

Abstract
Herpes simplex virus 1 (HSV-1) causes a chronic, lifelong infection in >60% of adults. Multiple recent vaccine trials have failed, with viral diversity likely contributing to these failures. To understand HSV-1 diversity better, we comprehensively compared 20 newly sequenced viral genomes from China, Japan, Kenya, and South Korea with six previously sequenced genomes from the United States, Europe, and Japan. In this diverse collection of passaged strains, we found that one-fifth of the newly sequenced members share a gene deletion and one-third exhibit homopolymeric frameshift mutations (HFMs). Individual strains exhibit genotypic and potential phenotypic variation via HFMs, deletions, short sequence repeats, and single-nucleotide polymorphisms, although the protein sequence identity between strains exceeds 90% on average. In the first genome-scale analysis of positive selection in HSV-1, we found signs of selection in specific proteins and residues, including the fusion protein glycoprotein H. We also confirmed previous results suggesting that recombination has occurred with high frequency throughout the HSV-1 genome. Despite this, the HSV-1 strains analyzed clustered by geographic origin during whole-genome distance analysis. These data shed light on likely routes of HSV-1 adaptation to changing environments and will aid in the selection of vaccine antigens that are invariant worldwide.
INTRODUCTION
Herpes simplex virus 1 (HSV-1; species Human herpesvirus 1, genus Simplexvirus, subfamily Alphaherpesvirinae, family Herpesviridae, order Herpesvirales) is among the most successful human pathogens in terms of its global distribution, longevity in the host, and mild symptoms among the great majority of those exposed (1–4). HSV-1 is a large, enveloped DNA virus that infects lytically at epithelial surfaces and establishes a lifelong, latent infection in sensory neurons. HSV-1 infection produces a wide range of symptoms, ranging from few or none in many seropositive individuals to periodic lesions on epithelial surfaces in a significant proportion of people and to lethal encephalitis as an extreme manifestation in a few. There is no vaccine at present (5, 6). Studies in animal models have characterized the ways in which genetic variation between viral strains can influence the symptoms of pathology, including lesion severity and rates of reactivation from latency. The most recent phase III vaccine trial for HSV failed to provide protection from infection (7, 8), and one contributing factor to this failure may well be variation among HSV isolates found in the field.
Based on early restriction fragment length polymorphism (RFLP) analyses, HSV-1 has been described as more diverse than HSV-2 (9–11). In contrast to both HSV-1 and HSV-2, the related human alphaherpesvirus, varicella-zoster virus (VZV), has relatively low interstrain diversity (12–15). Decades of research comparing RFLP bands, polypeptide size, and PCR-based sequence analysis have revealed that HSV-1 strains vary between individuals, over sequential isolates from the same individual, and by geographic region (10, 16–28). However, these approaches have limitations. RFLP analysis can be applied on a genome-wide basis to collections of strains but only reveals major differences in fragment size. More sensitive techniques, such as PCR and polypeptide analysis, provide greater detail about individual genes and proteins but are not practical for whole genomes or large strain collections. However, the application of high-throughput sequencing can rapidly increase our knowledge of sequence diversity among HSV-1 strains.
An analysis of genome-wide variation in two sequenced HSV-1 strains (F and H129) (29) in comparison to the reference strain 17 revealed much greater coding diversity in the 152-kb genome than had been found for VZV (14, 15). However, the small number of strains analyzed (n = 3) was limiting. Although research on the genomes of two slightly larger collections (n = 7 [30] and n = 9 [13]) has since been published, the sequences are either incomplete in a substantial number of coding sequences or are not available publicly. Without further data, we have been unable to answer questions raised about the amount of diversity among strains, the frequency of recombination, and the potential for rapid evolution in key targets of the immune system. Further knowledge of these areas will aid the development of a broadly effective vaccine and may also lead to improved treatments to control HSV infection.
To address these questions, we have determined the genome sequences of 20 HSV-1 strains, which, together with published data, create a sizeable collection of sequences (n = 26) and provide a global view of circulating strains. We observed large deletions and frameshifts that are deleterious for growth in vivo, suggesting in vitro expansion of these mutations. Various mechanisms of natural variation were evident, including single nucleotide polymorphisms (SNPs) and small insertions and deletions due to length variation in short sequence repeats (SSRs). The least divergent open reading frames (ORFs) tended to be those that are conserved among herpesviruses and are involved in basic aspects of replication. Within individual proteins, a small number of amino acid residues exhibited signs of positive selection. These data create a framework for future analyses of clinical HSV-1 isolates and provide a context for the development of intervention strategies and vaccine candidates that embrace the breadth of circulating viral diversity.
MATERIALS AND METHODS
Viruses and sequencing.
Following the standard nomenclature of microbiology, we refer to the viruses used as strains, because these have been isolated, grown as pure cultures in vitro, and characterized to the point of genome sequencing (31). Twenty passaged HSV-1 strains were provided by Hiroshi Sakaoka, as DNA isolated from infected cell lysates. In all cases, the isolates were selected from epidemiologically unrelated cases, from patients who were ethnically native to each country. Sakaoka and colleagues cultured isolates on Vero cells at a low multiplicity of infection (MOI), without plaque purification, and isolated viral DNA from high-titer stocks. These approaches are described similarly in all of Sakaoka's multiple publications about these strain collections (10, 18, 20, 32–34), but the procedures are particularly clearly delineated in several papers (21, 35, 36). Since Sakaoka and colleagues performed multiple analyses using overlapping sets of strains, we list here the first publication that described each set of sequenced strains. The Chinese strain (CR38) was part of a collection of strains isolated in Shenyang between 1980 and 1988 (20). The Japanese strains were isolated in Sapporo (on Hokkaido Island) between 1980 and 1983 (36). The Kenyan strains were isolated in Nairobi from 1981 to 1984 (32).The South Korean strains were isolated in Seoul between 1980 and 1988 (20).
Details on sequencing the strains are summarized in Table 1. Eight strains (CR38, E03, E14, E19, S23, S25, R11, and R62) were recovered from the original DNA stocks by transfection of baby hamster kidney clone 21 (BHK-21) cells, and fresh DNA stocks were prepared from purified virions, without plaque purification and with the minimum expansion required to prepare virions. Six of these recovered strains (CR38, E03, E14, E19, S23, and S25) were sequenced by using the Sanger approach, via random DNA plasmid clones. The other two recovered strains (R11 and R62) were sequenced by using an Illumina GAIIx instrument, via direct sonication of viral DNA. The remaining 14 strains (Table 1) were also sequenced by using an Illumina GAIIx instrument, using the original DNA stocks received from Hiroshi Sakaoka. Data on read length, total number of reads, proportion of reads matching virus versus host, and average coverage of the finished sequence are listed in Table S1 in the supplemental material.
TABLE 1.
Sources of HSV-1 strains, sequencing methods used, and accession numbers
| Strain | Country (city), time (yr) of strain collection/isolation (reference[s]) | Sequencing method (reference[s]) | Accession no. |
|---|---|---|---|
| 17 | UK (Glasgow), 1973 (51) | Prior (44, 45) | JN555585, NC_001806 |
| CR38a | China (Shenyang), 1980-1988 (20) | Sanger | HM585508 |
| E03a | Kenya (Nairobi), 1981-1984 (32) | Sanger | HM585509 |
| E06 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585496 |
| E07 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585497 |
| E08 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585498 |
| E10 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585499 |
| E11 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585500 |
| E12 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585501 |
| E13 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585502 |
| E14a | Kenya (Nairobi), 1981-1984 (32) | Sanger | HM585510 |
| E15 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585503 |
| E19a | Kenya (Nairobi), 1981-1984 (32) | Sanger | HM585511 |
| E22 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585504 |
| E23 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585505 |
| E25 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585506 |
| E35 | Kenya (Nairobi), 1981-1984 (32) | Illumina | HM585507 |
| R11a | South Korea (Seoul), 1980-1988 (20) | Illumina | HM585514 |
| R62a | South Korea (Seoul), 1980-1988 (20) | Illumina | HM585515 |
| S23a | Japan (Sapporo), 1980-1983 (36) | Sanger | HM585512 |
| S25a | Japan (Sapporo), 1980-1983 (36) | Sanger | HM585513 |
| F | USA (Chicago), 1968 (52) | Prior (29) | GU734771 |
| H129 | USA (San Francisco), 1983 (53) | Prior (29) | GU734772 |
| McKrae | USA (Gainesville), 1965 (55) | Prior (49) | JQ730035, JX142173 |
| KOS | USA (Houston), 1964 (54) | Prior (48) | JQ673480, JQ780693 |
| HF10 | USA (New York),b 1925 (56, 57) | Prior (47) | DQ889502 |
Assembly and annotation of genome sequences.
Sanger sequence reads were assembled and edited by using the Staden software (37). Illumina sequence reads were assembled de novo by using Velvet (38) as described previously (39). The resulting contigs were oriented and assembled by alignment against the genome sequence of HSV-1 reference strain 17, producing a draft genome sequence. The reads were aligned against this draft by using Maq (40), and the output was quality checked by visualizing the alignment in Tablet (41). Improvements to the sequence were made by iterative alignment and visualization. In addition to providing the final sequences, this approach was capable of revealing major deleted or defective DNA populations, which were evident from regions of unusually high- or low-read coverage in the high-throughput sequencing data.
A DNA sequence alignment containing the 20 genome sequences along with those of the six previously analyzed genomes was created by using Gap4 (37) and manually curated to improve the alignment around sequence gaps. As described in Results and Discussion below, the alignment consisted of trimmed versions of the genome (lacking terminal repeat long [TRL] and terminal repeat short [TRS] regions). Annotations from reference strain 17 were transferred to the other strains on the basis of the alignment, which was used as the input for dendrogram generation and analyses of positive selection. The alignment is available at http://szparalab.psu.edu/hsv-diversity/. After annotation, full-length versions of each genome were created by placing inverted copies of internal repeat long (IRL) and internal repeat short (IRS) regions at the appropriate termini. The GenBank record for each strain (Table 1) presents this full-length genome version; the trimmed-format genome is available under the Revision History for each record (42). Both genome formats are available at the URL mentioned above.
The finished and annotated data for the new strains are listed in GenBank as partial genome sequences (Table 1), since all contain gaps at several major SSRs or reiterations. Table S1 in the supplemental material lists which strains have indeterminate SSR lengths at these locations. These are marked as gaps of 100 (N100) in the finished GenBank sequences. The indeterminate SSRs are listed here with the designations used herein and their locations in the full genome of reference strain 17 (JN555585): SSRUL (positions 71604 to 71814), SSRRL1 (9033 to 9213 and 117160 to 117341), SSRRL4 (5732 to 5878 and 120496 to 120642), SSRRL6 (988 to 1040 and 125334 to 125386), SSRa' (1 to 399, 125975 to 126373, and 151824 to 152222), SSRRS1 (126573 to 126712 and 151486 to 151624), SSRRS2 (126813 to 127145 and 151052 to 151384), SSRRS3 (132391 to 132516 and 145681 to 145806), SSRUS1 (143716 to 143868), and SSRUS2 (144787 to 145003). These SSRs affect three protein-coding sequences, where they form repeating amino acid tracts. The length of the repeating amino acid tract is not known for a majority of the newly sequenced strains or for several published strains (see Table S1; see also the GenBank records). VP1-2 (UL36) contains SSRUL and is indeterminate in 17 genomes from the 26-strain collection. ICP34.5 (RL1) contains SSRRL6 and is indeterminate in 10 genomes. gI (US7) contains SSRUS1 and is indeterminate in 14 genomes. One strain, S23, has a sequencing gap in ICP4 (RS1); this is noted in the GenBank record and in Table S1.
Previously sequenced genomes.
Six previously sequenced HSV-1 strains were included in the analysis, with data derived from each strain's GenBank record and associated publication(s). The publications describing these sequenced genomes are as follows, and the accession numbers for each sequence are listed in Table 1: 17 (43–46), HF10 (47), F (29), H129 (29), KOS (48), and McKrae (49, 50). Since the geographic origin of HSV-1 strains is considered in our analyses, the publications describing the first isolation and geographic origin of each strain are as follows: 17 (51), F (52), H129 (53), KOS (54), and McKrae (55). Strain HF10 was isolated by a laboratory in Japan (56) as a spontaneous in vitro derivative of the U.S. strain HF (57), and therefore, we list the country of this strain's origin as the United States. Four additional representatives of the wider herpesvirus family were included for comparison of G+C content and prevalence of tandem repeats. Data for these were drawn from the respective RefSeq records: VZV strain Dumas (NC_001348), human cytomegalovirus (HCMV) Merlin (NC_006273), Epstein-Barr virus (EBV) Raji (NC_007605), and Kaposi's sarcoma-associated herpesvirus (KSHV) GK18 (NC_009333).
We updated the genome sequence of reference strain 17 (GenBank accession number NC_001806) on the basis of data from a whole-genome clone (GenBank accession number FJ593289) and a transcriptomic study (A. Davison, unpublished data), depositing the annotation under GenBank accession number JN555585. This corrected known errors in the reference genome (NC_001806), such as minor double frameshifts in UL2 and UL17. We reassembled the high-throughput read data for strains F and H129, to validate the prior assembly and replace with N100 the eight indeterminate SSRs whose sequences had originally been copied from reference strain 17 (see Results; see also Table S1 in the supplemental material) (29). We did not have access to sequence read data for strains HF10, KOS, and McKrae, and we used these genome sequences as recorded in GenBank. Both records for KOS are listed in Table 1; we used the record published under accession number JQ673480 for the genome-wide alignment (48). For McKrae, we used the genome sequence from the record published under accession number JQ730035 (49). Three coding frameshifts deleterious for growth in vivo (in UL36, UL56, and US10) were found in the record under accession number JQ730035 for McKrae but not the alternate record under accession number JX142173; we used the latter amino acid sequence in these cases (49, 50). Seven substantially incomplete genomes (of U.S. origin) were not included in the comparisons because of a large number of gaps in both coding and noncoding regions (30). Partial genome sequences for nine strains (of U.S. and Swedish origin) were also not included, because the data are not publicly available and intergenic regions were not completed (13).
DNA variation analysis.
The transition/transversion ratio was calculated for the 26-genome alignment, using the MEGA software package (58). The transition/transversion ratio calculation used the formula R = [(A · G · k1) + (T · C · k2)]/[(A + G) · (T + C)], where k1 and k2 are transition/transversion ratios for purines (k1 = 3.53) and pyrimidines (k2 = 3.845), respectively. The number of nucleotide polymorphisms was measured using the Jukes-Cantor model in MEGA (59, 60). Variation in nucleotide polymorphisms across the alignment was plotted using DNAsp (61), with a sliding window of 500 bp (nongapped positions). These windows on the strain 17 genome are displayed to show the localization of DNA polymorphisms relative to HSV-1 coding sequences. Because each window represents 500 bp of ungapped residues, regions with large numbers of gapped positions in the multiple-genome alignment show a single window stretching over an area of >500 bp (e.g., see Fig. 3, kb 107 to 111). Mean pairwise identity was calculated using Geneious (version 6.1.6; Biomatters), which examines all pairs of bases in a column, assigning a score of 100% if identical, and then computes the mean of all pairs across all columns.
FIG 3.

Overview of the HSV-1 genome depicting coding regions, noncoding features, polymorphisms, and SSRs. (A) Locations of UL, US, IRL, and IRS in the genome of HSV-1 reference strain 17 (TRL and TRS are omitted). (B) Graph plotting the number of DNA polymorphisms per 500 bp (nongapped columns) in a whole-genome alignment of 26 HSV-1 sequences (from Table 1). (C) Well-known features of the HSV-1 reference genome are shown mapped to the two DNA strands. These include ORFs, the latency-associated transcript (LAT), untranslated regions (UTRs), origins of DNA replication (OriL and two copies of OriS), and microRNAs (miRNA or mir). Widely recognized protein names (e.g., gB, encoded by UL27) are included. (D) Locations of SSRs plotted along the reference genome, with homopolymers (the same nucleotide repeated ≥6 times in a row) plotted separately from larger microsatellites (repeating unit of 2 to 9 bp) and minisatellites (repeated unit of ≥10 bp). SSRs are color coded to distinguish those for which length is conserved in at least half of the 26 strains (green) versus those for which length is variable in a majority of strains (orange). Gray SSRs (marked by gray arrowheads) were not coded for conservation, since their length could not be determined by high-throughput sequencing in a majority of strains.
SSRs.
SSRs in the reference strain 17 genome were mapped by using MsatFinder and Tandem Repeat Finder (TRF) (62, 63), employing the approach described previously (64–66). First, MsatFinder was used to detect perfect repeats with units of 1 to 6 bp. To be counted by MsatFinder, homopolymer repeats (i.e., a repeating unit of 1 bp) were required to be >5 bp, and all other small repeats were required to be >9 bp (e.g., >4 copies of a dinucleotide repeat, >3 copies of a trinucleotide repeat, etc.). Next, TRF was used to locate larger or imperfect repeats (specifically, TRF version 4.04 with parameters as follows: match, 2; mismatch, 5; delta, 5; PM, 80; PI, 10; minScore, 40; and maxPeriod, 500). An alignment score of >39 was required for the TRF output, in order to reduce the chances of counting nonrepeats (65, 66). Finally, the MsatFinder and TRF results were combined, and overlapping repeats were removed, retaining the SSR with the higher alignment score in each case. Three microsatellites were detected by both TRF and MsatFinder; the duplicates were removed before calculating the overall SSR counts.
Orthologous SSRs were mapped in the genome sequence collection as described previously (65, 66), utilizing the multiple-genome alignment. This alignment was screened for SSRs comparable to those found in reference strain 17. The copy numbers of orthologous SSRs were recorded for all 26 strains, along with their locations in coding, promoter (<500 bp upstream from a coding sequence), or other intergenic regions. This list is available at http://szparalab.psu.edu/hsv-diversity/. Any SSR that was not present in more than half of the strains was excluded from further analysis; these occurred at the indeterminate SSRs described above. SSRs were then scored as conserved if more than half of the collection had an SSR that matched that of the reference strain 17 in both position and length (i.e., matched the sequence and copy number of the repeating unit).
Amino acid sequence alignments.
Corresponding amino acid sequences for each strain were grouped into multiline fasta files and aligned using T-Coffee (67). Individual alignments for each HSV-1 protein are available in text and color-coded format (as in Fig. 5) at http://szparalab.psu.edu/hsv-diversity/. Graphical presentations of these alignments were colored using Geneious. To extend the useful lifetime of these amino acid alignments, a working set of HSV-1 amino acid alignments, which will be updated regularly with new strain data (69), has been created on the Virus Pathogen Resource (ViPR) website at http://www.viprbrc.org/brc/workbench_landing.do?decorator=herpes&method=WorkbenchDetail&public=true. The ViPR amino acid alignments also include additional data available on a per-protein basis in GenBank, e.g., thymidine kinase has been sequenced from hundreds of strains.
FIG 5.

Amino acid (AA) sequence conservation in the RNA-binding protein and PKR antagonist US11. The amino acid alignment of US11 shows it to be 89.5% identical across the 26 strains analyzed. Gray in the bar across the top indicates identical residues in all strains; orange indicates nonidentity. The median divergence of all strains versus the consensus (top line) is 1.2% (98.8% similarity to the consensus) (see Table 3; see also Table S3 in the supplemental material). Green-shaded blocks above the alignment indicate known functional regions of the protein (146–152). The variations in US11 illustrate those commonly seen among HSV-1 strains. Boxes indicate strain-specific SNPs (e.g., P12S, V22M, V66I, and P112S), variations shared by a group of strains (e.g., G13C, R40H, P45S, and E162K), and SSR-related indels (PRX repeat beginning at residue 130).
Indeterminate SSRs occur in three protein-coding sequences (see “Assembly and annotation of genome sequences” above). The indeterminate SSR regions of these amino acid sequences were excised computationally before calculating protein divergence; the number of columns excised for each protein is listed in Table S3 in the supplemental material. For each protein alignment, the Henikoff and Henikoff algorithm was used to calculate weights for each sequence in the alignment, to reduce redundancy and emphasize diversity (70). A consensus sequence was built by choosing the amino acid with the largest sum of weights per column of the alignment. Insert columns were identified as those with gaps in a majority of strains by weight, e.g., the tail end of the US8A alignment exists only to accommodate a C-terminal extension unique to the KOS strain. The insert columns were excised and are listed in Table S3.
The remaining columns were used for calculations of diversity. To discriminate among the most highly conserved proteins, we counted all mutations in the amino acid alignment. Because this measure of perfect identity will flag as divergent all alignment columns that follow a frameshift or deletion in just one strain, it overlooks conservation among other strains in these regions. Therefore, we used another metric to assess the most divergent proteins, based on the median divergence from the amino acid consensus. We computed a weighted median divergence for each protein (71) by determining the median divergence per strain versus the consensus and then taking the median across weighted strains. To test whether the weighted median was performing the desired goal of reducing the impact of outliers, we repeated the above-described computation using the mean value instead of the median. We computed a weighted average divergence for each protein by the same approach, where we determined the average (mean) divergence per sequence versus the consensus and then averaged across sequences using their weights. Only proteins with known frameshift and deletion issues that occur misalignment (US9, UL55, and PK [UL13]) have mean and median divergences that differ by more than 1% (see Table S3 in the supplemental material).
For each divergence metric, we computed statistical significance as follows. The median value of the divergence was selected across all alignments, and this value was taken as the expected rate of divergence (pdiv), which we assume to be the same as the null model for all proteins. For each protein alignment, we counted the number of conserved columns (N) and the number of mutations observed (n, rounded to the nearest integer for the weighted median and mean metrics). We then computed the two-tailed P value of n using the Poisson distribution with the parameter N · pdiv (the expected number of mutations from the consensus). If the P value was smaller than 0.01 and n was greater than N · pdiv, we considered the protein to have larger than expected divergence. Likewise, if the P value was smaller than 0.01 and n was less than N · pdiv, we considered the protein to have a smaller than expected divergence (see Table S3 in the supplemental material).
Positive selection.
Table S4 in the supplemental material gives a summary of the positive-selection analysis on a gene-by-gene basis. For this analysis, amino acid alignments containing all 26 strains were curated to infer ancestral sequences for those with frameshifts and/or missing stop codons. This preserved the largest possible number of sequences (n) for calculation of positive selection. No sequence could be inferred for deleted regions, and trailing codons were removed. As in the calculations of amino acid identity, indeterminate SSR regions of the proteins VP1-2 (UL36), ICP34.5 (RL1), and gI (US7) were removed from the alignments due to a lack of data for more than half of the strain collection (see “Assembly and annotation of genome sequences” above).
A combination of published software and in-house scripts were used to analyze the protein collection for evidence of positive selection. Additional details on these methods are included at http://szparalab.psu.edu/hsv-diversity/. The multiple genome alignment described above was sliced based on annotated coding regions from the HSV-1 reference strain 17 genome. EMBASSY fdnadist, PHYLIP neighbor, and Codeml (part of the Parsimony Analysis by Maximum Likelihood [PAML] software package) were used to produce input and tree files and then to perform pairwise dN/dS ratio (ratio of nonsynonymous to synonymous evolutionary substitutions) analysis (72–74; PHYLIP version 3.52c; J. Felsenstein, University of Washington, Seattle, WA [http://evolution.genetics.Washington.edu/phylip.html]). Table S4 in the supplemental material lists the maximum, minimum, mean, and median values for both pairwise dN and pairwise dS for each alignment. The same files were used for positive-selection analysis, assuming either the M0 model (single value of Ω [dN/dS]) or M8 model (multiple values of Ω) for each alignment (106, 107). This gave an overall value for Ω and kappa (κ, transition/transversion bias) across the alignment and flagged sites detected as being under statistically significant positive selection. We also used the alternative sitewise likelihood ratio (Slr) (76) model to test for positive selection per protein and per amino acid residue. The outputs of all analyses are available at http://szparalab.psu.edu/hsv-diversity/.
The Protein Database (PDB) was interrogated using Molecular Operating Environment (MOE; Chemical Computing Group, Inc), employing amino acid sequences from reference strain 17 as input. Where protein matches were found, these were classified as “exact” if the input sequence matched a solved structure of an HSV-1 protein and as “model” if there was no exact match but another solved structure was sufficiently close to the HSV-1 protein to allow a homology model to be constructed. Homology modeling of HSV-1 gH (UL22) was carried out on the solved structure of the HSV-2 gH ectodomain (PDB ID 3M1C) (77) using MOE. Protein structures were visualized in MOE. Additional details on modeling are included at http://szparalab.psu.edu/hsv-diversity/.
Dendrograms and recombination analysis.
Tree diagrams of the genetic distances between HSV-1 strains were constructed using the multiple genome alignment. We calculated genetic distance by using the unweighted-pair group method using average linkages (UPGMA) method in MEGA (59, 78), with 1,000 bootstrap replicates (79), and we used the maximum composite likelihood method (80) for distance estimation. Bootscan analysis from the SimPlot (similarity plot) package (81) was used to test for similarity of DNA sequence between a test genome (e.g., reference strain 17) and other related genomes (e.g., other members of the 26-strain collection).
Nucleotide sequence accession numbers.
The newly determined sequences were submitted to GenBank under the accession numbers listed in Table 1.
RESULTS AND DISCUSSION
Determining genome sequences.
To understand the range of genome diversity among circulating HSV-1 strains, the sequences of 20 strains from various parts of the world were determined (Table 1). The strains chosen originated from China, Japan, Kenya, and South Korea. These strains were part of a much larger collection isolated by Sakaoka and colleagues, which were described in a series of RFLP-based studies on genome diversity (10, 18, 20, 32–34). Sakaoka and colleagues repeatedly found greater within-country diversity among Kenyan strains than in any other country under study, using RFLP pattern analysis (10, 20, 32), estimations of nucleotide diversity (20), and Sanger sequence comparisons (33). For this reason, a greater number of strains from Kenya than from other countries was included for sequencing. To augment the global diversity in our analyses, these 20 genomes were supplemented by six previously published sequences from the United States and United Kingdom (Table 1) (29, 43, 45, 47–50).
All strains used in these studies originated from patients who were ethnically native to their respective countries. Sakaoka and colleagues reported minimal passage of these strains (10, 18, 20, 32–34). The new genome sequences were determined from purified, randomly fragmented viral DNA by either the Illumina high-throughput sequencing approach or the Sanger method (Table 1). Coverage was 516 to 1,591 reads per nucleotide for the former method and 8 to 13 reads per nucleotide for the latter (see Table S1 in the supplemental material). The sequence read data were assembled de novo, and the resulting contigs were then ordered by alignment to the sequence of the HSV-1 reference strain 17.
The HSV-1 genome consists of two unique regions (unique long [UL], 108 kb, and unique short [US], 13 kb), each flanked by large inverted repeats (TRL/IRL, 9.2 kb, and TRS/IRS, 6.6 kb) (Fig. 1A). Protein-coding genes are named with a prefix that indicates the region in which they are located, followed by a number (e.g., UL1, RL1, RS1, and US1); some proteins also have common names, e.g., UL1 is also known as glycoprotein L (gL). Since sequence reads from one copy of an inverted repeat could not generally be distinguished from those from the other copy, the data were assembled into a trimmed version of the genome, which contained only one copy of each inverted repeat (IRL and IRS) (Fig. 1B). These genome sequences begin at the left end of UL, proceed through IRL+IRS, and finish at the right-hand end of US. A full-length genome was also generated for each strain by creating terminal copies of IRL and IRS; these were deposited in GenBank. For the analyses described here, we used the trimmed version of all genomes (Fig. 1B) to avoid double contributions from the G+C content, SSRs, and genes contained in the internal and terminal repeats. The nucleotide composition of the HSV-1 reference strain 17 (67.5% G+C) is mirrored in the newly sequenced strains (67.3 to 67.5%). As noted previously (44), the distribution of G+C residues is biased, with overall higher values in IRL+IRS (74.7% in strain 17) than UL (66.9%) or US (64.3%) (Fig. 2A). This is a feature of all sequenced HSV-1 strains (see Fig. S1 in the supplemental material; also data not shown). Enrichment of G+C residues in repeat regions is a general characteristic of alpha-, beta-, and gammaherpesvirus genomes, and it occurs even in the overall A+T-rich genome of VZV (Fig. 2B to E).
FIG 1.

The complete HSV-1 genome includes two unique regions and two sets of large inverted repeats. (A) The full structure of the HSV-1 genome includes a unique long region (UL) and a unique short region (US), each of which is flanked by inverted copies of large repeats known as the terminal and internal repeats of the long region (TRL and IRL) and the short region (TRS and IRS). The gene content of each region (UL, US, TRL/IRL, and TRS/IRS) is distinct, as shown in Fig. 3. The length of each region is marked; the regions are drawn approximately to scale. A short cleavage and packaging sequence called a is located as a direct repeat at both genome termini (in TRL and TRS) and as an inverted repeat (a') where IRS and IRL overlap. (B) Since sequences originating from one copy of an inverted repeat could not be distinguished from sequences originating from the other copy, the data were assembled into a trimmed form lacking the terminal repeats TRL and TRS. The GenBank records contain both a full-length and a trimmed version for each genome (see Materials and Methods for details).
FIG 2.

Nucleotide compositional bias toward G+C residues in repeat regions of herpesvirus genomes. (A) A line graph overlay of G+C versus A+T distribution in the HSV-1 genome (JN555585; human herpesvirus 1 [HHV-1]). A diagram beneath the line graph depicts the locations of UL and US (gray), as well as TRL/IRL and TRS/IRS (orange). SSRs are also marked in orange. (B) Another human alphaherpesvirus, VZV, is A+T rich in the UL and US regions (56%) but G+C rich in the inverted repeat regions (59% G+C). (C to E) Similar plots depict nucleotide distribution in unique versus repeated regions of human beta- (human cytomegalovirus [HCMV]) and gammaherpesviruses (Epstein-Barr virus [EBV] and Kaposi's sarcoma-associated herpesvirus [KSHV]). Note that each genome is drawn to an individual scale, as marked below each line graph. The KSHV genome has 35 to 45 copies of a terminal repeat (TR) on its termini; we show 40 here. The genome diagram follows the NCBI Refseq annotation in displaying the EBV and KSHV TRs only on the right-hand side. These TRs join together in circularized genomes. Nucleotide sequences and annotations of unique and repeated regions are derived from NCBI RefSeq records as follows: VZV strain Dumas (accession number NC_001348), HCMV strain Merlin (NC_006273), EBV strain B95-8 (NC_007605), and KSHV strain GK18 (NC_009333).
In addition to the large inverted repeats, the HSV-1 genome contains a large number of SSRs, also known as variable number tandem repeats (VNTRs) or reiterations (82–86). We previously analyzed all classes of SSR in the HSV-1 reference strain 17, which in the trimmed format contains 87 minisatellites (≥10-nucleotide repeating unit), 60 microsatellites (2- to 10-nucleotide repeating unit), and 499 homopolymers (≥6 nucleotides long) (Fig. 3D) (64). These SSRs are not distributed evenly throughout the genome. Thus, although 84% of the HSV-1 genome consists of protein-coding regions, only about 60% of SSR loci are located therein (Fig. 4A). In contrast, although the inverted repeats (IRL+IRS) occupy only 11% of the trimmed genome sequence, they contain >50% of all SSR loci (Fig. 4B).
FIG 4.

Localization and conservation of SSRs in HSV-1 strains. (A) SSRs in reference strain 17 are overrepresented in noncoding regions. The pie chart on the left shows the distribution of all nucleotides in the trimmed genome alignment among protein-coding and noncoding (promoter and intergenic) regions. The pie chart on the right shows the distribution of SSR-encoding nucleotides among protein-coding and noncoding regions. (B) SSRs in reference strain 17 are more common in the large repeat regions. The pie chart on the left shows the distribution of all nucleotide bases in the trimmed genome (Fig. 2B) among the unique long (UL), unique short (US), and internal repeat (IRL+IRS) regions. The pie chart on the right shows the distribution of SSR bases in UL, US, and IRL+IRS. (C) Although protein-coding SSRs outnumber noncoding repeats, they are more likely to be conserved in length. An SSR was counted as conserved if it had the same position and length (same number of repeated units) in a majority of strains. Coding SSRs are largely conserved in length (pale blue versus dark blue); the number of SSRs in each group is shown to the right of the histograms. In contrast, there are approximately equivalent numbers of conserved versus nonconserved SSRs in noncoding regions. SSRs with incomplete sequences in more than half the strains (gray in Fig. 3) were excluded.
A few of the SSRs in the HSV-1 genome can reach several hundred base pairs in length (Fig. 2, orange arrows) and have previously been shown to vary in length within a virus strain population, as well as among strains (64, 85–89). This makes determining their sizes a challenge, since a population of viral DNA used for sequencing may contain genomes with different lengths of a given SSR and high-throughput sequencing reads rarely span the full length of the larger SSRs. Originally, the lengths of these SSRs were determined in the HSV-1 reference genome by Sanger sequencing of cloned genome fragments that spanned each SSR (44, 45); all high-throughput-sequenced genomes since then have left one or more of these SSRs indeterminate (13, 29, 30, 48–50). In the newly sequenced genomes, we marked the subset of these SSRs that cannot be determined by high-throughput sequencing as gaps (Fig. 3D, gray arrowheads; see also Table S1 in the supplemental material, where they are listed per strain) and excluded from amino acid variation analysis any sites affected by these gaps (see Materials and Methods for details).
Characterizing DNA variation.
To compare differences among the newly sequenced strains, we aligned the finished genome sequences with those of the six strains sequenced previously (Table 1). This alignment is available at http://szparalab.psu.edu/hsv-diversity/. We used this alignment to assess variation at the DNA level across the genome collection, as well as to map coding and noncoding regions of the DNA (Fig. 3A to C). First, we examined the frequency of nucleotide polymorphisms in nongapped bases across the genome. As noted previously (90) and now revealed in greater detail, the occurrence of SNPs is higher in US than UL (Fig. 3A and B). Of 138,334 columns in the alignment, 5,239 columns contain single-nucleotide variations (3.8%), with approximately half of these columns (1.7%) showing variation in only one strain. The mean pairwise identity between strains is 96.8%. The ratio of transitions to transversions is 1.62 (see Materials and Methods for details).
In addition to polymorphisms, the 26-genome alignment contains many small gaps due to variations in length at SSRs (i.e., differences in copy number of the repeating unit). We characterized SSR-based variation in the DNA by mapping homologous SSRs among strains and then characterizing each SSR as being variable or conserved in length. Variation in SSR length has been shown to generate phenotypic diversity for organisms from bacteria to yeast and humans (91–94). Of the SSRs in the trimmed version of the HSV-1 reference strain 17 genome, 90% (584 of 646) occur in equivalent positions in a majority of other strains; we classified these as conserved if they had the same length in a majority of strains. Using this criterion, there were five times as many conserved as nonconserved SSRs (487 versus 97), indicating selection or preservation of genetic stability (Fig. 4C). In noncoding regions, the ratio of conserved versus nonconserved SSRs was almost equal; in contrast, in coding regions, the conserved SSRs greatly outnumbered the nonconserved ones (Fig. 4C). Thus, although SSRs occur frequently in the HSV-1 genome, their length is conserved in a majority of strains.
During inspection of the genome sequence alignment, we noted that certain SNPs were clustered at the tips of potential hairpin-forming sequences, which consist of a short sequence flanked by an inverted repeat (see Table S2 in the supplemental material). The SNP-hairpin association was observed in both noncoding and coding sequences and led to minor coding variations. These hairpins may have occurred via inversions prompted by errors in DNA replication or recombination. The association of DNA hairpins with variation has been previously reported in eukaryotic cells but not, to our knowledge, in herpesvirus genomes (94).
Large deletions and insertions.
Substantial mutations have been reported previously in several HSV-1 strains propagated in vitro, specifically, in the right-hand side of UL, containing UL55 and UL56 (Fig. 3C). These genes are not required for growth in vitro, but UL56 appears to be important for virulence and the establishment of latency in vivo (95–99). Deletions in one or both of these genes have been noted in two attenuated strains, namely, HF10, which harbors a deletion in UL56 and a homopolymer-based frameshift in UL55 (47), and HFEM, where restoration of the UL56 gene restored virulence in vivo (97, 99). Moreover, HSV-1 strains can harbor mixed populations of mutations in this region, as demonstrated by the isolation of single-plaque variants of HSV-1 strains 17 and K52 that harbor deletions of different sizes (96, 101).
Large deletions in the UL55-UL56 region were apparent in several of the newly sequenced strains (E10, E13, E23, and E35), as detected by a severe underrepresentation in sequence read coverage in this region (see Fig. S2 and Table S1 in the supplemental material). For three of these strains (E10, E13, and E23), sufficient nondeleted genomes were present to make it possible to determine the sequence of the affected region. However, this was not possible for E35, in which a very low proportion of nondeleted genomes was present. Also, in place of a deletion in the UL55-UL56 locus, two strains (E23 and E35) appear to have insertions from the left end of UL, as evidenced by an overrepresentation in sequence read coverage in this region (see Fig. S2 and Table S1). This type of insertion, from the left side of UL into the UL56 locus, has also been reported in single-plaque isolates of strain 17 (96).
Finally, three strains (E12, E13, and E15) contain an overrepresentation of sequence reads mapping to an origin of DNA replication, DNA packaging signals, and other small regions of the genome (shown in Fig. S2 in the supplemental material; data are listed in Table S1). Previous work has shown that HSV strains grown in vitro can form defective interfering particles (DIPs), which consist of an origin of DNA replication, a packaging signal for encapsidation, and frequently, fragments of variable length from the US region of the genome (102–104). This naturally occurring genome fragmentation has served as the basis for widely used HSV amplicon vectors (104, 105). Although the regions of disproportionately high coverage are not definitive proof of DIPs in these stocks, the data suggest this as a likely possibility. High-throughput sequencing reveals the nonhomogeneity of HSV-1 stocks more easily than techniques such as RFLP analysis, which suggests that routine sequencing may be a useful way to screen stocks for future studies.
Frameshifts at homopolymer tracts.
Several HSV strains have been reported to contain homopolymer-based frameshift mutations (HFMs), which occur as a result of variation in the length of homopolymeric tracts. HFMs have been observed in HSV-1 strains HF10, MP, and KOS321, HSV-2 strain HG52, and subpopulations or clones of HSV-1 strains 17, F, and KOS (29, 47, 106–109). The affected ORFs, listed with the protein name followed by the genetic locus, include protein kinase (PK; UL13), VP1-2 (UL36), virion host shutoff (vhs; UL41), glycoprotein C (gC; UL44), VP11-12 (UL46), VP22 (UL49), UL55, ICP34.5 (RL1), and glycoprotein I (gI; US7). HFMs are major contributors to the development of viral resistance to acyclovir, where they cause loss of function in thymidine kinase (TK; UL23), which normally activates acyclovir by phosphorylation (110–114). HFMs in the major secreted glycoprotein G (gG; US4) of HSV-1 and HSV-2 allow antibody escape, facilitating viral evasion of the host immune response (115, 116). Of the 20 newly sequenced genomes, seven contain HFM variations (Table 1).
The affected ORFs in the new strain collection encode the tegument protein UL11 (UL11), a protein kinase (PK; UL13), and glycoprotein H (gH; UL22). Details and the effects of these mutations are discussed below. We noticed that, whereas the mutations in UL13 would severely truncate PK and are likely to ablate function, those in UL11 and UL22 affect only a few codons near the 3′ end of the ORF (see below). Since the polyadenylation sites for UL11 and UL22 are located close to the stop codon, the predicted outcome of these HFMs is a transcript of normal length lacking a stop codon. In eukaryotic cells, nonstop mRNAs such as these lead to ribosome stalling, poor translation, and non-stop-mediated mRNA decay (117, 118). Recently, ribosomal frameshifting has been shown to allow the recovery of a low level of protein product from TK (UL23) transcripts that lack a stop codon due to HFM (119). The discovery of these HFMs in UL11 and UL22 will allow broader exploration of the role of ribosomal frameshifting in HSV biology. If confirmed, the combination of HFMs, nonstop mRNA decay, and ribosomal frameshifting will greatly increase the number of ways that HSV-1 can vary its protein sequences and levels of protein production. This likewise may provide greater diversity for the virus to adapt to new cells and new hosts.
HSV encodes three kinases, with PK (UL13) being the only one found across alpha-, beta-, and gammaherpesviruses (120). This distinction earned it the alternative name of conserved herpesvirus-encoded protein kinase (CHPK). Loss of PK function attenuates viral growth in vitro and is deleterious for spread in vivo (121–127). The UL13 frameshift mutations in strains E06 and E25 are identical to each other and to one described previously in a subpopulation of HSV-1 strain F (29). This C6-to-C5 homopolymer mutation creates a frameshift at codon 118, terminating the protein at 166 residues instead of the usual 518 and removing the entirety of the kinase domain (128). Strain E11 contains an alternate A4-to-A3 mutation in UL13, which creates a frameshift within codon 319 and spares a portion of the kinase domain. Loss of PK due to a homopolymer-based frameshift in UL13 has also been observed in a bacterial artificial chromosome (BAC) clone of the fowl alphaherpesvirus Marek's disease virus (MDV) (125, 126). Remarkably, the truncation of PK in MDV (at residue 176 of 513) occurs at a site similar to the truncation site in strains E06 and E25, again removing the kinase domain. This truncation allows growth in culture but limits MDV transmission in vivo (125, 126), suggesting that these PK truncations are selected for or amplified during passage in vitro.
UL11 is conserved across alpha-, beta-, and gammaherpesviruses and plays an essential role in virion envelopment and egress (129–132). The C terminus of UL11 normally interacts with glycoprotein E (gE; US8) (133, 134). Strains S23 and S25 harbor a C-terminal C6-to-C8 mutation in UL11, which causes a frameshift and ablates the normal stop codon. Transcription of an extended ORF is likely blocked by the nearby polyadenylation site, resulting in an mRNA lacking a stop codon, with an alternate C terminus (MSDSE* to PCPIANK, where the asterisk indicates a stop codon). Despite the presence of other C homopolymers in UL11, the same C-terminal site is affected in strain R62, albeit with a longer extension of the homopolymer (C6 to C10, resulting in MSDSE* to PHVR). Further work will be needed to determine whether the mutant UL11 transcripts are subject to nonstop mRNA decay or are rescued by ribosomal frameshifting (117–119). If UL11 is translated in these strains, it will be relevant to explore whether the altered C terminus affects UL11-gE protein interactions.
gH (UL22) is also conserved across alpha-, beta-, and gammaherpesviruses, and it interacts with glycoproteins B (gB, UL27), D (gD, US6), and L (gL, UL1) to drive viral fusion with host membranes. Strain E07 harbors a T7-to-T8 mutation in the sequence encoding the C-terminal end of gH that causes a frameshift and ablates the stop codon. As with UL11, the close proximity of the polyadenylation site minimizes the effects of the frameshift, changing only the final few residues (WRRE* to LETRIK). The lack of a stop codon creates a potential target for nonstop mRNA decay or ribosomal frameshifting (117–119). The fusion machinery of gH lies in its extensive N-terminal ectodomain (800 amino acid residues), whereas the observed mutation is present in the short internal tail of 14 amino acid residues (77). The effects of this mutation are unknown, although the sequence of the internal tail is completely conserved in all other strains in the collection and is well conserved between HSV-1 and HSV-2. These homopolymeric repeats, in addition to those described above, add diversity to the HSV-1 coding potential.
All HSV-1 strains sequenced to date have been passaged in cell culture, including the 20 newly sequenced ones in the present study. Their passage histories are obscure and likely include a variety of opportunities for viral strains to evolve or expand mutations, including multiple passages in vitro, potential plaque purifications, and shifts in the host species or type of cell used for culture. Culturing a virus necessarily removes a variety of selection pressures and bottlenecks present in vivo and introduces new selection pressures unique to cell growth in vitro. The HFMs and deletions described above are very likely to be deleterious for virus spread in humans (95–99, 121–127). This is likewise true for frameshifts and deletions recognized in previously sequenced strains (e.g., US9 in KOS or glycoprotein N [UL49.5] and UL56 in HF10) (47, 135). Without access to uncultured material from the original human hosts, it is not possible to determine whether these mutations were present as minority populations in vivo or arose later during culture. Regardless of their starting point, we surmise that these mutations probably expanded during passage in vitro. Further investigation of the attenuating mutations in UL55, UL56, UL11, PK (UL13), and gH (UL22) is warranted to deduce how prevalent these deletions and HFMs are in vivo and what role they play in HSV-1 spread in humans.
Diversity of protein-coding sequences.
To estimate the global diversity of the HSV-1 proteome, we used conceptually translated proteins and generated amino acid sequence alignments for all 74 canonical proteins encoded by the HSV-1 genome (13, 25, 30, 34, 107, 136). All alignments are provided at http://szparalab.psu.edu/hsv-diversity/ and are mirrored with additional HSV strain data on the Virus Pathogen Resource website at http://www.viprbrc.org/ (see Materials and Methods for details) (69). We used a variety of measures to assess protein conservation, identity, and mutations across the 26-strain collection. Because these strains are not evenly distributed in geographic origin, we weighted the sequences to reduce redundancy and emphasize diversity (70). We examined protein-coding diversity relative to a consensus built for each protein (see Materials and Methods for details). Columns containing sequencing gaps or small insertions/deletions (indels) in a majority of strains were excluded from further analysis (1% of total alignment columns) (see Table S3 in the supplemental material). We found that, on average, 94% of the columns in these amino acid alignments agreed perfectly with the consensus and an average of 5.4% contained differences. The exact proportions varied on a per-protein basis (listed in full in Table S3).
The 15 most conserved HSV-1 proteins by percent identity are those involved in essential aspects of herpesvirus reproduction, including DNA replication, capsid formation, and nuclear egress (Table 2). Eleven of these proteins are conserved among alpha-, beta-, and gammaherpesviruses, and nine of these are absolutely required for viral growth in cell culture (Table 2) (137). Four proteins unique to alphaherpesviruses are also highly conserved (UL20, UL45, VP13-14 [UL47], and gK [UL53]). The functions of two of these, UL20 and UL45, are not well known, and their conservation indicates that further investigation of their function is warranted (138–142). A third protein, VP13-14 (UL47) modulates transactivator VP16 (UL48) to induce high levels of immediate early gene expression (143, 144). The proteins UL20, UL45, and gK have been proposed to interact with each other and with the herpesvirus fusion machinery (138–142).
TABLE 2.
The most conserved proteins encoded by 26 HSV-1 genomes
| Protein (locus) | % Identityc | Function |
|---|---|---|
| VP26 (UL35)a | 99.1 | Small capsid protein |
| UL15a,b | 98.8 | DNA packaging terminase subunit 1 |
| UL29a,b | 98.7 | Single-stranded DNA-binding protein |
| UL20 | 98.6 | Envelope protein of unknown function |
| UL28a,b | 98.5 | DNA packaging terminase subunit 2 |
| UL33a,b | 98.5 | DNA packaging protein, interacts with UL28 |
| VP13-14 (UL47) | 98.4 | Tegument protein, modulates transactivating protein VP16 (UL48) |
| UL45 | 98.3 | Membrane protein of unknown function |
| gK (UL53) | 98.2 | Envelope glycoprotein involved in entry |
| VP5 (UL19)a,b | 97.9 | Major capsid protein, forms hexons and pentons |
| RR2 (UL40)a | 97.9 | Ribonucleotide reductase subunit 2 |
| Pol (UL30)a,b | 97.9 | DNA polymerase catalytic subunit |
| UL18a,b | 97.8 | Capsid triplex subunit 2, with capsid triplex subunit 1 (UL38), connects capsid hexons/pentons |
| UL25a,b | 97.8 | DNA packaging tegument protein, stabilizes capsid vertices |
| UL31a,b | 97.7 | Nuclear egress lamina protein |
Protein is conserved across the family Herpesviridae.
Protein is essential for growth in culture, as described by McGeoch et al. (137).
Percentage of amino acid alignment columns that are identical (without mutations). See Materials and Methods for details; see also Table S3 in the supplemental material.
Since percent identity is highly sensitive to outliers, such as frameshifts and deletions, we used a median divergence metric to identify proteins with the greatest overall amino acid diversity across all strains. Table 3 lists the most divergent proteins in the 26-strain collection, although it should be noted that these values underestimate diversity in the proteins discussed above that contain deletions, frameshifts, and indeterminate SSRs (e.g., PK [UL13], UL56, and ICP34.5 [RL1]). The 15 most divergent proteins include many that are unique to alphaherpesviruses (ICP0 [RL2], ICP4 [RS1], gI [US7], US9, US10, and UL43) or are present only in a subset of alphaherpesviruses (ICP34.5 [RL1], ICP22 [US1], gG [US4], gJ [US5], ICP47 [US12], US8A, US11, and UL55) (137, 145). One-third of these are found on both the virion envelope and the surface of infected cells; their divergence could result from selection to evade host immune surveillance. Several additional proteins function in transcriptional regulation (ICP4 [RS1], ICP22 [UL54], and VP11/12 [UL46]) or blockade of the host immune response (ICP0 [RL2], US11, and ICP34.5 [RL1]). Using a Poisson model that takes protein length into account, we found that gL (UL1), gG (US4), and ICP34.5 (RL1) all have significantly more divergent columns than expected for their length (P < 0.01) (see Materials and Methods for details; see also Table S3 in the supplemental material).
TABLE 3.
The most divergent proteins encoded by 26 HSV-1 genomes
| Protein (locus) | Median % divergencec | Function |
|---|---|---|
| ICP34.5 (RL1) | 2.8 | Inhibits translational arrest, role in neurovirulence |
| gG (US4) | 2.3 | Envelope glycoprotein, major antibody target |
| gL (UL1)a,b | 2.2 | Envelope glycoprotein, complexed with gH (UL22), role in fusion |
| UL11a | 1.9 | Egress, interaction with UL16 |
| gJ (US5) | 1.5 | Envelope glycoprotein |
| US10 | 1.4 | Unknown function |
| gC (UL44) | 1.3 | Envelope glycoprotein, binds heparan sulfate for cell attachment |
| US11 | 1.2 | Binds double-stranded RNA, acts as a PKR antagonist |
| UL43 | 1.1 | Envelope protein |
| ICP22 (US1) | 1.1 | Regulatory protein required for expression of a subset of late genes |
| VP11/12 (UL46) | 0.9 | Modulates transactivating protein VP16 (UL48) |
| ICP4 (RS1) | 0.9 | Transcriptional regulator |
| UDG (UL2)a | 0.8 | Uracil-DNA glycosylase |
| gI (US7) | 0.8 | Envelope glycoprotein, complexed with gE (US8) to form an Fc receptor |
| ICP0 (RL2) | 0.7 | Ubiquitin E3 ligase, disrupts ND10, triggers protein degradation |
Protein is conserved across the family Herpesviridae.
Protein is essential for growth in culture, as described by McGeoch et al. (137).
Median percentage of amino acid alignment columns that diverge from the consensus (number of columns with mutations divided by total number of conserved columns). Divergence in gI (US7) and ICP34.5 (RL1) does not include indeterminate SSR regions. See Materials and Methods for details; see also Table S3 in the supplemental material.
The amino acid diversity in US11 illustrates that seen in most HSV-1 proteins in Table 3, which include SNPs, indels, and SSR-based variations that are typical of those in other alignments (Fig. 5). US11 is an RNA-binding protein that resembles and can partially substitute for the transactivating Rex and Rev proteins of human T-lymphotropic virus 1 (HTLV-1) and HIV, respectively (146, 147). During HSV-1 infection, US11 inhibits protein kinase R (PKR) activation, preventing the defensive shutoff of host translation machinery that PKR would otherwise trigger (148–152). The C-terminal region of US11 contains a coding SSR (specifying a repeated PRX motif), which is proposed to form a polyproline helix that mediates RNA binding, nucleolar localization, and antagonism of PKR; this SSR varies in length across strains (Fig. 5). The US11 alignment includes strain-specific SNPs at P12S (strain H129), V22M and P122S (strain HF10), and V66I (strain E03), as well as variations observed in multiple strains, such as the four amino acid variations shared by strains S23, S25, and R62.
Positive selection.
We examined the ratio of nonsynonymous (dN) versus synonymous (dS) substitutions (dN/dS, or Ω) for evidence of purifying, neutral, or positive selection (Fig. 6; see Table S4 in the supplemental material for full details). Weak evolutionary constraint appears to be the predominant mode of evolution at the level of whole genes, based on the average Ω value of 0.27 (see Materials and Methods for details). Several proteins are closer to neutral selection or drift—for example, UL11, UL14, and US12 (Ω > 0.7) (Fig. 6). In contrast, five proteins have an Ω value of <0.1, indicating a strong selective constraint against nonsynonymous mutations (Fig. 6); these include UL15, ICP8 (UL29), VP26 (UL35), ribonucleotide reductase (RR2 [UL40]), and VP13-14 (UL47). These findings mirror the summary of protein conservation and diversity shown in Tables 2 and 3.
FIG 6.
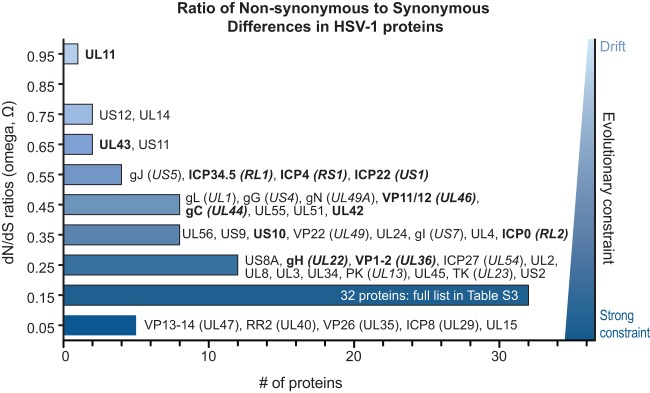
Distribution of Ω substitution rates for all HSV-1 proteins. Histogram of the number of HSV-1 proteins at each Ω substitution rate (in bins of 0.1, centered around the values shown). An Ω value of 1 indicates neutral selection or drift (light blue), whereas an Ω value of 0 indicates absolute constraint (dark blue). Protein names are listed next to each bin for all but the largest bin (0.1 to 0.19; see Table S3 in the supplemental material for list of all values). Protein names in boldface show signs of positive selection of individual amino acid residues (see Table 4). The average Ω value of 0.27 indicates that weak evolutionary constraint is the most common mode of protein evolution, while a few proteins approach levels indicating drift (UL11, US12, and UL14) and several others show strong selective constraint (UL15, VP26 [UL35], VP13-14 [UL47], RR2 [UL40], and ICP8 [UL29]). The Ω values reflect the overall amino acid sequence conservation values listed in Table 2.
In addition to analyzing evolutionary constraints in whole proteins, we investigated whether particular amino acid residues showed signs of positive selection. Using a conservative model for detection of positive selection (72, 73), we identified 13 proteins with a total of 54 positively selected residues (Table 4; see Materials and Methods for details). Full reports of positively selected residues for each protein, along with amino acid alignments highlighting the positions of these 54 positively selected residues, are included at http://szparalab.psu.edu/hsv-diversity/. A less conservative analysis (76) identified 41 proteins with a total of 113 positively selected residues (see Table S4 in the supplemental material; per-protein analyses are included at the URL mentioned above).
TABLE 4.
Positively selected residues in HSV-1 proteins
| HSV-1 protein (in genome order) | No. of positively selected residues | Amino acid position(s)a |
|---|---|---|
| UL11 | 1 | 8 |
| gH (UL22) | 4 | 119, 284, 369, 370 |
| Pol (UL30) | 1 | 1124 |
| VP1-2 (UL36) | 20 | 279, 342, 355, 367, 370, 373, 453, 483, 900, 1003, 1127, 1222, 1707, 1866, 2523, 2624, 2726, 2834, 2872, 2981 |
| UL42 | 1 | 13 |
| UL43 | 1 | 216 |
| gC (UL44) | 6 | 23, 75, 132, 300, 306, 421 |
| VP11-12 (UL46) | 2 | 593, 639 |
| ICP34.5 (RL1) | 3 | 56, 96, 121 |
| ICP0 (RL2) | 2 | 599, 653 |
| ICP4 (RS1) | 6 | 705, 798, 800, 899, 918, 1256 |
| ICP22 (US1) | 1 | 116 |
| US10 | 5 | 79, 82, 148, 167, 207 |
Positively selected residues are shown in highlighted amino acid alignments at http://szparalab.psu.edu/hsv-diversity/.
Although relatively few crystal or solution structures are available for HSV-1 proteins, we modeled two examples of positively selected residues: gH (UL22), which is a component of the viral fusion machinery, and the DNA polymerase processivity subunit UL42 (Fig. 7). For gH, the HSV-1 protein was modeled using the available crystal structure for the HSV-2 gH ectodomain (Fig. 7A, C, and D) (77). Two of the positively selected residues are surface exposed, where their variation may influence interactions with other proteins. For UL42, we used a crystal structure of the N-terminal portion of this protein, which encompasses all of UL42's functions as a DNA-binding protein and processivity factor (153). The positively selected residue 284 (P > 99%) lies adjacent to residues proposed to interact with DNA (R279, R280, and Q282) and with HSV polymerase (K289 and V296) (Fig. 7B and E) (153). These examples illustrate the future directions suggested by the analysis of positively selected residues in the HSV proteome.
FIG 7.

Coding diversity and positive selection of residues in the HSV-1 entry protein gH and the DNA-binding protein UL42. (A and B) Amino acid sequence alignments of gH (UL22) (A) and UL42 (B) from 26 HSV-1 strains, showing only those positions where a residue varies in one or more strains compared to the residue in reference strain 17 (top line). Positions in the sequence are shown along the top. Yellow shading denotes residues exhibiting positive selection (Codeml, P > 99%; see Materials and Methods for details), and asterisks (*) mark those visible as red spheres on the 3-dimensional models in the panels below. (B) UL42 alignment from 26 HSV-1 strains, in which positive selection was detected for 829 residue 13 (P > 99%) and 284 (P > 95%). (C) Ribbon diagram of the HSV-1 gH ectodomain, homology modeled using the crystal structure of HSV-2 gH (77). Highlighted residues fall into the H1 and H2 domains described previously (77). (D) Surface interactions (surface indicated by mesh, color coded as follows: green, hydrophobic; pink, H bonding; blue, polar) are low for residue 284 (top) and are greater for the exposed pair of positively selected residues 369 and 370. (E) The available structure of UL42 (153) captures only residue 284, which lies adjacent to residues proposed to interact with DNA and with HSV-1 DNA polymerase (Pol [UL30]).
Relationships among circulating HSV-1 strains.
Our determination of 20 new genome sequences provides the first real opportunity to assess the relatedness of geographically diverse HSV-1 strains. Together with previously published sequences, this collection spans six nations around the globe. The two most recent analyses of multiple HSV-1 genome sequences included strains from the United States or Sweden only (13, 30). Also, in contrast to previous studies that focused on selected regions of the genome or excluded intergenic sequences, we used the full genome alignment to assess relationships among strains. The dendrogram built from the matrix of genetic distances contains subgroups that reflect geographic zones of origin (Fig. 8). As Sakaoka found in his RFLP- and PCR-based studies on intra- and intercountry diversity (10, 20, 32, 33), the African strains reveal deep structure and separation into multiple subgroups. The North American strain KOS presents one exception to the geographic groupings, as it clusters with Asian strain CR38. A similar exception of one U.S. VZV strain coclustering with the Asian clade of VZV has been observed (12); these isolated examples may reflect the effects of human migration, travel, and interactions. Overall, the genome-wide dendrogram reflects the outcome of similar clustering approaches that used smaller DNA segments or coding regions (data not shown; 13, 25, 30, 154), though the branch points have higher confidence than those typically found in trees based on shorter DNA inputs.
FIG 8.

Dendrogram of genetic distances among HSV-1 genomes reveals broad geographic clustering. The multiple-genome alignment of 26 strains of HSV-1 was used to generate a genetic distance matrix under a maximum composite likelihood substitution model. A dendrogram was then calculated using UPGMA in MEGA, with 1,000 bootstrap replicates. Numbers indicate branch confidence. The majority of strains cluster into four groupings that reflect their geographic origins, with the large collection of African strains splitting into two groups or, potentially, three groups (i.e., E03 as a third singleton group).
In prior analyses of smaller sequence collections, recombination was described as “widespread” and “extremely frequent” among HSV-1 genomes (13, 34). We checked this conclusion in the 26-strain collection using Simplot Bootscan recombination analysis, as described previously (13, 81). Bootscan plots illustrate the statistical (bootstrap) support for close clustering of one sequence with each member of a set of comparison sequences for each of many small segments of a sequence alignment. Switches in the identity of the most similar sequence as one moves along the alignment are interpreted as support for multiple recombination events in the history of the sampled sequences. Thus, for example, we found that the HSV-1 reference strain 17 is most similar to a variety of different strains in different parts of the genome (Fig. 9A). Similarity switches were visible even among the subgroups of closely related strains described above (Fig. 8; see also Fig. S3 in the supplemental material), suggesting that recombination events are both historical and ongoing (Fig. 9B). These results confirm the previous findings and suggest that HSV-1 strains are highly recombinogenic throughout their genomes. Future experiments are needed to address both the potential and the actual frequency of recombination between HSV strains in modern human populations, because the rate and extent of human movement and interaction has increased substantially since many of these strains were collected (Table 1).
FIG 9.

Bootscan analyses of similarity between HSV-1 strains contain breakpoints suggesting frequent recombination. (A) Similarity plot of HSV-1 reference strain 17 versus all other strains, demonstrating that recombination occurs throughout the tree. The longest colinear area of similarity (between strains 17 and McKrae; rose line) is about 30 kb. The trimmed format of the HSV-1 strain 17 genome (Fig. 1B) was used as the query sequence. (B) Similarity plot of the European subgroup (as shown in Fig. 8) of the HSV-1 collection, with HF10 used as the query sequence. There is extensive recombination even within genetically similar geographical clusters. The longest colinear area of similarity (between HF10 and F; green line) is about 20 kb. Bootscan parameters were as follows: 3-kb window, 200-bp step size, GapStrip on, 100 repetitions, Kimura (2 parameter), T/t = 2.0, neighbor joining.
Comments on HSV-1 evolution.
This paper presents the first survey of HSV-1 variation that has near-global coverage and incorporates both coding and noncoding features of the genome. Geographical clustering prevails despite signs of recombination (Fig. 8 and 9). During the coevolution of humans and HSV-1, spatial segregation of ancestral host populations is likely to have generated some geographic isolation of viral lineages. Similar impacts of geographic separation on the results of viral genome clustering have been observed for VZV (12, 15, 155, 156). The current study, in combination with prior work, indicates extensive recombination between HSV-1 genomes (Fig. 9), at a level that far exceeds that observed for VZV (13, 15, 156). While dating of VZV evolution has been proposed based on similar data, we find that the prevalence of recombination in HSV-1 makes it unwise to add a time scale to internal nodes or to interpret these clusters as clades (13, 155, 157).
Although past geographical separation may well have impacted the currently available sequences, it is worth noting that all of the currently sequenced genomes were isolated more than 25 years ago (Table 1). The degree and extent of human global movement and interaction have increased at a pace that begs future projects to encompass currently circulating strains. Progress in VZV genome sequencing and comparison has moved more rapidly in this regard (158, 159). It has been proposed that the geographically linked clade structure of VZV may be fading under the impact of host mixing (12, 155, 160), whereas the signs of recombination suggest that even decades ago, when these strains were collected, HSV-1 strains were already a single, panmictic population (Fig. 8). Future sequencing efforts will be needed to assess whether the rate of mixing among HSV strains has increased due to human movement and to assess the modern extent of HSV-1 interstrain diversity as a result. These data will be crucial to our understanding of the efficacy of vaccine candidates and to the development of widely effective therapeutics.
The preponderant mode of protein evolution appears to be moderate constraint, although the outliers range from tight constraint to near neutrality (Fig. 6). We found evidence for a few residues being under positive selection using stringent tests (Table 3; see also Table S4 in the supplemental material), and borderline signals were obtained for many more residues. As further sequences become available, more extensive sampling of protein variability will provide a clearer picture of selective pressures. Likewise, testing of the alternative versions of positively selected residues, such as those found in gH (UL22) and UL42 (Fig. 7), will determine their relative efficacies and how these variations affect viral spread between hosts.
These data present a rich resource for mining information about the coevolution of HSV and the human immune response, which will in turn be relevant for the development of highly stimulatory vaccine antigens. For instance, gB (UL27) has been a key vaccine target in several recent trials (5). These data reveal that more than half of the variation among HSV strains in this 904-amino-acid protein occurs in the first 80 residues (see Table S3 in the supplemental material; see also reference 161). This N-terminal fragment lies outside the gB crystal structure (162). The functions of this region are not well defined but include a binding site for major histocompatibility complex (MHC) class II molecules (163). Some of the most variable gB residues (residues 59 to 80, of which one-third are variable) are also highly antigenic and capable of stimulating immunity (164, 165). Knowledge of interstrain variations in gB and other viral proteins will allow the refinement of vaccine antigens to create a strong and broadly effective immune response.
As future studies add to our knowledge of HSV genome diversity, it will be important to maintain as much clinical and passage history data as possible. The current sequences reveal a wealth of data about genetic diversity among HSV-1 strains, but we now need to enrich these data by capturing information about specific strain origins, disease presentation, immune status, passage history for cultured strains, and related metadata. Community-wide resources, such as GenBank and the Virus Pathogen Resource, provide avenues to include these data alongside newly produced sequences (69). These approaches will aid future analyses of genetic links to phenotype, host status, and disease progression.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for the tremendous contributions of two retired colleagues: Hiroshi Sakaoka, who characterized hundreds of HSV-1 strains during his career, and Duncan McGeoch, who sequenced the first HSV-1 genome and provided the driving force to sequence the collection described here. We thank Lance Parsons for feedback on analyses and construction of the website at http://szparalab.psu.edu/hsv-diversity/. Yolanda Tafuri assisted in strain propagation, and Derrick Dargan, Clare Addison, and Tracey Neilson recovered HSV-1 strains by transfection. High-throughput sequencing was carried out at the Sir Henry Wellcome Functional Genomics Facility (University of Glasgow), the Gene Pool (University of Edinburgh), and the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (University of Oxford).
This work was supported by the NIH-NIAID Virus Pathogen Resource (ViPR) Bioinformatics Resource Center (BRC), NIH-NIGMS Center (grant P50 GM071508), and NIH (grant P40 RR 018604 [M.L.S. and L.W.E.]), by the UK Medical Research Council (A.J.D., D.G., and A.D.), by the Eric and Wendy Schmidt Member in Biology fund (B.G.), and by the Wellcome Trust (grant 090532/Z/09/Z) and MRC Hub (grant G0900747 91070 [R.J.B.]).
Footnotes
Published ahead of print 13 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01987-13.
REFERENCES
- 1.Davison AJ, Eberle R, Ehlers B, Hayward GS, McGeoch DJ, Minson AC, Pellett PE, Roizman B, Studdert MJ, Thiry E. 2009. The order Herpesvirales. Arch. Virol. 154:171–177. 10.1007/s00705-008-0278-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitley RJ, Roizman B. 2001. Herpes simplex virus infections. Lancet 357:1513–1518. 10.1016/S0140-6736(00)04638-9 [DOI] [PubMed] [Google Scholar]
- 3.Taylor TJ, Brockman MA, McNamee EE, Knipe DM. 2002. Herpes simplex virus. Front. Biosci. 7:d752–d764. 10.2741/taylor [DOI] [PubMed] [Google Scholar]
- 4.Roizman B, Sears E. 1996. Herpes simplex viruses and their replication, p 1043–1107 InIn Fields BN, Knipe DM, Howley PM. (ed), Fundamental virology, 3rd ed. Lippincott-Raven, Philadelphia, PA [Google Scholar]
- 5.Johnston C, Koelle DM, Wald A. 2011. HSV-2: in pursuit of a vaccine. J. Clin. Invest. 121:4600–4609. 10.1172/JCI57148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koelle DM, Corey L. 2008. Herpes simplex: insights on pathogenesis and possible vaccines. Annu. Rev. Med. 59:381–395. 10.1146/annurev.med.59.061606.095540 [DOI] [PubMed] [Google Scholar]
- 7.Belshe RB, Leone PA, Bernstein DI, Wald A, Levin MJ, Stapleton JT, Gorfinkel I, Morrow RL, Ewell MG, Stokes-Riner A, Dubin G, Heineman TC, Schulte JM, Deal CD, Herpevac Trial for Women 2012. Efficacy results of a trial of a herpes simplex vaccine. N. Engl. J. Med. 366:34–43. 10.1056/NEJMoa1103151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen J. 2010. Immunology. Painful failure of promising genital herpes vaccine. Science 330:304. 10.1126/science.330.6002.304 [DOI] [PubMed] [Google Scholar]
- 9.Sakaoka H, Kawana T, Grillner L, Aomori T, Yamaguchi T, Saito H, Fujinaga K. 1987. Genome variations in herpes simplex virus type 2 strains isolated in Japan and Sweden. J. Gen. Virol. 68:2105–2116. 10.1099/0022-1317-68-8-2105 [DOI] [PubMed] [Google Scholar]
- 10.Sakaoka H, Saito H, Sekine K, Aomori T, Grillner L, Wadell G, Fujinaga K. 1987. Genomic comparison of herpes simplex virus type 1 isolates from Japan, Sweden and Kenya. J. Gen. Virol. 68:749–764. 10.1099/0022-1317-68-3-749 [DOI] [PubMed] [Google Scholar]
- 11.Norberg P, Kasubi MJ, Haarr L, Bergstrom T, Liljeqvist JA. 2007. Divergence and recombination of clinical herpes simplex virus type 2 isolates. J. Virol. 81:13158–13167. 10.1128/JVI.01310-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chow VT, Tipples GA, Grose C. 2013. Bioinformatics of varicella-zoster virus: single nucleotide polymorphisms define clades and attenuated vaccine genotypes. Infect. Genet. Evol. 18:351–356. 10.1016/j.meegid.2012.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norberg P, Tyler S, Severini A, Whitley R, Liljeqvist JA, Bergstrom T. 2011. A genome-wide comparative evolutionary analysis of herpes simplex virus type 1 and varicella zoster virus. PLoS One 6:e22527. 10.1371/journal.pone.0022527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tyler SD, Peters GA, Grose C, Severini A, Gray MJ, Upton C, Tipples GA. 2007. Genomic cartography of varicella-zoster virus: a complete genome-based analysis of strain variability with implications for attenuation and phenotypic differences. Virology 359:447–458. 10.1016/j.virol.2006.09.037 [DOI] [PubMed] [Google Scholar]
- 15.Peters GA, Tyler SD, Grose C, Severini A, Gray MJ, Upton C, Tipples GA. 2006. A full-genome phylogenetic analysis of varicella-zoster virus reveals a novel origin of replication-based genotyping scheme and evidence of recombination between major circulating clades. J. Virol. 80:9850–9860. 10.1128/JVI.00715-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umene K, Sakaoka H. 1991. Homogeneity and diversity of genome polymorphism in a set of herpes simplex virus type 1 strains classified as the same genotypic group. Arch. Virol. 119:53–65. 10.1007/BF01314323 [DOI] [PubMed] [Google Scholar]
- 17.Umene K, Yoshida M. 1993. Genomic characterization of two predominant genotypes of herpes simplex virus type 1. Arch. Virol. 131:29–46. 10.1007/BF01379078 [DOI] [PubMed] [Google Scholar]
- 18.Umene K, Sakaoka H. 1997. Populations of two eastern countries of Japan and Korea and with a related history share a predominant genotype of herpes simplex virus type 1. Arch. Virol. 142:1953–1961. 10.1007/s007050050213 [DOI] [PubMed] [Google Scholar]
- 19.Sakaoka H, Aomori T, Gouro T, Kumamoto Y. 1995. Demonstration of either endogenous recurrence or exogenous reinfection by restriction endonuclease cleavage analysis of herpes simplex virus from patients with recrudescent genital herpes. J. Med. Virol. 46:387–396. 10.1002/jmv.1890460416 [DOI] [PubMed] [Google Scholar]
- 20.Sakaoka H, Kurita K, Iida Y, Takada S, Umene K, Kim YT, Ren CS, Nahmias AJ. 1994. Quantitative analysis of genomic polymorphism of herpes simplex virus type 1 strains from six countries: studies of molecular evolution and molecular epidemiology of the virus. J. Gen. Virol. 75:513–527. 10.1099/0022-1317-75-3-513 [DOI] [PubMed] [Google Scholar]
- 21.Sakaoka H, Aomori T, Ozaki I, Ishida S, Fujinaga K. 1984. Restriction endonuclease cleavage analysis of herpes simplex virus isolates obtained from three pairs of siblings. Infect. Immun. 43:771–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warren KG, Koprowski H, Lonsdale DM, Brown SM, Subak-Sharpe JH. 1979. The polypeptide and the DNA restriction enzyme profiles of spontaneous isolates of herpes simplex virus type 1 from explants of human trigeminal, superior cervical and vagus ganglia. J. Gen. Virol. 43:151–171. 10.1099/0022-1317-43-1-151 [DOI] [PubMed] [Google Scholar]
- 23.Roizman B, Tognon M. 1983. Restriction endonuclease patterns of herpes simplex virus DNA: application to diagnosis and molecular epidemiology. Curr. Top. Microbiol. Immunol. 104:273–286. 10.1007/978-3-642-68949-9_17 [DOI] [PubMed] [Google Scholar]
- 24.Buchman TG, Simpson T, Nosal C, Roizman B, Nahmias AJ. 1980. The structure of herpes simplex virus DNA and its application to molecular epidemiology. Ann. N. Y. Acad. Sci. 354:279–290. 10.1111/j.1749-6632.1980.tb27972.x [DOI] [PubMed] [Google Scholar]
- 25.Norberg P, Bergstrom T, Rekabdar E, Lindh M, Liljeqvist JA. 2004. Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J. Virol. 78:10755–10764. 10.1128/JVI.78.19.10755-10764.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruyechan WT, Morse LS, Knipe DM, Roizman B. 1979. Molecular genetics of herpes simplex virus. II. Mapping of the major viral glycoproteins and of the genetic loci specifying the social behavior of infected cells. J. Virol. 29:677–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pereira L, Cassai E, Honess RW, Roizman B, Terni M, Nahmias A. 1976. Variability in the structural polypeptides of herpes simplex virus 1 strains: potential application in molecular epidemiology. Infect. Immun. 13:211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heine JW, Honess RW, Cassai E, Roizman B. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol. 14:640–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szpara ML, Parsons L, Enquist LW. 2010. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 84:5303–5313. 10.1128/JVI.00312-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kolb AW, Adams M, Cabot EL, Craven M, Brandt CR. 2011. Multiplex sequencing of seven ocular herpes simplex virus type-1 genomes: phylogeny, sequence variability, and SNP distribution. Invest. Ophthalmol. Vis. Sci. 52:9061–9073. 10.1167/iovs.11-7812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dijkshoorn L, Ursing BM, Ursing JB. 2000. Strain, clone and species: comments on three basic concepts of bacteriology. J. Med. Microbiol. 49:397–401 [DOI] [PubMed] [Google Scholar]
- 32.Sakaoka H, Aomori T, Saito H, Sato S, Kawana R, Hazlett DT, Fujinaga K. 1986. A comparative analysis by restriction endonucleases of herpes simplex virus type 1 isolated in Japan and Kenya. J. Infect. Dis. 153:612–616. 10.1093/infdis/153.3.609 [DOI] [PubMed] [Google Scholar]
- 33.Bowden R, Sakaoka H, Ward R, Donnelly P. 2006. Patterns of Eurasian HSV-1 molecular diversity and inferences of human migrations. Infect. Genet. Evol. 6:63–74. 10.1016/j.meegid.2005.01.004 [DOI] [PubMed] [Google Scholar]
- 34.Bowden R, Sakaoka H, Donnelly P, Ward R. 2004. High recombination rate in herpes simplex virus type 1 natural populations suggests significant co-infection. Infect. Genet. Evol. 4:115–123. 10.1016/j.meegid.2004.01.009 [DOI] [PubMed] [Google Scholar]
- 35.Ueno T, Suzuki N, Sakaoka H, Fujinaga K. 1982. A simple and practical method for typing and strain differentiation of herpes simplex virus using infected cell DNAs. Microbiol. Immunol. 26:1159–1170. 10.1111/j.1348-0421.1982.tb00265.x [DOI] [PubMed] [Google Scholar]
- 36.Sakaoka H, Aomori T, Honda O, Saheki Y, Ishida S, Yamanishi S, Fujinaga K. 1985. Subtypes of herpes simplex virus type 1 in Japan: classification by restriction endonucleases and analysis of distribution. J. Infect. Dis. 152:190–197. 10.1093/infdis/152.1.190 [DOI] [PubMed] [Google Scholar]
- 37.Staden R, Beal KF, Bonfield JK. 2000. The Staden package, 1998. Methods Mol. Biol. 132:115–130. 10.1385/1-59259-192-2:115 [DOI] [PubMed] [Google Scholar]
- 38.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829. 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cunningham C, Gatherer D, Hilfrich B, Baluchova K, Dargan DJ, Thomson M, Griffiths PD, Wilkinson GW, Schulz TF, Davison AJ. 2010. Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J. Gen. Virol. 91:605–615. 10.1099/vir.0.015891-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Ruan J, Durbin R. 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18:1851–1858. 10.1101/gr.078212.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. 2010. Tablet—next generation sequence assembly visualization. Bioinformatics 26:401–402. 10.1093/bioinformatics/btp666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davison AJ. 2011. Evolution of sexually transmitted and sexually transmissible human herpesviruses. Ann. N. Y. Acad. Sci. 1230:E37–E49. 10.1111/j.1749-6632.2011.06358.x [DOI] [PubMed] [Google Scholar]
- 43.Perry LJ, McGeoch DJ. 1988. The DNA sequences of the long repeat region and adjoining parts of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:2831–2846. 10.1099/0022-1317-69-11-2831 [DOI] [PubMed] [Google Scholar]
- 44.McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, Perry LJ, Scott JE, Taylor P. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531–1574. 10.1099/0022-1317-69-7-1531 [DOI] [PubMed] [Google Scholar]
- 45.McGeoch DJ, Dolan A, Donald S, Brauer DH. 1986. Complete DNA sequence of the short repeat region in the genome of herpes simplex virus type 1. Nucleic Acids Res. 14:1727–1745. 10.1093/nar/14.4.1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGeoch DJ, Dolan A, Donald S, Rixon FJ. 1985. Sequence determination and genetic content of the short unique region in the genome of herpes simplex virus type 1. J. Mol. Biol. 181:1–13. 10.1016/0022-2836(85)90320-1 [DOI] [PubMed] [Google Scholar]
- 47.Ushijima Y, Luo C, Goshima F, Yamauchi Y, Kimura H, Nishiyama Y. 2007. Determination and analysis of the DNA sequence of highly attenuated herpes simplex virus type 1 mutant HF10, a potential oncolytic virus. Microbes Infect. 9:142–149. 10.1016/j.micinf.2006.10.019 [DOI] [PubMed] [Google Scholar]
- 48.Macdonald SJ, Mostafa HH, Morrison LA, Davido DJ. 2012. Genome sequence of herpes simplex virus 1 strain KOS. J. Virol. 86:6371–6372. 10.1128/JVI.00646-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watson G, Xu W, Reed A, Babra B, Putman T, Wick E, Wechsler SL, Rohrmann GF, Jin L. 2012. Sequence and comparative analysis of the genome of HSV-1 strain McKrae. Virology 433:528–537. 10.1016/j.virol.2012.08.043 [DOI] [PubMed] [Google Scholar]
- 50.Macdonald SJ, Mostafa HH, Morrison LA, Davido DJ. 2012. Genome sequence of herpes simplex virus 1 strain McKrae. J. Virol. 86:9540–9541. 10.1128/JVI.01469-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown SM, Ritchie DA, Subak-Sharpe JH. 1973. Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mutants, their arrangement into complementation groups and recombination analysis leading to a linkage map. J. Gen. Virol. 18:329–346 [DOI] [PubMed] [Google Scholar]
- 52.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357–364. 10.1099/0022-1317-2-3-357 [DOI] [PubMed] [Google Scholar]
- 53.Dix RD, McKendall RR, Baringer JR. 1983. Comparative neurovirulence of herpes simplex virus type 1 strains after peripheral or intracerebral inoculation of BALB/c mice. Infect. Immun. 40:103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith KO. 1964. Relationship between the envelope and the infectivity of herpes simplex virus. Proc. Soc. Exp. Biol. Med. 115:814–816. 10.3181/00379727-115-29045 [DOI] [PubMed] [Google Scholar]
- 55.Williams LE, Nesburn AB, Kaufman HE. 1965. Experimental induction of disciform keratitis. Arch. Ophthalmol. 73:112–114. 10.1001/archopht.1965.00970030114023 [DOI] [PubMed] [Google Scholar]
- 56.Takakuwa H, Goshima F, Nozawa N, Yoshikawa T, Kimata H, Nakao A, Nawa A, Kurata T, Sata T, Nishiyama Y. 2003. Oncolytic viral therapy using a spontaneously generated herpes simplex virus type 1 variant for disseminated peritoneal tumor in immunocompetent mice. Arch. Virol. 148:813–825. 10.1007/s00705-002-0944-x [DOI] [PubMed] [Google Scholar]
- 57.Flexner S, Amoss HL. 1925. Contributions to the pathology of experimental virus encephalitis. II. Herpetic strains of encephalitogenic virus. J. Exp. Med. 41:233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar S, Tamura K, Nei M. 1994. MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Comput. Appl. Biosci. 10:189–191 [DOI] [PubMed] [Google Scholar]
- 60.Jukes T, Cantor C. 1969. Evolution of protein molecules, p 21–132 In Munro H. (ed), Mammalian protein metabolism. Academic Press, New York, NY [Google Scholar]
- 61.Rozas J, Rozas R. 1995. DnaSP, DNA sequence polymorphism: an interactive program for estimating population genetics parameters from DNA sequence data. Comput. Appl. Biosci. 11:621–625 [DOI] [PubMed] [Google Scholar]
- 62.Thurston MI, Field D. 2005. MsatFinder: detection and characterization of microsatellites. CEH Oxford, Oxford, United Kingdom [Google Scholar]
- 63.Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, Enquist LW. 2011. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 7:e1002282. 10.1371/journal.ppat.1002282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Legendre M, Pochet N, Pak T, Verstrepen KJ. 2007. Sequence-based estimation of minisatellite and microsatellite repeat variability. Genome Res. 17:1787–1796. 10.1101/gr.6554007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vinces MD, Legendre M, Caldara M, Hagihara M, Verstrepen KJ. 2009. Unstable tandem repeats in promoters confer transcriptional evolvability. Science 324:1213–1216. 10.1126/science.1170097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217. 10.1006/jmbi.2000.4042 [DOI] [PubMed] [Google Scholar]
- 68. Reference deleted.
- 69.Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, Liu M, Kumar S, Zaremba S, Gu Z, Zhou L, Larson CN, Dietrich J, Klem EB, Scheuermann RH. 2012. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 40:D593–D598. 10.1093/nar/gkr859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Henikoff S, Henikoff JG. 1994. Position-based sequence weights. J. Mol. Biol. 243:574–578. 10.1016/0022-2836(94)90032-9 [DOI] [PubMed] [Google Scholar]
- 71.Edgeworth FY. 1888. On a new method of reducing observations relating to several quantities. Philos. Mag. Ser. 5:184–191 [Google Scholar]
- 72.Yang Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13:555–556 [DOI] [PubMed] [Google Scholar]
- 73.Adachi J, Hasegawa M. 1996. MOLPHY version 2.3: programs for molecular phylogenetics based o-n maximum likelihood. Computer science monographs no. 28. Institute of Statistical Mathematics, Tokyo, Japan [Google Scholar]
- 74.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16:276–277. 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- 75. Reference deleted.
- 76.Massingham T, Goldman N. 2005. Detecting amino acid sites under positive selection and purifying selection. Genetics 169:1753–1762. 10.1534/genetics.104.032144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882–888. 10.1038/nsmb.1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sneath PHA, Sokal RR. 1973. Numerical taxonomy. Freeman, San Francisco, CA [Google Scholar]
- 79.Felsenstein J. 1985. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39:783–791. 10.2307/2408678 [DOI] [PubMed] [Google Scholar]
- 80.Tamura K, Nei M, Kumar S. 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U. S. A. 101:11030–11035. 10.1073/pnas.0404206101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73:152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deback C, Boutolleau D, Depienne C, Luyt CE, Bonnafous P, Gautheret-Dejean A, Garrigue I, Agut H. 2009. Utilization of microsatellite polymorphism for differentiating herpes simplex virus type 1 strains. J. Clin. Microbiol. 47:533–540. 10.1128/JCM.01565-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deback C, Luyt CE, Lespinats S, Depienne C, Boutolleau D, Chastre J, Agut H. 2010. Microsatellite analysis of HSV-1 isolates: from oropharynx reactivation toward lung infection in patients undergoing mechanical ventilation. J. Clin. Virol. 47:313–320. 10.1016/j.jcv.2010.01.019 [DOI] [PubMed] [Google Scholar]
- 84.Maertzdorf J, Remeijer L, Van Der Lelij A, Buitenwerf J, Niesters HG, Osterhaus AD, Verjans GM. 1999. Amplification of reiterated sequences of herpes simplex virus type 1 (HSV-1) genome to discriminate between clinical HSV-1 isolates. J. Clin. Microbiol. 37:3518–3523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Umene K, Watson RJ, Enquist LW. 1984. Tandem repeated DNA in an intergenic region of herpes simplex virus type 1 (Patton). Gene 30:33–39. 10.1016/0378-1119(84)90102-1 [DOI] [PubMed] [Google Scholar]
- 86.Umene K, Kawana T. 2003. Divergence of reiterated sequences in a series of genital isolates of herpes simplex virus type 1 from individual patients. J. Gen. Virol. 84:917–923. 10.1099/vir.0.18809-0 [DOI] [PubMed] [Google Scholar]
- 87.Simon A, Mettenleiter TC, Rziha HJ. 1989. Pseudorabies virus displays variable numbers of a repeat unit adjacent to the 3′ end of the glycoprotein gII gene. J. Gen. Virol. 70:1239–1246. 10.1099/0022-1317-70-5-1239 [DOI] [PubMed] [Google Scholar]
- 88.Locker H, Frenkel N. 1979. BamI, KpnI, and SalI restriction enzyme maps of the DNAs of herpes simplex virus strains Justin and F: occurrence of heterogeneities in defined regions of the viral DNA. J. Virol. 32:429–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Umene K, Oohashi S, Yoshida M, Fukumaki Y. 2008. Diversity of the a sequence of herpes simplex virus type 1 developed during evolution. J. Gen. Virol. 89:841–852. 10.1099/vir.0.83467-0 [DOI] [PubMed] [Google Scholar]
- 90.Brown J. 2004. Effect of gene location on the evolutionary rate of amino acid substitutions in herpes simplex virus proteins. Virology 330:209–220. 10.1016/j.virol.2004.09.020 [DOI] [PubMed] [Google Scholar]
- 91.Verstrepen KJ, Jansen A, Lewitter F, Fink GR. 2005. Intragenic tandem repeats generate functional variability. Nat. Genet. 37:986–990. 10.1038/ng1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. 2010. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 44:445–477. 10.1146/annurev-genet-072610-155046 [DOI] [PubMed] [Google Scholar]
- 93.van Belkum A, Scherer S, van Alphen L, Verbrugh H. 1998. Short-sequence DNA repeats in prokaryotic genomes. Microbiol. Mol. Biol. Rev. 62:275–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Richard GF, Kerrest A, Dujon B. 2008. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol. Mol. Biol. Rev. 72:686–727. 10.1128/MMBR.00011-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nash TC, Spivack JG. 1994. The UL55 and UL56 genes of herpes simplex virus type 1 are not required for viral replication, intraperitoneal virulence, or establishment of latency in mice. Virology 204:794–798. 10.1006/viro.1994.1595 [DOI] [PubMed] [Google Scholar]
- 96.MacLean AR, Brown SM. 1987. Deletion and duplication variants around the long repeats of herpes simplex virus type 1 strain 17. J. Gen. Virol. 68:3019–3031. 10.1099/0022-1317-68-12-3019 [DOI] [PubMed] [Google Scholar]
- 97.Kehm R, Rösen-Wolff A, Darai G. 1996. Restitution of the UL56 gene expression of HSV-1 HFEM led to restoration of virulent phenotype; deletion of the amino acids 217 to 234 of the UL56 protein abrogates the virulent phenotype. Virus Res. 40:17–31. 10.1016/0168-1702(96)80248-6 [DOI] [PubMed] [Google Scholar]
- 98.Berkowitz C, Moyal M, Rösen-Wolff A, Darai G, Becker Y. 1994. Herpes simplex virus type 1 (HSV-1) UL56 gene is involved in viral intraperitoneal pathogenicity to immunocompetent mice. Arch. Virol. 134:73–83. 10.1007/BF01379108 [DOI] [PubMed] [Google Scholar]
- 99.Rösen-Wolff A, Frank S, Raab K, Moyal M, Becker Y, Darai G. 1992. Determination of the coding capacity of the BamHI DNA fragment B of apathogenic Herpes simplex virus type 1 strain HFEM by DNA nucleotide sequence analysis. Virus Res. 25:189–199. 10.1016/0168-1702(92)90133-T [DOI] [PubMed] [Google Scholar]
- 100. Reference deleted.
- 101.Umene K, Fukumaki Y. 2011. DNA genome of spontaneously occurring deletion mutants of herpes simplex virus type 1 lacking one copy of the inverted repeat sequences of the L component. Arch. Virol. 156:1305–1315. 10.1007/s00705-011-0983-2 [DOI] [PubMed] [Google Scholar]
- 102.Frenkel N, Jacob RJ, Honess RW, Hayward GS, Locker H, Roizman B. 1975. Anatomy of herpes simplex virus DNA. III. Characterization of defective DNA molecules and biological properties of virus populations containing them. J. Virol. 16:153–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Denniston KJ, Madden MJ, Enquist LW, Vande Woude G. 1981. Characterization of coliphage lambda hybrids carrying DNA fragments from Herpes simplex virus type 1 defective interfering particles. Gene 15:365–378. 10.1016/0378-1119(81)90180-3 [DOI] [PubMed] [Google Scholar]
- 104.Frenkel N, Locker H, Vlazny DA. 1980. Studies of defective herpes simplex viruses. Ann. N. Y. Acad. Sci. 354:347–370. 10.1111/j.1749-6632.1980.tb27977.x [DOI] [PubMed] [Google Scholar]
- 105.Cuchet D, Potel C, Thomas J, Epstein AL. 2007. HSV-1 amplicon vectors: a promising and versatile tool for gene delivery. Expert Opin. Biol. Ther. 7:975–995. 10.1517/14712598.7.7.975 [DOI] [PubMed] [Google Scholar]
- 106.Everett RD, Fenwick ML. 1990. Comparative DNA sequence analysis of the host shutoff genes of different strains of herpes simplex virus: type 2 strain HG52 encodes a truncated UL41 product. J. Gen. Virol. 71:1387–1390. 10.1099/0022-1317-71-6-1387 [DOI] [PubMed] [Google Scholar]
- 107.Norberg P, Olofsson S, Tarp MA, Clausen H, Bergstrom T, Liljeqvist JA. 2007. Glycoprotein I of herpes simplex virus type 1 contains a unique polymorphic tandem-repeated mucin region. J. Gen. Virol. 88:1683–1688. 10.1099/vir.0.82500-0 [DOI] [PubMed] [Google Scholar]
- 108.Draper KG, Costa RH, Lee GT, Spear PG, Wagner EK. 1984. Molecular basis of the glycoprotein-C-negative phenotype of herpes simplex virus type 1 macroplaque strain. J. Virol. 51:578–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dolan A, McKie E, MacLean AR, McGeoch DJ. 1992. Status of the ICP34.5 gene in herpes simplex virus type 1 strain 17. J. Gen. Virol. 73:971–973. 10.1099/0022-1317-73-4-971 [DOI] [PubMed] [Google Scholar]
- 110.Wang K, Mahalingam G, Hoover SE, Mont EK, Holland SM, Cohen JI, Straus SE. 2007. Diverse herpes simplex virus type 1 thymidine kinase mutants in individual human neurons and ganglia. J. Virol. 81:6817–6826. 10.1128/JVI.00166-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sasadeusz JJ, Tufaro F, Safrin S, Schubert K, Hubinette MM, Cheung PK, Sacks SL. 1997. Homopolymer mutational hot spots mediate herpes simplex virus resistance to acyclovir. J. Virol. 71:3872–3878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grey F, Sowa M, Collins P, Fenton RJ, Harris W, Snowden W, Efstathiou S, Darby G. 2003. Characterization of a neurovirulent aciclovir-resistant variant of herpes simplex virus. J. Gen. Virol. 84:1403–1410. 10.1099/vir.0.18881-0 [DOI] [PubMed] [Google Scholar]
- 113.Sauerbrei A, Deinhardt S, Zell R, Wutzler P. 2010. Phenotypic and genotypic characterization of acyclovir-resistant clinical isolates of herpes simplex virus. Antiviral Res. 86:246–252. 10.1016/j.antiviral.2010.03.002 [DOI] [PubMed] [Google Scholar]
- 114.Hwang CB, Chen HJ. 1995. An altered spectrum of herpes simplex virus mutations mediated by an antimutator DNA polymerase. Gene 152:191–193. 10.1016/0378-1119(94)00712-2 [DOI] [PubMed] [Google Scholar]
- 115.Liljeqvist JA, Svennerholm B, Bergstrom T. 1999. Herpes simplex virus type 2 glycoprotein G-negative clinical isolates are generated by single frameshift mutations. J. Virol. 73:9796–9802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rekabdar E, Tunback P, Liljeqvist JA, Lindh M, Bergstrom T. 2002. Dichotomy of glycoprotein g gene in herpes simplex virus type 1 isolates. J. Clin. Microbiol. 40:3245–3251. 10.1128/JCM.40.9.3245-3251.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Klauer AA, van Hoof A. 2012. Degradation of mRNAs that lack a stop codon: a decade of nonstop progress. Wiley Interdiscip. Rev. RNA 3:649–660. 10.1002/wrna.1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Inada T. 2013. Quality control systems for aberrant mRNAs induced by aberrant translation elongation and termination. Biochim. Biophys. Acta 1829:634–642. 10.1016/j.bbagrm.2013.02.004 [DOI] [PubMed] [Google Scholar]
- 119.Pan D, Coen DM. 2012. Net1 frameshifting on a noncanonical sequence in a herpes simplex virus drug-resistant mutant is stimulated by nonstop mRNA. Proc. Natl. Acad. Sci. U. S. A. 109:14852–14857. 10.1073/pnas.1206582109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gershburg E, Pagano JS. 2008. Conserved herpesvirus protein kinases. Biochim. Biophys. Acta 1784:203–212. 10.1016/j.bbapap.2007.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tanaka M, Nishiyama Y, Sata T, Kawaguchi Y. 2005. The role of protein kinase activity expressed by the UL13 gene of herpes simplex virus 1: the activity is not essential for optimal expression of UL41 and ICP0. Virology 341:301–312. 10.1016/j.virol.2005.07.010 [DOI] [PubMed] [Google Scholar]
- 122.Coulter LJ, Moss HW, Lang J, McGeoch DJ. 1993. A mutant of herpes simplex virus type 1 in which the UL13 protein kinase gene is disrupted. J. Gen. Virol. 74:387–395. 10.1099/0022-1317-74-3-387 [DOI] [PubMed] [Google Scholar]
- 123.Coller KE, Smith GA. 2008. Two viral kinases are required for sustained long distance axon transport of a neuroinvasive herpesvirus. Traffic 9:1458–1470. 10.1111/j.1600-0854.2008.00782.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Klopfleisch R, Klupp BG, Fuchs W, Kopp M, Teifke JP, Mettenleiter TC. 2006. Influence of pseudorabies virus proteins on neuroinvasion and neurovirulence in mice. J. Virol. 80:5571–5576. 10.1128/JVI.02589-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Blondeau C, Chbab N, Beaumont C, Courvoisier K, Osterrieder N, Vautherot JF, Denesvre C. 2007. A full UL13 open reading frame in Marek's disease virus (MDV) is dispensable for tumor formation and feather follicle tropism and cannot restore horizontal virus transmission of rRB-1B in vivo. Vet. Res. 38:419–433. 10.1051/vetres:2007009 [DOI] [PubMed] [Google Scholar]
- 126.Jarosinski KW, Margulis NG, Kamil JP, Spatz SJ, Nair VK, Osterrieder N. 2007. Horizontal transmission of Marek's disease virus requires US2, the UL13 protein kinase, and gC. J. Virol. 81:10575–10587. 10.1128/JVI.01065-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Moffat JF, Zerboni L, Sommer MH, Heineman TC, Cohen JI, Kaneshima H, Arvin AM. 1998. The ORF47 and ORF66 putative protein kinases of varicella-zoster virus determine tropism for human T cells and skin in the SCID-hu mouse. Proc. Natl. Acad. Sci. U. S. A. 95:11969–11974. 10.1073/pnas.95.20.11969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Smith RF, Smith TF. 1989. Identification of new protein kinase-related genes in three herpesviruses, herpes simplex virus, varicella-zoster virus, and Epstein-Barr virus. J. Virol. 63:450–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Baines JD, Roizman B. 1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J. Virol. 66:5168–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.MacLean CA, Dolan A, Jamieson FE, McGeoch DJ. 1992. The myristylated virion proteins of herpes simplex virus type 1: investigation of their role in the virus life cycle. J. Gen. Virol. 73:539–547. 10.1099/0022-1317-73-3-539 [DOI] [PubMed] [Google Scholar]
- 131.Loomis JS, Courtney RJ, Wills JW. 2003. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 77:11417–11424. 10.1128/JVI.77.21.11417-11424.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yeh PC, Meckes DG, Jr, Wills JW. 2008. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J. Virol. 82:10693–10700. 10.1128/JVI.01230-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Han J, Chadha P, Meckes DG, Jr, Baird NL, Wills JW. 2011. Interaction and interdependent packaging of tegument protein UL11 and glycoprotein e of herpes simplex virus. J. Virol. 85:9437–9446. 10.1128/JVI.05207-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Han J, Chadha P, Starkey JL, Wills JW. 2012. Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc. Natl. Acad. Sci. U. S. A. 109:19798–19803. 10.1073/pnas.1212900109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Negatsch A, Mettenleiter TC, Fuchs W. 2011. Herpes simplex virus type 1 strain KOS carries a defective US9 and a mutated US8A gene. J. Gen. Virol. 92:167–172. 10.1099/vir.0.026484-0 [DOI] [PubMed] [Google Scholar]
- 136.Kolb AW, Schmidt TR, Dyer DW, Brandt CR. 2011. Sequence variation in the herpes simplex virus U(S)1 ocular virulence determinant. Invest. Ophthalmol. Vis. Sci. 52:4630–4638. 10.1167/iovs.10-7032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McGeoch DJ, Rixon FJ, Davison AJ. 2006. Topics in herpesvirus genomics and evolution. Virus Res. 117:90–104. 10.1016/j.virusres.2006.01.002 [DOI] [PubMed] [Google Scholar]
- 138.Baines JD, Ward PL, Campadelli-Fiume G, Roizman B. 1991. The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress. J. Virol. 65:6414–6424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chouljenko VN, Iyer AV, Chowdhury S, Kim J, Kousoulas KG. 2010. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J. Virol. 84:8596–8606. 10.1128/JVI.00298-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Melancon JM, Luna RE, Foster TP, Kousoulas KG. 2005. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J. Virol. 79:299–313. 10.1128/JVI.79.1.299-313.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dollery SJ, Lane KD, Delboy MG, Roller DG, Nicola AV. 2010. Role of the UL45 protein in herpes simplex virus entry via low pH-dependent endocytosis and its relationship to the conformation and function of glycoprotein B. Virus Res. 149:115–118. 10.1016/j.virusres.2010.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Haanes EJ, Nelson CM, Soule CL, Goodman JL. 1994. The UL45 gene product is required for herpes simplex virus type 1 glycoprotein B-induced fusion. J. Virol. 68:5825–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhang Y, Sirko DA, McKnight JL. 1991. Role of herpes simplex virus type 1 UL46 and UL47 in alpha TIF-mediated transcriptional induction: characterization of three viral deletion mutants. J. Virol. 65:829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang Y, McKnight JL. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67:1482–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Davison AJ. 2010. Herpesvirus systematics. Vet. Microbiol. 143:52–69. 10.1016/j.vetmic.2010.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schaerer-Uthurralt N, Erard M, Kindbeiter K, Madjar JJ, Diaz JJ. 1998. Distinct domains in herpes simplex virus type 1 US11 protein mediate post-transcriptional transactivation of human T-lymphotropic virus type I envelope glycoprotein gene expression and specific binding to the Rex responsive element. J. Gen. Virol. 79:1593–1602 [DOI] [PubMed] [Google Scholar]
- 147.Diaz JJ, Dodon MD, Schaerer-Uthurralt N, Simonin D, Kindbeiter K, Gazzolo L, Madjar JJ. 1996. Post-transcriptional transactivation of human retroviral envelope glycoprotein expression by herpes simplex virus Us11 protein. Nature 379:273–277. 10.1038/379273a0 [DOI] [PubMed] [Google Scholar]
- 148.Poppers J, Mulvey M, Khoo D, Mohr I. 2000. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type I Us11 protein J. Virol. 74:11215–11221. 10.1128/JVI.74.23.11215-11221.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cassady KA, Gross M, Roizman B. 1998. The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 72:8620–8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Roller RJ, Roizman B. 1992. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J. Virol. 66:3624–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Roller RJ, Roizman B. 1991. Herpes simplex virus 1 RNA-binding protein US11 negatively regulates the accumulation of a truncated viral mRNA. J. Virol. 65:5873–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Roller RJ, Roizman B. 1990. The herpes simplex virus Us11 open reading frame encodes a sequence-specific RNA-binding protein. J. Virol. 64:3463–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zuccola HJ, Filman DJ, Coen DM, Hogle JM. 2000. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase. Mol. Cell 5:267–278. 10.1016/S1097-2765(00)80422-0 [DOI] [PubMed] [Google Scholar]
- 154.McGeoch DJ, Dolan A, Ralph AC. 2000. Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J. Virol. 74:10401–10406. 10.1128/JVI.74.22.10401-10406.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Grose C. 2012. Pangaea and the out-of-Africa model of varicella-zoster virus evolution and phylogeography. J. Virol. 86:9558–9565. 10.1128/JVI.00357-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.McGeoch DJ. 2009. Lineages of varicella-zoster virus. J. Gen. Virol. 90:963–969. 10.1099/vir.0.007658-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kolb AW, Ane C, Brandt CR. 2013. Using HSV-1 genome phylogenetics to track past human migrations. PLoS One 8:e76267. 10.1371/journal.pone.0076267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Depledge DP, Palser AL, Watson SJ, Lai IY, Gray ER, Grant P, Kanda RK, Leproust E, Kellam P, Breuer J. 2011. Specific capture and whole-genome sequencing of viruses from clinical samples. PLoS One 6:e27805. 10.1371/journal.pone.0027805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zell R, Taudien S, Pfaff F, Wutzler P, Platzer M, Sauerbrei A. 2012. Sequencing of 21 varicella-zoster virus genomes reveals two novel genotypes and evidence of recombination. J. Virol. 86:1608–1622. 10.1128/JVI.06233-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Norberg P. 2010. Divergence and genotyping of human alpha-herpesviruses: an overview. Infect. Genet. Evol. 10:14–25. 10.1016/j.meegid.2009.09.004 [DOI] [PubMed] [Google Scholar]
- 161.Kosovsky J, Vojvodova A, Oravcova I, Kudelova M, Matis J, Rajcani J. 2000. Herpes simplex virus 1 (HSV-1) strain HSZP glycoprotein B gene: comparison of mutations among strains differing in virulence. Virus Genes 20:27–33. 10.1023/A:1008104006007 [DOI] [PubMed] [Google Scholar]
- 162.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. 10.1126/science.1126548 [DOI] [PubMed] [Google Scholar]
- 163.Sievers E, Neumann J, Raftery M, SchOnrich G, Eis-Hubinger AM, Koch N. 2002. Glycoprotein B from strain 17 of herpes simplex virus type I contains an invariant chain homologous sequence that binds to MHC class II molecules. Immunology 107:129–135. 10.1046/j.1365-2567.2002.01472.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Becker Y. 1992. Computer prediction of antigenic and topogenic domains in HSV-1 and HSV-2 glycoprotein B (gB). Virus Genes 6:131–141. 10.1007/BF01703062 [DOI] [PubMed] [Google Scholar]
- 165.Mester JC, Highlander SL, Osmand AP, Glorioso JC, Rouse BT. 1990. Herpes simplex virus type 1-specific immunity induced by peptides corresponding to an antigenic site of glycoprotein B. J. Virol. 64:5277–5283 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.