

Abstract
The United Nations has declared 2014 the International Year of Crystallography, and in commemoration, this review features a selection of 54 notable macromolecular crystal structures that have illuminated the field of biophysics in the 54 years since the first excitement of the myoglobin and hemoglobin structures in 1960. Chronological by publication of the earliest solved structure, each illustrated entry briefly describes key concepts or methods new at the time and key later work leveraged by knowledge of the three-dimensional atomic structure.
1. Myoglobin
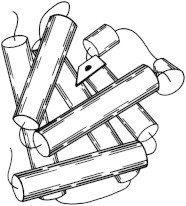
Myoglobin was the very first atomic-detail x-ray crystal structure of a biological macromolecule (Protein Databank (PDB) PDB:1MBN (1)). It showed us that the 3.6-residue/turn Pauling α-helix was indeed true, although the helices interacted at unexpected angles. Myoglobin became, and still is, a model system for many biophysical methods (2) including neutron diffraction (PDB:1MB5 (3)) and the kinetics of binding O2 and other small ligands (4). (Fig. 1. Myoglobin helix cylinders (PDB:1MBN)).
2. Hemoglobin
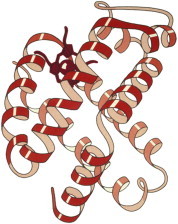
The hemoglobin structure was the culmination of Max Perutz’s 18-year quest. It appeared along with myoglobin, at lower resolution but clearly with the same fold for all chains of its α2/β2 tetramer (PDB:1MHB, PDB:1DHB (5)). The idea that breathing required a protein channel to open for O2 to access the heme (6), as in myoglobin, was startling. The coupled change of local and quaternary conformations between oxy and deoxy forms explained the cooperativity of O2 binding, and for decades hemoglobin was both the main teaching example and also a focus of research on allostery. (Fig. 2. Hemoglobin β-chain ribbons (PDB:1MHB)).
3. Hen-egg lysozyme
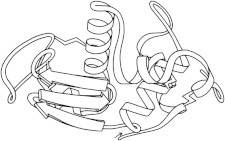
Hen egg-white lysozyme (PDB:1LYZ (7)) was the first β-sheet seen, the first enzyme structure solved (8), and the first to have coordinates refined (in real-space; PDB:2LYZ, PDB:3LYZ, PDB:4LYZ, PDB:5LYZ, PDB:6LYZ (9)). It forms large, high-resolution crystals very readily in many space groups, and has been the control case for nearly every new crystallographic method and instrument, most recently nanocrystals for the free-electron laser (PDB:4ET8 (10)) and microcrystals for micro electron diffraction (PDB:3J4G (11)). (Fig. 3. Hen-egg-white lysozyme (PDB:2LYZ)).
4. Ribonuclease A
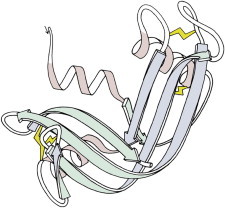
Ribonuclease A (PDB:2RSA (12)), stabilized by SS bonds, had prompted Anfinsen’s original work on protein folding. The cleaved ribonuclease S form (PDB:1RNS (13)) showed a nearly identical structure, both overall and in the active site, and was shown to be enzymatically active in the crystal, addressing doubts about the relevance of protein crystal structures to biological function. (Fig. 4. SS in ribonuclease A (PDB:2RSA)).
5. Chymotrypsin
The crystal structures of chymotrypsin (PDB:2CHA (14)), chymotrypsinogen (PDB:1CHG (15)), and trypsin (PDB:1PTN (16)) illuminated the catalytic mechanism of serine proteases, their differing substrate specificities, and the activation process that buries a cleaved chain end to properly rearrange the active site. [Note: Insertion-code residue numbering (hated by programmers) made Ser195 and His57 intuitive and textbook-worthy.] (Fig. 5. Ser protease: elastase (PDB:1EST (138))).
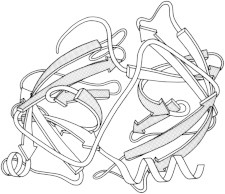
6. Carboxypeptidase
Carboxypeptidase was the first look at an exopeptidase that cleaves at the C-terminal rather than internal residues, and at a metalloprotease, with Zn at the active site (PDB:1CPA (17)). It also showed parallel β-sheet for the first time, which twists through the hydrophobic core, flanked by layers of helices in right-handed β−α−β connections. (Fig. 6. Carboxypeptidase A (PDB:1CPA)).
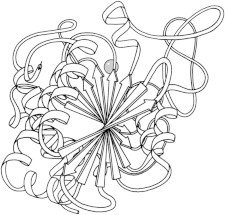
7. Subtilisin
Subtilisin (PDB:1SBT (18)) was a second type of serine protease with a near-identical active site to the trypsin relatives, but with an entirely different overall fold. This was our first demonstration of convergent evolution at the atomic level. Later, an intensive mutational study on subtilisin documented the effects of all 19 other amino acids at each individual position (19). (Fig. 7. Subtilisin BPN′ (PDB:1SBT)).
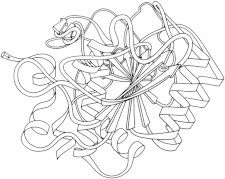
8. Lactate dehydrogenase
Lactate dehydrogenase (PDB:2LDH (20)) was the largest protein crystal structure until the 1980s, a tetramer of two-domain, 334-residue chains. Domain 1 of lactate dehydrogenase showed what was to become the classic version of the fold it shares with carboxypeptidase, subtilisin, and many other α/β-proteins: a doubly-wound topology known as the Rossmann fold. (Fig. 8. Rossmann fold: two views of LDH domain 1 (PDB:2LDH)).
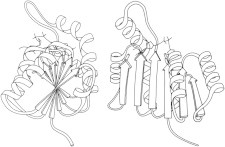
9. Trypsin inhibitor
The basic pancreatic trypsin inhibitor (BPTI) (PDB:2PTI (21)) uses SS bonds to hold a lysine and its backbone in the right conformation to form an essentially irreversible complex with trypsin (PDB:1PTC (22)). BPTI has been an important model system for protein folding, NMR (23), and computations such as the early molecular dynamics calculations of flip rates for buried aromatic rings (24). (Fig. 9. SS in trypsin inhibitor (PDB:2PTI)).
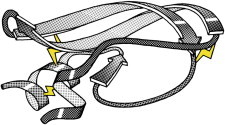
10. Rubredoxin
Rubredoxin (PDB:2RXN (25)) was the first redox structure solved, a minimalist protein with the iron bound by four Cys side chains from two loops at the top of β-hairpins. It diffracted to 1.2 Å, enabling reciprocal-space refinement of a protein (PDB:4RXN, PDB:5RXN (26)). [Note: beware PDB:4RXN, done without geometry restraints!] Archaeal rubredoxins account for many of the highest-resolution small structures in the PDB. (Fig. 10. Fe in rubredoxin (PDB:2RXN)).
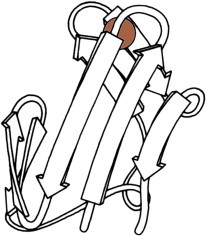
11. Insulin
Insulin (PDB:1INS (27)) is biophysically as well as medically interesting for its propensity to crystallize within the cell, its threefold Zn site and monomer-dimer-hexamer equilibrium, and its two disulfide-linked chains that require a long intervening Pro piece to fold up correctly. (Fig. 11. Insulin 2-chain monomer (PDB:1INS)).

12. Staphylococcal nuclease
Staphylococcal nuclease (PDB:1SNS (28)) was desired by Christian B. Anfinsen as a disulfide-free second model for protein folding to complement his original ribonuclease system. The nuclease has indeed been used extensively in folding studies (29,30). It was also the first structure for which multiresidue disordered regions were described, at both termini and in a loop near the active site. (Fig. 12. Staphylococcal nuclease (PDB:1SNS)).
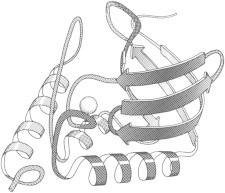
13. Cytochrome c
Cytochrome c (PDB:1CYT (31)) uses its heme Fe to play important roles in the electron-transport chain of photosynthesis. Structural detail determines how the local protein environment tunes redox potential, and how quantum tunneling can help an electron jump long distances such as in the cytochrome c/cytochrome c peroxidase complex (PDB:2PCB (32)). (Fig. 13. Cytochrome c (PDB:1CYT)).
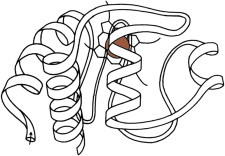
14. T4 lysozyme
The T4 phage lysozyme work was undertaken to understand what makes a mutation temperature-sensitive. The original structure was PDB:1LZM (33), and there are now more than 500 entries in the PDB, nearly all with deposited structure factors. This one molecule has been central to our improved understanding of the energetic and structural effects of packing quality, hydrogen bonding, secondary structure, solvent exposure, entropy, etc., as altered by sequence substitutions (34). Movie S1: T4 lysozyme domain motions: the upper domain first nods, in a classic domain-hinge motion, shown in two views. Then it shakes its head back and forth, in a torsional motion. (Fig. 14. Domain motion in T4 lysozyme mutants (PDB:150L (139), PDB:1KNI (140))).

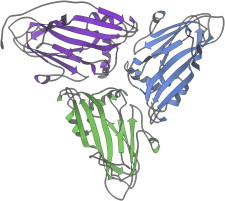
15. Immunoglobulins
Immunoglobulin fragments such as Fab’s (PDB:1FAB (35)) and VL dimers (PDB:1REI (36)) were very important but hard to crystallize, relative to earlier x-ray structures. The domains, with their Greek-key topology and their buried SS and Trp bracing the center of the β-barrel, explained the reliable fold of the framework. Some of the hypervariable loops could be classified into conformational categories that allowed prediction of binding-region shape from sequence (37,38). (Fig. 15. Immunoglobulin VL domain (PDB:1REI)).
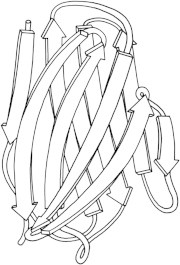
16. Superoxide dismutase
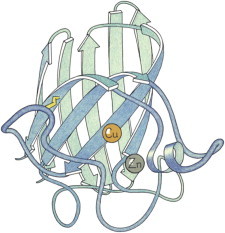
Cu,Zn superoxide dismutase (PDB:2SOD (39)) is a very efficient enzyme that protects against the cellular damage caused by O2- radicals; its dimer was seen to cover much of the surface by placing active-site funnels back to back, with electrostatic guidance inward. Superoxide dismutase and immunoglobulin domains together led to definition of the Greek-key β-barrel (40), one of the commonest all-β folds. (Fig. 16. Cu,Zn superoxide dismutase (PDB:2SOD (141))).
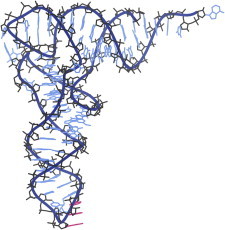
17. Transfer RNA
tRNA Phe was the first crystal structure of a large RNA (PDB:1TNA (41), PDB:3TNA (42)). It showed that the A-form stems of the classic cloverleaf basepair diagram actually stack in pairs on one another, forming an L-shape with well-ordered loops interacting at the corner and the anticodon triplet and CCA acceptor end 70 Å apart. These days we see how tRNAs move, bend, and interact in the ribosome, through the dynamic dance of translation (43,44). (Fig. 17. Transfer RNA Phe (PDB:1EHZ (142))).
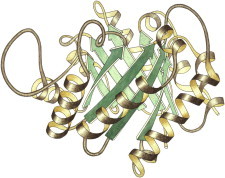
18. Triose phosphate isomerase
Triose phosphate isomerase (PDB:1TIM (45)) introduced the amazing TIM barrel fold, with a central cylinder of eight β-strands surrounded by a ring of eight helices, each joining adjacent strands with a right-handed β−α−β connection to produce a singly-wound fold. A sequence-conserved loop over the active site is disordered but folds down to protect catalysis, trading entropy with enthalpy to provide both specific binding and product release (46). (Fig. 18. Triose phosphate isomerase (PDB:1TIM)).
19. Icosahedral virus
Icosahedral virus structures, first tomato bushy stunt (PDB:2TBV (47)) and then Southern bean mosaic (PDB:2SBV (48)), were a huge step up in size, symmetry, and complexity, and required the development of new crystallographic methodology. There were many surprises, in how a single sequence accommodates five- versus sixfold neighbors, how protruding spikes are formed, and how the geometry changes during capsid development. (Fig. 19. Southern bean mosaic virus, three chains (PDB:2SBV)).
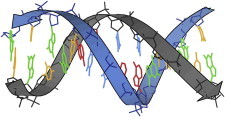
20. Dickerson DNA dodecamer
The Dickerson dodecamer of complementary DNA sequence CGCGAATTCGCG (PDB:1BNA (49)) provided the first in-depth analysis of structure and variation in double-helical B-form DNA, one of the most fundamental and ubiquitous atomic structures in biology. The details of AT versus GC basepairs and of the possible steps between stacked basepairs were analyzed by parameters such as rise, tilt, and roll. (Fig. 20. B-form DNA dodecamer (PDB:1BNA)).
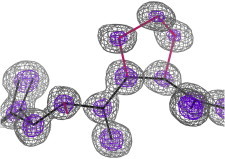
21. Crambin
Crambin, a tiny hydrophobic protein, was solved at 1.5 Å resolution from native S anomalous data (PDB:1CRN (50)), showing detailed water structure, then to a record 0.54 Å (PDB:1EJG (51)), with visible electron density in bond orbitals. Schmidt et al. (52) at 0.48 Å (PDB:3NIR) tried unrestrained refinement of H atoms, and crambin gives very high resolution neutron diffraction (53). (Fig. 21. Crambin electron density detail (PDB:1EJG)).
22. Calmodulin
Calmodulin (PDB:1CLN (54)) and troponin C (PDB:2TNC (55)) are Ca++ modulated regulatory systems with a pair of EF hand Ca++ binding domains connected in dumbbell shape by a startlingly noncompact long α-helix. It was later found by both NMR (56) and crystallography (57) that binding of a helical-peptide ligand produces a huge domain-hinge motion, closing down around the ligand into a compact, globular structure. Movie S2: Calmodulin closing around a ligand: the long helix between the separate Ca++-binding domains locally unfolds and bends in the middle, to let the domains form a compact globular domain around the helical-peptide ligand. (Fig. 22. Calmodulin: open + closed around ligand (PDB:1CLN, PDB:1CM1).
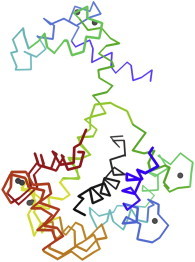
23. DNA polymerase
The architecture of DNA polymerase was first shown by the structure of Klenow fragment (PDB:1DPI (58)), with the growing DNA held across a palm and manipulated by fingers and thumb domains, illuminating processivity. Later structures demonstrated catalysis in the crystal, and showed how local distortion toward A-form could reliably distinguish correct Watson-Crick basepairing from nearly all other possibilities (PDB:2BDP (59)). (Fig. 23. DNA polymerase/DNA (PDB:2HHV (143))).
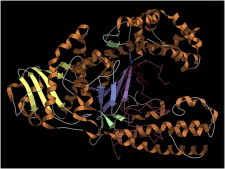
24. Photosynthetic reaction center
Photosynthetic reaction center (PDB:1PRC (60)) was the landmark, hard-earned first crystal structure of a membrane protein, showing us the trademark hydrophobic transmembrane helices, the associated globular domains, and the odd aromaticity and charge distribution of the layers coplanar with the membrane surface. For spectroscopy, it showed the light-capturing special pair of chlorophylls and the complex, nonequivalent pair of electron transfer pathways. (Fig. 24. Photosynthetic reaction center in membrane (PDB:1PRC)).
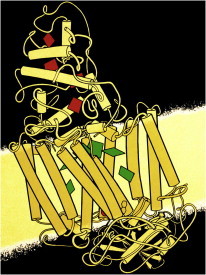
25. Repressor/DNA complexes
Repressor/DNA interactions were first visualized in detail by a series of complexes solved in the late 1980s, including Cro (PDB:1CRO (61)), Lambda (PDB:1LRD (62)), Trp (PDB:1TRO (63)), CAP (PDB:1CGP (64)), and Zn fingers (PDB:1ZAA (65)). They set expectations for nonspecific charged contacts to DNA backbone, mostly for dimeric, palindromic sites, and sometimes bending the DNA. Sequence specificity mostly used base edges in the major groove, but was not simple except for the Zn fingers, which had a neat modular system readily used for design. (Fig. 25. CAP protein dimer on bent DNA (PDB:1CGP)).
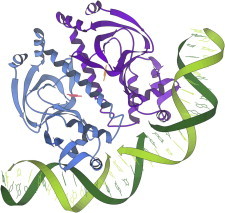
26. Histocompatibility antigen
Histocompatibility antigens (PDB:1HLA (66)) provided a new paradigm for a universal system that achieves specific binding of short peptides. They are presented on a flat β-sheet platter flanked by two helices, and the end result is a combination of somewhat specific interactions for peptide backbone and side chains in that groove, plus specificity from interaction of T-cell receptors with the total surface of HLA and peptide together. (Fig. 26. Histocompatibility antigen (PDB:3HLA (144))).
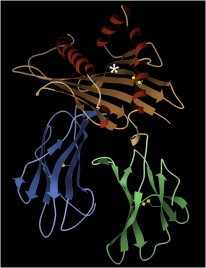
27. Ubiquitin
Ubiquitin (PDB:1UBQ (67)) has enormous biological significance, and has recently been much studied as the hub of an outstandingly large and varied protein-protein interaction network. The human protein is also small, stable, and tractable for genetic, physical, and computational studies, the results of which are synergistic: for example, no other protein supports collecting as much experimental NMR data, providing a testbed for calculations (PDB:2NR2 (68)). (Fig. 27. Ubiquitin computational models (PDB:2NR2)).
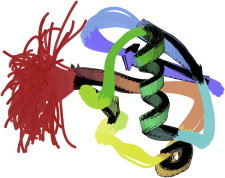
28. Bacteriorhodopsin
Trimers of bacteriorhodopsin protein form two-dimensional crystals in the bacterial purple membrane, and their structure (PDB:1BRD (69)) established electron diffraction as a feasible route to membrane protein structures. It showed the up-and-down topology of the seven trans-membrane helices and the retinol positioning and ligands, while PDB:2BRD (70) located the connecting loops and many lipids. (Fig. 28. Bacteriorhodopsin 3-mer, with lipids (PDB:2BRD)).
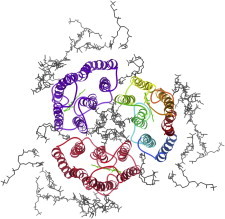
29. GCN4 coiled-coil
The GCN4 dimer is a prototypical coiled-coil structure with the heptad repeat that produces alternating layers of Leu and Val/Ile between the two α-helices. The crystal structure (PDB:2ZTA (71)) showed how the buried H-bonding of the repeat-busting Asn controls specificity of register and chain pairing. Later studies established rules for sequence variants to form trimers or tetramers (72) and for reliable design of coiled-coil dimerization domains. (Fig. 29. Core of GCN4 coiled-coil (PDB:2ZTA)).
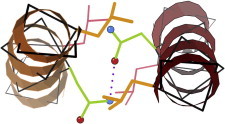
30. Beta-helix
The beta-helix was a new superfold of repeating wide spiral turns, each with three parallel β-strands. Very unusually for protein structure, it comes in two versions of opposite handedness. Pectate lyase (PDB:1PEC (73)), with a kidney-bean-shaped cross section, was first of the right-handed examples; an archaeal carbonic anhydrase (PDB:1THJ (74)), with an equilateral-triangle cross-section, was first of the left-handed examples. (Fig. 30. Left-handed β-helix, end view (PDB:1QRE (145))).
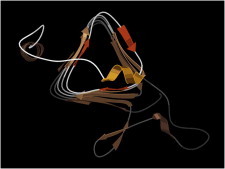
31. Collagen
The versatile toughness of collagen depends on the properties conferred by a triple helix of repeating Gly-Pro-hydroxyPro sequence (PDB:1CAG (75)). Pro keeps the strands extended, with high pitch, and prevents formation of most other conformations, while the OH group aids solubility. Gly lets the chains come close enough for an interstrand backbone H-bond. Gly→Ala, or other sequence variants, cause irregularity in the fibers. (Fig. 31. Collagen triple helix (PDB:1CAG)).
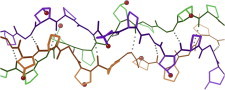
32. Barnase/barstar
The barnase/barstar complex (PDB:1BRS (76)) is a high-affinity, well-behaved, midsized enzyme-inhibitor pair that has served as a principal model system for study of protein/protein interactions by genetic, biophysical, and computational methods (77). (Fig. 32. Barnase/barstar interface (PDB:1BRS)).
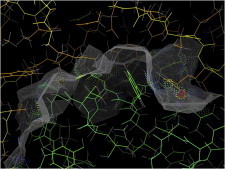
33. F1 ATPase
F1/F0 ATPase is a spectacularly elegant rotary molecular motor (or generator) studied by many biophysical techniques. The crystal structure of F1 (PDB:1BMF (78)) is essential to understanding the succession of large conformational changes within the trimer, and the atomic details of how those are coupled to each other, to the ATP hydrolysis or synthesis, and to rotation around the central γ-chain that connects with the F0 motor ring. Movie S3: F1 ATPase conformational changes: the 3 catalytic β (green, red, and blue), 3 α (cyan), and central γ (yellow) chains are rotated in 120° steps around the pseudo-3fold axis (green, tilted from vertical), then the conformation and state of one β chain is featured in a still. (Fig. 33. F1 ATPase (PDB:1BMF)).
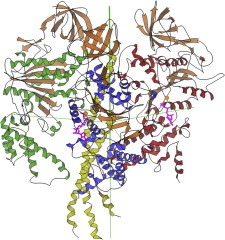
34. Heterotrimeric G proteins
Heterotrimeric G proteins translate the receptor messages from a great variety of external signals into downstream cascades that direct cellular responses. Crystal structures (PDB:1GG2 (79), PDB:1GOT (80)) showed how the catalytic α-subunit with its mobile switch I and II loops relates to the membrane, the accessibility of phosphorylation, and especially to binding with or dissociation from the large β-subunit with its sevenfold β-propeller architecture (81). (Fig. 34. G protein: α with GDP, β with propeller rainbow (PDB:1GG2)).
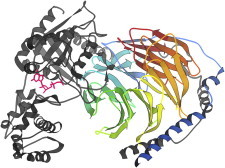
35. Green fluorescent protein
Green fluorescent protein is a biophysically interesting system in its own right—a stable cylindrical-can shape of β-sheet enclosing a helix and loop apparently poised just right for the unaided polypeptide to catalyze a covalently linked ring that is a strong green fluorophore. The structure (PDB:1EMA (82)) enabled study of that process and construction of variants with other colors, for multiple fluorescent labeling. (Fig. 35. GFP and fluorophore (PDB:1EMA).
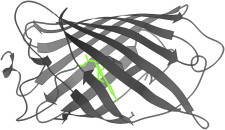
36. CDK2/cyclin A complex
CDK/cyclin complexes control cell cycle progression by regulating activity of the kinase partner. The classic CDK2/cyclin A structure (PDB:1FIN (83)) showed that in this case the cyclin reorients catalytic Glu51 on the PSTAIRE loop and pulls out the T-loop into a binding pocket in the cyclin, exposing and thus activating the kinase catalytic site. Further activation occurs on phosphorylation at the active site (84). Movie S4: Cyclin-A activation of CDK2: first the unbound CDK2 is shown, with red PSTAIRE and yellow T-loop, then their movement on cyclin binding to unblock the kinase active site and swing Glu51 into position. (Fig. 36. CDK/cyclin, PSTAIRE and T loops (PDB:1FIN, PDB:1HCK (146)).
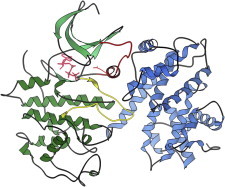
37. Kinesin
Kinesin and dynein are similar molecular motors with two ATPase heads at one end of a long coiled-coil and a cargo attachment at the other. Dynein brings cargo inward while kinesin moves it outward in the cell. Crystal structures of heads and neck (PDB:1BG2; (85), PDB:3KIN (86)) help explain mechanisms that leverage differing motions. Single-molecule visualization and force measurements show it walking, or limping along microtubules (87). (Fig. 37. Kinesin heads and neck (PDB:3KIN)).
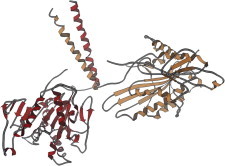
38. GroEL/ES
GroEL (PDB:1AON (88)) is the type example of a class of folding chaperones with paired cavities formed by two asymmetric seven-chain rings that open and close in alternation by a concerted ATP-driven conformational change. Partly folded proteins bind on the inner surface of an open GroEL ring. Upon ATP cleavage and conformational change, the bound protein is released to try folding inside the protected cavity, the top of which may be closed by a 7-mer cap of GroES. (Fig. 38. GroEL rings with GroE cap (PDB:1AON)).
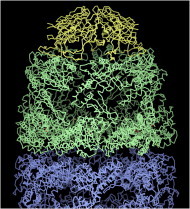
39. Nucleosome
Nucleosomes organize and sequester DNA but must make it accessible for replication when needed. The crystal structure (PDB:1AOI (89)) shows the tight two-turn, spiral wrap of DNA around the core of histone proteins, the interaction regions, and the largely disordered histone tails whose enzymatic modifications code for the level of DNA access. That access seems to involve histone variant incorporation, accessory proteins, and probably some large-scale opening of the nucleosome (90). (Fig. 39. Nucleosome DNA and histones (PDB:1AOI)).
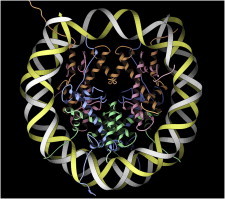
40. Group I intron
Group I self-splicing introns are ribozymes that self-catalyze their own splice-out and rejoining of the messenger RNA. The crystal structure of the Tetrahymena example (PDB:1GRZ (91)) was the largest RNA structure that preceded the ribosome, and it showed the complex architecture of double-helical junctions and tertiary contacts we now consider typical of RNA active sites. (Fig. 40. Tetrahymena group I intron (PDB:1GRZ)).
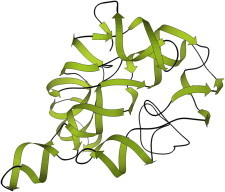
41. DNA topoisomerase
DNA topoisomerases take care of the entanglements that routinely result from basic processes such as replicating a coiled DNA double helix. Topoisomerase type I relaxes overtwisted supercoils, and the structures (PDB:1A31, PDB:1A35 (92) show that they cleave one strand, hold on to an end, let the helix twist relax, and then reconnect it. Type II topoisomerases (PDB:1BGW (93)) cut both strands and pass a separate DNA helix through the gap, to decatenate DNA rings. (Fig. 41. Topoisomerase II composite (PDB:1BGW, PDB:1EI1 (147))).
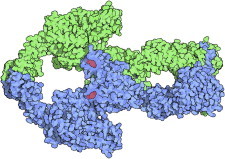
42. Tubulin α/β-dimer
The dimer of tubulin α- and β-subunits is the basic building block of microtubules. The electron crystallography structure (PDB:1TUB (94)) shows their detailed interactions and geometrical relationships, aiding modeling of both the straight assembled form and the curved, dissociated form of individual tubulin fibers. Ensuing studies combine x-ray, electron microscopy (EM), and single-molecule methods to analyze the complex dynamic control of assembly and disassembly at the plus (+) and minus (−) microtubule ends (95). (Fig. 42. Tubulin α/β dimer (PDB:1TUB)).
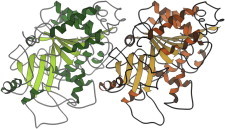
43. K+ channel
The potassium channel (PDB:1BL8 (96)) was the first ion channel crystal structure. Most unexpected and interesting was the specificity region, which transports K+ but not the slightly smaller Na+. Each of the four chains contributes a TVGYG sequence that uses glycine’s expanded conformational range to orient successive backbone carbonyls inward, forming rings of four liganding oxygen atoms at just the right diameter for optimal fit to the K+ ion. (Fig. 43. Selectivity filter of K+ channel (PDB:1BL8)).
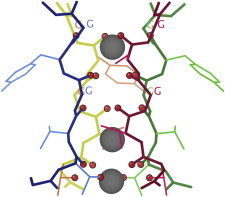
44. Holliday junction
Holliday junctions are the compact four-way junctions that allow helical crossover and migration during DNA replication, and the many DNA recombination processes such as crossover, integration, and transposition. The first crystal structures were solved in complex with Cre (PDB:2CRX (97)) or RuvA proteins (PDB:1C7Y (98)). A series of model systems followed, stabilized in various ways and in various geometries (e.g., PDB:1L6B (99)). (Fig. 44. Holliday junction DNA (PDB:2CRX)).
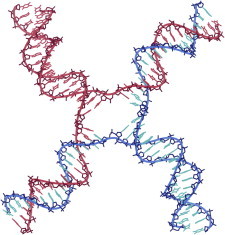
45. Ribosome
Ribosome structure in atomic detail was first achieved for entire ribosomal subunits in 2000 (PDB:1JJ2 (100), PDB:1FJG (101), PDB:1FKA (102)), and for the full Escherichia coli 70S in 2005 (PDB:2AVY, PDB:2AW4 (103)). Those structures showed that RNA is indeed the catalytic agent of protein synthesis, leveraged the extensive cryoEM work on the ribosome, and tripled the data for structural bioinformatics of RNA. They enabled mechanistic studies of drug binding or translation factor interactions, and especially single-molecule experiments to follow the large coupled motions and forces of the dynamic translation cycle (43). (Fig. 45. Escherichia coli 70S ribosome (PDB:4GD1, PDB:4GD2 (40))).

46. AAA+ ATPase
AAA+ ATPases comprise a versatile, ATP-powered motor system that acts on protein or DNA chains threaded through a ring of subunits. For instance, HslU (PDB:1G3I (104)) is a 6-mer ring that caps the bacterial proteasome and feeds proteins targeted for destruction into the central protease cavity, while SV40 LTag is a helicase that melts DNA replication forks (PDB:1SVM (105)). The asymmetrical clamp-loader, in contrast (PDB:3U5Z (106)), spirals around DNA to bind the sliding-clamp trimer in an open lock-washer form, pushes the clamp closed, then uses ATP hydrolysis to dissociate. Movie S5: Functional cycle of the clamp-loader AAA+ ATPase: the asymmetrical clamp-loader pentamer (green, with pink ATP) spirals around a DNA double helix (yellow) and binds the sliding-clamp trimer (brown) around the DNA in an open lock-washer position. (Fig. 46. Clamp (brown), clamp-loader, and DNA (PDB:3U5Z)).
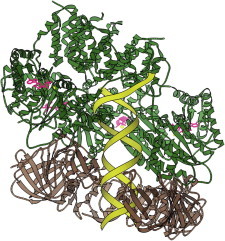
47. Ankyrin repeats
Ankyrin repeats provide versatile protein-binding elements for the cytoskeleton, and are representative of other modular α/α or α/β sequence repeats such as RNA-binding pumilio domains (PDB:1M8Y (107)) or Leu-rich repeats (PDB:1DFJ (108)). The ankyrin crystal structure (PDB:1N11 (109)) showed the principles of its modular architecture, which has also turned out to be readily co-opted for design or selection of new, specific protein-protein binding (110). Movie S6: Structure of 12 ankyrin cytoskeletal repeats: rocking emphasizes the long, open spiral curl of the ankyrin helix-pair and loop repeats, colored blue to red from N- to C-terminus, then the structure is chipped away and rebuilt to show the regular, simple repeat. (Fig. 47. Twelve ankyrin repeats (PDB:1N11)).
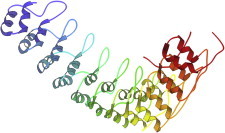
48. TOP7 designed protein
TOP7 was the first fully de novo design of a novel globular protein topology to be proven correct in detail by a high-resolution structure of the experimental construct (PDB:1QYS (111)). It was a proof-of-concept milestone: protein design is still far from repeatably foolproof, but TOP7 confirms the growing utility of the ROSETTA software system both for design and prediction, and now also as an important aid in experimental structure determination by NMR (112), cryoEM (113), and even crystallography (114). (Fig. 48. TOP7 designed protein (PDB:1QYS)).
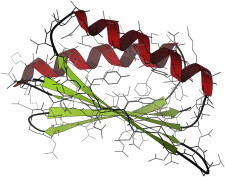
49. Circadian clock
The circadian clock from cyanobacteria (KaiA PDB:1R8J (115), KaiB PDB:1R5P (116), KaiC PDB:2GBL (117)) is the simplest such system known, with just three component proteins. KaiA and KaiC compete to phosphorylate and dephosphorylate buried sites on the large hexameric KaiB, acting slowly enough to produce an approximate 24-h cycle that is further synchronized by light. (Fig. 49. Space-fill drawing of KaiB (PDB:1R5P)).
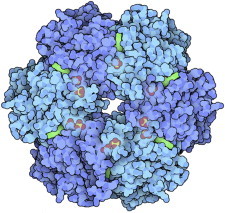
50. Riboswitch
Riboswitches show another recently appreciated function of RNA in addition to catalysis, aptamer binding, RNA interference, etc. Some messenger RNAs, such as the initial G and A riboswitch examples (PDB:1U8D (118), PDB:1Y26 (119)), form tertiary structures that specifically bind small-molecule ligands and change conformation to up- or downregulate the expression of their own gene. The change involves switches in basepairing, but is not as simple as first thought and constitutes an ongoing biophysical puzzle. (Fig. 50. G riboswitch with ligand (PDB:1U8D)).

51. Exosome
Exosomes are large, dynamic assemblies central to the highly regulated process of mRNA decay. The crystal structure of a nine-subunit human exosome (PDB:2NN6 (120)) will serve as a textbook from which to learn why evolution chose to use related but distinct proteins for such a job, and more generally, to learn the subtle rules for redesigning protein-protein interfaces to make many separate pairs that each interact with reliably high specificity. Movie S7: The eukaryotic 9-subunit exosome, a machine for mRNA decay: the exosome is rotated from a 3-fold top view to a side view, then rocked to show the shell of related but quite distinct chains, with only a single catalytic site inside. (Fig. 51. Top view of nine-subunit exosome (PDB:2NN6).
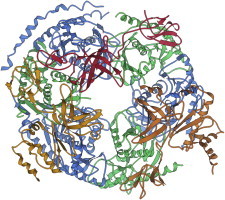
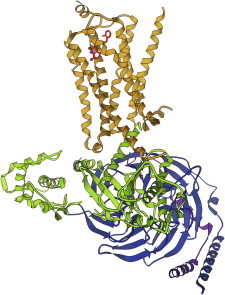
52. β2-Adrenergic receptor
β2-adrenergic receptor, first by itself (PDB:2RH1 (121)) and then in complex with its G protein (PDB:3SN6 (122)), are the first, long-sought structures for one of the numerous and critically important G-protein-coupled receptors. The receptor transmits the message that binding has occurred, in through the membrane to the G-protein, which initiates a signaling cascade to control cell response. Novel methodologies were used to solve these structures, including crystallization in the lipid cubic phase (123), a viscous and totally intermeshed topological oddity. (Fig. 52. β2-adrenergic receptor (gold) with its G protein (PDB:3SN6)).
53. Vault particle
Vaults are very big molecular containers common inside eukaryotic cells. Each chain of 12 domains is nearly 400 Å long, and 39 of them swirl together to enclose each half of the vault (PDB:2ZUO, PDB:2ZV4, PDB:2ZV5 (124), PDB:4HL8 (125)). Several different biological functions have been suggested for vaults, but they already provide us with a provocative example of an unusual large-scale architecture. (Fig. 53. Thirty-nine chains in half a vault (PDB:2ZUO, PDB:2ZV4, PDB:2ZV5)).

54. Free-electron laser crystallography
Free-electron laser crystallography is without question the most spectacular new technical development in the field. A jet of droplets with nanocrystals is sent past a beam of femtosecond x-ray laser pulses, and diffraction is recorded before the crystal has time to explode. This has the potential for determining otherwise intractable molecular structures and for accessing rapid time steps. The many challenging problems with data and analysis appear to be solvable. After demonstration structures of photosystems and lysozyme (e.g., PDB:3PCQ (126), PDB:4FBY (127), PDB:4ET8 (10)), there is now a novel structure of propeptide-inhibited trypanosomal cathepsin B (PDB:4HWY (128)) potentially useful for drug design. (Fig. 54. Cα splines and chromophores (pink) in Photosystem II (PDB:4FBY)).
Technical Notes
All 54 of these structures are protein or nucleic acid macromolecules solved by crystallography, with atomic coordinates deposited in the Protein Data Bank (PDB), and chosen for their importance to biophysics. The chronological list from 1960 to 2013 is ordered by publication year of the article describing the earliest atomic-detail structure; coordinate deposition date could not be used because many were solved before the PDB started taking entries in 1972 (129). In each case, at least one notable, biophysically relevant aspect is briefly described, such as being the first of its kind, introducing a new concept, or enabling an important set of later work. In the on-line version a subset of the PDB files are tagged for interactive 3D visualization, providing added value such as an overview if the image is a detail or the second example of a comparison noted in the text.
Most of the early figures are hand-drawn ribbons (130) and most of the later ones are computer-graphics ribbons or stick figures made in the softwares MAGE (131) or KING (132), with some further processing in the software PHOTOSHOP (Adobe Systems, Mountain View, CA). Figs. 41 and 49 are space-fill drawings courtesy of David Goodsell, from the RCSB Molecule of the Month feature (134). Fig. 45 was made in QUTEMOL (135). All images are open-license with attribution (CC BY 3.0 US) and are available on Wikimedia Commons (133). Frames and rotations for movies were made in the softwares MAGE or KING, and include no interpolations between experimental structures.
This list will be available on Wikipedia as part of the Biophysical Society-sponsored WikiProject Biophysics (http://en.wikipedia.org/wiki/Wikipedia:WikiProject_Biophysics, under “New articles”), to encourage discussion, editing, and continuation of the compendium. Comments, edits, and additions are welcome, at the List of Biophysically Important Macromolecular Crystal Structures on the English Wikipedia (136).
Seven movies are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(14)00033-2.
Acknowledgments
We especially thank the crystallographers who have continually pushed for success on increasingly difficult and important structures and kept the field exciting, and the Protein Data Bank, which, from nearly the beginning, has made the coordinates, derived information, and now the experimental data easily available to anyone in the world (137). The referees of this review made several good additions of structures for inclusion.
The National Institutes of Health supported our own contributions over the years with grant No. R01-GM15000 and our current overviews and methods development with grant Nos. R01-GM73930, R01-GM73919, and P01-GM63210.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
- 1.Kendrew J.C., Dickerson R.E., Shore V.C. Structure of myoglobin: a three-dimensional Fourier synthesis at 2 Å resolution. Nature. 1960;185:422–427. doi: 10.1038/185422a0. [DOI] [PubMed] [Google Scholar]
- 2.Frauenfelder H., McMahon B.H., Fenimore P.W. Myoglobin: the hydrogen atom of biology and a paradigm of complexity. Proc. Natl. Acad. Sci. USA. 2003;100:8615–8617. doi: 10.1073/pnas.1633688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanson J.C., Schoenborn B.P. Real space refinement of neutron diffraction data from sperm whale carbonmonoxymyoglobin. J. Mol. Biol. 1981;153:117–146. doi: 10.1016/0022-2836(81)90530-1. [DOI] [PubMed] [Google Scholar]
- 4.Ruscio J.Z., Kumar D., Onufriev A.V. Atomic level computational identification of ligand migration pathways between solvent and binding site in myoglobin. Proc. Natl. Acad. Sci. USA. 2008;105:9204–9209. doi: 10.1073/pnas.0710825105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perutz M.F., Rossmann M.G., North A.C.T. Structure of hemoglobin: a three-dimensional Fourier synthesis at 5.5-Å resolution, obtained by x-ray analysis. Nature. 1960;185:416–422. doi: 10.1038/185416a0. [DOI] [PubMed] [Google Scholar]
- 6.Perutz M.F., Mathews F.S. An x-ray study of azide Met-hemoglobin. J. Mol. Biol. 1966;21:199–202. doi: 10.1016/0022-2836(66)90088-x. [DOI] [PubMed] [Google Scholar]
- 7.Blake C.C.F., Koenig D.F., Sarma V.R. Structure of hen egg-white lysozyme. A three-dimensional Fourier synthesis at 2 Ångstrom resolution. Nature. 1965;206:757–761. doi: 10.1038/206757a0. [DOI] [PubMed] [Google Scholar]
- 8.Warshel A., Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 9.Diamond R. Real-space refinement of the structure of hen egg-white lysozyme. J. Mol. Biol. 1974;82:371–391. doi: 10.1016/0022-2836(74)90598-1. [DOI] [PubMed] [Google Scholar]
- 10.Boutet S., Lomb L., Schlichting I. High-resolution protein structure determination by serial femtosecond crystallography. Science. 2012;337:362–364. doi: 10.1126/science.1217737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi D., Nannenga B.L., Gonen T. Three-dimensional electron crystallography of protein microcrystals. Elife. 2013;2:e01345. doi: 10.7554/eLife.01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kartha G., Bello J., Harker D. Tertiary structure of ribonuclease. Nature. 1967;213:862–865. doi: 10.1038/213862a0. [DOI] [PubMed] [Google Scholar]
- 13.Wyckoff H.W., Hardman K.D., Richards F.M. The structure of ribonuclease-S at 3.5 Å resolution. J. Biol. Chem. 1967;242:3984–3988. [PubMed] [Google Scholar]
- 14.Matthews B.W., Sigler P.B., Blow D.M. Three-dimensional structure of tosyl-α-chymotrypsin. Nature. 1967;214:652–656. doi: 10.1038/214652a0. [DOI] [PubMed] [Google Scholar]
- 15.Freer S.T., Kraut J., Xuong N.H. Chymotrypsinogen: 2.5-Ångstrom crystal structure, comparison with α-chymotrypsin, and implications for zymogen activation. Biochemistry. 1970;9:1997–2009. doi: 10.1021/bi00811a022. [DOI] [PubMed] [Google Scholar]
- 16.Fehlhammer H., Bode W. The refined crystal structure of bovine β-trypsin at 1.8 Å resolution. II. Crystallographic refinement, calcium binding site, benzamidine binding site and active site at pH 7.0. J. Mol. Biol. 1975;98:693–697. doi: 10.1016/s0022-2836(75)80005-2. [DOI] [PubMed] [Google Scholar]
- 17.Lipscomb W.N., Hartsuck J.A., Coppola J.C. Brookhaven Symposia in Biology: Structure, Function, and Evolution in Proteins No. 21. Brookhaven National Laboratory; Upton, NY: 1969. The structure of carboxypeptidase A, VII. The 2.0-Å resolution studies of the enzyme and of its complex with glycyltyrosine, and mechanistic deductions; pp. 24–90. [PubMed] [Google Scholar]
- 18.Wright C.S., Alden R.A., Kraut J. Structure of subtilisin BPN′ at 2.5 Ångström resolution. Nature. 1969;221:235–242. doi: 10.1038/221235a0. [DOI] [PubMed] [Google Scholar]
- 19.Wells J.A., Estell D.A. Subtilisin—an enzyme designed to be engineered. Trends Biochem. Sci. 1988;13:291–297. doi: 10.1016/0968-0004(88)90121-1. [DOI] [PubMed] [Google Scholar]
- 20.Adams M.J., Ford G.C., Wonacott A.J. Structure of lactate dehydrogenase at 2–8 Å resolution. Nature. 1970;227:1098–1103. doi: 10.1038/2271098a0. [DOI] [PubMed] [Google Scholar]
- 21.Huber R., Kukla D., Formanek H. The basic trypsin inhibitor of bovine pancreas. I. Structure analysis and conformation of the polypeptide chain. Naturwissenschaften. 1970;57:389–392. doi: 10.1007/BF00599976. [DOI] [PubMed] [Google Scholar]
- 22.Ruehlmann A., Kukla D., Huber R. Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor. Crystal structure determination and stereochemistry of the contact region. J. Mol. Biol. 1973;77:417–436. doi: 10.1016/0022-2836(73)90448-8. [DOI] [PubMed] [Google Scholar]
- 23.Dubs A., Wagner G., Wuthrich K. Individual assignments of amide proton resonances in the proton NMR spectrum of the basic pancreatic trypsin inhibitor. Biochem. Biophys. Acta. 1979;577:177–194. doi: 10.1016/0005-2795(79)90020-5. [DOI] [PubMed] [Google Scholar]
- 24.McCammon J.A., Karplus M. Dynamics of tyrosine ring rotations in a globular protein. Biopolymers. 1980;19:1375–1405. [Google Scholar]
- 25.Herriott J.R., Sieker L.C., Lovenberg W. Structure of rubredoxin: an x-ray study to 2.5 Å resolution. J. Mol. Biol. 1970;50:391–406. doi: 10.1016/0022-2836(70)90200-7. [DOI] [PubMed] [Google Scholar]
- 26.Watenpaugh K.D., Sieker L.C., Jensen L.H. Crystallographic refinement of rubredoxin at 1 × 2 Å degrees resolution. J. Mol. Biol. 1980;138:615–633. doi: 10.1016/s0022-2836(80)80020-9. [DOI] [PubMed] [Google Scholar]
- 27.Blundell T.L., Cutfield J.F., Vijayan M. Atomic positions in rhombohedral 2-zinc insulin crystals. Nature. 1971;231:506–511. doi: 10.1038/231506a0. [DOI] [PubMed] [Google Scholar]
- 28.Arnone A., Bier C.J., Yonath A. A high resolution structure of an inhibitor complex of the extracellular nuclease of Staphylococcus aureus. I. Experimental procedures and chain tracing. J. Biol. Chem. 1971;246:2302–2316. [PubMed] [Google Scholar]
- 29.Tucker P.W., Hazen E.E., Jr., Cotton F.A. Staphylococcal nuclease reviewed: a prototypic study in contemporary enzymology. IV. The nuclease as a model for protein folding. Mol. Cell. Biochem. 1979;23:131–141. doi: 10.1007/BF00219452. [DOI] [PubMed] [Google Scholar]
- 30.Shortle D. Staphylococcal nuclease: a showcase of m-value effects. Adv. Protein Chem. 1995;46:217–247. doi: 10.1016/s0065-3233(08)60336-8. [DOI] [PubMed] [Google Scholar]
- 31.Dickerson R.E., Takano T., Margoliash E. Ferricytochrome c. I. General features of the horse and bonito proteins at 2.8 Å resolution. J. Biol. Chem. 1971;246:1511–1535. [PubMed] [Google Scholar]
- 32.Pelletier H., Kraut J. Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 33.Matthews B.W., Remington S.J. The three-dimensional structure of the lysozyme from bacteriophage T4. Proc. Natl. Acad. Sci. USA. 1974;71:4178–4182. doi: 10.1073/pnas.71.10.4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baase W.A., Liu L., Matthews B.W. Lessons from the lysozyme of phage T4. Protein Sci. 2010;19:631–641. doi: 10.1002/pro.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poljak R.J., Amzel L.M., Saul F. The three-dimensional structure of the Fab′ fragment of a human myeloma immunoglobulin at 2.0-Ångstrom resolution. Proc. Natl. Acad. Sci. USA. 1974;71:3440–3444. doi: 10.1073/pnas.71.9.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Epp O., Lattman E.E., Palm W. The molecular structure of a dimer composed of the variable portions of the Bence-Jones protein REI refined at 2.0-Å resolution. Biochemistry. 1975;14:4943–4952. doi: 10.1021/bi00693a025. [DOI] [PubMed] [Google Scholar]
- 37.Chothia C., Lesk A.M. Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol. 1987;196:901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 38.North B., Lehmann A., Dunbrack R.L., Jr. A new clustering of antibody CDR loop conformations. J. Mol. Biol. 2011;406:228–256. doi: 10.1016/j.jmb.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richardson J.S., Thomas K.A., Richardson D.C. Crystal structure of bovine Cu,Zn superoxide dismutase at 3 Å resolution: chain tracing and metal ligands. Proc. Natl. Acad. Sci. USA. 1975;72:1349–1353. doi: 10.1073/pnas.72.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richardson J.S. β-Sheet topology and the relatedness of proteins. Nature. 1977;268:495–500. doi: 10.1038/268495a0. [DOI] [PubMed] [Google Scholar]
- 41.Sussman J.L., Kim S.-H. Three-dimensional structure of a transfer RNA in two crystal forms. Science. 1976;192:853–858. doi: 10.1126/science.775636. [DOI] [PubMed] [Google Scholar]
- 42.Jack A., Ladner J.E., Klug A. Crystallographic refinement of yeast phenylalanine transfer RNA at 2–5Å resolution. J. Mol. Biol. 1976;108:619–649. doi: 10.1016/s0022-2836(76)80109-x. [DOI] [PubMed] [Google Scholar]
- 43.Tinoco I., Jr., Gonzalez R.L., Jr. Biological mechanisms, one molecule at a time. Genes Dev. 2011;25:1205–1231. doi: 10.1101/gad.2050011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dunkle J.A., Wang L., Cate J.H. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science. 2011;332:981–984. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banner D.W., Bloomer A., Wilson I.A. Atomic coordinates for triose phosphate isomerase from chicken muscle. Biochem. Biophys. Res. Commun. 1976;72:146–155. doi: 10.1016/0006-291x(76)90972-4. [DOI] [PubMed] [Google Scholar]
- 46.Lolis E., Petsko G.A. Crystallographic analysis of the complex between triosephosphate isomerase and 2-phosphoglycolate at 2.5-Å resolution: implications for catalysis. Biochemistry. 1990;29:6619–6625. doi: 10.1021/bi00480a010. [DOI] [PubMed] [Google Scholar]
- 47.Harrison S.C., Olson A.J., Bricogne G. Tomato bushy stunt virus at 2.9 Å resolution. Nature. 1978;276:368–373. doi: 10.1038/276368a0. [DOI] [PubMed] [Google Scholar]
- 48.Abad-Zapatero C., Abdel-Meguid S.S., Tsukihara T. Structure of Southern bean mosaic virus at 2.8 Å resolution. Nature. 1980;286:33–39. doi: 10.1038/286033a0. [DOI] [PubMed] [Google Scholar]
- 49.Drew H.R., Wing R.M., Dickerson R.E. Structure of a B-DNA dodecamer: conformation and dynamics. Proc. Natl. Acad. Sci. USA. 1981;78:2179–2183. doi: 10.1073/pnas.78.4.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hendrickson W.A., Teeter M.M. Structure of the hydrophobic protein crambin determined directly from the anomalous scattering of sulfur. Nature. 1981;290:107–113. doi: 10.1038/290107a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jelsch C., Teeter M.M., Lecomte C. Accurate protein crystallography at ultra-high resolution: valence electron distribution in crambin. Proc. Natl. Acad. Sci. USA. 2000;97:3171–3176. doi: 10.1073/pnas.97.7.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmidt A., Teeter M., Lamzin V.S. Crystal structure of small protein crambin at 0.48 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011;67:424–428. doi: 10.1107/S1744309110052607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J.C.-H., Fisher Z., Langan P. Room-temperature ultrahigh-resolution time-of-flight neutron and x-ray diffraction studies of H/D-exchanged crambin. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012;68:119–123. doi: 10.1107/S1744309111051499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Babu Y.S., Sack J.S., Cook W.J. Three-dimensional structure of calmodulin. Nature. 1985;315:37–40. doi: 10.1038/315037a0. [DOI] [PubMed] [Google Scholar]
- 55.Herzberg O., James M.N.G. Structure of the calcium regulatory muscle protein troponin-C at 2.8 Å resolution. Nature. 1985;313:653–659. doi: 10.1038/313653a0. [DOI] [PubMed] [Google Scholar]
- 56.Ikura M., Clore G.M., Bax A. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- 57.Wall M.E., Clarage J.B., Phillips G.N., Jr. Motions of calmodulin characterized using both Bragg and diffuse x-ray scattering. Structure. 1997;5:1599–1612. doi: 10.1016/s0969-2126(97)00308-0. [DOI] [PubMed] [Google Scholar]
- 58.Ollis D.L., Brick P., Steitz T.A. Structure of large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature. 1985;313:762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 59.Kiefer J.R., Mao C., Beese L.S. Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature. 1998;391:304–307. doi: 10.1038/34693. [DOI] [PubMed] [Google Scholar]
- 60.Deisenhofer J., Epp O., Michel H. Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3Å resolution. Nature. 1985;318:618–624. doi: 10.1038/318618a0. [DOI] [PubMed] [Google Scholar]
- 61.Takeda Y., Kim J.G., Matthews B.W. Different interactions used by Cro repressor in specific and nonspecific DNA binding. J. Biol. Chem. 1986;261:8608–8616. [PubMed] [Google Scholar]
- 62.Jordan S.R., Pabo C.O. Structure of the λ-complex at 2.5 Å resolution: details of the repressor-operator interactions. Science. 1988;242:893–899. doi: 10.1126/science.3187530. [DOI] [PubMed] [Google Scholar]
- 63.Otwinowski Z., Schevitz R.W., Sigler P.B. Crystal structure of Trp repressor/operator complex at atomic resolution. Nature. 1988;335:321–329. doi: 10.1038/335321a0. [DOI] [PubMed] [Google Scholar]
- 64.Schultz S.C., Shields G.C., Steitz T.A. Crystal structure of a CAP-DNA complex: the DNA is bent by 90°. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- 65.Pavletich N.P., Pabo C.O. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 Å. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 66.Bjorkman P.J., Saper M.A., Wiley D.C. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329:506–512. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- 67.Vijay-Kumar S., Bugg C.E., Cook W.J. Structure of ubiquitin refined at 1.8 Å resolution. J. Mol. Biol. 1987;194:531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- 68.Richter B., Gsponer J., Vendruscolo M. The MUMO (minimal under-restraining minimal over-restraining) method for the determination of native state ensembles of proteins. J. Biomol. NMR. 2007;37:117–135. doi: 10.1007/s10858-006-9117-7. [DOI] [PubMed] [Google Scholar]
- 69.Henderson R., Baldwin J.M., Downing K.H. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 70.Grigorieff N., Ceska T.A., Henderson R. Electron-crystallographic refinement of the structure of bacteriorhodopsin. J. Mol. Biol. 1996;259:393–421. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 71.O’Shea E.K., Klemm J.D., Alber T. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science. 1991;254:539–544. doi: 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- 72.Harbury P.B., Zhang T., Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 73.Yoder M.D., Keen N.T., Jurnak F. New domain motif: the structure of pectate lyase C, a secreted plant virulence factor. Science. 1993;260:1503–1507. doi: 10.1126/science.8502994. [DOI] [PubMed] [Google Scholar]
- 74.Kisker C., Schindelin H., Rees D.C. A left-hand β-helix revealed by the crystal structure of a carbonic anhydrase from the archaeon Methanosarcina thermophila. EMBO J. 1996;15:2323–2330. [PMC free article] [PubMed] [Google Scholar]
- 75.Bella J., Eaton M., Berman H.M. Crystal and molecular structure of a collagen-like peptide at 1.9 Å resolution. Science. 1994;266:75–81. doi: 10.1126/science.7695699. [DOI] [PubMed] [Google Scholar]
- 76.Buckle A.M., Schreiber G., Fersht A.R. Protein-protein recognition: crystal structural analysis of a barnase-barstar complex at 2.0-Å resolution. Biochemistry. 1994;33:8878–8889. doi: 10.1021/bi00196a004. [DOI] [PubMed] [Google Scholar]
- 77.Wang T., Tomic S., Wade R.C. How optimal are the binding energetics of barnase and barstar? Biophys. J. 2004;87:1618–1630. doi: 10.1529/biophysj.104.040964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abrahams J.P., Leslie A.G.W., Walker J.E. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 79.Wall M.A., Coleman D.E., Sprang S.R. The structure of the G protein heterotrimer Giα1β1γ2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 80.Lambright D.G., Sondek J., Sigler P.B. The 2.0 Å crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 81.Lefkowitz R.J. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 2004;25:413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 82.Ormö M., Cubitt A.B., Remington S.J. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 83.Jeffrey P.D., Russo A.A., Pavletich N.P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 84.Russo A.A., Jeffrey P.D., Pavletich N.P. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 1996;3:696–700. doi: 10.1038/nsb0896-696. [DOI] [PubMed] [Google Scholar]
- 85.Kull F.J., Sablin E.P., Vale R.D. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380:550–555. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kozielski F., Sack S., Mandelkow E. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- 87.Asbury C.L., Fehr A.N., Block S.M. Kinesin moves by an asymmetric hand-over-hand mechanism. Science. 2003;302:2130–2134. doi: 10.1126/science.1092985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu Z., Horwich A.L., Sigler P.B. The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature. 1997;388:741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- 89.Luger K., Mäder A.W., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 90.Luger K., Dechassa M.L., Tremethick D.J. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012;13:436–447. doi: 10.1038/nrm3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Golden B.L., Gooding A.R., Cech T.R. A preorganized active site in the crystal structure of the Tetrahymena ribozyme. Science. 1998;282:259–264. doi: 10.1126/science.282.5387.259. [DOI] [PubMed] [Google Scholar]
- 92.Redinbo M.R., Stewart L., Hol W.G.J. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 93.Berger J.M., Gamblin S.J., Wang J.C. Structure and mechanism of DNA topoisomerase II. Nature. 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 94.Nogales E., Wolf S.G., Downing K.H. Structure of the α β tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 95.Nogales E.S., Wang H.-W. Structural mechanisms underlying nucleotide-dependent self-assembly of tubulin and its relatives. Curr. Opin. Struct. Biol. 2006;16:221–229. doi: 10.1016/j.sbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 96.Doyle D.A., Morais Cabral J., MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 97.Gopaul D.N., Guo F., van Duyne G.D. Structure of the Holliday junction intermediate in Cre-loxP site-specific recombination. EMBO J. 1998;17:4175–4187. doi: 10.1093/emboj/17.14.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ariyoshi M., Nishino T., Morikawa K. Crystal structure of the Holliday junction DNA in complex with a single RuvA tetramer. Proc. Natl. Acad. Sci. USA. 2000;97:8257–8262. doi: 10.1073/pnas.140212997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vargason J.M., Ho P.S. The effect of cytosine methylation on the structure and geometry of the Holliday junction: the structure of d(CCGGTACm5CGG) at 1.5 A resolution. J. Biol. Chem. 2002;277:21041–21049. doi: 10.1074/jbc.M201357200. [DOI] [PubMed] [Google Scholar]
- 100.Ban N., Nissen P., Steitz T.A. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 101.Carter A.P., Clemons W.M., Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 102.Schluenzen F., Tocilj A., Yonath A. Structure of functionally activated small ribosomal subunit at 3.3 Ångstroms resolution. Cell. 2000;102:615–623. doi: 10.1016/s0092-8674(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 103.Schuwirth B.S., Borovinskaya M.A., Cate J.H. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 104.Sousa M.C., Trame C.B., McKay D.B. Crystal and solution structures of an HslUV protease-chaperone complex. Cell. 2000;103:633–643. doi: 10.1016/s0092-8674(00)00166-5. [DOI] [PubMed] [Google Scholar]
- 105.Gai D., Zhao R., Chen X.S. Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell. 2004;119:47–60. doi: 10.1016/j.cell.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 106.Kelch B.A., Makino D.L., Kuriyan J. How a DNA polymerase clamp loader opens a sliding clamp. Science. 2011;334:1675–1680. doi: 10.1126/science.1211884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang X., McLachlan J., Hall T.M.T. Modular recognition of RNA by a human pumilio-homology domain. Cell. 2002;110:501–512. doi: 10.1016/s0092-8674(02)00873-5. [DOI] [PubMed] [Google Scholar]
- 108.Kobe B., Deisenhofer J. A structural basis of the interactions between leucine-rich repeats and protein ligands. Nature. 1995;374:183–186. doi: 10.1038/374183a0. [DOI] [PubMed] [Google Scholar]
- 109.Michaely P., Tomchick D.R., Anderson R.G.W. Crystal structure of a 12 ANK repeat stack from human ankyrinR. EMBO J. 2002;21:6387–6396. doi: 10.1093/emboj/cdf651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kramer M.A., Wetzel S.K., Grütter M.G. Structural determinants for improved stability of designed ankyrin repeat proteins with a redesigned C-capping module. J. Mol. Biol. 2010;404:381–391. doi: 10.1016/j.jmb.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 111.Kuhlman B., Dantas G., Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 112.Raman S., Lange O.F., Baker D. NMR structure determination for larger proteins using backbone-only data. Science. 2010;327:1014–1018. doi: 10.1126/science.1183649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.DiMaio F., Tyka M.D., Baker D. Refinement of protein structures into low-resolution density maps using ROSETTA. J. Mol. Biol. 2009;392:181–190. doi: 10.1016/j.jmb.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Adams P.D., Baker D., Terwilliger T.C. Advances, interactions, and future developments in the CNS, PHENIX, and ROSETTA structural biology software systems. Annu. Rev. Biophys. 2013;42:265–287. doi: 10.1146/annurev-biophys-083012-130253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ye S., Vakonakis I., Sacchettini J.C. Crystal structure of circadian clock protein KaiA from Synechococcus elongatus. J. Biol. Chem. 2004;279:20511–20518. doi: 10.1074/jbc.M400077200. [DOI] [PubMed] [Google Scholar]
- 116.Garces R.G., Wu N., Pai E.F. Anabaena circadian clock proteins KaiA and KaiB reveal a potential common binding site to their partner KaiC. EMBO J. 2004;23:1688–1698. doi: 10.1038/sj.emboj.7600190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pattanayek R., Williams D.R., Egli M. Analysis of KaiA-KaiC protein interactions in the cyano-bacterial circadian clock using hybrid structural methods. EMBO J. 2006;25:2017–2028. doi: 10.1038/sj.emboj.7601086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Batey R.T., Gilbert S.D., Montange R.K. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004;432:411–415. doi: 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- 119.Serganov A., Yuan Y.R., Patel D.J. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem. Biol. 2004;11:1729–1741. doi: 10.1016/j.chembiol.2004.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu Q., Greimann J.C., Lima C.D. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell. 2006;127:1223–1237. doi: 10.1016/j.cell.2006.10.037. [DOI] [PubMed] [Google Scholar]
- 121.Cherezov V., Rosenbaum D.M., Stevens R.C. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rasmussen S.G., DeVree B.T., Kobilka B.K. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Landau E.M., Rosenbusch J.P. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA. 1996;93:14532–14535. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tanaka H., Kato K., Tsukihara T. The structure of rat liver vault at 3.5 Ångstrom resolution. Science. 2009;323:384–388. doi: 10.1126/science.1164975. [DOI] [PubMed] [Google Scholar]
- 125.Casañas A., Querol-Audí J., Fita I. New features of vault architecture and dynamics revealed by novel refinement using the deformable elastic network approach. Acta Crystallogr. D Biol. Crystallogr. 2013;69:1054–1061. doi: 10.1107/S0907444913004472. [DOI] [PubMed] [Google Scholar]
- 126.Chapman H.N., Fromme P., Spence J.C. Femtosecond x-ray protein nanocrystallography. Nature. 2011;470:73–77. doi: 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kern J., Alonso-Mori R., Yachandra V.K. Room temperature femtosecond x-ray diffraction of photosystem II microcrystals. Proc. Natl. Acad. Sci. USA. 2012;109:9721–9726. doi: 10.1073/pnas.1204598109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Redecke L., Nass K., Chapman H.N. Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an x-ray laser. Science. 2013;339:227–230. doi: 10.1126/science.1229663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bernstein F.C., Koetzle T.F., Tasumi M.J. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 130.Richardson J.S. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 131.Richardson D.C., Richardson J.S. Mage, probe, and kinemages. In: Rossmann M.G., Arnold E., editors. IUCr’s International Tables of Crystallography, Vol. F: Crystallography of Biological Macromolecules. Kluwer Academic Press; Dordrecht, the Netherlands: 2001. [Google Scholar]
- 132.Chen V.B., Davis I.W., Richardson D.C. KING (Kinemage, Next Generation): a versatile interactive molecular and scientific visualization program. Protein Sci. 2009;18:2403–2409. doi: 10.1002/pro.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Commons, Wikimedia. http://commons.wikimedia.org/wiki/User:Dcrjsr.
- 134.Goodsell, D. Molecule of the Month. http://www.rcsb.org/pdb/101/motm_archive.do.
- 135.Tarini M., Cignoni P., Montani C. Ambient occlusion and edge cueing to enhance real time molecular visualization. IEEE Trans. Vis. Comput. Graph. 2006;12:1237–1244. doi: 10.1109/TVCG.2006.115. [DOI] [PubMed] [Google Scholar]
- 136.Wikipedia. English. http://en.wikipedia/wiki/List_of_biophysically_important_macromolecular_crystal_structures.
- 137.Burley S.K. PDB40: the Protein Data Bank celebrates its 40th birthday. Biopolymers. 2013;99:165–169. doi: 10.1002/bip.22182. [DOI] [PubMed] [Google Scholar]
- 138.Sawyer L., Shotton D.M., Watson H.C. The atomic structure of crystalline porcine pancreatic elastase at 2.5 Å resolution: comparisons with the structure of α-chymotrypsin. J. Mol. Biol. 1978;118:137–208. doi: 10.1016/0022-2836(78)90412-6. [DOI] [PubMed] [Google Scholar]
- 139.Zhang X.J., Matthews B.W. Conservation of solvent-binding sites in 10 crystal forms of T4 lysozyme. Protein Sci. 1994;3:1031–1039. doi: 10.1002/pro.5560030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jacobson R.H., Matsumura M., Matthews B.W. Structure of a stabilizing disulfide bridge mutant that closes the active-site cleft of T4 lysozyme. Protein Sci. 1992;1:46–57. doi: 10.1002/pro.5560010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tainer J.A., Getzoff E.D., Richardson D.C. Determination and analysis of the 2 Å-structure of copper, zinc superoxide dismutase. J. Mol. Biol. 1982;160:181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 142.Shi H., Moore P.B. The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: a classic structure revisited. RNA. 2000;6:1091–1105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Warren J.J., Forsberg L.J., Beese L.S. The structural basis for the mutagenicity of O6-methyl-guanine lesions. Proc. Natl. Acad. Sci. USA. 2006;103:19701–19706. doi: 10.1073/pnas.0609580103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Saper M.A., Bjorkman P.J., Wiley D.C. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 Å resolution. J. Mol. Biol. 1991;219:277–319. doi: 10.1016/0022-2836(91)90567-p. [DOI] [PubMed] [Google Scholar]
- 145.Iverson T.M., Alber B.E., Rees D.C. A closer look at the active site of γ-class carbonic anhydrases: high-resolution crystallographic studies of the carbonic anhydrase from Methanosarcina thermophila. Biochemistry. 2000;39:9222–9231. doi: 10.1021/bi000204s. [DOI] [PubMed] [Google Scholar]
- 146.Schulze-Gahmen U., De Bondt H.L., Kim S.-H. High-resolution crystal structures of human cyclin-dependent kinase 2 with and without ATP: bound waters and natural ligand as guides for inhibitor design. J. Med. Chem. 1996;39:4540–4546. doi: 10.1021/jm960402a. [DOI] [PubMed] [Google Scholar]
- 147.Brino L., Urzhumtsev A., Moras D. Dimerization of Escherichia coli DNA-gyrase B provides a structural mechanism for activating the ATPase catalytic center. J. Biol. Chem. 2000;275:9468–9475. doi: 10.1074/jbc.275.13.9468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.