0) |
| Cav1−/− | 2/2 (100) | 2/2 (100) | 3/3 (100) | 2/5 (40) |
| Cav1−/− × Nos3−/− | 2/2 (100) | 2/2 (100) | 3/3 (100) | 1/4 (25) |
| Hrh1−/− | 0/3 | 0/3 | 0/3 | 0/3 |
WT, Cav1-deficient, Cav1/Nos3 doubly deficient, and Hrh1-deficient mice were analyzed for Bphs. Deaths were recorded at 30 min, and the results expressed as the number dead/number studied.
Individual PKC isoforms do not mediate Bphs.
In most mammalian tissues, H1R induces the activation of phospholipase C (PLC) via PTX-resistant Gαq/11-proteins to generate inositol-1,4,5-triphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol-4,5-bisphosphate (43). IP3 leads to increased intracellular Ca2+, and DAG leads to activation of specific isoforms of protein kinase C (PKC). We therefore examined the requirements for activation of PKC downstream of H1R in eliciting Bphs. There are at least 12 PKC isoforms (46), and we surveyed selected gene-deficient mice representing representative members of the different isozyme subclasses for their roles in susceptibility to Bphs. We selected three “conventional” DAG/Ca2+-dependent isozymes (PKC-α, -β, and -γ), two DAG-dependent and Ca2+-independent “novel” class members (PKC-δ and -ε), and one DAG- and Ca2+-independent “atypical” member (PKC-ζ) for PTX sensitization and subsequent HA challenge. All of these gene-deficient mice are on the B6 background, but all carry 129 strain DNA because of gene targeting in strain 129 embryonic stem cells. However, we found no difference in Bphs in the background control strains over a 16-fold HA dose range (Table 8). As expected, H1RKO mice were Bphs resistance, again demonstrating the critical role of Hrh1 in eliciting Bphs. Lastly, we observed that all B6.129 PKC KO mice were susceptible to Bphs (Table 8). Thus, irrespective of the mode of activation or tissue expression patterns, neither PKC-α, -β, -γ, -δ, -ε, nor -ζ individually appears to control Bphs.
TABLE 8.
Bphs in PKC KO micea
| Strain | No. of mice dead/total after challenge with HA at: |
||
|---|---|---|---|
| 50 mg/kg | 12.5 mg/kg | 3.13 mg/kg | |
| 129XlISvJ | 10/10 | 10/10 | 4/6 |
| C57BLl6J | 6/6 | 7/8 | 4/6 |
| B6129PF2/J | 4/4 | 4/4 | 5/6 |
| Hrh1−/− | 0/4 | 0/4 | 0/4 |
| Prkca (PKCα)−/− | 3/3 | 3/3 | 3/3 |
| Prkcb (PKCβ)−/− | 3/3 | 3/3 | 3/3 |
| Prkcc (PKCγ)−/− | 6/6 | 8/8 | 9/9 |
| Prkcd (PKCδ)−/− | 4/4 | 3/3 | 3/3 |
| Prkce (PKCε)−/− | 11/11 | 3/3 | 3/3 |
| Prkcz (PKCζ)−/− | 4/4 | 3/3 | 1/2 |
WT controls and PKC-α, -β, -γ, -δ, -ε, and -ζ singly deficient mice were analyzed for Bphs. Deaths were recorded at 30 min, and the results expressed as the number dead/number studied.
Bphs is not controlled by Tlr4.
Experiments with murine splenocytes using radiolabeled PTX and lipopolysaccharide (LPS) showed that both compounds bind to Toll-like receptor 4 (47). In 2004, Kerfoot et al. reported that induction of experimental autoimmune encephalomyelitis (EAE) with PTX as an ancillary adjuvant and PTX-mediated lymphocyte rolling and adhesion were diminished in the absence of Tlr4 (48). In conjunction with a recent report showing that commercial PTX preparations can contain LPS/endotoxin (49), we found that our source of PTX contained approximately 40 endotoxin units (EU) per PTX sensitization inoculum. This concentration is equivalent to 0.001 EU/kg of body weight (data not shown), which is far below the 5-EU/kg cutoff in preclinical animal models (50). In light of these considerations, we tested the hypothesis that the VAAS activity of PTX in the Bphs model was dependent on Tlr4. For these experiments, we sensitized WT C57BL/10J mice and C57BL/10ScNJ mice with active holotoxin and challenged them with HA 3 days later. C57BL/10ScNJ mice have a deletion of the Tlr4 gene that results in the absence of both mRNA and protein (Tlr4lps-del) and thus is defective in response to LPS stimulation (24). Tlr4lps-del differs from the Tlr4Lps-d mutation of C3H/HeJ mice, a point mutation that causes an amino acid substitution, resulting in a hypomorphic Tlr4 gene product (24). Studying the responsiveness of C57BL/10ScNJ mice therefore precludes the possibility that PTX can still mediate its effects through a non-LPS-responsive allele. We found that both C57BL/10J and C57BL/10ScNJ mice were Bphs (Table 9). These data are also consistent with our previous study showing that PTX-induced angiogenesis of brain microvascular ECs was not affected by the lack of Tlr4 signaling (51). Thus, our data suggest that while certain defined effects of PTX, such as leukocyte recruitment, may require Tlr4, the HA-sensitizing activity of PTX does not.
TABLE 9.
Bphs in Tlr4 mutant micea
| Strain | No. of mice dead/total after challenge with HA at: |
||
|---|---|---|---|
| 50 mg/kg | 12.5 mg/kg | 3.125 mg/kg | |
| C57BL/10J | 5/5 | 6/6 | 1/6 |
| C57BL/10ScNJ | 5/5 | 6/6 | 0/6 |
C57BL/10J (Tlr4+) and C57BL/10ScNJ (Tlr4-deficient) mice were analyzed for Bphs. Deaths were recorded at 30 min, and the results expressed as the number dead/number studied.
Gαi1/3 are critical PTX targets for elicitation of VAASH.
The ADP-ribosylating activity of PTX is a well-established mechanism of action whereby PTX inhibits the function of Gαi/o proteins (52) and is required to elicit the myriad physiological responses following in vivo intoxication, including VAAS and the immune potentiating effects in EAE (18). Conjugation of ADP-ribose to a key cysteine residue in the C terminus of Gαi/o (but not Gαs, Gαq, or Gα11) proteins prevents the association of the Gαi/oGβγ complex with the GPCR (5, 26). Therefore, PTX is a powerful tool to discriminate whether a given GPCR signals through Gαi/o-dependent or Gαi/o-independent mechanisms (19). However, there are several Gαi proteins (Gαi1, Gαi2, Gαi3, and Gαz) and Gαo proteins (Gαo1 and Gαo2, splice variant products of the Gnao gene) (28). To determine which Gαi/o proteins are targeted by PTX for HA sensitization, we directly assessed the responses of mice genetically deficient in specific Gαi or Gαo proteins to HA challenge. We did not test Gαz, since this protein lacks the key cysteine for PTX-mediated ADP-ribosylation (29) and is therefore not affected by PTX. Our hypothesis was that genetic deficiency in one or more Gαi/o proteins would mimic HA hypersensitivity; i.e., such mice would be Bphs without prior PTX sensitization.
We first addressed the role of Gαo proteins as targets for ADP-ribosylation in VAASH. Gαo1 and Gαo2 are different gene products because of alternative exon usage in the Gnao gene (30). Gnao-deficient mice lack both Gαo1 and Gαo2 splice products because of disruption of Gnao exon 6, which is common to both gene products (30). Neither Gnao-deficient mice nor mice specifically lacking either Gαo1 or Gαo2 were spontaneously sensitive to HA (Fig. 1). These results demonstrated that the HA-sensitizing activity of PTX was not due to its ADP-ribosylation of Gαo proteins.
FIG 1.
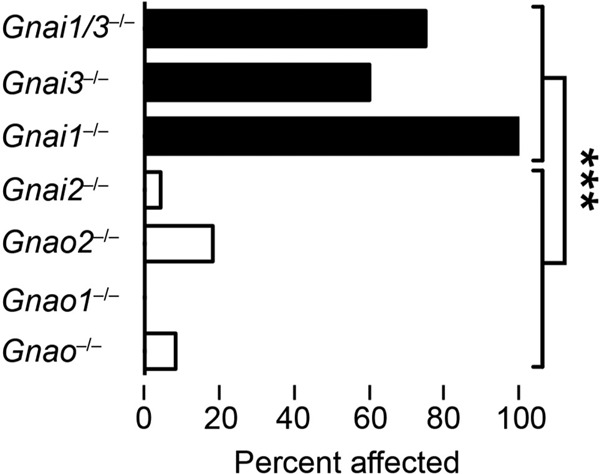
Loss of Gαi1 or Gαi3 mimics PTX to elicit HA sensitization. Mice deficient in Gαi/o subunits were analyzed for HA sensitivity in the absence of PTX exposure. Mice received 100 mg/kg HA i.v., and deaths were recorded within 30 min. Statistical significance was determined by one-way ANOVA using 8 to 23 mice per group. ***, P < 0.001.
Next we addressed the putative role of Gαi proteins as targets for PTX. Gαi1, Gαi2, and Gαi3 are encoded by distinct genes, Gnai1, Gnai2, and Gnai3, respectively (29), and are widely expressed (28). Compared to WT mice, Gαi1−/− (P = 0.0002) and Gαi3−/− (P = 0.008) mice were significantly more sensitive to HA, whereas Gαi2−/− mice were not (see Table S2 in the supplemental material). We also analyzed the response of Gαi1−/− Gαi3−/− double-KO mice and found them to also be significantly more susceptible to spontaneous HA sensitivity than WT mice are (P = 0.001) (see Table S2). Additionally, if one directly compares the susceptibility to spontaneous sensitivity to HA among the various KO strains, there is a significant overall effect of genotype (P < 0.0001) with no significant difference among Gnao−/−, Gαo1−/−, Gαo2−/−, and Gαi2−/− mice (P = 0.5) or among Gαi1−/−, Gαi3−/−, and Gαi1−/− Gαi3−/− mice (P = 0.3); however, there is a highly significant difference in susceptibility to spontaneous HA sensitivity between the two groups (P < 0.0001). Taken together, these results demonstrate that Gαi1 and Gαi3 are clear targets of ADP-ribosylation associated with PTX-induced VAASH.
DISCUSSION
We have focused on understanding the genetic basis for VAAS elicited by PTX sensitization. Changes in vascular permeability elicited during inflammation may promote antigen leakage and contribute to pathogen dissemination. Dysregulated vascular permeability may allow for inappropriate tissue access to immune cell surveillance, leading to tissue damage (13). In this regard, PTX has long been known to alter vascular permeability through its ability to promote VAAS (53). The vasoactive amines HA, 5-HT, and BDK have all been shown to influence vascular permeability, in particular, the BBB (16, 53, 54). Here we have shown that after PTX sensitization, (i) hypersensitivity to HA, 5-HT, or BDK (Bphs, Bpss, and Bpbs, respectively) is uniquely genetically controlled; (ii) unlike the genetic control of Bphs, that of Bpss and Bpbs and is not linked to any of the known 5-HT or BDK receptors; (iii) Bpbs is mediated by Bdkrb2; and (iv) Gαi1/3 proteins are the physiological in vivo targets of ADP-ribosylation by PTX associated with VAASH.
Under inflammatory conditions, vascular permeability is modulated by activated leukocytes acting on the vascular unit either through secreted or cell surface factors (13, 54). Mast cells are classically known to produce and store HA, 5-HT, and other amine/lipid/glycosylated mediators (55). More recently, mast cells have been shown to also produce BDK (14). Mast cells perfuse tissues and rapidly respond to external local stimuli which may act as secretagogues to stimulate the release of stored granular contents, including vasoactive amines. In fact, the critical role of mast cell-derived products in the control of vascular permeability was recently shown (14). Concomitant with the relaxed vascular permeability promoted by mast cell degranulation, neutrophils may gain tissue access to promote damage and promote subsequent recruitment of adaptive immune cells (13). From these data, one may then posit that environmental exposure may affect mast cells or the target vasculature to influence vascular permeability.
Mast cell number, function (56, 57), and susceptibility to VAAS vary across different genetic backgrounds (58, 59). Although Bphs was mapped by using the SJL/J-derived Hrh1 allele (11, 12), up to 75% of the available inbred mouse strains harbor this same allele (11; R. Noubade, unpublished data). We and others have indeed observed that SJL/J mice were also susceptible to Bpss (17, 53), though Bpss was not mediated by Hrh1 (17). Like HA and 5-HT, a BDK challenge after PTX intoxication also leads to death due to acute hypovolemic and hypotensive shock. Given these phenotypic similarities and the extensive genetic variation across inbred laboratory mouse strains, we tested whether a conserved genetic mechanism controls general VAAS by PTX. To address this possibility, we examined responses to HA, 5-HT, and BDK in a panel of inbred mice. Among these strains, we observed the full spectrum of VAAS to different individual mediators, indicating that the phenotypic variation in responsiveness to PTX reflects the genetic control of distinct intermediate phenotypes rather than allelic variation in genes controlling overall susceptibility to PTX intoxication.
Furthermore, using F1 hybrid crosses, we found that Bpss and Bpbs are autosomal recessive, while Bphs is autosomal dominant (12). The susceptibility pattern for Bpss and Bpbs in (SJL/J × B10.S) × SJL/J N2 crosses was consistent with a two-gene model of genetic control. Unlike Bphs, which showed genetic linkage to a key HA receptor, Hrh1 (11), for both Bpss and Bpbs, susceptibility was not linked to genes encoding the cognate ligand receptors. Furthermore, although Bpbs was not linked to Bdkrb1/2, this phenotype was abrogated in Bdkrb2-deficient mice, indicating that Bdkrb2 is nonetheless required for Bpbs. Taken together, our results confirm and extend the finding that the phenotypic variation in susceptibility to VAAS following PTX sensitization reflects the unique genetic control of specific intermediate phenotypes, i.e., Bphs, Bpss, and Bpbs, rather than susceptibility and resistance to intoxication in general.
Previously, we investigated the cell type requirements for H1R expression in Bphs. Using bone marrow chimeras, we found that the nonhematopoietic compartment determined susceptibility to Bphs (16). We found that EC-specific expression of H1R was not sufficient to mediate Bphs (16). Here we extended our analysis of ECs in VAASH. Inhibition of Gαi proteins by PTX has been shown to increase eNOS (Nos3) expression (60), yet Nos3-deficient mice were not protected from Bphs. Similarly, disruption of caveolin-1 either alone or in conjunction with Nos3 disruption did not affect Bphs. Another possibility that we have not analyzed here is that VAASH is due to PTX-mediated inhibition of sphingosine-1-phosphate (S1P) receptor signaling in EC. S1P regulates barrier function, and mice deficient in S1P in plasma were more susceptible to HA-induced vascular leakage (61). Nonetheless, administration of PTX to mice expressing EC-specific H1R did not render them sensitive to VAASH (16), which argues against this notion. Taken together, these results did not formally rule out the possibility that Hrh1 on EC is required but suggested that coexpression of H1R in another component of the vascular unit may be required for Bphs. Given the roles for H1R in vascular smooth muscle and neural function (62, 63), we hypothesize that H1R signaling in non-ECs of the vascular unit may be an important component of Bphs (16). Indeed, in the central nervous system (CNS), astrocytic end feet projecting into the EC/smooth muscle layers that wrap cerebral arterioles are important in relaying neural signals that control arterial tone in the brain (64).
Our results illustrate a central role for Gα protein activation and GPCRs in vasoregulation. The mast cell mediators HA, 5-HT, and BDK all promote vascular leakage/edema (65), and all signal through GPCRs. Nonetheless, diversity in molecular responses is controlled by receptor specificity and the activation of different effector Gα proteins (28). Forty-five years ago, Bergman and Munoz introduced the concept of counterbalanced signals controlling VAAS, with a special emphasis on the interplay between HA and the adrenergic-receptor (AR) pathways (66). These authors also suggested that PTX might sensitize to HA by targeting AR pathways. More recent observations are also consistent with the existence of opposing Gα protein effects in VAAS. Many ARs are coupled to Gαi proteins (α2A, α2B, α2C) (67), whereas key receptors for the vasoactive amines (H1R, 5-HT2, and the B2 BDK receptor) are all linked to Gαq/11 (28). It was recently shown that the anaphylactic shock response is controlled by endothelial Gαq/11 signaling (68). Our data suggest that inactivation of Gαi1/3 proteins by PTX is a key destabilizing effect that may allow unabated signaling through Gαq/11 or Gαs-linked GPCRs such as those for HA, 5-HT, and BDK.
PTX catalyzes the ADP-ribosylation of Gαi/o proteins associated in the Gα/β/γ heterotrimer and prevents the interaction of the trimeric signaling complex with GPCRs (6). In mice, there are eight Gαi/o genes (Gnai1 to -3, Gnao, Gnaz, and Gnat1 to -3) which, because of alternative splice forms of Gnao, encode at least 10 different protein products (28). PTX has been generally considered a broad-spectrum Gαi/o inhibitor with only limited insight into either Gαi/o specificity or specific function. Here we have identified Gαi1 and Gαi3 as the targets of ADP-ribosylation associated with Bphs. Mice lacking Gαi1 and Gαi3 were spontaneously susceptible to Bphs, a response normally observed in intact mice only after intoxication with active PTX holotoxin. It is possible that PTX targets other than Gαi1/3 contribute to Bphs. This would be detected only if we treated Gαi1/3-deficient mice with PTX and observed 100% penetrance. We did not formally address this possibility because we are not aware of any WT or KO mouse strain besides Gαi1- or Gαi1/3-deficient mice that exhibit spontaneous HA sensitivity at 100% penetrance (17, 36). Therefore, we interpret our present results to be due to incomplete penetrance possibly due in part to HA dosing (17, 36). Furthermore, given that Bphs was seen in all other Gαi/o-deficient mice, our results show the specificity of PTX for Gαi1/3 with regard to HA sensitization.
We hypothesize that H1R signaling (which uses Gαq/11) in the vascular unit is counterbalanced by Gαi1/3-dependent signals to maintain proper vascular tone and permeability. The mice used here were ubiquitously deficient in Gαi1/3, precluding the assignment of Gαi1/3 function to a specific cell type. The GPCR(s) that links to Gαi1/3 in the control of Bphs is also not known, although ARs remain a possibility. Since the identification of Hrh1 as Bphs in 2002 (11), more has been learned about the other three HA receptors, two of which—H3R and H4R—are linked to Gαi/o and are therefore sensitive to PTX (63). H4R expression is restricted to the hematopoietic system, but our finding that Bphs is controlled by the nonhematopoietic system (16) rules out this receptor as a Gαi/o-linked candidate for PTX inhibition in Bphs. Expression of H3R, however, is found in the CNS and in the autonomic nervous system (69) and has been found to regulate BBB permeability and susceptibility to both CNS autoimmune disease (70) and CNS infection in the case of cerebral malaria (71). Accordingly, another potential explanation for the physiological Bphs response could involve PTX-mediated inhibition of H3R to destabilize baseline vascular permeability and allow HA to signal unabated through another of its receptors (H1R) to induce acute vasodysregulation.
Collectively, our data reveal that Gαi1/3 proteins play an important role in the sensing of PTX and the regulation of vascular permeability in response to vasoactive amines. While H1R signaling in the nonhematopoietic compartment appears to ultimately control Bphs, the hematopoietic compartment is likely to serve as a source of vasoactive amines, for example, with mast cells as major producers of HA. Thus, it is important to understand Gα function in specific cell types at the interface of vasculature and tissue. Under inflammatory conditions, Gαi2 expression in ECs, but not in leukocytes, is important in the extravasation of leukocytes into inflamed lung tissue (72). PTX exposure strongly impairs immune cell migration due to inhibition of Gαi/o-linked chemokine receptor signaling (73). Deletion of Gαi2 affects T and B cell homing under steady-state conditions (33, 74, 75) and leads to T cell hyperreactivity (76). However, not all functions of Gαi2 are unique to this molecule, since competition for CXCR3-mediated signaling between Gαi2 and Gαi3 occurs, where Gαi3 may compete for and quench Gαi2-mediated chemotaxis in in vitro migration assays (77). Extravasation of immune cells into tissues under inflammatory conditions involves a combination of chemokines (78, 79) and hence may involve different Gαi members. Our data linking PTX to Gαi1/3 function and the ability of PTX to enhance the development of inflammatory disease (10) implicate Gαi1- and Gαi3-linked GPCRs in this process.
Our data may also give insight into the molecular mechanisms discriminating immunity to B. pertussis provided through exposure to whole-cell pertussis (wP) via infection or vaccination or via vaccination with the acellular pertussis (aP) vaccine (which replaced wP in 1997). aP refers to a vaccine in which PTX is inactivated by chemical fixation or by genetic means. This modification decreases the incidence of side effects, but data showing that aP may exhibit less durability and protection than wP are mounting (80). Taken with our data, it is possible that the Gαi1/3-linked GPCRs inhibited by PTX are critical for durable immunity to B. pertussis. Indeed, production of inflammatory cytokines was increased in stimulated splenocytes from Gαi1/3-deficient mice while that of regulatory cytokines was decreased (81), lending support to the notion that Gαi1/3 damping may contribute to a robust systemic immune response. Identification of these critical GPCRs may lead to strategies for their blockade in order to maximize the benefits of aP while increasing vaccine durability. The hypersensitivity to vasoactive amines released during the course of inflammation, as we have modeled here with Bphs, may be an indicator of such an effective strategy.
Supplementary Material
ACKNOWLEDGMENTS
We thank Michael Leitges (Max Planck Institute, Hannover, Germany) and Lutz Birnbaumer (National Institute for Environmental Health Sciences, Research Triangle Park, NC) for kinds gifts of PKC- and Gαi/o-deficient mice, respectively. We thank the staff of the University of Vermont Animal Facility for expert care.
This research was supported by grants from the NIH (NS061014, NS069628, and NS36526).
Footnotes
Published ahead of print 9 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00971-13.
REFERENCES
- 1.von König CH, Halperin S, Riffelmann M, Guiso N. 2002. Pertussis of adults and infants. Lancet Infect. Dis. 2:744–750. 10.1016/S1473-3099(02)00452-8 [DOI] [PubMed] [Google Scholar]
- 2.Yajima M, Hosoda K, Kanbayashi Y, Nakamura T, Nogimori K, Mizushima Y, Nakase Y, Ui M. 1978. Islets-activating protein (IAP) in Bordetella pertussis that potentiates insulin secretory responses of rats. Purification and characterization. J. Biochem. 83:295–303 [DOI] [PubMed] [Google Scholar]
- 3.Yajima M, Hosoda K, Kanbayashi Y, Nakamura T, Takahashi I, Ui M. 1978. Biological properties of islets-activating protein (IAP) purified from the culture medium of Bordetella pertussis. J. Biochem. 83:305–312 [DOI] [PubMed] [Google Scholar]
- 4.Tamura M, Nogimori K, Murai S, Yajima M, Ito K, Katada T, Ui M, Ishii S. 1982. Subunit structure of islet-activating protein, pertussis toxin, in conformity with the A-B model. Biochemistry 21:5516–5522. 10.1021/bi00265a021 [DOI] [PubMed] [Google Scholar]
- 5.Hoshino S, Kikkawa S, Takahashi K, Itoh H, Kaziro Y, Kawasaki H, Suzuki K, Katada T, Ui M. 1990. Identification of sites for alkylation by N-ethylmaleimide and pertussis toxin-catalyzed ADP-ribosylation on GTP-binding proteins. FEBS Lett. 276:227–231. 10.1016/0014-5793(90)80548-W [DOI] [PubMed] [Google Scholar]
- 6.Katada T, Oinuma M, Ui M. 1986. Two guanine nucleotide-binding proteins in rat brain serving as the specific substrate of islet-activating protein, pertussis toxin. Interaction of the alpha-subunits with beta gamma-subunits in development of their biological activities. J. Biol. Chem. 261:8182–8191 [PubMed] [Google Scholar]
- 7.Kaslow HR, Burns DL. 1992. Pertussis toxin and target eukaryotic cells: binding, entry, and activation. FASEB J. 6:2684–2690 [DOI] [PubMed] [Google Scholar]
- 8.Morse SI, Morse JH. 1976. Isolation and properties of the leukocytosis- and lymphocytosis-promoting factor of Bordetella pertussis. J. Exp. Med. 143:1483–1502. 10.1084/jem.143.6.1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furman BL, Walker E, Sidey FM, Smith M. 1988. Metabolic disturbances produced by pertussis toxin, p 147–172 In Wardlaw AC, Parton R. (ed), Pathogenesis and immunity in pertussis. John Wiley and Sons, Chichester, United Kingdom [Google Scholar]
- 10.Baxter AG. 2007. The origin and application of experimental autoimmune encephalomyelitis. Nat. Rev. Immunol. 7:904–912. 10.1038/nri2190 [DOI] [PubMed] [Google Scholar]
- 11.Ma RZ, Gao J, Meeker ND, Fillmore PD, Tung KS, Watanabe T, Zachary JF, Offner H, Blankenhorn EP, Teuscher C. 2002. Identification of Bphs, an autoimmune disease locus, as histamine receptor H1. Science 297:620–623. 10.1126/science.1072810 [DOI] [PubMed] [Google Scholar]
- 12.Sudweeks JD, Todd JA, Blankenhorn EP, Wardell BB, Woodward SR, Meeker ND, Estes SS, Teuscher C. 1993. Locus controlling Bordetella pertussis-induced histamine sensitization (Bphs), an autoimmune disease-susceptibility gene, maps distal to T-cell receptor beta-chain gene on mouse chromosome 6. Proc. Natl. Acad. Sci. U. S. A. 90:3700–3704. 10.1073/pnas.90.8.3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker ME, Hatfield JK, Brown MA. 2012. New insights into the role of mast cells in autoimmunity: evidence for a common mechanism of action? Biochim. Biophys. Acta 1822:57–65. 10.1016/j.bbadis.2011.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oschatz C, Maas C, Lecher B, Jansen T, Bjorkqvist J, Tradler T, Sedlmeier R, Burfeind P, Cichon S, Hammerschmidt S, Muller-Esterl W, Wuillemin WA, Nilsson G, Renne T. 2011. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity 34:258–268. 10.1016/j.immuni.2011.02.008 [DOI] [PubMed] [Google Scholar]
- 15.Noubade R, Milligan G, Zachary JF, Blankenhorn EP, del Rio R, Rincon M, Teuscher C. 2007. Histamine receptor H1 is required for TCR-mediated p38 MAPK activation and optimal IFN-gamma production in mice. J. Clin. Invest. 117:3507–3518. 10.1172/JCI32792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu C, Diehl SA, Noubade R, Ledoux J, Nelson MT, Spach K, Zachary JF, Blankenhorn EP, Teuscher C. 2010. Endothelial histamine H1 receptor signaling reduces blood-brain barrier permeability and susceptibility to autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U. S. A. 107:18967–18972. 10.1073/pnas.1008816107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao JF, Call SB, Fillmore PD, Watanabe T, Meeker ND, Teuscher C. 2003. Analysis of the role of Bphs/Hrh1 in the genetic control of responsiveness to pertussis toxin. Infect. Immun. 71:1281–1287. 10.1128/IAI.71.3.1281-1287.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Black WJ, Munoz JJ, Peacock MG, Schad PA, Cowell JL, Burchall JJ, Lim M, Kent A, Steinman L, Falkow S. 1988. ADP-ribosyltransferase activity of pertussis toxin and immunomodulation by Bordetella pertussis. Science 240:656–659. 10.1126/science.2896387 [DOI] [PubMed] [Google Scholar]
- 19.Milligan G, Carr C, Gould GW, Mullaney I, Lavan BE. 1991. Agonist-dependent, cholera toxin-catalyzed ADP-ribosylation of pertussis toxin-sensitive G-proteins following transfection of the human alpha 2-C10 adrenergic receptor into rat 1 fibroblasts. Evidence for the direct interaction of a single receptor with two pertussis toxin-sensitive G-proteins, Gi2 and Gi3. J. Biol. Chem. 266:6447–6455 [PubMed] [Google Scholar]
- 20.Inoue I, Yanai K, Kitamura D, Taniuchi I, Kobayashi T, Niimura K, Watanabe T. 1996. Impaired locomotor activity and exploratory behavior in mice lacking histamine H1 receptors. Proc. Natl. Acad. Sci. U. S. A. 93:13316–13320. 10.1073/pnas.93.23.13316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borkowski JA, Ransom RW, Seabrook GR, Trumbauer M, Chen H, Hill RG, Strader CD, Hess JF. 1995. Targeted disruption of a B2 bradykinin receptor gene in mice eliminates bradykinin action in smooth muscle and neurons. J. Biol. Chem. 270:13706–13710. 10.1074/jbc.270.23.13706 [DOI] [PubMed] [Google Scholar]
- 22.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. 1996. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U. S. A. 93:13176–13181. 10.1073/pnas.93.23.13176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. 2001. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 276:38121–38138. 10.1074/jbc.M105408200 [DOI] [PubMed] [Google Scholar]
- 24.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088. 10.1126/science.282.5396.2085 [DOI] [PubMed] [Google Scholar]
- 25.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. 2004. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat. Med. 10:248–254. 10.1038/nm1000 [DOI] [PubMed] [Google Scholar]
- 26.West RE, Jr, Moss J, Vaughan M, Liu T, Liu TY. 1985. Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site. J. Biol. Chem. 260:14428–14430 [PubMed] [Google Scholar]
- 27.Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S. 1993. Modified hippocampal long-term potentiation in PKC gamma-mutant mice. Cell 75:1253–1262. 10.1016/0092-8674(93)90613-U [DOI] [PubMed] [Google Scholar]
- 28.Wettschureck N, Offermanns S. 2005. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85:1159–1204. 10.1152/physrev.00003.2005 [DOI] [PubMed] [Google Scholar]
- 29.Wilkie TM, Gilbert DJ, Olsen AS, Chen XN, Amatruda TT, Korenberg JR, Trask BJ, de Jong P, Reed RR, Simon MI, Jenkins NA, Copeland NG. 1992. Evolution of the mammalian G protein alpha subunit multigene family. Nat. Genet. 1:85–91. 10.1038/ng0592-85 [DOI] [PubMed] [Google Scholar]
- 30.Jiang M, Gold MS, Boulay G, Spicher K, Peyton M, Brabet P, Srinivasan Y, Rudolph U, Ellison G, Birnbaumer L. 1998. Multiple neurological abnormalities in mice deficient in the G protein Go. Proc. Natl. Acad. Sci. U. S. A. 95:3269–3274. 10.1073/pnas.95.6.3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang M, Spicher K, Boulay G, Martin-Requero A, Dye CA, Rudolph U, Birnbaumer L. 2002. Mouse gene knockout and knockin strategies in application to alpha subunits of Gi/Go family of G proteins. Methods Enzymol. 344:277–298. 10.1016/S0076-6879(02)44721-0 [DOI] [PubMed] [Google Scholar]
- 32.Pineda VV, Athos JI, Wang H, Celver J, Ippolito D, Boulay G, Birnbaumer L, Storm DR. 2004. Removal of G(ialpha1) constraints on adenylyl cyclase in the hippocampus enhances LTP and impairs memory formation. Neuron 41:153–163. 10.1016/S0896-6273(03)00813-4 [DOI] [PubMed] [Google Scholar]
- 33.Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, Boulay G, Bradley A, Birnbaumer L. 1995. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat. Genet. 10:143–150. 10.1038/ng0695-143 [DOI] [PubMed] [Google Scholar]
- 34.Butterfield RJ, Sudweeks JD, Blankenhorn EP, Korngold R, Marini JC, Todd JA, Roper RJ, Teuscher C. 1998. New genetic loci that control susceptibility and symptoms of experimental allergic encephalomyelitis in inbred mice. J. Immunol. 161:1860–1867 [PubMed] [Google Scholar]
- 35.Blankenhorn EP, Butterfield R, Case LK, Wall EH, del Rio R, Diehl SA, Krementsov DN, Saligrama N, Teuscher C. 2011. Genetics of experimental allergic encephalomyelitis supports the role of T helper cells in multiple sclerosis pathogenesis. Ann. Neurol. 70:887–896. 10.1002/ana.22642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wardlaw AC. 1970. Inheritance of responsiveness to pertussis HSF in mice. Int. Arch. Allergy Appl. Immunol. 38:573–589. 10.1159/000230313 [DOI] [PubMed] [Google Scholar]
- 37.Weydert A, Cloez-Tayarani I, Fillion MP, Simon-Chazottes D, Guenet JL, Fillion G. 1992. Molecular cloning of two partial serotonin 5-HT1D receptor sequences in mouse and one in guinea pig. C. R. Acad. Sci. III 314:429–435 [PubMed] [Google Scholar]
- 38.Das S, Kumar KN. 1995. Nitric oxide: its identity and role in blood pressure control. Life Sci. 57:1547–1556. 10.1016/0024-3205(95)02130-B [DOI] [PubMed] [Google Scholar]
- 39.Francis SH, Corbin JD. 1999. Cyclic nucleotide-dependent protein kinases: intracellular receptors for cAMP and cGMP action. Crit. Rev. Clin. Lab Sci. 36:275–328. 10.1080/10408369991239213 [DOI] [PubMed] [Google Scholar]
- 40.Hussain MB, Hobbs AJ, MacAllister RJ. 1999. Autoregulation of nitric oxide-soluble guanylate cyclase-cyclic GMP signalling in mouse thoracic aorta. Br. J. Pharmacol. 128:1082–1088. 10.1038/sj.bjp.0702874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin S, Fagan KA, Li KX, Shaul PW, Cooper DM, Rodman DM. 2000. Sustained endothelial nitric-oxide synthase activation requires capacitative Ca2+ entry. J. Biol. Chem. 275:17979–17985. 10.1074/jbc.275.24.17979 [DOI] [PubMed] [Google Scholar]
- 42.Bongers G, de Esch I, Leurs R. 2010. Molecular pharmacology of the four histamine receptors. Adv. Exp. Med. Biol. 709:11–19. 10.1007/978-1-4419-8056-4_2 [DOI] [PubMed] [Google Scholar]
- 43.Hill SJ, Ganellin CR, Timmerman H, Schwartz JC, Shankley NP, Young JM, Schunack W, Levi R, Haas HL. 1997. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol. Rev. 49:253–278 [PubMed] [Google Scholar]
- 44.Leurs R, Brozius MM, Jansen W, Bast A, Timmerman H. 1991. Histamine H1-receptor-mediated cyclic GMP production in guinea-pig lung tissue is an l-arginine-dependent process. Biochem. Pharmacol. 42:271–277. 10.1016/0006-2952(91)90713-F [DOI] [PubMed] [Google Scholar]
- 45.Morais C, Ebrahem Q, Anand-Apte B, Parat MO. 2012. Altered angiogenesis in caveolin-1 gene-deficient mice is restored by ablation of endothelial nitric oxide synthase. Am. J. Pathol. 180:1702–1714. 10.1016/j.ajpath.2011.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mellor H, Parker PJ. 1998. The extended protein kinase C superfamily. Biochem. J. 332(Pt 2):281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lei MG, Morrison DC. 1993. Evidence that lipopolysaccharide and pertussis toxin bind to different domains on the same p73 receptor on murine splenocytes. Infect. Immun. 61:1359–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, Zanardo RC, Bonder C, James WG, Robbins SM, Kubes P. 2004. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J. Immunol. 173:7070–7077 http://www.jimmunol.org/content/173/11/7070.long [DOI] [PubMed] [Google Scholar]
- 49.Bache C, Spreitzer I, Becker B, Loeschner B, Rosskopf U, Hanschmann KM, Schwanig M, Schneider CK, Lieb B, Montag T. 2012. Bordetella pertussis toxin does not induce the release of pro-inflammatory cytokines in human whole blood. Med. Microbiol. Immunol. 201:327–335. 10.1007/s00430-012-0238-1 [DOI] [PubMed] [Google Scholar]
- 50.Malyala P, Singh M. 2008. Endotoxin limits in formulations for preclinical research. J. Pharm. Sci. 97:2041–2044. 10.1002/jps.21152 [DOI] [PubMed] [Google Scholar]
- 51.Lu C, Pelech S, Zhang H, Bond J, Spach K, Noubade R, Blankenhorn EP, Teuscher C. 2008. Pertussis toxin induces angiogenesis in brain microvascular endothelial cells. J. Neurosci. Res. 86:2624–2640. 10.1002/jnr.21716 [DOI] [PubMed] [Google Scholar]
- 52.Katada T, Ui M. 1982. Direct modification of the membrane adenylate cyclase system by islet-activating protein due to ADP-ribosylation of a membrane protein. Proc. Natl. Acad. Sci. U. S. A. 79:3129–3133. 10.1073/pnas.79.10.3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Linthicum DS, Munoz JJ, Blaskett A. 1982. Acute experimental autoimmune encephalomyelitis in mice. I Adjuvant action of Bordetella pertussis is due to vasoactive amine sensitization and increased vascular permeability of the central nervous system. Cell. Immunol. 73:299–310 [DOI] [PubMed] [Google Scholar]
- 54.Abbott NJ. 2000. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell. Mol. Neurobiol. 20:131–147. 10.1023/A:1007074420772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalesnikoff J, Galli SJ. 2008. New developments in mast cell biology. Nat. Immunol. 9:1215–1223. 10.1038/ni.f.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Becker M, Reuter S, Friedrich P, Doener F, Michel A, Bopp T, Klein M, Schmitt E, Schild H, Radsak MP, Echtenacher B, Taube C, Stassen M. 2011. Genetic variation determines mast cell functions in experimental asthma. J. Immunol. 186:7225–7231. 10.4049/jimmunol.1100676 [DOI] [PubMed] [Google Scholar]
- 57.Kitamura Y, Go S, Hatanaka K. 1978. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood 52:447–452 [PubMed] [Google Scholar]
- 58.Munoz J, Bergman RK. 1968. Histamine-sensitizing factors from microbial agents, with special reference to Bordetella pertussis. Bacteriol. Rev. 32:103–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oh H, Joung J, Kim BG, Nam KT, Hong SH, Song HC, Lee HL, Ahn BY. 2012. Improved protocols for histamine sensitization testing of acellular pertussis vaccines. Vaccine 30:7246–7252. 10.1016/j.vaccine.2012.10.005 [DOI] [PubMed] [Google Scholar]
- 60.Danson EJ, Zhang YH, Sears CE, Edwards AR, Casadei B, Paterson DJ. 2005. Disruption of inhibitory G-proteins mediates a reduction in atrial beta-adrenergic signaling by enhancing eNOS expression. Cardiovasc. Res. 67:613–623. 10.1016/j.cardiores.2005.04.034 [DOI] [PubMed] [Google Scholar]
- 61.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, Coughlin SR. 2009. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J. Clin. Invest. 119:1871–1879. 10.1172/JCI38575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim SF, Huang AS, Snowman AM, Teuscher C, Snyder SH. 2007. From the cover: antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc. Natl. Acad. Sci. U. S. A. 104:3456–3459. 10.1073/pnas.0611417104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parsons ME, Ganellin CR. 2006. Histamine and its receptors. Br. J. Pharmacol. 147(Suppl 1):S127–S135. 10.1038/sj.bjp.0706704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT. 2010. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc. Natl. Acad. Sci. U. S. A. 107:3811–3816. 10.1073/pnas.0914722107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Theoharides TC, Kalogeromitros D. 2006. The critical role of mast cells in allergy and inflammation. Ann. N. Y. Acad. Sci. 1088:78–99. 10.1196/annals.1366.025 [DOI] [PubMed] [Google Scholar]
- 66.Bergman RK, Munoz J. 1968. Efficacy of beta-adrenergic blocking agents in inducing histamine sensitivity in mice. Nature 217:1173–1174. 10.1038/2171173a0 [DOI] [PubMed] [Google Scholar]
- 67.Hein L. 2008. Alpha adrenergic system, p 42–45 In Offermanns S, Rosenthal W. (ed), Encyclopedia of molecular pharmacology, 2nd ed. Springer, Berlin, Germany [Google Scholar]
- 68.Korhonen H, Fisslthaler B, Moers A, Wirth A, Habermehl D, Wieland T, Schutz G, Wettschureck N, Fleming I, Offermanns S. 2009. Anaphylactic shock depends on endothelial Gq/G11. J. Exp. Med. 206:411–420. 10.1084/jem.20082150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, Jackson MR, Erlander MG. 1999. Cloning and functional expression of the human histamine H3 receptor. Mol. Pharmacol. 55:1101–1107 [PubMed] [Google Scholar]
- 70.Teuscher C, Subramanian M, Noubade R, Gao JF, Offner H, Zachary JF, Blankenhorn EP. 2007. Central histamine H3 receptor signaling negatively regulates susceptibility to autoimmune inflammatory disease of the CNS. Proc. Natl. Acad. Sci. U. S. A. 104:10146–10151. 10.1073/pnas.0702291104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beghdadi W, Porcherie A, Schneider BS, Morisset S, Dubayle D, Peronet R, Dy M, Louis J, Arrang JM, Mecheri S. 2009. Histamine H(3) receptor-mediated signaling protects mice from cerebral malaria. PLoS One 4:e6004. 10.1371/journal.pone.0006004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pero RS, Borchers MT, Spicher K, Ochkur SI, Sikora L, Rao SP, Abdala-Valencia H, O'Neill KR, Shen H, McGarry MP, Lee NA, Cook-Mills JM, Sriramarao P, Simon MI, Birnbaumer L, Lee JJ. 2007. Galphai2-mediated signaling events in the endothelium are involved in controlling leukocyte extravasation. Proc. Natl. Acad. Sci. U. S. A. 104:4371–4376. 10.1073/pnas.0700185104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ransohoff RM. 2009. Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity 31:711–721. 10.1016/j.immuni.2009.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Han SB, Moratz C, Huang NN, Kelsall B, Cho H, Shi CS, Schwartz O, Kehrl JH. 2005. Rgs1 and Gnai2 regulate the entrance of B lymphocytes into lymph nodes and B cell motility within lymph node follicles. Immunity 22:343–354. 10.1016/j.immuni.2005.01.017 [DOI] [PubMed] [Google Scholar]
- 75.Hwang IY, Park C, Kehrl JH. 2007. Impaired trafficking of Gnai2+/− and Gnai2−/− T lymphocytes: implications for T cell movement within lymph nodes. J. Immunol. 179:439–448 http://www.jimmunol.org/content/179/1/439 [DOI] [PubMed] [Google Scholar]
- 76.Huang TT, Zong Y, Dalwadi H, Chung C, Miceli MC, Spicher K, Birnbaumer L, Braun J, Aranda R. 2003. TCR-mediated hyper-responsiveness of autoimmune Galphai2(−/−) mice is an intrinsic naive CD4(+) T cell disorder selective for the Galphai2 subunit. Int. Immunol. 15:1359–1367. 10.1093/intimm/dxg135 [DOI] [PubMed] [Google Scholar]
- 77.Thompson BD, Jin Y, Wu KH, Colvin RA, Luster AD, Birnbaumer L, Wu MX. 2007. Inhibition of G alpha i2 activation by G alpha i3 in CXCR3-mediated signaling. J. Biol. Chem. 282:9547–9555. 10.1074/jbc.M610931200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bartholomäus I, Kawakami N, Odoardi F, Schlager C, Miljkovic D, Ellwart JW, Klinkert WE, Flugel-Koch C, Issekutz TB, Wekerle H, Flugel A. 2009. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 462:94–98. 10.1038/nature08478 [DOI] [PubMed] [Google Scholar]
- 79.Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. 2009. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 10:514–523. 10.1038/ni.1716 [DOI] [PubMed] [Google Scholar]
- 80.Witt MA, Arias L, Katz PH, Truong ET, Witt DJ. 2013. Reduced risk of pertussis among persons ever vaccinated with whole cell pertussis vaccine compared to recipients of acellular pertussis vaccines in a large US cohort. Clin. Infect. Dis. 56:1248–1254. 10.1093/cid/cit046 [DOI] [PubMed] [Google Scholar]
- 81.Fan H, Zingarelli B, Peck OM, Teti G, Tempel GE, Halushka PV, Spicher K, Boulay G, Birnbaumer L, Cook JA. 2005. Lipopolysaccharide- and Gram-positive bacteria-induced cellular inflammatory responses: role of heterotrimeric Galpha (i) proteins. Am. J. Physiol. Cell Physiol. 289:C293–C301. 10.1152/ajpcell.00394.2004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.