

Abstract
Gene therapy as a treatment for cancer is regarded as high in promise, but low in delivery, a deficiency that has become more obvious with ever-increasing reports of the successful correction of monogenic disorders by this approach. We review the commercial and scientific obstacles that have led to these delays and describe how they are progressively being overcome. Recent and striking successes and correspondingly increased commercial involvement suggest that gene transfer could finally become a powerful method for development of safe and effective cancer therapeutic drugs.
Introduction
Because cancer is an acquired genetic disorder, gene therapy promises to provide highly bespoke and effective treatment that is tailored to the precise genetic structure of each tumour and therefore produces low systemic toxicity. Unfortunately, however, the claim that the next 5 years will meet this promise has been repeated for more than 20 years. Most clinicians are therefore convinced that although gene therapy for cancer could be a bespoke option, the suit itself is essentially empty. In this Review, we discuss why we think the next 5 years will be different, provide examples of how present clinical studies are showing increasing benefit to patients with cancer, and discuss how combination gene therapies might begin to meet this promise.
Gene therapy for monogenic disorders is beginning to meet expectations. During the past few years, the successful treatment of severe immunodeficiencies, storage disorders, hereditary blindness, haemophilia B, hypercholesterolaemia, and other diseases has led to the licensing of one drug and the continued clinical development of many others by major pharmaceutical companies. Yet, although 1700 gene therapy studies have been done worldwide and more than 80% of patients entered have been treated for malignant disease, the level of success for cancer is far behind that obtained in the treatment of monogenic disorders.
To understand the reasons for this delay and to appreciate the changes in this discipline that we believe now predict success, we must understand the potential barriers to success, and the approaches taken to overcome them.
Commercial obstacles
The broad application of gene therapy in every context, including cancer, has been hampered by its lack of similarity to the traditional pharmaceutical model of drug development. Gene therapies are complex biological therapies, many of which, such as genetically modified stem cells, are made separately for each individual treated, which represents a challenge to the robust scalability needed for late-phase clinical studies. Moreover, the standard pharmaceutical business model is to recoup the costs of initial drug development by selling cheap-to-manufacture licensed drugs with very high margins. For many complex biological treatments, however, the cost of goods is high even after approval; an effect compounded by stacked licence fees for the many patents needed for the various intellectual properties of one product. The specificity of these therapies means that only a small subset of patients with any given cancer might be suited to treatment, making each gene therapy an orphan drug. In combination, these market issues can lead to an unaffordable pricing structure with little appeal to major pharmaceutical companies.
Beyond these commercial considerations, well designed clinical trials for cancer using complex biological drugs are difficult to undertake. Endpoints that are standard for most small-molecule therapeutic studies in cancer, such as tumour shrinkage at 4–6 weeks, are unsuited to some complex biological treatments that might induce initial tumour inflammation with apparent progression by imaging, or might produce extended stabilisation of the tumour without shrinkage or eradication, so that patients live with, rather than die from, their disease. Although such benefits to long-term survival could be substantial, they greatly increase the cost and time of clinical studies, as shown by the development of sipuleucel-T,1 so far the only gene-to-cell therapeutic to have received a product licence in the USA. Additionally, many gene therapies function by recruiting the immune system and could be particularly unsuited to assessment in the classic phase 1 setting of advanced disease, because immunity will probably be disrupted by both the disease and its treatment.
Potential limitations
Although investigators working with inherited disorders share most of the difficulties we have described, their achievements have clearly been in greater evidence. Other factors should therefore be invoked to explain the more moderate accomplishments of cancer gene therapy. Gene therapy can be used to attack the tumour directly or to modify the host to increase resistance to the disease or its treatment (table 1). Although the success of gene therapy to directly correct monogenic disorders seems translatable to a direct tumour attack for cancer as well, this success has proved very difficult to replicate. In the successfully treated inherited monogenic disorders, one transgenic construct can be used to replace or supplement a well defined single genetic abnormality. Although cancer is an (acquired) genetic disorder, the abnormalities are polygenic. Moreover, there is substantial genetic heterogeneity,3,4 not only between tumours in different individuals, but also between tumours at different sites within the same patient. Unless a universal corrective gene could be identified, efforts to use gene transfer to correct abnormal genetic function in cancer cells would need access to many transgenes, individually tailored for optimum use between, and even within, each individual. Moreover, clinical correction of most monogenic disorders often needs only a small proportion of the targeted cells to be successfully transduced with the relevant transgene. In disorders in which the missing gene produces a secreted protein (eg, haemophilia B), less than 10% of normal concentration of the product could restore health, and if the transgene is secreted from each cell at supranormal concentrations, then the number of cells transduced could be far lower than 10% of the targeted population.5 For treatment of malignant diseases, such low uptake of corrective or destructive gene transfer to tumour tissue could have negligible effect on the course of the disease. Thus, gene transfer alone cannot be a curative option, unless the bulk of tumour cells is genetically modified—eg, by a lethal viral vector that was selectively able to replicate only within tumour cells. Until now, and for the foreseeable future, such high efficiency is not straightforward. Most gene transfer vectors have insufficient infectivity or inadequately broad biodistribution to transfer the desired corrective or destructive genes to each cancer cell (table 2). The gene therapy community has taken a surprisingly long time to accept these limitations and to diminish the resources devoted to trials of direct gene therapy other than virotherapy.
Table 1.
Therapeutic applications of cancer gene therapy
| Comments | |
|---|---|
| Gene repair | Correction of genetic defects associated with the malignant process hampered by low efficiency of gene transfer |
| Prodrug-metabolising enzyme gene therapy | Renders the tumour cells sensitive to corresponding cytotoxic drug |
| Viral oncolysis | Delivery of viruses that selectively replicate in tumour cells |
| Modulation of the tumour microenvironment | Inhibition of angiogenesis or tumour cell proteinases |
| Drug resistance gene therapy | Prevention of toxic side-effects of chemotherapeutic drugs |
| Immunotherapy with genetically modified cells | Generation of or boosting of immune responses to tumour antigens by genetically modifying effector cells, antigen-presenting cells, or tumour cells |
Adapted from Gottschalk and coworkers2 with permission.
Table 2.
Advantages and disadvantages of cancer gene therapy vectors
| Advantages | Disadvantages | Cancer gene therapy application | |
|---|---|---|---|
| Adenovirus | Poxviruses and vaccinia virus | High titre, broad tropism, accepts large insert size | Complex viral genome, replicates in target cell, immunogenic* |
| Adeno-associated virus | High titre, broad tropism, efficient gene transfer, transduces non-dividing cells, limited immunogenicity | Limited insert size | PDME gene therapy, modulation of the tumour microenvironment, immunotherapy |
| Herpes virus | High titre, accepts large insert size | Complex viral genome, can be cytotoxic to cells | Viral oncolysis, PDME gene therapy |
| Lentivirus | Stable genome integration†, integrates into non-dividing cells, long-term gene expression, low immunogenicity | Risk of insertional mutagenesis, inefficient in-vivo gene delivery | Gene repair, PDME gene therapy, drug resistance gene therapy, gene marking, immunotherapy |
| Poxviruses and vaccinia virus | High titre, broad tropism, accepts large insert size | Complex viral genome, replicates in target cell, immunogenic* | Viral oncolysis, immunotherapy |
| Retrovirus | Stable genome integration†, long-term gene expression, low immunogenicity | Integration only in dividing cells, imited insert size, risk of insertional mutagenesis, inefficient in-vivo gene delivery | Gene repair, PDME gene therapy, drug resistance gene therapy, gene marking, immunotherapy |
| Non-viral DNA delivery | Non-viral, accepts large insert size, low immunogenicity | Transient gene expression, inefficient in-vivo gene delivery | Gene repair, modulation of the tumour microenvironment, immunotherapy |
PDME=prodrug-metabolising enzyme.
Immunogenicity is regarded as advantageous for many cancer immunotherapy applications.
Integration of vector sequences into the genome is a characteristic of retroviruses, lentiviruses, and to an extent, adeno-associated viruses; depending on the application this might be an advantage or disadvantage. Adapted from Gottschalk and coworkers2 with permission.
Much effort in cancer gene therapy is now directed not at the tumour itself, but at healthy host tissue, with the intent to disrupt the tumour microenvironment or niche, increase the resistance of healthy tissue to cytotoxic drugs or radiation, or transfer genes that augment the immune response against the tumour. This last approach is by far the most common, because the component parts of the immune system have individualised targeting capacity, active biodistribution, and a multiplicity of effector mechanisms by which even bulky tumours can be destroyed; all activities that could be enhanced by genetic engineering of the cell.
Overcoming the barriers
The multiplicity of practical and potential obstacles to successful gene therapy of cancer will be overcome only by continued and widespread economic commitment to research and development. However, to persuade the industrial and clinical community of the wisdom of this extended commitment, investigators need to show that gene therapy, alone, or in non-cross-resistant combination with conventional therapy, is qualitatively better than any other available therapies, and able to benefit, and ideally cure, cancers that are otherwise not amenable to conventional treatment. Yet, paradoxically, the resources necessary to develop truly effective gene therapy for cancer will be available only once truly effective gene therapies are developed. Fortunately, the prospects of escape from this dilemma have now greatly improved.
Acceptance is increasing among physicians and drug companies alike that the future of cancer drug development will consist of creating medicines, such as gene therapies, that are highly effective cures for identified subsets of patients rather than unpredictable palliatives that can be used with moderate benefit for most patients. Once these potent targeted therapies show improved therapeutic activities even in phase 1 (nominal safety) studies, pivotal (licensing) studies can be completed faster and with far smaller numbers of patients than will be needed for conventional drugs. Since these new targeted drugs will have vastly improved pharmacoeconomics in terms of quality and length of life, they can be priced at a premium and the reduced development costs readily recovered.
Gradual successes have been made, with sustained tumour regression noted in substantial numbers of patients, particularly when investigators use genetic modification of the immune system to achieve benefit. Although few of these treatments have yet progressed to pivotal trials, the results have been sufficiently striking to break down many of the barriers perceived by large pharmaceutical companies. Perhaps most importantly, many of these individual approaches are non-cross resistant and are therefore complementary to each other and can be integrated with conventional therapy, resulting in synergy and lack of overlapping toxicity.
Approaches that directly target the tumour
Prodrug metabolising enzymes
Modification of cancer cells with genes encoding enzymes that convert harmless prodrugs into lethal cytotoxins is an amplification step that could overcome the difficulties of inefficient gene transfer to cancer cells. More than 20 prodrug metabolising enzyme systems have been described, of which the herpes simplex virus-derived thymidine kinase (HSV-tk) gene is the most widely studied. The enzyme encoded by this gene phosphorylates prodrugs, such as valaciclovir, to toxic nucleosides.6 For selectivity to tumour cells, investigators could use a vector that is expressed only in dividing cells (eg, a gamma retroviral vector), an agent that is mainly active only in dividing cells (eg, HSV-tk plus valaciclovir), or a vector that is targeted to tumour cells by expression of a receptor to a target cell ligand of restricted distribution. Despite 20 years of effort initially using HSV-tk gene transfer trials for patients with CNS malignancies, and encouraging results of one randomised phase 3 clinical trial for patients with glioma,7 no HSV-tk gene-based therapy product has received approval from regulatory agencies. In many of these studies, low efficacy has probably been due to poor transduction, poor targeting, and low anti-tumour activity of the activated prodrug. Further clinical advances await improvements in all three approaches.
Virotherapy or viral oncolysis
Oncolytic viruses can selectively replicate in cancer cells, causing cell death while sparing healthy cells.8 Case reports documenting substantial regression of cancers after viral infections in the first half of the 20th century gave credence to the use of replicating viruses as treatment. More detailed knowledge of the molecular biology of viruses and cancer has allowed oncolytic viruses, which replicate only in tumour cells, to be developed from adenoviruses, herpes simplex, reo, mumps, West Nile, vaccinia, measles, and Newcastle disease viruses; all of which have been tested in human clinical studies.9,10 These viruses are engineered with a promoter for crucial viral replication genes that function only in malignant cells, or have a genetic deficiency (eg, a missing transcriptional regulator) that is complemented by an overabundance in malignant cells. For example, the oncolytic adenovirus dl1520 (Onyx-015) has a deletion of the viral E1B gene, which limits its replication to (malignant) cells with a defective P53 pathway,11 or that complement dl1520-defective viral RNA export.12 Onyx-015 was tested in several phase 1 and 2 clinical trials, either alone or in combination with chemotherapy, with improved response rates noted in patients with head and neck cancer after local injection in combination with systemic cisplatin and fluorouracil.13 Of 37 patients treated, eight had a complete response and 11 had partial responses.14 For various commercial reasons, however, development in the USA was not continued. But, in China, a closely related E1B-deleted adenovirus was successful in patients with head and neck cancer and in 2005 received marketing approval.15 Subsequent studies13,16 with other oncoviruses continue to show promising results, but none have yet successfully concluded late-phase or pivotal clinical studies. Most probably, there is little initial tumour transduction and only few subsequent replicative cycles occur before the host innate and adaptive immune responses terminate the process. Other oncolytic viruses try to exploit this otherwise unwanted immune response by incorporating immunostimulatory genes, such as granulocyte-macrophage colony-stimulating factor (GM-CSF) or interleukin 12, effectively turning infected target cells into tumour vaccines generating an immune response that recognises and kills even uninfected tumour targets.
The tumour environment
The tumour microenvironment provides a crucial cellular support matrix for sustained tumour growth. For example, angiogenesis is a prerequisite for the development of tumours, and drugs that block this process have shown therapeutic value. Gene therapy should be an excellent means of inhibiting angiogenesis, because it allows continuous delivery of the drug, avoiding the irregularity of delivery resulting from intermittent injection, and by targeting the tumour vasculature, it can increase local concentration of angiogenesis inhibitors.17 In animal models, injection of adeno-associated viruses containing angiostatin or endostatin, or both genes reduced tumour growth,18 and early-phase clinical studies with the human endostatin gene (E10A) have safely reduced proangiogenic factors in serum19 and are being assessed for clinical benefit.
Approaches that enhance host resistance to drug toxicity and cancer
Gene therapy to protect against toxicity
An increased understanding of the molecular mechanisms of cytotoxic drug resistance has allowed the development of gene therapy approaches to protect healthy tissues from the toxicities of chemotherapy. An effective increase in the therapeutic index of cytotoxic drugs or radiation might allow more patients to be cured, analogous to the rescue by haemopoietic stem cell transplantation of patients with cancer from the curative, but lethal, effects of ablative high-dose therapy. Although great effort has been expended on this approach, there have been few successes. In early trials,20,21 gene-modified cells were not selected, and hence there was no protection as a result of poor gene transfer and low expression. However, technical advances in gene transfer have enabled the limitations of low transfer or low expression to be overcome, but for the haemopoietic system, lack of gene transfer to other vital organs means that toxicity to these could rapidly supervene when increased marrow resistance has allowed intensification of cytotoxic drug dose. Drug resistance gene therapy also has the risk of transferring the genes to malignant cells, neutralising potential benefit. However, a clinical study22 has shown that transfer of 06-methylguanine DNA methyltransferase gene to the autologous marrow cells ex vivo confers resistance to alkylating agents such as carmustine, improving haematological recovery in patients with glioblastoma given carmustine, an outcome that could be associated with improved anti-tumour responses. However, use of this approach for cytotoxic drugs with a broader toxicity profile is not straightforward, and as targeted small molecules make ever greater progress in clinical practice, the need for such drug resistance genes could become moot.
Cancer vaccines
Vaccination is an effective strategy to protect animals and human beings from bacterial and viral infections, but few vaccines are effective against established disease.23 Not surprisingly therefore, the past 20 years have shown that use of gene transfer to make a successful vaccine for cancer is a futile task, with promising preclinical data often leading to failure in subsequent clinical studies, so that less than 10% of recipients of cancer vaccine show benefit.24–26
To be effective, a cancer vaccine should contain a tumour-associated antigen (TAA) that does not effectively activate the immune system, but can induce an immune response of sufficient potency to eradicate established tumours in a host whose immune system has often been severely compromised by the disease or its treatment. Although most investigators agree that these benefits require vaccines to induce cytotoxic T lymphocyte (CTL) rather than antibody or other responses, little agreement has been made on how this could best be obtained or which antigens are the most appropriate for targeting.27 Many different strategies are being investigated, and although one at least has led to a licensed gene therapy for advanced prostate cancer1 the hoped-for potential is yet to be reached.
To elicit CTL responses in vivo, either the TAA should be expressed by antigen-presenting cells (APCs), such as dendritic cells, or the tumour cell itself should be modified to become a de-facto APC. The simplest vaccine strategies use the DNA sequence of known tumour antigens, often in association with genes encoding immunostimulatory molecules such as cytokines. These vaccines are injected at distal sites and are taken up and expressed by APCs.28,29 DNA vaccines containing TAA genes, such as immunoglobulin idiotype for B-cell lymphomas, tyrosinase epitopes for melanoma, and cancer carcinoembryonic antigen (CEA) for colorectal cancer, have been investigated,30–32 but so far clinical potency is insufficient for licensing.
Investigators have tried to optimise presentation of TAAs by directly loading APCs, such as dendritic cells with antigen (DNA, RNA, or protein) ex vivo.33–37 To overcome immune unresponsiveness, many of these vaccines also incorporate transgenes for immunostimulatory molecules including cytokines (eg, GM-CSF38) or T-cell stimulatory ligands (eg, CD40L39). The use of dendritic cells as cancer vaccines has been tested in more than 175 reported clinical trials, most of which have been in melanoma, prostate cancer, colorectal cancer, myeloma, and pancreatic cancer.24,25 Although several of these vaccines have failed pivotal studies, an APC vaccine targeting prostate acid phosphatase produced 31.7% versus 23.0% 3-year survival in men with advanced disease and has received a product licence from the US Food and Drug Administration.1 Investigators hope that the next generation of dendritic cell vaccines with improved immunostimulatory properties in phase 1 and 2 clinical studies will have correspondingly greater anti-tumour activity. If, however, their potency is unchanged, then—irrespective of licensing—their cost will make them unlikely to become mainstays of therapeutic practice.
Tumour cells themselves can be genetically modified in vivo or in vitro to express genes that induce one or many phases of CTL activation and expansion directed to the specific target antigens expressed by the tumour (figure 1). These cells might be autologous or derived from an allogeneic tumour cell line, and are modified to express genes that could be intended to attract APCs,39,40 or to summon, activate, or expand T cells.41,42 Although undoubted and sustained tumour responses have been noted with genetically modified tumour vaccines in individual patients with diseases, including melanoma, prostate cancer, neuroblastoma, and chronic and acute lymphocytic leukaemias, these responses have been sporadic and unpredictable; thus, no successful phase 3 studies have been reported.
Figure 1. Genetic modification of tumour cells.
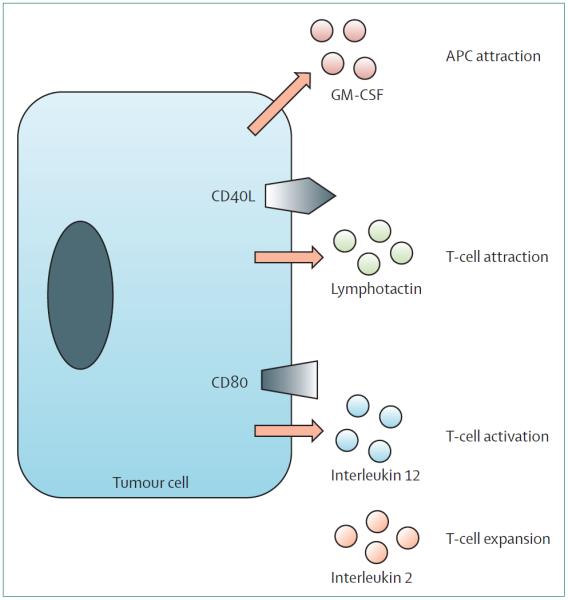
Tumour cells can be genetically modified to improve their antigen-presenting capacity or ability to attract APCs (CD40L, GM-CSF), to attract (lymphotactin) or activate T cells (CD80; interleukin-12), or to expand T cells (interleukin-2). GM-CSF=granulocyte-macrophage colony-stimulating factor.
APC=antigen-presenting cell. Adapted from Gottschalk and coworkers2 with permission.
Why has the tumour vaccination strategy proved so frustrating, with just enough success to maintain interest, but too little to allow definitive trials that lead to licensing? The success of vaccination with tumour cells depends on a multiplicity of factors, including tumour type, type of immune evasion strategy used by the individual tumour, level of transgene expression, challenge site, and vaccine schedule. These factors can vary between vaccine preparations and between patients. Although some variability might be reduced—eg, by using standardised, genetically enhanced tumour cell lines rather than the patient's own tumour cells as the source of antigenic stimulation—inherent complexity is a major barrier to identification of the best study group and the optimum trial design.
Despite these disappointments, two factors seem likely to ensure that tumour immunisation strategies will be resurgent over the next 5 years. The licensure of ipilimumab,43 the first of many potential checkpoint antibodies for T cells, means that investigators now have a method to augment and sustain anti-tumour immune responses by blocking the inhibitory or checkpoint signals that usually regulate the growth and activity of T cells. Although in a randomised trial43 in patients with melanoma, the combination of tumour immunisation and ipilimumab was not better than ipilimumab alone, the vaccine consisted of two HLA-A2-restricted peptides emulsified only in incomplete Freund's adjuvant. More potent vaccines in combination with one or more of these checkpoint antibodies could ensure a more uniformly potent immune response and produce more predictable tumour responses. Equally importantly, the success of adoptive T-cell therapies for an ever-broadening range of tumours will afford an opportunity for tumour vaccines to be used to boost these adoptive responses still further.
Genetic modification of T cells
Because of the intrinsic deficiencies in the immune systems of many patients with cancer, much effort has focused on the direct enhancement of the effector arm of the immune system ex vivo, and then returning these effector cells to the patient. Further, because this manipulation bypasses the need for antigen presentation and recruitment or activation of effector cells, it has potential for benefit, even if the afferent arm of the immune system is severely impaired by cancer or its treatment. Moreover, the effector cells could be genetically modified ex vivo to enhance their ability to target tumour cells and to counteract the numerous evasion strategies manifest by many potentially immunogenic tumours. These efforts have begun to show results, and the integration of adoptive-cell therapy with vaccination strategies and checkpoint antibodies will probably lead to effective cancer therapies that will exceed in breadth and in potency those already produced by any one component alone. So far, most clinical studies of adoptive transfer of effector cells use T lymphocytes, based on preclinical evidence that suggests that these will be the most efficacious, although clinical studies using natural killer or natural killer T cells have begun.
Enhancement of T-cell targeting
To reduce the difficulty and inherent interpatient variability of ex-vivo generation of tumour-reactive T cells directly from patients with cancer, investigators have transferred sequences encoding standardised TAA-specific receptors into bulk T-cell populations. Two types of receptor are used (figure 2). In transfer of the α and β T-cell receptor (TCR) genes, TRA and TRB, T cells are genetically modified to express transgenic TCR α and β chains chosen for their high specificity and affinity for the intended target antigen.44,45 Both TCR chains can be accommodated in one retroviral or lentiviral vector, and transfer of the tumour-specific TRA and TRB genes to mitogen-activated T cells allows rapid generation of large numbers of tumour-directed T cells;46 several clinical trials47–51 have investigated these. For example, investigators at the National Cancer Institute47 adoptively transferred autologous T cells transduced with a retroviral vector encoding the TCR α and β chains specific for an HLA-A2-restricted MART1 (also known as MLANA) peptide, and reported objective clinical responses in 12% of patients. To increase the frequency of pronounced tumour responses, the same investigators infused T cells that expressed higher affinity transgenic αβTCR specific for MLANA or glycoprotein 100 peptides. Disappointingly, this strategy only slightly increased the tumour response rate, but greatly increased toxicity, with most patients developing skin rash, uveitis, and hearing loss.48 Other studies49–51 have also reported severe toxicities from this approach. Parkhurst and colleagues49 infused autologous T cells expressing a murine TCR against human CEA to three patients with metastatic colorectal cancer. Although all patients had decreased serum CEA concentrations (>70%), with one objective regression of metastatic lesions, they all developed severe inflammatory colitis. T cells expressing TCR α and β chains, whose affinity for the TAA MAGE-A3 was artificially enhanced, have caused fatal encephalitis and cardiotoxicity.50,51 These results show that a high affinity transgenic TCR directed to a single tumour antigen epitope could have unexpected reactivity with the same antigen expressed at low level in healthy tissue (ie, on-target antigen, but off-target organ), or could cross-react with a related antigen on healthy tissue (off-target toxicity).48–51
Figure 2. Transgenic T-cell receptors.

(A) Characteristics of transgenic α and β (native) T-cell receptors and (B) corresponding features of chimeric antigen receptors, to compare these two most often used strategies for redirecting the T-cell response to cancer. TM=transmembrane domain. scFv=single-chain variable fragment.
Because T cells expressing genetically modified TCR recognise only a single short peptide sequence in association with a single HLA specificity, their use is limited to targeting protein-derived TAAs and only in patients with the appropriate HLA polymorphism. These limitations have increased interest in the use of chimeric antigen receptors (CARs), which are artificial receptors composed of an extracellular domain that causes antigen recognition, a transmembrane domain, and an intracellular signalling domain (figure 2). The extracellular domain is most commonly derived from the antigen-binding portion of a monoclonal antibody, whereas the intracellular signalling domain (endodomain) is most usually from the TCR. CAR expression provides the flexibility of a monoclonal antibody, in terms of HLA-independent recognition and the types of antigens that can be recognised, while retaining the ability of T cells to actively migrate to tumour sites, to expand in vivo, and to recruit a multiplicity of cellular and humoural effector mechanisms to destroy the tumour. Preclinical and clinical studies of CARs have been done (table 3). Although initial studies were disappointing, next-generation CARs have incorporated one or more additional components (derived from costimulatory molecules, such as CD27, CD28, 41BB, OX40, or ICOS), which enhance T-cell activation, proliferation, differentiation, and survival by providing the necessary stimulatory signals that allow T cells to pass through the many checkpoints that govern their fate after receptor engagement.36 Substantial success with this approach has now been independently reported by several teams of investigators,52–58 who have treated CD19-positive B-cell cancers with T cells expressing a next-generation CD19-CAR. They reported substantial in-vivo expansion and persistence for at least 6 months, and complete clinical responses in up to half the patients, including those with relapsed B-cell acute lymphoblastic leukaemia. Solid tumours might also be amenable to a similar approach. Pule and colleagues59,60 expressed CAR directed to disialoganglioside 2 (a non-protein TAA) on Epstein-Barr virus-specific T cells, and gave it to 11 children with advanced neuroblastoma. Three had complete responses (sustained in two), whereas an additional two with bulky tumours showed substantial tumour necrosis. Many other clinical studies are now in progress (table 3).
Table 3.
Chimeric antigen receptors in (or approaching) clinical development for cancer
| In vitro | Animal data | Clinical data | |
|---|---|---|---|
| Haematological malignant diseases | ✓ | ✓ | ✓ |
| CAR-CD19 | ✓ | ✓ | ✓ |
| CAR-CD2 | ✓ | ✓ | ✓ |
| CAR-CD30 | ✓ | ✓ | Ongoing |
| CAR-Kappa | ✓ | ✓ | Ongoing |
| CAR-CD70 | ✓ | ✓ | .. |
| CAR-NKG2D ligands | ✓ | ✓ | .. |
| Solid tumours | |||
| CAR-GD2 (neuroblastoma) | ✓ | ✓ | ✓ |
| CAR-HER2(colorectal/lung/sarcoma/glioblastoma) | ✓ | ✓ | ✓ |
| CAR-FBP (ovarian) | ✓ | ✓ | ✓ |
| CAR-CD171 (neuroblastoma) | ✓ | .. | ✓ |
| CAR-CAIX (kidney) | ✓ | .. | ✓ |
| CAR-PSMA (prostate) | ✓ | ✓ | Ongoing |
| CAR-IL13Rα2 (glioblastoma) | ✓ | ✓ | Ongoing |
| CAR-KDR (vasculature/melanoma) | ✓ | ✓ | Ongoing |
| CAR-EGFRvIII (glioblastoma) | ✓ | .. | Ongoing |
| CAR-Mesothelin (pleural mesothelioma/breast) | ✓ | ✓ | Ongoing |
| CAR-EphA2 (glioblastoma) | ✓ | ✓ | .. |
| CAR-CD44v6/v7 (cervical) | ✓ | ✓ | .. |
| CAR-TAG72 (colon) | ✓ | ✓ | .. |
| CAR-Lewis-Y (breast/ovarian/pancreatic) | ✓ | ✓ | .. |
| CAR-Muc1 (ovarian/breast/prostate) | ✓ | ✓ | .. |
| CAR-IL11Rα (osteosarcoma) | ✓ | ✓ | .. |
| CAR-FAR (rhabdomyosarcoma) | ✓ | .. | .. |
| CAR-NCAM (colorectal) | ✓ | .. | .. |
| CAR-EGP2 (carcinomas) | ✓ | .. | .. |
| CAR-EGP40 (colon) | ✓ | .. | .. |
| CAR-CEA (colorectal) | ✓ | .. | .. |
| CAR-ERBB3/4 (breast/ovarian) | ✓ | .. | .. |
| CAR-GD3 (melanoma) | ✓ | .. | .. |
| CAR-PSCA (prostate/pancreatic) | ✓ | .. | .. |
| CAR-HLA-A1+MAGE1 (melanoma) | ✓ | .. | .. |
Appendix provides complete references for this table.
Although potent, CAR-T cells have potential for lethal toxicities. Adverse events are predominantly associated with CAR-T-cell activation in vivo and tumour lysis,61–63 because they are often associated with high-plasma cytokine concentrations, but they could also occur as a result of cross reactivity with healthy tissue.64
Engineering to resist immune evasion strategies
Despite the successes of T-cell therapy, many tumours are likely to prove entirely resistant to this approach because of their possession of potent immune evasion strategies. Tumour-targeting T cells are therefore further genetically modified to incorporate countermeasures against tumour immune evasion mechanisms, thereby improving their in-vivo migration, proliferation, and survival (figure 3). For example, T cells targeted to Epstein-Barr virus antigens expressed by almost half of Hodgkin's and non-Hodgkin lymphomas can be further engineered to resist local secretion of transforming growth factor β (TGFβ). This is a widely used tumour immune evasion strategy because TGFβ can promote tumour growth while limiting effector T cell proliferation and function, and activating regulatory T cells. These detrimental effects of TGFβ can be negated by modifying T cells to express a dominant-negative TGFβ receptor type II (dnTGFβ-RII).65 Results of a clinical study66 in progress suggest that dnTGFβ-RII-modified tumour-specific CTL will benefit patients who do not respond to therapy with conventional tumour-targeted T cells.
Figure 3. Genetic modification of T cells.

T cells can be modified to enhance their activity against tumours by: (A) promoting migration—eg, by forcing expression of a transgenic chemokine receptor that is cognate for a chemokine produced by the tumour; (B) increasing their safety by expressing suicide genes that can be activated if needed; (C) increasing resistance to inhibitory signals and ligands in the tumour microenvironment by blocking FasL receptor expression, neutralising the inhibitory signal of TGFβ or converting the inhibitory signal of interleukin 4 into an immunostimulant; and (D) improving their response to growth signals or diminishing their rate of apoptosis.
CCR4=chemokine (C-C motif) receptor 4. FasL=Fas ligand. siRNA=small interfering RNA. TGFβ=transforming growth factor β. Bcl2=B-cell lymphoma 2. BCL2L1=Bcl-2-like protein 1. IL7Rα=interleukin 7 receptor α.
Gene transfer to improve safety
Because the aim of gene transfer is to modify the function of healthy or malignant cells, every approach has the risk of producing unwanted effects including uncontrolled proliferation, tissue damage, or the reported widespread cytokine release syndrome.67,68 Unlike toxicities from small-molecule drugs that might abate as the drug is cleared, toxicities due to gene transfer could persist or worsen if the modified cells remain and multiply in vivo. As cancer gene therapy becomes more potent and more widely used, there will be an ever-increasing need to integrate a safety switch or suicide gene to control any unwanted effects.
Expression of the gene encoding HSV-tk has shown great clinical promise as a safety switch and is now in phase 3 clinical trials as a means of controlling the effects of T-cell therapy after allogeneic stem-cell transplantation. In principle, this approach could be applied to a broad range of cell types and help to enhance the safety of almost any gene therapy. HSV-tk does, however, have potential limitations. Its mechanism of action requires interference with DNA synthesis, thus cell killing could take days or weeks to occur and could be incomplete; this could reduce the value if toxicities are acute or if the target cells are post-mitotic. Moreover, HSV-tk is a virus-derived gene and hence the enzyme is potentially immunogenic and could lead to premature and unintentional elimination of infused cells, thereby compromising the persistence and efficacy of the transferred cells.69–71
An alternative clinically validated suicide system is based on transgenic expression of inducible caspase 9, an engineered protein that can be activated with a synthetic chemical inducer of dimerisation, which is otherwise bioinert. Caspase 9 is of human origin (non-immunogenic) and clinical studies have shown that modified T cells are very sensitive to the drug, with elimination of greater than 95% of transgenic T cells achieved within 24 h, coincident with resolution of clinical signs and symptoms.72,73 Thus, incorporation of the inducible caspase 9 gene with other modifications that enhance T-cell specificity, proliferation, tumour resistance, or in-vivo persistence could be warranted as potency is increased and engineering strategies are combined.
Conclusions
Although only two licensed cancer gene therapy products have been developed during the past 20 years, there is now a confluence of forces that should ensure substantially greater success in the next 5 years. Commercial groups have recognised that advances in our understanding of cancer biology are needed to develop more individualised targeted therapies that have high efficacy in identifiable patient subpopulations, and whose improved pharmacoeconomics enable sufficient reimbursement to justify development and distribution. Increasing clinical successes with gene-modified cell therapies such as CAR-T cells, which are producing 25–50% complete and sustained responses in early-phase clinical trials in advanced malignant diseases, predict wider acceptance of this approach to enhance host resistance to cancer. These advances, coupled with an improved understanding of vaccine technologies (including virotherapies) and the availability of monoclonal antibodies able to block signals that downregulate the immune response, are a potent combination, in which genetically modified cells targeted to tumours are stimulated in vivo by genetically modified tumour vaccines and further enhanced by antibodies that block immune inhibition. As investigators progressively exploit the potential synergies of cancer gene therapy, together with one or more of the safety switches established in clinical practice, we can expect increased success in this discipline. The challenge then will be to ensure that a sufficient cadre of skilled therapists will be available to deliver these novel products as they emerge from clinical trials.
Supplementary Material
Acknowledgments
We are grateful for long-term support from the US National Institutes of Health (National Cancer Institute and National Institute of Health Heart, Lung and Blood Institute), the Department of Defense, and the Leukemia and Lymphoma Research Fund. We are also grateful to the foundations who have supported the centres investigators, including Alex's Lemonade Stand, Clayton Foundation for Research, V Foundation, Dana Foundation, James S McDonnell Foundation, Adrienne Helis Medical Research Foundation, and Bear Necessities Pediatric Cancer Foundation.
Footnotes
Contributors All authors contributed first drafts of components of the Review, and associated figures and tables. MKB assembled the individual components into a complete draft and wrote the introductory, linking, and concluding sections. All authors reviewed the final version of the report.
Conflicts of interest MKB is a scientific advisory board member of Jennerex and of Bluebird Bio. Present research in Center for Cell and Gene Therapy (of which he is director) receives support from CellMedica and the centre has a Research Collaboration with Celgene and Bluebird Bio. JFV is a scientific adviser for Wilson Wolf Manufacturing. All authors have patent applications in the specialty of T cell and gene-modified T-cell therapy for cancer.
References
- 1.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 2.Gottschalk S, Rooney CM, Brenner MK. Cell and gene therapy. In: Pizzo PA, Poplack DG, editors. Principles and Practice of Pediatric Oncology. Lippicott Williams & Wilkins; Philadelphia, PA, USA: 2011. pp. 426–45. [Google Scholar]
- 3.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13:795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–82. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–65. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niculescu-Duvaz I, Springer CJ. Introduction to the background, principles, and state of the art in suicide gene therapy. Mol Biotechnol. 2005;30:71–88. doi: 10.1385/MB:30:1:071. [DOI] [PubMed] [Google Scholar]
- 7.Westphal M, Ylä-Herttuala S, Martin J, et al. Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): a randomised, open-label, phase 3 trial. Lancet Oncol. doi: 10.1016/S1470-2045(13)70274-2. in press. [DOI] [PubMed] [Google Scholar]
- 8.Chiocca EA. Oncolytic viruses. Nat Rev Cancer. 2002;2:938–50. doi: 10.1038/nrc948. [DOI] [PubMed] [Google Scholar]
- 9.Lin E, Nemunaitis J. Oncolytic viral therapies. Cancer Gene Ther. 2004;11:643–64. doi: 10.1038/sj.cgt.7700733. [DOI] [PubMed] [Google Scholar]
- 10.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4:101–17. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 11.Heise C, Sampson-Johannes A, Williams A, McCormick F, Von HoffDD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3:639–45. doi: 10.1038/nm0697-639. [DOI] [PubMed] [Google Scholar]
- 12.O'Shea CC, Johnson L, Bagus B, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611–23. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 13.Patel MR, Kratzke RA. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl Res. 2013;161:355–64. doi: 10.1016/j.trsl.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 14.Khuri FR, Nemunaitis J, Ganly I, et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med. 2000;6:879–85. doi: 10.1038/78638. [DOI] [PubMed] [Google Scholar]
- 15.Yu W, Fang H. Clinical trials with oncolytic adenovirus in China. Curr Cancer Drug Targets. 2007;7:141–48. doi: 10.2174/156800907780058817. [DOI] [PubMed] [Google Scholar]
- 16.Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–36. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tandle A, Blazer DG, 3rd, Libutti SK. Antiangiogenic gene therapy of cancer: recent developments. J Transl Med. 2004;2:22. doi: 10.1186/1479-5876-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ponnazhagan S, Mahendra G, Kumar S, et al. Adeno-associated virus 2-mediated antiangiogenic cancer gene therapy: long-term efficacy of a vector encoding angiostatin and endostatin over vectors encoding a single factor. Cancer Res. 2004;64:1781–87. doi: 10.1158/0008-5472.can-03-1786. [DOI] [PubMed] [Google Scholar]
- 19.Lin X, Huang H, Li S, et al. A phase I clinical trial of an adenovirus-mediated endostatin gene (E10A) in patients with solid tumors. Cancer Biol Ther. 2007;6:648–53. doi: 10.4161/cbt.6.5.4004. [DOI] [PubMed] [Google Scholar]
- 20.Hanania EG, Giles RE, Kavanagh J, et al. Results of MDR-1 vector modification trial indicate that granulocyte/macrophage colony-forming unit cells do not contribute to posttransplant hematopoietic recovery following intensive systemic therapy. Proc Natl Acad Sci USA. 1996;93:15346–51. doi: 10.1073/pnas.93.26.15346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hesdorffer C, Ayello J, Ward M, et al. Phase I trial of retroviral-mediated transfer of the human MDR1 gene as marrow chemoprotection in patients undergoing high-dose chemotherapy and autologous stem-cell transplantation. J Clin Oncol. 1998;16:165–72. doi: 10.1200/JCO.1998.16.1.165. [DOI] [PubMed] [Google Scholar]
- 22.Adair JE, Beard BC, Trobridge GD, et al. Extended survival of glioblastoma patients after chemoprotective HSC gene therapy. Sci Transl Med. 2012;4:133ra57. doi: 10.1126/scitranslmed.3003425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berzofsky JA, Terabe M, Oh S, et al. Progress on new vaccine strategies for the immunotherapy and prevention of cancer. J Clin Invest. 2004;113:1515–25. doi: 10.1172/JCI21926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ridgway D. The first 1000 dendritic cell vaccinees. Cancer Invest. 2003;21:873–86. doi: 10.1081/cnv-120025091. [DOI] [PubMed] [Google Scholar]
- 25.Nencioni A, Grünebach F, Schmidt SM, et al. The use of dendritic cells in cancer immunotherapy. Crit Rev Oncol Hematol. 2008;65:191–99. doi: 10.1016/j.critrevonc.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Goldman B, DeFrancesco L. The cancer vaccine roller coaster. Nat Biotechnol. 2009;27:129–39. doi: 10.1038/nbt0209-129. [DOI] [PubMed] [Google Scholar]
- 27.Cheever MA, Allison JP, Ferris AS, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–37. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stevenson FK, Mander A, Chudley L, Ottensmeier CH. DNA fusion vaccines enter the clinic. Cancer Immunol Immunother. 2011;60:1147–51. doi: 10.1007/s00262-011-1042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu MA. DNA vaccines: an historical perspective and view to the future. Immunol Rev. 2011;239:62–84. doi: 10.1111/j.1600-065X.2010.00980.x. [DOI] [PubMed] [Google Scholar]
- 30.Timmerman JM, Singh G, Hermanson G, et al. Immunogenicity of a plasmid DNA vaccine encoding chimeric idiotype in patients with B-cell lymphoma. Cancer Res. 2002;62:5845–52. [PubMed] [Google Scholar]
- 31.Tagawa ST, Lee P, Snively J, et al. Phase I study of intranodal delivery of a plasmid DNA vaccine for patients with stage IV melanoma. Cancer. 2003;98:144–54. doi: 10.1002/cncr.11462. [DOI] [PubMed] [Google Scholar]
- 32.Conry RM, Curiel DT, Strong TV, et al. Safety and immunogenicity of a DNA vaccine encoding carcinoembryonic antigen and hepatitis B surface antigen in colorectal carcinoma patients. Clin Cancer Res. 2002;8:2782–87. [PubMed] [Google Scholar]
- 33.Boczkowski D, Nair SK, Synder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med. 1996;184:465–72. doi: 10.1084/jem.184.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Radford KJ, Jackson AM, Wang JH, Vassaux G, Lemoine NR. Recombinant E coli efficiently delivers antigen and maturation signals to human dendritic cells: presentation of MART1 to CD8+ T cells. Int J Cancer. 2003;105:811–19. doi: 10.1002/ijc.11149. [DOI] [PubMed] [Google Scholar]
- 35.Mackensen A, Herbst B, Chen JL, et al. Phase I study in melanoma patients of a vaccine with peptide-pulsed dendritic cells generated in vitro from CD34(+) hematopoietic progenitor cells. Int J Cancer. 2000;86:385–92. doi: 10.1002/(sici)1097-0215(20000501)86:3<385::aid-ijc13>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 36.Gottschalk S, Edwards OL, Sili U, et al. Generating CTL against the subdominant Epstein-Barr virus LMP1 antigen for the adoptive immunotherapy of EBV-associated malignancies. Blood. 2003;101:1905–12. doi: 10.1182/blood-2002-05-1514. [DOI] [PubMed] [Google Scholar]
- 37.Wei Y, Sticca RP, Holmes LM, et al. Dendritoma vaccination combined with low dose interleukin-2 in metastatic melanoma patients induced immunological and clinical responses. Int J Oncol. 2006;28:585–93. [PubMed] [Google Scholar]
- 38.Jinushi M, Hodi FS, Dranoff G. Enhancing the clinical activity of granulocyte-macrophage colony-stimulating factor-secreting tumor cell vaccines. Immunol Rev. 2008;222:287–98. doi: 10.1111/j.1600-065X.2008.00618.x. [DOI] [PubMed] [Google Scholar]
- 39.Biagi E, Rousseau R, Yvon E, et al. Responses to human CD40 ligand/human interleukin-2 autologous cell vaccine in patients with B-cell chronic lymphocytic leukemia. Clin Cancer Res. 2005;11:6916–23. doi: 10.1158/1078-0432.CCR-05-0484. [DOI] [PubMed] [Google Scholar]
- 40.Loskog A, Dzojic H, Vikman S, et al. Adenovirus CD40 ligand gene therapy counteracts immune escape mechanisms in the tumor microenvironment. J Immunol. 2004;172:7200–05. doi: 10.4049/jimmunol.172.11.7200. [DOI] [PubMed] [Google Scholar]
- 41.Dilloo D, Bacon K, Holden W, et al. Combined chemokine and cytokine gene transfer enhances antitumor immunity. Nat Med. 1996;2:1090–95. doi: 10.1038/nm1096-1090. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Ehtesham M, Samoto K, et al. In situ adenoviral interleukin 12 gene transfer confers potent and long-lasting cytotoxic immunity in glioma. Cancer Gene Ther. 2002;9:9–15. doi: 10.1038/sj.cgt.7700399. [DOI] [PubMed] [Google Scholar]
- 43.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li LP, Lampert JC, Chen X, et al. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat Med. 2010;16:1029–34. doi: 10.1038/nm.2197. [DOI] [PubMed] [Google Scholar]
- 45.Leisegang M, Wilde S, Spranger S, et al. MHC-restricted fratricide of human lymphocytes expressing survivin-specific transgenic T cell receptors. J Clin Invest. 2010;120:3869–77. doi: 10.1172/JCI43437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stauss HJ, Cesco-Gaspere M, Thomas S, et al. Monoclonal T-cell receptors: new reagents for cancer therapy. Mol Ther. 2007;15:1744–50. doi: 10.1038/sj.mt.6300216. [DOI] [PubMed] [Google Scholar]
- 47.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–29. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–26. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–51. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maus MV, Linette GP, Stadtmauer EA, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced TCR-engineered T cells against HLA-A1 restricted MAGE-A3 antigen. Mol Ther. 2013;21:S24. doi: 10.1182/blood-2013-03-490565. abstr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–20. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brentjens RJ, Rivière I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–26. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T Cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–56. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heslop HE. Safer CARS. Mol Ther. 2010;18:661–62. doi: 10.1038/mt.2010.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Büning H, Uckert W, Cichutek K, Hawkins RE, Abken H. Do CARs need a driver's license? Adoptive cell therapy with chimeric antigen receptor-redirected T cells has caused serious adverse events. Hum Gene Ther. 2010;21:1039–42. doi: 10.1089/hum.2010.131. [DOI] [PubMed] [Google Scholar]
- 63.Peinert S, Kershaw MH, Prince HM. Chimeric T cells for adoptive immunotherapy of cancer: using what have we learned to plan for the future. Immunotherapy. 2009;1:905–12. doi: 10.2217/imt.09.69. [DOI] [PubMed] [Google Scholar]
- 64.Lamers CH, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 65.Bollard CM, Rössig C, Calonge MJ, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179–87. doi: 10.1182/blood.v99.9.3179. [DOI] [PubMed] [Google Scholar]
- 66.Bollard CM, Dotti G, Gottschalk S, et al. Administration of TGF-beta resistant tumor-specific CTL to patienst with EBV-associated HL and NHL. Mol Ther. 2012;20:S22. [Google Scholar]
- 67.Hacein-Bey-Abina S, von Kalle C, Schmidt M, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–56. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 68.Cavazzana-Calvo M, Fischer A, Hacein-Bey-Abina S, Aiuti A. Gene therapy for primary immunodeficiencies: part 1. Curr Opin Immunol. 2012;24:580–84. doi: 10.1016/j.coi.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 69.Ciceri F, Bonini C, Gallo-Stampino C, Bordignon C. Modulation of GvHD by suicide-gene transduced donor T lymphocytes: clinical applications in mismatched transplantation. Cytotherapy. 2005;7:144–49. doi: 10.1080/14653240510018136. [DOI] [PubMed] [Google Scholar]
- 70.Ciceri F, Bonini C, Marktel S, et al. Antitumor effects of HSV-TK-engineered donor lymphocytes after allogeneic stem-cell transplantation. Blood. 2007;109:4698–707. doi: 10.1182/blood-2006-05-023416. [DOI] [PubMed] [Google Scholar]
- 71.Traversari C, Marktel S, Magnani Z, et al. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109:4708–15. doi: 10.1182/blood-2006-04-015230. [DOI] [PubMed] [Google Scholar]
- 72.Straathof KC, Pulè MA, Yotnda P, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–54. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.