

Abstract
A synonymous variant in the first exon of CYP2A6, rs1137115 (51G > A), defines the common reference allele CYP2A6*1A, and is associated with lower mRNA expression and slower in-vivo nicotine metabolism. Another common allele, CYP2A6*14, differs from CYP2A6*1A by a single variant, rs28399435 (86G > A, S29N). However, CYP2A6*14 shows in-vivo activity comparable with that of full-function alleles, and significantly higher than CYP2A6*1A. rs1137115A is predicted to create an exonic splicing suppressor site overlapping an exonic splicing enhancer (ESE) site in the first exon of CYP2A6, whereas rs28399435A is predicted to strengthen another adjacent ESE, potentially compensating for rs1137115A. Using an allelic expression assay to assess cDNAs produced from rs1137115 heterozygous liver biopsy samples, lower expression of the CYP2A6*1A allele is confirmed while CYP2A6*14 expression is found to be indistinguishable from that of rs1137115G alleles. Quantitative PCR assays to determine the relative abundance of spliced and unspliced or partially spliced CYP2A6 mRNAs in liver biopsy samples show that *1A/*1A homozygotes have a significantly lower ratio, due to both a reduction in spliced forms and an increase in unspliced or partially spliced CYP2A6. These results show the importance of common genetic variants that effect exonic splicing suppressor and ESEs to explain human variation regarding clinically-relevant phenotypes.
Keywords: CYP2A6, exonic splice enhancer, exonic splice suppressor, nicotine metabolism, splicing
Introduction
The cytochrome P450 2A6 (CYP2A6) enzyme, responsible for the majority of nicotine metabolism in most smokers (reviewed in Mwenifumbo and Tyndale [1]), is encoded by a highly heterogeneous gene including at least five relatively common reduced-function alleles among Europeans. In a previous study we determined that ~70% of the variance in oral nicotine metabolism can be explained by CYP2A6 diplotype. These experiments also allowed us to estimate the relative activities of different CYP2A6 alleles, and show that a frequent haplotype, CYP2A6*1A (~15% in European Americans), is associated with significantly slower nicotine metabolism compared with other common reference alleles (CYP2A6*1B/*1D) [2]. The *1A haplotype was previously associated with lower CYP2A6 mRNA and protein levels, and coumarin 7-hydroxylation activity in liver samples [3,4]; however, the mechanism of this difference was not obvious: the defining genetic variant, rs1137115A (51A), is a synonymous single nucleotide polymorphism (SNP) in the first exon in high linkage disequilibrium (D’ = 1) with many other SNPs throughout the locus [3]. An interesting footnote to our results was the apparently normal activity of a less common allele, CYP2A6*14 (4%), that differs from *1A by only one nucleotide, rs28399435 (86A, S29N), a nonsynonymous SNP 35 base pairs from rs1137115. A previous study of nicotine metabolism that also found *14 activity to be equivalent to that of *1 alleles did not take the rs1137115 genotype into account [5]. Thus, the proximity of rs28399435 and its apparently compensatory effect on CYP2A6 activity highlight the possible functional impact of rs1137115 itself, and point toward hypotheses about its mechanism.
Here we provide evidence that rs1137115 and rs28399435 influence CYP2A6 expression and nicotine metabolism by altering CYP2A6 mRNA splicing efficiency. Splicing of mRNA is a key regulatory point along the pathway to gene expression; the inclusion or exclusion of exons resulting in different protein isoforms allows organisms to increase the diversity of products derived from a limited number of genes (reviewed in Keren et al. [6]). The spliceosome, a complex of proteins and small RNAs assembled in the nucleus, recognizes conserved motifs in the exons of unspliced precursor mRNAs, including 3’ donor and 5’ acceptor sites, exonic splicing enhancer (ESE) and suppressor (ESS) motifs. Alternatively, spliced exons typically have weaker (nonconsensus) donor and acceptor sites that provide greater flexibility for regulation. As such, genetic variants that weaken donor and acceptor sites or change the balance of ESE and ESS motifs can lead to ‘exon skipping’. Rare variants that disrupt splicing or alter the inclusion of both constitutively spliced and alternatively spliced exons have been associated with disease [7–15], but common alleles that alter splicing efficiency provide an important portion of the genetically determined variance in clinically-relevant traits including drug metabolism [16]. The importance of maintaining the balance of ESE and ESS motifs is showed by the relatively low frequency of human SNPs that disrupt ESEs, especially near exon extremities [17,18]; and the identification of pairs of fixed differences in closely related species strongly suggests that ‘splicing positive’ mutations that compensate for ‘splicing negative’ events are positively selected during evolution [19]. In this study we reveal such a compensatory event among the extant haplotypes of CYP2A6 with a direct bearing on variation in nicotine metabolism and smoking behavior.
Materials and methods
This study complies with the Code of Ethics of the World Medical Association and obtained informed consent from participants and approval from the appropriate Washington University institutional review boards. Statistical analyses were performed using the software package ‘R’ (R Foundation for Statistical Computing, Vienna, Austria) and all t-tests performed were two sided.
Genotyping and haplotype determination
CYP2A6 nomenclature follows official recommendations (http://www.cypalleles.ki.se) except that CYP2A6*1A is always rs1137115A. Additional genotyping was performed using the KBioscience Competitive Allele Specific PCR genotyping system (KASPar; KBioscience, Hoddesdon, UK) following standard procedures with custom designed primers as previously described [2,20]. KASPar assays were set up as 8 μl reactions and measured with the 7900HT Fast Real-Time PCR System (Applied Biosystems, Carlsbad, California, USA).
Allelic expression study
DNA and RNA extracted from deidentified noncancerous liver biopsy samples were supplied by the Tissue Procurement Core, Laboratory for Translational Pathology at the Siteman Cancer Center, Washington University Medical Center. Frozen normal liver tissue blocks were cut into 25 μm sections using cryostat (Leica CM 1850; Leica Microsystems, Wetzlar, Germany), followed by DNA isolation [QIAamp DNA Mini Kit (250), Qiagen Cat# 51306; Qiagen, Valencia, California, USA], treated with RNase A (Qiagen Cat# 158922), quantitative analysis by NanoDrop (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and agarose gel electrophoresis; RNA was isolated by standard trizol reagent protocol, cleaned with Lithium Chloride Precipitation Solution (AM9480) (Invitrogen, Grand Island, New York, USA), followed by quantitative analysis using NanoDrop and Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, California, USA). cDNA was prepared from total RNA, treated with DNAse, using the Applied Biosystem High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). cDNA and genomic DNA were arrayed together in the same 384-well plate in triplicate, and were run on an ABI-7900 Real-Time PCR System (Applied Biosystems) under standard conditions. A custom assay was designed to recognize rs1137115 with the following primer and probe sequences, forward primer: CATCCCACTACCACCATGCT; reverse primer: TCTTCCTCTGCTGCCAAACAG; reporter 1: CCTGA CTGTAATGGTCT; reporter 2: CTGACTGTGATGGTCT.
The relative expression of both alleles for each expression marker was determined by subtracting the smaller cycle threshold (Ct, the number of cycles at which PCR products generated exceed a defined threshold) value of one allele PCR reaction from the larger Ct value of the other allele PCR reaction (ΔCt). For the statistical analysis, ΔCt values were obtained as an average of two or three reactions for each sample and data point. ΔCt values were also obtained from heterozygous genomic DNAs from the same biopsy samples and cDNA allelic ratios were normalized against the overall average ratio obtained for gDNA.
Quantitative real-time expression study
Single PCR products of the correct size were confirmed for all primer pairs by agarose gel electrophoresis (Supplemental Fig. 1, http://links.lww.com/FPC/A544). Reactions for pairs of assays to be compared in each experiment were arrayed together in the same 384-well plate in duplicate pairs, and run on an ABI-7900 real-time PCR system under standard conditions. A measure of 10 μl reactions included 2 × PerfeCTa SYBR Green FastMix ROX (Quant Biosciences Inc., Gaithersburg, Maryland, USA), 0.5 μmol/l each forward and reverse primer, and 1 μl cDNA. Dissociation curves for all primer pairs demonstrated single peaks consistent with single PCR products without contamination from primer dimers (Supplemental Fig. 2, http://links.lww.com/FPC/A545). Ct values were obtained as the average of two reactions for each sample and assay. The difference in relative quantity detected by each assay was determined by subtracting the smaller average Ct value of one reaction from the larger average Ct value of the other reaction (ΔCt).
Splicing construct cell transfection experiment
GoTaq Polymerase (Promega, Madison, Wisconsin, USA) and two primers incorporating HindIII and XhoI sites, respectively (5’-ATATaagcttGCCGTCACCATCTATCAT CC-3’ and 5’-ATATctcgagTTTGAAGACCCAGTCGAA GG-3‘) were used to amplify the majority of exons 1, 2 and the intervening first intron, using genomic DNA from multiple participants previously genotyped at SNPs rs1137115 and rs28399435. Fragments were cloned into the HindIII and XhoI sites of vector pcDNA3.1 (generously provided by Dr Virginia Lee). Clones were directly Sanger sequenced to verify the complete sequence of all cloned fragments and to select constructs corresponding to the three haplotypes.
Human embryonic kidney (HEK293-T) cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, 1% l-glutamine, and penicillin/ streptomycin. For transient transfection, HEK293-T cells were cultured in six-well lysine-coated plates. Upon reaching 90% confluence, cells were transfected with Lipofectamine 2000 (Life Technologies, Grand Island, New York, USA) and harvested after 24 h. cDNA was prepared from total RNA using the Applied Biosystem High Capacity cDNA Reverse Transcription Kit. cDNA from multiple cultures transfected with each construct were compared by PCR using the cloning primers to detect splicing.
Results
CYP2A6 variants rs1137115 and rs28399435 are associated with allelic expression in liver
rs1137115 was previously associated with reduced CYP2A6 protein level [3] and nicotine metabolism [2], but CYP2A6*1A does not differ from common fully-functional alleles (*1B/*1D) at the amino acid level [3]. Therefore, we hypothesized that the lower activity of the *1A allele, and possible compensation by rs28399435 resulting in normal *14 allele activity, act through effects upon CYP2A6 expression. To test this we acquired matching pairs of cDNA and genomic DNAs from 99 European American liver biopsy samples and genotyped them for CYP2A6 variants relevant to expression (Table 1). Forty samples were determined to be heterozygous at rs1137115, and among these nine were also heterozygous at rs28399435. Because primers associated with the commonly available rs1137115 Taqman assay overlap rs28399435, we ordered a custom rs1137115 assay from Applied Biosystems designed to avoid the other variant. Among the 40 rs1137115 heterozygotes, the assay failed to detect sufficient signal from either allele to measure an allelic differences in six samples, one of which was also an rs28399435 heterozygote; these samples were excluded from the analysis.
Table 1.
CYP2A6 haplotype definitions and frequencies
| Haplotype names |
rs61663607; -745 |
rs28399433, -48 (TATA box) |
rs1137115; 51 (V17V) |
rs28399435; 86 (S29N) |
rs1801272; 1799 (L160H) |
3' gene conversion |
Alleles | Frequency (%) |
|---|---|---|---|---|---|---|---|---|
| *1A | A | T | A | G | T | - | 39 | 19.7 |
| *1B | A | T | G | G | T | + | 69 | 34.8 |
| *1B12 | A | T | A | G | T | + | 1 | 0.5 |
| *1H | G | T | G | G | T | - | 15 | 7.6 |
| *2 | A | T | A | G | A | - | 2 | 1.0 |
| *9 | A | G | G | G | T | - | 6 | 3.0 |
| *14 | A | T | A | A | T | - | 12 | 6.1 |
| All others | A | T | G | G | T | - | 54 | 27.3 |
Polymorphic sites analyzed are given at the top of each column by rs number, gene position and further relevant description. Haplotype name indicates the common allele name. ‘ + ’ indicates the presence of the 5' UTR gene conversion.
Significantly different relative allelic expression was observed in rs28399435GG homozygotes compared with rs28399435AG (*14) heterozygotes (P = 7.4 10– 5). These results are consistent with lower expression of rs1137115A-rs28399435G haplotypes compared with other haplotypes, but similar expression of rs1137115A–rs28399435A (*14) and other haplotypes. The two lowest values among rs1137115AG–rs28399435GG heterozygotes were also the two heterozygotes for rs28399433 and rs61663607, defining the CYP2A6*9 and *1H alleles, respectively, both of which are associated with lower expression [21,22]. These single data points are consistent with much lower relative expression of *9 and similar expression of *1H and the rs1137115A haplotype (Fig. 1). The single *1H/*14 heterozygote is consistent with lower expression of *1H relative to *14 (Fig. 1). Excluding these three samples, the difference between the relative expression of rs1137115AG– rs28399435GG and rs28399435AG (*14) is also significant (Fig. 1, P = 1.7 × 10– 4).
Fig. 1.
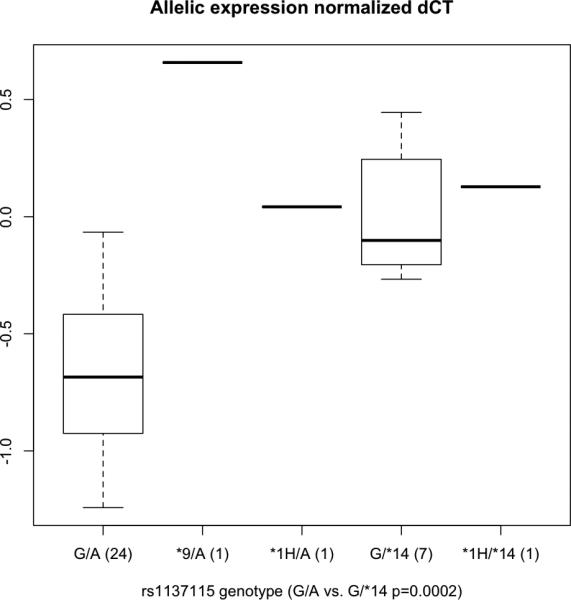
CYP2A6 relative allelic expression in rs1137115 heterozygous liver cDNAs. Data points are the difference in Ct (the number of cycles at which products generated exceed a defined threshold) between PCR reactions for the G minus the A allele for five diplotypes (larger Ct corresponds to lower expression) normalized against the average ratio obtained for all rs1137115 heterozygous gDNAs. 0.0 equals no difference. The boxplot summarizes the data distribution of (n) samples. The box represents the interquartile range, divided by a line indicating the median; whisker lines extend to the maximum and minimum values.
rs1801272A (CYP2A6*2) is known to disrupt enzyme function but not expression. The single rs1801272 (*2) heterozygote had a relative allelic expression less than 1 SD from the mean of rs1137115AG–rs28399435GG participants and was not excluded from the analysis. The CYP2A6*1B allele is defined by a 58 base-pair 3‘ untranslated region conversion associated with in-vitro mRNA perdurance [23]. No difference was seen between CYP2A6*1B heterozygotes and heterozygotes for other haplotypes (most likely CYP2A6*1D, see Haberl et al. [3]), whether compared with rs1137115A (*1A and *2) or *14, consistent with our previous result [2] demonstrating no difference in activity between CYP2A6*1B and *1D alleles.
Variants associated with allelic expression are predicted to alter exon splicing
Because of the close proximity of rs1137115 and rs28399435 in the same exon, we hypothesized that the variants might exert their effects upon CYP2A6 mRNA quantity through differences in splicing efficiency and nonsense-mediated decay of unprocessed message (reviewed in Chang et al. [24]). Web-based tools are available that recognize predicted ESE and ESS motifs in nucleotide sequence (RESCUE-ESE [25] at http://genes.mit.edu/burgelab/rescue-ese/ and FAS-ESS [26] at http://genes.mit.edu/fas-ess/) developed from hybrid computational/experimental methods to identify short sequences differentially represented in exons, especially exons with weak (nonconsensus) splice sites, and introns.
Consistent with the associated apparent reduction in CYP2A6 mRNA expression, the rs1137115A allele contains a predicted ESS not present in the rs1137115G allele that also overlaps a predicted ESE. In contrast, rs28399435A introduces new predicted ESE motifs overlapping a predicted ESS motif (Fig. 2). Splicing enhancer and suppressor motifs have a larger effect on exons with weak; that is, nonconsensus 5‘ and 3‘ splice sites, such as alternatively spliced exons (reviewed in Keren et al. [6]), and there is evidence that changes in ESE/ESS motifs can compensate for weak splice sites [19]. The first exon of CYP2A6, containing rs1137115 and rs28399435, has a donor splice site predicted with a confidence of 0.24, below the threshold (0.50) of nearly all true donor sites, making it a likely candidate to be influenced by changes in the number of ESE and ESS motifs [27].
Fig. 2.

Predicted exonic splicing suppressor (ESS) and exonic splicing enhancer (ESE) sites altered by rs1137115 and rs28399435. Single nucleotide polymorphism minor alleles are indicated in white on black. Predicted ESEs are shown above, and ESSs below the sequence of each haplotype.
CYP2A6*1A is associated with lower levels of spliced and higher levels of unspliced or partially spliced CYP2A6 mRNA in liver
On the basis of predictions of exon splicing motifs associated with rs1137115 and rs28399435, we sought evidence of differences in exon splicing between different haplotypes of CYP2A6. Effects of alterations in ESS and ESE motifs upon splicing have previously been studied with respect to internal exons wherein decreased splicing efficiency results in exon skipping. Because the motifs altered by the SNPs in question are located in the first exon, we were uncertain what kinds of alterations might result; therefore, we first attempted to detect differences in the relative abundance of spliced versus unspliced mRNAs associated with different CYP2A6 diplotypes, focusing on the first intron. Two pairs of primers were designed to specifically recognize either spliced (lacking the first intron) or unspliced (including the first intron) mRNAs, using different forward primers and a reverse primer in the second exon (Fig. 3). These sets were then compared in a quantitative real-time PCR experiment including 94 cDNAs from European American liver biopsy samples of various CYP2A6 diplotypes.
Fig. 3.
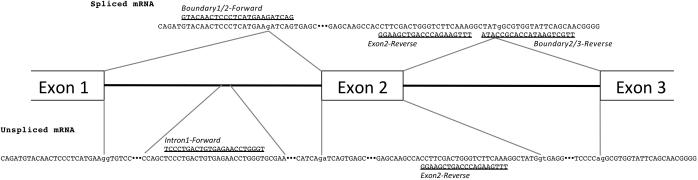
Diagram of PCR primers targeting CYP2A6 cDNA and gDNA. Introns 1 and 2 are indicated by horizontal lines connecting boxes representing exons 1–3, not to scale. Sequence of the spliced cDNA is above the exon–intron diagram, and gDNA or unspliced cDNA sequence is below. Forward primers are above sequences and reverse primers (represented by the reverse-complement of the actual primer sequence for clarity) are below.
The difference in cycles threshold (ΔCt), the number of cycles at which products generated from PCR reactions exceed a defined threshold, shows the relative ratio of the targets of the reactions – in this case cDNA produced from spliced or unspliced CYP2A6 mRNAs. Consistent with decreased splicing efficiency, the ΔCt (spliced – unspliced) is significantly lower among *1A (rs1137115A/rs28399435G) homozygotes compared with homozygotes for other common haplotypes (rs1137115G/ rs28399435G) (3.7 vs. 6.2, P = 8.7 × 10– 5, Fig. 4), equivalent to a 5.7-fold difference. *1A homozygotes also demonstrated significantly lower ΔCt than either *1A or *14 (rs1137115A/rs28399435A) heterozygotes (6.2 or 6.0, P = 5.7 × 10– 5 or 0.008, respectively). There is insufficient power to demonstrate a significant difference between *1A/*1A and *1A/*14 participants (3.7 vs. 6.2, p = 0.3) or between any other pair of diplotypes (Fig. 4). These differences are due to both lower amounts of the spliced form and significantly higher amounts of the unspliced forms (*1A/*1A vs. rs1137115GG, Ct = 20.8 vs. 22.5, P = 0.002, lower Ct is consistent with higher quantity).
Fig. 4.
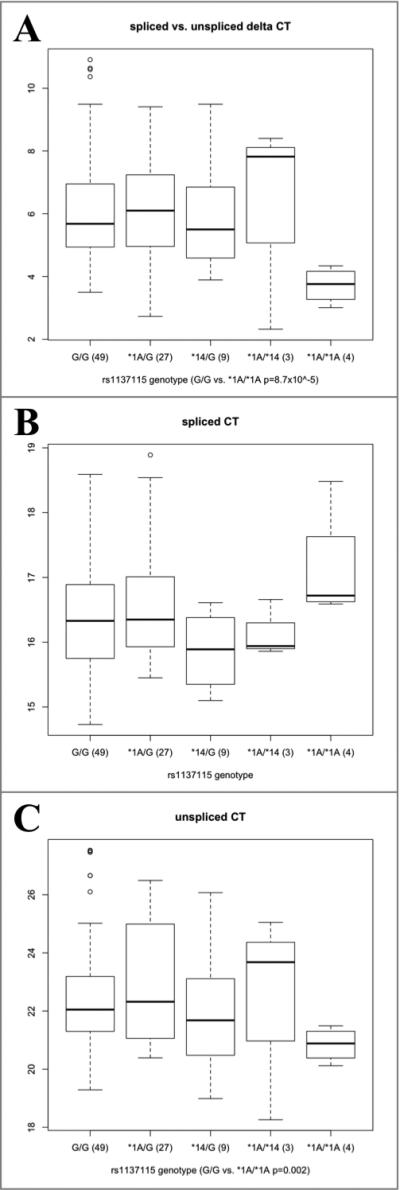
(a) Relative expression of spliced and unspliced CYP2A6 in liver cDNAs. Data points are the difference in Ct (the number of cycles at which products generated exceed a defined threshold) between PCR reactions for the unspliced minus the spliced form (larger Ct corresponds to lower expression). (b) Ct for total expression of spliced CYP2A6, (c) Ct for total expression of unspliced CYP2A6. Primers intron 1-forward and exon 2-reverse are used to detect the unspliced form; boundary 1/2-forward and exon 2-reverse are used to detect the spliced form. The boxplot summarizes the data distribution of (n) samples. The box represents the interquartile range, divided by a line indicating the median; whisker lines extend to the maximum and minimum values within ×1.5 the interquartile range and further outliers are marked with circles.
That both spliced and unspliced Ct differ by diplotype is evidence that the unspliced product is not an artifact of genomic DNA contamination. To further confirm this, we repeated the quantitative PCR experiment with the same two forward primers and a reverse primer overlapping the boundary of exons 2 and 3 that would only recognize spliced or partially spliced forms (Fig. 3). Initial PCR tests indicated the presence of the partially spliced form in all cDNA samples tested, regardless of genotype. gDNA template did not produce a PCR product with these primers (Supplemental Fig. 1, http://links.lww.com/FPC/A544). Consistent with the previous result, the ΔCt (spliced – partially spliced) was significantly lower among *1A homozygotes than among rs1137115G homozygotes or *1A heterozygotes (P = 0.03 and 0.01, respectively, Fig. 5). These differences were also because of both lower quantities of spliced forms and higher quantities of partially spliced forms, although neither difference reached statistical significance separately (data not shown). The difference in ΔCt values between this and the previous experiment also reflects the rarity of the partially spliced form compared with all unspliced forms regardless of genotype (Figs 4 and 5).
Fig. 5.
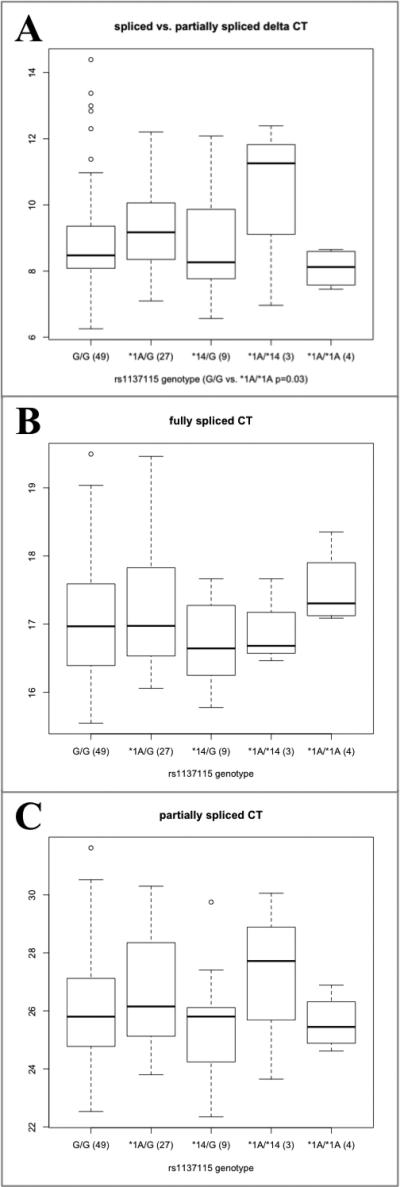
(a) Relative expression of fully spliced and partially spliced CYP2A6 in liver cDNAs. Data points are the difference in Ct between PCR reactions for the partially spliced minus the spliced form (larger Ct corresponds to lower expression). (b) Ct for total expression of fully spliced CYP2A6, (c) Ct for total expression of partially spliced CYP2A6. Primers intron 1-forward and boundary 2/3-reverse are used to detect the partially spliced form; boundary 1/2-forward and boundary 2/3-reverse are used to detect the fully spliced form. The boxplot summarizes the data distribution of (n) samples. The box represents the interquartile range, divided by a line indicating the median; whisker lines extend to the maximum and minimum values within × 1.5 the interquartile range and further outliers are marked with circles.
In an attempt to replicate the liver biopsy sample cDNA results in vitro, we also prepared three DNA constructs containing the first two exons and the entire intervening first intron of CYP2A6 representing haplotypes rs1137115– rs28399435 GG, AG and AA. These were transfected into human embryonic kidney 293 cells, mRNA from cultures were converted to cDNA, and these were tested for the presence of spliced and unspliced CYP2A6 message using primers designed to flank the first intron. Only two size fragments were detected by agarose gel electrophoresis, corresponding to the spliced and unspliced forms. The splicing efficiency of all three constructs appeared much lower than that of the native, presumably full-length mRNAs extracted from liver biopsy samples; that is, the large majority of PCR products from the transfected cell culture cDNAs were unspliced, whereas PCR products from liver biopsy cDNAs were overwhelmingly the spliced form (Supplemental Fig. 3, http://links.lww.com/FPC/A546). Real-time expression assays were carried out as above using cDNA from multiple cultures transfected with each construct, but a consistent difference in splice form ratio was not observed between different genotype constructs (data not shown).
Discussion
The association between nicotine metabolism and CYP2A6 exemplifies the difficulty in dissecting associations between variable human traits and complex heterogeneous genetic loci. The largest contributor to nicotine metabolism, CYP2A6 is highly polymorphic, including multiple common null-activity and intermediate-activity alleles in all studied populations, as well as less-frequent alleles with uncertain activities. SNPs near CYP2A6 are among the few loci identified as associated with consumption of cigarettes/ day with genome-wide significance in unbiased studies of Europeans [28,29]. We have since determined that these SNPs are proxies for several functionally important CYP2A6 haplotypes [20]; for example, the minor allele of rs4105144, the intergenic SNP identified by Thorgeirsson et al. [28] is in strong linkage disequilibrium (D‘ = 1) with most loss-of-function CYP2A6 alleles common in Europeans: *1A, *1H, *2, *4, and *12, along with CYP2A6*14. Such ‘synthetic’ associations, resulting from the coincidental linkage of common markers with multiple less-frequent causal variants, are proposed as a source of unexplained genome-wide association study findings [30]. However, a key corollary to this phenomenon is its reverse: causal variants will mask the phenotypic impacts of other variants occurring in the same gene hindering the detection of real associations by single SNP analyses [31]. Cis-interactions between variants that can occur together on the same chromosome, such as rs1137115 and rs28399435, are a particular problem; although the minor allele of rs1137115 certainly contributed to the synthetic association reported by Thorgeirsson and colleagues, its complete linkage disequilibrium with rs28399435 (the minor allele of rs28399435 only occurs on one allele of rs1137115, D’ = 1) also reduced the apparent association. In another population with different allele frequencies, or another complex locus with a different linkage disequilibrium structure between causative variants, such a serendipitous marker SNP might not be found. This is also a caution against over-interpreting negative results in unbiased genome-wide association studies.
The association between CYP2A6 genotype and in-vivo nicotine metabolism has been typically investigated by dividing experimental participants by CYP2A6 diplotype. However, this method lacks power to compare infrequently occurring haplotypes with modest effect sizes in limited samples. Recently, in developing a predictive genetic model of CYP2A6 activity, we used linear regression to analyze a metric of nicotine metabolism in terms of CYP2A6 haplotype [2]. This approach allowed us to determine that the activity of the *1A allele (~15% frequency) is both significantly less than that of other common reference haplotypes, and significantly greater than that of known null alleles, *2 and *4. It also allowed us to demonstrate that the *14 allele (~4% frequency) has significantly more activity than *1A, and is indistinguishable from other reference alleles (*1B and *1D). This difference between two haplotypes, *1A and *14, that differ by a single SNP [3], thus provided a hint at the functional effects of the variants leading to the distinct activity of each allele.
In addition to the CYP2A6/CYP2A7 hybrid alleles called CYP2A6*12 (frequency <3%), CYP2A6 haplotypes in Europeans are divided into three clades: *1A/*2/*14 (~29%), *1B/*1D/*9 (~58%), and *1H (~8%). Thorough sequencing has identified nine common variants (> 8% minor allele frequency), all in high linkage disequilibrium (D’) with each other within ~3 kb 5’ of CYP2A6, that define these clades [3]. In-vitro luciferase experiments using more than 1 kb fragments of the CYP2A6 5’ promoter region have demonstrated significantly lower expression of both the *1B/*1D and *1H promoters relative to *1A [4,22]; nevertheless, as confirmed here, *1A is associated with significantly lower in-vivo mRNA levels [3,4]. Beyond six variants in the 5’ promoter, *1A and *1B/*1D differ only by two base pairs, rs1137115, and a common SNP in the second intron (rs8192725). However, given the limitations of in-vitro expression assays, prior reports cannot exclude that the differences in allelic expression we see between *1A and *1B/*1D are due to variants in the promoter. Therefore, regarding this question, expression of the *14 allele is informative: complete sequencing of the locus in multiple studies including more than a dozen *14 haplotypes find that this allele differs from *1A by only a single base pair, rs28399435 [3,5]; that is, whatever variant or combination of variants results in lower levels of in-vivo CYP2A6*1A mRNA, the effect is reversed by a single SNP in the first exon located 35 base pairs away from rs1137115.
Prior in-vitro investigations of variation in ESE and ESS sites and splicing have focused on internal exons and the detection of aberrant exon skipping or exon inclusion; that is, production of novel splice-forms with greater or lesser numbers of exons, a relatively black and white result [11–15,32]. However, in the case of rs1137115 and rs28399435, which occur in the first exon of CYP2A6, the difference we detect in vivo is more subtle, a change in the efficiency of normal splicing. Such a difference is likely to be more difficult to robustly replicate in vitro, and given that our transfection constructs did not splice efficiently regardless of genotype (Supplemental Fig. 3, http://links.lww.com/FPC/A546), it was not surprising that we failed to detect a difference. To our knowledge, this is the first report of variation in ESE or ESS, in the first exon of a gene, associated with altered splicing. But the detection of differences in splicing efficiency in vivo is also not without its difficulties. Although we detected a significant difference between the ratios of spliced and unspliced CYP2A6 mRNAs between G/G and *1A/*1A homozygotes in vivo, levels of mRNA for *1A/G heterozygotes did not differ from G/G homozygotes (Figs 4 and 5). This may be due to the limits of detection in this experiment: even if the *1A allele produced zero spliced mRNA, G/G homozygotes would only produce twice as much spliced mRNA as *1A/G heterozygotes on average, which is equivalent to a difference of 1.0 Ct. Given the range of ratios for each genotype, this is a relatively small difference. It is also possible that in-vivo CYP2A6 expression is governed by feedback mechanisms that could compensate for poorer splicing efficiency and obscure a difference between *1A/G and G/G genotypes.
Sequence variation can influence gene product function through a wide variety of mechanisms. In general, synonymous changes, those that do not alter amino acid sequence, are given short shrift; this is reflected in standard cytochrome P450 nomenclature, which does not typically differentiate newly discovered alleles by number based on synonymous variants. But the evolutionary conservation of predicted splicing motifs [17–19] and rare variants found associated with disease [10] indicate the importance of variation in the coding region with effects more subtle than the protein level. With the investigation of rs1137115, we have demonstrated that the importance of variation affecting exon splicing motifs extends to common variation in a metabolic activity associated with a complex behavior, tobacco abuse. These results underscore the value of bioinformatics tools in prioritizing genetic variation for study and demonstrate the need to investigate gene function using the best possible in-vivo assays in the context of complete haplotypes.
Supplementary Material
Acknowledgements
The authors thank and mention the following: Investigators directing data collection for COGEND are Laura Bierut, Naomi Breslau, Dorothy Hatsukami, and Eric Johnson. Data management is organized by Nancy Saccone and John Rice. Laboratory analyses are led by Alison Goate. Data collection was supervised by Tracey Richmond.
This study was supported by NIH Grants CA089392, CA77598, DA021237, 5T32MH014677-33, AA015572, CA91842, 1RR024992, UL1 RR024992.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website (www.pharmacogeneticsandgenomics.com).
Conflicts of interest
Dr Goate is listed as an inventor on a patent (US 20070258898) covering the use of certain SNPs in determining the diagnosis, prognosis, and treatment of addiction. Dr McDonald is a former employee of Pfizer. For the remaining authors there are no conflicts of interest.
References
- 1.Mwenifumbo JC, Tyndale RF. Molecular genetics of nicotine metabolism. Handb Exp Pharmacol. 2009;192:235–259. doi: 10.1007/978-3-540-69248-5_9. [DOI] [PubMed] [Google Scholar]
- 2.Bloom J, Hinrichs AL, Wang JC, von Weymarn LB, Kharasch ED, Bierut LJ, et al. The contribution of common CYP2A6 alleles to variation in nicotine metabolism among European-Americans. Pharmacogenet Genomics. 2011;21:403–416. doi: 10.1097/FPC.0b013e328346e8c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haberl M, Anwald B, Klein K, Weil R, Fuss C, Gepdiremen A, et al. Three haplotypes associated with CYP2A6 phenotypes in Caucasians. Pharmacogenet Genomics. 2005;15:609–624. doi: 10.1097/01.fpc.0000171517.22258.f1. [DOI] [PubMed] [Google Scholar]
- 4.Pitarque M, von Richter O, Rodriguez-Antona C, Wang J, Oscarson M, Ingelman-Sundberg M. A nicotine C-oxidase gene (CYP2A6) polymorphism important for promoter activity. Hum Mutat. 2004;23:258–266. doi: 10.1002/humu.20002. [DOI] [PubMed] [Google Scholar]
- 5.Mwenifumbo JC, Al Koudsi N, Ho MK, Zhou Q, Hoffmann EB, Sellers EM, et al. Novel and established CYP2A6 alleles impair in vivo nicotine metabolism in a population of Black African descent. Hum Mutat. 2008;29:679–688. doi: 10.1002/humu.20698. [DOI] [PubMed] [Google Scholar]
- 6.Keren H, Lev-Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet. 2010;11:345–355. doi: 10.1038/nrg2776. [DOI] [PubMed] [Google Scholar]
- 7.Mukherjee O, Wang J, Gitcho M, Chakraverty S, Taylor-Reinwald L, Shears S, et al. Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum Mutat. 2008;29:512–521. doi: 10.1002/humu.20681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 9.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 10.McVety S, Li L, Gordon PH, Chong G, Foulkes WD. Disruption of an exon splicing enhancer in exon 3 of MLH1 is the cause of HNPCC in a Quebec family. J Med Genet. 2006;43:153–156. doi: 10.1136/jmg.2005.031997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suphapeetiporn K, Kongkam P, Tantivatana J, Sinthuwiwat T, Tongkobpetch S, Shotelersuk V. PTEN c.511C > T nonsense mutation in a BRRS family disrupts a potential exonic splicing enhancer and causes exon skipping. Jpn J Clin Oncol. 2006;36:814–821. doi: 10.1093/jjco/hyl107. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen KB, Sorensen S, Cartegni L, Corydon TJ, Doktor TK, Schroeder LD, et al. Seemingly neutral polymorphic variants may confer immunity to splicing-inactivating mutations: a synonymous SNP in exon 5 of MCAD protects from deleterious mutations in a flanking exonic splicing enhancer. Am J Hum Genet. 2007;80:416–432. doi: 10.1086/511992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet. 2007;16:3149–3159. doi: 10.1093/hmg/ddm276. [DOI] [PubMed] [Google Scholar]
- 14.Gaildrat P, Krieger S, Di Giacomo D, Abdat J, Revillion F, Caputo S, et al. Multiple sequence variants of BRCA2 exon 7 alter splicing regulation. J Med Genet. 2012;49:609–617. doi: 10.1136/jmedgenet-2012-100965. [DOI] [PubMed] [Google Scholar]
- 15.Burgess R, MacLaren RE, Davidson AE, Urquhart JE, Holder GE, Robson AG, et al. ADVIRC is caused by distinct mutations in BEST1 that alter pre-mRNA splicing. J Med Genet. 2009;46:620–625. doi: 10.1136/jmg.2008.059881. [DOI] [PubMed] [Google Scholar]
- 16.Hofmann MH, Blievernicht JK, Klein K, Saussele T, Schaeffeler E, Schwab M, et al. Aberrant splicing caused by single nucleotide polymorphism c.516G > T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CYP2B6 in liver. J Pharmacol Exp Ther. 2008;325:284–292. doi: 10.1124/jpet.107.133306. [DOI] [PubMed] [Google Scholar]
- 17.Fairbrother WG, Holste D, Burge CB, Sharp PA. Single nucleotide polymorphism-based validation of exonic splicing enhancers. PLoS Biol. 2004;2:E268. doi: 10.1371/journal.pbio.0020268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlini DB, Genut JE. Synonymous SNPs provide evidence for selective constraint on human exonic splicing enhancers. J Mol Evol. 2006;62:89–98. doi: 10.1007/s00239-005-0055-x. [DOI] [PubMed] [Google Scholar]
- 19.Ke S, Zhang XH, Chasin LA. Positive selection acting on splicing motifs reflects compensatory evolution. Genome Res. 2008;18:533–543. doi: 10.1101/gr.070268.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bloom AJ, Harari O, Martinez M, Madden PA, Martin NG, Montgomery GW, et al. Use of a predictive model derived from in vivo endophenotype measurements to demonstrate associations with a complex locus, CYP2A6. Hum Mol Genet. 2012;21:3050–3062. doi: 10.1093/hmg/dds114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshida R, Nakajima M, Nishimura K, Tokudome S, Kwon JT, Yokoi T. Effects of polymorphism in promoter region of human CYP2A6 gene (CYP2A6*9) on expression level of messenger ribonucleic acid and enzymatic activity in vivo and in vitro. Clin Pharmacol Ther. 2003;74:69–76. doi: 10.1016/S0009-9236(03)00090-0. [DOI] [PubMed] [Google Scholar]
- 22.von Richter O, Pitarque M, Rodriguez-Antona C, Testa A, Mantovani R, Oscarson M, et al. Polymorphic NF-Y dependent regulation of human nicotine C-oxidase (CYP2A6). Pharmacogenetics. 2004;14:369–379. doi: 10.1097/00008571-200406000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Mwenifumbo JC, Lessov-Schlaggar CN, Zhou Q, Krasnow RE, Swan GE, Benowitz NL, et al. Identification of novel CYP2A6*1B variants: the CYP2A6*1B allele is associated with faster in vivo nicotine metabolism. Clin Pharmacol Ther. 2008;83:115–121. doi: 10.1038/sj.clpt.6100246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 25.Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 27.Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24:3439–3452. doi: 10.1093/nar/24.17.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet. 2010;42:448–453. doi: 10.1038/ng.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42:441–447. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saccone NL, Culverhouse RC, Schwantes-An TH, Cannon DS, Chen X, Cichon S, et al. Multiple independent loci at chromosome 15q25.1 affect smoking quantity: a meta-analysis and comparison with lung cancer and COPD. PLoS Genet. 2010;6:e1001053. doi: 10.1371/journal.pgen.1001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Becker K, Braune M, Benderska N, Buratti E, Baralle F, Villmann C, et al. A retroelement modifies pre-mRNA splicing: the murine glrb(spa) allele is a splicing signal polymorphism amplified by long interspersed nuclear element insertion. J Biol Chem. 2012;287:31185–31194. doi: 10.1074/jbc.M112.375691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.