

Abstract
Chromosomal instability is central to the process of carcinogenesis. The genome-wide detection of somatic chromosomal alterations (SCAs) in small premalignant lesions remains challenging since sample heterogeneity dilutes the aberrant cell information. To overcome this hurdle, we focused on the B allele frequency data from single nucleotide polymorphism microarrays (SNP arrays). The difference of allelic fractions between paired tumor and normal samples from the same patient (delta-θ) provides a simple but sensitive detection of SCA in affected tissue. We applied the delta-θ approach to small, heterogeneous clinical specimens including endobronchial biopsies and brushings. Regions identified by delta-θ were validated by FISH and qPCR in heterogeneous samples. Distinctive genomic variations were successfully detected across the whole genome in all invasive cancer cases (6/6), carcinoma in situ (3/3), and high grade dysplasia (severe or moderate) (3/11). Not only well-described SCAs in lung squamous cell carcinoma, but also several novel chromosomal alterations were frequently found across the pre-invasive dysplastic cases. Within these novel regions, losses of putative tumor suppressors (RNF20 and SSBP2) and an amplification of RASGRP3 gene with oncogenic activity were observed. Widespread sampling of the airway during bronchoscopy demonstrated that field cancerization reflected by SCAs at multiple sites was detectable. SNP arrays combined with delta-θ analysis can detect SCAs in heterogeneous clinical sample and expand our ability to assess genomic instability in the airway epithelium as a biomarker of lung cancer risk.
Keywords: chromosomal alteration, SNP microarray, biomarker, lung cancer risk
Introduction
Lung cancer is the leading cause of cancer-related death worldwide and in the United States (1, 2). This high mortality is due to the late diagnosis at a symptomatic advanced stage when surgical cure is impossible. To improve the outcome for lung cancer, new approaches for prevention and early detection are required (3–5).
In lung squamous cell carcinogenesis, the stepwise histopathologic changes in the bronchial epithelia that precede cancer development in heavy smokers, starting with normal epithelium, progressing through hyperplasia, squamous metaplasia, dysplasia, and carcinoma in situ (CIS), have been well documented (6). However, the rate and risk of progression of squamous dysplasia to CIS and ultimately to invasive cancer remains controversial and poorly understood (4, 7). Several publications have supported the concept that somatic chromosomal alterations (SCAs) are better prognostic biomarkers than premalignant histology alone (8–13). The detection of these SCAs in small biopsies with significant cellular heterogeneity has been limited by dilution with normal cells, as well as the inability of in situ techniques, such as multiprobe fluorescence in situ hybridization (FISH), to interrogate the majority of the genome.
The loss of genome integrity is regarded as the most prominent “enabling characteristic” in the development of cancers, by which certain mutant genotypes expand and evolve (14). The consequences of genomic instability are SCAs such as amplifications and deletions in genome copy number (15, 16). Array-based comparative genomic hybridization (aCGH) is one tool employed to identify some of these alterations across the whole genome (17, 18). More recently, single nucleotide polymorphism microarray (SNP array) technology has been used widely because it enables high-density genotyping, leading to more comprehensive SCA detection (19–23). Unlike aCGH, SNP arrays generate intensity differences as well as allelic ratios, and allow for analysis of not only copy number change, but also loss of heterozygosity (LOH) (including copy-neutral LOH) at high resolution. Furthermore, the power to detect SCAs is greatly increased when paired with normal samples, since the differences between subject and reference directly reflect somatic events (20, 24–26). The within-patient, paired analysis removes unrelated germline copy number changes that occur normally in all individuals.
Meanwhile, the cellular heterogeneity from typical clinical samples remains a basic obstacle to the sensitive and accurate genomic analysis of any type of cancer (20, 27, 28). The use of laser capture microdissection (LCM) technology may improve the ability to collect more affected homogeneous cell populations from formalin-fixed slides. However, collecting an adequate amount of DNA from these microscopic sections to perform efficient SNP array analysis remains problematic (29, 30). To overcome these issues, a number of analytical methods which enable the detection and evaluation of SCAs even in heterogeneous specimens are under development (25, 31–36).
In this report, we initially describe and validate subtraction of allelic fraction (delta-θ) for SCA detection utilizing the quantitative measurement of allelic imbalances in paired sample analysis, which controls for natural chromosomal variations in copy number. Validation experiments show SCAs can be detected in a background of ~90% normal cell content. Next, we demonstrate that when using delta-θ, even heterogeneous specimens such as bronchial biopsies and brushings can be reliable and informative sources for SNP array analyses. Using this strategy, we have found novel SCAs in preneoplastic lesions. Detection of SCAs in small, heterogeneous clinical samples will enable novel insights into the pathobiology of premalignant lesions.
Materials and Methods
Samples and experimental data sets
Genomic DNA from the non-small cell lung cancer cell line (CRL-5868D; NCI-H1395, adenocarcinoma) and its matched lymphoblastoid cell line (CRL-5957D: NCI-BL1395, B lymphoblast cell) were obtained from the American Type Culture Collection (ATCC; Manassas, VA). The genomic DNA, purchased from the ATCC, was prepared from cell lines grown by the ATCC and authenticated by using the Promega PowerPlex Systems STR profiling kit. A titration series (100.0%, 25.0%, 12.5%, 6.3%, 3.1%, 1.6% and 0% tumor DNA content) was made by mixing the cancer and the normal cell DNA. DNA concentrations {diluted to 50 ng/μl, and 200 ng (4 μl) for each sample} were processed according to the manufacturer’s protocol and hybridized to the HumanOmni2.5-Quad BeadChips (Illumina; San Diego, CA).
Clinical specimens
Autofluorescence and white-light bronchoscopy as well as blood collection were carried out on current or former smokers with a > 20 pack year smoking history after obtaining written informed consent in an IRB approved protocol. Endobronchial biopsy and/or brushing (after biopsy) was performed at the same visually concerning area. Multiple endobronchial sites were biopsied throughout the airway according to study protocol.
These multiple biopsy specimens, including the biopsy at the concerning area, were formalin fixed, paraffin embedded, stained with hematoxylin and eosin, and histologically graded by the study pathologist (WAF) as described (3). They were classified into 8 categories as defined by WHO classification and assigned a score according to the following system: 1, normal; 2, reserve cell hyperplasia; 3, squamous metaplasia; 4, mild dysplasia; 5, moderate dysplasia; 6, severe dysplasia; 7, CIS; 8, invasive carcinoma. All non-malignant lesions, including normal histology, hyperplasia, squamous metaplasia and dysplasia, are referred to as premalignant. We use the term “preinvasive” for lesions up to and including CIS lesions. Since some patients had multiple bronchoscopies, some bronchial lesions were biopsied more than once. We define a biopsy sample as “persistent” when the histology grade score does not change or declines only 1 grade score in the next bronchoscopy (at least more than 12 months interval in between two bronchoscopies). “Regressive” samples are defined when the grade score declines 2 or more in the next bronchoscopy.
For DNA isolation, fresh biopsies from each concerning site were homogenized in TRIzol reagent (Life Technologies; Carlsbad, CA) and after chloroform addition, the interface layer was saved. Protein was precipitated from the interface using sodium citrate, followed by ethanol precipitation of the supernatant. The DNA pellet was dissolved in TE buffer (10mM Tris-HCl pH 8.0, 0.1mM EDTA). For brushings, after overnight digestion with Proteinase-K, DNA was isolated from protein using sodium chloride (salting out), precipitated by ethanol, and dissolved in TE buffer. For reference blood samples, the column-based extraction protocol was conducted using QuickGene-610 (AutoGen; Holliston, MA). Extracted DNA was quantified, verified to be of high molecular weight by agarose gel, diluted to 50 ng/μl, and labeled for SNP array analysis (HumanOmni 2.5-Quad BeadChip and Human 660W-Quad BeadChip; Illumina). Obtained call rates for all samples run were 98.8 ± 1.6% (mean ± SD). For one brushing specimen, we tested two DNA isolation methods: one is an extraction from saline in which the brush was vigorously vortexed to detach the epithelial cells. The other is a direct DNA extraction from residual cells adhering to the brush after vortexing. The same amounts of high quality genomic DNA in these 2 conditions were processed and hybridized to the arrays.
Data analysis
Fluorescent signals were imported into GenomeStudio software (Illumina), where the genotype data were generated and transformed to normalized intensity (R), and allelic ratio (theta; θ) through the calculations below (20),
where XA and YB denote transformed normalized signal intensities from A and B alleles for a particular SNP locus. In paired sample analysis, these two parameters are conventionally transformed into two outputs: Log2(Rsubject/Rreference) referred to as Log2 R Ratio (LRR), and B Allele Frequency shown individually as BAFsubject and BAFreference. In LRR, any deviations from zero are evidence for copy number change, whereas BAF refers to a normalized measure of relative signal intensity ratio of the B and A alleles. Deviations from the expected values (0.0, 0.5 and 1.0 representing AA, AB and BB alleles, respectively) are indicative of chromosomal alterations. To obtain a transformed BAF profile in which genomic segmentation strategy can be applied (see next section), non-informative homozygous alleles (AA and BB) in the reference (normal) sample were removed by comparison of genotype calls between the subject and the reference. Then, BAF profile was reflected into transformed BAF along the 0.5 axes, named “modified BAF” in our study. This approach is derived from the mirrored BAF method (25).
Subtraction of allelic fractions (delta-θ) is generated through the following calculation between θsubject and θreference:
Homozygous alleles in the reference sample were also removed. If there is no somatic alteration at a locus, the delta-θ value is near zero. However, once any somatic change occurs in subject, delta-θ shows any positive value (up to 0.5). In a rare case of balanced biallelic amplification (e.g., copy number is 4 with AABB alleles), delta-θ as well as BAF show normally distributed plots since there is no allelic imbalance.
Delta-θ and modified BAF are both based on θ values, but are composed of different concepts. Delta-θ is a direct, intra-patient comparison of θsubject and θreference, which represents only somatic alterations. In modified BAF, the reference sample is used only for selecting heterozygous alleles to exclude uninformative homozygous alleles. This implies the aberrant regions detected by modified BAF could include not only somatic changes but also germline ones. An example is shown in Supplementary Figure S1. Subtraction of allelic fractions (delta-θ) is not a unique use of BAF or θ measures, but works efficiently to detect somatic changes in heterogeneous samples.
To efficiently identify SCA, we first started with the review of the delta-θ plot (SCAs are detected in this first step), then we referred to the LRR and BAF plots to identify the nature of the detected SCA. The SNP array data have been deposited at the Gene Expression Omnibus (GSE43168).
Data visualization and Genomic Segmentation methods
Visualization of the data was performed using Partek Genomic Suite 6.6 software (Partek Inc., St. Louis, MO). For visualizing delta-θ and modified BAF, each gray dot represents gross delta-θ or modified BAF values from heterozygous alleles in the reference, whereas red (or black in black and white figures) dots represent the smoothed values for every 30 gray dots. For visualizing LRR, each gray dot comes from all individual SNP markers, and blue (or black) dots indicate the smoothed values for every 30 gray dots. In BAFsubject visualization, gray dots reflect all θsubject values.
To statistically delineate each SCA region and its breakpoints, genomic segmentation was conducted. Genomic segmentation results are labelled “GS” in the figures. Partek has implemented the Circular Binary Segmentation (CBS) algorithm (37) in their product. Detailed information is available at the Partek website (38). We applied this algorithm for LRR, delta-θ and modified BAF profiles setting the following three parameters: minimum genomic markers, p-value threshold, and signal to noise ratio. Practically, multiple parameter corrections and iterative optimizing processes were performed for each case to achieve or approach the most convincing segmentation data. However, conducting genomic segmentation especially for LRR is occasionally challenging in heterogeneous samples since the noise hinders the effective parameter setting, and the samples have variable noise in their SNP arrays. Thus, to achieve a balance of sensitivity and specificity, data from these 3 profiles were calculated using the same parameter settings for a given sample. In the comparative studies of titration cell line series, minimum markers, p-value, and signal to noise ratio were set to 100, 0.001, and 0.3, respectively. In clinical samples, those parameters were decided individually after iterative optimizations (shown in Supplementary Table S1). Genomic position is based on human genome assembly (hg19).
Fluorescence in situ hybridization (FISH)
To validate selected SNP array results, FISH was performed using sections of endobronchial biopsy specimens obtained at the same location prior to the arrayed biopsy/brushing sample. Unstained slides with paraffin-embedded sections were hybridized with a number of FISH probes including sequences of PIK3CA, TP63, D5S721-D5S23 (encompassing SEMA5A), CDKN2A, and NKX2-1, according to the previously published protocols (12). They are located at 3q26.32, 3q28, 5p15.2, 9p21, and 14q13.3, respectively. Centromere probes (CEP3 at 3p11.1-3q11.1 and CEP9 at 9p11-9q11) were used as references.
Copy number qPCR assay
Quantitative PCR (qPCR) assays were used to validate the copy number status of the genes located within aberrant chromosomal regions. The corresponding blood sample DNA was used as a calibrator control. The gene copy numbers of RARB (located at 3p24.2), SEMA3B (3q21.3), DNAH5 (5p15.3), GDNF (5p13.2), RNF20 (9q31.1), RASGRP3 (2p22.3), and SSBP2 (5q14.1) were determined by a duplex Taqman copy number assay (Life Technologies) with RNase P (14q11.2) as the reference assay. Assays were performed according to the manufacturer’s protocol. Copy numbers were called by relative quantification methods in 4–6 replicate measurements through CopyCaller software version 2.0.
Results
Delta-θ detects SCA regions
The Illumina genotyping assay generates two independent values at each SNP locus: R and θ (22). R is a representation of normalized signal intensity, while θ indicates the allelic ratio at a given locus. These two parameters are conventionally visualized as LRR and BAF plots, respectively. Our study focused on the detection of somatic alterations in the comparison of subject (e.g., bronchial biopsy or brushing) and reference (e.g., blood) using a paired sample set from the same individual. Whereas LRR is represented as Log2(Rsubject/Rreference) comparing Rsubject to Rreference, BAF is plotted separately as BAFsubject or BAFreference (see Materials and Methods for additional detail). To apply the pairwise concept to a θ-based plot, we used delta-θ, the difference of θreference and θsubject (subtraction of allelic fractions). Delta-θ can be analyzed through genomic segmentation and is a sensitive parameter to detect somatic chromosomal rearrangements including copy-neutral events. The basic concept of delta-θ is illustrated using a cancer cell line genomic DNA (NCI-H1395; H1395) as subject and the lymphoblastoid cell line DNA (NCI-BL1395) as reference, which both originated from the same individual (Figure 1A). The genome-wide SCA profile of the NCI-H1395 cell line is also found at the Wellcome Trust Sanger Institute website. In Figure 1B, the delta-θ plot for this pair is visualized together with conventional LRR and BAFsubject plots. Each plot represents a variety of different SCAs across chromosome 6. Delta-θ is an easily interpretable one-band plot in which differences between normal and abnormal regions are clearly discernible, including copy-neutral LOH regions, which are undetected using LRR.
Figure 1. The visualization of delta-θ and overlap rates of detected segments in the genomic DNA titration series.

A. The process of delta-θ generation is illustrated in polar coordinate plot: SNP markers in the NCI-H1395 cancer cell line (subject) result from the somatic change of the same markers compared to the NCI-BL1395 lymphoblastoid cell line (reference). 100 SNP markers are randomly selected from a hemizygous deletion region at chromosome 6q.
B. Schematic diagram of chromosome 6 showing the range of SCA determined in NCI-H1395 as annotated at the Wellcome Trust Sanger Institute: delta-θ (upper), LRR (middle), and BAFsubject (lower) represent a variety of different SCAs including various degrees of amplifications and regions with LOH as well as normal regions.
C. The overall segments detected by each plot across the entire autosomal chromosomes in titrated tumor contents (25.0%, 12.5%, and 6.25%) are shown as concordance rates to the total detected segments in 100% tumor content. modified BAF was used to apply the genomic segmentation strategy to BAFsubject plot.
Delta-θ yields improved sensitivity in heterogeneous cancer models
Next, we modeled the effect of tumor heterogeneity to detect various types of SCAs by mixing the cancer cell line DNA with its matched lymphoblastoid cell line DNA, creating a titration series with the following cancer cell DNA content: 100.0%, 25.0%, 12.5%, 6.3%, 3.1%, 1.6% and 0.0% (100.0% lymphoblastoid cell line DNA). Each sample in this series containing cancer cell DNA was paired for analysis with 100.0% lymphoblastoid cell line DNA as the reference sample.
For each tumor content case, LRR, BAFsubject as well as delta-θ plots were generated. The CBS algorithm, which is implemented as genomic segmentation strategy (GS) in Partek Genomics Suite 6.6, was used to computationally define SCA regions (see Methods). BAFsubject was transformed into “modified BAF” profile borrowing the mirrored BAF concept, in which GS was applied (see Methods). We evaluated whether these 3 plots correctly detected SCA regions regardless of whether they identified the nature of SCA (amplification, deletion, or copy-neutral LOH). Figure 1C shows the concordance rates of detected SCA segments overlapping between 100% tumor content and other diluted tumor contents across all autosomal chromosomes. The genome-wide view of all detected segments represented from 100%, 25.0%, and 12.5% tumor contents are also shown (Supplementary Figure S2).
In the 100.0% tumor content sample, almost identical aberrant segments were detected among those 3 approaches, except for copy-neutral LOH regions in the LRR. However, in the 25% tumor content sample, over 50% of true SCA segments were missed by LRR, whereas almost all SCA regions were sensitively segmented by θ-based approaches (delta-θ and modified BAF). In the 12.5% tumor content sample, θ-based approaches were still able to correctly call ~50% of SCA segments. In samples with less than 12.5% tumor content, conducting accurate segmentation became more difficult. However, regions large in size or extensively amplified were still detected. These results imply that when investigating heterogeneous samples using Illumina’s SNP microarrays, θ-based approaches can produce more reliable segmentation data than an R-based approach.
Genome-wide SNP array analysis of heterogeneous clinical samples
To explore the practical utility of SNP array analysis combined with delta-θ for heterogeneous clinical samples, we investigated bronchial biopsies and brushings containing cancer or dysplastic cells which were likely to be contaminated with normal stromal cells. In total, 30 whole bronchial biopsies/brushes from 18 patients with heavy smoking histories were investigated {6 invasive cancer, 3 CIS, 15 dysplasia (4 severe, 7 moderate, and 4 mild dysplasia), 3 hyperplasia, and 3 normal histology} (Table 1, Supplementary Figure S3). In spite of each specimen’s heterogeneity, delta-θ sensitively detected SCAs across the entire genome (Supplementary Table S1 for complete listing). Whereas SCAs were detected in all 6 invasive cancer cases, SCAs were detected in 6 out of 14 samples with higher grade dysplasias (grades 5–7). None of the 10 samples with histology grade less than moderate dysplasia (grade < 5) showed SCAs.
Table 1.
Clinical features and SNP array results of 18 patients
| histology grade | sample name | bronchial location | biopsy/brush | patient number | SCA detection | smoking history | histology stability |
|---|---|---|---|---|---|---|---|
| SQ | IC-1 | LUL | brush | 1 | Yes | former | - |
| SQ | IC-2 | RLL | brush | 1 | Yes | former | - |
| SQ | IC-3 | RUL | brush | 1 | Yes | former | - |
| SQ | IC-4 | RUL | brush | 2 | Yes | current | - |
| SQ | IC-5 | RUL | biopsy | 3 | Yes | current | - |
| LC | IC-6 | LUDB | biopsy | 4 | Yes | current | - |
|
| |||||||
| CIS | CIS-1 | LUL | biopsy | 5 | Yes | current | persistent |
| CIS | CIS-2 | LUDB | biopsy | 6 | Yes | current | persistent |
| CIS | CIS-3 | LUL | brush | 7 | Yes | current | persistent |
|
| |||||||
| SD | SD-1 | RML | biopsy | 5 | Yes | current | - |
| SD | SD-2 | LLL | brush | 8 | No | former | - |
| SD | SD-3 | RUL | brush | 9 | No | current | regressive |
| SD | SD-4 | RUL | brush | 10 | No | former | - |
|
| |||||||
| MD | MD-1 | LUL | brush | 6 | Yes | current | persistent |
| MD | MD-2 | RUL | brush | 6 | No | current | - |
| MD | MD-3 | LUL(Li) | brush | 7 | Yes | current | persistent |
| MD | MD-4 | RLL | brush | 7 | No | current | - |
| MD | MD-5 | RUL | brush | 11 | No | current | regressive |
| MD | MD-6 | LUL | brush | 12 | No | former | persistent |
| MD | MD-7 | RML | brush | 12 | No | former | regressive |
|
| |||||||
| MiD | MiD-1 | LUL | brush | 12 | No | former | persistent |
| MiD | MiD-2 | LUDB | brush | 13 | No | current | - |
| MiD | MiD-3 | RUL | brush | 14 | No | current | regressive |
| MiD | MiD-4 | RML | brush | 15 | No | former | regressive |
|
| |||||||
| HP | HP-1 | RLL | biopsy | 5 | No | current | - |
| HP | HP-2 | LUL | brush | 16 | No | current | - |
| HP | HP-3 | RUL | brush | 15 | No | former | persistent |
|
| |||||||
| NH | NH-1 | RML | brush | 11 | No | current | - |
| NH | NH-2 | LUDB | brush | 17 | No | former | persistent |
| NH | NH-3 | RML | brush | 18 | No | former | persistent |
The results of Fisher’s exact tests in arrayed patients/lesions with 5–7 histology grade; p = 0.20 in current vs. former smokers with SCA detection compared to no SCA detection, and p = 0.048 in persistence vs. regression of lesions with SCA detection compared to no SCA detection. Disease stability: persistent change; ≤ 1 histology grade, regressive change; ≥ 2 grades, -; no repeated bronchoscopy available. Definition of abbreviations: SQ, squamous cell lung cancer; LC, large cell lung cancer; CIS, carcinoma in situ; SD, severe dysplasia; MD, moderate dysplasia; MiD, mild dysplasia; HP, hyperplasia; NH, normal histology; LUL, left upper lung; LUDB, left upper divisional bronchus; LUL(Li), left upper lung (lingula); LLL, left lower lung; RUL, right upper lung; RML, right middle lung; RLL, right lower lung
We identified genomic regions by delta-θ with frequently overlapping SCAs in the 6 preinvasive samples (Table 2). Among them, SCAs at chromosome 3p (3p26.3 to 3p12.3, 76.8Mbps), 5p (5p15.33 to 5p11, 47.8Mbps), 8p (8p23.3 to 8p11.21, 39.9Mbps), 9p (9p24.3 to 9p21.2, 26.6Mbps), and 13q (13q11 to 13q34, 95.8Mbps) are known to be common genetic events observed in preinvasive bronchial dysplasias (4, 39). Other frequently overlapping regions, which have never been previously reported in the bronchial preneoplasia, were also discovered. Some of these novel regions are relatively short, but affect several genes (e.g., a minimum SCA overlap shared at 2p22.3 (3.1Mbp) contains 13 genes).
Table 2.
Frequently overlapping regions among preinvasive cases
| chromosome | cytoband | length (bp) | potential gene of interest | previous report | estimated SCA type | SCA detected (n = 6) |
|---|---|---|---|---|---|---|
|
| ||||||
| 2p | 2p22.3 | 3,112,242 | RASGRP3 | no | amplification | 3 |
| 3p | 3p26.3 - 12.3 | 76,829,014 | RARB, SEMA3B | yes | deletion/CN-LOH | 4 |
| 5p | 5p15.33 - 11 | 47,814,582 | TERT, GDNF | yes | amplification | 3 |
| 5q | 5q14.1 | 613,020 | SSBP2 | no | deletion/CN-LOH | 3 |
| 7p | 7p15.3 | 531,270 | DFNA5 | no | deletion/CN-LOH | 4 |
| 7q | 7q36.3 | 858,066 | PTPRN2 | no | deletion/CN-LOH | 3 |
| 8p | 8p23.3 - 11.21 | 39,942,807 | MTUS1 | yes | complex | 3 |
| 9p | 9p24.3 - 21.2 | 26,562,586 | CDKN2A | yes | deletion/CN-LOH | 5 |
| 9q | 9q31.1 - 31.2 | 5,599,950 | RNF20 | no | deletion/CN-LOH | 5 |
| 13q | 13q11 - 34 | 95,827,527 | RB1, BRCA2 | yes | deletion/CN-LOH | 3 |
| 14q | 14q21.1 - 21.2 | 2,530,363 | FANCM | no | deletion/CN-LOH | 3 |
| 21q | 21q22.2 - 22.3 | 6,154,199 | TFF1 | no | deletion/CN-LOH | 3 |
SCA regions shared by at least 2 patients are shown. The trend SCA type was estimated by investigating the mean value of LRR in detected segment. “complex” indicates both deleted and amplified segments exist in the region. Boldface types are the genes selected for FISH and qPCR assays to confirm the estimated trend of SCA type (shown in Figure 2 and Figure 3).
In order to validate the regions detected by delta-θ, several SCA regions containing the sequences of known cancer-related genes were selected and investigated by FISH and copy number qPCR assays (Figure 2A, 2B for FISH, and Figure 2C for copy number qPCR). The regions identified as SCAs by θ-based approaches were validated by those assays. On the other hand, the LRR plot was not necessarily useful as a reliable way to identify SCAs. In a sample with a noisy or deviated LRR plot, fragmented segments that resulted from highly variable LRR values failed to detect true SCA regions (Supplementary Figure S4).
Figure 2. Detection and validation of SCAs by FISH and qPCR assays.
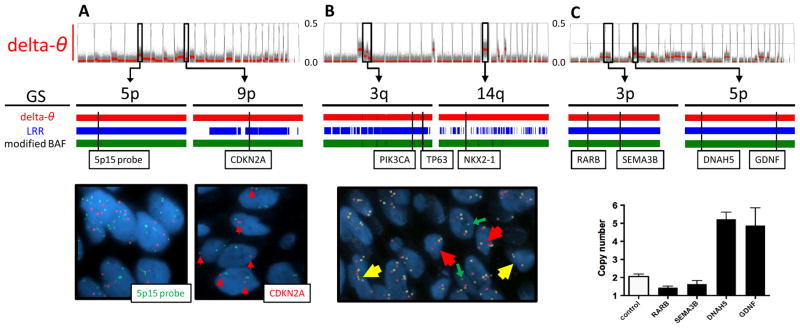
A. In a brushing sample with CIS (CIS-3 in Table 1), contaminated with normal stromal cells, several SCA regions were suggested by delta-θ (upper). Among them, the 2 regions at 5p and 9p were chosen (middle), and confirmed as amplification (copy number = 5.90 ± 2.31) and deletion (copy number = 1.36 ± 0.48), respectively, by FISH using 5p15 (green) and CDKN2A probes (red) (lower). LRR and modified BAF also detected those two regions.
B. In a biopsy sample with CIS (CIS-1), which is relatively homogeneous, several SCA regions were detected by delta-θ (upper). Using tri-color FISH probes (PIK3CA in red, TP63 in yellow at 3q, and NKX2-1 in green at 14q), 3q and 14q were confirmed as amplification (3.52 ± 1.07 and 4.30 ± 1.05) and deletion (0.48 ± 0.58), respectively. Modified BAF detected both regions, whereas fragmented segments by LRR missed the deletion at 14q.
C. In a brushing sample with moderate dysplasia (MD-3), which is relatively homogeneous, several SCA regions were suggested by delta-θ. qPCR assays predicted copy numbers at 4 locus (1.44 ± 0.08, 1.63 ± 0.20, 5.28 ± 0.40, and 4.92 ± 0.98, shown as mean ± SD in quadruplicate measurements). Control indicates reference blood (predicted copy number is 2.03 ± 0.14).
GS: genomic segmentation
Next, we used copy number qPCR assays to confirm the delta-θ results in several novel SCA regions identified. The following genes were selected for this study: Ring Finger Protein 20 (RNF20) located at 9q31.1, RAS guanyl releasing protein 3 (RASGRP3) at 2p22.3, and single-stranded DNA binding protein 2 (SSBP2) at 5q14.1 (Table 2). Although heterogeneity in some of the preinvasive samples made LRR plots uninterpretable, qPCR assay results showed a trend of amplification or deletion/copy-neutral LOH, in each SCA detected by delta-θ (Figure 3).
Figure 3. The trend of SCA types estimated by qPCR at the regions containing 3 selected genes in preinvasive samples.

Using the copy number probes for 3 different genes (RNF20, RASGRP3, and SSBP2), the common trend of amplification or deletion(/copy-neutral LOH) was estimated in each gene. In delta-θ plots, the regions of 90–120Mbp at 9q, 0–50Mbp at 2p, and 50–100Mbp at 5q were illustrated. Mean values of delta-θ, LRR from SNP array and copy number by qPCR in each segmented SCA region are shown. The direction of either amplification or deletion was inferred by considering those mean values. qPCR resulted in showing the concordant trend of copy number estimation to SNP array data in spite of various degrees of sample heterogeneity.
*p-values resulted from two-sample t-tests in the comparison of individual preinvasive sample and blood in 4–6 replicate measurements
Among the 14 cases with histology grade 5–7 (preinvasive), there was no significant difference in the average histology grades of the actual clinical samples prepared and run on the SNP array (p = 0.07) (filled dots in Supplementary Figure S3). However, an individual patient’s overall average histology (filled and open circles) was significantly higher in SCA-positive patients versus SCA-negative patients (p = 0.021) (Figure 4). The biopsies were obtained from an individual patient in a broad sampling of the airway mucosa (at ≥ 4 well-distributed bronchial areas biopsied besides concerning areas). Patients whose bronchial mucosa showed any SCA had highly severe lesions across the bronchus.
Figure 4. The correlation between individual patient’s overall average histology grade and SCA detection.

Average histology grade (from all biopsies for each patient) were compared among 3 different groups: 3 patients whose arrayed samples with CIS/SD/MD (histology grade 5–7) showed positive SCAs (left), 5 patients whose arrayed samples with SD/MD (histology grade 5–6) resulted in negative SCA detection (middle), and 6 patients whose samples with less than MD (histology grade < 5) were arrayed, but resulted in negative SCA detection (right). Each dot represents the average histology grade of a patient (see Supplementary Figure S3), and each horizontal bar shows the average value of each group’s average histology (4.5, 3.0, and 2.6, respectively). *p = 0.021 in t-test between left and middle groups; **p = 0.010 in t-test between left and middle + right groups; ns = not significant
Additionally, we compared two different DNA isolation methods from the brushing specimen shown above (MD-3) (see Methods). The sample obtained directly from brushing resulted in higher delta-θ values, suggesting that an adequate amount of more homogeneously affected epithelial cells can be obtained directly from a brush after vortexing (Supplementary Figure S5).
Discussion
The advances made in high-density chip technology have improved sensitivity and specificity of detection of aberrant chromosomal rearrangements. Nevertheless, the heterogeneous nature of relatively smaller premalignant specimens compared with solid invasive cancer tissues has made the analysis and interpretation of data more challenging. By using blood genomic DNA from the same patient as a reference, we demonstrate how delta-θ (subtraction of allelic fraction) helps alleviate this problem.
Theta-based approaches are very sensitive at detecting regions of chromosomal abnormality, but do not provide information about the type of genomic change. Computationally integrated algorithms, like Genome Alteration Print (GAP) (40), consider both the LRR and BAF data to infer copy number gain or loss. In the analysis of relatively homogeneous samples, such integrated algorithms can generate more information including copy number estimation and take less time for completing comprehensive analysis than delta-θ. However, in heterogeneous samples, delta-θ provides greater sensitivity for detecting alterations between 10%-25% tumor/abnormal cell content, depending on the type of SCA. In cancer genome studies, finding somatically derived SCAs is critical in order to identify truly carcinogenic variants (26).
Initially, we were unsure as to whether biopsies or brushings would be better suited for SNP array analysis. Although both specimens can be used, brushing samples contain a higher proportion of epithelial cells. In general, genomic DNA isolated directly from the brush showed the highest signal to noise ratio (Supplementary Figure S5). Overall, brushings appear more attractive than biopsies for SNP array-based genome research. We have not assessed microdissected biopsies.
In the analysis of preinvasive lesions, delta-θ revealed previously characterized SCAs as well as novel SCA regions which were highly overlapping among preinvasive lesions. Some of these novel regions are small in size, but do contain at least one gene which may have a cancer-related function. For example, RNF20 deficiency has been recently reported to trigger genomic instability (41, 42). SSBP2 stabilizes transcriptional cofactor protein and regulates malignant transformation (43). Both of these genes are reported to act as putative tumor suppressors. Meanwhile, another study indicated that RASGRP3 has a tumorigenic function by activating the RAS signaling pathway (44) (Table 2). Analysis of over 200 lung squamous cell carcinoma samples (Cancer Genome Workbench) (45) showed the genomic regions containing RNF20 and SSBP2 are frequently deleted (35% and 66%, respectively), and the region containing RASGRP3 is frequently amplified (45%). These novel overlapping SCAs across preinvasive cases can be added to the frequent somatic genomic rearrangements associated with the development of cancer. We speculate that these SCAs were not previously discovered due to their relative small size and low signal, particularly in heterogeneous samples. Our findings demonstrate, even at the preinvasive dysplastic stage in bronchial epithelium, more instances of genomic alteration are occurring across the genome than was previously appreciated.
The preinvasive specimens with positively detected SCAs were shown to be accompanied by multifocal and advanced histologic changes throughout the airway (Figure 4), referred to as “field cancerization” (46, 47). Although a wide variety of chromosomal alterations are thought to be important in the development of invasive cancer, these changes are identified in the airway of current or former smokers without known lung cancer. The delta-θ’s consistency implies that, even if a sample contains only a small portion of cytogenetically affected epithelial cells (~10%), SCA regions are detectable in SNP array analysis. Considering these findings together, advanced preinvasive lesions including high grade dysplasia and CIS with detectable SCAs may identify early stage patients prior to the dominant and clonal expansion observed in late stage invasive tumors.
While the number of samples analyzed is limited, several patients in our study have undergone repeated bronchoscopies to monitor dysplastic lesions. SCAs are more likely to be detected in moderate or worse dysplasias that are persistent over multiple bronchoscopies (Table 1). Meanwhile, no significant difference was seen between current and former smokers whose specimens showed positive or negative SCA.
Since SNP arrays are able to sensitively monitor these emerging SCAs, testing early stage lesions by this approach may identify patients at higher risk of developing invasive cancer and these subjects may be excellent candidates for chemoprevention studies. As our study is cross-sectional and of limited size, both larger cross-sectional and longitudinal studies will be needed to prove this hypothesis.
More and more studies are now using next generation sequencing (NGS) technology. NGS not only provides efficient mutation analysis, but also can detect copy number variations under specific circumstances (48). Specific gene mutations can be detected by NGS technology in highly heterogeneous cases (49, 50). However, the high cost and analytical complexity currently limit this approach (51). In addition, concise and efficient analytical methods seem to be needed to derive copy number estimation in heterogeneous cases (52). We believe that a microarray-based approach, supported with established analytical methodologies, can be a more cost-effective approach for screening small, pre-malignant bronchial lesions.
In conclusion, distinctive genomic variations were successfully detected across the whole genome by SNP arrays even in the heterogeneous cell population found in bronchial premalignancy, by using subtraction of allelic fractions, delta-θ. Using this strategy, we have demonstrated the occurrence of at least 3 SCAs previously undescribed in preinvasive lesions. The genes contained within these regions show losses of putative tumor suppressors (RNF20 and SSBP2) and an amplification of a gene with oncogenic activity (RASGRP3). SNP array technology can expand our ability to assess genomic instability in the airway epithelium as a biomarker of lung cancer risk.
Supplementary Material
Acknowledgments
The authors thank Okyong Cho (University of Colorado Denver Genomics and Microarray Core Lab) for technical assistance and the use of Cancer Center Shared Resource (P30 CA046934). The authors also thank Heather Malinowski (Department of Pathology, University of Colorado Denver) for technical assistance.
Grant support
The study was partially supported by NCI grants SPORE in Lung Cancer P50 CA058187 (M. Varella-Garcia, W. Franklin, Y. Miller, M. Geraci), Cancer Center Support Grant P30 CA046934 (M. Varella-Garcia, W. Franklin, Y. Miller, M. Geraci) and RO1 CA164780 (Y. Miller, M. Geraci), a NHLBI grant R21 HL094927 (C. Coldren), a LUNGevity Foundation Early Detection grant (Y. Miller), and Cancer League of Colorado research grant (Y. Miller, M. Geraci). This is also supported in part by the Biostatistics/Bioinformatics Shared Resource of Colorado’s NIH/NCI CCSG P30 CA046934 (M. Edwards).
Footnotes
Disclosure of Potential Conflicts of Interest
D. Merrick is in on the advisory board of Pfizer, Inc.. M. Varella-Garcia has received honoraria as an educational speaker for Abbott Molecular, Inc.. No potential conflicts were disclosed by the other authors.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Youlden DR, Cramb SM, Baade PD. The International Epidemiology of Lung Cancer: geographical distribution and secular trends. J Thorac Oncol. 2008;3:819–31. doi: 10.1097/JTO.0b013e31818020eb. [DOI] [PubMed] [Google Scholar]
- 3.Keith RL, Blatchford PJ, Kittelson J, Minna JD, Kelly K, Massion PP, et al. Oral iloprost improves endobronchial dysplasia in former smokers. Cancer Prev Res (Phila) 2011;4:793–802. doi: 10.1158/1940-6207.CAPR-11-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wistuba Genetics of preneoplasia: lessons from lung cancer. Curr Mol Med. 2007;7:3–14. doi: 10.2174/156652407779940468. [DOI] [PubMed] [Google Scholar]
- 5.Hirsch FR, Franklin WA, Gazdar AF, Bunn PA., Jr Early detection of lung cancer: clinical perspectives of recent advances in biology and radiology. Clin Cancer Res. 2001;7:5–22. [PubMed] [Google Scholar]
- 6.Nicholson AG, Perry LJ, Cury PM, Jackson P, McCormick CM, Corrin B, et al. Reproducibility of the WHO/IASLC grading system for pre-invasive squamous lesions of the bronchus: a study of inter-observer and intra-observer variation. Histopathology. 2001;38:202–8. doi: 10.1046/j.1365-2559.2001.01078.x. [DOI] [PubMed] [Google Scholar]
- 7.Breuer RH, Pasic A, Smit EF, van Vliet E, Vonk Noordegraaf A, Risse EJ, et al. The natural course of preneoplastic lesions in bronchial epithelium. Clin Cancer Res. 2005;11:537–43. [PubMed] [Google Scholar]
- 8.van Boerdonk RA, Sutedja TG, Snijders PJ, Reinen E, Wilting SM, van de Wiel MA, et al. DNA copy number alterations in endobronchial squamous metaplastic lesions predict lung cancer. Am J Respir Crit Care Med. 2011;184:948–56. doi: 10.1164/rccm.201102-0218OC. [DOI] [PubMed] [Google Scholar]
- 9.Ishizumi T, McWilliams A, MacAulay C, Gazdar A, Lam S. Natural history of bronchial preinvasive lesions. Cancer Metastasis Rev. 2010;29:5–14. doi: 10.1007/s10555-010-9214-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lockwood WW, Chari R, Coe BP, Thu KL, Garnis C, Malloff CA, et al. Integrative genomic analyses identify BRF2 as a novel lineage-specific oncogene in lung squamous cell carcinoma. PLoS Med. 2010;7:e1000315. doi: 10.1371/journal.pmed.1000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massion PP, Zou Y, Uner H, Kiatsimkul P, Wolf HJ, Baron AE, et al. Recurrent genomic gains in preinvasive lesions as a biomarker of risk for lung cancer. PLoS One. 2009;4:e5611. doi: 10.1371/journal.pone.0005611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jonsson S, Varella-Garcia M, Miller YE, Wolf HJ, Byers T, Braudrick S, et al. Chromosomal aneusomy in bronchial high-grade lesions is associated with invasive lung cancer. Am J Respir Crit Care Med. 2008;177:342–7. doi: 10.1164/rccm.200708-1142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varella-Garcia M, Chen L, Powell RL, Hirsch FR, Kennedy TC, Keith R, et al. Spectral karyotyping detects chromosome damage in bronchial cells of smokers and patients with cancer. Am J Respir Crit Care Med. 2007;176:505–12. doi: 10.1164/rccm.200609-1329OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Aguilera A, Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–17. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 16.Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nat Genet. 2003;34:369–76. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- 17.Pinkel D, Albertson DG. Array comparative genomic hybridization and its applications in cancer. Nat Genet. 2005;37 (Suppl):S11–7. doi: 10.1038/ng1569. [DOI] [PubMed] [Google Scholar]
- 18.Albertson DG, Pinkel D. Genomic microarrays in human genetic disease and cancer. Hum Mol Genet. 2003;12(Spec No 2):R145–52. doi: 10.1093/hmg/ddg261. [DOI] [PubMed] [Google Scholar]
- 19.Kim S, Misra A. SNP genotyping: technologies and biomedical applications. Annu Rev Biomed Eng. 2007;9:289–320. doi: 10.1146/annurev.bioeng.9.060906.152037. [DOI] [PubMed] [Google Scholar]
- 20.Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–48. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steemers FJ, Gunderson KL. Whole genome genotyping technologies on the BeadArray platform. Biotechnol J. 2007;2:41–9. doi: 10.1002/biot.200600213. [DOI] [PubMed] [Google Scholar]
- 22.Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS. A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet. 2005;37:549–54. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
- 23.Huang J, Wei W, Zhang J, Liu G, Bignell GR, Stratton MR, et al. Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum Genomics. 2004;1:287–99. doi: 10.1186/1479-7364-1-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamy P, Andersen CL, Dyrskjot L, Torring N, Wiuf C. A Hidden Markov Model to estimate population mixture and allelic copy-numbers in cancers using Affymetrix SNP arrays. BMC Bioinformatics. 2007;8:434. doi: 10.1186/1471-2105-8-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Staaf J, Lindgren D, Vallon-Christersson J, Isaksson A, Goransson H, Juliusson G, et al. Segmentation-based detection of allelic imbalance and loss-of-heterozygosity in cancer cells using whole genome SNP arrays. Genome Biol. 2008;9:R136. doi: 10.1186/gb-2008-9-9-r136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinrichs S, Li C, Look AT. SNP array analysis in hematologic malignancies: avoiding false discoveries. Blood. 2010;115:4157–61. doi: 10.1182/blood-2009-11-203182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng HT, Peng ZH, Li S, He L. Loss of heterozygosity analyzed by single nucleotide polymorphism array in cancer. World J Gastroenterol. 2005;11:6740–4. doi: 10.3748/wjg.v11.i43.6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dumur CI, Dechsukhum C, Ware JL, Cofield SS, Best AM, Wilkinson DS, et al. Genome-wide detection of LOH in prostate cancer using human SNP microarray technology. Genomics. 2003;81:260–9. doi: 10.1016/s0888-7543(03)00020-x. [DOI] [PubMed] [Google Scholar]
- 29.Rook MS, Delach SM, Deyneko G, Worlock A, Wolfe JL. Whole genome amplification of DNA from laser capture-microdissected tissue for high-throughput single nucleotide polymorphism and short tandem repeat genotyping. Am J Pathol. 2004;164:23–33. doi: 10.1016/S0002-9440(10)63092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinard R, de Winter A, Sarkis GJ, Gerstein MB, Tartaro KR, Plant RN, et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 2006;7:216. doi: 10.1186/1471-2164-7-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez JR, Rodriguez-Santiago B, Caceres A, Pique-Regi R, Rothman N, Chanock SJ, et al. A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics. 2011;12:166. doi: 10.1186/1471-2105-12-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parisi F, Ariyan S, Narayan D, Bacchiocchi A, Hoyt K, Cheng E, et al. Detecting copy number status and uncovering subclonal markers in heterogeneous tumor biopsies. BMC Genomics. 2011;12:230. doi: 10.1186/1471-2164-12-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun W, Wright FA, Tang Z, Nordgard SH, Van Loo P, Yu T, et al. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic Acids Res. 2009;37:5365–77. doi: 10.1093/nar/gkp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Attiyeh EF, Diskin SJ, Attiyeh MA, Mosse YP, Hou C, Jackson EM, et al. Genomic copy number determination in cancer cells from single nucleotide polymorphism microarrays based on quantitative genotyping corrected for aneuploidy. Genome Res. 2009;19:276–83. doi: 10.1101/gr.075671.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Assie G, LaFramboise T, Platzer P, Bertherat J, Stratakis CA, Eng C. SNP arrays in heterogeneous tissue: highly accurate collection of both germline and somatic genetic information from unpaired single tumor samples. Am J Hum Genet. 2008;82:903–15. doi: 10.1016/j.ajhg.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamamoto G, Nannya Y, Kato M, Sanada M, Levine RL, Kawamata N, et al. Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays. Am J Hum Genet. 2007;81:114–26. doi: 10.1086/518809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–72. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 38.Partek. [cited; Available from: http://www.partek.com/Resources]
- 39.Garnis C, Davies JJ, Buys TP, Tsao MS, MacAulay C, Lam S, et al. Chromosome 5p aberrations are early events in lung cancer: implication of glial cell line-derived neurotrophic factor in disease progression. Oncogene. 2005;24:4806–12. doi: 10.1038/sj.onc.1208643. [DOI] [PubMed] [Google Scholar]
- 40.Popova T, Manie E, Stoppa-Lyonnet D, Rigaill G, Barillot E, Stern MH. Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome Biol. 2009;10:R128. doi: 10.1186/gb-2009-10-11-r128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chernikova SB, Razorenova OV, Higgins JP, Sishc BJ, Nicolau M, Dorth JA, et al. Deficiency in mammalian histone H2B ubiquitin ligase Bre1 (Rnf20/Rnf40) leads to replication stress and chromosomal instability. Cancer Res. 2012;72:2111–9. doi: 10.1158/0008-5472.CAN-11-2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shema E, Tirosh I, Aylon Y, Huang J, Ye C, Moskovits N, et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev. 2008;22:2664–76. doi: 10.1101/gad.1703008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Klumpp S, Amin HM, Liang H, Li J, Estrov Z, et al. SSBP2 is an in vivo tumor suppressor and regulator of LDB1 stability. Oncogene. 2010;29:3044–53. doi: 10.1038/onc.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang D, Tao J, Li L, Kedei N, Toth ZE, Czap A, et al. RasGRP3, a Ras activator, contributes to signaling and the tumorigenic phenotype in human melanoma. Oncogene. 2011;30:4590–600. doi: 10.1038/onc.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Workbench CG. [cited; Available from: http://cgwb.nci.nih.gov]
- 46.Auerbach O, Stout AP, Hammond EC, Garfinkel L. Changes in bronchial epithelium in relation to cigarette smoking and in relation to lung cancer. N Engl J Med. 1961;265:253–67. doi: 10.1056/NEJM196108102650601. [DOI] [PubMed] [Google Scholar]
- 47.Gomperts BN, Spira A, Massion PP, Walser TC, Wistuba, Minna JD, et al. Evolving concepts in lung carcinogenesis. Semin Respir Crit Care Med. 2011;32:32–43. doi: 10.1055/s-0031-1272867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loewe RP. Combinational usage of next generation sequencing and qPCR for the analysis of tumor samples. Methods. 2012;59:126–31. doi: 10.1016/j.ymeth.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 49.Curry JL, Torres-Cabala CA, Tetzlaff MT, Bowman C, Prieto VG. Molecular platforms utilized to detect BRAF V600E mutation in melanoma. Semin Cutan Med Surg. 2012;31:267–73. doi: 10.1016/j.sder.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 50.Buttitta F, Felicioni L, Del Grammastro M, Filice G, Di Lorito A, Malatesta S, et al. Effective assessment of egfr mutation status in bronchoalveolar lavage and pleural fluids by next-generation sequencing. Clin Cancer Res. 2012;19:691–8. doi: 10.1158/1078-0432.CCR-12-1958. [DOI] [PubMed] [Google Scholar]
- 51.Chiang DY, Getz G, Jaffe DB, O’Kelly MJ, Zhao X, Carter SL, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Magi A, Tattini L, Pippucci T, Torricelli F, Benelli M. Read count approach for DNA copy number variants detection. Bioinformatics. 2011;28:470–8. doi: 10.1093/bioinformatics/btr707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.