

Abstract
Sequence diversity and the ages of the deepest nodes of the MSY phylogeny remain largely unexplored due to the severely biased collection of SNPs available for study. We characterized 68 worldwide Y chromosomes by high-coverage next-generation sequencing, including 18 deep-rooting ones, and identified 2386 SNPs, 80% of which were novel. Many aspects of this pool of variants resembled the pattern observed among genome-wide de novo events, suggesting that in the MSY, a large proportion of newly arisen alleles has survived in the phylogeny. Some degree of purifying selection emerged in the form of an excess of private missense variants. Our tree recapitulated the previously known topology, but the relative lengths of major branches were drastically modified and the associated node ages were remarkably older. We found significantly different branch lengths when comparing the rare deep-rooted A1b African lineage with the rest of the tree. Our dating results and phylogeography led to the following main conclusions: (1) Patrilineal lineages with ages approaching those of early AMH fossils survive today only in central-western Africa; (2) only a few evolutionarily successful MSY lineages survived between 160 and 115 kya; and (3) an early exit out of Africa (before 70 kya), which fits recent western Asian archaeological evidence, should be considered. Our experimental design produced an unbiased resource of new MSY markers informative for the initial formation of the anatomically modern human gene pool, i.e., a period of our evolution that had been previously considered to be poorly accessible with paternally inherited markers.
Analyses of genetic diversity at nonrecombining uniparental loci (mitochondrial DNA [mtDNA] and the male-specific portion of the Y chromosome [MSY]) have provided important clues regarding human evolutionary events (Underhill and Kivisild 2007). Although mtDNA and the MSY each represent only a single realization of the evolutionary path, they share three crucial advantages in order to obtain a full phylogeographic analysis. All three factors are linked to their lack of interallelic recombination and haploidy: (1) Their evolution can be described through an unequivocal phylogenetic tree, i.e., a cladistic description of the affinities among extant molecular types that might also accommodate extinct ones obtained from ancient DNA (aDNA); (2) the pattern of their geographic diversity can be described in terms of the spread of monophyletic lineages; and (3) the antiquity of their lineages can be estimated assuming that mutations at a variety of loci, evolving in different modes and at different rates, are sequentially accumulated.
The phylogeography of the MSY in all continents has been the subject of intense research (for a review, see Chiaroni et al. 2009). However, based on dating estimates obtained from microsatellite and single-nucleotide variation (Pritchard et al. 1999; Thomson et al. 2000; Wilder et al. 2004; Shi et al. 2010), the entire phylogeny of the MSY, to date, has been considered to be rather young and of null or limited informativeness for a time horizon comparable to the age of anatomically modern humans (AMHs) in Africa or findings of early AMHs outside Africa. However, recent updates to the MSY tree topology and new figures for the substitution rate have led to much older dates than previously thought for the root of the MSY tree (Cruciani et al. 2011b; Mendez et al. 2013) and nodes immediately downstream (Francalacci et al. 2013; Poznik et al. 2013).
A comprehensive description of the features of the MSY phylogeny requires an unbiased search across lineages, considering widely divergent lineages and reaching a low error rate in variant calling. Under these conditions, all nodes can be appropriately dated, heterogeneity among branch lengths can be tested, and evidence for purifying selection can be evaluated. Single-nucleotide substitutions (SNSs) and small indels have often been the markers chosen to define the branches of the MSY tree (Underhill et al. 2000; Karafet et al. 2008), due to their evolutionary stability and low rates of recurrent mutations. The possibility of revealing numerous SNSs by resequencing large portions of the genome using next-generation technology has also led to the re-evaluation of SNSs as the optimal tool for age estimation. Recently, low-depth, whole-genome sequencing studies produced thousands of single-nucleotide polymorphisms (SNPs) of the MSY from a large set of males (The 1000 Genomes Project Consortium 2010, 2012). A refinement of the phylogenetic relationships in a portion of the MSY tree with these SNPs has been attempted, but due to the likely abundance of false negatives and biases resulting from low-depth sequencing, not all of these SNPs could be used to confidently reconstruct and date the MSY phylogeny (Rocca et al. 2012). High-depth, whole-genome sequences have been generated and made publicly available by Complete Genomics. Wei et al. (2013) used this information to extract the MSY genotypes of 35 males, which were analyzed together with a single haplogroup A3 subject as an outgroup in order to obtain a time-calibrated phylogeny of the MSY based on 6662 high-confidence variants. However, due to the ancestry of males in the above studies, deep branches of the MSY tree were underrepresented or not represented at all.
To gain insights into the timing of early human evolutionary processes, through high-coverage next-generation sequencing, we characterized 18 deep-rooting Y chromosomes selected from thousands of worldwide Y chromosomes already genotyped for known markers (Cruciani et al. 2004, 2007, 2010, 2011a,b; Trombetta et al. 2011; Scozzari et al. 2012; present study). We analyzed these chromosomes in the wider context of 68 Y chromosomes that represent major branches of the entire MSY phylogeny. In this way, we were able to interpret our findings from the 18 deep-rooted chromosomes in light of the mutational pattern and branch divergence observed across the entire tree. More specifically, in the present study we discuss our reconstructed phylogeny and the antiquity of its branches, and how they relate to the population genetics of Africa over a time horizon that begins well before the exit from the continent.
Results
A high-depth resequencing (average 50×) of ∼1.5 Mb of the MSY was performed in 68 unrelated males representing major Y chromosome haplogroups (Supplemental Table S1). Here we limit the description of the results to single-nucleotide substitutions, focusing on deep-rooted lineages.
Single-nucleotide mutational pattern
We identified 2386 positions that display a nucleotide substitution among the 68 unrelated males under study (Supplemental Table S2). These do not include 13 nucleotide positions that were invariant in the entire sample but different from the reference sequence (Supplemental Table S3), a finding that can be interpreted as either reference-specific mutations or sequencing errors. Two of the 2386 variant positions were triallelic.
Figure 1 shows the distribution of the variant positions across the five selected MSY regions. The apparent uneven density of variant positions can be explained by the different occurrence of repetitive elements, which were largely excluded from targeted sequences (see Methods). For example, the region that is most densely populated by SNPs (chrY:8,400,000–8,650,000) is particularly devoid of repetitive elements. When the occurrence of the 2386 variants in each of the 5274 sequenced DNA fragments (see Methods) was considered, no evidence of any uneven distribution was obtained. The linear slope (0.001591) closely matched the overall rate of occurrence (2386/1,495,512 = 0.001595), and all points but two fell within the 0.999 confidence interval (CI) estimated according to the Poisson distribution (Supplemental Fig. S1). Of the 2386 positions, 12.1% were inferred to be located at ancestral CpG dinucleotides (see Methods), a proportion similar to that reported by Kong et al. (2012).
Figure 1.

Regions of the Y chromosome analyzed and distribution of variants discovered. The different tracks from top to bottom report the following features: Y chromosome ideogram, Y chromosome position according to the human Y chromosome reference GRCh37/hg19, the five regions targeted for capture (black bars), variant positions (thin marks), and UCSC genes.
Six protein-coding genes (RPS4Y1, ZFY, USP9Y, DDX3Y, UTY, and TMSB4Y) were covered in our capture design. Nineteen variant positions were located within codons (Supplemental Table S4). Overall, the number of variant positions we found in coding regions (19/15,397 bases) was proportionally lower than that residing in noncoding regions (2367/1,480,115), though not significantly (P = 0.30, Fisher exact test). As negatively selected mutations can be expected to be younger than neutral noncoding mutations, we only analyzed those mutations that are present in older branches (i.e., >30 kyr ago [kya]). No evidence indicating any enrichment of noncoding variants was obtained. In our data, 10 of the substitutions were predicted to produce amino acid changes, of which eight were private (i.e., found in a subject only) and two were shared between at least two subjects. This compares with three private and six shared synonymous variants. A similar imbalance was present after removing four long terminal branches, each represented by a single individual (S07, S08, S09, and S38) (Supplemental Fig. S2), in which some of the private mutations can be relatively old (seven private vs. two shared, and three private vs. five shared for nonsynonymous and synonymous mutations, respectively). Though these differences were not nominally significant (Fisher exact test, P = 0.07, and P = 0.15, respectively), they suggested that purifying selection might have caused the slight underrepresentation of missense variants we observed in shared lineages. Two previously unreported missense variants (chrY:14834046 G>A, USP9Y R84Q; chrY:14870505 G>T, USP9Y V567L) were predicted to be damaging (PolyPhen-2 scores 0.927 and 0.955, respectively). Both of these mutations were private and found in conserved positions (phyloP scores 0.974 and 0.983, respectively) of the USP9Y gene, a member of the peptidase C19 family.
MSY phylogeny
We used the 2386 variable positions to reconstruct a maximum parsimony (MP) tree using two independent methods (see Methods). These methods yielded trees with identical topologies, with substitutions at the same positions in each branch, and indicated recurrent mutational events in only four positions, for a total of 2392 distinct mutational events (including double hits at the two triallelic positions). The proportion of recurrent events (four out of 2386 positions, 0.2%) was significantly lower (P = 2.2 × 10−16, Fisher exact test) than that reported by Wei et al. (2013) (172 out of 5865 mutations, 2.9%), a discordance that can be attributed to differences between the two studies in both the regions analyzed and the strategies adopted to infer the ancestral states (see Methods). The overall transition/transversion ratio (1513/879 = 1.72) was within the range of genome-wide estimates for de novo events (Campbell et al. 2012; Kong et al. 2012; Michaelson et al. 2012) with an excess of G>A and C>T compared with the opposite changes (Supplemental Table S5).
A condensed version of the MP tree, with a particular emphasis on the deeply rooted African lineages, is shown in Figure 2. First, all Y chromosomes in our data set previously known to belong to different major haplogroups partitioned into distinct clades in the tree, with the same phyletic relationships reported in previous studies (for reviews, see Karafet et al. 2008; Batini et al. 2011; Cruciani et al. 2011b; Scozzari et al. 2012; Francalacci et al. 2013; Poznik et al. 2013). Second, new features emerged in the internal topology of some clades. The polyphyletic nature of “haplogroup” A was confirmed (Cruciani et al. 2011b), with A1b being the most deeply rooted clade in our set. Furthermore, while the previous topology of haplogroup A1b consisted of a single lineage defined by terminal markers plus three paragroups (Scozzari et al. 2012), here we describe markers for each of four A1b lineages. We confirm that A1a is the deepest branch in the clade that groups all other haplogroups. Within A2-F, a major bifurcation grouped A2 and A3 together, which stemmed from a short branch (branch 11 in Supplemental Fig. S2), previously defined by markers PK1 (Batini et al. 2011) and V249 (Scozzari et al. 2012). Within A3b2, a small clade grouped together two A-M13 European subjects (S10 and TV20) that differed by 17 mutations, and separated them from two African A-M13 chromosomes. Such loose affinity between the two European A-M13 chromosomes denotes a more remote relatedness than recently reported for seven A-M13 chromosomes from Sardinia (Francalacci et al. 2013).
Figure 2.
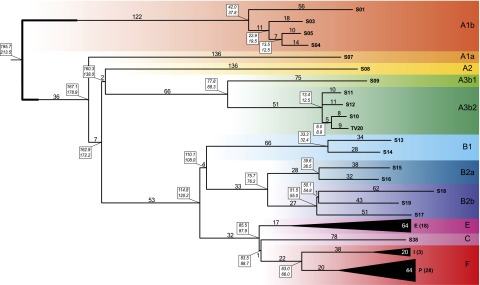
Maximum parsimony tree obtained with 2386 variable positions. The number of mutational events defining each branch is shown above it. For the collapsed haplogroups E, I, and P, the average number of mutations is shown. Dating estimates are reported in boxes near each node (upper and lower values obtained with BEAST and the rho method, respectively). Colored belts indicate major haplogroups according to current nomenclature (Karafet et al. 2008; Scozzari et al. 2012).
The other samples, belonging to haplogroup B-F, shared a long branch (branch 21), with B as a monophyletic clade sister to E-F. Haplogroup B was confirmed to consist of two deep clades, corresponding to B1 and B2, with the latter in turn consisting of B2a and B2b. Compared to the previous topology (Scozzari et al. 2012), we found markers for each of the paragroups B1*, B2a* and B2b*. While a remarkable advancement in the phylogenetic structure of haplogroup B2b was obtained by Poznik et al. (2013), we detected a new haplogroup-defining node for B2a, which is deeper than that reported in previous studies.
The remaining haplogroups (E, C, and F) were arranged according to the previously known topology (Karafet et al. 2008). In particular, a single mutation (branch 37 in Supplemental Fig. S2), which is phylogenetically equivalent to P143, defines a sister clade of E comprising haplogroups F and C, the latter of which has never been covered in other large-scale resequencing studies (Francalacci et al. 2013; Poznik et al. 2013; Wei et al. 2013).
A remarkable aspect of our tree was that the relative lengths of major branches (in number of mutations) differed greatly from previously reported values. Some striking examples include branches 1, 9, and 23 (defining A1b, A1a, and B1, respectively), in which the number of mutations increased by at least six times compared with previous studies (Karafet et al. 2008; Scozzari et al. 2012). A notable increase in length was also observed for branches 21 and 35 (defining B-F and E-F, respectively). Conversely, branches basal to haplogroup P did not show the same increase in length compared with previously known markers. This can be partially attributed to the asymmetry involved when using a sequence that is mainly derived from haplogroup P DNA as a reference, and to the intense sequencing and search for mutations carried out on haplogroup P subjects (Underhill et al. 2010; Myres et al. 2011).
In order to further confirm the increase in branch length compared with previous studies, we considered dbSNP (build 135) as an alternative source of variants, though not necessarily phylogenetically assigned. Only 407 of the 2386 variant positions (17.1%) detected here were reported in dbSNP (Supplemental Table S2; Supplemental Fig. S2). dbSNP polymorphisms were underrepresented in deep-rooting African-specific branches of the phylogeny and in haplogroup C. A paucity of known SNPs was evident for 17 of the 18 terminal branches of African-specific haplogroups. Conversely, dbSNP markers almost saturated branches leading to haplogroups E and F.
The above results dramatically modified one major feature of the tree, i.e., the proportions of mutations that mark the phylogeny for the periods prior to and after the exit out of Africa. Haplogroup E-F is informative for this event because it includes, among others, all the lineages that are found out of Africa (for a thorough discussion, see Underhill and Kivisild [2007]). The tree by Karafet et al. (2008) depicted a close proximity between the root and the MRCA of E-F. Conversely, in our tree 128 mutations separated the root from the node defining E-F, whereas 84.2 mutations per lineage (on average) were downstream from the same node.
While our results corrected an evident underdetection of variants for deep-rooted branches, a short length of haplogroups A1b, A1a, and A2-A3 (158, 172, and 177 average mutations from the root, respectively) compared with the rest of the tree (haplogroup B-F, 211 average mutations from the root) was nevertheless apparent. This added to the finding of Wei et al. (2013), who noticed a short branch for A3, represented by a single individual in their study. We tested whether rate heterogeneity among branches of the entire tree could explain the data better than a strict clock model. We compared the distributions of tree log(likelihoods) generated by BEAST (Drummond and Rambaut 2007) under both models, and found that the difference between the harmonic means was 2.3, corresponding to positive evidence (Nylander et al. 2004) in favor of rate heterogeneity. Thus, compared with two recent large screenings (Francalacci et al. 2013; Poznik et al. 2013), deep branches of the Y phylogeny reveal an appreciable heterogeneity in the accumulation of mutations. In particular, in our tree, A1b was by far the shortest branch (158 average mutations from the root). When the length of A1b was compared with the rest of the tree (A1a-F, 207 average mutations from the root), the difference turned out to be statistically significant (χ2 = 6.72, P = 0.0095). The corresponding tests for A1a (vs. A2-F) and A2-A3 (vs. B-F) produced nominally significant P-values, which, however, did not resist the Bonferroni correction for multiple tests. We investigated whether structural rearrangements could be responsible for the reduction in the number of countable positions, but on short branches we only detected a 6.2-kb deletion (0.42% of the total sequence scored), shared by all A1b chromosomes. Additionally, we had no evidence (Supplemental Table S6) of an excess of clustering of variants on long branches, where an excess may indicate a structural rearrangement and alignment of paralogous sequences.
Dating and phylogeography
We used sequence data and two independent methods to estimate the age of the nodes in the tree. In both methods, we used a substitution rate obtained by adjusting the rate of autosomal de novo mutations from recent genome-wide screens to the MSY, as independently worked out by Mendez et al. (2013) with minor differences (see Methods). The results are shown as boxes in Figure 2. The two methods produced highly concordant values (Supplemental Fig. S3). Hereafter we refer to the results obtained with BEAST (Drummond and Rambaut 2007), which averages the influence of many parameters over the entire tree, also accounting for rate heterogeneity among branches. The consensus tree showing the node ages with associated CIs is reported in Supplemental Figure S4. The TMRCA of the samples examined here (equivalent to haplogroup A0-T in the nomenclature of Mendez et al. [2013]) was dated at 196 kya (95% CI 147–248 kya), in agreement with the value obtained by Mendez et al. (2013) with a different method. Our estimate was much older than the previous one based on a similar topology but a different substitution rate (Cruciani et al. 2011b). Three nodes basal to A1a-F, A2-F, and A2-A3 clustered in the narrow interval between 167 and 160 kya. The node basal to A2-F coincides with the MRCA in the data sets by Francalacci et al. (2013) and Poznik et al. (2013), and our estimated date (162.9 kya; 95% CI 125–207 kya) is comparable to both studies (185 and 138 kya, respectively). We observed no other node until 115 kya, a date that marks the separation between African-specific and all remaining haplogroups. An age of as much as 110 kya was estimated for haplogroup B, corresponding to the split between chromosomes currently found only in central-western Africa (B1) and chromosomes spread all over sub-Saharan Africa (B2) (Fig. 3; Supplemental Table S7). Such an old date could not be highlighted in recent large-scale resequencing studies (Francalacci et al. 2013; Poznik et al. 2013; Wei et al. 2013), due to the lack of B1 representatives.
Figure 3.

Geographic distribution of deep-rooting haplogroups in the African continent. Map of Africa showing schematically the present-day home ranges of the MSY haplogroups discussed in the text (redrawn from Chiaroni et al. [2009], with modifications and updates based on haplogroup frequencies reported in Supplemental Table S7). Colors are as in Figure 2, and their intensity does not reflect haplogroup frequencies in the corresponding populations. Haplogroup B2, ubiquitous in sub-Saharan Africa, was omitted.
In the time frame between 85.5 and 75.7 kya, four splits were observed: (1) The node within haplogroup A3b, which separates southern (A3b1) from eastern (A3b2) African lineages; (2) the node within haplogroup B2, separating clades B2a and B2b, which are frequently observed among present day African food-producers and hunter-gatherers, respectively; and (3) two nodes that are highly informative for the exit out of Africa that are basal to E-F and C-F, respectively. In fact, haplogroup E has representatives both within and out of Africa, whereas haplogroup C-F (83.5 kya, CI 64.3–106.6 kya) encompasses chromosomes found virtually only outside of Africa.
We used two discrete phylogeographic analyses (Lemey et al. 2009; Yu et al. 2010) to associate each node of the tree to each of four broad geographic regions, with emphasis on sub-Saharan Africa. First, we used a Bayesian analysis (Supplemental Fig. S5) in which, when starting with an even prior, the posterior probabilities (0.44–0.45) favored a central-western African placement for the four deepest nodes in the tree, i.e., from 196 to 160 kya. Southern and eastern African locations were favored for the nodes defining haplogroups A3b and A3b2, respectively. The emergence of new diversity out of Africa was captured in this analysis by a shift in location assignment along the branch leading to E-F, with all nodes downstream assigned to non-sub-Saharan African locations with high confidence (Supplemental Fig. S5). Finally, a further shift in location assignments was observed within haplogroup A3b2, from eastern Africa to non-sub-Saharan Africa. Second, we used a maximum parsimony approach (Yu et al. 2010) that similarly predicted a 100% probability of a central-western African location for the two deepest nodes. In this analysis, however, the oldest node unambiguously assigned to non-sub-Saharan African locations was the MRCA of haplogroup C-F (Supplemental Fig. S6).
Discussion
In the present study, we applied next-generation sequencing coupled with sequence capture to obtain a large number of variable positions that represent an independent test for and improvement of the MSY phylogeny. Two aspects of our experimental design enabled us to obtain high-quality data, i.e., the selection of segments with little or no homology with the X chromosome and a high depth. The first reduced the rate of errors attributable to the presence of gametologous sequences in the captured material, and the second allowed reliable SNP calling also for below-average enriched segments.
Implications of the new MSY chronology
For a long time, the MSY tree has suffered from a lower level of resolution than that of the mtDNA phylogeny. However, with the advent of new technologies now allowing for high-throughput identification of Y chromosome SNSs, these markers can now be used to characterize the MSY tree at a greater level of resolution as well as to improve age estimates. Because the accumulation of MSY SNSs over the course of human evolutionary history would not have plateaued, these markers provide a nearly unlimited resource for refining and dating the phylogeny, given that an appropriately long sequence is evaluated. In contrast, diversity at microsatellite loci is not only confounded by recurrent mutations, but may reach a maximum (Busby et al. 2012). This limits their use in resolving the phylogeny and has often given rise to unreliable dating results, especially for deep-rooted lineages. The accuracy of SNS-based dating methods relies on the equally efficient discovery of new SNSs across all lineages. Our results show that a single targeted next-generation run can produce a highly reliable and informative phylogeny with a uniformly intense search for markers across all lineages included in the study.
Knowledge of the deepest branches in the MSY tree has long been incomplete, and the phyletic relationships between lineages have often been reordered, including the placement of the root (Batini et al. 2011; Cruciani et al. 2011b; Scozzari et al. 2012; Mendez et al. 2013). The representation of deep lineages at low frequencies and often from small remote populations has also made their study difficult. Yet, deep-rooted lineages are particularly informative in the reconstruction of the scenario of an ancient population structuring in Africa. This is currently considered to be the source of global patterns of genome-wide diversity under the generally held view of an exit out of Africa originating from the eastern portion of the continent (Campbell and Tishkoff 2010; Henn et al. 2012; Scally and Durbin 2012).
The subjects entered in this work were intentionally selected in order to represent a wide range of diversity and antiquity among MSY lineages to resolve and date the deepest branches in parallel with more widely studied lineages. The resulting tree, with a TMRCA of 196 kya, displays extraordinary deep ancestry for most of the early branches within Africa, a fact that has not been fully acknowledged in previous works.
The first two splits in our tree, dated at 196 kya and 167 kya, separate branches (A1b and A1a) that are currently found at low frequencies in central-western Africa (Fig. 3; Supplemental Table S7) but have not been detected elsewhere in the African continent. This geographical confinement of deep lineages is at odds with the mainly eastern African position of sites providing fossils of comparable ages (McDougall et al. 2005; but see Tattersall and Schwartz 2008). The question then becomes: When did these lineages reach central-western Africa? Two hypotheses can be put forward: (1) ancient residence of A1b and A1a in eastern Africa, followed by relocating to central-western Africa and extinction in the motherland eastern Africa (possibly together with other yet unknown deep-rooted branches), or (2) ancient residence of A1b and A1a in central-western Africa, with loss of fossil record there. The finding of the oldest lineage recorded so far (A00, 338 kya) in Cameroon (Mendez et al. 2013) adds to our phylogeographic results in suggesting central-western Africa as a broad region populated by deep MSY lineages earlier than 160 kya.
In discussing the implications of these findings, we note that the tens of thousands of years separating some of the consecutive branching events dramatically reduce the power of the Bayesian phylogeographic inference (Supplemental Fig. S5), resulting in decreasing statistical support from the tips toward the root of the tree. It should also be considered that the demography of past populations, characterized by small effective sizes with intense drift, and possibly subsequent expansions, may have caused lineages to wander over vast geographic regions, also in response to climate pressures (Burroughs 2005; Castañeda et al. 2009), with the potential for generating an altered phylogeographic signal. Finally, we note that the sampling scheme used here is geographically uneven, and is constrained by current knowledge on the distribution of extremely rare deep lineages (Supplemental Table S7). This calls for a more even sampling coverage of MSY diversity in Africa, which should be also compared with the conclusions of recent autosomal genetic and craniometric data (Ramachandran et al. 2005; Manica et al. 2007; Tishkoff et al. 2009; Pagani et al. 2012; Schlebusch et al. 2012).
Our data place the TMRCA of haplogroup B at 110 kya, a date that is unexpectedly old if one considers the previous length of this branch. The current distribution of chromosomes and dating of the two B subclades (Fig. 3) also testify to early dispersals followed by partial isolation. In particular, haplogroup B2a-M150, which has been associated with the expansion of Bantu speakers (Beleza et al. 2005; Berniell-Lee et al. 2009) and dated at 6.0 kya on the basis of its STR diversity (Batini et al. 2011), turned out to be a very ancient lineage (40 kya), long predating the alleged timing of the Bantu expansion. Beyond the disparity of the microsatellite- and SNP-derived ages, these data indicate that only a small subset of the overall B2a diversity became incorporated into the male gene pool of Bantu speakers. As for B2b, Tishkoff et al. (2007) reported that south African Khoe speakers harbor a highly divergent subset of these chromosomes, with a STR-based TMRCA of 69.9 kya, suggesting that we possibly did not sample the most divergent lineages of this clade.
Two main routes for the AMH dispersal out of Africa are still widely debated: the northern route through Egypt to the Levant, where AMH fossils dated prior to 100 kya have been found (Grün et al. 2005), and the southern route through the Bab-el-Mandab strait to the Arabian peninsula at 125 kya as argued by Armitage et al. (2011) based on archaeological records. As far as genetic evidence is concerned, mtDNA data (Soares et al. 2011; Fernandes et al. 2012) favor this latter route, but not before ∼70 kya. Our phylogeographic analyses suggest that the nodes basal to haplogroups E-F and C-F provide information regarding the exit out of Africa. In fact, these are the oldest nodes for which a non-sub-Saharan ancestral state received statistical support, with a level of probability that was two times higher than any of the alternatives in the Bayesian analysis (Supplemental Fig. S5). Three main scenarios are compatible with our phylogeographic analyses (Supplemental Figs. S5, S6), dating results (Fig. 2), the known geographic distribution of patrilineages (Underhill and Kivisild 2007; Chiaroni et al. 2009), and the possibility that lineages were driven to extinction or to exceedingly low frequencies by drift. In the first one, the exit of carriers of a precursor of haplogroup E-F occurred anytime between 114.8 and 85.5 kya (overall window 145 to 65 kya, corresponding to the length of branch 35 and CIs of its defining nodes) (see Supplemental Figs. S2, S4), followed by the diversification of E-F in Eurasia. This scenario requires the re-entry of a single lineage (haplogroup E) in Africa, as originally proposed by Hammer et al. (1998). In the second scenario, the node basal to E-F originated in Africa and the exit of a precursor of C-F took place between 85.5 and 83.5 kya (overall window 108–64 kya, corresponding to the length of branch 37 and CIs of its defining nodes) (see Supplemental Figs. S2, S4), together or separately from E, and followed by the extinction of the early C-F in Africa. In the third scenario, three or more lineages left Africa after 83.5 kya; this would require the not remote possibility that multiple lineages went extinct or are yet to be found in Africa.
One of the implications of the first scenario is that the AMH occupation of the Middle East and/or the Arabian peninsula before 100 kya could no longer be regarded as a “temporary excursion” (Scally and Durbin 2012) but rather the seeding event for the MSY diversity found today in Eurasia. The dates from the second and third scenario are similar to estimates from mtDNA variation that date the exit of matrilineages based on the topology and TMRCA of haplogroup L3 (Atkinson et al. 2009; Soares et al. 2011; Fu et al. 2013a). Neither MSY scenario excludes an out-of-Africa exit before a major event marking AMH occupation further east, i.e., the Toba eruption at ∼74 kya (Chesner et al. 1991; Mellars et al. 2013). Moreover, they all open up the possibility of a temporal gap in which an intermediate bottlenecked population existed in the Middle East/Arabian peninsula, and whose genetic signature is now visible in the genome pool of Eurasians. They also fit with the finding of deep-rooted Eurasian Y haplogroups in the southern Arabian peninsula (Abu-Amero et al. 2009) and Lebanon (Zalloua et al. 2008).
In summary, inferences regarding the ancestral relationships and timing of movements of human populations in the exit out of Africa based on extant MSY diversity remain rather imprecise, due to the reduced topological structure at branches leading to haplogroups E-F and C-F through the time window 145–64 kya (Fig. 2). The array of new markers generated here strongly prompts the typing of haplogroup D (not represented among our males) as well as rare African and non-African carriers of E*, C* (Weale et al. 2003; Zalloua et al. 2008; Abu-Amero et al. 2009), or even older paragroups, in search for lineages that could modify the topology of the MSY tree with new informative nodes. Reconciling archaeological and genetic dates for the two uniparental systems is also linked to the finding of rare old lineages for the mtDNA, as recently suggested (Rose et al. 2011). Strong evidence may also derive from aDNA collected in appropriate archaeological layers of eastern Africa and south-western Eurasia.
One remarkable aspect of our results regards the statistically significant low number of mutations on the branch corresponding to haplogroup A1b. The reason for this is yet to be clarified. In a genome-wide analysis (Conrad et al. 2011), a lower number of de novo mutations was found in a Yoruban compared with a non-African trio, suggesting the need for more extensive analyses to assess possible population-specific effects. As far as selection is concerned, the low number of coding variants observed prevented the testing of their differential occurrence across branches, and we cannot exclude different selective pressures acting on different lineages. Two studies (Lohmueller et al. 2008; Fu et al. 2013b) showed a higher number of deleterious variants in Europeans compared with Africans, which is most likely due to the combined effects of a long lasting bottleneck and re-expansion of Europeans. The reduction of the male effective population size, which is most likely to be associated with the exit out of Africa, may have provided enough opportunity for deleterious variants to appear and increase in frequency. In this case, the net effect would be an extra load of mildly deleterious mutations that elongated the branches that were involved in the bottleneck.
Further developments
Approximately 80% of the markers reported here are novel and open new perspectives for the refinement of the phylogeny through the study of additional subjects, including A00 and possibly undiscovered deep lineages. In fact, the resolution attained will enable a search for highly specific lineages with PCR-based approaches, which will be eventually also applicable to ancient DNA and help shed light on the possible historical continuity between individuals of the past and current populations.
We endorse the need for a reference sequence that incorporates ancestral alleles at all known variable positions (Wei et al. 2013), which would greatly facilitate further work in this field.
Estimates of the substitution rate for the MSY (Xue et al. 2009; Francalacci et al. 2013; Mendez et al. 2013; Poznik et al. 2013; Wei et al. 2013; present work) currently suffer from an appreciable uncertainty. Major improvements in dating may derive from taking into account the complexity of the mutational process (Michaelson et al. 2012). For example, the possibility that clusters of mutations may hit the MSY seriously challenges the concept of linear accumulation with time. We highly recommend that, in the future, mutation rates be worked according to the local features of the MSY sequences, and that they then be used on appropriately partitioned data sets as previously suggested (Fu et al. 2013a). We see deep-rooted pedigrees (Xue et al. 2009) as the material that should be chosen in order to work out robust estimates of these rates, given the low chances of observing mutational events in such a small portion of the genome in a single generation.
Methods
Samples
Human Y chromosomes to be sequenced (Supplemental Table S1) were selected on the basis of their SNP/STR genotype, which had been determined in the present or previous studies (Cruciani et al. 2004, 2007, 2010, 2011a,b; Trombetta et al. 2011; Scozzari et al. 2012). Most samples were chosen in order to represent as many deep branches of the Y phylogeny as possible. In the vast majority of cases, DNA was prepared from fresh venous blood, with no cell culturing. The study was approved by the “Policlinico Umberto I, Sapienza Università di Roma” ethical committee (document number 496/13), and informed consent was obtained from all participants.
Selection, targeting, and alignment of MSY unique regions
We selected five regions (Fig. 1; Supplemental Table S8) of the X-degenerate portion of the MSY, which showed a low degree of similarity with X gametologous sequences, for a total of 3,768,982 bp.
A custom sequence capture array was manufactured by Roche Nimblegen for the target enrichment of the indexed genomic library. A set of unique and overlapping probes was designed to capture unique sequences at the five MSY regions under study. Probe uniqueness was assessed using Sequence Search and Alignment by Hashing Algorithm (SSAHA) (Ning et al. 2001). Probe tiling of the target regions excluded most of the repetitive interspersed elements. The capture probe set covered a total of 1,495,512 bases of the target region, distributed into 5274 fragments (Supplemental Table S9).
The captured library was loaded onto an Illumina HiSeq 2000 platform to produce a 50× mean depth sequence for the 1.5-Mb targeted region.
SNP calling, filtering, and annotation
Candidate variant nucleotide positions (compared to the human reference sequence) were identified by using the SOAPsnp software (Li et al. 2009), with haploid specific parameters for variant calling. Among the candidate mutations from the SOAPsnp analysis, we only considered those found in the 1.5-Mb target region and those that fulfilled all of the following criteria: (1) quality score of consensus (QS) ≥ 90; (2) depth ≥ 4×; and (3) difference between the depth and the total number of reads for the two best bases ≤4. This latter criterion was adopted to identify false SNP calls due to misalignments in proximity of insertions/deletions. The filtering was then refined by visual inspection of the .sam files using the Integrative Genomics Viewer (IGV) software (Thorvaldsdóttir et al. 2013). In particular, we inspected variant calls falling in these categories: 90 ≤ QS ≤ 98; 4× ≤ depth ≤ 10×; distance from the closest SNP in the same sample ≤20 bases; and depth for the second best base ≥3. One well-known complication associated with estimating sequence divergence is that mapping quality for a read depends on the number of differences between the read and the reference. We then inspected the alignments of all subjects, in sliding windows of 25 kb (using IGV), searching for extreme variations in sequence coverage among samples, which may be indicative of structural rearrangements and the inability to detect variants. We also used GASVPro (Sindi et al. 2012) to identify structural variants from paired-end mapping data. Since such rearrangements can also lead to the unscheduled capture of paralogous divergent sequences, we checked a posteriori (after constructing the tree) the clustering of variants in short stretches of DNA on each tree branch.
To assess the accuracy of our set of filtered variants, we performed a series of quality controls using both resequencing and literature data (for details, see Supplemental Text).
The program wANNOVAR (Chang and Wang 2012) was used to identify and note exonic variants found in this and other (Rozen et al. 2009; Wei et al. 2013) works. The program also returns conservation levels (phyloP scores) and predicted functional importance (PolyPhen-2 scores). The UCSC Known Gene and Ensembl gene definitions were used.
Tree construction
A contingency table of alternative bases by subject (rows) and chromosome position (columns) was converted into .rdf and .meg files, to be handled with the programs Network (Bandelt et al. 1999) and MEGA (Tamura et al. 2011), respectively. Network was used to obtain a median joining network for rho calculations, a complete listing of mutated positions along each branch and a precise count of inferred recurrent mutations at the same position. This tiny subset of positions (four recurrent mutations) (Supplemental Table S2) was rechecked and confirmed in the original alignment files. MEGA was used to obtain a maximum parsimony tree. Note that both methods ignore the information on the ancestral versus derived state for the particular allele observed in each subject, as they only consider state changes.
Mutation rate
In order to model the substitution process at the surveyed positions, we took into account the careful measurements of the genome-wide de novo mutation rates recently obtained from parent–child transmissions and deep-rooted pedigrees (The 1000 Genomes Project Consortium 2010; Awadalla et al. 2010; Roach et al. 2010; Campbell et al. 2012; Kong et al. 2012; Michaelson et al. 2012). Remarkable findings in this field include the effect of the sex of the transmitting parent (summarized by the alpha ratio) and paternal age at conception.
We used the repeatedly confirmed genome-wide value of 1.2 × 10−8/position/gamete/generation to infer an MSY-specific value of 0.64 × 10−9/position/gamete/year (for details, see Supplemental Text).
Time estimates and phylogeographic analyses
We applied two independent methods for dating the tree nodes. The first is based on the rho statistic, i.e., the average number of differing sites between a set of sequences and a specified common ancestor (which needs not be among the sampled sequences) (Forster et al. 1996). This statistic is linearly related to time and mutation rate (rho = μ × t) (Jobling et al. 2004), assuming constancy of the rate across the tree branches. The statistic and associated confidence interval were computed with the program Network (Bandelt et al. 1999). The mutation rate was as reported above, corresponding to a substitution every 1044 yr over the 1.5-Mb sequence scored.
We also applied a Bayesian estimation of node ages, through BEAST (Drummond and Rambaut 2007), using the entire set of 2386 variable positions. This makes it possible to consider complex models, including different substitution matrices, a relaxed clock for heterogeneous rates across the tree branches, and different dynamics of population growth (for details, see Supplemental Text). The heterogeneity of substitution rates in different tree branches was tested by repeating the same runs under a strict clock model with identical priors, and comparing the tree likelihoods by means of Bayes factors (Nylander et al. 2004). A test to compare rates between selected branches of the tree was performed by using a χ2 test as reported in equation 10 in Kumar and Filipski (2001), taking into account only nonrecurrent mutations, as recommended.
In order to make inferences on the most likely locations for ancestors corresponding to nodes in the tree, the 68 subjects were assigned to four geographic macroregions, i.e., central-western Africa, southern Africa, eastern Africa, and the rest of the world (comprising northern Africa and other continents). A discrete phylogeographic model was examined by both Bayesian search and maximum parsimony. A run of BEAST using geographic categories as a discrete trait (Lemey et al. 2009) was performed. The maximum parsimony approach implemented in the program RASP (Yu et al. 2010) was applied to the maximum parsimony tree of Figure 2, allowing ancestral ranges to include no more than two of the four geographic macroregions. For each of the two phylogeographic analyses, the inference on ancestral locations for each node was represented as a pie chart and overlaid on the BEAST tree.
Data access
Variant positions are deposited in dbSNP (www.ncbi.nlm.nih.gov/SNP/; handle: HUMGEN, ssid ss778077189–ss778079576) and are available as supporting information online.
Supplementary Material
Acknowledgments
This work was supported by grants PRIN-MIUR 2009P2CNKK_003 to A.N. and 2009P2CNKK_004 to R.S., and by grant “Ricerche Universitarie 2012” to F.C.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.160788.113.
References
- The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Amero KK, Hellani A, Gonzalez AM, Larruga JM, Cabrera VM, Underhill PA 2009. Saudi Arabian Y-chromosome diversity and its relationship with nearby regions. BMC Genet 10: 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage SJ, Jasim SA, Marks AE, Parker AG, Usik VI, Uerpmann HP 2011. The southern route “out of Africa”: Evidence for an early expansion of modern humans into Arabia. Science 331: 453–456 [DOI] [PubMed] [Google Scholar]
- Atkinson QD, Gray RD, Drummond AJ 2009. Bayesian coalescent inference of major human mitochondrial DNA haplogroup expansions in Africa. Proc Biol Sci 276: 367–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, Griffing AR, Coté M, Henrion E, Spiegelman D, Tarabeux J, et al. 2010. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am J Hum Genet 87: 316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A 1999. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48 [DOI] [PubMed] [Google Scholar]
- Batini C, Ferri G, Destro-Bisol G, Brisighelli F, Luiselli D, Sánchez-Diz P, Rocha J, Simonson T, Brehm A, Montano V, et al. 2011. Signatures of the preagricultural peopling processes in sub-Saharan Africa as revealed by the phylogeography of early Y chromosome lineages. Mol Biol Evol 28: 2603–2613 [DOI] [PubMed] [Google Scholar]
- Beleza S, Gusmão L, Amorim A, Carracedo A, Salas A 2005. The genetic legacy of western Bantu migrations. Hum Genet 117: 366–375 [DOI] [PubMed] [Google Scholar]
- Berniell-Lee G, Calafell F, Bosch E, Heyer E, Sica L, Mouguiama-Daouda P, van der Veen L, Hombert JM, Quintana-Murci L, Comas D 2009. Genetic and demographic implications of the Bantu expansion: Insights from human paternal lineages. Mol Biol Evol 26: 1581–1589 [DOI] [PubMed] [Google Scholar]
- Burroughs WJ. 2005. Climate change in prehistory. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Busby GBJ, Brisighelli F, Sánchez-Diz P, Ramos-Luis E, Martinez-Cadenas C, Thomas MG, Bradley DG, Gusmão L, Winney B, Bodmer W, et al. 2012. The peopling of Europe and the cautionary tale of Y chromosome lineage R-M269. Proc Biol Sci 279: 884–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MC, Tishkoff SA 2010. The evolution of human genetic and phenotypic variation in Africa. Curr Biol 20: R166–R173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CD, Chong JX, Malig M, Ko A, Dumont BL, Han L, Vives L, O'Roak BJ, Sudmant PH, Shendure J, et al. 2012. Estimating the human mutation rate using autozygosity in a founder population. Nat Genet 44: 1277–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañeda IS, Mulitza S, Schefuss E, Lopes dos Santos RA, Sinninghe Damsté JS, Schouten S 2009. Wet phases in the Sahara/Sahel region and human migration patterns in North Africa. Proc Natl Acad Sci 106: 20159–20163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X, Wang K 2012. wANNOVAR: Annotating genetic variants for personal genomes via the web. J Med Genet 49: 433–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesner CA, Rose WI, Deino A, Drake R, Westgate JA 1991. Eruptive history of Earth's largest quaternary caldera (Toba, Indonesia) clarified. Geology 19: 200–203 [Google Scholar]
- Chiaroni J, Underhill PA, Cavalli-Sforza LL 2009. Y chromosome diversity, human expansion, drift and cultural evolution. Proc Natl Acad Sci 106: 20174–20179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JEM, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani F, La Fratta R, Santolamazza P, Sellitto D, Pascone R, Moral P, Watson E, Guida V, Beraud Colomb E, Zaharova B, et al. 2004. Phylogeographic analysis of haplogroup E3b (E-M215) Y chromosomes reveals multiple migratory events within and out of Africa. Am J Hum Genet 74: 1014–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani F, La Fratta R, Trombetta B, Santolamazza P, Sellitto D, Beraud Colomb E, Dugoujon JM, Crivellaro F, Benincasa T, Pascone R, et al. 2007. Tracing past human male movements in northern/eastern Africa and western Eurasia: New clues from Y-chromosomal haplogroups E-M78 and J-M12. Mol Biol Evol 24: 1300–1311 [DOI] [PubMed] [Google Scholar]
- Cruciani F, Trombetta B, Sellitto D, Massaia A, Destro-Bisol G, Watson E, Beraud Colomb E, Dugoujon JM, Moral P, Scozzari R 2010. Human Y chromosome haplogroup R-V88: A paternal genetic record of early mid Holocene trans-Saharan connections and the spread of Chadic languages. Eur J Hum Genet 18: 800–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani F, Trombetta B, Antonelli C, Pascone R, Valesini G, Scalzi V, Vona G, Melegh B, Zagradisnik B, Assum G, et al. 2011a. Strong intra- and inter-continental differentiation revealed by Y chromosome SNPs M269, U106 and U152. Forensic Sci Int Genet 5: e49–e52 [DOI] [PubMed] [Google Scholar]
- Cruciani F, Trombetta B, Massaia A, Destro-Bisol G, Sellitto D, Scozzari R 2011b. A revised root for the human Y chromosomal phylogenetic tree: The origin of patrilineal diversity in Africa. Am J Hum Genet 88: 814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes V, Alshamali F, Alves M, Costa MD, Pereira JB, Silva NM, Cherni L, Harich N, Cerny V, Soares P, et al. 2012. The Arabian cradle: Mitochondrial relicts of the first steps along the southern route out of Africa. Am J Hum Genet 90: 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster P, Harding R, Torroni A, Bandelt HJ 1996. Origin and evolution of Native American mtDNA variation: A reappraisal. Am J Hum Genet 59: 935–945 [PMC free article] [PubMed] [Google Scholar]
- Francalacci P, Morelli L, Angius A, Berutti R, Reinier F, Atzeni R, Pilu R, Busonero F, Maschio A, Zara I, et al. 2013. Low-pass DNA sequencing of 1200 Sardinians reconstructs European Y-chromosome phylogeny. Science 341: 565–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Mittnik A, Johnson PL, Bos K, Lari M, Bollongino R, Sun C, Giemsch L, Schmitz R, Burger J, et al. 2013a. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr Biol 23: 553–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Altshuler D, Shendure J, Nickerson DA et al. 2013b. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493: 216–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün R, Stringer C, McDermott F, Nathan R, Porat N, Robertson S, Taylor L, Mortimer G, Eggins S, McCulloch M 2005. U-series and ESR analyses of bones and teeth relating to the human burials from Skhul. J Hum Evol 49: 316–334 [DOI] [PubMed] [Google Scholar]
- Hammer MF, Karafet T, Rasanayagam A, Wood ET, Altheide TK, Jenkins T, Griffiths RC, Templeton AR, Zegura SL 1998. Out of Africa and back again: Nested cladistic analysis of human Y chromosome variation. Mol Biol Evol 15: 427–441 [DOI] [PubMed] [Google Scholar]
- Henn BM, Cavalli-Sforza LL, Feldman MW 2012. The great human expansion. Proc Natl Acad Sci 109: 17758–17764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling MA, Hurles ME, Tyler-Smith C 2004. Human evolutionary genetics. Garland Science, New York [Google Scholar]
- Karafet TM, Mendez FL, Meilerman MB, Underhill PA, Zegura SL, Hammer MF 2008. New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res 18: 830–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Filipski AJ 2001. Molecular clock: Testing. In Encyclopedia of life sciences. Macmillan, London. doi: 10.1038/npg.els.0001803. [Google Scholar]
- Lemey P, Rambaut A, Drummond AJ, Suchard MA 2009. Bayesian phylogeography finds its roots. PLoS Comput Biol 5: e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, Wang J 2009. SNP detection for massively parallel whole-genome resequencing. Genome Res 19: 1124–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, Indap AR, Schmidt S, Boyko AR, Hernandez RD, Hubisz MJ, Sninsky JJ, White TJ, Sunyaev SR, Nielsen R, et al. 2008. Proportionally more deleterious genetic variation in European than in African populations. Nature 451: 994–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manica A, Amos W, Balloux F, Hanihara T 2007. The effect of ancient population bottlenecks on human phenotypic variation. Nature 448: 346–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall I, Brown FH, Fleagle JG 2005. Stratigraphic placement and age of modern humans from Kibish, Ethiopia. Nature 433: 733–736 [DOI] [PubMed] [Google Scholar]
- Mellars PA, Gori KC, Carr M, Soares PA, Richards MB 2013. Genetic and archaeological perspectives on the initial modern human colonization of southern Asia. Proc Natl Acad Sci 110: 10699–10704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez FL, Krahn T, Schrack B, Krahn AM, Veeramah KR, Woerner AE, Fomine FLM, Bradman N, Thomas MG, Karafet TM, et al. 2013. An African American paternal lineage adds an extremely ancient root to the human Y chromosome phylogenetic tree. Am J Hum Genet 92: 454–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, et al. 2012. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151: 1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myres NM, Rootsi S, Lin AA, Järve M, King RJ, Kutuev I, Cabrera VM, Khusnutdinova EK, Pshenichnov A, Yunusbayev B, et al. 2011. A major Y-chromosome haplogroup R1b Holocene era founder effect in Central and Western Europe. Eur J Hum Genet 19: 95–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Z, Cox AJ, Mullikin JC 2001. SSAHA: A fast search method for large DNA databases. Genome Res 11: 1725–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylander JAA, Ronquist F, Huelsenbeck JP, Nieves-Aldrey JL 2004. Bayesian phylogenetic analysis of combined data. Syst Biol 53: 47–67 [DOI] [PubMed] [Google Scholar]
- Pagani L, Kivisild T, Tarekegn A, Ekong R, Plaster C, Gallego Romero I, Ayub Q, Mehdi SQ, Thomas MG, Luiselli D, et al. 2012. Ethiopian genetic diversity reveals linguistic stratification and complex influences on the Ethiopian gene pool. Am J Hum Genet 91: 83–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poznik GD, Henn BM, Yee M-C, Sliwerska E, Euskirchen GM, Lin AA, Snyder M, Quintana-Murci L, Kidd JM, Underhill PA, et al. 2013. Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science 341: 562–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Seielstad MT, Perez-Lezaun A, Feldman MW 1999. Population growth of human Y chromosomes: A study of Y chromosome microsatellites. Mol Biol Evol 16: 1791–1798 [DOI] [PubMed] [Google Scholar]
- Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL 2005. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci 102: 15942–15947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, et al. 2010. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328: 636–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca RA, Magoon G, Reynolds DF, Krahn T, Tilroe VO, Op den Velde Boots PM, Grierson AJ 2012. Discovery of Western European R1b1a2 Y chromosome variants in 1000 Genomes project data: An online community approach. PLoS ONE 7: e41634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JI, Usik VI, Marks AE, Hilbert YH, Galletti CS, Parton A, Geiling JM, Cerný V, Morley MW, Roberts RG 2011. The Nubian Complex of Dhofar, Oman: An African middle stone age industry in Southern Arabia. PLoS ONE 6: e28239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Marszalek JD, Alagappan RK, Skaletsky H, Page DC 2009. Remarkably little variation in proteins encoded by the Y chromosome's single-copy genes, implying effective purifying selection. Am J Hum Genet 85: 923–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally A, Durbin R 2012. Revising the human mutation rate: Implications for understanding human evolution. Nat Rev Genet 13: 745–753 [DOI] [PubMed] [Google Scholar]
- Schlebusch CM, Skoglund P, Sjödin P, Gattepaille LM, Hernandez D, Jay F, Li S, De Jongh M, Singleton A, Blum MG, et al. 2012. Genomic variation in seven Khoe-San groups reveals adaptation and complex African history. Science 338: 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scozzari R, Massaia A, D'Atanasio E, Myres NM, Perego UA, Trombetta B, Cruciani F 2012. Molecular dissection of the basal clades in the human Y chromosome phylogenetic tree. PLoS ONE 7: e49170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Ayub Q, Vermeulen M, Shao RG, Zuniga S, van der Gaag K, de Knijff P, Kayser M, Xue Y, Tyler-Smith C 2010. A worldwide survey of human male demographic history based on Y-SNP and Y-STR data from the HGDP-CEPH populations. Mol Biol Evol 27: 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindi S, Onal S, Peng H, Wu H, Raphael BJ 2012. An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol 13: R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P, Alshamali F, Pereira JB, Fernandes V, Silva NM, Afonso C, Costa MD, Musilová E, Macaulay V, Richards MB, et al. 2011. The expansion of mtDNA haplogroup L3 within and out of Africa. Mol Biol Evol 29: 915–927 [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattersall I, Schwartz JH 2008. The morphological distinctiveness of Homo sapiens and its recognition in the fossil record: Clarifying the problem. Evol Anthropol 17: 49–54 [Google Scholar]
- Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW 2000. Recent common ancestry of human Y chromosomes: Evidence from DNA sequence data. Proc Natl Acad Sci 97: 7360–7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP 2013. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Gonder MK, Henn BM, Mortensen H, Knight A, Gignoux C, Fernandopulle N, Lema G, Nyambo TB, Ramakrishnan U, et al. 2007. History of click-speaking populations of Africa inferred from mtDNA and Y chromosome genetic variation. Mol Biol Evol 24: 2180–2195 [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, Hirbo JB, Awomoyi AA, Bodo JM, Doumbo O, et al. 2009. The genetic structure and history of Africans and African Americans. Science 324: 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta B, Cruciani F, Sellitto D, Scozzari R 2011. A new topology of the human Y chromosome haplogroup E1b1 (E-P2) revealed through the use of newly characterized binary polymorphisms. PLoS ONE 6: e16073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill PA, Kivisild T 2007. Use of Y chromosome and mitochondrial DNA population structure in tracing human migrations. Annu Rev Genet 41: 539–564 [DOI] [PubMed] [Google Scholar]
- Underhill PA, Shen P, Lin AA, Jin L, Passarino G, Yang WH, Kauffman E, Bonné-Tamir B, Bertranpetit J, Francalacci P, et al. 2000. Y chromosome sequence variation and the history of human populations. Nat Genet 26: 358–361 [DOI] [PubMed] [Google Scholar]
- Underhill PA, Myres NM, Rootsi S, Metspalu M, Zhivotovsky LA, King RJ, Lin AA, Chow CET, Semino O, Battaglia V, et al. 2010. Separating the post-Glacial coancestry of European and Asian Y chromosomes within haplogroup R1a. Eur J Hum Genet 18: 479–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weale ME, Shah T, Jones AL, Greenhalgh J, Wilson JF, Nymadawa P, Zeitlin D, Connell BA, Bradman N, Thomas MG 2003. Rare deep-rooting Y chromosome lineages in humans: Lessons for phylogeography. Genetics 165: 229–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ayub Q, Chen Y, McCarthy S, Hou Y, Carbone I, Xue Y, Tyler-Smith C 2013. A calibrated human Y-chromosomal phylogeny based on resequencing. Genome Res 23: 388–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder JA, Mobasher Z, Hammer MF 2004. Genetic evidence for unequal effective population sizes of human females and males. Mol Biol Evol 21: 2047–2057 [DOI] [PubMed] [Google Scholar]
- Xue Y, Wang Q, Long Q, Ng BL, Swerdlow H, Burton J, Skuce C, Taylor R, Abdellah Z, Zhao Y, et al. 2009. Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree. Curr Biol 19: 1453–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Harris AJ, He X 2010. S-DIVA (Statistical Dispersal-Vicariance Analysis): A tool for inferring biogeographic histories. Mol Phylogenet Evol 56: 848–850 [DOI] [PubMed] [Google Scholar]
- Zalloua PA, Xue Y, Khalife J, Makhoul N, Debiane L, Platt DE, Royyuru AK, Herrera RJ, Hernanz DFS, Blue-Smith J, et al. 2008. Y-chromosomal diversity in Lebanon is structured by recent historical events. Am J Hum Genet 82: 873–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.