

Abstract
Epigenetic information is available from contemporary organisms, but is difficult to track back in evolutionary time. Here, we show that genome-wide epigenetic information can be gathered directly from next-generation sequence reads of DNA isolated from ancient remains. Using the genome sequence data generated from hair shafts of a 4000-yr-old Paleo-Eskimo belonging to the Saqqaq culture, we generate the first ancient nucleosome map coupled with a genome-wide survey of cytosine methylation levels. The validity of both nucleosome map and methylation levels were confirmed by the recovery of the expected signals at promoter regions, exon/intron boundaries, and CTCF sites. The top-scoring nucleosome calls revealed distinct DNA positioning biases, attesting to nucleotide-level accuracy. The ancient methylation levels exhibited high conservation over time, clustering closely with modern hair tissues. Using ancient methylation information, we estimated the age at death of the Saqqaq individual and illustrate how epigenetic information can be used to infer ancient gene expression. Similar epigenetic signatures were found in other fossil material, such as 110,000- to 130,000-yr-old bones, supporting the contention that ancient epigenomic information can be reconstructed from a deep past. Our findings lay the foundation for extracting epigenomic information from ancient samples, allowing shifts in epialleles to be tracked through evolutionary time, as well as providing an original window into modern epigenomics.
Ancient DNA research started in the mid-1980s with the successful cloning and sequencing of a short mitochondrial DNA fragment from the quagga zebra, a species that became extinct in the early 20th century (Higuchi et al. 1984). Soon after, the invention of PCR unlocked access to this fragmented and degraded DNA material (Pääbo 1989), making it possible to amplify short gene markers of interest and compare their sequence to that from extant organisms. This illuminated a range of topics ranging from the reconstruction of the evolutionary origins of several now-extinct iconic mammals (Orlando et al. 2003; Krause et al. 2006), the evaluation of the possible role played by major past climatic changes in driving mega-fauna extinctions (Shapiro et al. 2004; Campos et al. 2010; Lorenzen et al. 2011), to the identification of the pathogens responsible for massive historical outbreaks (Taubenberger et al. 1997).
However, before the advent of next-generation sequencing (NGS) platforms, the amount of ancient sequence information one had access to was limited to several tens of thousands of nucleotides at best (Noonan et al. 2005, 2006), and until very recently, sequencing whole ancient mitochondrial genomes was considered a major achievement (Cooper et al. 2001; Krause et al. 2006). Parallel sequencing of millions to billions of short DNA fragments has revolutionized ancient DNA research, and today a series of ancient genomes has been reconstructed from humans (Rasmussen et al. 2010, 2011; Keller et al. 2012; Raghavan et al. 2013), archaic hominins (Green et al. 2010; Reich et al. 2010; Meyer et al. 2012), the woolly mammoth (Miller et al. 2008), and several microbial pathogens (Bos et al. 2011; Martin et al. 2013; Schuenemann et al. 2013; Yoshida et al. 2013). Those mainly date back to recent historical periods or the Late Pleistocene, but most recently, the characterization of a 560,000- to 780,000-yr-old horse draft genome revealed that genomic information could be retrieved over much longer evolutionary time scales, probably up until the last million years (Orlando et al. 2013).
Ancient genomes have provided important new insights into human evolution and dispersals (Rasmussen et al. 2010, 2011; Keller et al. 2012; Raghavan et al. 2013), revealing an admixture between contemporary human ancestors and archaic hominins (Green et al. 2010; Reich et al. 2010; Meyer et al. 2012) and multiple early human expansions into both Asia and North America (Rasmussen et al. 2010, 2011). The information gained from these samples has largely been limited to nucleotide polymorphisms. Unlike mutations, epigenetic modifications do not alter the underlying DNA sequence, but can be inherited across cell divisions and from parents to offspring and can control gene expression by reshaping cytosine methylation landscapes, nucleosome organization, and histone modification patterns. The range of biological processes that depend on some level of epigenetic regulation is diverse and includes imprinting (Bird 2002), transposition (Hollister and Gaut 2009), cell differentiation (Meissner et al. 2008), and cancer (Teschendorff et al. 2011). In this study, we use the Saqqaq genome that was retrieved from an ∼4000-yr-old tuft of hair of a Paleo-Eskimo from Greenland and sequenced to an average depth of 20× (Rasmussen et al. 2010). We demonstrate that NGS data can be used in the absence of bisulfite or further experimental treatment to directly infer genome-wide nucleosome organization and regional methylation levels, thereby providing the first survey of an ancient epigenome.
Results
Nucleosome occupancy signal
A striking variation in read depth is apparent in the sequence data that underlies the Saqqaq genome. This variation correlates with functional regions, ranging from below genomic average (GA) in intergenic regions (0.9 × GA) to far above the average in coding regions (2.8 × GA) and 5′ UTRs (4.0 × GA) (Fig. 1A; Supplemental Table S2.1). Strikingly, CpG islands (CGIs) stand out genome-wide as highly defined regions with extreme read depth (6.5 × GA) (Fig. 1A). Read depth also varies dramatically on a local scale, showing a strong tendency to peak in regions of defined width (∼200 bp) and at regular intervals (Fig. 1B,C). We hypothesized that instead of resulting from alignment or sequencing artifacts, these patterns could stem from protection of DNA by nucleosome binding, with preferential degradation of linker regions between nucleosomes (Fig. 1D) either by DNases that enter the nucleus during cell death or by post-mortem strand breaks (Nagata et al. 1998). Such cleavage patterns are one of the hallmarks of apoptosis, which happens during the final stages of hair formation (Botchkavera et al. 2006), and are exploited in standard micrococcal nuclease (MNase) assays for mapping nucleosome location (Schones et al. 2008; Gaffney et al. 2012). In this scenario, the observed read depth would reflect nucleosome occupancy.
Figure 1.
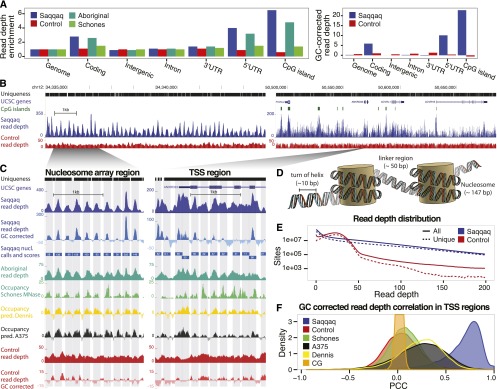
Paleo-Eskimo read depth reflects nucleosome occupancy. (A, left) Regional variation in read depth relative to genomic average (enrichment) for Saqqaq, Control, Aboriginal, and an experimental occupancy map (‘Schones’) (Schones et al. 2008). (Right) Saqqaq and Control regional read-depth variation after GC-correction. (B) Read-depth variation in a centromeric region known to harbor a 200-kb array of well-positioned nucleosomes (Gaffney et al. 2012) (left) and a region with genes (right). CpG islands (green bars) correlate with elevated read depth in the Saqqaq. The variation is also observed in genomically unique regions (black bars), where reads down to length 25 can map. The read depth of the Control exhibits lower variance. (C) Examples of Saqqaq read-depth variation, GC corrected read-depth variation, Saqqaq nucleosome predictions, and experimental (Schones, from CD4+ cells) as well as computational (Dennis and A375) (Dennis et al. 2007; Ozsolak et al. 2007) occupancy maps in ∼2-kb regions of the nucleosome array (left) and a transcription start site (TSS) region (right). Light gray denotes the 147-bp-long nucleosome predictions. Saqqaq read depth correlates with both the read depth of the ancient Aboriginal genome and the occupancy maps, but not with the Control. (D) DNA packaged around nucleosomes. We hypothesize that DNA wound around nucleosomes to be better protected from degradation. (E) The Saqqaq shows more variation in read depth than Control, with more genomic sites showing extremely low or high read depth. (F) Distribution of correlations for Saqqaq versus other sets across all promoter regions.
To rule out mapping biases, potentially exacerbated by the short read lengths of ancient DNA (aDNA), we simulated a control data set (‘Control’) with the same number of reads and the same read-length distribution as the Saqqaq genome data set (‘Saqqaq’). Control reads were randomly sampled and truncated to match Saqqaq read lengths from a panel of sequencing runs from modern human genomes based on lymphoblastoid cell lines of the Human Genome Diversity Project (HGDP) (Green et al. 2010; Reich et al. 2010; Supplemental Material SI2.2). This control displayed less variation in read depth (Fig. 1A–C), with fewer sites showing extreme values and with an overall distribution very different from that of Saqqaq (Fig. 1E). When restricting the comparison to unique regions of the genome, unaffected by short-read mappability issues (Supplemental Material SI2.2), the difference becomes even more pronounced, with Saqqaq showing much greater read-depth variation than Control (Saqqaq SD = 32.1; Control SD = 9.1).
The sequencing reaction and fragment length-dependent GC biases introduced while amplifying NGS libraries (Dabney and Meyer 2012) could also potentially be responsible for the observed variations in read depth. We corrected for this second source of bias by making use of pre-existing methods that are proficient in accounting for base compositional and mappability biases (Benjamini and Speed 2012). As anticipated, the GC-corrected read depth correlates strongly with the original read depth (unique regions: Pearson correlation coefficient, PCC = 0.47; P < 1 × 10−16) (see Supplemental Material S2.3 for conservative P-value estimation) and is uncorrelated with GC content (PCC = 0.003; P < 0.75). Even though the level of read-depth variation decreased slightly for both Saqqaq and Control after GC-correction (unique regions: Saqqaq SD = 14.4; Control SD = 5.4), both the regional and the local-scale variation remained (Fig. 1A,C). This suggests that sequencing bias could explain part, but not all, of the original read-depth variation, in agreement with our hypothesis of nucleosome protection. All following analyses are based on the GC-corrected read depth unless otherwise noted.
As neither mapping nor sequencing biases could account for the observed patterns, we proceeded to compare read-depth variations to existing nucleosome occupancy maps (Supplemental Material SI2.3). We first evaluated the correlation coefficients between data sets across the unique regions of a 20-kb subsection of a known nucleosome array region (Fig. 1B; Supplemental Fig. S2.1a), where nucleosomes are thought to be consistently and specifically positioned independently of tissue type (Gaffney et al. 2012). Saqqaq correlates positively with both computational predictions (PCC = 0.47 and PCC = 0.43; P < 2 × 10−4) (Dennis et al. 2007; Ozsolak et al. 2007) and experimental MNase-based maps (PCC = 0.23 and PCC = 0.43; P < 3 × 10−3) (Schones et al. 2008, Gaffney et al. 2012). The two MNase-based maps show comparable levels of correlation between them (PCC = 0.38; P < 1 × 10−4), despite being based on related cell types (CD4+ and lymphoblastoid cells), and slightly lower correlations against the computational maps (PCC from 0.16 to 0.28; P < 7 × 10−2). These relatively low levels of correlation for the same experimental technique across well-positioned nucleosomes could suggest some level of noise in the state-of-the-art MNase-based occupancy maps, potentially from the cutting biases of the MNase enzyme (Gaffney et al. 2012). The uncorrected Saqqaq read depth correlates equally well with the experimental data sets and at even higher levels with the computational predictions (PCC = 0.77 and PCC = 0.74; P < 1 × 10−16).
Transcription start sites (TSSs), generally impose stronger positioning of nucleosomes in their vicinity, though the positioning depends on expression level and tissue type (Valouev et al. 2011). Across these regions, Saqqaq correlates less strongly, albeit still positively, with both the computational predictions (median PCC of 0.25; P < 1 × 10−16) and to the experimental maps (median PCC of 0.02 and 0.07; P < 1 × 10−16). In contrast, the two MNase-based maps show a comparable level of correlation to the nucleosome array regions (PCC = 0.35; P < 1 × 10−16) (Supplemental Fig. S2.1c), consistent with the similarity of their originating cells. Again, the uncorrected Saqqaq read depth correlates more strongly to both the computational predictions (median PCC = 0.79 and PCC = 0.76; P < 1 × 10−16) (Supplemental Fig. S2.1b) as well as to the MNase-based maps (median PCC = 0.17 and 0.06; P < 1 × 10−16), which is not unexpected given the importance of GC-rich sequence signals in determining nucleosome positions and the absence of GC-correction for the MNase-based data sets (Collings et al. 2010; Valouev et al. 2011).
At actively transcribed genes, the region upstream of the TSS is depleted to facilitate access to the transcriptional machinery, while the downstream nucleosomes are strongly phased with high occupancy at the +1 position (Schones et al. 2008; Valouev et al. 2011). The read-depth profile across TSS regions closely recapitulated this pattern (Fig. 2A). Consistently, the accumulated read depth also matched the known occupancy patterns for splice sites (Supplemental Fig. S2.2), in agreement with our nucleosome protection hypothesis.
Figure 2.

Read depth and fragment length periodicity. (A) Read-depth variation at TSS. Spectrogram around TSS (top) showing the strength of the periodicity signal at different wavelengths. Nucleosome abundance (bottom) summed over aligned transcription start sites. High occupancy at the +1 nucleosome position is characteristic of transcriptional activity. (B) Spectral density (periodogram) for TSS regions. The frequency spectrum shows a peak in relative signal at 193 bp corresponding to the expected inter-nucleosome distance. (C) 5′ read-end phasograms showing the distribution of distances between reads in gene bodies. A clear ∼200-bp periodicity is apparent, consistent with the presence of nucleosomes (right). A short-range periodicity of ∼10 bp is also apparent (left), corresponding to a turn of the DNA helix as it winds around the nucleosome. (D) Distribution of fragment sizes from ancient samples of horse (top), polar bear (middle), and Saqqaq (bottom) are consistent with preferential cleavage of exposed nucleosome-wrapped DNA strands every 10 bp.
We subsequently tested whether we could find evidence in the Saqqaq data set for the ∼200-bp read-depth periodicity characteristic of nucleosome protection (147 bp) and linker region (50 bp) cleavage (Fig. 1D). Short-time Fourier transformation revealed a strong signal at 200 bp at TSS regions, where nucleosomes are strongly phased, and downstream (Fig. 2A). Using Fourier transform periodograms (Welch method), we found the overall peak periodicity to be 193 bp for TSS regions (Fig. 2B). Similarly, strong signals around 200 bp were also observed for other regions, including CpG islands, gene bodies, and sequences known to be bound by CTCF (Supplemental Figs. S2.3–S2.6). Nucleosome protection and extensive cleavage of linker regions would also predict NGS reads to predominantly start at the edge of nucleosomes. This bias should result in a periodicity of 5′ read starts proportional to the nucleosome inter-distance. Phasograms, illustrating the distance between 5′ read starts (Valouev et al. 2011), revealed the presence of periodicity in gene bodies at the expected length of ∼200 bp (Fig. 2C; Supplemental Fig. S2.8).
Interestingly, phasograms also revealed a short-range periodicity of 10 bp, coinciding with the length of a turn of the DNA helix. This could reflect preferential shifts in nucleosome positioning every 10 bp (Brogaard et al. 2012) and/or preferential cleavage of the DNA backbone facing away from nucleosome protection. The size distribution of full-length Saqqaq aDNA fragments (Supplemental Material SI1.7), which shows three peaks at 38, 48, and 58 bp (Fig. 2D; Supplemental Fig. S1.1) and an inter-peak distance mirroring both the phasograms and the length of one DNA helix turn, could be indicative of preferential cleavage of the DNA backbone facing away from nucleosome protection. However, size distribution profiles could also be affected by other factors, in particular, the gel size selection performed on libraries post-amplification. We therefore investigated the fragment length distribution of libraries prepared without gel selection; first, from a permafrost-preserved 4400-yr-old horse bone and second, from a much older (110,000- to 130,000-yr-old) polar bear bone (Fig. 2D; Supplemental Material SI1.7; Miller et al. 2012). This revealed a striking pattern, with fragment abundance increasing by up to a factor of two across periods of 10.3 bp until fragment sizes compatible with nucleosomal length (147 bp) are reached. This ruled out size selection as a possible driving factor and supported protection of DNA strands facing the nucleosome. It also suggests that nucleosome protection footprints could be present in tissues other than hair and survive over extremely long periods. Interestingly, the ∼10-bp periodicity was also observed in modern hair but not in modern blood sequencing data (Supplemental Fig. S1.1). This is in line with apoptosis taking part in the hair differentiation process (Botchkavera et al. 2006) and the known short-range size periodicity in DNA fragmentation following apoptosis (Aruscavage et al. 2010).
Finally, we asked whether the read-depth signal was consistent between ancient hair samples by taking advantage of the ∼100-yr-old Aboriginal Australian genome sequence data set (Rasmussen et al. 2011). The Aboriginal read depth clearly showed the same periodic pattern of variation as Saqqaq (Fig. 1C), with a strikingly high degree of correlation in unique regions unaffected by mapping issues (PCC = 0.87; P < 1 × 10−16), even though a different procedure was used for preparing DNA libraries (Supplemental Material SI1.3). Consistent regional variation and a 200-bp periodic signal were also observed in DNA reads from modern hair (Supplemental Table S2.1; Supplemental Fig. S2.7), suggesting again that DNase-dependent cleavage during apoptotic processes inherent to hair differentiation, rather than post-mortem degradation, drives the nucleosomal protection pattern observed in ancient samples. The fragment-length signal in the bone-derived samples from horse and polar bear, however, shows that some nucleosome protection signal may also stem from necrotic tissues, in line with previous findings (Dong et al. 1997).
Nucleosome map and positioning patterns
As the variations in read depth observed in the Saqqaq sequence data could not be explained by experimental artifacts or alignment biases, but matched with all patterns reflecting nucleosome protection, we defined a genome-wide human nucleosome map for the Saqqaq based on GC-corrected read-depth variations. Peaks of the GC-corrected read depth were called as nucleosome centers when showing a maximal positive value within a nucleosome-wide window (147 bp). Each nucleosome call was scored by the difference in read depth between the peak and flanking (linker) regions in order to capture information about both occupancy (peak read depth) and positioning (depletion in linker regions) (Fig. 3A; Supplemental Material SI2.7). We then compared the Saqqaq scores to the Control scores and estimated the false discovery rate (FDR) by assuming that calls in the Control are all false (Fig. 3B; Supplemental Material SI2.8). We found that the 25% top-scoring calls (2.66M; score ≥ 22.5), spanning 13% of the genome, have an FDR of 5% (Supplemental Fig. S2.10).
Figure 3.
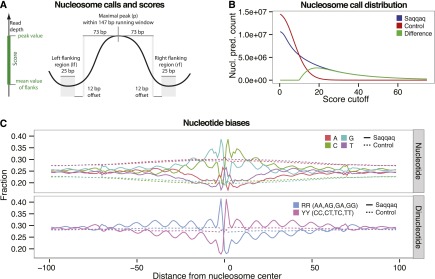
Nucleosome calls and positioning patterns. (A) Nucleosome center positions (dyads) are called as read-depth peaks if maximal at the center of a running window of nucleosome length 147 bp. Calls are scored by the difference in read depth between the peak (p) and the average read depth of the left (lf) and right (rf) flanking regions [score = p − (lf + rf)/2]. (B) Nucleosome call abundance is shown as a function of quality score cutoff for the Saqqaq (blue) and the Control (red), which lacks the nucleosome signal. The difference (green) gives the expected number of true positive calls at a given score cutoff and, indirectly, the FDR (<1% for the 1.9M calls with a score cutoff >29). (C) Base composition and distribution of purine/pyrimidine sequence dimers across the top 25% of called nucleosomes.
DNA sequence determinants of nucleosome positioning are well-described in model organisms (Collings et al. 2010; Ioschikhes et al. 2011; Brogaard et al. 2012; Struhl and Segal 2013) such as yeast and Caenorhabditis elegans, but are only beginning to be deciphered in human (Kogan et al. 2006; Valouev et al. 2011; Gaffney et al. 2012). We took advantage of our Saqqaq nucleosome map to gain insights into the sequence patterns that drive nucleosome positioning in the human genome (Supplemental Material SI2.9). We first looked at the single nucleotide distribution across the top quartile of nucleosome calls in unique regions (n = 1.37M, FDR = 8.5%). Base frequencies show the expected 10.2-bp periodicity corresponding to the turn of the slightly stretched DNA helix around nucleosomes (Brogaard et al. 2012), with phases offset and pairwise opposite of each other (C vs. A and G vs. T) (Fig. 3C; Supplemental Fig. S2.11). This pattern is reverse-complemented and symmetric across the center position, which is located at the nucleosome dyad, as double-stranded DNA can match in either direction. Strikingly, the G and C frequencies vary dramatically across the center, with a spike of 35% G at position −2 (+2 for C) and a drop to 14% at position +1 (−1 for C). Such localized changes in nucleotide frequencies are only possible if the nucleosome sequences are accurately aligned, suggesting nucleotide-level precision for a large fraction of the calls, as recently obtained for yeast (Brogaard et al. 2012).
We next focused on the distribution of dinucleotides, which is known to influence nucleosome positioning (Ioschikhes et al. 2011; Struhl and Segal 2013). Interestingly, the characteristic strong/weak dinucleotide pattern found in yeast (Segal et al. 2006) is absent in Saqqaq nucleosome calls (Supplemental Fig. S2.12), in line with recent human studies (Ioschikhes et al. 2011; Valouev et al. 2011; Gaffney et al. 2012). Instead, we detect dramatic 10-bp periodic variations in pyrimidine vs. purine dinucleotide frequencies, with increasing amplitudes toward the center, where they are reverse-complemented (Fig. 3C). This pattern has previously been observed with a limited human nucleosome sequence set (Kogan et al. 2006), and is confirmed here with unprecedented precision and at the genome-wide scale.
These patterns are also recovered when analyzing nucleosome calls based on the uncorrected Saqqaq read depth, in which case the GC enrichment at the nucleosome center becomes more pronounced (Supplemental Material SI2.9; Supplemental Fig. S2.13). They likely reflect the thermodynamically optimal packaging of DNA around nucleosomes and thereby participate in defining the equilibrium location of the nucleosome (Ioschikhes et al. 2011). Sliding DNA away from the center would have accumulated effects across the nucleosome on the match to (1) the strong positional preferences near the dyad, (2) the 10-bp periodic variation in dinucleotide frequencies (Struhl and Segal 2013), (3) the increasing amplitude of this variation, and (4) the highly strand-specific signal, which is reverse-complemented at the center position. Additionally, the sequence patterns observed here predict that nucleosomes should be repelled from linker regions, where strand specificity must switch again when approaching the next nucleosome. We conclude that the combined effect of these individually weak compositional biases may determine a precise, thermodynamically optimal positioning of individual nucleosomes.
Cytosine methylation signal
We next focused on detecting cytosine methylation at CpG dinucleotides, as experimental evidence supports the long-term survival of methylated cytosines (5mC) in aDNA fragments (Briggs et al. 2010; Llamas et al. 2012). Genome-wide methylation maps of the human genome have already been reconstructed using high-throughput sequencing in combination with a variety of approaches. In the case of bisulfite sequencing (Krueger et al. 2012), unmethylated cytosines are chemically converted into uracils, generating cytosine to thymine (C→T) mutations that can be located in the human genome at the single-nucleotide level.
Similar chemical conversions are known to occur naturally post-mortem, through the hydrolytic deamination of cytosines into uracils (Hofreiter et al. 2001). Some Taq DNA polymerases such as Platinum Taq DNA Polymerase High Fidelity (HiFi) can replicate through uracils, copying them as native thymine residues and thereby introducing C→T misincorporations in the pool of molecules amplified. Such misincorporation rates increase toward sequence starts where deamination rates are inflated due to the presence of single-stranded overhangs (Briggs et al. 2007). However, with Pfu DNA polymerases that cannot bypass uracils (e.g., Phusion), such misincorporations should vanish (Rasmussen et al. 2010) except at 5mC sites where post-mortem deamination transforms 5mC residues into thymines, which represents a native template for all DNA polymerases. We therefore hypothesized that C→T misincorporation events observed in the Saqqaq sequence reads generated following library amplification with Phusion could be used to track ancient 5mC residues and reveal genome-wide information about ancient DNA methylation levels (Supplemental Fig. S3.1; Supplemental Material SI3.1).
We first tested this prediction by analyzing mismatch patterns in reads starting at CpGs, which are the main targets of methylation in mammals (Fig. 4A; Lister and Ecker 2009). Focusing on the first position where deamination rates are maximal (Briggs et al. 2007), we observed a 5.04-fold increase in C→T errors for Phusion reads starting at CpG. Reads sequenced with HiFi also showed an increase, albeit smaller (1.74-fold), suggesting a higher fidelity for T than U. This pattern was absent (1) for Phusion reads starting with CpA, CpT, and CpC (Fig. 4B), (2) at inner sequence positions known to be affected by lower post-mortem deamination rates (Supplemental Fig. S3.2), and (3) among Phusion mitochondrial reads in agreement with the absence or low levels of methylation present in this genome (Rebelo et al. 2009).
Figure 4.

Substitution rates at CpG reveal methylation of DNA. (A) C→T mismatch rates (gray) versus rate of other mismatches (black) between a random subset of 1,000,000 Phusion (left) or HiFi (right) reads mapping uniquely. Reads are split by those starting with CpG (top; 26,864 Phusion and 25,568 HiFi reads) and other dinucleotides (bottom). (B) Mismatch frequencies for Phusion (left) and HiFi (right) for reads aligned to various genomic locations starting with the dinucleotides: CpG (top) and Cp[ACT] (bottom). (C) Distribution of Ms values for three classes of promoters with low, medium, and high CpG densities (Supplemental Material SI3.2). (D) Methylation profile (Ms, top) and read depth variation (bottom) at CTCF regions. Read depth provides a proxy for nucleosome occupancy. (E) Distribution of Ms values across nucleotide positions covered with nucleosomes, showing a depletion in methylation levels within a core region (20 nt before and after the nucleosome center) that is particularly marked at the nucleosome center.
We next investigated whether C→T mismatch rates were lower in known hypomethylated regions than in hypermethylated regions. CGIs are well-characterized hypomethylated regions (Deaton and Bird 2011). We therefore compared CpG→TpG rates within and outside CGIs (Illingworth et al. 2010). Introns and exons residing outside CGIs (Fig. 4B) showed higher CpG→TpG mismatch frequencies (4.8% and 5.1%) than both CGIs and the genomic average (1.8% and 4.2%), supporting methylation as the source of the elevated mismatch frequencies observed at CpG sites in the Saqqaq Phusion sequence data.
Our ability to detect cytosine methylation depends on the extent of post-mortem deamination rates and ultimately on sequencing depth. The 5mCpG sites that did not experience a post-mortem deamination event are copied as regular CpGs, and therefore do not leave any methylation footprint in the Phusion sequence data set. Consequently, the absence of CpG→TpG mismatches at a given location of the Saqqaq genome cannot be taken as proof that this locus was devoid of methylation. This could only be demonstrated with extensive depth-of-coverage, as in this situation the chances that at least one of the templates sequenced was deaminated are increased. An easy way to increase sequence coverage is to go beyond the level of single dinucleotides and record CpG→TpG mismatches within a full genomic region including multiple CpG sites. Similarly, since post-mortem cytosine deamination rates decline rapidly from sequencing ends (Briggs et al. 2010), focusing on read starts should increase our ability to capture deamination events in the pool of molecules sequenced, thus, to detect CpG→TpG mismatches.
We therefore defined a measure of regional methylation level based on the average CpG→TpG mismatch frequencies observed within a given region at read starts (Ms) (Supplemental Material SI3.2; Supplemental Fig. S3.3) and applied this to a series of genomic regions in order to demonstrate its ability to capture genuine methylation information (Supplemental Material SI3.3). CpG→TpG conversions at read ends and other positions within reads were disregarded given (1) the drop in sequence quality toward read ends, (2) the drop in post-mortem cytosine deamination from read starts, and (3) the presence of a significant fraction of inserts not sequenced over their full length. Interestingly, at promoter regions, we recovered the previously observed negative correlation between CpG site density and methylation levels (Fig. 4C; Ball et al. 2009). Ms also reproduced the expected methylation pattern at CGI promoters showing significant strand asymmetry in the distribution of guanines and cytosines (Ginno et al. 2012), with increasing methylation levels from the TSS toward the 2 kb located upstream and downstream (Supplemental Fig. S3.4). In addition, we found a 7.4-fold reduction in Ms at CGIs predicted to be undermethylated compared to ubiquitously methylated CGIs (Supplemental Fig. S3.5; Straussman et al. 2009). We also observed the expected reduction in methylation levels between exons and introns across splice sites (Supplemental Fig. S2.2; Laurent et al. 2010), with 5′ splice sites showing average methylation levels higher than at 3′ splice sites. Finally, we compared our methylation estimates to experimental methylation measurements gathered across a variety of modern individuals and somatic tissues (Supplemental Material SI3.4; Slieker et al. 2013). For all tissues and individuals investigated, we found significant and high correlations between normalized modern methylation levels and regional Ms values for regions spanning 750 bp or 1000 bp around the CpGs from the Illumina 450K array (Supplemental Table S3.1, S3.2). Adjusted R-squareds were, however, found maximal with hair methylation levels (adjusted R-squared = 0.620–0.785 depending on the coverage threshold considered). Selecting CpGs from the Illumina 450K array with at least a twofold average difference in methylation levels between hair, blood, buccal, and saliva, we found greater Ms values for Saqqaq at CpGs showing higher average methylation in modern hairs and lower Ms values at CpGs showing higher methylation in other tissues (Supplemental Figs. S3.6, S3.7). Finally, we used this set of CpG and Ms values calculated on the Saqqaq data to perform unsupervised hierarchical clustering based on individual methylation profiles and found that the Saqqaq grouped together with modern hair tissues (Fig. 5; Supplemental Figs. S3.9–S3.13). This holds true regardless of the coverage threshold implemented during data filtering, suggesting that our approach is largely robust to regional variation in sequence coverage (Supplemental Material SI3.4). Overall, this supports the validity of Ms and the methylation information recovered from the Saqqaq Phusion sequence data.
Figure 5.

Unsupervised hierarchical clustering of tissue methylation profiles. Ms-based methylation levels of the Saqqaq individual are compared to the methylation profiles of five modern donors (PT1, PT2, PT3, PT4, and PT5) across four tissues (blood, buccal, saliva, and hair). Ms calculations were based on 2000-bp-wide genomic regions centered on each locus from the Illumina 450k array, disregarding those that showed less than 100 CpG sites at read starts (Supplemental Material SI3.4). The final set includes a total number of 7383 CpG sites.
Correlation of nucleosome and methylation signals
We next compared our nucleosome calls and methylation proxy at CTCF binding sites. CTCF-bound sites provide anchor points for arrays of well-positioned nucleosomes stretching over ∼4 kb in the human genome (Fu et al. 2008) and play a key role in the regulation of gene expression (Bell et al. 2001) and in shaping the 3D structure of chromosomes (Handoko et al. 2011). Importantly, our map displays the characteristics of in vivo nucleosome positioning around CTCF sites (Valouev et al. 2011), indicating that nucleosomes do not revert to a more in vitro–like positioning post-mortem (Fig. 4D). Welch's FFT analysis (Supplemental Fig. S2.5) showed a sharp 182-bp spacing signal in CTCF flanking regions, consistent with the ∼185-bp spacing reported by Fu and colleagues (Fu et al. 2008). Strikingly, we found nucleosome calls out-of-phase with Ms (Fig. 4D), in agreement with recent surveys reporting that nucleosome occupancy and accessibility to GpC methyltransferase are anti-correlated in human IMR90 cells (Kelly et al. 2012). Significantly negative correlations were found following Ms calculation on sequence data sets down-sampled to even depth, suggesting that this analysis was not affected by the greater power achieved to detect methylation at positions with greater read depth (Supplemental Table S3.7). Minimal Ms values at anchor points (<0.024) provided a direct measure of 63 bp for the footprint of the insulator CTCF protein that cannot bind to methylated DNA. This is consistent with the range of 32–64 nt recovered from direct DNAse treatment (Fu et al. 2008). Our findings add hair shafts to the empirical evidence available for characterizing the features of chromatin organization at CTCF flanking regions that is so far limited to a number of cell lines in humans.
Based on the strong consistency between our nucleosome and methylation data at CTCF regions, we decided to survey methylation variations across nucleosomes interspersed across the whole genome. We evaluated Ms positionally across our top quartile of nucleosome calls from unique regions. Methylation levels were found to vary dramatically across nucleosomes, with a depletion (position −20 to +20) and a sharp drop at the center position (Fig. 4E). Intriguingly, the zone depleted for methylation is enriched in CpG dinucleotides (and strong dinucleotides in general, CC, CG, GC, GG; position −30 to +30), suggesting that the presence of nucleosomes protects DNA from methylation in vivo, in agreement with recent experimental results in HeLa cells (Felle et al. 2011). Additionally, the drop in methylation levels could ease steric constraints while wrapping DNA around the nucleosome, consistent with the known reduction in CpG deformability following methylation (Perez et al. 2012) and the relatively increased difficulty to assemble nucleosomes in vitro on methylated templates compared to methylation-free templates (Buttinelli et al. 1998).
Gene expression inference
Since gene expression is influenced by epigenetic marks, we reasoned that our data could indirectly reveal information about transcriptional regulation in ancient cells. DNA methylation is often, but not always, associated with gene silencing. Methylation at the first exon within gene bodies hinders further elongation by the transcriptional machinery and is tightly linked to gene expression down-regulation (Brenet et al. 2011). We therefore ranked all hg18 gene annotations showing sufficient sequence coverage (Supplemental Material SI3.5) according to Ms values at the first exon (Supplemental Table S3.6). This provided a list of candidate accessions with low, if any, expression levels. Following Ball et al. (2009), we further calculated the ratio Rs of gene body to promoter methylation as a proxy for gene expression, with low (high) Rs values indicating low (high) expression levels. The vast majority of genes showing highest Ms values at the first exon was found in the first quartile of Rs values (100.0% for the genes with top-99% Ms values at the first exon, and 88.3% for top-95%) (Supplemental Material SI4), suggesting strong consistency across both methylation-based expression proxies. We next recovered gene accessions for a set of proteins known to be expressed in hair shafts (Lee et al. 2006) and found that they represented a group of transcripts with greater Rs values than the overall distribution of all genes annotated (Kolmogorov-Smirnov test, P-value = 0.00152) (Supplemental Material SI4). This is in line with the latter being a mixture of expressed and silenced genes.
High Rs values predicted expression for a range of keratin transcripts, including keratins 71 and 85 (Supplemental Tables S4.1–S4.3). Keratins 71 and 85 are known components of the inner root sheath of hair follicles and hair shaft cortex and medulla, respectively (Moll et al. 2008; Langbein et al. 2010). We also found low Rs values, suggesting low expression, if any, for nonhair-specific keratins, such as keratin 79 (Supplemental Tables S4.1–S4.2). Moderate to high Rs values also confirmed the presence of a number of proteins involved in cellular adhesion and cytoskeleton organization, including plakophilin 1, plakophilin 3, desmoplakin, periplakin, and plectin, in agreement with the importance of desmosomes, hemi-desmosomes, and/or adherens junctions in hair biology (Bazzi et al. 2009). We also predicted high levels of trichohyalin (TCHH), a protein known to confer mechanical strength to the hair follicle inner root sheath. Overall, our Rs predictions are in line with the biology of hair formation.
As a further validation of our expression predictions, we used expression data from modern hair follicles (Supplemental Material SI4) to rank genes into 10 groups of increasing expression, looking for correlation with Rs. We found a significant positive correlation with the expression groups, indicating that our approach can provide information about the transcriptional state of ancient cells (Fig. 6A). We then selected two additional measures known to correlate with expression, and calculated those using the Saqqaq data: (1) the presence of a strongly positioned +1 nucleosome (Fig. 2A; Valouev et al. 2011); and (2) the level of downstream regularly spaced phasing of nucleosomes (Schones et al. 2008). Those measures also showed significant positive correlation with the expression groups (Supplemental Material SI4; Fig. 6B,C). When extending to a more fine-grained division of expression, a weaker, albeit more significant correlation was observed, as both the measure and ranking become more sensitive to noise. We further selected the top 1000 genes showing maximal Rs values and the top 1000 genes showing minimal Rs values as candidates of genes with high and low levels of expression in the Saqqaq hairs. Functional enrichment analyses (Huang et al. 2009) of those down-regulated candidates revealed chymotrypsin and trypsin-like enzymes, homeobox, and genes involved in signalization, cell adhesion, ionic channels, glycoproteins, muscle proteins, and proteins integral to plasma membrane. Up-regulated candidates were enriched in categories involving the ubiquitin ligase complex, phosphorus metabolic process, ligase, inorganic anion transport, and genes associated with metal-binding activities (Supplemental Material SI4).
Figure 6.
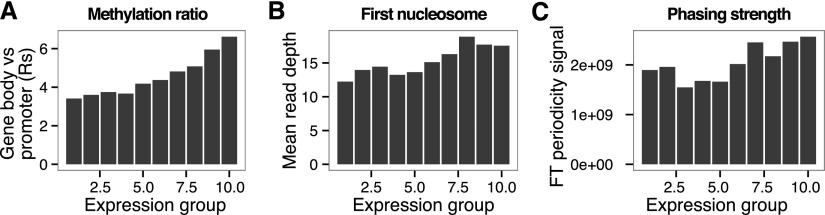
Nucleosome and methylation maps as proxies for ancient gene expression. Relationship between three measures assessing gene expression. (A) Methylation ratio (Rs), a measure of methylation in promoter versus gene bodies. (B) First nucleosome occupancy, average read depth over the TSS +1 nucleosome region. (C) Phasing strength, a measure of strength of the periodicity between neighboring nucleosomes across the TSS region by Fourier transform analysis. All display a significant correlation with expression as measured by microarrays in modern hair follicles.
Prediction of the age at death
Aging is increasingly recognized as a developmentally regulated program involving epigenetic modifications at different stages of our lives (Boyd-Kirkup et al. 2013). A number of CpG sites in the human genome have been shown to undergo age-associated changes in methylation (Alisch et al. 2012), and linear models relating age and methylation levels have even been described (Koch and Wagner 2011; Johansson et al. 2013). Such models provide a unique opportunity to predict the age of a given individual based on cytosine methylation levels, although with limited precision. We used the methylation information recovered from the Phusion sequence data at several CpG sites undergoing age-associated changes to propose an age at death for the Saqqaq individual. Following the framework presented by Koch and Wagner (2011), we focused on four particular CpGs for which age-methylation linear models have been established across a variety of tissues and cell types (Supplemental Material SI3.6). As the latter did not include hair samples, we first tested whether such models could accurately predict the age of five living donors based on their hair methylation levels (Slieker et al. 2013). Two CpGs (cg23571857 and cg25148589) showed large differences between predicted and real age (SD = 9.4 and 12.4 yr, respectively) and were disregarded (Supplemental Fig. S3.14). However, two other CpGs (cg07533148 and cg01530101) provided reliable age estimates, with differences between predicted and real ages within 1.7–12.4 yr, in agreement with the error margin originally reported for such approaches (Supplemental Fig. S3.14; Supplemental Material SI3.6; Koch and Wagner 2011). We therefore used those loci to predict the age at death of the Saqqaq individual. Estimating Ms for a 2000-bp-wide region centered on each CpG from the Illumina 450k array, we built a linear model relating Ms and the methylation levels measured in hairs of five modern donors (adjusted R-squared = 0.620–0.785) (Supplemental Table S3.1) in order to convert Ms into absolute methylation levels for Saqqaq at the two loci of interest (Supplemental Material SI3.6). Following Koch and Wagner (2011), the absolute levels were used to infer age. Both CpGs considered provided strikingly similar age estimates ranging from 44.1 to 69.3 yr and 52.1 to 64.1 yr, respectively (95% CI across all analyses) (Supplemental Fig. S3.15). Considering the prediction error measured in our five modern donors, this indicates that the Saqqaq individual was probably among the elderly when he died and was likely at least in his late thirties.
Discussion
Epigenetics complements genetics in determining the phenotypic state of cells and organisms (Bird 2002; Meissner et al. 2008; Hollister and Gaut 2009; Teschendorff et al. 2011). The extraction of epigenetic information from ancient samples can therefore both elucidate the ancient phenotypic state as well as the evolutionary changes of the epigenome. Yet, to our knowledge, ancient epigenomic information has not been extracted genome-wide previously, and our study is the first to report both nucleosome occupancy and methylation levels. The long-term survival of 5mC in ancient DNA has previously been reported using either sophisticated enzymatic reactions (Briggs et al. 2010) or bisulfite treatments (Llamas et al. 2012). In contrast, our approach does not require any extra treatment and simply relies on deep-sequencing following standard protocols. Taking advantage of a series of DNA degradation steps preceding cell death, it provides both genomic and epigenomic information from a single sequence library. Accurate estimates of regional levels of methylation from high depth (≥20×) whole-genome sequencing data of the 4000-yr-old Saqqaq individual (Rasmussen et al. 2010) was achieved despite low levels of cytosine deamination (Fig. 4A; Ginolhac et al. 2011). We anticipate that similar methylation profiles could be gathered at lower coverage in cases where DNA is affected by extensive post-mortem damage.
Importantly, our framework can also deliver methylation information in cases where other molecular tools than the Phusion are used. In particular, UNG-EndoVIII treatment of ancient DNA extracts prior to library construction has become a standard procedure for limiting the extent of artifactual mutations in final ancient genome assemblies (Briggs et al. 2010; Bos et al. 2011; Meyer et al. 2012; Schuenemann et al. 2013). This method shows great efficiency at eliminating post-mortem deamination by-products at regular cytosines (i.e., uracils) (Briggs et al. 2010) but cannot detect deaminated 5mC, as the latter are nothing but regular thymines (Supplemental Fig. S3.1). Therefore, similar to our use of Phusion, CpG→TpG misincorporations observed at read starts following UNG-EndoVIII treatment offer an opportunity to track ancient CpG methylation footprints. One proximate perspective from our work is, therefore, to apply our framework to the high-quality Neandertal and Denisovan genomes where such UNG-EndoVIII treatments have been used (Meyer et al. 2012) in order to contrast genome-wide ancient bone methylation profiles in archaic hominins and modern humans.
Our ancient nucleosome map is of similar accuracy to modern MNase-based maps, but is unlikely to be subject to the same set of sequence biases that result from MNase cutting preferences (Chung et al. 2010). In our case, the fragmentation happens post-mortem, likely by a combination of cleavage by endogenous DNases and spontaneous de-purination processes (Dong et al. 1997; Botchkavera et al. 2006; Briggs et al. 2007). It therefore offers a unique view of nucleosome occupancy. The distinct positioning patterns with strong nucleotide preferences for individual positions have not been reported genome-wide for humans before and show that a large fraction of the nucleosome calls are at nucleotide-level resolution. Their strand-specific, oscillating nature coupled with reverse complementation at the dyad show how optimal nucleosome positioning may be specified by the accumulated effect of individually weak compositional biases. In particular the strand-specific purine versus pyrimidine patterns, which are reverse-complemented at the dyad, would contribute strongly to this. Furthermore, the observed hypomethylation of DNA spanning nucleosome cores might also partake in nucleosome positioning by increasing DNA deformability (Buttinelli et al. 1998; Perez et al. 2012). All in all, our findings illustrate how aDNA can offer an original source of information to advance our understanding of nucleosome biology.
Our results show that both methylation tracts and nucleosome occupancy patterns can be preserved for significant time periods after death in both hairs and bones. How long such signals survive in a full range of environments and tissues remains to be determined; however, the sequence data underlying the ancient Aborigine genome demonstrate that genome-wide epigenetic information could be recovered after one century in warm environments (Rasmussen et al. 2011). This age limit is extended by at least three orders of magnitude, to over 100,000 yr, in cold environments, as revealed by the polar bear shotgun sequence data (Miller et al. 2012). This time range opens the possibility to track major epigenomic modifications at different time scales: over a few generations, by following major changes in diet and epidemics using medical archives, potentially including the extensive collections of formalin-fixed and paraffin-embedded biopsies (Kerick et al. 2011); but also over thousands of years by following epigenomic changes over long-term environmental changes such as those from the Last Glacial Maximum. Epigenomic analysis of ancient DNA therefore paves the way for charting shifts in the frequency of epialleles over time, providing a direct way of detecting epigenetic adaptations to environmental conditions in analogy to how positive selection is detected with genomic data. In addition to investigating the full spectrum of possible changes driving adaptation of human populations to their environment, these data will more generally contribute to evaluating the potential of epigenetic modifications, besides mutations, as a major evolutionary force.
Interestingly, the Saqqaq methylation profile was found to cluster together with modern hair shafts to the exclusion of other tissue types (Fig. 5; Supplemental Figs. S3.9–S3.13). Such information can be used to demonstrate the absence of major contamination from other sources, and therefore can prove essential for authenticating ancient human sequence data sets in a number of cases. One important authentication criterion when working on ancient humans is the ability to demonstrate that the ancient genetic signature does not match that of any of the coworkers, from field archeologists and anthropologists to molecular biologists performing ancient DNA analyses in the lab (Gilbert et al. 2005). Yet, such analyses are not always possible (e.g., in cases where archeological remains were discovered a long time ago and where not all persons who have been in direct contact with the material could be tracked). In such cases, methylation-based clustering of the ancient specimen with expected profiles of the fossil material (mostly bones, teeth, and/or hair) can provide evidence supporting the absence of contamination, as the latter will most likely originate from different types of tissues, such as skin, saliva, and possibly blood. This information can be lined up with post-mortem DNA damage signatures (Krause et al. 2010) to further demonstrate the absence of contamination by fresh DNA material. In cases where fossil specimens and coworkers originate from the same geographical region (e.g., ancient European specimens studied by European researchers), SNP variation will likely support that the ancient specimen could belong to the same population background as the coworkers, leaving again epigenetic signatures as an invaluable complement to the analysis of DNA damage patterns in the final authentication. It is noteworthy that particular sample decontamination procedures such as bleaching prior to DNA analyses can also introduce DNA modifications mimicking bona fide ancient fragmentation patterns (Garcia-Garcera et al. 2011). In such cases, modern contaminants cannot easily be falsified by the analysis of DNA damage patterns, and methylation profiles might reveal an important line of evidence for authenticating the sequence data and confirming the absence of contamination.
Taking advantage of age-associated changes in the methylation levels observed at particular CpGs, we proposed an estimate for the age at death of the Saqqaq individual (Supplemental Material SI3.6). Our approach relies on linear models available from the literature that relate age and the methylation levels measured at two loci in contemporary humans across a range of tissues. We estimate the accuracy of our method using five modern donors and predicted ages closely matching expectations within 1.7–12.4 yr (Supplemental Fig. S3.14). Central to our predictions is the assumption that similar age-associated changes in methylation levels are at play in ancient and contemporary human populations. Yet, recent evolutionary changes in human diet, health conditions, and environment could have shifted the methylation clock, with slower or faster methylation rates at different CpGs in ancient and contemporary populations. Testing the robustness of the age-methylation models used in this study to a variety of environmental and temporal contexts will require further work. Current genome-wide analyses of age-associated methylation changes (Johansson et al. 2013) will likely extend the list of loci that could be used for estimating the age at death of ancient individuals well beyond the sole two CpGs investigated here. Assuming a constant methylation clock of past and ancient populations, it is likely that including multiple and large numbers of loci within a single analysis will enable achieving better age predictions.
Finally, our approach opens the possibility of predicting ancient gene expression levels and functional interpretation. Together with ancient proteomics (Cappellini et al. 2012), it provides additional phenotypic information from ancient individuals, which can complement functional SNP genotyping. Our study also shows that nucleosome protection can cause dramatic variation in read depth on a local scale across the genome, which dramatically increases the amount of sequencing needed to uniformly call SNPs with high confidence. Taken together with the observation that nucleosome-associated DNA fragments are more prone to be preserved than linker regions, this study has implications for designing aDNA studies, genome-wide target-enrichment procedures (Fu et al. 2013), and forensic analyses, and for the expanding field of sequencing preserved clinical samples.
Methods
Sequence data sets
Sequencing and mapping of the Saqqaq and Aborigine genomes are described in full detail in their respective publications (Rasmussen et al. 2010, 2011). Importantly, for evaluating the robustness of the nucleosome signal, the protocols differ in several respects, including the method used for constructing DNA libraries, the DNA polymerases used for amplifying DNA libraries (Phusion, Finnzymes, vs. HiFi, Life Technologies), the sequencing platform used (Illumina GA IIx vs. Illumina HiSeq 2000), and the mapping software used (SESAM vs. BWA) (see Supplemental Material SI1.1 and SI1.3 for details). For the methylation analysis, Saqqaq reads were remapped using BWA with default parameters for refined adapter handling and for allowing indels (Li and Durbin 2009). The modern hair data set was generated following the same procedure as Saqqaq (Supplemental Material SI1.6). The ancient polar bear reads were available from the literature (Miller et al. 2012), trimmed for adapter sequences, and mapped against de novo assembled scaffolds of the polar bear genome using BWA and standard parameters, except that the seed was disabled. The ancient horse sample was provided by one of us (A.T.) and sequenced at the Center for GeoGenetics on a HiSeq 2000 and mapped using BWA with standard parameters and disabling the seed (see Supplemental Material SI1.4 for details).
The Control set was constructed from modern sequencing libraries to have the same number of reads and length distribution as the Saqqaq library. Reads were randomly sampled and truncated to match Saqqaq reads (Supplemental Material SI1.2). The sequence data recently released for a series of modern horse genomes generated from fresh blood (Orlando et al. 2013) was also used to investigate possible short-range periodicities in the size distribution of library inserts. Those analyses were based on full-length DNA inserts that were obtained by collapsing overlapping paired-end reads before mapping (Supplemental Material SI1.7).
GC correction
GCcorrect (Benjamini and Speed 2012) was used to calculate the association of GC-content with read depth and, in turn, to estimate the expected read depth for all unique positions over all relevant read lengths in the human genome. The observed read depth at a given position was normalized for GC content effects by subtracting the expected read depth, summed across all read lengths, to yield the final GC-corrected read depth (Supplemental Material SI2.2).
Anchor site analysis
Nucleosome occupancy, represented by the GC-corrected read depth, was plotted around multiple anchor points in the genome: UCSC TSSs, CTCF sites (Supplemental Material SI3.3), and UCSC splice sites (Supplemental Material SI2.4). All instances of each group of anchor points were aligned and the mean GC-corrected read-depth of the surrounding regions was plotted.
Read-depth periodicity
Spectral density plots (periodograms) across CpG islands, TSSs ± 1000 bp, gene bodies, and CTCF sites ± 1000 bp were made using Fourier transform (Welch's method). To remove low-frequency variations and constant offsets, we subtracted the background signal estimated by exponential curve modeling. Short-time Fourier transform was used to make spectrograms of the spectral decomposition across anchor sites (Supplemental Material SI2.5).
Gene body (UCSC Genes) phasograms (Valouev et al. 2011) were produced using raw uncorrected reads and counting the distance between pairs of 5′ ends on the same strand at positions with at least five reads. Background signal caused by local variation in read depth was subtracted using exponential curve modeling (Supplemental Fig. S2.5a). Modes of the autocorrelation were used to infer dominant long-range and short-range phasing.
Nucleosome calls
Nucleosomes were called using a sliding window of 147 bp over regions with a positive GC-corrected read depth. A given position was called as the center of a nucleosome when showing the highest GC-corrected read depth within a 147-bp window centered on that position (Supplemental Fig. S2.9). A score incorporating both occupancy and positioning was then calculated for each nucleosome defined as the read depth over the peak (occupancy) minus the mean read depth of the flanking regions (positioning) (see Supplemental Fig. S2.9). Repeating the same procedure over the Control set enabled us to calculate the FDR for any given score threshold.
Nucleotide patterns across nucleosomes
Mono- and dinucleotide distributions across nucleosomes were produced by ranking and stratifying nucleosome calls by score. All nucleosomes within each group were aligned at the center (dyad) position, and the average usage across each position was calculated (see Supplemental Material SI2.9 for details).
Methylation signal
Nucleotide misincorporations were identified using mapDamage (Ginolhac et al. 2011) over genomic regions for reads produced using either HiFi or Phusion DNA polymerases. While both polymerases are capable of bypassing deaminated methylated cytosine residues (thymine), only HiFi is able to bypass deaminated unmethylated cytosines (uracil), providing a distinct signature for the methylation state of cytosines in the genome. We defined a proxy for regional methylation levels, Ms as the fraction of CpG dinucleotides giving rise to TpG misincorporations at read starts (Supplemental Fig. S3.3; see Supplemental Material SI3.1 for a detailed description). Ms was measured in a variety of genomic regions, including splice sites (Supplemental Fig. S2.2); promoters stratified across three classes of CpG density (Fig. 4C,E); CGIs showing significant strand asymmetry in the distribution of guanines and cytosines immediately downstream from their TSS (GC skew) (Supplemental Fig. S3.4); undermethylated and ubiquitously methylated CGIs (Supplemental Fig. S3.5); and 1500- and/or 2000-bp-wide regions centered on each CpG site from the Illumina 450k array (Supplemental Material SI3.4). Unsupervised hierarchical clustering was performed using normalized methylation data from Slieker et al. (2013) and Ms-based methylation estimates derived from linear models relating Ms and the methylation levels observed at each CpG from the Illumina 450k array on five modern human donors (Supplemental Material SI3.4).
The CTCF nucleosomes patterns are derived from a set of 12,864 published CTCF binding sites (Fu et al. 2008). We calculated (1) the Ms score, and (2) the nucleosome occupancy (GC-corrected read depth) of 25-bp sliding windows within 1 kb of the sites (Fig. 4D).
Estimates of age at death were derived using linear models from the literature (Koch and Wagner 2011) that relate age and methylation levels at given CpG sites (Supplemental Material SI3.6).
Expression analysis
Three proxies for expression were defined: (1) the level of gene body to promoter methylation, Rs; (2) occupancy of the +1 nucleosome; and (3) strength of nucleosome phasing (see Supplemental Material SI4 for details). Their respective performance was evaluated using expression data from 10 samples of modern hair (Kim et al. 2006) (GSE3058), in the absence of direct measurement of ancient expression levels. The modern expression data was used to define groups of genes with increasing expression levels (using 10, 20, or 50 quantiles), which were ranked by each of the three proxies, and the Spearman correlation coefficient evaluated. Functional enrichment analyses were performed in DAVID (Huang et al. 2009) using standard parameters. Categories showing enrichment scores lower than 1.2 and Benjamini-Hochberg P-values greater than 0.05 were disregarded.
Data access
All nucleosome related data sets are publicly available from our mirror of the UCSC Genome Browser, hg18 assembly, “Ancient Epigenomics” section (http://genome-mirror.moma.ki.au.dk/). The ancient horse sequence data have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRA105533.
Supplementary Material
Acknowledgments
We thank laboratory technicians at the Centre for GeoGenetics and staff at the Danish High-throughput DNA Sequencing Centre for technical assistance, members of the paleomix group for discussions, Andrea Pauli for useful comments, and Ole Jacob Kielland for illustrating Figure 1D. This work was supported by the Danish Councils for Independent Research, Natural Sciences (FNU) and Medical Sciences (FSS); the Danish National Research Foundation (DNRF94); the Lundbeck Foundation; a Marie-Curie Career Integration Grant (CIG-293845); the Novo Nordisk Foundation; and the Human Frontier Science Program (HFSP).
Author contributions: J.S.P. initiated and led the nucleosome analysis. J.S.P., E.V., and B.J.P. conducted the nucleosome analysis, with FFT analyses by B.J.P., including assistance and advice from S.L., B.L., D.T., S.V., R.A., A.S., and A.K. M.R. and M.T.P.G. sequenced the modern hair. L.O. generated the ancient horse data, with input from C.A.H. L.O. initiated and led the methylation analysis. A.M.V.V. and L.O. conducted the methylation analysis with input from J.S.P. E.V., A.M.V.V., B.J.P., L.O., and J.S.P. conducted the expression and functional analysis and the comparison between nucleosome and methylation maps. A.T. provided samples. E.R., E.W., and L.O. contributed reagents and molecular methods. All authors participated in discussing and interpreting results. J.S.P., E.V., E.W., and L.O. wrote the paper with input from all other authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.163592.113.
Freely available online through the Genome Research Open Access option.
References
- Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST 2012. Age-associated DNA methylation in pediatric populations. Genome Res 22: 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruscavage PJ, Hellwig S, Bass BL Small DNA pieces in C. elegans are intermediates of DNA fragmentation during apoptosis. 2010. PLoS ONE 5: e11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM 2009. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol 27: 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzi H, Demehri S, Potter CS, Barber AG, Awgulewitsch A, Kopan R, Christiano AM 2009. Desmoglein 4 is regulated by transcription factors implicated in hair shaft differentiation. Differentiation 78: 292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AC, West AG, Felsenfeld G 2001. Insulators and boundaries: Versatile regulatory elements in the eukaryotic genome. Science 291: 447–450 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Speed TP 2012. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res 40: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21 [DOI] [PubMed] [Google Scholar]
- Bos KI, Schuenemann VJ, Golding GB, Burbano HA, Waglechner N, Coombes BK, McPhee JB, DeWitte SN, Meyer M, Schmedes S, et al. 2011. A draft genome of Yersinia pestis from victims of the Black Death. Nature 478: 506–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botchkavera NV, Ahluwalia G, Shander D 2006. Apoptosis in the hair follicle. J Invest Dermatol 126: 258–264 [DOI] [PubMed] [Google Scholar]
- Boyd-Kirkup JD, Green CD, Wu G, Wang D, Han JD 2013. Epigenomics and the regulation of aging. Epigenomics 5: 205–227 [DOI] [PubMed] [Google Scholar]
- Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, Scandura SM 2011. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 6: e14524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs AW, Stenzel U, Johnson PL, Green RE, Kelso J, Prufer K, Meyer M, Krause J, Ronan MT, Lachman M, et al. 2007. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci 104: 14616–14621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs AW, Stenzel U, Meyer M, Krause J, Kircher M, Paabo S 2010. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res 38: e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K, Xi L, Wang JP, Widom J 2012. A map of nucleosome positions in yeast at base-pair resolution. Nature 486: 496–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttinelli M, Minnock A, Panetta G, Waring M, Travers A 1998. The exocyclic groups of DNA modulate the affinity and positioning of the histone octamer. Proc Natl Acad Sci 95: 8544–8549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos PF, Willerslev E, Sher A, Orlando L, Axelsson E, Tikhonov A, Aaris-Sørensen K, Greenwood AD, Kahlke RD, Kosintsev P, et al. 2010. Ancient DNA analyses exclude humans as the driving force behind late Pleistocene musk ox (Ovibos moschatus) population dynamics. Proc Natl Acad Sci 107: 5675–5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellini E, Jensen LJ, Szklarczyk D, Ginolhac A, de Fonseca RA, Stafford TW, Holen SR, Collins MJ, Orlando L, Willerslev E, et al. 2012. Proteomic analysis of a Pleistocene mammoth femur reveals more than one hundred ancient bone proteins. J Proteome Res 11: 917–926 [DOI] [PubMed] [Google Scholar]
- Chung HR, Dunkel I, Heise F, Linke C, Krobitsch S, Ehrenhofer-Murray AE, Sperling SR, Vingron M 2010. The effect of micrococcal nuclease digestion on nucleosome positioning data. PLoS ONE 5: e15754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collings CK, Fernandez AG, Pitschka CG, Hawkins TB, Anderson JN 2010. Oligonucleotide sequence motifs as nucleosome positioning signals. PLoS ONE 5: e10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A, Lalueza-Fox C, Anderson S, Rambaut A, Austin J, Ward R 2001. Complete mitochondrial genome sequences of two extinct moas clarifies ratite evolution. Nature 409: 704–707 [DOI] [PubMed] [Google Scholar]
- Dabney J, Meyer M 2012. Length and GC-biases during sequencing library amplification: A comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques 52: 87–94 [DOI] [PubMed] [Google Scholar]
- Deaton AM, Bird A 2011. CpG islands and the regulation of transcription. Genes Dev 25: 1010–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis JH, Fan HY, Reynolds SM, Yuan G, Meldrim JC, Richter DJ, Peterson DG, Randon OJ, Noble WS, Kingston RE 2007. Independent and complementary methods for large-scale structural analysis of mammalian chromatin. Genome Res 17: 928–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Saikumar P, Weinberg JM, Venkatachalam MA 1997. Internucleosomal DNA cleavage triggered by plasma membrane damage during necrotic cell death. Involvement of serine but not cysteine proteases. Am J Pathol 151: 1205. [PMC free article] [PubMed] [Google Scholar]
- Felle M, Hoffmeister H, Rothammer J, Fuchs A, Exler JH, Langst G 2011. Nucleosomes protect DNA from DNA methylation in vivo and in vitro. Nucleic Acids Res 39: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sinha M, Peterson CL, Weng Z 2008. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet 4: e1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Meyer M, Gao X, Stenzel U, Burbano A, Kelso J, Paabo S 2013. DNA analysis of an early modern human from Tianyuan Cave, China. Proc Natl Acad Sci 110: 2223–2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney DJ, McVicker G, Pai AA, Fondufe-Mittendorf YN, Lewellen N, Michelini K, Widom J, Gilad Y, Pritchard JK 2012. Controls of nucleosome positioning in the human genome. PLoS Genet 8: e1003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Garcera M, Gigli E, Sanchez-Quinto F, Ramirez O, Calafell F, Civit S, Lalueza-Fox C 2011. Fragmentation of contaminant and endogenous DNA in ancient samples determined by shotgun sequencing: Prospects for human palaeogenomics. PLoS ONE 6: e24161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert MT, Bandelt HJ, Hofreiter M, Barnes I 2005. Assessing ancient DNA studies. Trends Ecol Evol 20: 541–544 [DOI] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F 2012. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginolhac A, Rasmussen M, Gilbert MT, Willerslev E, Orlando L 2011. mapDamage: Testing for damage patterns in ancient DNA sequences. Bioinformatics 27: 2153–2155 [DOI] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, et al. 2010. A draft sequence of the Neandertal genome. Science 328: 710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Muladawi F, et al. 2011. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 43: 630–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi R, Bowman B, Freiberger M, Ryder OA, Wilson AC 1984. DNA sequences from the quagga, an extinct member of the horse family. Nature 312: 282–284 [DOI] [PubMed] [Google Scholar]
- Hofreiter N, Jaenicke V, Serre D, von Haeseler A, Paabo S 2001. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res 29: 4793–4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister JD, Gaut BS 2009. Epigenetic silencing of transposable elements a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res 19: 1419–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA 2009. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, Bird AP 2010. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet 6: e1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioschikhes I, Hosid S, Pugh BF 2011. Variety of genomic DNA patterns for nucleosome positioning. Genome Res 21: 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson A, Enroth S, Gyllensten U 2013. Continuous aging of the human DNA methylome throughout the human lifespan. PLoS ONE 8: e67378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Graefen A, Ball M, Matzas M, Boisguerin V, Maixner F, Leidinger P, Backes C, Khairat R, Forster M, et al. 2012. New insights into the Tyrolean Iceman's origin and phenotype as inferred by whole genome sequencing. Nat Comm 3: 698. [DOI] [PubMed] [Google Scholar]
- Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones P 2012. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res 22: 2497–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerick M, Isau M, Timmermann B, Sultmann H, Herwig R, Krobitsch S, Schaefer G, Verforder I, Bartsch G, Klocker H, et al. 2011. Targeted high throughput sequencing in clinical cancer settings: Formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med Genet 4: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Dix DJ, Thompson KE, Murrell RN, Schmid JE, Gallagher JE, Rockett JC 2006. Gene expression in head hair follicles plucked from men and women. Ann Clin Lab Sci Spring 36: 115–126 [PubMed] [Google Scholar]
- Koch CM, Wagner W 2011. Epigenetic-aging-signature to determine age in different tissues. Aging 3: 1018–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan SB, Kato M, Kiyana R, Trifonov EN 2006. Sequence structure of human nucleosome DNA. J Biomol Struct Dyn 24: 43–48 [DOI] [PubMed] [Google Scholar]
- Krause J, Dear PH, Pollack JL, Slatkin M, Spriggs H, Barnes I, Lister AM, Ebersberger I, Paabo S, Hofreiter M 2006. Multiplex amplification of the mammoth mitochondrial genome and the evolution of Elephantidae. Nature 439: 724–727 [DOI] [PubMed] [Google Scholar]
- Krause J, Briggs AW, Kircher M, Maricic T, Zwyns N, Derevianko AP, Paabo S 2010. A complete mtDNA gneome of an early modern human from Kostenski, Russia. Curr Biol 20: 231–236 [DOI] [PubMed] [Google Scholar]
- Krueger F, Kreck B, Franke A, Andrews SR 2012. DNA methylome analysis using short bisulfite sequencing data. Nat Methods 9: 145–151 [DOI] [PubMed] [Google Scholar]
- Langbein L, Yoshida H, Praetzel-Wunder S, Parry DA, Schweizer J 2010. The keratins of the human beard hair medulla: The riddle in the middle. J Invest Dermatol 130: 55–73 [DOI] [PubMed] [Google Scholar]
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. 2010. Dynamic changes in the human methylome during differentiation. Genome Res 20: 320–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Rice RH, Lee YM 2006. Proteome analysis of human hair shaft. Mol Cell Proteomics 5: 789–800 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Ecker JR 2009. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res 19: 959–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llamas B, Holland ML, Chen K, Cropley JE, Cooper A, Suter CM 2012. High-resolution analysis of cytosine methylation in ancient DNA. PLoS ONE 7: e30226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzen E, Nogues-Bravo D, Orlando L, Weinstock J, Binladen J, Marske KA, Ugan A, Borregaard MK, Gilbert MT, Nielsen R, et al. 2011. Species-specific responses of Late Quaternary megafauna to climate and humans. Nature 479: 359–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MD, Cappellini E, Samaniego JA, Zepeda ML, Campos PF, Seguin-Orlando A, Wales N, Orlando L, Ho SY, Dietrich FS, et al. 2013. Reconstructing genome evolution in historic samples of the Irish potato famine pathogen. Nat Commun 4: 2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prufer K, de Filippo C, et al. 2012. A high-coverage genome sequence from an archaic Denisovan individual. Science 338: 222–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W, Drautz DI, Ratan A, Pusey B, Qi J, Lesk AM, Tomsho LP, Packard MD, Zhao F, Sher A, et al. 2008. Sequencing the nuclear genome of the extinct woolly mammoth. Nature 456: 387–390 [DOI] [PubMed] [Google Scholar]
- Miller W, Schuster SC, Welch AJ, Ratan A, Bedoya-Reina OC, Zhao F, Kim HL, Burhans RC, Drautz DI, Wittekindt NE, et al. 2012. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc Natl Acad Sci 109: E2382–E2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll R, Divo M, Langbein L 2008. The human keratins: Biology and pathology. Histochem Cell Biol 129: 705–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S, Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A 1998. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391: 43–50 [DOI] [PubMed] [Google Scholar]
- Noonan JP, Hofreiter M, Smith D, Priest JR, Rohland N, Rabeder G, Krause J, Detter JC, Paabo S, Rubin EM 2005. Genomic sequencing of Pleistocene cave bears. Science 309: 597–599 [DOI] [PubMed] [Google Scholar]
- Noonan JP, Coop G, Kudaravalli S, Smith D, Krause J, Alessi J, Chen F, Platt D, Paabo S, Pritchard JK, et al. 2006. Sequencing and analysis of Neanderthal genomic DNA. Science 314: 1113–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando L, Leonard JA, Thenot A, Laudet V, Guerin C, Hanni C 2003. Ancient DNA analysis reveals woolly rhino evolutionary relationships. Mol Phylogenet Evol 28: 485–499 [DOI] [PubMed] [Google Scholar]
- Orlando L, Ginolhac A, Zhang G, Froese D, Albrechtsen A, Stiller M, Schubert M, Cappellini E, Petersen B, Moltke I, et al. 2013. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499: 74–78 [DOI] [PubMed] [Google Scholar]
- Ozsolak F, Song JS, Liu XS, Fisher DE 2007. High-throughput mapping of the chromatin structure of human promoters. Nat Biotechnol 25: 244–248 [DOI] [PubMed] [Google Scholar]
- Pääbo S 1989. Ancient DNA: Extraction, characterization, molecular cloning, and enzymatic amplification. Proc Natl Acad Sci 86: 1939–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez A, Castellazzi CL, Battistini F, Collinet K, Flores O, Deniz O, Ruiz ML, Torrents D, Eritja R, Soler-Lopez M, et al. 2012. Impact of methylation on the physical properties of DNA. Biophys J 102: 2140–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan M, Skoglund P, Graf K, Metspalu M, Albrechtsen A, Moltke I, Rasmussen S, Stafford TW, Orlando L, Metspalu E, et al. 2013. Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature. 10.1038/nature12736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Li Y, Lindgreen S, Pedersen JS, Albrechtsen A, Moltke I, Metspalu M, Metspalu E, Kivisild T, Gupta R, et al. 2010. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 463: 757–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Guo X, Wang Y, Lohmueller KE, Rasmussen S, Albrechtsen A, Skotte L, Lindgreen S, Metspalu M, Jombart T, et al. 2011. An Aboriginal Australian genome reveals separate human dispersals into Asia. Science 334: 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebelo AP, Williams SL, Moraes CT 2009. In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res 37: 6701–6715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PL, et al. 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468: 1053–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K 2008. Dynamic regulation of nucleosome positioning in the human genome. Cell 132: 887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuenemann VJ, Singh P, Mendum TA, Krause-Koyra B, Jager G, Bos KI, Herbig A, Economou C, Benjak A, Busso P, et al. 2013. Genome-wide comparison of medieval and modern Mycobacterium leprae. Science 341: 179–183 [DOI] [PubMed] [Google Scholar]
- Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J 2006. A genomic code for nucleosome positioning. Nature 442: 772–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B, Drummond AJ, Rambaut A, Wilson MC, Matheus PE, Sher AV, Pybus OG, Gilbert MT, Barnes I, Binladen J, et al. 2004. Rise and fall of the Beringian steppe bison. Science 306: 1561–1565 [DOI] [PubMed] [Google Scholar]
- Slieker RC, Bos SD, Goeman JJ, Bovée JV, Talens RP, van der Breggen R, Suchiman HE, Lameijer EW, Putter H, van den Akker EB, et al. 2013. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straussman R, Nejman D, Roberts D, Steinfeld I, Blum B, Benvenisty N, Simon I, Yakhini Z, Cedar H 2009. Developmental programming of CpG island methylation profiles in the human genomes. Nat Struct Mol Biol 16: 564–571 [DOI] [PubMed] [Google Scholar]
- Struhl K, Segal E 2013. Determinants of nucleosome positioning. Nat Struct Mol Biol 20: 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Reid AH, Krafft AE, Bijwaard KE, Fanning TG 1997. Initial genetic characterization of the 1918 “Spanish” influenza virus. Science 275: 1794–1796 [DOI] [PubMed] [Google Scholar]
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, et al. 2011. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 20: 440–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A 2011. Determinants of nucleosome organization in primary human cells. Nature 474: 516–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Schuenemann VJ, Cano LM, Pais M, Mishra B, Sharma R, Lanz C, Martin FN, Kamoun S, Krause J, et al. 2013. The rise and fall of the Phytophthora infestans lineage that triggered the Irish potato famine. eLife 2: e00731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.