

Abstract
Glucocorticoid action on target tissues is determined by the density of “nuclear” receptors and intracellular metabolism by the two isozymes of 11β-hydroxysteroid dehydrogenase (11β-HSD) which catalyze interconversion of active cortisol and corticosterone with inert cortisone and 11-dehydrocorticosterone. 11β-HSD type 1, a predominant reductase in most intact cells, catalyzes the regeneration of active glucocorticoids, thus amplifying cellular action. 11β-HSD1 is widely expressed in liver, adipose tissue, muscle, pancreatic islets, adult brain, inflammatory cells, and gonads. 11β-HSD1 is selectively elevated in adipose tissue in obesity where it contributes to metabolic complications. Similarly, 11β-HSD1 is elevated in the ageing brain where it exacerbates glucocorticoid-associated cognitive decline. Deficiency or selective inhibition of 11β-HSD1 improves multiple metabolic syndrome parameters in rodent models and human clinical trials and similarly improves cognitive function with ageing. The efficacy of inhibitors in human therapy remains unclear. 11β-HSD2 is a high-affinity dehydrogenase that inactivates glucocorticoids. In the distal nephron, 11β-HSD2 ensures that only aldosterone is an agonist at mineralocorticoid receptors (MR). 11β-HSD2 inhibition or genetic deficiency causes apparent mineralocorticoid excess and hypertension due to inappropriate glucocorticoid activation of renal MR. The placenta and fetus also highly express 11β-HSD2 which, by inactivating glucocorticoids, prevents premature maturation of fetal tissues and consequent developmental “programming.” The role of 11β-HSD2 as a marker of programming is being explored. The 11β-HSDs thus illuminate the emerging biology of intracrine control, afford important insights into human pathogenesis, and offer new tissue-restricted therapeutic avenues.
I. INTRODUCTION
A. The Historical Context
The story of 11β-hydroxysteroid dehydrogenase (11β-HSD) begins with the isolation, synthesis, and therapeutic exploitation of adrenal corticosteroids. In the late 1930s, the laboratories of Kendall and Reichstein independently isolated and characterized the structures of steroids from the adrenal cortex, a triumph of 20th century chemistry (444, 445, 578, 579, 665). A number of preclinical studies were undertaken with the tiny amounts of these compounds that could be obtained by tissue extraction methods, but it was only in 1949 that Kendall's compound E, soon known as cortisone, was synthesized in sufficient quantities for its detailed investigation to begin. Hench used this first in patients with rheumatoid arthritis and then in rheumatic fever and showed remarkable efficacy in what had hitherto been inexorable inflammatory disorders (273–275). Immediately there followed a plethora of studies trying out the new “miracle cure” on almost every known disorder. Cortisone effected dramatic improvements in inflammatory and adrenal deficiency disorders (57); produced beneficial responses in certain malignancies, such as lymphoma, though not in many solid tumours; but often caused deterioration in infections like syphilis and tuberculosis. In 1950 alone, over 300 papers were published on the use of cortisone, a remarkable level of activity, mirroring some of the initial hope and hype surrounding gene and cell therapies in the current era. This early enthusiasm is now tempered in the light of contemporary understanding of the extraordinary range of benefits and harms attributable to glucocorticoids. Nonetheless, glucocorticoids remain among the most prescribed medicines (758). In recognition of their scientific tour-de-force, the 1950 Nobel Prize for Physiology or Medicine was awarded to Kendall, Reichstein, and Hench. However, as early as 1951 when Kendall's compound F or cortisol (hydrocortisone) became available, it was increasingly clear that this was more active than cortisone (286). It is salutary to reflect that the remarkable therapeutic responses Hench observed involved cortisone, a steroid which is intrinsically inert. Explaining this paradox reflects one major role for 11β-HSD.
In all the kerfuffle over clinical efficacy and side effects of cortisone (Hench carefully reported Cushing's disease-like features in his initial patient population; Ref. 273), a few groups began to address metabolism of the new glucocorticoids using chromatographic techniques to separate steroid moieties. First, Burton et al. (106) demonstrated cortisone metabolism to other steroids including cortisol in humans. Then in 1953, Amelung et al. (21) in Frankfurt administered cortisone to rats in vivo and incubated cortisone with homogenates of various tissues in vitro and found conversion to cortisol. The activity, localized to microsomes, was highest in liver with lower amounts in kidney and muscle. This was 11β-HSD (FIGURE 1).
Figure 1.
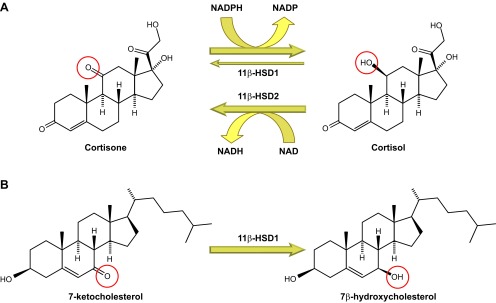
Reactions catalyzed by 11β-hydroxysteroid dehydrogenase (11β-HSD) isozymes. A: interconversion of cortisol and cortisone by 11β-HSD1 and -2. In intact cells and in vivo, 11β-HSD1 is predominantly a reductase, catalyzing NADPH-dependent reduction of cortisone to cortisol, predominantly in the liver. Under some circumstances and in some cells, it may act as an NADP-dependent dehydrogenase, inactivating cortisol. 11β-HSD2, in contrast, catalyzes the NAD+-dependent inactivation of cortisol, converting it to cortisone, predominantly in the kidney. B: conversion of 7-ketocholesterol to 7β-hydroxycholesterol by 11β-HSD1. Other reactions catalyzed by 11β-HSD1, including oxysterol metabolism, are probably of physiological importance. The 11β- and 7α-positions of the steroid nucleus show rotational symmetry, probably explaining the metabolism of 7-ketocholesterol to 7β-hydroxycholesterol by 11β-HSD1 as well as its metabolism of other 7-oxygenated sterols and steroids.
In the 1960s, 11β-HSD activity was reported in a variety of organs, predominantly detected using histochemical techniques of poor specificity (47, 102, 353). For more than 30 years whilst the biology and pharmacology of various glucocorticoids was intensively explored and their intracellular receptors characterized, 11β-HSD remained an arcane biochemical detail, a bidirectional shuttle catalyzing interconversion of cortisol and corticosterone (a minor glucocorticoid in most mammals, but the only form in rats and mice) to cortisone and 11-dehydrocorticosterone, respectively. This was considered of little interest to physiologists or clinicians, merely one of several pathways of metabolism of glucocorticoids. Nonetheless, this otherwise obscure enzyme reaction has added a novel strand to the 60-year-old story of glucocorticoids, revealing that prereceptor glucocorticoid metabolism is a critical control of physiological steroid action, underpins pathogenesis of rare and common disorders, and affords the possibility of tissue-targeted therapeutic manipulation of glucocorticoid action.
B. Glucocorticoid and Mineralocorticoid Receptors
In mammals, the adrenal cortex synthesizes aldosterone, the major physiological mineralocorticoid, from the zona glomerulosa and glucocorticoids (cortisol, corticosterone) from the zona fasciculata-reticularis. Circulating levels of cortisol and corticosterone vary widely from low nanomolar during sleep, to low micromolar during severe stress/illness. They are tightly bound to a high-affinity, limited capacity protein carrier, corticosteroid-binding globulin (CBG), and more loosely to albumin such that around 5% is “free” basally, although stress levels may exceed CBG's capacity. Inert 11-keto corticoids bind poorly to plasma proteins, as does aldosterone, which circulates at picomolar concentrations, perhaps 2–3 logs lower than glucocorticoids.
Corticosteroids are highly lipophilic and are thought readily to diffuse across biological membranes to access their intracellular receptors. Inward glucocorticoid carriers analogous to the recently discovered monocarboxylate transporter-8 that facilitates thyroid hormone access to cells (284) are, as yet, unreported, although some glucocorticoids are partly excluded from tissues such as the CNS by members of the ABC transporter family, notably ABCB1 (Mdr1) (460).
At the cellular level, the myriad effects of corticosteroids are largely a consequence of transcriptional actions mediated via binding to two types of intracellular receptor: the high-affinity mineralocorticoid receptor (MR) and the lower affinity glucocorticoid receptor (GR) (208, 452). 11β-Hydroxycorticosteroids are ligands for these receptors, but binding of their 11-keto forms is negligible. On binding ligand, GR and MR dissociate from complexes with chaperone proteins, translocate to the nucleus, and bind directly or indirectly to the regulatory regions of target genes: ∼2% of the human genome is regulated by glucocorticoids (577), although few if any genes are exclusively controlled by corticosteroids. In addition, a role for membrane receptors is emerging at least in the brain (143). Mice lacking MR lose such membrane effects, suggesting that products of the same gene underpin both nuclear/transcriptional and surface/rapid actions, although GPR30 may also be involved (249). MR show a rather restricted tissue distribution, with high expression confined to classical aldosterone target organs such as the kidney, colon, and salivary glands, as well as specific brain regions (notably the hippocampus) and more modest expression in vascular tissues, adipocytes (803), and specific immune cell populations (722). In contrast, GR are widely expressed, at higher levels than MR at most sites.
In vitro, MR have a high and very similar affinity (Kd ∼0.5–1 nM) for corticosterone, cortisol, and aldosterone (37, 370, 637), whereas potent synthetic glucocorticoids such as dexamethasone dissociate rapidly and only activate MR at high concentrations (416). In contrast, GR have a lower Kd for physiological glucocorticoids (typically ∼10–25 nM in cytosolic extracts), although a much higher affinity for synthetic glucocorticoids such as dexamethasone (∼1–5 nM), but barely bind mineralocorticoids (287). GR are therefore little occupied by basal levels of glucocorticoids (free tissue levels are likely to be subnanomolar), but become progressively activated as glucocorticoid levels rise during ultradian pulses, the diurnal maximum, a stress response or pharmacotherapy (144, 406, 580).
C. 11β-HSD Renaissance
In the mid 1980s, an 11β-HSD activity was characterized biochemically in the laboratory of Carl Monder, who subsequently purified the enzyme from rat liver and isolated the encoding cDNA (4, 377–379). The enzyme activity was bidirectional in tissue homogenates, containing both 11β-dehydrogenase (glucocorticoid inactivating) and 11β-reductase (glucocorticoid regenerating) activities, with NADP(H) as cosubstrate (FIGURE 1). This rat liver enzyme had a modest affinity (micromolar Km) for glucocorticoids (379).
A number of clinical case reports described deficiency in the interconversion of cortisol to cortisone (as evidenced by an elevated urinary ratio of cortisol to cortisone metabolites; Ref. 720) in association with a very rare disease, the syndrome of “apparent mineralocorticoid excess” (AME). AME presents in childhood with severe hypertension, sodium retention, potassium loss, metabolic alkalosis, and suppressed plasma renin activity, findings compatible with mineralocorticoid excess. However, it is accompanied by undetectable levels of all known mineralocorticoids, notably aldosterone and deoxycorticosterone. The condition is normally fatal in childhood (483, 631, 632, 719). In the late 1980s, Stewart, Edwards, and colleagues in Edinburgh investigated a unique adult patient with AME (668). Their elegant clinical experiments showed that the mineralocorticoid excess was due to cortisol. Suppression of endogenous cortisol with dexamethasone reversed mineralocorticoid excess, whilst concurrent “replacement” with physiological doses of cortisol recapitulated mineralocorticoid excess, an effect not seen in healthy controls. These investigators also recognized that the syndrome was analogous to the effects of licorice, long known to cause hypertension and hypokalemia (128), and showed that ingestion of licorice in humans produced AME only in the presence of cortisol (672).
Meanwhile, Evans and his colleagues at the Salk Institute had just cloned a cDNA encoding human MR. In vitro the recombinant MR bound and was activated by cortisol, corticosterone, and aldosterone with similar affinity (37), confirming data from earlier biochemical studies (637). Indeed, it seemed remarkable that MR in the distal nephron are activated in vivo selectively by picomolar levels of aldosterone but not the higher nanomolar levels of cortisol, whereas structurally identical MR in the hippocampus are occupied by glucocorticoids in vivo (124, 473, 580). Thus the paradox was that renal MR are selectively activated by aldosterone in vivo, but both physiological glucocorticoids and mineralocorticoids in vitro.
The solution was provided in the late 1980s by the Edinburgh group (181, 672), as well as Funder and colleagues in Melbourne (210), who recognized that selectivity of MR in the kidney in vivo was not due to any intrinsic specificity for aldosterone but to the activity of 11β-HSD. In the kidney, 11β-HSD catalyzes the rapid inactivation of cortisol and corticosterone to inert 11-keto forms (cortisone, 11-dehydrocorticosterone) which do not bind MR. Only aldosterone, which is not a substrate for 11β-HSD, was able to activate renal MR. 11β-HSD inhibition by licorice or deficiency in AME allowed cortisol to bypass the enzymic “barrier” and bind to and activate MR causing sodium retention, potassium loss, and hypertension. For the glucocorticoid system this was the first example of prereceptor metabolism apparently gating steroid access to receptors. Analogous biology had been described for thyroid hormone receptors with mono-deiodinase isozymes inactivating or activating thyroid hormones in a cell-specific manner (228).
D. One Enzyme or Two (or Three)?
The rat liver 11β-HSD isolated and characterized and subsequently cloned by Monder and colleagues was found to be widely expressed, including in rat kidney, consistent with the idea that it underpinned MR selectivity and AME. However, several lines of evidence militated against the “one enzyme” hypothesis. Notably, 1) there are few MR in liver, the highest site of expression of this enzyme; 2) no mutations in the encoding gene, hsd11b1, were found in AME patients (695); 3) the enzyme is in the proximal tubule in the rat kidney and thus does not colocalise with MR in the distal nephron (181); 4) the enzyme is bidirectional in liver yet apparently a unidirectional dehydrogenase in kidney (321); and 5) there are marked discrepancies between “liver” 11β-HSD mRNA levels and enzyme activity in kidney (but not liver) (418), suggesting the existence of a distinct isozyme in kidney.
In 1993, a novel enzyme with 11β-hydroxysteroid dehydrogenase activity was isolated and characterized from human placenta (90) by Seckl, Chapman and colleagues and from rat kidney (602) by Naray-Fejes-Toth et al. This 41-kDa protein was distinct in physicochemical characteristics and apparent molecular weight from Monder's 34 kDa enzyme, and acted as a high affinity (low nM Km) exclusive 11β-dehydrogenase which used NAD+ as cosubstrate rather than NADP(H). The next year, Krozowski's group (15) isolated a cDNA encoding the “renal” 11β-HSD isozyme from human kidney and White and colleagues cloned the analogous gene product from sheep kidney (6). The enzyme was purified from human placenta, and the encoding cDNA was found to be identical to the kidney version (91, 92). The rodent homologs were also cloned (127, 583). The new enzyme was called 11β-HSD type 2 to distinguish it from Monder's 11β-HSD type 1 (FIGURE 1). Mutations in hsd11b2 encoding 11β-HSD2 were soon identified in patients with AME (141, 498, 777). Stewart and Edward's original adult patient is a compound heterozygote with modest activity encoded by one allele, presumably responsible for his survival (388). Mice homozygous for targeted disruption of the Hsd11b2 gene faithfully recapitulate AME (363) (FIGURE 2).
Figure 2.
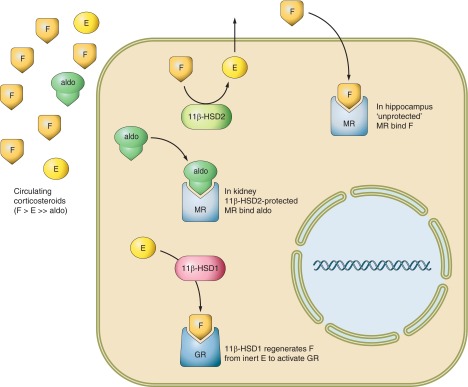
Diagrammatic representation of the reactions catalyzed by 11β-HSDs. The adrenal cortex secretes nanomolar concentrations of cortisol (F) and picomolar concentrations of aldosterone (Aldo) into the circulation. While mineralocorticoid receptors (MR) in the kidney only bind Aldo in vivo, identical MR in the hippocampus are occupied by F in vivo and MR bind F and Aldo with similar affinity in vitro. The solution to this conundrum lies in the collocation of renal MR with 11β-HSD2, which catalyzes the rapid inactivation of cortisol to inert cortisone (E) thus only allowing the nonsubstrate Aldo to access MR. 11β-HSD2 is absent from hippocampus so MR bind F. In tissues such as the liver, adipose, and adult brain, 11β-HSD2 is absent, but there is abundant 11β-HSD1. This catalyzes the reverse reaction in intact cells and organs and thus regenerates active F from inert E, amplifying the local glucocorticoid signal particularly at glucocorticoid receptors (GR). GR have 10-fold lower affinity for F than MR and are thus partially unoccupied by F at physiological concentrations allowing a dynamic range for 11β-HSD1 amplification inside cells to impact on signaling. In contrast, MR are largely occupied by physiological F concentrations where 11β-HSD2 is absent so 11β-HSD1 may make less impact on signaling via MR.
This left open the question of the function of Monder's original enzyme, 11β-HSD1, which is widespread but most highly expressed in liver. Some suggested it might be a lower affinity 11β-dehydrogenase. However, studies in clonal amphibian and mammalian cells transfected with rat 11β-HSD1 cDNA showed that, whilst bidirectional in homogenates, surprisingly it acts as a predominant 11β-reductase in most intact cells and thus functionally amplifies glucocorticoid action (174, 420). Indeed, in cells expressing 11β-HSD1, cortisone (or 11dehydrocorticosterone) is equipotent with or even more potent than cortisol (or corticosterone) (206, 231, 646). 11β-Reductase predominance was also indicated in humans by in vivo observations of a high cortisol-to-cortisone ratio across the hepatic circulation (liver only expresses 11β-HSD1) (743). Thereafter, 11β-HSD1 action in a host of primary cell types was shown to be predominantly glucocorticoid regeneration and dependent on NADP(H) cosubstrate (FIGURE 2). Located inside the inner leaflet of the endoplasmic reticulum, 11β-HSD1 interacts with hexose-6-phosphate dehydrogenase (H6PDH), the major generator of endoplasmic reticulum NADPH, which in turn drives 11β-reduction (40, 41, 54, 97, 177) (FIGURE 3, and see below). Loss of H6PDH leads to reaction reversal of 11β-HSD1, although the physiological relevance remains uncertain (389, 391).
Figure 3.

Cartoon of the possible intracellular relationships of 11β-HSDs. Despite having a lower affinity for F (or corticosterone) than MR, 11β-HSD2 is able to successfully exclude glucocorticoids from MR. While the basis for this is unknown, a possible scenario invokes lipophilic steroids preferentially localizing to the membranes inside cells including those of the endoplasmic reticulum (ER). 11β-HSD2 is located on the cytosolic surface of the ER and is in close association with the MR complex. Therefore, F may perhaps have to pass via 11β-HSD2 before it can gain access to MR. If there is sufficient enzyme and its turnover is suitably fast, it may successfully form the biochemical equivalent of an anatomical moat around MR. In addition, some data suggest that the 11-dehydrocorticosteroid products of 11β-HSD2 (cortisone/E; 11-dehydrocorticosterone) functionally antagonize aldosterone activation of MR. 11β-HSD1 is bidirectional in homogenates and microsomal preparations, but a predominant reductase in intact cells and in vivo. 11β-HSD1 is located inside the inner leaflet of the ER in close association with hectose-6-phosphate dehydrogenase (H6PDH), a powerful generator of NADP(H). NADP(H) drives the 11β-reductase direction of 11β-HSD1 and maintains this in many cell types. Other redox active processes may be important in organs such as brain where H6PDH may be at low levels.
More recently, the existence of a third 11β-HSD isozyme has been suggested. An NADP+-dependent dehydrogenase activity in sheep kidney was first dubbed 11β-HSD3 (238), but this has not been further characterized and may represent 11β-HSD1 or -2. It is unlikely that this is the same as the discovered evolutionary ancestor of 11β-HSD1, confusingly called 11β-HSD3 (51), but also SDR26C2 according to accepted nomenclature (557), HSD11B1L and SCDR10. 11β-HSD3/SDR26C2 is present in humans, with orthologs in other species including fish, but is absent from rats and mice (299). 11β-HSD3/SDR26C2 only poorly catalyzes conversion of cortisol to cortisone (in an NADP+-dependent manner) (299). Indeed, it predates the glucocorticoid receptor in evolution (49), implying a primary function distinct from glucocorticoid biology.
E. Intracrine Versus Endocrine Effects
It is helpful to consider the effects of enzymes such as 11β-HSDs in two contexts: intracrine and endocrine. Intracrine effects are exemplified by the intracellular “gating” of glucocorticoid action by 11β-HSD2 within cells of the distal nephron. 11β-HSD2 modulates the activation of intracellular receptors by a ubiquitous ligand but only where receptors and enzyme are colocalized (395). This occurs without altering circulating levels of cortisol, itself largely determined by activation of, and glucocorticoid feedback upon, the hypothalamic-pituitary-adrenal (HPA) axis. Thus patients with AME have intense MR activation by cortisol within specific cells of the kidney but without change in systemic cortisol levels, an intracrine effect. Similarly, 11β-HSD1 acting as a reductase appears to amplify glucocorticoid levels inside expressing cells in adipose tissue and brain (494, 791), increasing local glucocorticoid levels and thus the signal via GR and, if present, MR.
However, these enzymes also contribute to the bulk turnover of glucocorticoids, with 11β-HSD1 in the splanchnic bed generating ∼30–40% of the total daily production of cortisol in humans (26, 62) and 11β-HSD2 in the kidney inactivating a similar proportion. These actions affect the turnover of glucocorticoids and may per se alter HPA axis function under some circumstances. Moreover, expression of 11β-HSDs in cells involved in glucocorticoid feedback control of the HPA axis could also alter circulating hormones (179, 622, 624). These are endocrine effects.
F. Measurement of 11β-HSDs
Differentiating 11β-HSD isozymes at the level of mRNA is straightforward, given their distinct sequences. In contrast, estimating 11β-HSD1 protein and activity can be more challenging yet is crucial to explore their biology, especially in humans. As discussed below, antisera for 11β-HSD1 and -2 are often species-specific (not surprising given the sequence and substrate differences) and probably cross-react with related members of the extensive short-chain dehydrogenase/reductase (SDR) protein family, although some sera perform well in practice. Ex vivo bioassays are well-established; both isozymes can readily be measured in tissue homogenates or microsomal preparations (418, 479). These are uncomplicated for measuring 11β-HSD in relevant samples and should be even more specific when coupled with selective 11β-HSD1 inhibition to distinguish isozyme activities in dehydrogenase assays. However, because 11β-HSD1 is bidirectional in tissue homogenates and microsomes, assays in semi-purified preparations will determine only the amount of active protein and not its reaction direction, unlike 11β-HSD2 which is essentially a unidirectional dehydrogenase with cortisol or corticosterone as substrates. Assays in intact cells can distinguish isozymes (91), but do not necessarily determine 11β-HSD1 reaction direction in vivo if this is dynamically regulated by intracellular energy levels, as detailed below.
In humans, some tissues (leukocytes, adipose, skeletal muscle, sometimes liver) can be directly assayed ex vivo from blood samples/biopsies, but for most organs indirect techniques have been needed to determine 11β-HSD activity and the reaction catalyzed by 11β-HSD1 in vivo. Initially, radioimmunoassay and then mass spectrometry approaches have been developed to estimate corticosteroid metabolites in urine (typically over 24 h to remove any distortion from circadian rhythms, etc.) (719). Cortisol (Kendal's compound F) is metabolized by 11β-HSD2 to cortisone (E). Both cortisol and cortisone are then subject to 5β-reduction (by 5β-reductase largely in liver) to form dihydrocortisol (DHF) and dihydrocortisone (DHE). Cortisol is also metabolized by 5α-reductase type 1 to allo-dihydrocortisol (aDHF), again largely but not exclusively in liver. The dihydro-metabolites are then further reduced by near ubiquitous 3α-hydroxysteroid dehydrogenases to form the tetrahydro metabolites (THF, aTHF, and THE) (745). Urinary ratios of THF+aTHF/THE give a convenient measure of combined 11β-HSD1 and -2 activity. A normal ratio of near unity becomes markedly elevated in AME patients with some correlation between the ratio and phenotype (491).
There are some caveats to the widespread use of urinary steroid ratios: 1) it measures both 11β-HSDs; 2) the ratio also has components of 5α- and 5β-reductase activities which, if altered, will impact upon the ratio; and 3) the ratio does not address tissue-specific impacts. In a refinement, urinary free cortisol (UFF):urinary free cortisone (UFE) have been advocated as selective for 11β-HSD2 (542), leaving the THF+aTHF/THE to reflect 11β-HSD1 when UFF:UFE is unaltered, but this assumption requires comprehensive validation. Whilst selective 11β-HSD1 inhibitors reduce the THF+aTHF/THE ratio within subjects, using this as an absolute measure of whole body 11β-HSD1 activity in population samples is a blunt and probably ineffective tool. Thus the ratio correlates positively (24), negatively (574), and not at all (23, 762) with obesity. Obesity and ageing also alter 5α- and 5β-reductases adding to the complexity and well-illustrating the limitations of this approach (745).
These indirect estimates have long been supplemented with administration of radio-labeled cortisol tracer to estimate bodily conversion to cortisone (668, 719). Again this works well for 11β-HSD2 mutations, but is otherwise complicated when trying to take into account recycling of cortisone back to cortisol by 11β-reductase.
To directly estimate 11β-reductase in the splanchnic bed (gut, mesentery, liver), oral cortisone (315, 574, 671) and prednisone (712) tests have been developed. These use first pass hepatic metabolism to measure the rate of appearance of cortisol and prednisolone in peripheral blood. This appears linear over 15–30 min, although with longer sampling, cycling through both 11β-HSDs and downstream metabolism complicate interpretation. Rigorous determination of the relationship between the output of some of these bioassays and the actual level of 11β-HSD1 in the splanchnic bed has not been reported as yet and may not be linear, a problem when using such data to assess the degree of inhibition by novel agents.
To estimate 11β-reductase and 11β-dehydrogenase more directly, quadruply deuterated (D4)-cortisol (9,11,12,12-[2H4]cortisol) has been administered. 11β-HSD2 removes the deuterium on C11 creating D3-cortisone. This is a substrate for 11β-HSD1 which reduces D3-cortisone to D3-cortisol which can be distinguished from D4-cortisol by mass spectrometry (25, 62, 612). In addition to whole-body 11β-HSD activity, this can be used, if somewhat more invasively, to determine turnover of cortisol and cortisone in individual organs (59, 303, 674).
Finally, 11β-reductase can also be assessed in human subcutaneous adipose tissue by microdialysis, infusing [3H]cortisol and measuring conversion to [3H]cortisone in the dialysate. While attractive conceptually, and responsive to acute regulation (459, 612, 712, 740, 741), this is a challenging technique.
Overall, with care and attention to detail and controls, 11β-HSD activities can be measured with some accuracy and consistency in humans in vivo.
G. Investigation of the Physiological Functions of 11β-HSDs
Study of the physiological function of 11β-HSDs began with nonselective, licorice-based inhibitors such as glycyrrhetinic acid and its hemisuccinate carbenoxolone (FIGURE 7), a drug historically used to treat peptic ulcers (79). These compounds inhibit both isozymes with a low nanomolar Ki in vitro. Potency is lower in vivo, and the drugs also inhibit related enzymes such as 15-hydroxyprostaglandin dehydrogenase (697) and gap junctions (235, 252, 254), although with lower potency. Why the root of the licorice plant, Glycyrrhiza glabra and related species, produces a triterpenoid which so potently inhibits 11β-HSDs is unclear, but its production is sensitive to the environment (356), so perhaps these substances promote plant survival and parasite resistance.
Figure 7.
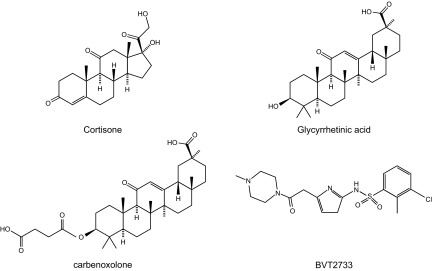
Structures of nonselective licorice-based 11β-HSD inhibitors, glycyrrhetinic acid and carbenoxolone, BVT2733, a synthetic selective 11β-HSD1 inhibitor, and the natural cortisone substrate for 11β-HSD1.
Dissection of 11β-HSD function came of age with the development of knockout mouse models, 11β-HSD1 in 1997 and 11β-HSD2 in 1999 (363, 364). Subsequently, a variety of transgenic lines and more recently selective 11β-HSD1 inhibitors and tissue-specific genetic manipulations have expanded the weaponry to dissect function and pathophysiology. These studies are described below in an organ and system approach.
Here we review the intriguing biology of 11β-HSDs and highlight their role in pathogenesis through the lifespan.
II. 11β-HSD1 GENE STRUCTURE, ENZYMOLOGY, AND REGULATION
A. Gene Structure and Control
11β-HSD1 is found in land vertebrates and sharks, but is absent from fish, likely arising from an ancestral 11β-HSD3/SDR26C2 gene (51). The encoding gene, HSD11B1, is located close to the end of the long arm of chromosome 1 in mice and humans and chromosome 13 in rats. It comprises seven exons; exons 2–7 encode the full-length protein (FIGURE 4). The gene is transcribed from three promoters: P1, P2, and P3 (95, 475); P2-initiated transcripts predominate in most tissues including liver, brain, and adipose (95, 663). P1-initiated transcripts are the majority in mouse lung, but the minority in human lung (663) and kidney (our unpublished data). The P3 transcript, discovered in rat kidney (and negligible in other tissues), initiates within the intron between exons 2 and 3 (475). It encodes an expressed NH2-terminally truncated 11β-HSD1 protein that lacks 11β-HSD activity (462). Its biological relevance (if any) is unclear.
Figure 4.

Structure of the human HSD11B1 gene, associated promoters and relevant transcription factor binding sites. Schematic representation of the HSD11B1 gene (not to scale). Exonic sequences are shown as boxes; white boxes encode the open reading frame with red boxes and a blue box indicating the 5′ leader (dependent on the promoter used) and the 3′ untranslated sequence, respectively. Arrows indicate the positions of the 3 promoters. The gene spans 30 kb with a large intron (∼25 kb) between exons 5 and 6. The position of 2 conserved C/EBP binding sites, located between −112 and −160 in the promoter (244, 775), are shown. These sites are bound by C/EBPα and/or C/EBPβ in hepatocytes and adipocytes and are implicated in HSD11B1 regulation by glucocorticoids, cAMP, ceramide, AMPK, and DHEA. See text for details.
The predominant P2 promoter of hsd11b1 contains several binding sites for the CCAAT/enhancer binding protein (C/EBP) family of transcription factors (775). Two C/EBP sites close to the transcription start and highly conserved between human, rat, and mouse (244, 775) are critical for basal and regulated expression (775), either alone or in concert with other transcription factors including GR (789).
In vivo, transcription from P2, at least in liver and brown adipose tissue, requires C/EBPα (95, 775), whereas transcription from P1 in lung is independent of C/EBPα (95). While C/EBPβ is an activator of Hsd11b1 expression in adipocytes and C/EBPβ knockout mice have reduced adipose 11β-HSD1 expression (552), C/EBPβ is only a weak activator in hepatoma cells and acts as a relative repressor of 11β-HSD1 expression in liver, possibly because of heterodimerization with the strong activator C/EBPα (775). A decreased C/EBPα-β ratio is implicated in dehydroepiandrosterone (DHEA) repression of Hsd11b1 in liver and adipose tissue (30). Involvement of other C/EBP family members has not been found as yet (190, 604).
Importantly, C/EBPβ mediates 11β-HSD1 (P2) regulation by proinflammatory cytokines, glucocorticoids, diet, and other regulators (cAMP, ceramide, AMP-activated protein kinase) in a variety of cell types including adipocytes, fibroblasts, and lung epithelial cells (31, 244, 312, 604, 790), although C/EBPα mediates glucocorticoid effects in human amnion fibroblasts (789). C/EBPβ is a central regulator of inflammation and metabolism (35), suggesting that HSD11B1 might play an important downstream role in these pleiotropic effects. C/EBPβ is itself glucocorticoid induced, including during inflammation (64, 807) and adipocyte differentiation (708). This indicates a possible feed-forward loop-inflammation stimulating the HPA axis to secrete glucocorticoids, both of which induce C/EBPβ in turn increasing 11β-HSD1, further amplifying local glucocorticoid signaling and shaping the trajectory of an inflammatory response via pro-resolution macrophage polarization (600). A recent intriguing twist is added by the discovery that the C/EBPβ posttranslational isoforms, liver inhibitory protein (LIP) and liver activator protein (LAP), play distinct physiological roles (647, 763) and oppose each other in mediating 11β-HSD1 regulation in adipose tissue by high-fat diet (190), itself linked to adipose inflammation (750).
Beyond C/EBPs, indirect regulatory effects appear to predominate (see regulation below). Thus HNF1α occupies the HSD11B1 promoter in human hepatocytes (532) and is functionally relevant since mice lacking HNF1α have negligible hepatic 11β-HSD1 mRNA (639). However, this could be an indirect effect mediated via alterations in bile acids; bile duct ligation reduces hepatic 11β-HSD1 activity and mRNA levels in rats (2, 189), and HNF1α is a key regulator of bile acid and HDL-cholesterol metabolism. In contrast, although HNF4α occupies the HSD11B1 promoter in human hepatocytes (532), any function is unclear. The wealth of data emerging from chromatin immunoprecipitation experiments, coupled to microarray or RNA sequencing, will clarify the molecular mechanisms regulating HSD11B1.
B. 11β-HSD1: Protein Structure
In most species, including humans and mice, 11β-HSD1 (also called SDR26C1; Ref. 557) comprises 292 amino acids with a predicted molecular mass of 34 kDa. 11β-HSD1, like 11β-HSD2, is a member of the large family of SDR (557, 778). These show relatively low overall identity (18% sequence identity between 11β-HSD1 and 11β-HSD2) but share a structurally conserved nucleotide cofactor-binding Rossmann-fold in the NH2-terminal region (Thr-Gly-[Xaa]3-Gly-Xaa-Gly) and an invariant Tyr-[Xaa]3-Lys motif in the active site (Tyr183 and Lys187 in human 11β-HSD1 and most other species). Tyr183 and Lys187 sit with the conserved Asn143 and Ser170 in the catalytic site and are essential for the proton transfer between substrate and NAD(P)H cofactor: mutation of either inactivates 11β-HSD1 (526), whereas mutation of nearby serines has little effect on Km but markedly reduces the rate of catalysis (528).
Within cells, tagged fusion proteins have localized 11β-HSD1 to the inner leaflet of the endoplasmic reticulum (ER) (507), confirming earlier structural analyses showing 11β-HSD1 is a high mannose glycoprotein typical of the ER lumen (541). A single transmembrane helix in the NH2 terminus of 11β-HSD1 spans the ER membrane and dictates the orientation of the protein within the membrane, with the NH2-terminal end in the cytoplasm and the bulk of the protein within the ER lumen (204, 530). The luminal orientation is maintained by Lys5 and/or Glu25/Glu26 (204, 530). Surprisingly, mutant 11β-HSD1 with cytoplasmic orientation retained reductase activity with cortisone, but not with 7-ketocholesterol (204, 530), yet cortisol oxidation (low in most intact cells) was abolished. The source of cytoplasmic cofactor for reductase activity was not clear, although it should be noted that these experiments were performed in intact HEK293 cells which, when transfected with 11β-HSD1, show both reductase and dehydrogenase activity due to low levels of endogenous H6PDH (41).
Functional 11β-HSD1 most probably comprises a homodimer (442, 533, 804), although tetrameric structures are possible (295). The two subunits may be linked by a disulfide bond (541), although this is not supported by the crystal structures (533, 804). The human enzyme is glycosylated at three asparagine residues (Asn123, Asn162, and Asn207) (81). Asn207 is invariant in 11β-HSD1 sequences, Asn162 is highly conserved, and Asn123 shows little conservation. Thus there are only two N-linked glycosylation sites in rat and mouse 11β-HSD1 (Asn158 and Asn203 in rat, equivalent to Asn162 and Asn207, respectively, in the human enzyme) (9) and just one in guinea pig. Despite an early study suggesting glycosylation is important for activity (5), the current consensus is that it plays little or no role in activity or reaction direction: enzymatic removal of glycan chains has no effect (9, 541) and human 11β-HSD1 expressed in yeast retained full activity despite lacking glycosylation (81). Similarly, our own experiments have shown no effect of mutation of Asn162 or Asn207 to glutamine on either activity or reaction direction of human 11β-HSD1 in intact cells (unpublished data).
Two human patients with cortisone reductase deficiency have been reported. These subjects are heterozygous for mutations in the coding sequence of HSD11B1. One mutation (Arg137Cys) disrupts salt bridges at the dimerization interface and reduces enzyme activity, and the other (Lys187Asn) interrupts the active site and not surprisingly abolishes 11β-HSD1 activity altogether (393). The mutants appear to exert dominant negative effects on the normal allelic product.
C. Crystal Structure of 11β-HSD1
To date, 26 11β-HSD1 crystal structures have been deposited in the Protein Data Bank (see Ref. 704 for a recent review). Of these, three are mouse 11β-HSD1 (without substrate, with corticosterone bound, and with selective inhibitor bound), three are guinea pig 11β-HSD1 (one without substrate and two with inhibitor bound), and the remainder are human 11β-HSD1 with a variety of inhibitors bound, reflecting the strong pharmaceutical interest in 11β-HSD1 inhibition. All have NADP(H) bound in the cofactor binding site, consistent with the ordered bi-bi sequential enzymatic mechanism proposed by Monder (481), in which NADPH cofactor binds first, followed by substrate binding and catalysis, with cofactor leaving last, confirmed in more recent studies (603).
The 11β-HSD1 structures are similar overall, although the hydrophobic substrate binding pocket, predominantly lined by nonpolar residues and with lower sequence conservation than other regions, shows some variability between species, accounting for the species specificity of inhibitors (32, 626, 704). The two subunits of the 11β-HSD1 homodimer are related by a pseudo-twofold axis. All structures show a central seven-stranded parallel β-sheet sandwiched between six parallel α-helices (the Rossmann-fold; FIGURE 5). The NH2 termini of the two subunits are on the same face of the dimer and point in the same direction, anchoring the protein within the membrane.
Figure 5.
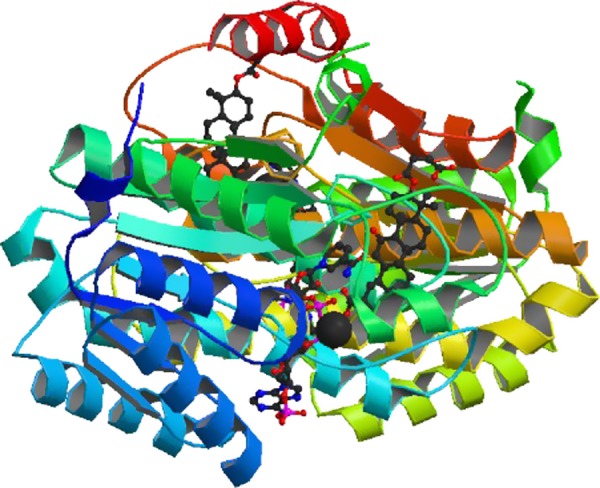
Structure of human 11β-hydroxysteroid dehydrogenase in complex with NADP and carbenoxolone. Structure of a dimer of human 11β-HSD1 with NADP cosubstrate (bottom middle) and the inhibitor carbenoxolone (top right) bound. [Image from the RCSB PDB (www.pdb.org) of PDB ID 2BEL (339).]
The COOH-terminal part of 11β-HSD1, unique among SDR proteins, is involved in dimer interactions, contributes to substrate binding and active site architecture, and is important for membrane lipid bilayer interactions through a large hydrophobic surface (533, 804). The crystal structures suggest it funnels substrate from the lipid bilayer into the active site cleft, driving catalysis. This facilitated entry into the active site may explain the discrepancy between the relatively high Km values measured in vitro with hydrophobic glucocorticoid or oxysterol substrates and the efficient substrate conversion at low concentrations by intact cells (140).
Both ends of the substrate binding pocket are open to solvent, allowing ligands that exceed the 12 Å length of the pocket to extend out of it (704). Interactions with Ser170 and Tyr183 stabilize the 11-keto group of the substrate, anchoring it in the active site (331, 533). Protonation of the reactive keto oxygen of the substrate is catalyzed by Tyr183, which functions as a catalytic acid, with Lys187 lowering the pKa of the Tyr183 hydroxyl. Concomitantly, hydride transfer takes place from the nicotinamide ring to the C11 position of steroid substrate. Hydrogen exchange with bulk solvent reprotonates Tyr183 via a conserved relay system which includes Asn143, the 2'-OH of the nicotinamide ribose, and a water molecule (533).
D. 11β-HSD1: Reaction Direction and Cosubstrate
11β-HSD1 is predominantly an oxo-reductase in most intact cells and in vivo (FIGURE 1) and is the only enzyme able to reduce 11-keto-glucocorticoids, at least in mice (364). The reported Km of 11β-HSD1 for its glucocorticoid substrates ranges from ∼150 nM to almost 20 μM (379, 442), higher than the low nanomolar free circulating substrate levels. However, in intact cells, 11β-HSD1 is clearly functionally relevant even at low (e.g., 2 nM) substrate levels (231). In mice, there is a circadian rhythm in plasma 11-dehydrocorticosterone levels that mirrors corticosterone, with basal levels 2–7 nM, but rising markedly (>25 nM) following acute stress (265). Humans show a similar diurnal rhythm in cortisone levels, varying from a nadir of 10–20 nM to a peak of ∼60 nM in men (519). As cortisol, cortisone levels are increased and the diurnal rhythm flattened in patients with depression (757); such elevated cortisone levels imply increased glucocorticoid action in brain cells expressing 11β-HSD1, potentially contributing to pathogenesis.
In tissue homogenates, 11β-HSD1 is bidirectional. Initially it was postulated that liver 11β-HSD (11β-HSD1) comprised two distinct enzymes (377, 379). The literature was further confused by the inability adequately to distinguish 11β-HSD1 and 11β-HSD2 activities; 11β-HSD1 can use either NAD(H) or NADP(H) as cofactor (36, 437). This was resolved by two discoveries: 1) there are two 11β-HSD isozymes, and 2) the predominant oxo-reductase activity of 11β-HSD1 in intact cells is due to its colocalization with H6PDH (41, 97).
1. H6PDH determines 11β-HSD1 reaction direction
H6PDH physically and functionally interacts with 11β-HSD1 within the ER lumen (54), and the two enzymes copurify (541) (FIGURE 3). H6PDH catalyzes the first two steps of the ER pentose-phosphate pathway (distinct from the cytosolic pathway), converting glucose-6-phosphate to 6-phosphogluconolactone and concomitantly reducing NADP+ to NADPH (693). H6PDH thus generates the high NADPH/NADP+ ratio inside the ER lumen required for 11β-HSD1 to function efficiently as a reductase (177). In human omental preadipocytes which lack H6PDH, 11β-HSD1 is a dehydrogenase (101), whereas in mouse visceral preadipocytes which express some H6PDH, reductase activity predominates (146).
In mice and humans, inactivating mutations in H6PDH switch 11β-HSD1 activity from oxo-reduction to dehydrogenation (389, 390) and are causal in the human disorder “apparent cortisone reductase deficiency” (390). Interestingly, mutations which inactivate 11β-HSD1 itself, cortisone reductase deficiency (393), produce a milder phenotype because, although 11β-reductase activity is lost, there is no “gain” of dehydrogenase activity which occurs with H6PDH deficiency.
The ER is impermeable to NADP(H). Thus its generation within the ER by H6PDH is dependent on the intraluminal concentration of glucose-6-phosphate which readily enters the ER via the glucose-6-phosphate transporter (693). 11β-HSD1 reaction direction is therefore coupled to cellular energy (glucose) levels. Indeed, 11β-HSD1 reductase activity is decreased in humans and mice with mutations in the glucose-6-phosphate transporter (glycogen storage disease, type 1b) in whom ER glucose-6-phosphate levels are reduced (748). Conversely, 11β-reductase activity is markedly increased in patients with glycogen storage disease type 1a caused by deficiency of glucose-6-phosphatase-α that, in liver, competes with H6PDH for glucose-6-phosphate (748). Fructose-6-phosphate converted to glucose-6-phosphate by an ER hexose-6-phosphate isomerase (628), also increases 11β-reductase activity in liver microsomes (449, 628).
Glucose levels probably regulate 11β-HSD1 activity, but whether they alter reaction direction is uncertain. Lowering extracellular glucose levels (from 25 mM to a more physiological 6 mM) switches 11β-HSD1 activity from reductase to dehydrogenase in transfected cells. However, in hepatoma and adipocyte cell lines which express endogenous H6PDH, 11β-reductase activity predominates irrespective of extracellular glucose levels (177). Conversely, rat Leydig cells, which express ample H6PDH (241), switch 11β-HSD1 activity from reductase to dehydrogenase when glucose concentrations fall (196). The extent to which this happens in vivo is unclear. In the perfused rat liver (with 11 mM glucose), 11β-reductase activity predominates (319). Even in starvation, which reduces intraluminal NADPH/NADP+ in rat liver, reductase activity still predominates though 11β-dehydrogenase activity is increased (341). Clearly understanding of the coupling between glucose, ER luminal redox, and 11β-HSD1 reaction direction is incomplete.
2. But there may be more than H6PDH?
Although H6PDH is often coexpressed with 11β-HSD1 (241), there is an apparent lack of complete congruity even in cells clearly having 11β-reductase. In kidney, 11β-HSD1 but not H6PDH is expressed in medullary interstitial cells. In the CNS, the expression of H6PDH and 11β-HSD1 generally differs (241), contrasting with the exclusive 11β-reductase activity in brain (572) that is indeed unusually stable (377, 379, 380). Finally, as mentioned above, an 11β-HSD1 mutant directed to the cytoplasm retains 11β-reductase activity with cortisone and loses cortisol oxidation altogether (204, 530). Thus whether or not H6PDH is the exclusive supplier of reducing equivalents to 11β-HSD1 in all tissues is currently unclear; it is possible that ER-localized isocitrate dehydrogenase may also provide NADPH to 11β-HSD1 (430). Dissecting the relative importance of this and other possible pathways generating luminal NADPH in brain, adipose, and other tissues is of key interest, especially as drugs inhibiting 11β-reductase are in clinical trials.
3. 11β-HSD1: nonglucocorticoid substrates
11β-HSD1 is a promiscuous enzyme. It has broad substrate (and inhibitor) specificity (TABLES 1 AND 2). Moreover, differences in the substrate-binding pocket result in considerable species differences in 11β-HSD1 substrate specificity and reaction kinetics. In addition to its role in glucocorticoid metabolism, which includes the synthetic glucocorticoids prednisolone and betamethasone, but not dexamethasone (154), 11β-HSD1 catalyzes other steroid and sterol interconversions. It also detoxifies xenobiotics (440).
Table 1.
Selected substrates of 11β-HSD1
| Substrate | Apparent Km, μM | Species | Source | Reference Nos. |
|---|---|---|---|---|
| Cortisol | 10–50 | Human | Purified or expressed in yeast | 305 |
| Cortisol | 300 | Mouse | Purified liver protein | 437 |
| Corticosterone | 6–40 | Human | Purified liver protein | 305 |
| Corticosterone | 2–7 | Mouse | Liver or lung microsomes | 127, 435 |
| Cortisone | 2–40 | Human | Purified, liver microsomes or expressed in yeast | 305 |
| Cortisone | 10 | Mouse | Purified liver protein | 438, 443 |
| 11-Dehydrocorticosterone | 0.3–20 | Human | Purified liver protein, transfected cell lysate, or expressed in yeast | 305 |
| 11-Dehydrocorticosterone | 1–40 | Mouse | Purified liver protein or liver/lung microsomes | 127, 435, 438, 443 |
| NNK | 630 | Mouse | Lung microsomes | 435 |
| 7-Ketocholesterol | 0.4–50 | Human, mouse | Transfected cell lysates, purified recombinant proteins from yeast | 304, 621 |
| 7-KetoDHEA | 1 | Human | Expressed in yeast | 497 |
| 7-Keto-5α-androstane-3β,17β-diol | 7 | Human | Expressed in yeast | 277 |
| 7-Ketoepiandrosterone | 0.5 | Human | Expressed in yeast | 278 |
| Metyrapone | 370 | Human | Expressed in yeast | 306 |
| Ketoprofen | 20 | Human | Expressed in yeast | 306 |
| Prednisone | 21 | Human | Expressed in yeast | 305 |
Table 2.
Selected inhibitors of 11β-HSD1
| Inhibitor (of Glucocorticoid Metabolism) | Ki, μM | Species | Source | Reference Nos. |
|---|---|---|---|---|
| Carbenoxolone | 0.02–0.3 | Human | Liver microsomes or expressed in yeast | 305 |
| Carbenoxolone | 0.1 | Mouse | Expressed in yeast | 55 |
| Glycyrrhetinic acid | 0.04–2 | Human | Liver microsomes, transfected cells, or expressed in yeast | 305 |
| Progesterone | 2 | Human | Liver microsomes | 155 |
| 11β-Hydroxyprogesterone | 0.4 | Human | Liver microsomes | 155 |
| 7-Ketocholesterol | 8 | |||
| 7-Ketopregnenolone | 0.7 | Human | Transfected cells | 515 |
| Deoxycorticosterone | 4 | Human | Liver microsomes | 155 |
| Dexamethasone | 8 | Human | Expressed in yeast | 305 |
| Budesonide | 60 | Human | Expressed in yeast | 305 |
| 7-Keto-DHEA | 1 | Human | Human skin microsomes | 276, 515 |
| CDCA | 4 | Human | Liver microsomes | 155 |
| Lithocholic acid | 3 | Human | Liver microsomes | 155 |
| Metyrapone* | 3,000* | Human | Liver microsomes | 155 |
| Ketoconazole* | >10* | Human | Liver microsomes | 155 |
| Flavanone | 18–21 | Human | Transfected cells | 620 |
| Abietic acid | 5–27 | Human | Transfected cells | 620 |
| BVT14255 | 0.052 | Human | Transfected cells | 55 |
| BVT14255 | 0.28 | Mouse | Transfected cells | 55 |
| BVT2733 | 3.34 | Human | Transfected cells | 55 |
| BVT2733 | 0.096 | Mouse | Transfected cells | 55 |
| Merck-544/T0504 | 0.008+–0.015 | Human | Transfected cells | 282 |
| Merck-544/T0504 | 0.08–0.097+ | Mouse | Transfected cells | 282 |
For additional inhibitors, see Reference 811. Ki values are in μM and normally measured for inhibition of cortisone reduction. Values for transfected cells are ascertained in lysates.
Inhibits reductase only. +IC50 values. Merck-544 is the same compound as T0504.
A) 7-STEROLS.
The metabolism of 7-oxygenated sterols and steroids by 11β-HSD1 probably reflects the rotational symmetry of the 11β- and 7α- positions of the steroid nucleus (386) (FIGURE 1). 11β-HSD1 interconverts 7-keto- and 7-hydroxy-DHEA (both bind with higher affinity to 11β-HSD1 than do glucocorticoid substrates; Refs. 497, 515), 7-keto- and 7-hydroxy-pregnenolone (515), and 5α-androstane-3β-ol-7,17-dione and 5α-androstane-3β-ol-7-hydroxy,17-one (515). Additionally, 11β-HSD1 may act as an epimerase, interconverting 7α- and 7β-hydroxy-steroids via a 7-keto intermediate (277, 278, 497). Epimerase activity requires oxidative and reductive activities and therefore may be determined by H6PDH levels and glucose availability (515).
In humans, rats and mice (though not dog or guinea pig; Ref. 32), 11β-HSD1 interconverts 7-ketocholesterol and 7β-hydroxycholesterol (304, 621) (hamster 11β-HSD1 produces both 7β- and 7α-hydroxycholesterol; Ref. 32). The 7-keto-sterol substrates competitively inhibit glucocorticoid metabolism, and vice versa, with glucocorticoid substrates inhibiting oxysterol reduction (53, 515, 582, 749), suggesting the oxysterols could be important modulators of intracellular glucocorticoid levels in tissues where they accumulate to significant levels, with liver, brain and adipose tissue being the most obvious sites. This also has the potential to impact 11β-HSD1 reaction direction when H6PDH is limiting; 7-ketocholesterol represses whereas 7β-hydroxycholesterol enhances cellular glucocorticoid activity at GR, an effect alleviated by increasing NADPH supply to 11β-HSD1 through over-expression of H6PDH (749).
B) BILE ACIDS.
Other substrates are emerging. Human 11β-HSD1 unidirectionally converts the secondary bile acid 7-oxo-lithocholic acid (generated by gut micro-organisms) to chenodeoxycholic acid and, to a lesser extent, ursodeoxycholic acid (531). Whether this reaction is relevant to bile acid hepatotoxicity and cholestasis merits attention, but the potential for ursodeoxycholic acid in therapy of primary biliary cirrhosis and as an anti-inflammatory (543) supports some involvement.
C) CARBONYL REDUCTION.
Carbonyl reductases play key roles in the detoxification of reactive intermediary metabolites and xenobiotics (e.g., drugs, insecticides, carcinogens) containing carbonyl (aldehyde or ketone) groups (537). In mouse liver, 11β-HSD1 shows carbonyl reductase activity with metyrapone, an inhibitor of 11β-hydroxylase and thus adrenocortical glucocorticoid synthesis (436, 439). As other nonglucocorticoid substrates, metyrapone competes with glucocorticoids for metabolism by 11β-HSD1 (155, 609). Other xenobiotics metabolized by 11β-HSD1 include, in lung, nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (441), a potent nitrosamine carcinogen in tobacco. 11β-HSD1 also metabolizes triadimefon, a broad-spectrum conazole fungicide used extensively in agriculture (340). In liver, 11β-HSD1 metabolizes the anti-inflammatory drug ketoprofen and activates the pro-drug DFU-lactol (306). Activity on other drugs and xenobiotics is probable.
This broad substrate specificity of 11β-HSD1, with its role in detoxification reactions as well as potential activation of neurosteroids such as DHEA and pregnenolone, has implications for the use of inhibitors. Indeed, some of the beneficial cognitive (613, 660, 791, 794) and atheroprotective (282, 493) effects of 11β-HSD1 deficiency or inhibition may, in part, reflect decreased metabolism of nonglucocorticoid substrates (792).
E. 11β-HSD1: Distribution
In adult mammals, 11β-HSD1 is widely distributed. Roughly similar distributions are reported in rodents, non-human primates, and humans (4, 478, 695). Expression is highest in the liver (478, 695). In addition, 11β-HSD1 mRNA and protein/enzyme activity occur in adipose tissues (100), vasculature (747), ovary (68), testis (in some species including human and rat; Refs. 558, 695), but not mouse or squirrel monkey (486, 571), brain (380, 477, 478), uterus (103), placenta (notably in the decidua) (734), immune and inflammatory cells (231), skeletal muscle (767), and heart (747). 11β-HSD1 is not expressed in fetal life until late in gestation when levels are greatest in organs where glucocorticoid activity is required for late maturation prior to birth, notably lung and liver (662).
A problem with tissue localization of 11β-HSD1 reflects the difficulties in generating specific antisera. Early efforts gave a major 34-kDa band on western blots, compatible with the cDNA-predicted molecular weight plus known glycosylation (9, 541). A 68-kDa band is reasonably assumed to be a dimer. However, other higher and lower molecular weight bands were prominent and were not unequivocally 11β-HSD1 (380). More recent antisera also yield additional bands (738). Their use to localize 11β-HSD1 by immunocytochemistry remains complicated by issues of specificity, although preabsorption with purified antigen suggests this may be feasible in specific tissues (477). Similar caveats apply to commercially available antisera, generally raised to peptides rather than the purified or recombinant proteins. Allowing this caveat, 11β-HSD1 mRNA and immunoreactivity have been localized in a variety of cell types. Usefully, such localization can be coupled with 11β-HSD1 activity assays to show functional protein, crucial given the presence of alternative transcripts encoding inactive protein in some organs (kidney) and the sensitivity of PCR which may detect low levels of transcripts in almost any cell, whether or not this “expression” is of biological significance.
F. 11β-HSD1 Regulation
Many factors, endocrine, inflammatory, and metabolic, regulate 11β-HSD1. This is often in a temporal and tissue-specific manner. For example, proinflammatory cytokines, notably interleukin (IL)-1 and tumor necrosis factor (TNF)-α, potently upregulate 11β-HSD1 mRNA in a variety of cells, although interestingly not in immune cells (discussed in sect. V).
Glucocorticoids themselves increase 11β-HSD1 expression in many cells (99, 130, 183, 259, 316, 572, 680, 681, 768, 789), probably via C/EBPβ (604). In vivo, adrenalectomy lowers liver and hippocampal 11β-HSD1 expression in the short-term, but enzyme activity subsequently recovers (317), much like GR itself (202, 292). Conversely, elevated glucocorticoids in intact animals increase 11β-HSD1 in some tissues (adipose tissue, perinatal lung) but not in others (liver) (52, 465, 650).
The effects of insulin on 11β-HSD1 are less clear. In vivo, insulin-induced perturbations of metabolism complicate interpretation. Acutely, in human adipose tissue insulin increases 11β-HSD1 activity, perhaps via changes in glucose availability (194), without changes in 11β-HSD1 mRNA (355, 612, 740). Insulin has no effect on renal 11β-HSD1 in diabetic rats (411). In vitro, high levels of insulin downregulate 11β-HSD1 expression (259, 316, 410, 731) probably through insulin-like growth factor I (IGF-1) receptors, whereas lower concentrations of insulin (too low to bind IGF-1 receptors) have no effect or modestly increase 11β-HSD1 expression in adipocytes (52, 510). Interpretation is confounded by the use of high levels of glucose, which mimic an insulin-resistant state, and the mitogenic effects of insulin in culture which may decrease 11β-HSD1: there is negligible 11β-HSD1 in most proliferating cell lines.
Hepatic 11β-HSD1 is sexually dimorphic in rodents, with lower levels in females. In rats, this is underpinned by sex-specific patterns of pituitary growth hormone (GH) secretion (419). How GH secretion patterns alter 11β-HSD1 remains unclear. It is probably not a direct effect of GH on hepatocytes (316), but could be mediated by IGF-I (402, 410, 510, 711, 731, 768). Repressive effects of GH on 11β-HSD1 in humans have been inferred from patients with hypopituitarism or acromegaly (225, 487) but may be mediated via other hormones or intermediary metabolism.
Testosterone has no effect on 11β-HSD1, at least in liver and kidney (239, 418). However, estradiol, at high (pharmacological) levels, strongly represses 11β-HSD1 in rodent liver and kidney (though not in hippocampus) (239, 318, 394, 418), probably indirectly (316). In rats, repression of 11β-HSD1 by thyroid hormone is similarly tissue specific (354, 771), and disturbances in glucocorticoid metabolism in patients with thyroid dysfunction are also consistent with thyroid hormone repression of 11β-HSD1 (272, 813). The pleiotropic effects of thyroid hormone on metabolism are likely to underlie this tissue-specific regulation, rather than direct effects on the 11β-HSD1 gene.
Like GR, the peroxisome proliferator-activated receptors (PPARs) and the liver X receptors (LXRs), intersect metabolism and inflammation. Agonists of both classes of nuclear receptor are reported to downregulate 11β-HSD1 in liver and brown adipose tissue (69, 283, 679). However, others report no effect of LXRα agonism on adipocyte 11β-HSD1 (749). The time course and sensitivity to cycloheximide suggest that any changes following PPAR or LXR activation is unlikely to involve binding to the Hsd11b1 gene (and hepatic 11β-HSD1 is unaltered in PPARα or LXRα -β knockout mice) (283, 679). In vitro, a PPARγ binding site in the human HSD11B1 gene promoter mediates induction in alternatively activated macrophages (differentiated with IL-4/IL-13) (117), but this site is not conserved in rodents.
To date, the C/EBPs, critical integrators of inflammatory and metabolic signaling, remain the only factors directly implicated in binding to the gene promoter to effect alterations in 11β-HSD1 transcription following endocrine, inflammatory, and metabolic stimuli, possibly also mediating at least some of the effects of nuclear receptors, as recently suggested in rat vascular smooth muscle cells (725).
III. 11β-HSD2 GENE STRUCTURE, ENZYMOLOGY, AND REGULATION
A. The HSD11B2 Gene and Its Transcriptional Control
11β-HSD2 is present in sharks and land vertebrates, and an ancestral gene has been identified in the nematode worm Caenorhabditis elegans (50). This may catalyze retinol interconversions, thus potentially controlling ligand access to nematode nuclear receptors (471) in analogy to its mammalian functions. Orthologs of HSD11B2 have been found in 36 species to date (http://www.ensembl.org). Unlike 11β-HSD1, 11β-HSD2 is present in fish (51) where it may confer selective access of the potent mineralocorticoid deoxycorticosterone to MR (324, 373). In fish, 11β-HSD2 is also important in testicular androgen activation and sex determination (197) by catalyzing conversion of 11β-hydroxy-testosterone to active 11-keto-testosterone (470).
11β-HSD2 cDNA was first cloned from sheep kidney (6), although a sequencing error wrongly predicted the COOH terminus of the protein sequence (111). This was followed by 11β-HSD2 cDNAs from human, rat, rabbit, mouse (15, 91, 127, 512, 812), and other species (591).
The human HSD11B2 gene is located on chromosome 16q22 and comprises five exons spanning just 6 kb (8) (FIGURE 6). Transcription starts at multiple sites (8), typical of its TATA-less, CpG island-containing promoter. The 5′-flanking region and exon 1 of HSD11B2 are encompassed within a CpG island (8), conserved in baboon (556) and rodents (17). This contains binding sites for members of the Sp1 transcription factor family (10), NF1 (17), NF-κB, and Egr-1 (362) (FIGURE 6). Occupancy of the Sp1 sites correlates with expression levels of 11β-HSD2, although their relative importance differs between cell lines (10, 516). Sp1 also plays a key role in the induction of 11β-HSD2 that occurs with syncytialization of human placental trophoblasts (400). In colon carcinoma cells, HSD11B2 repression by TNF-α or phorbol ester (mimicking the effects of inflammation upon colon; see below) is associated with Egr-1 and NF-κB (p50 homodimers) binding to the HSD11B2 promoter, with Sp1/Sp3 constitutively bound (362). Selective PPARδ activation in primary human placental trophoblast cells also decreases HSD11B2 promoter activity (332), although whether this is direct or via Sp1 is unknown. A polymorphism in the proximal HSD11B2 promoter that decreases NF1 and Sp1 binding and reduces promoter activity associates with salt-sensitive blood pressure excursions in normotensive subjects (18). The molecular basis of tissue and disease-associated regulation of HSD11B2 is an important goal and has potential therapeutic implications.
Figure 6.

Structure of the human HSD11B2 gene showing positions of relevant transcription factor binding sites. Schematic representation of the human HSD11B2 gene (not to scale) showing positions of relevant transcription factor binding sites. The gene spans 6.4 kb. Exonic sequences are shown as boxes; white boxes encode the open reading frame, with a red box and a blue box indicating the 5′ leader and the 3′ untranslated sequence, respectively. An arrow indicates the position of the promoter, although it should be noted that transcription starts at a number of sites clustered around the site shown (see text for details). Sp1/Sp3 sites are shown as green ovals (these overlap with Egr1 sites), an NF1 site is shown as a pink hexagon, and an NF-κB binding site is shown as a purple diamond. The positions of 2 polymorphisms that reduce transcription factor binding and promoter activity (18) are indicated by asterisks. See text for details.
The HSD11B2 CpG island is subject to epigenetic regulation. Greater cytosine methylation within the CpG island associates with lower 11β-HSD2 expression across a range of human cells and tissues (17). Intrauterine growth retardation in the rat is linked to increased Hsd11b2 CpG island methylation in kidney and with reduced adult renal 11β-HSD2 expression (58), although overall methylation levels are low. However, fetal exposure to glucocorticoids in rats, which also reduces adult renal 11β-HSD2 levels, had no effect on methylation of the Hsd11b2 CpG island (782). Increased methylation of the HSD11B2 promoter in peripheral blood mononuclear cells has been reported in subjects with essential hypertension (205), although 11β-HSD2 is not normally expressed in leukocytes (compatible with the relatively high levels of methylation observed); any relevance to kidney 11β-HSD2 is moot. Nevertheless, maternal consumption of a highly unbalanced diet in pregnancy, which associates with hypertension and hyperglycaemia in her adult children, has also been linked to increased methylation of the HSD11B2 promoter in leukocytes (164). Thus leukocyte methylation may represent an echo of changes in kidney and, if so, implies that the early life environment impacts on this promoter, as others including GR (755) in the HPA axis system.
B. 11β-HSD2 Protein
11β-HSD2 (SDR9C3; Ref. 557) is a member of the SDR family (557, 778). Its nearest relative is 17β-HSD2 (35% identical) (15, 48). Translation starts in the first exon (8). Human 11β-HSD2 comprises 405 amino acids, with a predicted molecular mass of 44 kDa (15, 91), whereas mouse is shorter, with 386 amino acids and a predicted mass of 42 kDa (http://www.ensembl.org) due to a truncated COOH terminus. Like all other members of the SDR family, 11β-HSD2 contains a nucleotide cofactor-binding Rossmann-fold in the NH2-terminal region (Thr88 to Gly95) and a Tyr-[Xaa]3-Lys motif in the active site (Tyr232 to Lys236 in human 11β-HSD2).
C. Structure and Mutations
11β-HSD2 is a very basic (pI 9.9) and stable intrinsic membrane protein (91) that loses activity once extracted from membranes (91). Within cells, it is anchored in the ER membrane, probably via three transmembrane helices predicted in the NH2 terminus (91). Although the NH2-terminal five amino acids are likely to reside in the ER lumen, the COOH-terminal bulk of the protein following Ala73 sits within the cytoplasm (an orientation opposite to 11β-HSD1) (511, 530). Intriguingly, 11β-HSD2 may physically interact with cytoplasmic MR (529). Like 11β-HSD1, 11β-HSD2 is probably active as a dimer (527).
There are no three-dimensional crystal structures of 11β-HSD2 yet available. Homology models (715) put the conserved active site Tyr232 and Lys236 close to the nicotinamide group of NAD+, with the adenosine moiety of NAD+ bound in the Rossmann fold in the NH2 terminus (36). The preference of 11β-HSD2 for NAD+ over NADP+ depends primarily on Glu115, which sterically and electrically repulses the phosphate on NADP+ (36) and is conserved among SDR enzymes using NAD+.
The three-dimensional models have offered structural insights into AME (36). Most AME-causing mutations lie in the COOH-terminal half of 11β-HSD2 and affect conserved regions (491). Mutations that affect catalysis, cofactor binding, or dimerization produce the most severe phenotypes. These include mutation of the active site Tyr232 in a compound heterozygote (388) and deletion of Leu114 and Glu115, predicted to constrain the cofactor binding site (36). Mutation of Tyr226 in another compound heterozygote AME patient (388) is predicted to disrupt the dimer interface (714, 715) and may also affect substrate binding that occurs close to the side chain of Tyr226 (429). A mutation in the adjacent Pro227, which may normally position Tyr226 close to substrate (429), produces a mild form of AME with ∼50% reduced 11β-HSD2 activity and low-renin hypertension but without hypokalaemia or metabolic alkalosis (776). AME mutations at Tyr338 or Arg337 in the COOH terminus cause thermodynamic instability and misfolding, reducing protein half-life (39), consistent with reduced stability following deletion of the nonconserved COOH terminus of 11β-HSD2 (527). Mutations at Arg359 and Leu376 in the COOH terminus may similarly affect protein folding and stability (388, 429). Likewise, heterologous expression systems suggest that AME mutations at Ser180, Ala237, and Ala 328, which partially reduce 11β-HSD2 activity, do so by reducing stability (520). A homozygous mutation in Arg279 (399), predicted to lie on the surface of 11β-HSD2 (429), reduces Vmax but has little effect on substrate affinity, causing the milder “type II” AME, probably due to minor structural disruption. Overall, AME patients show clear relationships between loss of 11β-HSD2 activity and phenotype (491, 765).
D. 11β-HSD2 Enzymology
In all species examined, 11β-HSD2 acts exclusively as an NAD+-dependent, high-affinity but low turnover dehydrogenase with physiological glucocorticoid substrates (Km corticosterone ∼10 nM; cortisol ∼50 nM), showing little activity with NADP+ (15, 90–92, 511–513, 527, 812). It utilizes an ordered bi-bi sequential enzymatic mechanism in which cofactor binds prior to and leaves after the steroid (92).
1. Substrates and inhibitors
In addition to cortisol and corticosterone, dexamethasone is metabolized to some extent by 11β-HSD2 (91), with a Km of ∼100 nM (91). Moreover, while 11β-HSD2 shows exclusively oxidative activity with physiological glucocorticoid substrates, in microsomes and whole cell assays it reduces 9-fluorinated glucocorticoids, thus converting 11-dehydrodexamethasone to dexamethasone. This regeneration contributes to the long half-life of dexamethasone in vivo (154, 156, 157, 527) (TABLE 3).
Table 3.
Selected substrates of 11β-HSD2
| Substrate | Apparent Km, μM | Species | Source | Reference Nos. |
|---|---|---|---|---|
| Cortisol | 0.025–0.055 | Human | Purified placental protein, kidney microsomes, transfected cells | 90, 91 |
| Cortisol | Mouse | |||
| Corticosterone | 0.010–0.012 | Human | Purified placental protein, transfected cells | 90, 91 |
| Corticosterone | 0.01 | Rat | Transfected cells | 812 |
| Corticosterone | 100 | Mouse | Mouse kidney microsomes | 127 |
| Dexamethasone | 0.119 | Human | Transfected cells | 91 |
| 11-Dehydrodexamethasone | 0.068 | Human | Kidney microsomes | 157 |
Values for transfected cells are ascertained in lysates.
The classical licorice-derived 11β-HSD inhibitors glycyrrhetinic acid and carbenoxolone (see FIGURE 7) are more potent inhibitors of 11β-HSD2 than 11β-HSD1, with Ki or IC50 typically severalfold lower for 11β-HSD2 than 11β-HSD1 (658) (TABLE 4). Modification of glycyrrhetinic acid at the carboxyl and/or 3-hydroxyl positions creates more selective potent 11β-HSD2 inhibitors (367). Other 11β-HSD2 inhibitors, with varying degrees of selectivity, have been identified. Gossypol, a natural phenol isolated from cotton seeds and originally proposed as a male contraceptive, inhibits 11β-HSD2 (116), conceivably accounting for the hypokalemic paralysis it can cause (739). Synthetic endocrine disrupters, including some phthalate derivatives widely used in plasticisers and solvents, organotins used in agriculture, and alkylphenols used as industrial surfactants, also inhibit 11β-HSD2 (424), although the importance of this is uncertain.
Table 4.
Selected inhibitors of 11β-HSD2
| Inhibitor (of Glucocorticoid Metabolism) | Ki, μM | Species | Source | Reference Nos. |
|---|---|---|---|---|
| Carbenoxolone | 0.010–0.083 | Human | Kidney microsomes, transfected cells | 91 |
| Glycyrrhetinic acid | 0.006–0.028 | Human | Kidney microsomes, transfected cells | 91 |
| Progesterone | 0.048 | Human | Kidney microsomes | 155 |
| 11β-Hydroxyprogesterone | 0.007 | Human | Kidney microsomes | 155 |
| Deoxycorticosterone | 0.1 | Human | Kidney microsomes | 155 |
| CDCA | 20/none | Human | Kidney microsomes, transfected cells | 155 |
| Lithocholic acid | >10 | Human | Kidney microsomes | 155 |
| Metyrapone | >1,000 | Human | Kidney microsomes | 155 |
| Ketoconazole | 10 | Human | Kidney microsomes | 155 |
| Flavanone | 200 | Human | Transfected cells | 32 |
| Abietic acid | 11–12 | Human | Transfected cells | 620 |
| BVT14225, BVT2733 | >10 | Human | Transfected cells | 55 |
| Merck-544/T0504 | 1.8–>3.3 | Human | Transfected cells | 282 |
For additional inhibitors, see Reference 620. Values for transfected cells are ascertained in lysates. Merck-544 is the same compound as T0504.
E. 11β-HSD2: Tissue Distribution
In adults, 11β-HSD2 is largely restricted to the classical aldosterone (mineralocorticoid)-target tissues including distal nephron, sweat and salivary glands, and colonic epithelium (6, 15, 285, 368, 512, 588, 589, 652, 654, 770). It is also expressed in exocrine pancreas (15) and adrenal cortex (6, 588) and at more modest levels in the ileum, female and male reproductive systems (15, 588), and other epithelia including skin, lung (7, 654), and vascular endothelium (84). 11β-HSD2 expression in kidney and colon is sexually dimorphic, at least in mice (127). There are species differences in adult 11β-HSD2 expression. 11β-HSD2 is highly expressed in adrenal in sheep and rat, but less in mouse (127). 11β-HSD2 is expressed in testis in humans (15, 695), but not mice (127, 488) or rats (4, 588). Expression in some sites, including vascular smooth muscle cells, remains controversial (84, 120, 653), although a functional role in the nongenomic effects of aldosterone in the vasculature has been suggested (19).
During early to mid-gestation, 11β-HSD2 is widely expressed in the fetus and placenta (673), localized to syncytial trophoblasts (91, 368) (see developmental programming below). Many neoplastic cells express 11β-HSD2 perhaps reflecting their less differentiated state.
F. Regulation of 11β-HSD2
Estrogen upregulates 11β-HSD2 in kidney (239, 267, 418), the opposite of its effect on 11β-HSD1. This reciprocal control is generally true of other factors. Thus, whereas 11β-HSD1 is increased by inflammation and proinflammatory cytokines, such as TNF-α (129, 271, 362, 692), expression of 11β-HSD2 is usually reduced (discussed below). Hypoxia downregulates 11β-HSD2 in rat kidney, probably through Egr-1 (270). Beyond cytokines, protein kinases A and C stimulate and repress 11β-HSD2 mRNA and protein, respectively, in renal cells. The former is a pathway plausibly activated by vasopressin (599), affording a route to minimize renal retention of sodium alongside water, thus attenuating the risk of hypertension. Finally glucocorticoids upregulate 11β-HSD2 in lung cells (685), but apparently not in fetal kidney, and downregulate placental 11β-HSD2 (122, 343). More understanding is needed of this complex age- and organ-specific regulation. Even the widespread silencing of 11β-HSD2 in fetal tissues at the point of entry into terminal maturation is poorly understood. Such questions are of substantial importance, notably in dissecting mechanisms of developmental programming of offspring physiology and pathology.
IV. 11β-HSDs, METABOLISM, BLOOD PRESSURE, AND CARDIOVASCULAR FUNCTION
11β-HSD1 is highly expressed in the key metabolic organs liver, adipose tissue, skeletal muscle, and the islets of Langerhans, while 11β-HSD2 is crucial to blood pressure regulation. Both isozymes are expressed in the vasculature, and 11β-HSD1 at least is present in the heart and inflammatory cells (see below). Given this distribution and the striking metabolic disease seen in Cushing's syndrome of circulating glucocorticoid excess, it is not surprising that the role of 11β-HSDs in cardiometabolic function in health and disease has been studied in some detail. Although this is discussed below at the level of individual organs, of course in vivo these are highly integrated physiological systems.
A. Liver
11β-HSD1 was initially discovered, characterized, purified, cloned, and most highly expressed in liver (4, 21, 378, 478, 695). Despite this head start, and the absence of 11β-HSD2, the function of 11β-HSD1 in hepatocytes and especially in liver in vivo is far from completely understood.
In primary rat hepatocytes, 11β-HSD1 acts predominantly as a reductase (316). The in situ perfused rat liver also shows substantial and predominant 11β-reductase activity ex vivo (319), as does the dog liver in vivo (60, 178). In humans, the liver exhibits net regeneration of cortisol from cortisone (743). This activity is lower in simple obesity but unaltered in obesity complicated by type 2 diabetes (59, 61, 675), an observation suggesting maintenance of this “target” for selective 11β-HSD1 inhibitors in a patient population.
1. Endocrine effects of hepatic 11β-HSD1
Overall, the human liver/splanchnic bed contributes between 20 and 40% of daily cortisol production (25, 62), thus making a major impact on the half-life of cortisol. This affects the HPA axis. On some genetic backgrounds 11β-HSD1 null mice show elevated basal corticosterone levels and exaggerated responses to stress (265). This was originally attributed to deficient glucocorticoid regeneration in critical central sites of glucocorticoid negative feedback, such as the pituitary, paraventricular nucleus of the hypothalamus (PVN), and hippocampus, all of which express 11β-HSD1 (380, 478, 624). However, transgenic “rescue” of 11β-HSD1 solely in liver of null mice restores HPA axis function to normal (547). Conversely, liver-specific deletion of 11β-HSD1 increases adrenal size, suggesting HPA axis activation, consistent with a role for peripheral 11β-HSD1 in control of the HPA axis (392). While the underlying basis for this is not fully established, it is known that sugars and perhaps other fuel substrates impact on the HPA axis (139, 374, 387), and an afferent hepatic pathway via the vagus nerve also modulates HPA axis function (375). The notion that liver metabolism of glucocorticoids controls the stress axis is intriguing, plausibly representing a key node in the intimate association between the key organ determining metabolic fuel availability and the major neuroendocrine control of fuel homeostasis. This concept merits dissection.
2. Intracrine effects of hepatic 11β-HSD1
In general, 11β-HSD1 deficiency has rather little effect on basal hepatic function in the fed state. Thus 11β-HSD1 null mice fed ad libitum have modestly lowered triglyceride levels associated with less triglyceride-associated apoCIII in the HDL fraction (493). This associates with a modest increase in overall HDL cholesterol. The changes in triglyceride appear driven by increased hepatic expression of key enzymes of fat metabolism such as carnitine-palmitoyl transferase 1 (CPT-1), acyl-CoA oxidase, and uncoupling protein 2 (UCP2), themselves plausibly induced by their controlling transcription factor, PPARα, which is elevated in liver in 11β-HSD1 deficiency (493). Similar effects to increase β-oxidation of fats are seen with short-term selective 11β-HSD1 inhibition, which lowers hepatic very-low-density lipoprotein-triglyceride secretion (74).
On fasting, the normal elevation of PPARα is lost in mice deficient in 11β-HSD1, consistent with reduced glucocorticoid action (493). However, the oxidative response to fasting (induction of CPT-1) is maintained at wild-type values. Refeeding 11β-HSD1 knockout mice reveals exaggerated induction of enzymes of lipogenesis and suppression of enzymes of fat metabolism, effects again consistent with reduced glucocorticoid action and hence increased hepatic insulin sensitivity. Indeed, glucose and triglyceride levels on refeeding are lower in 11β-HSD1 null mice or with selective inhibitor treatment (73, 493).
Beyond the liver, 11β-HSD1 inhibition shifts the uptake of derived fatty acids to oxidative tissues, notably muscle and heart where increased lipid oxidation minimizes deleterious fat accumulation (74). These tisue-specific complex effects underline potential multi-organ benefits of 11β-HSD1 inhibition/deficiency.
In obesity or diabetes, fasting glucose levels are lower with 11β-HSD1 deficiency, consistent with reduced glucocorticoid action in the liver, particularly reduced gluconeogenesis. Specifically hepatic phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) are lower with 11β-HSD1 deficiency or inhibition (12–14, 364, 494). Interestingly, in lean animals, there is no fasting hypoglycemia (364), despite reduced G6Pase, presumably reflecting intact counterregulatory responses. In dogs, selective 11β-HSD1 inhibition attenuates liver glucose production in the face of insulinopenia (178), underpinned by reduced hepatic PEPCK and G6Pase mRNA expression. Gluconeogenic flux is unaffected, implying an effect on glycogenolysis, mirroring similar findings in humans given carbenoxolone (27), which inhibits 11β-HSD1 in liver but poorly in adipose tissue in humans (27) and rodents (413), and improves hepatic insulin sensitivity in humans in vivo, albeit modestly (744).
Overall, the data suggest that 11β-HSD1 increases glucocorticoid action within hepatocytes, stimulating gluconeogenesis and inhibiting β-oxidation of fats. Inhibition of 11β-HSD1 in the liver is predicted to be therapeutically beneficial in obesity and its associated metabolic disease, although the magnitude of the hepatic contribution remains uncertain. Indeed, the metabolic phenotype in liver-specific knockout of 11β-HSD1 in mice is relatively mild with modestly improved glucose tolerance (392). Perhaps the interplay between organs is of greater importance.
Hepatic 11β-HSD1 is regulated by diet, gender, and hormones. In most cases of metabolic syndrome, 11β-HSD1 expression in liver is either maintained or modestly lowered (in contrast to the consistent finding of elevated adipose tissue 11β-HSD1 in obesity). This perhaps represents a homeostatic attempt to minimize glucocorticoid-mediated insulin resistance. However, 11β-HSD1 is increased in liver in myotonic dystrophy (327), which is characterized by marked insulin resistance and dyslipidemia. Modeling hepatic overexpression in transgenic mice creates an insulin-resistant phenotype without obesity (548), but with increased hepatic PPARα and LXRα and reduced lipid clearance. Transgenic overexpression of hepatic 11β-HSD1 also causes hypertension, plausibly driven by increased angiotensinogen expression in liver.
Overall, therefore, despite high expression, the importance of hepatic 11β-HSD1 in human pathogenesis remains uncertain, and even its major physiological functions are essentially unresolved. Important questions for the future include the role of liver 11β-HSD1 in xenobiotic and oxysterol metabolism, determining its importance in starvation when glucocorticoids may be key to survival (although plasma glucocorticoids are not elevated in chronic malnutrition; Ref. 427), dissecting the relative importance of the various sources of 11β-HSD1 in the splanchnic bed in the overall metabolic and endocrine effects of 11β-HSD1, and resolving its function in the link between the liver and HPA axis activity. An interesting question is the importance of alternative substrates of 11β-HSD1 (7-ketocholesterol, bile acids) in liver. 7-Ketocholesterol in particular may regulate de novo cholesterol biosynthesis (89) which could also impact bile acid pathways. Bile acids are competitive inhibitors of 11β-HSD1 (2, 155, 189) (TABLE 2), but also potentially substrates (531). Glucocorticoids regulate bile acid homeostasis (593), so it is important to determine whether any metabolic effects of 11β-HSD1-deficiency are mediated through altered bile acid homeostasis.
B. Adipose Tissue
11β-HSD1 is expressed in all white and brown adipose tissue depots with higher levels in subcutaneous than visceral (intra-abdominal) fat in mice (75, 495), but either higher (234) or similar (550) levels in human visceral than subcutaneous fat. Interestingly, 11β-HSD1 mRNA and protein/activity can become dissociated in visceral but not subcutaneous adipose tissue in rodents (146) and humans (101, 397), implying posttranscriptional controls in this depot. This might explain discrepancies in the human visceral adipose tissue literature and calls into question studies based solely on mRNA estimations.
11β-HSD1 is found in both preadipocytes and adipocytes. It is a predominant reductase in adipocytes and murine preadipocytes (146, 494), but in human omental preadipocytes is a dehydrogenase as H6PDH is limiting (101). Glucocorticoids promote adipocyte differentiation. In murine and human cells, 11β-HSD1 activity is induced during adipogenesis, assisting the later stages of maturation (98, 510).
The detailed biology of glucocorticoids in adipocytes is imperfectly understood. Both GR and MR are expressed in adipocytes (434), with MR mediating proinflammatory effects (253) whereas GR are anti-inflammatory. This raises the possibility of inverted U-shaped relationships between glucocorticoid concentrations and function, analogous to the hippocampus, the archetypal tissue expressing both GR and MR as well as 11β-HSD1. Moreover, the adipose effects of glucocorticoids depend on context, differing in the fed (insulin high), fasted (insulin low), and obese/insulin-resistant states and differing by adipose depot.
With these caveats, glucocorticoids alone stimulate fatty acid uptake via lipoprotein lipase and lipolysis via induction of hormone-sensitive lipase and adipose triglyceride lipase (643, 730) and thus promote free fatty acid synthesis which increases metabolic fuel provision. However, at least in vitro, low doses of exogenous glucocorticoids suppress adipocyte lipogenesis (217). In line with this, 11β-HSD1 inhibition decreases lipolysis and increases lipogenesis in vitro (217). This conforms with increased subcutaneous adipose depots in high-fat-fed 11β-HSD1 knockout mice, but not with their attenuated visceral depots (494, 750).
In contrast, in the presence of insulin, glucocorticoids augment insulin-stimulated lipogenesis in vitro (217). Ex vivo, adipocytes taken from 11β-HSD1-deficient mice show reduced intra-adipose glucocorticoid levels and greater insulin-mediated glucose uptake and triglyceride hydrolysis (494). 11β-HSD1 inhibition decreases lipid synthesis and fatty acid cycling gene expression and increases fatty acid oxidation via CPT-1 in mesenteric adipose, but increases lipid synthesis and fatty acid cycling and decreases CPT-1 in epididymal fat (72). Interestingly, in humans, access of circulating glucocorticoids to adipose tissue is restricted and slow. This implies that 11β-HSD1 exerts a more substantial impact on local adipocyte steroid levels (303), and hence biology in vivo, than in other tissues where plasma glucocorticoids gain more rapid access. Overall, the effects of 11β-HSD1 on adipose tissues are depot and dietary state dependent. In general, the effects are slight in lean, insulin-sensitive states but, as discussed below, become marked with obesity-insulin resistance.
1. Adipose tissue 11β-HSD1 in obesity
A striking and consistent finding is a tissue-selective two- to threefold increase in 11β-HSD1 activity in subcutaneous adipose tissue in obese men and women (234, 472, 549, 550, 574, 575, 612). The effect is found both ex vivo in enzyme activity assays in adipose biopsies or by in vivo microdialysis in abdominal adipose (612) and is accompanied by lowering of hepatic 11β-HSD1 activity in simple obesity (574), but maintenance of hepatic activity if obesity is accompanied by type 2 diabetes (675). These effects are probably driven by increased transcription since 11β-HSD1 mRNA levels are also elevated (549). Such data have been reported in distinct human populations and appear to be a robust measure of an expanded adipose depot. Similar elevations of 11β-HSD1 in adipose tissue are found in obese rodents with deficits in leptin signaling, sometimes with a predominant effect in visceral adipose depots (413).
In humans, the literature on the effect of obesity on visceral adipose tissue 11β-HSD1 is discordant, with reports of both elevation (150) and no change (234). A possible confounding factor is the upregulation of 11β-HSD1 by cytokines (711), which are elevated in obesity, infection, and perhaps following surgery. Of course, human visceral adipose tissue can only be obtained during operation and if the indication was an inflammatory condition or perhaps a long bariatric procedure then measured 11β-HSD1 may not reflect the underlying state. Two studies of 11β-HSD1 in omental adipose tissue in healthy women undergoing routine minimally invasive surgery show 11β-HSD1 activity and mRNA levels correlate directly with adiposity, its metabolic complications (472) and adipocyte size (larger adipocytes are less favorable metabolically) (466), but in two other studies visceral 11β-HSD1 did not correlate with obesity (234). Thus, unlike the clear consensus in subcutaneous adipose tissue, the jury remains out on whether or not visceral adipose 11β-HSD1 is increased in human obesity.
2. Manipulation of 11β-HSD1 in adipose tissue
Modeling the increased 11β-HSD1 in adipose tissue in obesity, Masuzaki et al. (446) generated a mouse overexpressing 11β-HSD1 in adipocytes under the fatty acid binding protein-4 (aP2) promoter. This produced an ∼2.5-fold overexpression of 11β-HSD1 in adipose depots, mirroring changes in human obesity. This increased intra-adipose corticosterone levels twofold without changing circulating glucocorticoid levels. There was, however, an increase in corticosterone concentration in the portal vein and thus glucocorticoid supply to the liver. Adult aP2-HSD1 transgenic mice show predominantly visceral obesity, attributed to the higher GR expression in visceral than subcutaneous adipose (446). The mice faithfully replicate metabolic syndrome with insulin resistance/impaired glucose tolerance (exacerbated by high fat feeding), dyslipidemia, hypertension (plausibly caused by increased adipose expression of angiotensinogen and reversed by angiotensin 2 receptor antagonism), and hyperphagia (446, 447). These data suggest that elevated 11β-HSD1 in adipose tissue may be pathogenic in metabolic disease associated with obesity. Since plasma cortisol levels are normal or modestly reduced in simple obesity without metabolic disease (745), fat-specific 11β-HSD1 excess has been dubbed “Cushing's disease of the omentum” (100). The prominent involvement of subcutaneous fat suggests this is more accurately “Cushing's Syndrome of adipose tissue.”
Conversely, knockout or inhibition of 11β-HSD1 produces a “favorable” metabolic state, particularly in the presence of obesity. 11β-HSD1 gene disruption in mice produces insulin sensitization and lowers fasting blood glucose without hypoglycemia (364). Importantly, insulin sensitivity and cardiometabolic “health” are improved in diet-induced and in genetic models of obesity (493, 494). Analogous effects are seen with short- to medium-term administration of selective 11β-HSD1 inhibitors in a variety of rodent models of obesity/type 2 diabetes (12, 13, 282, 751, 752). The current consensus, that these beneficial effects are due to lowered intra-adipose glucocorticoid action, is supported by mice with ectopic transgenic expression of 11β-HSD2 in adipose tissue (decreasing intra-adipose levels of corticosterone) which show insulin sensitization and resistance to metabolic disease with obesity (342).
Intriguingly, on a high-fat diet, 11β-HSD1 deficiency leads to preferential fat loss from metabolically disadvantageous visceral and accumulation in “safer” subcutaneous depots (494). Early in the adaption to high-fat diet, subcutaneous adipose tissue from 11β-HSD1-deficient mice shows enhanced insulin and beta-adrenergic signaling associated with the expansion of smaller, more metabolically active adipocytes, rather than the large adipocytes seen in similarly fed controls (750). In contrast, 11β-HSD1 deficiency does not produce insulin sensitization in visceral adipose tissue, but associates instead with maintained “lean mouse” levels of AMPK, although these are reduced in controls. 11β-HSD1-deficient visceral adipose tissue also exhibits marked suppression of inflammatory signaling with reduced expression of proinflammatory chemokines and cytotoxic T-cell and macrophage infiltration (750). Both fat depots show higher levels of PPARγ mRNA and evidence of enhanced PPARγ activation. The altered adipose distribution seen in rodents lacking 11β-HSD1 resembles to some extent the effects of PPARγ agonists (788), but whether PPARγ activation is critical to any of the effects of 11β-HSD1 deficiency/inhibition remains to be clarified. Overall, 11β-HSD1 deficiency engenders anti-inflammatory, AMPK-activated visceral adipose tissue and insulin-sensitized, “safer” metabolic storage subcutaneous depots. This subtle partitioning of metabolism and inflammation between the different adipose depots underlies a potential for beneficial therapeutic effect if it applies to humans.
Of course, 11β-HSD1 is not expressed in adipose tissues merely for contemporary therapeutic benefit. Its evolutionary importance has been little addressed but may reflect needs for rapid storage of excess calories when conditions allow in an environment of alternating feast and famine. Higher adipose 11β-HSD1 would perhaps support expanded visceral adipose depots to allow rapid delivery of calories to the liver. This may have optimized local storage and then release during sleep, dormancy, and the next period of starvation.
3. Regulation of 11β-HSD1 in adipose tissue
Mice or rats fed a high-fat diet show unexpected downregulation of 11β-HSD1 in adipose tissue (165). This is greatest in obesity and metabolic disease-resistant strains and has been hypothesized to reflect an attempt to attenuate some of the adverse metabolic consequences of chronic dietary excess (495).
So how is 11β-HSD1 regulated in adipose tissue? In vivo and in vitro evidence in rodents suggests upregulation by glucocorticoids (604), proinflammatory cytokines (711) and weight loss/stearate feeding (426), and downregulation by high-fat diet/weight gain (495), PPARγ (283), and perhaps LXR agonists (679), although the latter is contested (749). Humans differ in some aspects (740).
The main factors directly regulating 11β-HSD1 transcription in adipose tissues appear to be C/EBPs, notably C/EBPβ (244) and its processed isoforms LIP (inhibitor) and LAP (activator). Indeed, the LIP-to-LAP ratio is key to downregulation of 11β-HSD1 in adipose with high-fat feeding (191). Whether or not this also underpins the upregulation in human obesity remains uncertain, yet crucial.
4. 11β-HSD1, adipose tissue hypoxia, and fibrosis
The expansion in adipose tissue accompanying dietary obesity is associated with a poor angiogenic response that contributes to adipose tissue hypoxia (296). Consequently, adipose tissue synthesizes hypoxia-inducible factor-1α (HIF-1α), potentially precipitating inflammation and fibrosis (258). 11β-HSD1 deficiency attenuates adipose tissue hypoxia, with reduced induction of HIF-1α and less fibrogenesis, associated with reduced activation of the transforming growth factor (TGF)-β/Smad3/α-smooth muscle actin signaling pathway (467). Underlying this is greater angiogenesis in 11β-HSD1-deficient adipose tissue, plausibly driven by PPARγ-stimulated expression of the proangiogenic factors VEGF-A, apelin, and angiopoietin-like protein 4. Thus, with 11β-HDSD1 deficiency, increased angiogenesis may underpin the nonpathological expansion of fat depots allowing safe weight gain. Increased angiogenesis is also seen in the ischemic 11β-HSD1-deficient myocardium and aorta (646) and indeed may contribute to other cardiometabolic benefits of 11β-HSD1 deficiency.
5. 11β-HSD1 deficiency/inhibition and human adipose tissue biology
Does adipose 11β-HSD1 matter in human obesity? Whilst splanchnic 11β-HSD1 contributes substantially (20–40%) to total daily glucocorticoid production rates, this is largely ascribed to the liver, so the adipose effects are likely to be mainly intracrine (674). The nonselective 11β-HSD inhibitor carbenoxolone increases insulin sensitivity (744) by an action largely mediated on the liver (27, 413), although some adipose tissue effects on glycerol production occur (712). Acutely (over hours), enzyme activity may be regulated: lipid infusion increases adipose 11β-HSD1 activity (740). However, over the longer term (days or weeks), PPARα and PPARγ agonists have no impact (741), unlike their repressive effects in rodents.
Sadly, no data on the metabolic phenotype, if any, of subjects with HSD11B1 mutations in cortisone reductase deficiency are available (393). However, a unique patient with “apparent cortisone reductase deficiency” who developed pituitary ACTH-dependent Cushing's disease presenting with secondary amenorrhea illustrates well the importance of 11β-reductase. Despite unequivocally elevated plasma and urinary free cortisol levels (2- to 3-fold above the reference range), the patient showed a normal habitus with a normal fat distribution, no obesity, no hirsutism, myopathy, bruising, striae, or hypertension (709). She was deficient in regeneration of cortisol from oral cortisone and her urinary THF+aTHF/THE ratio was approximately twofold lower than normal and approximately threefold lower than usual for Cushing's patients. Although no mutation was found in HSD11B1, so molecular proof of this effect is missing, the case supports the critical importance of the intracrine amplification of glucocorticoids in their effects on tissues. It is interesting to speculate whether the corticotroph adenoma was coincidental or a tertiary hyperplastic process in response to cortisone reductase deficiency causing chronic HPA axis activation. If the latter, this is likely a rare event given the high frequency of chronic HPA activation in affective disorders apparently without corticotroph adenoma formation.
Selective 11β-HSD1 inhibitors have entered Phase II clinical trials in patients with type 2 diabetes. One agent, ICNB13739, administered in a double-blind and placebo controlled study to patients taking but suboptimally controlled by metformin, significantly lowered fasting plasma glucose and haemoglobin A1c levels, plausibly via increased insulin sensitivity (594). There was a small but significant fall in body weight. The drug also reduced plasma cholesterol and triglyceride levels in hyperlipidaemic subjects. Therapeutic effects were greatest in those with the poorest metabolic control. In a similar trial, another drug, MK-0916, modestly reduced haemoglobin A1c, LDL-cholesterol, blood pressure, and body weight, without altering fasting glucose levels (195). A related agent, MK-0736, reduced ambulatory blood pressure, weight and LDL-cholesterol in nondiabetic obese hypertensive subjects (633).
Thus, in humans, as in mice and rats, 11β-HSD1 deficiency/inhibition improves multiple metabolic disease risk factors. To date, the effects on individual indices such as HbA1c are relatively modest, although probably in keeping with other insulin-sensitizing agents. Perhaps coadministration of 11β-HSD1 inhibitors with metformin has masked their impact, since the metformin target AMPK appears to be a core pathway downstream of 11β-HSD1 in visceral fat, at least in mice (750). While one study was in patients the majority of whom had been withdrawn from metformin (195), the drug-free duration was quite short. Studies in unmedicated (perhaps in metformin-intolerant) subjects may be revealing.
More importantly, 11β-HSD1 inhibition is a multifaceted therapeutic target and probably changes individual components of metabolic disease risk modestly; after all, glucocorticoid levels are being reduced, not obliterated, in target cells. Indeed, the advantage over GR antagonists is that receptors remain functional so any life-threatening stress can induce the HPA axis to provide glucocorticoids for survival. Crucial will be the impact of 11β-HSD1 inhibitors on cardiovascular events, which are the main cause of mortality in the obesity-type 2 diabetes-metabolic syndrome continuum (88).
6. Steatohepatitis: adipose-liver interactions
A potential pathogenic process involving 11β-HSD1 in liver is the deposition of ectopic lipid that underlies the spectrum of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. This affects around one-third of Americans (690) and is increasingly prevalent globally. Transgenic overexpression of 11β-HSD1 in liver causes fatty liver and insulin resistance without obesity (548), suggesting a potential pathogenic role for the enzyme. While most obese individuals have reduced 11β-HSD1 levels in liver (574, 575), liver 11β-HSD1 is increased in metabolic syndrome associated with myotonic dystrophy (327). Moreover, short-term 11β-HSD1 inhibition reduces liver triglyceride levels without altering food intake or weight (73). However, hepatic lipid is also increased with adipose-specific 11β-HSD1 overexpression in mice, plausibly reflecting increased glucocorticoid and fatty acid delivery to liver via the portal blood (446). Clinical studies show no association between liver 11β-reductase and liver fat content (357, 762), whereas visceral adipose tissue 11β-HSD1 is associated with hepatic steatosis (112). Thus there is, as yet, no strong evidence that hepatic 11β-HSD1 contributes to fatty liver disease, although 11β-HSD1 in visceral adipose tissue may well play a role.
7. Polycystic ovary syndrome
This syndromic disorder incorporates the common coincidence of reproductive (secondary oligomenorrhea/amenorrhea with reduced or absent ovulation, polycystic or enlarged ovaries on ultrasonography, reduced fertility), hyperandrogenism (hirsutism, male-pattern hair loss, acne, sebaceous hyperactivity), and sometimes metabolic (obesity with or without insulin resistance) features in women. There are many hypotheses of causation, but genetics and aberrant metabolism are commonly implicated. Lower 11β-HSD1 activity has been advocated based on the prominent hyperandrogenism seen in apparent cortisone reductase (H6PDH) deficiency (390) and cortisone reductase (HSD11B1) deficiency (393).
A functional HSD11B1 SNP (rs12086634 encoding a third intron variant that links with reduced 11β-HSD1 activity; Ref. 170) has been associated with hyperandrogenism in 102 patients with polycystic ovary syndrome (PCOS), a relationship seen in both obese and lean patients (suggesting the link is not merely secondary to obesity) (213). Indeed, lean PCOS patients show enhanced cortisol clearance and compensatory adrenal hyperandrogenism and interestingly appear partly to resist obesity and metabolic disease. These findings build on data showing increased adrenal cortisol and androgen production in lean PCOS with a lower ratio of cortisone to cortisol metabolites compatible with less 11β-HSD1 reductase and/or more 11β-HSD2 dehydrogenase (584). However, a recent larger study in 300 PCOS patients failed to replicate any link between HSD11B1 SNPs and PCOS features, although one combination of rs846910 and rs12086634 associated with increased enzyme activity and a substantially elevated risk of metabolic syndrome (odds ratio 2.77) (212).
Polymorphisms in H6PDH may also affect risk of PCOS and cardiometabolic disease (611), plausibly through effects on 11β-HSD1 activity (e.g., via increased adrenal androgen production secondary to altered hepatic 11β-HSD1 activity; Ref. 390). However, another study found no association of the same polymorphism (R453Q) in H6PD with metabolic parameters or adrenal androgen production (648) and a somewhat larger study of ∼450 patients also showed no linkage between SNPs in HSD11B1 or H6PD and PCOS (169). These discordances may reflect the uncertainties of genetic studies in relatively small populations. Overall, while an attractive hypothesis, a causative role for lower 11β-HSD1 activity in PCOS is far from established.
C. Skeletal Muscle
Skeletal muscle is a major target for insulin action and glucose storage/catabolism. 11β-HSD1 is expressed in skeletal muscle, albeit at lower levels than liver (767, 768). 11β-HSD2 is not expressed in muscle. In humans, 11β-HSD1 levels ex vivo do not appear to correlate with body weight or adiposity, at least under standard culture conditions (767, 768), although other evidence suggests that skeletal muscle 11β-HSD1 mRNA and protein levels are increased in obesity with diabetes in rodents (805) and in myotubes grown from humans with type 2 diabetes (1). These data require extension. Selective 11β-HSD1 inhibition increases biochemical markers of muscle insulin sensitivity in diabetic rodents in vivo (490). However, the contribution of muscle 11β-HSD1 to overall glucose-insulin homeostasis remains uncertain. Although effects of 11β-HSD1 on metabolism and glucocorticoid-induced muscle atrophy have been suggested (80), any biological importance, especially in the sarcopenia of ageing, remains undetermined.
Another potential approach to reduce intracellular glucocorticoid action is to reverse the reaction direction of 11β-HSD1 (389). Mice lacking H6PDH are relatively insensitive to glucocorticoids, exhibiting increased insulin sensitivity presumably due, in part, to the reversed reaction direction of 11β-HSD1 reducing glucocorticoid levels in key metabolic tissues (391). These mice also have a striking skeletal myopathy and show fasting hypoglycemia not seen in 11β-HSD1-deficient mice, but these features are independent of 11β-HSD1 and probably relate to loss of other NADPH-dependent processes (627).
D. Pancreas
11β-HSD1 mRNA and activity are present in mouse and human pancreatic islets (140). It remains contentious whether this localizes to alpha cells (689) or beta cells (140, 716) with evidence in support of both based on immunohistochemistry and effects of inhibitors on insulin or glucagon secretion. Probably therefore both alpha and beta cells have 11β-HSD1.
11β-HSD1 activity is increased in islets from diabetic rodents (175). Since glucocorticoids, albeit in pharmacological concentrations, directly inhibit beta-cell insulin secretion (248, 381), it has been proposed that increased islet 11β-HSD1 is part of the pathogenesis of beta cell failure (140). However, modest transgenic overexpression of 11β-HSD1 in beta cells alone, driven by the insulin promoter, to a level comparable to that observed in some type 2 diabetes models, is associated with enhanced glucose-stimulated insulin secretion both in vivo and in isolated islet cells in vitro (716). On the C57BL/KsJ mouse strain background, which develops beta-cell failure on high-fat diet, elevated 11β-HSD1 in beta cells maintains their survival and function. The underlying mechanisms appear to reflect protection from apoptosis and cellular stress, as well as facilitated secretion. Biochemically, the beta cells show induction of heat shock proteins, protein kinase A, ERK, and p21 signaling pathways. In contrast, greater beta-cell overexpression of 11β-HSD1 or lifelong deletion of 11β-HSD1 associates with poorer glucose-stimulated insulin secretion. Although these may be developmental effects, as glucocorticoids play critical roles in pancreatic islet formation and perinatal maturation of endocrine function, they also raise the possibility of an inverted U-shaped relationship between 11β-HSD1 in beta cells and insulin secretion with the modest elevation seen in early diabetes associated with improved insulin secretory function. A similar relationship between glucocorticoid dose and beta-cell function is also observed in vitro. Intriguingly, high-fat diet induces a similar modest elevation of 11β-HSD1 in islets of mouse strains that can compensate for the caloric challenge, while strains that are susceptible to beta-cell failure downregulate islet 11β-HSD1 and those susceptible to obesity-driven diabetes (Lepdb/db) have very high islet 11β-HSD1 levels, both of which cause failure of insulin secretion.
This is perhaps the first evidence of a physiological function for 11β-HSD1 in metabolic control. Normal mice given a high-fat diet exhibit a robust compensatory insulin secretion plausibly driven by the modest upregulation of beta-cell 11β-HSD1. At the same time, healthy mice substantially downregulate adipose tissue 11β-HSD1, driving subcutaneous fat cell insulin sensitization and “safe” calorie storage while AMPK-mediated beneficial metabolic and noninflammatory processes occur in visceral adipose (495, 750). In contrast, in severe obesity, excessive pancreatic 11β-HSD1 and hence glucocorticoid regeneration contributes to the failure of insulin secretion and perhaps beta cell failure, and increased adipose tissue 11β-HSD1 causes visceral obesity, insulin resistance, and metabolic syndrome (FIGURE 8).
Figure 8.

Cartoon of potential role of 11β-HSD1 in metabolic organ interrelationships in obesity. A: in normal weight health, 11β-HSD1 acts largely as a reductase. This performs an important endocrine role in the splanchnic bed by regenerating glucocorticoids, thus contributing ∼40% of daily glucocorticoid production. In addition, it has intracrine actions, amplifying the glucocorticoid signal inside hepatocytes, pancreatic islets (beta and alpha cells), and adipocytes. B: with modest obesity, 11β-HSD1 is elevated in adipocytes and beta cells but not hepatocytes. This increases insulin resistance in adipose tissues, increases release of proinflammatory/antimetabolic adipokines, and portal blood glucocorticoid and fatty acid deliver to the liver, but also increases islet insulin release to glucose, plausibly without changing hepatic insulin sensitivity. This may support storage of calories in adipose tissue in preparation for less bountiful nutritional circumstances. C: with severe obesity, greater rises in adipose and beta cell 11β-HSD1 lead to failure of pancreatic insulin release, worsening peripheral insulin resistance and metabolic disease despite potentially “compensatory” declines in hepatic glucocorticoid regeneration, which might even contribute to HPA axis activation due to loss of bulk glucocorticoid regeneration. D: 11β-HSD1 inhibition maximizes subcutaneous adipose and liver insulin sensitivity and visceral adipose AMPK signaling and reduces its inflammation and portal glucocorticoid and fatty acid release. Despite reduced beta cell insulin release and HPA axis activation (without elevation of glucocorticoid levels), the balance of metabolism favors reduced hepatic gluconeogenesis, increase beta-oxidation of fats, “safe” calorie storage in subcutaneous adipose depots, reduction of visceral adipose mass, and metabolic health.
The partitioning of metabolism effected by alterations in 11β-HSD1 in beta cells and adipocytes in response to high-fat diet appears advantageous in an ancestral environment where feast and famine alternated. Increased insulin secretion during early calorie excess appears to facilitate fat depot-specific storage that might aid survival in the occurrence of famine. With marked calorie excess, increased adipose tissue 11β-HSD1 may have allowed even more fat storage in rapidly accessible visceral depots. Of course in the modern continuous calorie-dense environment, elevated adipose tissue 11β-HSD1 causes metabolic disease, but this unusual environment is unlikely to have driven the evolution of this system. These data, while fascinating, also illustrate our paucity of understanding of the physiological (as opposed to pathological) roles of 11β-HSD1 in metabolic tissues.
E. Kidney
11β-HSD2 is highly expressed in the distal nephron that allows aldosterone selectivity in vivo. 11β-HSD1 is also expressed in the kidney, primarily in proximal tubule cells (478, 562, 571, 589, 617, 695), although any role remains largely unexplored.
In the distal nephron, 11β-HSD2 is concentrated in cortical collecting duct cells (91), largely overlapping with MR in principal cells (512, 640) which mediate aldosterone actions on salt-water homeostasis. An unresolved paradox lies in the relative affinities of MR and 11β-HSD2 for glucocorticoids. Human MR binds cortisol, corticosterone, and aldosterone with a Kd of ∼0.5 nM (37, 637), whereas the affinity of human 11β-HSD2 for cortisol (∼50 nM) and corticosterone (∼5 nM) is more than an order of magnitude lower (15, 92). Even allowing for molar excess of 11β-HSD2 over MR in distal nephron cells (514), these affinities suggest 11β-HSD2 is unlikely to completely exclude glucocorticoids from MR. However, modeling the cellular context by transfecting distal nephron cells with MR and an MR reporter gene with or without 11β-HSD2 showed that coexpression of 11β-HSD2 is sufficient to prevent glucocorticoid activation of MR, abrogating target gene activation, an effect reversed by carbenoxolone inhibition of 11β-HSD2 (395). Transfection with 11β-HSD2 cDNA carrying mutations that cause SAME fails to protect MR from activation by cortisol (529). Perhaps the cellular microanatomy involved may be sufficient to overcome the differences in affinity: lipophilic glucocorticoids may preferentially track through intracellular membranes such as the ER and exit past cytoplasmically directed 11β-HSD2 (530), with inactivation before the cytoplasmic MR can “compete” for ligand. Indeed, MR and 11β-HSD2 coassociate, at least in transfected cells (529), and it has been suggested that this is crucial to prevent activation of MR by bound cortisol through an, as yet unknown, mechanism (209), perhaps involving the apparent functional antagonism by physiologically relevant concentrations of cortisone and 11-dehydrocorticosterone of aldosterone activation and nuclear translocation of MR (529) (FIGURE 3).
Patients with AME are homozygous or compound heterozygous for mutations in the HSD11B2 gene (141, 498, 777). 11β-HSD2 knockout mice also show apparent mineralocorticoid excess (363). Interestingly, the mouse model suggests that the high early mortality of children with AME may relate to irreversible developmental effects on renal structure (distal nephron hyperplasia and hypertrophy) and the aorta, which shows a propensity to dissection associated with disorganised elastic laminae. These effects are partly responsive to lowering of blood pressure (363). The background genotype impacts substantially on lethality (288); how is uncertain.
Mice heterozygous for a gene disruption in Hsd11b2 have normal basal blood pressure which, in contrast to control C57BL/6 mice, is elevated on sodium loading. Unexpectedly this is reversed by GR rather than MR antagonism (46). The mechanism is unknown. It may reflect developmental programming of the kidney following fetal exposure to excess glucocorticoids due to feto-placental 11β-HDSD2 deficiency in utero (see below).
Such data suggest HSD11B2 as a candidate gene/gene product for hypertension beyond rare AME (198). Indeed, a subset of subjects with essential hypertension show a prolonged half-life of [3H]cortisol, implying 11β-HSD2 deficiency (661, 746), although not all studies find this (313). Measures of 11β-HSD2 activity (UFF-to-UFE ratio) show a decline in enzyme activity with ageing, perhaps correlating with the rise in blood pressure with advancing years (281). More directly, HSD11B2 polymorphisms have been associated with blood pressure in black men with renal failure (754). However, other studies in black subjects have been negative (764), and the gene has not emerged as a general candidate for hypertension from genome-wide association studies (182). In white males HSD11B2 polymorphisms associate with sodium-sensitive changes in blood pressure (417), reminiscent of the phenotype of Hsd11b2+/− heterozygous mice. Specifically, shorter alleles of a CA repeat polymorphism in the first intron of the HSD11B2 gene that associate with reduced 11β-HSD2 activity measured in vivo also associate with greater rises in blood pressure between sodium-depleted and loaded states (3). A molecular mechanism linking this intronic CA repeat length to enzyme function remains questionable. Perhaps the CA repeat is in linkage disequilibrium with a functional polymorphism.
A more functional connection is afforded by the G209A polymorphism affecting an NF-1 binding site in the HSD11B2 gene promoter, which reduces association of both NF-1 and GR with the promoter (18). The G209A polymorphism, which appears in linkage disequilibrium with shorter CA repeat alleles, is more common in salt-sensitive subjects (18). Intriguingly, methylation of a CpG island in the HSD11B2 promoter (see above), an epigenetic event that in cancer is associated with gene silencing (330), is associated with lower 11β-HSD2 expression and higher blood pressure (205) in keeping with epigenetic control and a key role for this gene in developmental programming of blood pressure. However, given the absolute difference in promoter methylation between “low enzyme function” hypertensive and controls was only ∼8%, any causal role remains uncertain.
11β-HSD1 is also expressed in kidney, predominantly in the proximal tubule, interstitial cells, and medulla (601). Interestingly, HSD11B1 polymorphisms associate with blood pressure in some populations (172, 201), and even with pressure changes to potassium challenge (266), although separating effects on blood pressure from metabolic syndrome as a whole is challenging and the low levels of 11β-HSD1 mRNA in human kidney mitigate against a major role in this tissue. Studies with selective 11β-HSD1 inhibitors in animals and humans suggest these reduce blood pressure, at least in subjects with metabolic syndrome (195, 633), although there is no primary reason to invoke a renal mechanism. Intriguingly, in Dahl salt-sensitive rats, renal medullary 11β-HSD1 levels fail to decrease normally on salt loading, although forced reduction (siRNA-mediated knock-down) keeps blood pressure low during salt loading (409). Importantly, the siRNA studies suggest 11β-HSD1 acts as a reductase in the renal medulla. The molecular mechanisms by which lowering active glucocorticoids in the renal medulla attenuate blood pressure responses to dietary sodium merit attention. In addition, the proximal nephron is an important site for gluconeogenesis, and indeed, renal PEPCK appears predominantly sensitive to glucocorticoids rather than insulin (114). Any importance of 11β-HSD1 in this key metabolic function is unexplored.
F. The Vasculature and Heart
Both 11β-HSD1 and 11β-HSD2 are expressed in the vasculature (747). In the vascular wall, 11β-HSD2 is mainly located in the endothelium (257), whereas 11β-HSD1 is highest in the vascular smooth muscle in both rodents and humans where it catalyzes 11β-reduction (747). 11β-HSD1 is expressed in neonatal heart (638), perhaps contributing to the perinatal maturational effects of glucocorticoids in this tissue (586). Expression of 11β-HSD1 in adult heart is also very modest and confined to cardiac fibroblasts (86). Any expression of 11β-HSD2 in heart is low and probably restricted to the coronary vasculature.
1. Vasculature
Expression of 11β-HSD1 in vascular smooth muscle suggests actions on vasoconstrictor function, but such effects have not been found in ex vivo preparations (257). However, 11β-HSD1 does play a key role in angiogenesis and vascular remodeling. Glucocorticoids inhibit angiogenesis in animals and humans. In line with this, 11β-HSD1-deficient mice show increased angiogenesis from aortic rings in vitro and into implanted sponges and in the infarcted myocardium following coronary artery ligation in vivo (646). The latter associates with improved ventricular ejection fraction despite a similar initial infarct size (454), implying facilitated recovery. The greater angiogenic response in the infarcted Hsd11b1−/− myocardium is perhaps driven by angiogenic factors such as IL-8, which is concomitantly increased. The Hsd11b1−/− infarct also shows enhanced early infiltration of neutrophils, followed by increased numbers of macrophages, plausibly attracted by locally increased MCP-1 expression. These are predominantly YM1+ (M2) macrophages, implying a reparative, anti-inflammatory phenotype. M2-like polarization is not a general feature of 11β-HSD1−/− macrophages (750), implying a vascular-specific effect.
2. Atherosclerosis
11β-HSD1 deficiency/inhibition improves multiple cardiovascular risk factors, anticipated to be atheroprotective. However, atherosclerosis has an important inflammatory component. 11β-HSD1 is expressed in differentiated/activated macrophages, and its deficiency exacerbates some models of acute inflammation (231). Nevertheless, 11β-HSD1 deficiency/inhibition indeed appears atheroprotective. A selective 11β-HSD1 inhibitor strikingly reduces circulating and intra-aortic cholesterol levels in atherosclerosis-prone Apoe−/− mice (282), although another selective inhibitor has no effect on atherosclerotic lesions in Ldlr−/− mice (415). Carbenoxolone, which inhibits both 11β-HSD1 and 11β-HSD2, reduces atherosclerosis in susceptible Ldlr−/− mice (521). Moreover, Apoe−/− mice crossed with Hsd11b1−/− mice show a substantial reduction of atherosclerosis on cholesterol-rich “western diet,” in association with reduced lesional inflammation perhaps due to reduced vascular cell adhesion molecule (VCAM)-1-mediated recruitment of inflammatory cells to the plaque (347). Such findings suggest direct atheroprotective effects as a result of 11β-HSD1 inhibition, over and above reduction of cardiovascular risk factors. Indeed, recent data show atheroprotection can be conferred by transplanting 11β-HSD1-deficient bone marrow cells into irradiated Apoe−/− hosts (346), implying a key role for 11β-HSD1 in macrophages and/or related leukocytes. If pertinent to humans such effects could be of substantial therapeutic importance (FIGURE 9).
Figure 9.

Possible impacts of 11β-HDSD1 inhibition/deficiency on atherosclerosis. Known effects include reductions of multiple risk factors and reduced atherosclerotic lesion size. If a myocardial infarction occurs, the angiogenic effects of 11β-HSD1 deficiency/inhibition promote angiogenesis and recovery of myocardial function for the same initial infarct size.
11β-HSD2 is also expressed in the vasculature, notably in the endothelium (84). Hsd11b2−/− mice show endothelial dysfunction with enhanced norepinephrine-mediated contraction and reduced relaxation associated with impaired NO-related function (120, 257). Mice lacking 11β-HSD2 crossed with Apoe−/− mice develop accelerated atherosclerosis even on chow diet, with increased lesional inflammation and reduced lesion-stabilising collagen (151). The accelerated atherogenesis is attenuated by the MR antagonist epleronone at a dose without effects on blood pressure implying, but not proving, actions via MR in the vasculature. A possible mechanism is via increased chemotaxis mediated by the observed increase in endothelial VCAM-1. The latter results from enhanced glucocorticoid activation of MR as it is mimicked by co-incubation of corticosterone with carbenoxolone (inhibiting 11β-HSD2) in mouse aortic endothelial cells (151). Thus loss of 11β-HSD2 leads to striking atherogenesis associated with activation of MR stimulating proinflammatory processes in the vessel wall. Tissue-specific manipulations are needed to dissect the cells involved in this process with potential implications for therapy.
3. Vascular injury
Glucocorticoids inhibit neointimal proliferation (729). 11β-HSD1 is expressed in the crucial vascular smooth muscle cells, and 11β-HSD1 deletion or inhibition reduces neointimal formation to wire injury. However, this is only observed in western diet-fed, atherosclerosis-prone Apoe−/− mice, suggesting effects mediated primarily via impacts on metabolic risk factors. 11β-HSD2 deficiency had no effect (314). Such data do not support local vascular effects, at least in this model, on injury responses.
4. The heart
A single nucleotide polymorphism (SNP) in the HSD11B1 locus associates with left ventricular mass (568), apparently independent of any effect on blood pressure or body weight. Moreover, heart size of Hsd11b1−/− mice is reduced (453). However, any function of the relatively low levels of either isozyme in the heart is unclear, as is the mechanism underlying the influence on heart mass. Cardiac MR appear to be occupied and tonically inhibited by physiological glucocorticoids (468). Transgenic overexpression of 11β-HSD2 in cardiac muscle induces fibrosis, supporting the notion that aldosterone-selective access to cardiac MR activates pathways leading to pathology, a concept supported by its improvement with the selective MR antagonist epleronone (561). Such molecular pharmacology is intriguing but hardly relevant to physiology. However, 11β-HSD2 mRNA levels selectively increase in hypoxic cardiomyocytes in association with inflammation, perhaps in part a consequence of reduced local glucocorticoids (352). Whether the benefits of 11β-HSD1 deficiency on recovery after experimental myocardial infarction reflect actions within the heart, over angiogenic and inflammatory actions, remains unexplored.
5. Genetics of HSD11B1 and cardiometabolic disease
Strong supporting evidence that 11β-HSD1 is causative in cardiometabolic disease (this is of course a distinct argument from its role as a drug target) could come from genetics. One study reported a weak association between HSD11B1 microsatellite sequence variation in intron 4 and central obesity (168) and another linked an intron 3 polymorphism to obesity, insulin resistance, and body composition in children (226), data echoed by the reported linkage of rs3753519 to obesity in children (536). The same intron 3 variant is linked to protection against type 2 diabetes, lower plasma insulin and insulin sensitivity independent of obesity in more than 700 Pima Native Americans (508). In the same population, the rs846910 SNP in the HSD11B1 promoter associates with blood pressure and hypertension, again independent of obesity (201), findings also reported in other populations (489). Also, combination of rs846910 GA heterozygosity and rs12086634 TT homozygosity that links to increased enzyme activity, associates with metabolic syndrome risk in southern European women (212). However, others (371) found effects on promoter activity without corresponding effects on metabolic phenotype in Koreans, and HSD11B1 has also not emerged from relevant genome-wide studies, although representation of this gene on many GWAS chips may be inadequate (212). The positive reports therefore either represent isolated examples in particular populations or may perhaps be false positives. Given the importance of the 11β-HSD1 hypothesis of metabolic disease, the genetics merit further consideration. Overall, however, the balance of current evidence suggests it is 11β-HSD1 dysregulation rather than HSD11B1 genetic variation that underlie the preponderance of its link to metabolic and other disorders.
V. 11β-HSD1 AND 11β-HSD2 IN IMMUNITY AND INFLAMMATION
Exogenous glucocorticoids in pharmacological doses are generally immunosuppressive. Endogenous glucocorticoids have a more nuanced effect, exerting permissive, suppressive, or stimulatory effects depending on concentration and the cellular and environmental context, thus modulating and shaping inflammatory and immune responses (reviewed in Refs. 135, 407, 452, 615, 655, 773, 774, 796). Perhaps unsurprisingly, a primary anti-inflammatory role for 11β-HSD1 has been demonstrated (136, 231, 808). However, the effects are complex and evidence, reviewed above, suggests that 11β-HSD1 deficiency/inhibition attenuates “metabolic inflammation” (obesity, atherosclerosis). Additionally, under some circumstances, 11β-HSD2 has an anti-inflammatory role, perhaps through its ability to control glucocorticoid activation of MR (151).
A. Expression of 11β-HSD1 and 11β-HSD2 in Immune Cells
Early studies suggesting immune effects of 11β-HSD largely failed to distinguish 11β-HSD1 and -2 or used nonselective licorice-based 11β-HSD inhibitors (199). The observed antiviral and anti-inflammatory effects could reflect renal 11β-HSD2 inhibition, delaying clearance of glucocorticoids (294), rather than inhibition of 11β-HSDs or effects on other targets. Recent studies unambiguously show isozyme expression in immune cells.
1. 11β-HSD1
A) MYELOID CELLS.
Glucocorticoids affect macrophage differentiation and function (135) which, in vivo, is highly diverse. During inflammation, macrophages predominantly derive from blood monocytes, which differentiate under instruction from the cytokine and chemokine microenvironment. 11β-HSD1 is not expressed in nonstimulated human monocytes but is induced, albeit to relatively low levels, following activation with the “Th2” cytokines IL-4, IL-13, or if monocytes are differentiated to macrophages in vitro (703). 11β-HSD1 expression is similarly modest in tissue-resident or bone marrow-derived macrophages of mice, the latter resembling human macrophages differentiated from monocytes in vitro (146, 231).
In macrophages, 11β-HSD1 expression is dynamically regulated depending on stimulus and context. Whereas incubation of human monocytes with bacterial lipopolysaccharide (LPS) has no effect on 11β-HSD1, in macrophages 11β-HSD1 is increased following LPS activation (termed “classical activation”) (326, 433, 703) (FIGURE 10), which polarizes macrophages to a pro-inflammatory M1 phenotype. Similarly, LPS induces 11β-HSD1 mRNA in microglia, the brain's resident macrophages, although curiously this is not accompanied by changes in protein (243).
Figure 10.
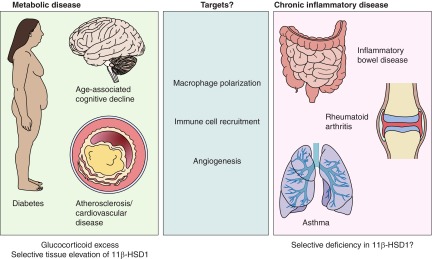
11β-HSD1 in macrophages reflects activation state and shapes the subsequent response towards resolution. Monocytes have negligible levels of 11β-HSD1 (703). After differentiation of human peripheral blood monocytes to macrophages, 11β-HSD1 expression is increased (703). Polarization to M1 with LPS and IFN-γ further increases 11β-HSD1 mRNA levels, whereas polarization to M2 with IL-4 and/or IL-13 has no further effect (326). Treatment of monocytes with IL-4 or IL-13 also increases 11β-HSD1 mRNA and activity levels (703), which are further increased by PPARγ activation (117). Differentiation of monocytes in the presence of glucocorticoid produces a highly phagocytic macrophage phenotype (230). Similarly, glucocorticoid treatment of macrophages promotes a phagocytic phenotype (412). High expression of 11β-HSD1 in M1 macrophages may promote the subsequent transition to a phagocytic phenotype, thus promoting resolution of inflammation. 11β-HSD1 is markedly downregulated following phagocytosis of apoptotic leukocytes (115). Dashed lines indicate speculative connections, whereas solid lines have been demonstrated.
“Alternative activation” of unstimulated macrophages with IL-4 or IL-13, which induces a pro-resolution M2 phenotype, has no effect on 11β-HSD1 (326, 433). This contrasts with the situation in monocytes (above). However, if monocytes are differentiated into macrophages in the presence of IL-4/IL-13, then levels of 11β-HSD1 are even higher than in M1-polarized macrophages and are further induced by PPARγ agonists, probably via a PPAR binding site exclusive to the human promoter (117). Chronic cold stress, which induces a regulatory (immunosuppressive) macrophage phenotype, also increases macrophage 11β-HSD1 (630). The relevance of these in vitro defined phenotypes to the situation in vivo is unclear (171), although the dynamic regulation of 11β-HSD1 by various stimuli suggests it shapes macrophage phenotype during the inflammatory response (231, 454). Phagocytosis of apoptotic neutrophils by macrophages which promotes resolution of inflammation, decreases 11β-HSD1, suggesting an active process (115).
Glucocorticoids suppress dendritic cell maturation and induce a tolerogenic phenotype eliciting T-cell hyporesponsiveness and regulatory T-cell formation (reviewed in Ref. 135). 11β-HSD1 induction accompanies dendritic cell differentiation. It is maintained or increased following maturation by innate immune activating signals (TNF-α, LPS) but decreased by CD40 ligation mimicking Th cell engagement, an adaptive immune activation signal (203). The latter occurs without alteration in 11β-HSD1 mRNA or protein levels. These data suggest 11β-HSD1 inhibition might enhance immunity in some circumstances, a notion requiring exploration.
Other myeloid cell types express 11β-HSD1. Again levels depend on cell state. Neutrophil expression of 11β-HSD1 (336) is increased during inflammation (137) but is absent in apoptosis (115). Neutrophil number and apoptosis depend on glucose shuttling between the cytoplasm and ER (119), and perhaps 11β-HSD1 alters because of coupling to H6PDH. 11β-HSD1 is expressed in mast cells, where it limits degranulation (134), with implications for inflammation, infection and allergy.
B) LYMPHOCYTES.
Although 11β-HSD1 activity and protein have been demonstrated in lymphoid organs, most is localized to stromal cells (280), consistent with low 11β-HSD1 activity in mouse splenic lymphocytes and thymocytes (809) and negligible 11β-HSD1 mRNA in human blood leukocytes (703). Activation of lymphocytes, by T-cell receptor activation or cytokine-induced polarization, increases 11β-HSD1 activity (809). The biological role in lymphocytes remains unclear, although it appears to dampen both pro- and anti-inflammatory cytokine release (809).
11β-HSD1 (but not 11β-HSD2) mRNA has also been reported in uterine natural killer cells (451), important in preparing the uterus for pregnancy and in placental development (162). Expression may not be high in other NK cells (http://biogps.gnf.org), again emphasizing dependence on differentiation and/or activation state of the cell.
2. 11β-HSD2
Except in some human diseases, immune cells do not express 11β-HSD2 (134, 203, 703). 11β-HSD2 appears transiently expressed in peripheral blood mononuclear cells of patients with early rheumatoid arthritis (535), and 11β-HSD2 expression is higher in transformed B cell lines established from rheumatoid arthritis compared with those of nonarthritic twins (256) (levels in nonarthritic controls are probably extremely low). Immunohistochemistry suggests 11β-HSD2, as well as 11β-HSD1, in synovial macrophages in both osteoarthritis and rheumatoid arthritis (256, 264, 618). 11β-HSD2-immunoreactive macrophages are found in lungs of patients dying with acute respiratory distress syndrome (686). The significance and generality of these findings remain to be established, but it is noteworthy that 11β-HSD2, in contrast to 11β-HSD1, is widely expressed during early embryonic development and is frequently expressed in transformed cell lines (see below), but shows a highly restricted distribution in mature tissues and primary cells. Macrophage 11β-HSD2 in the rheumatoid arthritis synovium may reflect an adaptation to allow increased cell proliferation or an altered differentiation programme in the chronically inflamed microenvironment. These findings raise the possibility that intracellular glucocorticoid action may be very different in chronic compared with acute inflammatory responses.
B. 11β-HSDs in Acute Inflammation and Infection
1. Acute inflammation and 11β-HSD1
Neutrophil recruitment is a critical early determinant of inflammation and tightly coupled to monocyte recruitment (656). In Hsd11b1−/− mice, more neutrophils and monocytes-macrophages are present early in acute inflammation in the peritoneal or pleural cavities (136). In contrast, 11β-HSD2 deficiency has no effect (136). Neutrophil density is also increased in the injured myocardium of Hsd11b1−/− mice early following myocardial infarction (454), reflecting greater recruitment and/or delayed clearance. In support of the latter, Hsd11b1−/− mice show delayed macrophage acquisition of in vivo phagocytic capacity for apoptotic neutrophils during sterile peritonitis, although surprisingly, the peritonitis resolves at the same time (231). 11β-HSD1 activity may promote neutrophil survival in vitro, although this is dependent on coupling of cortisol oxidation to glucose metabolism/NADPH generation in the ER rather than glucocorticoid metabolism per se (336). The significance of these findings merits further investigation.
Hsd11b1−/− mice are also more susceptible to endotoxemia (808), classically restrained by glucocorticoids (76, 78), with higher serum pro-inflammatory cytokines TNF-α, IL-6, and IL-12p40 due to increased activation of NF-κB- and MAPK-signaling pathways in macrophages (808). This was attributed to attenuated PI3K-dependent Akt activation in 11β-HSD1-deficient macrophages, secondary to elevated levels of Src homology 2-containing-inositol 5′-phosphatase (SHIP)-1 (808), which hydrolyzes the PI3K-generated second messenger PIP3 to PIP2. Importantly, there are no differences between the glucocorticoid responsiveness and functional properties of Hsd11b1−/− and wild-type macrophages differentiated in vitro, but Hsd11b1−/− macrophages ex vivo are hyperresponsive to LPS (808), suggesting altered myeloid cell differentiation in vivo in Hsd11b1−/− mice. Thus 11β-HSD1 plays an early constraining role in the inflammatory response, shaping the subsequent trajectory, probably through regulation of cytokine and chemokine production (231, 454, 808) or local glucocorticoid-mediated changes in cellular differentiation in inflamed tissues (135).
2. 11β-HSDs in the response to infection
There are few direct data on regulation of 11β-HSD1 and/or -2 during infection. What exists suggests the overall balance favors increased glucocorticoid generation. Whether 11β-HSD1 (or -2) affects resistance to pathogens will be of great interest.
Downregulation of 11β-HSD2 without alteration of 11β-HSD1 mRNA occurs in skin lesions of leprosy patients compared with unaffected skin (22), although this could be a response to damage rather than infection (707). In placenta, 11β-HSD1 increases and 11β-HSD2 decreases in chorioamnionitis (328). Carbenoxolone increases susceptibility to progressive disease in Listeria monocytogenes infection; as dexamethasone has a similar effect (279), this could reflect decreased glucocorticoid clearance by renal 11β-HSD2. A role for 11β-HSDs in shaping the course of tuberculosis has also been suggested (592), but is based largely on changes in urinary steroid profiles and remains speculative.
3. Regulation of 11β-HSDs in inflammation
Increased whole body conversion of cortisone to cortisol suggests 11β-HSD balance is altered in favor of 11β-HSD1 in patients with inflammatory disease (311). Increasing evidence suggests reciprocal regulation of 11β-HSD1 and 11β-HSD2 by pro-inflammatory cytokines at sites of inflammation, with upregulation of 11β-HSD1 and downregulation of 11β-HSD2. This regulation, particularly of 11β-HSD2, which is not expressed under most circumstances in leukocytes, is probably attributable primarily to 11β-HSD1 and -2 in nonimmune cells. This balance is predicted to increase local glucocorticoid concentrations, but direct evidence of this in inflamed tissues is lacking. There are also important exceptions to this generalization.
4. Reciprocal regulation of 11β-HSD1 and 2 by pro-inflammatory mediators
Although coexpressed within tissues, 11β-HSD1 and -2 are not usually colocalized within a cell. If this occurs, reciprocal regulation appears to minimize simultaneous expression. In vitro, in most cell types the pro-inflammatory cytokines TNF-α and IL-1 up regulate 11β-HSD1 (109, 188, 260, 263, 403, 567, 681, 700, 711, 798) and downregulate 11β-HSD2 (271), the latter mediated through a switch from active p65/p50 NF-κB heterodimers bound to the HSD11B2 promoter to inactive p50/p50 homodimers which coordinately bind with Egr-1 (362). This affords a plausible basis for the generalization that in vivo 11β-HSD1 is upregulated at sites of inflammation and 11β-HSD2 downregulated. Indeed, TNF-α-expressing transgenic mice have elevated hepatic 11β-HSD1 (312) and reduced renal 11β-HSD2 (362). However, 11β-HSD1 was not among mRNAs increased in liver, muscle, or adipose tissue following 1- or 4-day TNF-α infusion in rats (598), nor was it altered in murine vasculature following LPS (163). Importantly, TNF-α and LPS effects on 11β-HSD1 in immune cells themselves are highly context dependent and probably require a costimulus (203).
5. Glucocorticoids amplify 11β-HSD1 induction by inflammation
The actions of proinflammatory cytokines and glucocorticoids are usually mutually antagonistic. However, 11β-HSD1 expression is synergistically induced by TNF-α or IL-1 and glucocorticoids in a range of cell types (338, 403, 567, 681). The purpose might be to amplify intracellular glucocorticoid-mediated attenuation of proinflammatory cytokine action in preparation for recovery in key tissues.
6. Molecular mechanisms
IL-1β and TNF-α induce 11β-HSD1 in some human-derived cells (fetal lung fibroblasts, hepatoma) via C/EBPβ binding to the proximal HSD11B1 P2 promoter (790). However, these cytokines induce 11β-HSD1 via NF-κB in mouse mesenchymal cells (11). Curiously, in adipocytes, TNF-α decreases 11β-HSD1 expression through NF-κB, although this may have been related to decreased C/EBPα (597). This complexity probably underpins the cell specific effects of pro-inflammatory cytokines on 11β-HSD1 expression. Induction of 11β-HSD1 by TNF-α in adipose stromal cells is inhibited by insulin (260), suggesting insulin counters the effect of TNF-α and that TNF-α induction of 11β-HSD1 predominates in insulin resistance (e.g., obesity).
Interestingly, hypoxia downregulates 11β-HSD2 in rat kidney (270) and in placenta (262). Translational and transcriptional mechanisms (293), the latter perhaps via Egr-1 (270), have been invoked, although hypoxia causes inflammation (499), suggesting proinflammatory cytokine actions. Reduced glucocorticoid catabolism by 11β-HSD2, and hence increased local steroid action in placenta, may engender adaptation to hypoxia and its associated inflammation (300) by increasing fetoplacental maturational signals and suppressing inflammation. However, in adipose tissue, higher 11β-HSD1 (and hence increased local glucocorticoid levels) maximizes susceptibility to hypoxic damage via inhibition of compensatory angiogenesis (467). Clearly the balance of isozymes and tissues is complex.
C. 11β-HSDs in Disorders of Immunity, Inflammation, and Neoplasia
1. Bone and joint: inflammatory arthritis
The first clinical glucocorticoid therapy used cortisone to elicit a remarkable improvement in rheumatoid arthritis (275). While inert cortisone is largely activated to cortisol by the liver, 11β-HSD1 also plays a local role in controlling inflammation during rheumatoid arthritis. In the K/BxN serum transfer model of self-resolving experimental arthritis, Hsd11b1−/− mice show an earlier onset and delayed resolution of inflammation (136). They also exhibit greater reactive bone growth (exostosis), which could result from greater inflammation within the joint or a greater reaction of the tissue to inflammatory mediators. Hsd11b2−/− mice show a “wild-type” response (136). A role for endogenous glucocorticoids in bone turnover in arthritis has been suggested in transgenic mice expressing 11β-HSD2 specifically in osteoblasts and osteocytes (108). In these, the initial phase of K/BxN serum transfer arthritis is identical to wild-type, so presumably does not involve glucocorticoid action in osteoblasts. However, in the subsequent subacute phase, inflammation and cartilage destruction are attenuated indicating these processes depend on glucocorticoid action in osteoblasts (108). This contrasts with the phenotype of the Hsd11b1−/− mice in the same experimental model and suggests that glucocorticoids exert opposing effects in different cells/tissues during inflammation.
Patients with rheumatoid arthritis have increased 11β-HSD1 activity in synovial tissues (618) which correlates with the severity of inflammation (618). Within the rheumatic joint, 11β-HSD1 is mainly expressed in synovial fibroblasts where it is highly inducible by IL-1 or TNF-α (263, 264). In a rat adjuvant arthritis model, levels of 11β-HSD1 mRNA in the inflamed synovium and in immune cells of the draining lymph nodes increase markedly and are reduced by anti-inflammatory therapy by IL-1 receptor or TNF-α antagonism (185). 11β-HSD2 is restricted to the nonimmune cells of lymph nodes and is decreased in arthritis (185).
Dysregulation of 11β-HSD isozymes may contribute to the failure of resolution of chronic inflammation. 11β-HSD2 is expressed in synovial macrophages in rheumatoid arthritis (618) and, transiently, in peripheral blood mononuclear cells (535). Moreover, whereas IL-10 rapidly increases 11β-HSD1 mRNA in normal human macrophages, this fails in synovial macrophages in rheumatoid arthritis (29). This suggests that during chronic inflammation, a maladaptive mechanism may be engaged in which the balance of 11β-HSD activities is unfavorably altered, resulting in reduced intracellular glucocorticoid action in synovial macrophages and possibly local resistance to glucocorticoids that are substrates for 11β-HSD (e.g., prednisolone).
A) BONE.
In bone, 11β-HSD1 is mainly expressed in osteoblasts (131). Young and middle-aged Hsd11b1−/− mice have normal bone mass, bone formation, and bone resorption (333). However, 11β-HSD1 levels in bone increase with age or glucocorticoid exposure (130, 759), suggesting that endogenous glucocorticoids may contribute to age-related osteoporosis. Consistent with this notion, transgenic expression of 11β-HSD2 in osteoblasts and osteocytes protects against the age-related decline in bone strength and mass (759). The underlying mechanism may be alleviation of glucocorticoid inhibition of angiogenesis, mediated through suppression of HIF1α and VEGF-α production by osteoblasts and osteocytes (759). The effects of 11β-HSD1 inhibitors on bone structure and function with ageing remain to be established.
2. Lung
A) 11β-HSDS ARE EXPRESSED IN LUNG.
Both 11β-HSD isozymes are expressed in human lung. 11β-HSD2 is present in airway epithelia in adults (bronchiole to trachea) and the vascular endothelium (538, 687). 11β-HSD1 is found in alveoli. Levels vary greatly between individuals (687), possibly as a consequence of disease or glucocorticoid therapy (686). The adult rat lung barely expresses 11β-HSD2 (17). 11β-HSD1 is expressed (369), although more in interstitial fibroblasts than alveolar cells (86). Any function in adult lung remains uncertain, although the presence of fibrous adhesions in inflamed lungs of Hsd11b1−/−, but not wild-type mice (136), suggests 11β-HSD1 limits fibrosis during lung inflammation.
Late fetal lung maturation is dependent on, and accelerated by glucocorticoids (200, 405), which induce the synthesis and release of lung surfactant (43) and regulate epithelial cell differentiation (125). This is exploited clinically in the administration of synthetic glucocorticoids to women suffering threatened preterm labor to improve neonatal survival, especially respiratory distress syndrome (28). Human and rodent fetal lung have a developmental switch from 11β-HSD2 in early and mid-gestation (93, 126, 502) to 11β-HSD1 activity in late gestation (502, 662, 705). The timing of this “switch” is advanced by glucocorticoid administration in rodents (717) and in humans, by a pituitary factor (502), probably ACTH, driving fetal glucocorticoid secretion. Increased oxoreductase (11β-HSD1) activity at term parallels increased phospholipid biosynthesis (518) consistent with glucocorticoid stimulation of surfactant production (43). Deficiency or inhibition of 11β-HSD1 reduces fetal lung phospholipid and surfactant synthesis (309, 310, 649) and increases lung weight (649), the latter consistent with a permanent reduction in lung-to-body weight ratio following prenatal glucocorticoid exposure (667). Thus 11β-HSD1 inhibition in late pregnancy seems ill-advised. 11β-HSD1 induction in the late gestation lung may help its maturation without unwanted systemic glucocorticoid effects such as developmental programming (625).
Little definitive is known of any role for 11β-HSDs in asthma susceptibility or progression, although 11β-HSD2 immunoreactivity is inversely related to the dose of inhaled corticosteroid required for efficacy in asthma (538), perhaps reflecting intact downregulation of 11β-HSD2 by glucocorticoids in otherwise glucocorticoid-resistant airways. In human airway smooth muscle cells, the pro-asthmatic Th2 cytokine IL-13 increases 11β-HSD1 expression (via MAPK signaling through ERK1/2 and JNK pathways). This is anticipated to increase local glucocorticoid regeneration and may be a normal mechanism to enhance protection against the pro-asthmatic effects of IL-13 on bronchoconstrictor pathways (298). Not normally expressed in alveoli, in acute respiratory distress syndrome, 11β-HSD2 is induced in alveolar macrophages and type II cells (686), raising the possibility that dysregulated 11β-HSD2 contributes to the glucocorticoid resistance of this severe disorder (431). These interesting disparate observations emphasize the lack of any mechanistic understanding of 11β-HSDs in the lung, a topic worthy of considerable study.
B) LUNG CANCER.
Beyond glucocorticoid metabolism, 11β-HSD1 also has carbonyl reductase activity, metabolizing key carcinogenic nitrosamines in tobacco smoke (657). Levels of the 11β-HSD1 substrate 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and its active products predict lung cancer risk (800). The striking variation in lung 11β-HSD1 may associate with individual susceptibility to tobacco-induced cancers. At least some of this variation is genetic and associates with SNPs in the hsd11b1 promoter that are in linkage disequilibrium with a functional variant, rs13306401 (698). This genetic variance of 11β-HSD1 may be compounded by inhibition of 11β-HSD1 by licorice added to smoking tobacco (438). However, there is some lack of clarity as to whether the enzyme activates or inactivates carcinogens. Again, the biological processes underlying this idea remain to be tested, including straightforward studies of the susceptibility to tobacco carcinogenesis with 11β-HSD1-deficiency or inhibition.
3. The gastrointestinal tract and inflammatory bowel disease
In humans, 11β-HSD1 is expressed in the normal colonic epithelium as well as in nonepithelial cells of the lamina propria (654, 770). In rats, 11β-HSD1 is exclusively localized in the lamina propria (766), whereas 11β-HSD2 is confined to surface and crypt epithelial cells (770).
In both ulcerative colitis and in Crohn's disease, 11β-HSD1 mRNA is increased in the inflamed colon, whereas 11β-HSD2 mRNA and protein are decreased (802). These effects are replicated by TNF-α in colonic tissue explants (186). Interestingly, the 11β-HSD1 increase in patients with inflammatory bowel disease is greater in men (664). The same reciprocal regulation of 11β-HSD1 (increased) and 11β-HSD2 (decreased) is seen in rat experimental models of colitis (96, 184), with the increase in 11β-HSD1 in colonic intraepithelial lymphocytes consistent with migration of activated lymphocytes from the inflamed colon to mesenteric lymph nodes and spleen (184, 186). Mouse models of inflammatory bowel disease also show increased 11β-HSD1 in intestinal intraepithelial lymphocytes and mesenteric lymph nodes, but 11β-HSD2 is unchanged (724). The reciprocal regulation of 11β-HSD1 and -2 expression in colon may be mediated by the pro-inflammatory cytokines TNF-α and IL-1, which decrease 11β-HSD2 mRNA levels and enzyme activity in human SW620 colon carcinoma cells (362). Again, beyond the observations, the biological importance of these changes is unclear and requires investigation, especially as 11β-HSD1 inhibitors enter human clinical trials. Any clinical hazards of these agents in inflammatory bowel disease and other inflammatory disorders are unknown.
4. The reproductive system
A) THE FEMALE REPRODUCTIVE SYSTEM.
In most species, ovary (15, 68, 127, 581, 588) and uterus (103, 451) express both 11β-HSD1 and -2. 11β-HSD1 is also expressed in oocytes (68, 581), although any function is uncertain. In ovarian granulosa cells, a switch from 11β-HSD2 to 11β-HSD1 expression occurs immediately prior to ovulation, with the induction of 11β-HSD1 probably mediated by luteinizing hormone and augmented by IL-1β (700, 701) or prostaglandins (329), although the latter may be mediated posttranslationally. Interestingly, granulosa cell 11β-HSD1 activity is decreased by depletion of the leukocytes in human follicular aspirates, and restored by addition of IL-4 and IFN-γ, suggesting the source of cytokines in this regulation may be leukocytes (192). The presence of 11β-HSD2 activity (conversion of cortisol to cortisone) in cultured granulosa cells predicted pregnancy failure in women undergoing in vitro fertilization, whereas its absence was associated with successful pregnancy, although there was no association with fertilization per se (464). The underlying reason for this has not been elucidated. In rats, 11β-HSD2 is induced in the corpus luteum, coincident with luteal regression (735).
Ovulation is a natural inflammatory process in which the ovarian surface is cyclically ruptured and repaired following ovulation. The induction of 11β-HSD1 by IL-1 and other pro-inflammatory mediators in ovarian surface epithelial cells (567, 798) may quell inflammatory tissue damage following ovulatory rupture, supporting the nonscarring of this tissue despite cyclical injury.
There are conflicting reports on the localization and regulation of 11β-HSD1 and 11β-HSD2 in uterus. Some report mutually exclusive distributions with 11β-HSD1 in epithelial cells and 11β-HSD2 in endometrial stroma and myometrium (103, 706). In contrast, in humans, 11β-HSD2 is localized to the endometrial glandular epithelium, with little 11β-HSD1 in endometrium until menstruation (451). In rats, expression of both 11β-HSD1 and -2 is cycle dependent, maximal at proestrus, and minimal at diestrus, dependent on estrogen (103). During pregnancy in rats, 11β-HSD1 is dramatically upregulated in the myometrium shortly before parturition (736), an effect dependent on the placenta (736). In human endometrial cells, 11β-HSD1 mRNA levels markedly increase with decidualization (33, 34), suggesting a possible role for the enzyme in implantation. This has yet to be tested.
The biological relevance of these 11β-HSD expression patterns in ovary and uterus is unknown. Neither 11β-HSD1- nor 11β-HSD2-deficient mice show gross fertility defects. However, both 11β-HSD1 and -2 may play a role in fine-tuning uterine growth and remodeling, during menstrual/estrus cycling and in pregnancy. A role for 11β-HSD2 in promoting endometrial angiogenesis may underlie excessive menstrual bleeding in some women (566). Moreover, and consistent with reciprocal regulation of 11β-HSD1 and -2 during inflammation, 11β-HSD1 expression is increased and 11β-HSD2 decreased in endometriosis tissue, mimicked in the induction of 11β-HSD1 but repression of 11β-HSD2 by TNF-α in primary endometriotic stromal cells (484), suggesting increased glucocorticoid action in endometriosis. Again, the mechanism and relevance of this observation needs to be dissected.
B) THE MALE REPRODUCTIVE SYSTEM.
Glucocorticoids suppress Leydig cell production of testosterone by inhibiting steroidogenic enzyme expression. Colocalized 11β-HSD might be expected to influence this. However, although first described in the testis over 40 years ago (321, 353), the role of testicular 11β-HSD and indeed even its reaction direction remain contentious. Both 11β-HSD1 and 11β-HSD2 are expressed in the adult male reproductive tract, although there are substantial species differences (4, 369). In fish testis, 11β-HSD2 plays a glucocorticoid-independent role, converting 11β-hydroxytestosterone to 11-ketotestosterone, a major fish androgen important in fertility and sex determination (197, 324, 573). 11β-HSD2 is absent from the chicken testis (351). In mammals, 11β-HSD2 is expressed in human but not rat testis (15, 401). Neither 11β-HSD isozyme is found in the mouse testis (127, 488), and both 11β-HSD1 and 11β-HSD2 knockout male mice are normally fertile (363, 364), establishing that they are not essential for testicular function in mice, at least.
11β-HSD1 is expressed in rat Leydig cells (558). Some report it exhibits 11β-dehydrogenase activity (196, 214, 482), others 11β-reductase (396). There may be even be a switch from reductase in immature to dehydrogenase in mature Leydig cells (218). H6PDH is highly expressed in rat testis (241), so any oxidative activity of 11β-HSD1 cannot be attributed to lack of H6PDH. Rat studies with selective inhibitors will be particularly informative in establishing a role, if any, in the testis.
11β-HSD1 is present in prostate, principal epithelial cells of the epididymis, the epithelium of the vas deferens, seminal vesicle, and penile urethra as well as smooth muscle cells of the vas deferens and penile blood vessels (480, 738). 11β-HSD2 shows a complementary distribution, with expression in the clear epithelial cells of the epididymis and the corpora cavernous of the penis but not other tissues (738). The differential expression patterns of 11β-HSD1 and 11β-HSD2 throughout the male reproductive tract suggest they may modulate glucocorticoid and mineralocorticoid actions, particularly in the penile vasculature (738), but any function remains to be established.
Whole body glucocorticoid metabolism by 11β-HSD1 has endocrine consequences on gonads. Patients with apparent cortisone reductase deficiency due to H6PDH mutations (390) or the milder cortisone reductase deficiency due to HSD11B1 mutations (393) have hirsutism and often precocious pseudopuberty. The phenotype is plausibly due to overproduction of adrenal androgens, mainly DHEA and its sulfate, secondary to HPA axis activation to compensate for the lack of glucocorticoid regeneration by hepatic 11β-reductase.
5. Cancer, neoplasia, and cell proliferation
In accordance with the largely antiproliferative, pro-differentiation effects of glucocorticoids and their maturational role in preparation for birth (200), 11β-HSD2 is expressed in immature cells, protecting tissues from inappropriate glucocorticoid exposure during development (290, 780), whereas 11β-HSD1 is expressed only from late in development, apparently amplifying glucocorticoid maturational effects on fetal tissues (309). This ontogenic pattern is recapitulated in tumor-derived clonal cell lines, very few of which express much 11β-HSD1. Cells that do highly express 11β-HSD1, such as 3T3-L1 adipocytes (derived from normal cells; Refs. 245, 246), only do so once they stop proliferating and terminally differentiate (510). In contrast, many cell lines express 11β-HSD2. This has led to the concept that 11β-HSDs influence cell proliferation and neoplasia (308, 564, 565). Not surprisingly, transfection with 11β-HSD2 renders cells insensitive to the antiproliferative effects of cortisol, whereas cortisone is antiproliferative only in cells transfected with 11β-HSD1 (565). Similarly, inhibition of endogenous 11β-HSD2 by glycyrrhetinic acid in breast cancer or endometrial cell lines decreases the antiproliferative effect of glucocorticoids (308, 365, 366).
Consistent with this, the expression of 11β-HSD isozymes is altered in malignancy. 11β-HSD1 is decreased and 11β-HSD2 increased in pituitary tumors compared with normal pituitary (359, 563). In breast cancer, 11β-HSD1 expression is reduced compared with adjacent nonmalignant tissue (421) and, while 11β-HSD2 is not normally expressed in breast, it is found in two-thirds of tumor samples (366). Similarly, although 11β-HSD2 is not expressed in normal adult human adrenal gland (but is in fetal), it is expressed in adrenal cortical carcinoma and adenoma (133) and in colonic adenoma (806). However, the opposite regulation has been reported when adenomas have progressed to adenocarcinomas, with 11β-HSD1 increased and 11β-HSD2 decreased (801). Whether this regulation is causal in neoplastic transformation or a consequence (bystander effect) is unknown. The former is suggested by the effects of the nonselective 11β-HSD inhibitor glycyrrhizic acid, which decreases adenoma number and size in a mouse model of colorectal cancer. However, the anti-cancer effect apparently of 11β-HSD2 inhibition was attributed to enhanced glucocorticoid-suppression of COX-2 and PGE2 which drive neoplasia in this model (806) rather than a direct effect of glucocorticoids on cellular proliferation.
In childhood acute lymphoblastic leukemia (ALL) cells, 11β-HSD1 is upregulated by dexamethasone in cells sensitive to glucocorticoid-induced apoptosis in vitro, but downregulated in glucocorticoid-resistant cells (605). 11β-HSD2 is also expressed in leukemic cell lines, where inhibition increases the cytotoxic potency of cortisol (696) or prednisolone (606) (also readily catabolized by 11β-HSD2), to the equivalent of dexamethasone, poorly inactivated by 11β-HSD2, demonstrating that 11β-HSD2 contributes to prednisolone resistance in these cells, although it by no means accounts for it. Given that childhood ALL is treated primarily with dexamethasone in many countries, 11β-HSD2 is unlikely to contribute to resistance in a clinical setting. More probable, the dysregulation of 11β-HSDs reflects the hematopoietic differentiation stage (intrinsically sensitive or resistant to endogenous glucocorticoids; Ref. 87) at which ALL lymphoblasts are arrested. Overall, the data support the idea that 11β-HSD dysregulation is a consequence rather than cause of neoplastic transformation, but their expression pattern may be a useful biomarker of disease state and steroid sensitivity in relevant cancers such as ALL.
6. Skin
Glucocorticoids inhibit wound healing by reducing angiogenesis (via VEGF), cell motility, scar formation, and remodeling (via TGF-β and the matrix metalloproteinase system), suppressing differentiation and promoting terminal epidermal differentiation (677). 11β-HSD1 is widely expressed in human and mouse epidermis and dermis (276, 699), but is highest in keratinocytes in the suprabasal area of the epidermis and dermal fibroblasts where it is a reductase (121). Keratinocyte 11β-HSD1 increases with differentiation (699), parallelling the enzyme's pattern during differentiation of preadipocytes (510). 11β-HSD2 mRNA is also detectable by PCR in human skin (732), although whether this extends beyond known expression in the mineralocorticoid target cells of sweat glands (82) or if there is sufficient to influence glucocorticoid function elsewhere in the dermis/epidermis is unknown.
Skin 11β-HSD1 expression increases with aging and photodamage (707). Selective inhibition of 11β-HSD1 in mouse skin causes keratinocyte proliferation and enhances cutaneous wound healing. This effect was greatest in Lepob/ob obese and diabetic mice which have higher cutaneous 11β-HSD1 expression (although whether or not this reflects increased enzyme in subcutaneous adipose tissue is unclear). This suggests a mechanism by which increased 11β-HSD1 may contribute to loss of skin elasticity and poorer healing with age (699). The application of inhibitors in skin disease merits examination, especially to promote wound healing, notably in the obese, diabetics, and the elderly.
7. The eye
Both isozymes of 11β-HSD have been reported in the mammalian eye including in humans (678). 11β-HSD1 is most highly expressed in the nonpigmented epithelium of the ciliary body (576), but mRNA is also present in trabecular meshwork, corneal epithelium and endothelium, and anterior lens epithelium (678). Expression of 11β-HSD2 is less certain with both negative (576) and positive (678) reports of mRNA and activity in human and rodent ocular tissues, notably also in the ciliary body nonpigmented epithelium. In terms of function, location of 11β-HSD in the ciliary body implies a role in aqueous humor formation. Glucocorticoid excess/hypersensitivity is linked to open-angle glaucoma. In rabbits, 11β-HSD inhibition with the nonselective agent glycyrrhizin reduced intraocular pressure elevated by the synthetic glucocorticoid triamcinolone (659), although the mechanism(s) and isozyme(s) involved is uncertain. In humans, carbenoxolone reduces intraocular pressure, compatible with reduced glucocorticoid action via 11β-HSD1 inhibition (576). Such data require confirmation and dissection with isozyme selective drugs.
An additional role of 11β-HSD1 in the eye involves the substantial retro-orbital adipogenesis in thyroid-associated (Grave's disease) ophthalmopathy. Primary cultures of adipocytes from thyroid-associated ophthalmopathy patients have much higher expression of 11β-HSD1 than controls and produce a host of proinflammatory cytokines, which might underlie the increase in the enzyme, although this might merely reflect the more differentiated state of ophthalmopathy adipocytes. Cytokine production by these cells is attenuated by 11β-HSD inhibition (710), and the enzyme plays a role in adipogenesis in retro-ocular as other sites. Any role in therapy of this challenging disorder is uncertain, but merits exploration.
VI. 11β-HSDs AND THE BRAIN
While early studies reported 11β-dehydrogenase activity in glioma cell lines (250) and neonatal primate brain homogenates (251), the key reports of what turned out to be 11β-HSD2 in kidney failed to find 11β-HSD activity in the brain (181). This was fully in keeping with the presence of MR occupied by glucocorticoids in hippocampus (580), implying a lack of “protection” of such MR by 11β-dehydrogenase. However, the reason early studies failed to find ex vivo 11β-HSD activity in brain homogenates is probably due to lower levels of endogenous NADP(H) in brain than liver (215, 232). Exogenous cofactor reveals the activity (477, 478). 11β-HSD1 mRNA, immunoreactivity, and bioactivity have been repeatedly shown in hippocampus and myriad other CNS regions. 11β-HSD2 is highly expressed in the fetal CNS but is nearly absent from the adult brain. 11β-HSD1 is emerging as a key control of local glucocorticoid exposure with particular relevance to cognitive function in the ageing brain.
A. Distribution and Ontogeny
1. 11β-HSD1 in the adult CNS
11β-HSD1 is widely and unevenly distributed in the adult CNS. Although 11β-HSD1 is highly expressed in the cerebellum, hippocampus, and cortex, there is a discrete microdistribution within these areas. Highly expressing cells are seen in the Purkinje cell layer of the cerebellum, CA3 pyramidal cells of the hippocampus, and layer V neurons of the neocortex (478). There is also low expression of 11β-HSD1 generally throughout the brain and spinal cord, including the paraventricular nucleus of the hypothalamus, a key locus for glucocorticoid feedback control of the HPA axis (607). Intriguingly, 11β-HSD1 is found both in neurons and glia (607), notably in microglia (243) which express high levels of 11β-HSD1 particularly when activated, implicating this enzyme as a regulator of central inflammatory signals. Indeed, in humans, 11β-HSD1 levels in microglia appear increased in multiple sclerosis (268). 11β-HSD1 is also expressed in anterior pituitary cells including corticotrophs (359).
Rajan et al. (572) showed that primary cultures of rat hippocampal cells express 11β-HSD1 but not 11β-HSD2. Activity is exclusively 11β-reductase. In vitro, pretreatment with intrinsically inert 11-dehydrocorticosterone is equipotent with corticosterone in promoting death of hippocampal cells in the presence of high, but sublethal doses of the excitatory glutamatergic neurotransmitter kainic acid. Addition of carbenoxolone, itself without neurotoxic effects, attenuates the toxicity of 11-dehydrocorticosterone, but not corticosterone (572). These data support the 11β-reductase reaction direction of 11β-HSD1 in hippocampal cells and imply a role in amplifying intracellular glucocorticoid action.
2. 11β-HSD2 in the adult brain
11β-HSD2 expression in the adult nervous system is highly spatially restricted, much more so than MR. In the rat brain, in situ hybridization localized 11β-HSD2 mRNA to scattered cells of the ventromedial and paraventricular (PVN) nuclei of the hypothalamus, amygdala, locus ceruleus, subcommissural organ, and nucleus tractus solitarius (NTS) (583, 590, 810), areas associated with salt appetite and blood pressure regulation which are selectively affected by aldosterone rather than glucocorticoids. Distribution of 11β-HSD2 within the mouse brain is even more limited, confined to the NTS (290, 291), consistent with lesser aldosterone dependence on salt regulation in mice (596). Any 11β-HSD2 mRNA outside the NTS-sodium appetite/central cardiovascular control circuitry appears to reflect low-level expression without clear functional importance. A “knock-in” mouse in which Cre recombinase was driven by the endogenous Hsd11b2 gene promoter most likely reflects widespread fetal brain Hsd11b2-driven Cre-mediated activation of the ROSA26 reporter used, which remains expressed thereafter (221).
With the use of highly sensitive quantitative RT-PCR, some reports detect 11β-HSD2 mRNA in the adult human brain (799) but have yet to establish any functional significance of the low copy number transcripts. Within the PVN, 11β-HSD2 mRNA has been detected using RT-PCR (810), inhibition of 11β-HSD within the PVN increased sympathetic outflow and PVN activity (810), and the sympatho-excitory effects of carbenoxolone were blocked by intracerebroventricular spironolactone, an MR antagonist (810). However, the lack of specificity of the inhibitors used in this study [carbenoxolone and glycyrrhetinic acid also potently inhibit 11β-HSD1 which is expressed in the hypothalamus (478, 607, 624)] and the lack of spatial selectivity with intracerebroventricular injections, means any functional role for 11β-HSD2 mRNA in the PVN presently remains uncertain. In human brain, exploiting samples with very short post-mortem delays (<4 h), no 11β-HSD2 mRNA or 11β-dehydrogenase activity was found in cortex, hippocampus, or cerebellum (613).
In contrast, there is extensive 11β-HSD2 mRNA and activity in the fetal brain with complex age- and locus-specific patterns of silencing (discussed below).
B. The HPA Axis
Expression of 11β-HSD1 in brain regions responsible for the negative-feedback actions of glucocorticoids upon the HPA axis (cerebral cortex, hippocampus, hypothalamic PVN, and pituitary) suggests the enzyme might modulate feedback. Indeed, the original mice lacking 11β-HSD1 on a 129/MF1 strain background exhibit elevated basal and poststress corticosterone levels consistent with reduced glucocorticoid feedback (265). Interestingly, moving the disrupted 11β-HSD allele onto the C57BL/6J background normalized basal plasma corticosterone, with an efficient negative-feedback signal. The difference between mouse strains appears due to variability in plasticity in the key feedback sites, with most strains, including C57BL/6, showing a compensatory rise in GR expression in the hippocampus and PVN of the hypothalamus while the opposite occurs in the 129 strain of mice (113). However, all strains of 11β-HSD1-deficient mice have larger adrenals than their wild-type littermates with an exaggerated corticosteroid response to stress (113). Thus the genetic background is a crucial factor governing the consequences of 11β-HSD1 loss on the HPA axis.
Interindividual differences in vulnerability to disorders associated with HPA axis dysregulation raise the possibility that despite the fact that 11β-HSD1 inhibition does not inevitably activate the HPA axis and glucocorticoid levels, it may in certain susceptible individuals. However, to date, trials of 11β-HSD1 inhibitors in rodent models or clinical trials have failed to show elevation of basal glucocorticoid levels (594). Although adrenal enlargement is observed with 11β-HSD1 inhibition, this is thought to result from compensatory increased drive to the adrenals to replace the contribution of cortisol that normally derives from 11β-HSD1 regeneration. Peripheral 11β-HSD1 in the liver and splanchnic bed contributes substantially (20–40%) to total daily glucocorticoid production by regenerating cortisol from inert cortisone (62, 742). 11β-HSD1 inhibition in humans increases serum levels of ACTH and the ACTH-induced adrenal sex steroid precursor dehydroepiandrosterone, without changes in cortisol, presumably reflecting these compensatory processes (195, 594). The increased adrenal androgen production with short-term inhibition of 11β-HSD1 remains within the normal range (195). Whether or not longer term inhibition produces hirsutism, as reported in rare congenital HSD11B1 mutations in humans (393), remains to be seen, but dehydroepiandrosterone levels fall dramatically with ageing (71) so the side effect may be less in middle-aged and elderly subjects.
Intriguingly, on the mixed C57BL6/CBA/129 strain background in which Hsd11b1 gene disruption causes basal HPA axis activation, transgenic replacement of 11β-HSD1 selectively in the liver normalizes the basal and stress-induced elevations of plasma corticosterone levels and adrenal weight (547). This implies that peripheral (liver) 11β-HSD1 is key to the HPA abnormalities, a suggestion supported by the increased adrenal size of mice conditionally lacking hepatic 11β-HSD1 (392). No effects on circulating levels of corticosterone are observed in mice with overexpression of 11β-HSD1 in the liver (548), in fat (446), or in the forebrain (the hypothalamus does not express the transgene) (289). Furthermore, ectopic expression of 11β-HSD2 in fat reduces adipose corticosterone levels, but circulating glucocorticoid levels remain normal (342). It remains unknown whether the consequences of global deletion of 11β-HSD1 on HPA activity can be mimicked by loss of 11β-HSD1 solely in the brain. Currently, the data support the interpretation that bulk changes in glucocorticoid flux (largely mediated in liver) cause the observed adrenocortical hyperplasia with 11β-HSD1 deficiency. This also squares with emerging data on the role of carbohydrates and other energy substrates produced by the liver and of hepatic vagal afferents in HPA regulation (375, 387).
1. 11β-HSD1 and appetite regulation
Some findings suggest that glucocorticoids act within adipose tissues to control appetite. Mice overexpressing 11β-HSD1 under the aP2 promoter in adipose tissue show hyperphagia, whereas ectopic expression of 11β-HSD2 in adipose tissue induces hypophagia (342, 446) (a caveat is possible ectopic transgene expression). Paradoxically 11β-HSD1 knockout mice show increased transiently appetite for high-fat diet (493, 494), indicating a possible distinct central effect. Indeed, 11β-HSD1 mRNA and enzyme activity are expressed in the hypothalamic arcuate nucleus (478), a key locus for appetite control. Arcuate 11β-HSD1 is acutely induced by high-fat feeding (149), with the timing suggesting this may be part of the acute inflammatory hypothalamic response to high-fat diet (702). Furthermore, arcuate expression of appetite control neuropeptides is altered in 11β-HSD1-deficient mice with reduced anorexigenic cocaine and amphetamine-regulated transcript and melanocortin-4 receptor mRNAs, implying increased appetitive drive. Moreover, on high-fat diet, arcuate mRNA for orexigenic agouti-related peptide increases in Hsd11b1−/− mice, whereas it decreases in wild-type (149). This regulation appears mediated through μ-opioid receptor tone, a key control of appetitive reward. Hence, arcuate 11β-HSD1 appears to contribute to central adaption to dietary challenge, at least in rodents.
C. Cognition and Ageing
1. Cognition in young animals
Although 11β-HSD1 is widely expressed in areas of the brain involved in learning and memory (hippocampus and neocortex), young adult 11β-HSD1 knockout show normal acquisition and retention of spatial memory in tests of cognitive function such as the watermaze and Y-maze (791, 794). However, young mice are adept at these tasks and require fairly major cognitive deficits to alter behavior, an impact not anticipated with 11β-HSD1 deficiency. It is only as the animals age that major cognitive phenotypes associated with 11β-HSD1 and its manipulation emerge.
2. 11β-HSD1 and the ageing brain
People exposed to chronically high glucocorticoids (endogenous or exogenous) are susceptible to cognitive, affective and, rarely, psychotic disorders (688). Individual differences in cognitive function with aging associate with stressful events or rising cortisol levels across a lifespan in rodents and humans (423, 458). If, however, glucocorticoid levels are kept low throughout life, either via neonatal “programming” or adult treatment with antidepressant drugs, both of which increase GR and MR in hippocampus and provide tighter HPA axis feedback control (793), or by midlife adrenalectomy with low-dose glucocorticoid replacement (382, 456), the subsequent emergence of cognitive deficits with age is prevented. Why the brain becomes sensitive to what is probably relatively subtle chronic glucocorticoid excess with ageing is a key question to be addressed, but measurement of plasma cortisone, the substrate for 11β-HSD1, may be informative in this respect, as elevated substrate availability coupled with increased levels of enzyme (see below) could turn a subtle circulating glucocorticoid excess into a marked excess within sensitive cells.
The identification of 11β-reductase activity in hippocampus and other cognitive circuitry in rodents (478, 607) and humans (613) seeded the notion that it might amplify glucocorticoid action and, with ageing, adversely impact on cognitive function and the underlying biology (622). In this regard, Hsd11b1−/− mice (on two different genetic backgrounds) resist the expected high prevalence of age-associated cognitive decline measured in tests of hippocampus-associated spatial learning and memory such as the watermaze and Y-maze (791, 794). This is associated with reduced intrahippocampal glucocorticoid levels in the face of maintained or modestly elevated plasma glucocorticoid levels (791, 794). These data emphasize the importance of 11β-HSD1 in determining glucocorticoid action in the brain over and above any effect of circulating steroid levels. Importantly, short-term treatments with either nonselective (licorice derivatives) in young mice (152) or, crucially, with a selective 11β-HSD1 inhibitor (660) in already aged mice improves memory retention, suggesting that the action is on the adult brain rather than any notional developmental effect. These data endorse 11β-HSD1 inhibition as a potentially tractable approach to cognitive decline in aged humans. Indeed, treatment of elderly healthy men (aged 65–69 years) with carbenoxolone for 4 wk, plus amiloride to block the adverse effects of renal 11β-HSD2 inhibition, in a double-blind, placebo controlled, randomized study, led to cognitive improvements in verbal memory, a hippocampus-associated function (613). Similar effects were found in middle-aged subjects with type 2 diabetes. Moreover, a very short-term (1 h) effect of 11β-HSD1 inhibition on cognition in young rats has been reported (474); while the mechanisms are unclear, this raises again the intriguing possibility of rapid cognitive actions possibly mediated via membrane-associated corticosteroid receptors (337).
Cognitive alterations may be confounded by changes in mood, but, despite the adverse impacts of chronic glucocorticoid excess on affective function (123), 11β-HSD1 gene disruption does not noticeably alter anxiety-related behaviors (791, 794). Of course, gene disruption is life-long and affective behaviors are susceptible to developmental programming by glucocorticoids (623), potentially confounding these findings. Whether or not shorter-term 11β-HSD1 inhibition will alter mood, notably amygdala-associated fear and anxiety behaviors which are often sensitive to glucocorticoid manipulations (585), remains to be determined. While mood did not alter in healthy subjects and patients with type 2 diabetes treatment with carbenoxolone (613), it will be more pertinent to determine if 11β-HSD1 manipulations impact in individuals susceptible to depressive or anxiety-related illness.
3. Mechanisms of cognitive actions of 11β-HSD1
In terms of mechanism, improvement in peripheral metabolic factors, notably glucose homeostasis, might underlie the improvement in cognition (727). However, 11β-HSD1 deficiency did not alter glucose or lipid levels in lean, aged rodents, suggesting direct CNS effects (791). Indeed, long-term potentiation (LTP), a putative electrophysiological basis for synaptic “learning,” is increased in hippocampal slices from old 11β-HSD1-deficient mice compared with age-matched controls (791). Ectopic overexpression of 11β-HSD2 in the hippocampus improves learning, memory, and LTP in young animals (173), emphasizing the benefits of specifically reducing intrahippocampal glucocorticoid levels.
Neurogenesis is important in the hippocampus and its cognitive functions and is suppressed by glucocorticoids (110). However, while 11β-HSD1 deficiency increases hippocampal neurogenesis in young mice (as predicted), the negligible neurogenesis in old mice is unaltered by 11β-HSD1 deficiency (791).
The higher affinity of MR for glucocorticoids ensures it is largely occupied by physiological glucocorticoids even at the diurnal nadir, while GR are only activated as glucocorticoids rise to diurnal peak levels or during stress. The inverted-U-shaped relationship between glucocorticoid concentrations and both LTP in vitro and cognitive function in vivo (with both very low and high levels of steroids adversely impacting upon function) is underpinned by the procognitive effects of MR agonism and anticognitive effects of higher concentrations of glucocorticoids acting through GR (325). In line with this concept, intracerebroventricular infusion of the GR antagonist RU38486 (mifepristone) in old mice improves memory in the Y-maze, whereas the MR antagonist spironolactone has no effect (795). In contrast, despite similar age-associated elevation in plasma corticosterone, the maintained memory in elderly 11β-HSD1-deficient mice is attenuated by spironolactone, while RU38486 is without effect. These data suggest 11β-HSD1 deficiency sufficiently reduces intrahippocampal glucocorticoids to minimize GR activation by elevated plasma corticosterone with ageing, whilst maintaining optimal MR-mediated procognitive effects.
4. Does 11β-HSD1 cause age-related cognitive disorders?
A crucial issue beyond 11β-HSD1 as a therapeutic target in age-related cognitive dysfunction is whether or not it contributes to fundamental pathogenesis. Genetic linkage would suggest causation, and an intriguing report associates a rare (1:200) polymorphism in the promoter of the HSD11B1 gene with a sixfold increased risk of Alzheimer's disease (145), although the low frequency of the allele implies it is unlikely to make more than an occasional impact. More common variants of the gene do not associate with cognition in a healthy elderly population (148). However, in a prospective human study, an index of total body 11β-HSD1 activity “predicted” subsequent cerebral ventricular expansion (brain volume loss) and especially cognitive decline over 6 years (425). Whilst the locus of this effect is uncertain, the findings imply possible causal roles of elevated 11β-HSD1 in adverse cognitive and brain structural effects with ageing.
In the mouse, ageing associates with increased 11β-HSD1 mRNA in specific subfields of the hippocampus (CA3) and neocortex (layer V) (289) (FIGURE 11). Modelling this by transgenic overexpression of 11β-HSD1 in the forebrain causes cognitive impairments that only emerge with ageing (289). Such data suggest the enzyme may indeed be on the causal pathway, although the downstream processes are undetermined. Plausible mechanisms by which glucocorticoid excess might produce age-related cognitive decline are myriad and include changes in electrophysiological processes such as LTP (551), probably via changes in key target genes such as subunits of the NMDA and other glutamate receptors (509); second messenger systems such as intracellular calcium release (383); neuronal integrity and cytoskeleton; cellular energetics; cellular cholesterol dynamics and thus membrane function; amyloid precursor protein synthesis, processing, and degradation (247), the last notably via glucocorticoid regulation of insulin degrading enzyme expression (372); tau assembly and degradation (247); glial function, including microglia, the brain's phagocytes, and antigen presenting cells which express 11β-HSD1 that is increased in inflammation in human CNS (268); cerebral vasculature; and neurotransmitter synthesis, release, and breakdown (461, 645). Alternatively, the potential neuroprotective effects of 11β-HSD1 deficiency might reflect its contribution to other steroid or oxysterol metabolic pathways, 7-hydroxylated steroids/oxysterols being one obvious possibility (see below).
Figure 11.
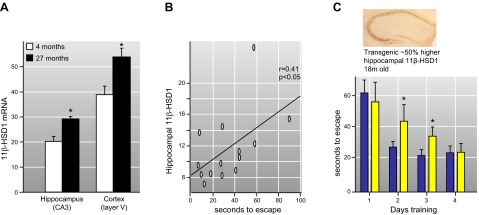
11β-HSD1 in the ageing brain. A: 11β-HSD1 is elevated in the ageing (27 mo old) mouse brain, specifically in regions of the hippocampus and cortex underpinning learning and memory. B: with ageing there is considerable interindividual variation in 11β-HSD1 that correlates with cognitive function (seconds to find a hidden platform after training in the watermaze). C: modeling this by transgenic overexpression of 11β-HSD1 in the forebrain from the age of weaning causes cognitive impairments that only emerge with ageing. [Adapted from Holmes et al. (289).]
Whether or not 11β-HSD1 inhibition will impact upon pathological brain aging, for example, in Alzheimer's disease, remains uncertain, but some preliminary data suggest that there may even be some reduction in beta-amyloid lesions in animal models of Alzheimer's disease (728). This “disease-modifying” effect is unexpected and, if confirmed, then dissection of the mechanism will be of great importance. In this regard, exogenous glucocorticoids increase Aβ and tau deposition in mouse models of Alzheimer's disease (247), raising the intriguing possibility that the elevated cortisol levels seen in patients (688) may be more than merely a reflection of neuronal loss and the stress of ill health.
Overall, accepting that translation from preclinical to clinical circumstances is tortuous, 11β-HSD1 inhibition in the brain is emerging as a target in the treatment for age-related memory and other cognitive disorders with functional and perhaps even disease-modifying effects.
5. Oxysterols
As discussed above, 7-ketocholesterol is a substrate for 11β-HSD1 (582, 621). Oxysterols such as 7-ketocholesterol rise with excitotoxicity (158, 345), suggesting it may perhaps be neurotoxic, although it is unknown whether 7-keto and 7-hydroxy cholesterol differ. Additionally, 7-keto- and 7β-hydroxy derivatives of the neurosteroids dehydroepiandrosterone (DHEA) and pregnenolone can be metabolized by 11β-HSD1 (515). Intriguingly, DHEA and pregnenolone modified in the 7-position potentiate neurosteroid activity, producing cognitive enhancement with ageing (792). Any involvement of 11β-HSD1 in these reactions is unaddressed.
D. Central Regulation of Blood Pressure and Salt Appetite
While most MR in the CNS are not linked to 11β-HSD2 and thus bind glucocorticoids under physiological conditions (144), some central effects appear aldosterone-sensitive but corticosterone-insensitive (219). Intracerebroventricular (icv) administration of aldosterone causes arterial hypertension (220, 223, 242). Moreover, salt-sensitive hypertension is mitigated by intracerebroventricular administration of MR antagonists (236, 237). Analogous studies suggest similar aldosterone-specific effects on salt appetite (534). It has therefore been postulated that aldosterone-selective central MR modulate blood pressure and salt appetite. In support of this hypothesis, in the rat brain, MR are expressed in loci that regulate blood pressure and salt appetite, namely, the nucleus of the tractus solitarius (NTS) and circumventricular organs such as subfornical organ and hypothalamic regions (153, 320). However, intracerebroventricular corticosterone antagonizes aldosterone-mediated hypertension (242), implying either distinct MR, some “protected” from corticosterone elevating blood pressure, whereas other MR bind corticosterone and exert opposing actions, or GR (locus unclear) may counter MR effects.
Although 11β-HSD2 has very limited expression within the adult brain, in the rat this is congruent with MR expression in loci relevant to blood pressure and salt appetite control (583, 590). Furthermore, intracerebroventricular administration of 11β-HSD inhibitors increases blood pressure, although no effect has been observed on sodium appetite (240). In the adult mouse brain, 11β-HSD2 colocalization with MR is only evident in the NTS (222, 290). Whether or not this locus impacts on salt appetite has been unclear.
Addressing this issue, recent data show that mice lacking 11β-HSD2 solely in the brain (exploiting the Cre-lox system) exhibit a dramatic immediate drive to ingest proffered saline, even when the animals are sodium replete. This salt appetite is reversed by the MR antagonist spironolactone, showing unequivocally that 11β-HSD2-protected MR, presumably in the NTS, control salt appetite in mice (193). Indeed, even though mice lacking 11β-HSD2 in the brain ingest large quantities of sodium, they maintain normal salt balance by increasing natriuresis and polyuria (193).
Curiously, dietary sodium deprivation still causes activation of 11β-HSD2 neurons within the NTS in adrenalectomized animals (219), which suggests that these 11β-HSD2 positive neurons are activated by other factors or pathways in addition to adrenal aldosterone and other corticosteroids. Angiotensin II has emerged as one potential regulator (222). It is also possible that corticosteroids locally produced within the brain could be responsible. Indeed, 11β-hydroxylase and aldosterone synthase activity (and their encoding mRNAs, Cyp11b1 and Cyp11b2) have been found within the brain (799). In addition, 11β-HSD2 positive neurons within the NTS are innervated by the amygdala, PVN, dorsomedial NTS, and components of the vagus (222, 223, 629, 641), which could provide a neural rather than hormonal regulation of the system. It is likely that all of these inputs may act in conjunction with circulating aldosterone to modulate NTS control of sodium appetite. Intriguingly, the 11β-HSD2 neurons in the NTS innervate both relay nuclei within the brain stem that project to the forebrain as well as directly to the forebrain itself (224), areas associated with behavioral changes related to sodium appetite, reward, arousal, and mood (222). Indeed, salt appetite is gratified very rapidly by drinking saline, implicating a direct association of salt appetite with the reward pathways (358), a hypothesis supported by pharmacological modulation of the reward pathways (404) and MR antagonism (492).
The importance of this 11β-HSD2-mediated aldosterone-sensitive blood pressure and salt-appetite control circuitry in the brain merits detailed investigation, especially in the light of the importance of the CNS in pressor control including in human hypertension (691).
VII. FETO-PLACENTAL 11β-HSD AND DEVELOPMENTAL “PROGRAMMING”
In the early 1950s it was noted that human placenta contained considerable amounts of cortisone (70, 142). High 11β-HSD activity was subsequently found in placenta (539). This was suggested to catalyze conversion of maternal cortisol to cortisone before the active steroid could reach the umbilical vein (544). Indeed, in placental mammals, cortisol (corticosterone) levels in the maternal circulation are 5- to 10-fold higher than in the fetus (65, 138, 485), a gradient thought to be maintained by high placental 11β-HSD2 (90, 93, 368). Indeed, placental 11β-HSD2 inactivates the majority of maternal glucocorticoids passing to the fetus in rodents (132) and humans (66). Prior to mid-gestation, there is also widespread expression of 11β-HSD2 in fetal tissues in rodents and humans (93, 153, 670). Importantly, the fetus, unlike the adult, has little or no 11β-HSD1 in its tissues to regenerate cortisol from cortisone, at least until near term when 11β-HSD1 becomes expressed in organs, such as lung and liver, sensitive to prenatal maturation by glucocorticoids (662).
Glucocorticoids are potent regulators of fetal growth; high levels restrict fetal growth and prime tissues for maturation in anticipation of extrauterine life. Placental 11β-HSD2 is thus thought to form a “protective barrier” to the much higher glucocorticoid levels in the maternal circulation. A longer-term importance of this barrier function has more recently emerged.
A. Glucocorticoid Programming
During development, the environment can change a growing organism's structure and function. Such effects may exert persisting influences for the lifespan, a phenomenon called “developmental programming.” Programming stimuli can impact from before conception to adolescence, acting during critical developmental “windows” of sensitivity to alter cell number and/or cellular gene expression and thus tissue function (625). In humans, a plethora of recent epidemiological studies have associated lower birth weight, albeit in the normal range, with a substantial increase in the prevalence of cardiometabolic and neuropsychiatric disorders in later life (56). These data imply programming of disease risk by factors acting on growth before birth. Such developmental programming phenomena appear near ubiquitous in the animal kingdom, and the contemporary linkage with adult disease may mask the biological importance to optimize an offspring's chances to survive long enough to reproduce in adverse environments (the thrifty phenotype hypothesis) and hence its evolutionary conservation. It has been suggested that when there is a mismatch between prenatal “predictions” of the postnatal environment and its reality that the offspring is placed at a disadvantage, largely of disorders of the postreproductive age (233).
The two major etiological hypotheses of developmental programming invoke early life exposure to malnutrition or stress/glucocorticoids. Indeed, in many mammalian species, maternal undernutrition (total calorie or protein deficiency) (56) or exposure to stress/glucocorticoid excess (625) reduce birth weight and cause persisting hypertension, hyperglycemia, affective dysfunction, and neuroendocrine abnormalities in the offspring.
1. Role of 11β-HSD2 in developmental programming
A) ANIMAL MODELS OF PLACENTAL 11β-HSD2 IN PROGRAMMING.
Prenatal exposure to endogenous or exogenous glucocorticoids accelerates fetal organ maturation in a trade-off against overall growth. Physiological glucocorticoids are catabolized by fetal and placental 11β-HSD2 (FIGURE 12). From this emerged the hypothesis that variations in feto-placental 11β-HSD2 may underlie prenatal glucocorticoid programming (180). In rodents, 11β-HSD2 activity in the placenta correlates with pup birth weight (67), a phenomenon reproduced in most (673), but not all (587), human studies, suggesting that normal variation in 11β-HSD2 activity and the consequent degree of exposure of the fetus to maternal glucocorticoids impacts on fetal growth.
Figure 12.
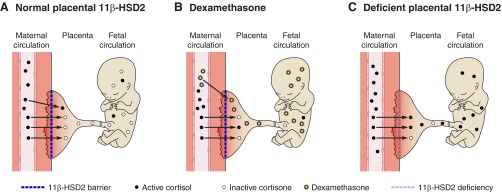
Glucocorticoid programming and placental 11β-HSD2. Placental 11β-HSD2 debulks the much higher levels of active glucocorticoids in the maternal blood. A: normally the enzyme oxidizes cortisol to inert cortisone that cannot be regenerated in the fetus which lacks 11β-HSD1 until near term. Thus the major source of active cortisol in the fetus is its own adrenal glands. B: maternal treatment with dexamethasone, which is a poor substrate for 11β-HSD2 and thus passes the placenta intact, increases glucocorticoid action on the fetus and placenta reducing growth and altering the developmental trajectory of specific tissues. C: similarly inhibition or relative deficiency of placental 11β-HSD2 allows increased passage of active maternal glucocorticoids to the fetus and placental receptors. Lowering of placental 11β-HSD2 occurs with genetic mutations, consumption of licorice, maternal malnutrition, infection, or stress.
Beyond such observational studies, inhibition, genetic deficiency, or bypass (using poor substrate steroids such as dexamethasone or betamethasone) of placental 11β-HSD2 in model species and humans associates with reduced pregnancy duration, lower birth weight, and programmed outcomes in the offspring (67, 141, 288, 408, 498, 517, 522, 525, 651, 760, 761, 781, 782). Thus administration of dexamethasone or the 11β-HSD inhibitor carbenoxolone to pregnant rats reduces birth weight and programs hypertension, hyperglycemia, HPA axis hyperactivity, and anxiety behaviors in the offspring (67, 105). Similar glucocorticoid/stress programming effects occur in the guinea pig (335), sheep (159–161, 216, 323), and non-human primates (522), including singleton-bearing old world species with physiology and placental structure similar to humans (147, 560).
B) HUMAN DATA ON THE ROLE OF 11β-HSD2 IN PROGRAMMING.
Human with homozygous/compound heterozygous mutations in HSD11B2 have decreased birth weight compared with their siblings, who are mostly heterozygous (141, 348, 498). Similarly, 11β-HSD2 knockout mice have decreased weight at birth (288). Furthermore, Finnish women who voluntarily eat larger amounts of licorice-containing foodstuffs in pregnancy have shorter gestations and their children show poorer cognitive function and behavioral disturbances (increases in attention seeking, rule breaking, and aggression) coupled with hyperactivity of their HPA axis (569, 570). Licorice directly inhibits human placental 11β-HSD2 immediately ex vivo (66), increasing passage of cortisol to the fetal circulation. The relationships between maternal licorice intake and offspring cognitive function and behavior are continuous, suggesting no threshold is completely safe.
2. Mechanisms: epigenetics
The mechanisms of these programmed effects are tissue-specific and involve glucocorticoid-driven changes in target organ structure, gene expression, and function. Epigenetic mechanisms to maintain such effects have been advocated (167, 523, 524, 623, 755, 760, 761, 781, 782). Indeed, the key HPA axis drivers, corticotropin releasing hormone (CRH) and arginine vasopressin (AVP), and feedback receptor, glucocorticoid receptor (GR), show altered expression in models of programming and the promoters of all three encoding genes show persisting changes in methylation in response to early life environmental challenges (496, 500, 756). Methylation of DNA during gestation is in part determined by the availability of methyl donors (753), and placental 11β-HSD2 impacts on placental transport of key methyl donors (779), implying a possible additional role in determining fetal epigenetic status.
Intriguingly, 11β-HSD2 itself appears to be a target for fetal programming with persisting reductions in renal 11β-HSD2 mRNA levels and increased mineralocorticoid activity of glucocorticoids in rats exposed prenatally to glucocorticoids (694) and sheep exposed to undernutrition (769). The HSD11B2 gene promoter is subject to methylation (17). Children exposed to the horrors of the Nazi Holocaust have reduced renal 11β-HSD2 activity in old age with the greatest effect seen in those youngest at traumatization (797). These data suggest 11β-HSD2 is a programming target in humans, possibly to maximize renal sodium retention in what may be anticipated to be a starvation-associated sodium-poor environment. This adaptation does not necessarily optimize physiology for the modern sodium-replete world.
11β-HSD1 is also programmed. Prenatal glucocorticoids (522) and malnutrition (769) permanently increase the isozyme in adipose tissue, liver, pancreas (522), and hippocampus (642). As outlined above, this may contribute to the risk of metabolic and cognitive disorders with ageing.
B. 11β-HSDs in the Placenta
1. Localization and ontogeny of placental 11β-HSD2
Placental 11β-HSD2 has been reported in many mammalian species including human (90, 92, 503, 553), non-human primates (44, 45), pig (350), sheep (785, 787), guinea pig (608), rat (67, 104, 105, 588), and mouse (93, 127). Placental 11β-HSD2 in the rat (588, 734) and human (368, 682) is localized in the syncytiotrophoblast, precisely where maternal and fetal circulations are in close proximity for transfer of nutrients and other substances. Thus 11β-HSD2 is strategically positioned to function as a “barrier” to glucocorticoid access not only to placental cells but also to the fetus.
The ontogeny of placental 11β-HSD2 is species-specific. In humans, there is a steady rise in placental 11β-HSD2 activity through most of gestation (229, 455, 634). A similar gestational rise occurs in the baboon placenta (554). In humans, 11β-HSD2 activity (but not mRNA) decreases in the last 2 wk prior to parturition (504). Fetal membranes also express 11β-HSD. In the human amnion, 11β-HSD1 is concentrated in fibroblasts where expression is increased by glucocorticoids and cytokines (681), providing a feed-forward loop (506). Here, glucocorticoids increase prostaglandin E2 production, which is central to the induction of parturition. 11β-HSD1 is also expressed in human chorion cells where glucocorticoids inhibit prostaglandin catabolism by repressing the gene encoding 15-hydroxy-PG dehydrogenase (546). Thus 11β-HSD1 amplification of glucocorticoid action reduces prostaglandin breakdown, further increasing the parturition signal. This fetal membrane system affords a possible link between maternal infection, cytokine production, local glucocorticoid production, and the induction of preterm labor. Moreover, increased amniotic fluid cortisol levels amplified by local 11β-HSD1 will be swallowed by the fetus and thus may contribute to accelerated fetal organ maturation at labor (681).
C. 11β-HSDs in Fetal Tissues
The 11β-HSDs are not only expressed in placenta and fetal membranes but also in the fetus itself where they shows complex stage- and organ-specific patterns implying exquisite control of cellular exposure to glucocorticoids as each tissue matures (93, 662). Most work on this topic addresses the developing brain.
1. 11β-HSDs in the fetal brain
A) 11β-HSD2.
The developing brain is extremely sensitive to glucocorticoids, which regulate cellular and biochemical development (360, 457) by initiating terminal maturation and remodeling of axons and dendrites, as well as inducing programmed cell death (463). The high expression of 11β-HSD2 in the fetal brain may be crucial to protect immature neurons from maturation through untimely exposure to fetal and/or maternal physiological glucocorticoids. A reduction in brain weight is observed in sheep at birth following antenatal glucocorticoid treatment (301), associated with delayed maturation of neurons, glia, and vasculature (302). In rhesus monkeys, antenatal glucocorticoid exposure causes degeneration of hippocampal neurons and reduces subsequent hippocampal volume (721). The hippocampus is particularly sensitive to perinatal glucocorticoid exposure, which can cause life-long changes in memory and behavior (85, 636, 666). In the rat, prenatal stress (repeated maternal restraint) reduces the number of proliferating hippocampal cells and feminizes the adult hippocampus (428). Prenatal stress similarly reduces hippocampal neurogenesis, with reduced placental 11β-HSD2 implicated in the mechanism (422).
GR, MR, and 11β-HSDs show intricate temporal and regional patterns in the developing brain (153, 211, 349). GR is highly expressed in the neuroepithelium of the fetal rat brain, whereas MR is restricted the epithelium of the septal-hippocampal system, areas of the anterior hypothalamus, pituitary, deep layers of the superior colliculus, piriform cortex, and lateral septum (153). Interestingly, neuronal 11β-HSD2 does not coincide with MR in the fetal brain but with GR, suggesting the enzyme protects immature neurons from premature exposure to glucocorticoids via GR. Throughout midgestation, 11β-HSD2 is abundant in neuroepithelium, but its expression rapidly declines as each brain area approaches the terminal stage of neurogenesis (93, 153). Perhaps analogously, 11β-HSD2 expression in the human fetal brain is silenced between gestational weeks 19–26 (669). This postulated protective role for 11β-HSD2 in fetal brain is akin to the proposed role of placental 11β-HSD2 in protecting the fetus from overexposure to maternal glucocorticoids (153, 625, 734) and may provide an additional barrier to protect the developing CNS from excess glucocorticoids derived from the mother or the fetal adrenal itself.
11β-HSD2 expression is restricted after birth to the proliferating external granular layer (EGL) of the cerebellum and several central, late-maturing nuclei of the thalamus (583, 589). Glucocorticoids and stress are known to inhibit the production of cells from the EGL resulting in decreased size and cell content of the molecular and/or granular layers of the mature cerebellum, depending on the timing of exposure (414, 469, 772). As anticipated, cerebellar size is also reduced in Hsd11b2−/− mice early in postnatal life with a decrease in the molecular and internal granule layers (290), associated with a delay in attainment of cerebellum-associated neurodevelopmental landmarks such as negative geotaxis and eye opening (290). Hence, the temporal and spatial expression of 11β-HSD2 in the developing brain tightly regulates the timing of its exposure to glucocorticoids which, in turn, dictate the timing of terminal maturation.
But does this relate to fetal programming of persisting effects? To address this, mice with brain-specific deletion of 11β-HSD2 have been made. These exhibit depressive-like behavior as adults, even though placental and fetal growth and circulating corticosterone levels are normal (Wyrwoll, Seckl, and Holmes, unpublished data). Thus 11β-HSD2 in the developing brain appears crucial to protect against glucocorticoid programming of behavior, even when there is normal placental expression of 11β-HSD2.
11β-HSD2 is significantly induced in cerebellar granular neuron precursor cells by the developmental patterning gene sonic hedgehog (269). Decreased 11β-HSD2 expression in frontal cortex and hippocampus of piglets follows early weaning or social isolation (559). In rodents, 11β-HSD2 is rapidly downregulated in the fetal brain at midgestation (94), perhaps due to epigenetic silencing of the promoter. The notion that bypass or mistiming of the shut-off of fetal brain 11β-HSD2 engenders developmental abnormalities or programming merits investigation.
B) 11β-HSD1.
In the hippocampus of the fetal sheep, 11β-HSD1 mRNA rises from mid to late gestation but rapidly falls prior to parturition (644). In the rat (153, 476) and mouse (662) fetal brain, 11β-HSD1 mRNA is not observed until the last third of gestation, a time when 11β-HSD2 is declining. Thereafter, 11β-HSD1 increases in the fetal brain with age. Expression is first seen in the hippocampus, precerebellar area, and medulla at E16.5. At term, highest expression is observed in the thalamus, neocortex, hippocampus, and hindbrain, and levels continue to rise through the postnatal period (153, 476). No neurodevelopmental abnormalities or related functional phenotypes have been associated with 11β-HSD1 deletion. However, the observation that 11β-HSD1-deficient mice are heavier at birth (780) suggests that these might have been overlooked.
2. Fetal kidney
The developing mammalian kidney highly expresses 11β-HSD2. In early to mid gestation, 11β-HSD2 is found throughout the kidney in the mouse (93), sheep (385), and human (285, 673), notably in Bowman's capsule and the vascular tufts of the developing glomeruli as they migrate from the surface to the inner cortex (126). In late gestation, the expression pattern becomes more discrete with very high levels in the distal convoluted tubules and collecting ducts (93, 126, 127, 307), resembling the adult pattern. Indeed, in the later stages of gestation, MR becomes colocalized with 11β-HSD2, suggesting 11β-HSD2 is protecting inappropriate activation of MR at this time, whereas at earlier time points 11β-HSD2 may modulate glucocorticoid signaling through GR (93, 126). Interestingly, 11β-HSD2 levels are low at birth, despite maintained MR expression (432). Plausibly, this could be a mechanism for glucocorticoid maturation of renal structure and function, although this remains to be tested. Conversely, high glucocorticoid levels in the perinatal period cause renal cysts (450).
Fetal kidney 11β-HSD2 is itself a target of programming. In the ovine fetus, renal 11β-HSD2 is strikingly downregulated by hypoxia (38, 501, 786) or maternal undernutrition (769). In rat offspring, maternal undernutrition, diabetes, or placental vasculature insufficiency (uterine artery ligation) all reduce 11β-HSD2 in the kidney (58, 207, 540). Such changes could, if persistent, contribute to the programmed hypertension seen in these models. Indeed, maternal glucocorticoid treatment in pregnancy in the rat reduces offspring renal 11β-HSD2 that persists from neonatal to mid-adult life and associates with increased mineralocorticoid actions of corticosterone compatible with mild AME (694), in essence a form of low renin hypertension. In terms of molecular mechanisms, intrauterine growth retardation exerts sex-specific effects on transcription factor binding to the Hsd11b2 gene promoter in the kidneys of offspring (58). These changes are linked to increased CpG methylation of the Hsd11b2 promoter compatible with reduced transcription of 11β-HSD2 mRNA.
Such effects may be hardwired. Thus sodium retention in 11β-HSD2 knockout mice can be readily reversed by suppression of corticosterone with dexamethasone, but hypertension and renal structural abnormalities persist (363), emphasizing the key importance of 11β-HSD2 in renal development.
D. Feto-placental 11β-HSD Regulation and Function
1. Placental functions of 11β-HSD2
A core issue in fetal programming is to determine the locus of action of environmental challenges, specifically distinguishing indirect effects on the mother from more direct impacts on the placenta and/or the fetus. Progress on this issue has been achieved by crossing male and female mice heterozygous for a null allele of Hsd11b2. This gives wild-type, heterozygous, and homozygous null Hsd11b2 offspring in the same dam. Since placental 11β-HSD2 in the labyrinth zone is derived from the fetal genotype, this design affords a clear differentiation of feto-placental from maternal effects. In this model, birth weight and offspring affective behavior follow the feto-placental genotype (288), excluding maternal effects from this manipulation of 11β-HSD2. Of course, the Hsd11b2−/− offspring also lack the enzyme in kidney and are thus hypertensive and hypokalemic (363), which complicates assessment of cardiometabolic programming. Studies of tissue-specific knockout of 11β-HSD2 in fetal organs will allow distinction of the relative importance of the placental and individual fetal tissue “barriers” to glucocorticoids.
An additional and important level of impact of 11β-HSD2 is on placental function per se. In the heterozygous cross model, growth of placentas completely lacking 11β-HSD2 is restricted in late gestation, even before fetal growth is retarded (783). In mid- to late-gestation, placental amino acid transport to Hsd11b2−/− offspring is increased, suggesting compensation for placental dysfunction. However, near term the Hsd11b2−/− placenta is failing with markedly reduced placental glucose transport and decreased glucose transporter 3 expression. The late-gestation Hsd11b2−/− placenta lacks the normal increase in density of fetally derived blood vessels and shows reduced expression of the key angiogenic factor VEGF (normally downregulated by glucocorticoids; Ref. 322), and a key angiogenic transcription factor, PPARγ. Glucocorticoids and 11β-HSD inhibitors advance apoptosis in the term placenta (737). Such underweight, undervascularized, and underfunctioning placentas are poorly equipped to supply the rapidly growing and maturing near-term fetus. In terms of molecular mechanisms, placenta expresses both GR and MR. Some data suggest that mineralocorticoids acting via MR promote trophoblast division and placental growth in vitro and in vivo, while glucocorticoid inhibit these effects (227). Moreover, glucocorticoids inhibit 15-hydroxyprostaglandin dehydrogenase, a key degradative enzyme. In human trophoblast cells which express 11β-HSD2, inhibition is greater with cortisol plus carbenoxolone than the GR-selective agonist dexamethasone, implying MR are involved in this control (546). The impact of the placental balance of 11β-HSD2 and 11β-HSD1 on local GR and MR remains unclear, although glycyrrhetinic acid (inhibiting both isozymes) enhances glucocorticoid-mediated placental cell hypoplasia in vitro (227), suggesting 11β-HSD2 predominates.
2. Pathogenesis and placental 11β-HSD2
Placental 11β-HSD2 is modified in disease. Activity is reduced in human pregnancies complicated by preeclampsia (42, 448), in association with increased cortisol in cord blood, suggesting increased fetal exposure to maternal (and/or fetal) glucocorticoids. Similarly, placental 11β-HSD2 is decreased in fetuses with intrauterine growth retardation (176, 455, 619, 634, 718, 733), perhaps a mechanism to increase fetal glucocorticoid exposure to optimize maturation in preparation for birth, trading off against growth (334). Low placental 11β-HSD2 in IUGR correlates inversely with catch-up growth (718), a marker of programming risk (187). Maternal inflammation (asthma) associates with reduced placental 11β-HSD2 and birth weight and increased fetal cortisol levels (505). Indeed proinflammatory cytokines, notably IL-1β, IL-6, and TNF-α, downregulate 11β-HSD2 activity and mRNA levels in primary cultures of human trophoblasts (118, 361).
Other regulators of placental 11β-HSD2 include nitric oxide, progesterone, estrogen, protein kinase A, retinoic acid, prostaglandins, catecholamines, oxygen, glucocorticoids, PPAR agonists, proinflammatory cytokines, and toxic heavy metals (16, 261, 262, 332, 545, 555, 616, 683, 684, 713, 726, 784). Furthermore, 11β-HSD2 expression is upregulated by the action of p38 MAPK on 11β-HSD2 mRNA stability in primary trophoblast cells (635). Thus a host of factors, many determined by the maternal environment and health, impact on placental 11β-HSD2, which may represent a key node between the maternal and feto-placental environments.
3. Is placental 11β-HSD2 the common link between glucocorticoid and nutritional programming?
Interestingly, low-protein diet in pregnancy causes an increase in maternal and fetal glucocorticoid levels (255, 398), together with a decrease in placental 11β-HSD2 activity (77, 384, 676). Conversely, exposure to dexamethasone during pregnancy decreases maternal food intake (376) and reduces maternal weight gain (525). These results suggest that there are common mechanisms underpinning both nutritional and glucocorticoid developmental programming. Feeding low-protein diet to the pregnant dams of heterozygous Hsd11b2 crosses shows that although both maternal undernutrition and 11β-HSD2 deficiency reduce fetal growth and both involve feto-placental overexposure to glucocorticoids, the mechanisms are distinct. Thus 11β-HSD2 insufficiency increases feto-placental exposure to maternal glucocorticoids, whereas low-protein diet activates the fetal HPA axis to produce increased fetal glucocorticoid levels (132). Consistent with this, prenatal undernutrition increases levels of adrenal steroidogenic enzymes at birth (344). With the obesity epidemic producing more babies born to obese mothers, it is a concern that that there is also adverse programming of cardiometabolic outcomes in these offspring, but at present, any role for 11β-HSD2 is unknown (166, 610).
VIII. CONCLUSIONS AND FUTURE PERSPECTIVES
In the six decades since the discovery of 11β-HSD, only in the last 25 years has interest burgeoned and understanding emerged. Summarizing the current state of play, there are two isozymes. 11β-HSD2 is a potent dehydrogenase that acts to “protect” MR from activation by glucocorticoids, thus engendering aldosterone specificity in the distal nephron and other classical mineralocorticoid target sites in vivo. Deficiency or inhibition in the kidney causes AME. This enzyme also protects GR and MR in the placenta and fetal tissues from premature activation by glucocorticoids. Deficiency or inhibition of feto-placental 11β-HSD2 (or its bypass with dexamethasone) causes developmental programming, increasing the risk of subsequent cardiometabolic and neuropsychiatric disorders. The biology is conserved from rodents to humans. In contrast, 11β-HSD1 is predominantly but not exclusively an 11β-reductase in intact cells, regenerating and thus amplifying glucocorticoid action. Enzyme levels are elevated in adipose tissue in obesity and appear to play a causal role in metabolic syndrome. Similarly, elevated 11β-HSD1 in the ageing CNS contributes to cognitive decline. 11β-HSD1 inhibition is an interesting drug target for metabolic syndrome and its complications, age-related cognitive disorders, and other less well validated indications, typically diseases associated with age-related physical decline.
A. Ageing
11β-HSD1 expression appears elevated in key glucocorticoid target organs with ageing or age-associated pathologies. Thus the aged hippocampus and neocortex overexpress 11β-HSD1 in mice (289). Similarly old bones in mice (759) and osteoblasts obtained from aged humans (130) have higher 11β-HSD1 levels. 11β-HSD1 is elevated in adipose tissue of obese individuals. Perhaps obesity mimics an age-related process in adipose tissues as it associates with hypoxia, fibrosis, and degenerative metabolic disease. Similarly, 11β-HSD1 is increased in skeletal muscle in type 2 diabetes (1), again a pathology associated with accelerated senescence of multiple organs. 11β-HSD1 and corticosterone levels are increased in the ageing obese mouse ovary (83), another organ that fails with senescence.
With aging, plasma glucocorticoids become elevated in a subset of individuals in rodents and humans. This coincides with and marks “unsuccessful” aging and its associated pathologies. Glucocorticoids may be causal, and attempts to keep circulating glucocorticoids low or reduce their tissue impacts can ameliorate age-related pathologies. Such findings have spawned the glucocorticoid hypothesis of age-related pathogenesis in the brain (614) as well as immune (63) and cardiovascular (595) systems.
The increase in expression of 11β-HSD1 in key tissues with aging and its amplification of glucocorticoid concentrations in organs sensitive to glucocorticoid pathogenesis suggests a component acting well beyond its physiological purpose. Perhaps the glucocorticoid-11β-HSD1 system is intended to configure medium-term tissue glucocorticoid responses, amplifying steroid action in some targets (after all 11β-HSD1 expression is generally increased by glucocorticoids) but not others, optimally to configure tissue effects to promote survival (increasing glucocorticoid-driven storage of excess calories in adipose tissue in preparation for the next period of starvation, facilitating fear conditioning to survive the inevitable succeeding mortal challenge, preventing overshoot of inflammatory responses, etc.). A lifetime of such stress hormone amplification and its trade-off consequences (reduced hippocampus-associated learning, insulin resistance, expanded and inflamed adipose tissue), beyond the age of peak reproductive fitness and thus Darwinian selection, or in the exceptional circumstances of chronic calorie excess beyond the range biology can be expected readily to handle, puts such subtle tuning of tissue steroid exposure beyond its purpose.
The mechanisms by which 11β-HSD1 becomes elevated in specific cells in the aging brain, bone, ovary, immune system, and perhaps adipose tissue is not fully understood, but proinflammatory cytokines and glucocorticoids themselves may be key. Indeed, proinflammatory cytokines also stimulate the HPA axis. Thus inflammation may drive both systemic (HPA axis) and local (11β-HSD1) glucocorticoid excess which, with chronicity and age-related vulnerability, mediates organ pathology. Whatever the mechanism and its biological importance, 11β-HSD1 and its elevation in specific tissues in association with pathogenesis offers an opportunity for therapy.
B. 11β-HSD1 as a Therapeutic Target
Inhibition of 11β-HSD1 is a plausible target for treating metabolic disease, atherosclerosis, and disorders of cognition with aging. The efficacy of this approach has been proven in rodents and other preclinical models of obesity-metabolic disease and age-related memory loss. The effects of selective 11β-HSD1 inhibitors in humans with diabetes and metabolic disease have been modest to date, at least for the chosen therapeutic target of lowering markers of glycemic control or blood pressure, and the utility of such inhibitors in clinical therapy remains to be established. Perhaps the main indication might be in early metabolic disease to ameliorate multiple risk factors, a contention indicated by the clinical trials published to date. Crucial is the effect on atherosclerosis, the main final cause of mortality in the obesity-diabetes-metabolic syndrome continuum. Whilst preclinical studies are rather encouraging, such end-points are long term and thus expensive for clinical trials and require a degree of confidence in the target to persuade funding.
Trials in other indications such as hepatic steatosis, wound healing, mild cognitive impairment, Alzheimer's disease, other cognitive disorders, glaucoma, and thyroid eye disease are being explored but are unreported as yet. In the era of stratified medicine, subgroups of subjects with elevated 11β-HSD1 in key target organs may represent the main beneficiaries. Indeed, the greatest glucose- and lipid-lowering actions in one phase II trial were seen in subjects with the highest plasma glucose and lipid levels (594).
However, the efficacy and indeed even the most promising therapeutic target of 11β-HSD1 inhibition are not yet clear. The preclinical data suggest that adipose tissue is the most important metabolic organ target, yet some agents access this poorly, as exemplified by carbenoxolone. A subtlety is that an inhibitor should probably preferentially inhibit 11β-reductase without impact on the reverse 11β-dehydrogenase direction. Such considerations militate for direction-specific screening assays in intact cells and tissues.
C. Side Effects of 11β-HSD1 Inhibition
An inevitable effect of 11β-HSDS1 inhibition is activation of the HPA axis merely to maintain circulating cortisol levels against a reduced glucocorticoid half-life because of the lack of regeneration in splanchnic tissues, etc. The elevation of ACTH drives cortisol to normal levels, but elevates DHEA and other adrenal androgens potentially causing hirsutism in sensitive women. This is an unwanted, but not usually a serious, side effect and may be readily reversible as ACTH and DHEA levels fall to normal levels on cessation of 11β-HSD1 inhibition (594). However, adrenal hypertrophy/hyperplasia are also likely. While this is a physiological response and is observed in other causes of HPA axis activation such as depression, there is a theoretical concern about persistence and even the possibility of tertiary tumor formation. No data support this eventuality which is not seen in other HPA activation states. Hirsutism is the main adult phenotype of cortisone reductase deficiency, also supporting the general safety of this approach.
Other adverse effects include delayed maturation of the fetal lung at term, and such agents should probably be avoided in pregnancy. Effects on the immune/inflammatory systems are complex, but it will be important to know the impact on clearance of acute and chronic infections and inflammatory states such as rheumatoid arthritis and connective tissue disorders. An important caveat to the inflammation studies in preclinical models is that most studies have been conducted in young, lean mice. Obesity, type 2 diabetes, and metabolic syndrome associate with dysregulated immune responses (297) and impaired resistance to bacterial infection, with increased mortality due to sepsis (20, 723). It will be important to dissect the complex relationships between obesity-metabolic syndrome, 11β-HSD1, and the immune system in human trials. To date, no excess of side effects over placebo have been reported in short-term (months) clinical trials with selective 11β-HSD1 inhibitors. This is somewhat reassuring.
D. 11β-HSD2 as a Biomarker and Therapeutic Target
11β-HSD2 deficiency is pathogenic, but the isozyme may be a biomarker for developmental programming of the fetus, both in placental biopsies and in leukocytes in cord blood and in the adult. Epigenetic changes in this promoter, even in leukocytes, may be a persisting reflection of the quality of the prenatal environment marking those at risk of sequelae.
There is much about these enzymes still to be understood from fundamental biology, for example, simply understanding the major physiological roles of 11β-HSD1, its reaction direction in all tissues, the balance of intracrine and endocrine impacts, and its true value as a therapeutic target. For 11β-HSD2, it remains unclear exactly how biochemically it generates MR selectivity, its importance in developmental programming beyond licorice eaters, and its role as a biomarker of this process in placenta or offspring tissues. Much has been achieved since the biology of 11β-HSD2 was revealed 25 years ago; the next 25 years should see equal if not greater progress.
GRANTS
Work in the authors' laboratories is funded by a Wellcome Trust Programme Grant (to J. Seckl and K. Chapman), Wellcome Trust project grants (to K. Chapman, J. Seckl, and M. Holmes), as well as project grants from the MRC (to K. Chapman, J. Seckl, and M. Holmes), the British Heart Foundation (to K. Chapman and J. Seckl), and the European Union (to J. Seckl).
DISCLOSURES
J. Seckl holds patents on the use of inhibitors of 11β-HSD1.
ACKNOWLEDGMENTS
We thank all members of the Endocrinology Unit for many insightful discussions and sharing of data.
Address for reprint requests and other correspondence: J. Seckl, Endocrinology Unit, Centre for Cardiovascular Science, The Queen's Medical Research Institute, Univ. of Edinburgh, 47 Little France Crescent, Edinburgh EH16 4TJ, UK (e-mail: j.seckl@ed.ac.uk).
REFERENCES
- 1. Abdallah BM, Beck-Nielsen H, Gaster M. Increased expression of 11beta-hydroxysteroid dehydrogenase type 1 in type 2 diabetic myotubes. Eur J Clin Invest 35: 627–634, 2005 [DOI] [PubMed] [Google Scholar]
- 2. Ackermann D, Vogt B, Escher G, Dick B, Reichen J, Frey BM, Frey FJ. Inhibition of 11beta-hydroxysteroid dehydrogenase by bile acids in rats with cirrhosis. Hepatology 30: 623–629, 1999 [DOI] [PubMed] [Google Scholar]
- 3. Agarwal AK, Giacchetti G, Lavery G, Nikkila H, Palermo M, Ricketts M, McTernan C, Bianchi G, Manunta P, Strazzullo P, Mantero F, White PC, Stewart PM. CA-repeat polymorphism in intron 1 of HSD11B2: effects on gene expression and salt sensitivity. Hypertension 36: 187–194, 2000 [DOI] [PubMed] [Google Scholar]
- 4. Agarwal AK, Monder C, Eckstein B, White PC. Cloning and expression of rat cDNA encoding corticosteroid 11 beta-dehydrogenase. J Biol Chem 264: 18939–18943, 1989 [PubMed] [Google Scholar]
- 5. Agarwal AK, Mune T, Monder C, White PC. Mutations in putative glycosylation sites of rat 11-beta-hydroxysteroid dehydrogenase affect enzymatic-activity. Biochim Biophys Acta 1248: 70–74, 1995 [DOI] [PubMed] [Google Scholar]
- 6. Agarwal AK, Mune T, Monder C, White PC. NAD+-dependent isoform of 11 beta-hydroxysteroid dehydrogenase. Cloning and characterization of cDNA from sheep kidney. J Biol Chem 269: 25959–25962, 1994 [PubMed] [Google Scholar]
- 7. Agarwal AK, Rogerson FM, Mune T, White PC. Analysis of the human gene encoding the kidney isozyme of 11 beta-hydroxysteroid dehydrogenase. J Steroid Biochem Mol Biol 55: 473–479, 1995 [DOI] [PubMed] [Google Scholar]
- 8. Agarwal AK, Rogerson FM, Mune T, White PC. Gene structure and chromosomal localization of the human hsd11k gene encoding the kidney (type-2) isozyme of 11-beta-hydroxysteroid dehydrogenase. Genomics 29: 195–199, 1995 [DOI] [PubMed] [Google Scholar]
- 9. Agarwal AK, Tusie-Luna MT, Monder C, White PC. Expression of 11 beta-hydroxysteroid dehydrogenase using recombinant vaccinia virus. Mol Endocrinol 4: 1827–1832, 1990 [DOI] [PubMed] [Google Scholar]
- 10. Agarwal AK, White PC. Analysis of the promoter of the NAD+ dependent 11 beta-hydroxysteroid dehydrogenase (HSD11K) gene in JEG-3 human choriocarcinoma cells. Mol Cell Endocrinol 121: 93–99, 1996 [DOI] [PubMed] [Google Scholar]
- 11. Ahasan MM, Hardy R, Jones C, Kaur K, Nanus D, Juarez M, Morgan SA, Hassan-Smith Z, Benezech C, Caamano JH, Hewison M, Lavery G, Rabbitt EH, Clark AR, Filer A, Buckley CD, Raza K, Stewart PM, Cooper MS. Inflammatory regulation of glucocorticoid metabolism in mesenchymal stromal cells. Arthritis Rheum 64: 2404–2413, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alberts P, Engblom L, Edling N, Forsgren M, Klingstrom G, Larsson C, Ronquist-Nii Y, Ohman B, Abrahmsen L. Selective inhibition of 11beta-hydroxysteroid dehydrogenase type 1 decreases blood glucose concentrations in hyperglycaemic mice. Diabetologia 45: 1528–1532, 2002 [DOI] [PubMed] [Google Scholar]
- 13. Alberts P, Nilsson C, Selen G, Engblom LO, Edling NH, Norling S, Klingstrom G, Larsson C, Forsgren M, Ashkzari M, Nilsson CE, Fiedler M, Bergqvist E, Ohman B, Bjorkstrand E, Abrahmsen LB. Selective inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivity in hyperglycemic mice strains. Endocrinology 144: 4755–4762, 2003 [DOI] [PubMed] [Google Scholar]
- 14. Alberts P, Ronquist-Nii Y, Larsson C, Klingstrom G, Engblom L, Edling N, Lidell V, Berg I, Edlund PO, Ashkzari M, Sahaf N, Norling S, Berggren V, Bergdahl K, Forsgren M, Abrahmsen L. Effect of high-fat diet on KKAy and ob/ob mouse liver and adipose tissue corticosterone and 11-dehydrocorticosterone concentrations. Horm Metab Res 37: 402–407, 2005 [DOI] [PubMed] [Google Scholar]
- 15. Albiston AL, Obeyesekere VR, Smith RE, Krozowski ZS. Cloning and tissue distribution of the human 11 beta-hydroxysteroid dehydrogenase type 2 enzyme. Mol Cell Endocrinol 105: R11–17, 1994 [DOI] [PubMed] [Google Scholar]
- 16. Alfaidy N, Gupta S, DeMarco C, Caniggia I, Challis JR. Oxygen regulation of placental 11 beta-hydroxysteroid dehydrogenase 2: physiological and pathological implications. J Clin Endocrinol Metab 87: 4797–4805, 2002 [DOI] [PubMed] [Google Scholar]
- 17. Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM. Epigenetic regulation of 11 beta-hydroxysteroid dehydrogenase type 2 expression. J Clin Invest 114: 1146–1157, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alikhani-Koupaei R, Fouladkou F, Fustier P, Cenni B, Sharma AM, Deter HC, Frey BM, Frey FJ. Identification of polymorphisms in the human 11beta-hydroxysteroid dehydrogenase type 2 gene promoter: functional characterization and relevance for salt sensitivity. FASEB J 21: 3618–3628, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Alzamora R, Michea L, Marusic ET. Role of 11 beta-hydroxysteroid dehydrogenase in nongenomic aldosterone effects in human arteries. Hypertension 35: 1099–1104, 2000 [DOI] [PubMed] [Google Scholar]
- 20. Amar S, Zhou Q, Shaik-Dasthagirisaheb Y, Leeman S. Diet-induced obesity in mice causes changes in immune responses and bone loss manifested by bacterial challenge. Proc Natl Acad Sci USA 104: 20466–20471, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amelung D, Hubener HJ, Roka L, Meyerheim G. Conversion of cortisone to compound F. J Clin Endocrinol Metab 13: 1125–1126, 1953 [DOI] [PubMed] [Google Scholar]
- 22. Andersson AK, Atkinson SE, Khanolkar-Young S, Chaduvula M, Jain S, Suneetha L, Suneetha S, Lockwood DN. Alteration of the cortisol-cortisone shuttle in leprosy type 1 reactions in leprosy patients in Hyderabad, India. Immunol Lett 109: 72–75, 2007 [DOI] [PubMed] [Google Scholar]
- 23. Andersson Ts, Simonyte K, Andrew R, Strand M, Burton J, Walker BR, Mattsson C, Olsson T. Tissue-specific increases in 11β-hydroxysteroid dehydrogenase type 1 in normal weight postmenopausal women. PLoS ONE 4: e8475, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Andrew R, Phillips DI, Walker BR. Obesity and gender influence cortisol secretion and metabolism in man. J Clin Endocrinol Metab 83: 1806–1809, 1998 [DOI] [PubMed] [Google Scholar]
- 25. Andrew R, Smith K, Jones GC, Walker BR. Distinguishing the activities of 11β-hydroxysteroid dehydrogenases in vivo using isotopically labeled cortisol. J Clin Endocrinol Metab 87: 277–285, 2002 [DOI] [PubMed] [Google Scholar]
- 26. Andrew R, Westerbacka J, Wahren J, Yki-Jarvinen H, Walker BR. The contribution of visceral adipose tissue to splanchnic cortisol production in healthy humans. Diabetes 54: 1364–1370, 2005 [DOI] [PubMed] [Google Scholar]
- 27. Andrews RC, Rooyackers O, Walker BR. Effects of the 11 beta-hydroxysteroid dehydrogenase inhibitor carbenoxolone on insulin sensitivity in men with type 2 diabetes. J Clin Endocrinol Metab 88: 285–291, 2003 [DOI] [PubMed] [Google Scholar]
- 28. Anonymous Effect of corticosteroids for fetal maturation on perinatal outcomes. NIH Consensus Development Panel on the Effect of Corticosteroids for Fetal Maturation on Perinatal Outcomes. JAMA 273: 413–418, 1995 [DOI] [PubMed] [Google Scholar]
- 29. Antoniv TT, Ivashkiv LB. Dysregulation of interleukin-10-dependent gene expression in rheumatoid arthritis synovial macrophages. Arthritis Rheum 54: 2711–2721, 2006 [DOI] [PubMed] [Google Scholar]
- 30. Apostolova G, Schweizer RA, Balazs Z, Kostadinova RM, Odermatt A. Dehydroepiandrosterone inhibits the amplification of glucocorticoid action in adipose tissue. Am J Physiol Endocrinol Metab 288: E957–E964, 2005 [DOI] [PubMed] [Google Scholar]
- 31. Arai N, Masuzaki H, Tanaka T, Ishii T, Yasue S, Kobayashi N, Tomita T, Noguchi M, Kusakabe T, Fujikura J, Ebihara K, Hirata M, Hosoda K, Hayashi T, Sawai H, Minokoshi Y, Nakao K. Ceramide and adenosine 5′-monophosphate-activated protein kinase are two novel regulators of 11β-hydroxysteroid dehydrogenase type 1 expression and activity in cultured preadipocytes. Endocrinology 148: 5268–5277, 2007 [DOI] [PubMed] [Google Scholar]
- 32. Arampatzis S, Kadereit B, Schuster D, Balazs Z, Schweizer RA, Frey FJ, Langer T, Odermatt A. Comparative enzymology of 11beta-hydroxysteroid dehydrogenase type 1 from six species. J Mol Endocrinol 35: 89–101, 2005 [DOI] [PubMed] [Google Scholar]
- 33. Arcuri F, Battistini S, Hausknecht V, Cintorino M, Lockwood CJ, Schatz F. Human endometrial decidual cell-associated 11 beta-hydroxysteroid dehydrogenase expression: its potential role in implantation. Early Pregnancy 3: 259–264, 1997 [PubMed] [Google Scholar]
- 34. Arcuri F, Monder C, Lockwood CJ, Schatz F. Expression of 11 beta-hydroxysteroid dehydrogenase during decidualization of human endometrial stromal cells. Endocrinology 137: 595–600, 1996 [DOI] [PubMed] [Google Scholar]
- 35. Arizmendi C, Liu S, Croniger C, Poli V, Friedman JE. The transcription factor CCAAT/enhancer-binding protein β regulates gluconeogenesis and phosphoenolpyruvate carboxykinase (GTP) gene transcription during diabetes. J Biol Chem 274: 13033–13040, 1999 [DOI] [PubMed] [Google Scholar]
- 36. Arnold P, Tam S, Yan L, Baker ME, Frey FJ, Odermatt A. Glutamate-115 renders specificity of human 11beta-hydroxysteroid dehydrogenase type 2 for the cofactor NAD+. Mol Cell Endocrinol 201: 177–187, 2003 [DOI] [PubMed] [Google Scholar]
- 37. Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237: 268–275, 1987 [DOI] [PubMed] [Google Scholar]
- 38. Asano H, Shearman K, Darnel A, Richardson BS, Yang K. Effects of sustained hypoxaemia with 72 hours recovery on 11beta-hydroxysteroid dehydrogenase types 1 and 2 gene expression in near-term fetal sheep. Reprod Fertil Dev 9: 755–761, 1997 [DOI] [PubMed] [Google Scholar]
- 39. Atanasov AG, Ignatova ID, Nashev LG, Dick B, Ferrari P, Frey FJ, Odermatt A. Impaired protein stability of 11beta-hydroxysteroid dehydrogenase type 2: a novel mechanism of apparent mineralocorticoid excess. J Am Soc Nephrol 18: 1262–1270, 2007 [DOI] [PubMed] [Google Scholar]
- 40. Atanasov AG, Nashev LG, Gelman L, Legeza B, Sack R, Portmann R, Odermatt A. Direct protein-protein interaction of 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase in the endoplasmic reticulum lumen. Biochim Biophys Acta 1783: 1536–1543, 2008 [DOI] [PubMed] [Google Scholar]
- 41. Atanasov AG, Nashev LG, Schweizer RA, Frick C, Odermatt A. Hexose-6-phosphate dehydrogenase determines the reaction direction of 11β-hydroxysteroid dehydrogenase type 1 as an oxoreductase. FEBS Lett 571: 129–133, 2004 [DOI] [PubMed] [Google Scholar]
- 42. Aufdenblatten M, Baumann M, Raio L, Dick B, Frey BM, Schneider H, Surbek D, Hocher B, Mohaupt MG. Prematurity is related to high placental cortisol in preeclampsia. Pediatr Res 65: 198–202, 2009 [DOI] [PubMed] [Google Scholar]
- 43. Avery ME. Pharmacological approaches to the acceleration of fetal lung matturation. Br Med Bull 31: 13–17, 1975 [PubMed] [Google Scholar]
- 44. Baggia S, Albrecht E, Pepe G. Regulation of 11β-hydroxysteroid dehydrogenase activity in the baboon placenta by estrogen. Endocrinology 126: 2742–2748, 1990 [DOI] [PubMed] [Google Scholar]
- 45. Baggia S, Albrecht ED, Babischkin JS, Pepe GJ. Interconversion of cortisol and cortisone in baboon trophoblast and decidua cells in culture. Endocrinology 127: 1735–1741, 1990 [DOI] [PubMed] [Google Scholar]
- 46. Bailey MA, Craigie E, Livingstone DE, Kotelevtsev YV, Al-Dujaili EA, Kenyon CJ, Mullins JJ. Hsd11b2 haploinsufficiency in mice causes salt sensitivity of blood pressure. Hypertension 57: 515–520, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baillie AH, Ferguson MM, Calman KC, Hart DM. Histochemical demonstration of 11beta-hydroxysteroid dehydrogenase. J Endocrinol 33: 119, 1965 [DOI] [PubMed] [Google Scholar]
- 48. Baker ME. 11Beta-hydroxysteroid dehydrogenase-type 2 evolved from an ancestral 17beta-hydroxysteroid dehydrogenase-type 2. Biochem Biophys Res Commun 399: 215–220, 2010 [DOI] [PubMed] [Google Scholar]
- 49. Baker ME. Evolution of 11β-hydroxysteroid dehydrogenase-type 1 and 11β-hydroxysteroid dehydrogenase-type 3. FEBS Lett 584: 2279–2284, 2010 [DOI] [PubMed] [Google Scholar]
- 50. Baker ME. Evolution of mammalian 11 beta- and 17 beta-hydroxysteroid dehydrogenases-type 2 and retinol dehydrogenases from ancestors in Caenorhabditis elegans and evidence for horizontal transfer of a eukaryote dehydrogenase to E. coli. J Steroid Biochem Mol Biol 66: 355–363, 1998 [DOI] [PubMed] [Google Scholar]
- 51. Baker ME. Evolutionary analysis of 11beta-hydroxysteroid dehydrogenase-type 1, -type 2, -type 3 and 17beta-hydroxysteroid dehydrogenase-type 2 in fish. FEBS Lett 574: 167–170, 2004 [DOI] [PubMed] [Google Scholar]
- 52. Balachandran A, Guan H, Sellan M, van Uum S, Yang K. Insulin and dexamethasone dynamically regulate adipocyte 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology 149: 4069–4079, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Balazs Z, Nashev LG, Chandsawangbhuwana C, Baker ME, Odermatt A. Hexose-6-phosphate dehydrogenase modulates the effect of inhibitors and alternative substrates of 11beta-hydroxysteroid dehydrogenase 1. Mol Cell Endocrinol 301: 117–122, 2009 [DOI] [PubMed] [Google Scholar]
- 54. Banhegyi G, Benedetti A, Fulceri R, Senesi S. Cooperativity between 11β-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase in the lumen of the endoplasmic reticulum. J Biol Chem 279: 27017–27021, 2004 [DOI] [PubMed] [Google Scholar]
- 55. Barf T, Vallgarda J, Emond R, Haggstrom C, Kurz G, Nygren A, Larwood V, Mosialou E, Axelsson K, Olsson R, Engblom L, Edling N, Ronquist-Nii Y, Ohman B, Alberts P, Abrahmsen L. Arylsulfonamidothiazoles as a new class of potential antidiabetic drugs. Discovery of potent and selective inhibitors of the 11beta-hydroxysteroid dehydrogenase type 1. J Med Chem 45: 3813–3815, 2002 [DOI] [PubMed] [Google Scholar]
- 56. Barker DJ. The developmental origins of adult disease. J Am Coll Nutr 23: 588S–595S, 2004 [DOI] [PubMed] [Google Scholar]
- 57. Bartter FC, Albright F, Forbes AP, Leaf A, Dempsey E, Carroll E. The effects of adrenocorticotropic hormone and cortisone in the adrenogenital syndrome associated with congenital adrenal hyperplasia: an attempt to explain and correct its disordered hormonal pattern. J Clin Invest 30: 237–251, 1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Baserga M, Kaur R, Hale MA, Bares A, Yu X, Callaway CW, McKnight RA, Lane RH. Fetal growth restriction alters transcription factor binding and epigenetic mechanisms of renal 11β-hydroxysteroid dehydrogenase type 2 in a sex-specific manner. Am J Physiol Regul Integr Comp Physiol 299: R334–R342, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Basu R, Basu A, Grudzien M, Jung P, Jacobson P, Johnson M, Singh R, Sarr M, Rizza RA. Liver is the site of splanchnic cortisol production in obese nondiabetic humans. Diabetes 58: 39–45, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Basu R, Edgerton DS, Singh RJ, Cherrington A, Rizza RA. Splanchnic cortisol production in dogs occurs primarily in the liver: evidence for substantial hepatic specific 11 beta hydroxysteroid dehydrogenase type 1 activity. Diabetes 55: 3013–3019, 2006 [DOI] [PubMed] [Google Scholar]
- 61. Basu R, Singh R, Basu A, Johnson CM, Rizza RA. Effect of nutrient ingestion on total-body and splanchnic cortisol production in humans. Diabetes 55: 667–674, 2006 [DOI] [PubMed] [Google Scholar]
- 62. Basu R, Singh RJ, Basu A, Chittilapilly EG, Johnson CM, Toffolo G, Cobelli C, Rizza RA. Splanchnic cortisol production occurs in humans: evidence for conversion of cortisone to cortisol via the 11-beta hydroxysteroid dehydrogenase (11beta-hsd) type 1 pathway. Diabetes 53: 2051–2059, 2004 [DOI] [PubMed] [Google Scholar]
- 63. Bauer ME. Stress, glucocorticoids and ageing of the immune system. Stress 8: 69–83, 2005 [DOI] [PubMed] [Google Scholar]
- 64. Baumann H, Morella KK, Campos SP, Cao Z, Jahreis GP. Role of Caat-enhancer binding-protein isoforms in the cytokine regulation of acute-phase plasma-protein genes. J Biol Chem 267: 19744–19751, 1992 [PubMed] [Google Scholar]
- 65. Beitins IZ, Bayard F, Ances IG, Kowarski A, Migeon CJ. The metabolic clearance rate, blood production, interconversion and transplacental passage of cortisol and cortisone in pregnancy near term. Pediatr Res 7: 509–519, 1973 [DOI] [PubMed] [Google Scholar]
- 66. Benediktsson R, Calder AA, Edwards CR, Seckl JR. Placental 11 beta-hydroxysteroid dehydrogenase: a key regulator of fetal glucocorticoid exposure. Clin Endocrinol 46: 161–166, 1997 [DOI] [PubMed] [Google Scholar]
- 67. Benediktsson R, Lindsay RS, Noble J, Seckl JR, Edwards CR. Glucocorticoid exposure in utero: new model for adult hypertension. Lancet 341: 339–341, 1993 [DOI] [PubMed] [Google Scholar]
- 68. Benediktsson R, Yau JLW, Low S, Brett LP, Cooke BE, Edwards CRW, Seckl JR. 11-Beta-hydroxysteroid dehydrogenase in the rat ovary: high expression in the oocyte. J Endocrinol 135: 53, 1992 [DOI] [PubMed] [Google Scholar]
- 69. Berger J, Tanen M, Elbrecht A, Hermanowski-Vosatka A, Moller DE, Wright SD, Thieringer R. Peroxisome proliferator-activated receptor-gamma ligands inhibit adipocyte 11beta -hydroxysteroid dehydrogenase type 1 expression and activity. J Biol Chem 276: 12629–12635, 2001 [DOI] [PubMed] [Google Scholar]
- 70. Berliner CL, Jones JE, Salhanick HA. The isolation of adrenal-like steroids from the human placenta. J Biol Chem 223: 1043–1053, 1956 [PubMed] [Google Scholar]
- 71. Berr C, Lafont S, Debuire B, Dartigues JF, Baulieu EE. Relationships of dehydroepiandrosterone sulfate in the elderly with functional, psychological, and mental status, and short-term mortality: a French community-based study. Proc Natl Acad Sci USA 93: 13410–13415, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Berthiaume M, Laplante M, Festuccia W, Gelinas Y, Poulin S, Lalonde J, Joanisse DR, Thieringer R, Deshaies Y. Depot-specific modulation of rat intra-abdominal adipose tissue lipid metabolism by pharmacological inhibition of 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology 148: 2391–2397, 2007 [DOI] [PubMed] [Google Scholar]
- 73. Berthiaume M, Laplante M, Festuccia WT, Berger JP, Thieringer R, Deshaies Y. Preliminary report: pharmacologic 11beta-hydroxysteroid dehydrogenase type 1 inhibition increases hepatic fat oxidation in vivo and expression of related genes in rats fed an obesogenic diet. Metabolism 59: 114–117, 2010 [DOI] [PubMed] [Google Scholar]
- 74. Berthiaume M, Laplante M, Festuccia WT, Cianflone K, Turcotte LP, Joanisse DR, Olivecrona G, Thieringer R, Deshaies Y. 11beta-HSD1 inhibition improves triglyceridemia through reduced liver VLDL secretion and partitions lipids toward oxidative tissues. Am J Physiol Endocrinol Metab 293: E1045–E1052, 2007 [DOI] [PubMed] [Google Scholar]
- 75. Berthiaume M, Sell H, Lalonde J, Gelinas Y, Tchernof A, Richard D, Deshaies Y. Actions of PPAR gamma agonism on adipose tissue remodeling, insulin sensitivity, and lipemia in absence of glucocorticoids. Am J Physiol Regul Integr Comp Physiol 287: R1116–R1123, 2004 [DOI] [PubMed] [Google Scholar]
- 76. Bertini R, Bianchi M, Ghezzi P. Adrenalectomy sensitizes mice to the lethal effects of interleukin-1 and tumor necrosis factor. J Exp Med 167: 1708–1712, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bertram C, Trowern AR, Copin N, Jackson AA, Whorwood CB. The maternal diet during pregnancy programs altered expression of the glucocorticoid receptor and type 2 11beta-hydroxysteroid dehydrogenase: potential molecular mechanisms underlying the programming of hypertension in utero. Endocrinology 142: 2841–2853, 2001 [DOI] [PubMed] [Google Scholar]
- 78. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate Toll-like receptor-4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 109: 4313–4319, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bickel M, Kauffman GL., Jr Gastric gel mucus thickness: effect of distention, 16,16-dimethyl prostaglandin E2, and carbenoxolone. Gastroenterology 80: 770–775, 1981 [PubMed] [Google Scholar]
- 80. Biedasek K, Andres J, Mai K, Adams S, Spuler S, Fielitz J, Spranger J. Skeletal muscle 11beta-HSD1 controls glucocorticoid-induced proteolysis and expression of E3 ubiquitin ligases Atrogin-1 and MuRF-1. Plos One 6: 6, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Blum A, Martin HJ, Maser E. Human 11β-hydroxysteroid dehydrogenase type 1 is enzymatically active in its nonglycosylated form. Biochem Biophys Res Commun 276: 428–434, 2000 [DOI] [PubMed] [Google Scholar]
- 82. Bocchi B, Kenouch S, Lamarre-Cliche M, Muffat-Joly M, Capron MH, Fiet J, Morineau G, Azizi M, Bonvalet JP, Farman N. Impaired 11-beta hydroxysteroid dehydrogenase type 2 activity in sweat gland ducts in human essential hypertension. Hypertension 43: 803–808, 2004 [DOI] [PubMed] [Google Scholar]
- 83. Brannian J, Eyster K, Greenway M, Henriksen C, TeSlaa K, Diggins M. Progressive obesity leads to altered ovarian gene expression in the Lethal Yellow mouse: a microarray study. J Ovarian Res 2: 10, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Brem AS, Bina RB, King TC, Morris DJ. Localization of 2 11beta-OH steroid dehydrogenase isoforms in aortic endothelial cells. Hypertension 31: 459–462, 1998 [DOI] [PubMed] [Google Scholar]
- 85. Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM, Delaney RC, McCarthy G, Charney DS, Innis RB. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 152: 973–981, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Brereton PS, van Driel RR, Suhaimi F, Koyama K, Dilley R, Krozowski Z. Light and electron microscopy localization of the 11β-hydroxysteroid dehydrogenase type I enzyme in the rat. Endocrinology 142: 1644–1651, 2001 [DOI] [PubMed] [Google Scholar]
- 87. Brewer JA, Sleckman BP, Swat W, Muglia LJ. Green fluorescent protein-glucocorticoid receptor knockin mice reveal dynamic receptor modulation during thymocyte development. J Immunol 169: 1309–1318, 2002 [DOI] [PubMed] [Google Scholar]
- 88. Brown A, Reynolds LR, Bruemmer D. Intensive glycemic control and cardiovascular disease: an update. Nat Rev Cardiol 7: 369–375, 2010 [DOI] [PubMed] [Google Scholar]
- 89. Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell 10: 237–245, 2002 [DOI] [PubMed] [Google Scholar]
- 90. Brown RW, Chapman KE, Edwards CR, Seckl JR. Human placental 11 beta-hydroxysteroid dehydrogenase: evidence for and partial purification of a distinct NAD-dependent isoform. Endocrinology 132: 2614–2621, 1993 [DOI] [PubMed] [Google Scholar]
- 91. Brown RW, Chapman KE, Kotelevtsev Y, Yau JLW, Lindsay RS, Brett L, Leckie C, Murad P, Lyons V, Mullins JJ, Edwards CRW, Seckl JR. Cloning and production of antisera to human placental 11β-hydroxysteroid dehydrogenase type 2. Biochem J 313: 1007–1017, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Brown RW, Chapman KE, Murad P, Edwards CR, Seckl JR. Purification of 11 beta-hydroxysteroid dehydrogenase type 2 from human placenta utilizing a novel affinity labelling technique. Biochem J 313: 997–1005, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Brown RW, Diaz R, Robson AC, Kotelevtsev YV, Mullins JJ, Kaufman MH, Seckl JR. The ontogeny of 11 beta-hydroxysteroid dehydrogenase type 2 and mineralocorticoid receptor gene expression reveal intricate control of glucocorticoid action in development. Endocrinology 137: 794–797, 1996 [DOI] [PubMed] [Google Scholar]
- 94. Brown RW, Kotolevtsev Y, Leckie C, Lindsay RS, Lyons V, Murad P, Mullins JJ, Chapman KE, Edwards CRW, Seckl JR. Isolation and cloning of human placental 11β-hydroxysteroid dehydrogenase-2 cDNA. Biochem J 313: 1007–1017, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bruley C, Lyons V, Worsley AG, Wilde MD, Darlington GD, Morton NM, Seckl JR, Chapman KE. A novel promoter for the 11beta-hydroxysteroid dehydrogenase type 1 gene is active in lung and is C/EBPalpha independent. Endocrinology 147: 2879–2885, 2006 [DOI] [PubMed] [Google Scholar]
- 96. Bryndova J, Zbankova S, Kment M, Pacha J. Colitis up-regulates local glucocorticoid activation and down-regulates inactivation in colonic tissue. Scand J Gastroenterol 39: 549–553, 2004 [DOI] [PubMed] [Google Scholar]
- 97. Bujalska IJ, Draper N, Michailidou Z, Tomlinson JW, White PC, Chapman KE, Walker EA, Stewart PM. Hexose-6-phosphate dehydrogenase confers oxo-reductase activity upon 11β-hydroxysteroid dehydrogenase type 1. J Mol Endocrinol 34: 675–684, 2005 [DOI] [PubMed] [Google Scholar]
- 98. Bujalska IJ, Gathercole LL, Tomlinson JW, Darimont C, Ermolieff J, Fanjul AN, Rejto PA, Stewart PM. A novel selective 11 beta-hydroxysteroid dehydrogenase type 1 inhibitor prevents human adipogenesis. J Endocrinol 197: 297–307, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bujalska IJ, Kumar S, Hewison M, Stewart PM. Differentiation of adipose stromal cells: the roles of glucocorticoids and 11beta-hydroxysteroid dehydrogenase. Endocrinology 140: 3188–3196, 1999 [DOI] [PubMed] [Google Scholar]
- 100. Bujalska IJ, Kumar S, Stewart PM. Does central obesity reflect “Cushing's disease of the omentum”? Lancet 349: 1210–1213, 1997 [DOI] [PubMed] [Google Scholar]
- 101. Bujalska IJ, Walker EA, Hewison M, Stewart PM. A switch in dehydrogenase to reductase activity of 11β-hydroxysteroid dehydrogenase type 1 upon differentiation of human omental adipose stromal cells. J Clin Endocrinol Metab 87: 1205–1210, 2002 [DOI] [PubMed] [Google Scholar]
- 102. Burton AF. Inhibition of 11beta-hydroxysteroid dehydrogenase activity in rat and mouse tissues in vitro and in vivo. Endocrinology 77: 325, 1965 [DOI] [PubMed] [Google Scholar]
- 103. Burton PJ, Krozowski ZS, Waddell BJ. Immunolocalization of 11 beta-hydroxysteroid dehydrogenase types 1 and 2 in rat uterus: variation across the estrous cycle and regulation by estrogen and progesterone. Endocrinology 139: 376–382, 1998 [DOI] [PubMed] [Google Scholar]
- 104. Burton PJ, Smith RE, Krozowski ZS, Waddell BJ. Zonal distribution of 11 beta-hydroxysteroid dehydrogenase types 1 and 2 messenger ribonucleic acid expression in the rat placenta and decidua during late pregnancy. Biol Reprod 55: 1023–1028, 1996 [DOI] [PubMed] [Google Scholar]
- 105. Burton PJ, Waddell BJ. 11β-Hydroxysteroid dehydrogenase in the rat placenta: developmental changes and the effects of altered glucocorticoid exposure. J Endocrinol 143: 505–513, 1994 [DOI] [PubMed] [Google Scholar]
- 106. Burton RB, Keutmann EH, Waterhouse C. The conversion of cortisone acetate to other alpha-ketoloic steroids. J Clin Endocrinol Metab 13: 48–63, 1953 [DOI] [PubMed] [Google Scholar]
- 107. Bush IE, Hunter SA, Meigs RA. Metabolism of 11-oxygenated steroids. Metabolism in vitro by preparations of liver. Biochem J 107: 239–258, 1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Buttgereit F, Zhou H, Kalak R, Gaber T, Spies CM, Huscher D, Straub RH, Modzelewski J, Dunstan CR, Seibel MJ. Transgenic disruption of glucocorticoid signaling in mature osteoblasts and osteocytes attenuates K/BxN mouse serum-induced arthritis in vivo. Arthritis Rheumatism 60: 1998–2007, 2009 [DOI] [PubMed] [Google Scholar]
- 109. Cai T, Wong B, Mundt SS, Thieringer R, Wright SD, Hermanowski-Vosatka A. Induction of 11β-hydroxysteroid dehydrogenase type 1 but not -2 in human aortic smooth muscle cells by inflammatory stimuli. J Steroid Biochem Mol Biol 77: 117–122, 2001 [DOI] [PubMed] [Google Scholar]
- 110. Cameron HA, Gould E. Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience 61: 203–209, 1994 [DOI] [PubMed] [Google Scholar]
- 111. Campbell LE, Yu M, Yang K. Ovine 11 beta-hydroxysteroid dehydrogenase type 2 gene predicts a protein distinct from that deduced by the cloned kidney cDNA at the C-terminus. Mol Cell Endocrinol 119: 113–118, 1996 [DOI] [PubMed] [Google Scholar]
- 112. Candia R, Riquelme A, Baudrand R, Carvajal CA, Morales M, Solís N, Pizarro M, Escalona A, Carrasco G, Boza C, Pérez G, Padilla O, Cerda J, Fardella CE, Arrese M. Overexpression of 11β-hydroxysteroid dehydrogenase type 1 in visceral adipose tissue and portal hypercortisolism in non-alcoholic fatty liver disease. Liver Int 32: 392–399, 2012 [DOI] [PubMed] [Google Scholar]
- 113. Carter RN, Paterson JM, Tworowska U, Stenvers DJ, Mullins JJ, Seckl JR, Holmes MC. Hypothalamic-pituitary-adrenal axis abnormalities in response to deletion of 11beta-HSD1 is strain-dependent. J Neuroendocrinol 21: 879–887, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chakravarty K, Cassuto H, Reshef L, Hanson RW. Factors that control the tissue-specific transcription of the gene for phosphoenolpyruvate carboxykinase-C. Crit Rev Biochem Mol Biol 40: 129–154, 2005 [DOI] [PubMed] [Google Scholar]
- 115. Chapman KE, Coutinho AE, Gray M, Gilmour JS, Savill JS, Seckl JR. The role and regulation of 11β-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol Cell Endocrinol 301: 123–131, 2009 [DOI] [PubMed] [Google Scholar]
- 116. Chen BB, Lin H, Hu GX, Su Y, Zhou HY, Lian QQ, Cai H, Hardy DO, Gu DY, Ge RS. The (+)- and (−)-gossypols potently inhibit human and rat 11beta-hydroxysteroid dehydrogenase type 2. J Steroid Biochem Mol Biol 113: 177–181, 2009 [DOI] [PubMed] [Google Scholar]
- 117. Chinetti-Gbaguidi G, Bouhlel M, Copin C, Duhem C, Derudas B, Neve B, Noel B, Eeckhoute J, Seckl J, Staels B. Peroxizome proliferator-activated receptor g activation induces 11b-hydroxysteroid dehydrogenase type 1 activity in human alternative macrophages. Arteriosclerosis Thrombosis Vascular Biol 32: 677–685, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Chisaka H, Johnstone JF, Premyslova M, Manduch Z, Challis JR. Effect of pro-inflammatory cytokines on expression and activity of 11beta-hydroxysteroid dehydrogenase type 2 in cultured human term placental trophoblast and human choriocarcinoma JEG-3 cells. J Soc Gynecol Invest 12: 303–309, 2005 [DOI] [PubMed] [Google Scholar]
- 119. Chou JY, Jun HS, Mansfield BC. Neutropenia in type Ib glycogen storage disease. Curr Opin Hematol 17: 36–42, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Christy C, Hadoke PW, Paterson JM, Mullins JJ, Seckl JR, Walker BR. 11Beta-hydroxysteroid dehydrogenase type 2 in mouse aorta: localization and influence on response to glucocorticoids. Hypertension 42: 580–587, 2003 [DOI] [PubMed] [Google Scholar]
- 121. Cirillo N, Prime SS. Keratinocytes synthesize and activate cortisol. J Cell Biochem 112: 1499–1505, 2011 [DOI] [PubMed] [Google Scholar]
- 122. Clarke KA, Ward JW, Forhead AJ, Giussani DA, Fowden AL. Regulation of 11 beta-hydroxysteroid dehydrogenase type 2 activity in ovine placenta by fetal cortisol. J Endocrinol 172: 527–534, 2002 [DOI] [PubMed] [Google Scholar]
- 123. Cohen SI. Cushing's syndrome: a psychiatric study of 29 patients. Br J Psychiatry 136: 120–124, 1980 [DOI] [PubMed] [Google Scholar]
- 124. Coirini H, Magarinos AM, Denicola AF, Rainbow TC, McEwen BS. Further-studies of brain aldosterone binding-sites employing new mineralocorticoid and glucocorticoid receptor markers in vitro. Brain Res 361: 212–216, 1985 [DOI] [PubMed] [Google Scholar]
- 125. Cole TJ, Solomon NM, Van Driel R, Monk JA, Bird D, Richardson SJ, Dilley RJ, Hooper SB. Altered epithelial cell proportions in the fetal lung of glucocorticoid receptor null mice. Am J Respir Cell Mol Biol 30: 613–619, 2004 [DOI] [PubMed] [Google Scholar]
- 126. Condon J, Gosden C, Gardener D, Nickson P, Hewison M, Howie AJ, Stewart PM. Expression of type 2 11beta-hydroxysteroid dehydrogenase and corticosteroid hormone receptors in early human fetal life. J Clin Endocrinol Metab 83: 4490–4497, 1998 [DOI] [PubMed] [Google Scholar]
- 127. Condon J, Ricketts ML, Whorwood CB, Stewart PM. Ontogeny and sexual dimorphic expression of mouse type 2 11beta-hydroxysteroid dehydrogenase. Mol Cell Endocrinol 127: 121–128, 1997 [DOI] [PubMed] [Google Scholar]
- 128. Conn JW, Rovner DR, Cohen EL. Licorice-induced pseudoaldosteronism–hypertension hypokalemia aldosteronopenia and suppressed plasma renin activity. J Am Med Assoc 205: 492, 1968 [DOI] [PubMed] [Google Scholar]
- 129. Cooper MS, Bujalska I, Rabbitt E, Walker EA, Bland R, Sheppard MC, Hewison M, Stewart PM. Modulation of 11β-hydroxysteroid dehydrogenase isozymes by proinflammatory cytokines in osteoblasts: an autocrine switch from glucocorticoid inactivation to activation. J Bone Miner Res 16: 1037–1044, 2001 [DOI] [PubMed] [Google Scholar]
- 130. Cooper MS, Rabbitt EH, Goddard PE, Bartlett WA, Hewison M, Stewart PM. Osteoblastic 11beta-hydroxysteroid dehydrogenase type 1 activity increases with age and glucocorticoid exposure. J Bone Miner Res 17: 979–986, 2002 [DOI] [PubMed] [Google Scholar]
- 131. Cooper MS, Walker EA, Bland R, Fraser WD, Hewison M, Stewart PM. Expression and functional consequences of 11beta-hydroxysteroid dehydrogenase activity in human bone. Bone 27: 375–381, 2000 [DOI] [PubMed] [Google Scholar]
- 132. Cottrell EC, Holmes MC, Livingstone DE, Kenyon CJ, Seckl JR. Reconciling the nutritional and glucocorticoid hypotheses of fetal programming. FASEB J 26: 1866–1874, 2012 [DOI] [PubMed] [Google Scholar]
- 133. Coulter CL, Smith RE, Stowasser M, Sasano H, Krozowski ZS, Gordon RD. Expression of 11beta-hydroxysteroid dehydrogenase type 2 (11betaHSD-2) in the developing human adrenal gland and human adrenal cortical carcinoma and adenoma. Mol Cell Endocrinol 154: 71–77, 1999 [DOI] [PubMed] [Google Scholar]
- 134. Coutinho AE. Consequences of 11β-Hydroxysteroid Dehydrogenase Deficiency During Inflammatory Responses (PhD thesis) Edinburgh: Univ. of Edinburgh, 2009 [Google Scholar]
- 135. Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol 335: 2–13, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Coutinho AE, Gray M, Brownstein DG, Salter DM, Sawatzky DA, Clay S, Gilmour JS, Seckl JR, Savill JS, Chapman KE. 11β-Hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice. Endocrinology 153: 234–240, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Coutinho AE, Kipari T, Zhang Z, Esteves C, Gilmour JS, Cailhier JF, Hughes J, Seckl JR, Savill JS, Chapman KE. Dynamic regulation of 11β-hydroxysteroid dehydrogenase type 1 in neutrophils during an inflammatory response. Endocr Rev 32: P2–575, 2011 [Google Scholar]
- 138. Dalle M, Giry J, Gay M, Delost P. Perinatal changes in plasma and adrenal corticosterone and aldosterone concentrations in the mouse. J Endocrinol 76: 303–309, 1978 [DOI] [PubMed] [Google Scholar]
- 139. Dallman MF, Akana SF, Laugero KD, Gomez F, Manalo S, Bell ME, Bhatnagar S. A spoonful of sugar: feedback signals of energy stores and corticosterone regulate responses to chronic stress. Physiology Behavior 79: 3–12, 2003 [DOI] [PubMed] [Google Scholar]
- 140. Davani B, Khan A, Hult M, Martensson E, Okret S, Efendic S, Jornvall H, Oppermann UC. Type 1 11beta-hydroxysteroid dehydrogenase mediates glucocorticoid activation and insulin release in pancreatic islets. J Biol Chem 275: 34841–34844, 2000 [DOI] [PubMed] [Google Scholar]
- 141. Dave-Sharma S, Wilson RC, Harbison MD, Newfield R, Azar MR, Krozowski ZS, Funder JW, Shackleton CH, Bradlow HL, Wei JQ, Hertecant J, Moran A, Neiberger RE, Balfe JW, Fattah A, Daneman D, Akkurt HI, De Santis C, New MI. Examination of genotype and phenotype relationships in 14 patients with apparent mineralocorticoid excess. J Clin Endocrinol Metab 83: 2244–2254, 1998 [DOI] [PubMed] [Google Scholar]
- 142. De Courcy C, Gray CH, Lunnon JB. Adrenal cortical hormones in human placenta. Nature 170: 494, 1952 [DOI] [PubMed] [Google Scholar]
- 143. De Kloet ER, Karst H, Joels M. Corticosteroid hormones in the central stress response: quick-and-slow. Front Neuroendocrinol 29: 268–272, 2008 [DOI] [PubMed] [Google Scholar]
- 144. De Kloet ER, Reul JMHM. Feedback action and tonic influences of corticosteroids on brain function: a concept arising from heterogeneity of brain receptor systems. Psychoneuroendocrinology 12: 83–105, 1987 [DOI] [PubMed] [Google Scholar]
- 145. De Quervain DJ, Poirier R, Wollmer MA, Grimaldi LM, Tsolaki M, Streffer JR, Hock C, Nitsch RM, Mohajeri MH, Papassotiropoulos A. Glucocorticoid-related genetic susceptibility for Alzheimer's disease. Hum Mol Genet 13: 47–52, 2004 [DOI] [PubMed] [Google Scholar]
- 146. De Sousa Peixoto RA, Turban S, Battle JH, Chapman KE, Seckl JR, Morton NM. Preadipocyte 11β-hydroxysteroid dehydrogenase type 1 is a keto-reductase and contributes to diet-induced visceral obesity in vivo. Endocrinology 149: 1861–1868, 2008 [DOI] [PubMed] [Google Scholar]
- 147. De Vries A, Holmes MC, Heijnis A, Seier JV, Heerden J, Louw J, Wolfe-Coote S, Meaney MJ, Levitt NS, Seckl JR. Prenatal dexamethasone exposure induces changes in nonhuman primate offspring cardiometabolic and hypothalamic-pituitary-adrenal axis function. J Clin Invest 117: 1058–1067, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Deary IJ, Hayward C, Permana PA, Nair S, Whalley LJ, Starr JM, Chapman KE, Walker BR, Seckl JR. Polymorphisms in the gene encoding 11β-hydroxysteroid dehydrogenase type 1 (HSD11B1) and lifetime cognitive change. Neurosci Lett 393: 74–77, 2006 [DOI] [PubMed] [Google Scholar]
- 149. Densmore VS, Morton NM, Mullins JJ, Seckl JR. 11β-Hydroxysteroid dehydrogenase type 1 induction in the arcuate nucleus by high fat feeding: a novel constraint to hyperphagia? Endocrinology 147: 4486–4495, 2006 [DOI] [PubMed] [Google Scholar]
- 150. Desbriere R, Vuaroqueaux V, Achard V, Boullu-Ciocca S, Labuhn M, Dutour A, Grino M. 11β-Hydroxysteroid dehydrogenase type 1 mRNA is increased in both visceral and subcutaneous adipose tissue of obese patients. Obesity 14: 794–798, 2006 [DOI] [PubMed] [Google Scholar]
- 151. Deuchar GA, McLean D, Hadoke PWF, Brownstein DG, Webb DJ, Mullins JJ, Chapman K, Seckl JR, Kotelevtsev YV. 11β-Hydroxysteroid dehydrogenase type 2 deficiency accelerates atherogenesis and causes proinflammatory changes in the endothelium in Apoe−/− mice. Endocrinology 152: 236–246, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Dhingra D, Parle M, Kulkarni SK. Memory enhancing activity of Glycyrrhiza glabra in mice. J Ethnopharmacol 91: 361–365, 2004 [DOI] [PubMed] [Google Scholar]
- 153. Diaz R, Brown RW, Seckl JR. Distinct ontogeny of glucocorticoid and mineralocorticoid receptor and 11beta-hydroxysteroid dehydrogenase types I and II mRNAs in the fetal rat brain suggest a complex control of glucocorticoid actions. J Neurosci 18: 2570–2580, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Diederich S, Eigendorff E, Burkhardt P, Quinkler M, Bumke-Vogt C, Rochel M, Seidelmann D, Esperling P, Oelkers W, Bahr V. 11β-Hydroxysteroid dehydrogenase types 1 and 2: an important pharmacokinetic determinant for the activity of synthetic mineralo- and glucocorticoids. J Clin Endocrinol Metab 87: 5695–5701, 2002 [DOI] [PubMed] [Google Scholar]
- 155. Diederich S, Grossmann C, Hanke B, Quinkler M, Herrmann M, Bahr V, Oelkers W. In the search for specific inhibitors of human 11beta-hydroxysteroid- dehydrogenases (11beta-HSDs): chenodeoxycholic acid selectively inhibits 11beta-HSD-I. Eur J Endocrinol 142: 200–207, 2000 [DOI] [PubMed] [Google Scholar]
- 156. Diederich S, Hanke B, Bahr V, Oelkers W. The metabolism of 9 alpha-fluorinated steroids in the human kidney. Endocr Res 22: 803–810, 1996 [DOI] [PubMed] [Google Scholar]
- 157. Diederich S, Hanke B, Oelkers W, Bahr V. Metabolism of dexamethasone in the human kidney: nicotinamide adenine dinucleotide-dependent 11beta-reduction. J Clin Endocrinol Metab 82: 1598–1602, 1997 [DOI] [PubMed] [Google Scholar]
- 158. Diestel A, Aktas O, Hackel D, Hake I, Meier S, Raine CS, Nitsch R, Zipp F, Ullrich O. Activation of microglial poly(ADP-ribose)-polymerase-1 by cholesterol breakdown products during neuroinflammation: a link between demyelination and neuronal damage. J Exp Med 198: 1729–1740, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Dodic M, Hantzis V, Duncan J, Rees S, Koukoulas I, Johnson K, Wintour EM, Moritz K. Programming effects of short prenatal exposure to cortisol. FASEB J 16: 1017–1026, 2002 [DOI] [PubMed] [Google Scholar]
- 160. Dodic M, May CN, Wintour EM, Coghlan JP. An early prenatal exposure to excess glucocorticoid leads to hypertensive offspring in sheep. Clin Sci 94: 149–155, 1998 [DOI] [PubMed] [Google Scholar]
- 161. Dodic M, Peers A, Coghlan J, May C, Lumbers E, Yu Z, Wintour E. Altered cardiovascular haemodynamics and baroreceptor-heart rate reflex in adult sheep after prenatal exposure to dexamethasone. Clin Sci 97: 103–109, 1999 [PubMed] [Google Scholar]
- 162. Dosiou C, Giudice LC. Natural killer cells in pregnancy and recurrent pregnancy loss: endocrine and immunologic perspectives. Endocr Rev 26: 44–62, 2005 [DOI] [PubMed] [Google Scholar]
- 163. Dover AR, Hadoke PW, Macdonald LJ, Miller E, Newby DE, Walker BR. Intravascular glucocorticoid metabolism during inflammation and injury in mice. Endocrinology 148: 166–172, 2007 [DOI] [PubMed] [Google Scholar]
- 164. Drake A, McPherson R, Godfrey K, Cooper C, Lillycrop K, Hanson M, Meehan R, Seckl J, Reynolds R. An unbalanced maternal diet in pregnancy associates with offspring epigenetic changes in genes controlling glucocorticoid action and fetal growth. Clin Endocrinol 77: 808–815, 2012 [DOI] [PubMed] [Google Scholar]
- 165. Drake AJ, Livingstone DE, Andrew R, Seckl JR, Morton NM, Walker BR. Reduced adipose glucocorticoid reactivation and increased hepatic glucocorticoid clearance as an early adaptation to high-fat feeding in Wistar rats. Endocrinology 146: 913–919, 2005 [DOI] [PubMed] [Google Scholar]
- 166. Drake AJ, Reynolds RM. Impact of maternal obesity on offspring obesity and cardiometabolic disease risk. Reproduction 140: 387–398, 2010 [DOI] [PubMed] [Google Scholar]
- 167. Drake AJ, Walker BR, Seckl JR. Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am J Physiol Regul Integr Comp Physiol 288: R34–R38, 2005 [DOI] [PubMed] [Google Scholar]
- 168. Draper N, Echwald SM, Lavery GG, Walker EA, Fraser R, Davies E, Sorensen TI, Astrup A, Adamski J, Hewison M, Connell JM, Pedersen O, Stewart PM. Association studies between microsatellite markers within the gene encoding human 11β-hydroxysteroid dehydrogenase type 1 and body mass index, waist to hip ratio, and glucocorticoid metabolism. J Clin Endocrinol Metab 87: 4984–4990, 2002 [DOI] [PubMed] [Google Scholar]
- 169. Draper N, Powell BL, Franks S, Conway GS, Stewart PM, McCarthy MI. Variants implicated in cortisone reductase deficiency do not contribute to susceptibility to common forms of polycystic ovary syndrome. Clin Endocrinol 65: 64–70, 2006 [DOI] [PubMed] [Google Scholar]
- 170. Draper N, Walker EA, Bujalska IJ, Tomlinson JW, Chalder SM, Arlt W, Lavery GG, Bedendo O, Ray DW, Laing I, Malunowicz E, White PC, Hewison M, Mason PJ, Connell JM, Shackleton CH, Stewart PM. Mutations in the genes encoding 11β-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase interact to cause cortisone reductase deficiency. Nat Genet 34: 434–439, 2003 [DOI] [PubMed] [Google Scholar]
- 171. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115: 56–65, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Dujic T, Bego T, Mlinar B, Semiz S, Malenica M, Prnjavorac B, Ostanek B, Marc J, Causevic A. Association between 11beta-hydroxysteroid dehydrogenase type 1 gene polymorphisms and metabolic syndrome in Bosnian population. Biochem Med 22: 76–85, 2012 [PMC free article] [PubMed] [Google Scholar]
- 173. Dumas TC, Gillette T, Ferguson D, Hamilton K, Sapolsky RM. Anti-glucocorticoid gene therapy reverses the impairing effects of elevated corticosterone on spatial memory, hippocampal neuronal excitability, and synaptic plasticity. J Neurosci 30: 1712–1720, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Duperrex H, Kenouch S, Gaeggeler HP, Seckl JR, Edwards CRW, Farman N, Rossier BC. Rat liver 11β-hydoxysteroid dehydrogenase cDNA encodes oxoreductase activity in a mineralocorticoid-responsive toad bladder cell line. Endocrinology 132: 612–619, 1993 [DOI] [PubMed] [Google Scholar]
- 175. Duplomb L, Lee Y, Wang MY, Park BH, Takaishi K, Agarwal AK, Unger RH. Increased expression and activity of 11beta-HSD-1 in diabetic islets and prevention with troglitazone. Biochem Biophys Res Commun 313: 594–599, 2004 [DOI] [PubMed] [Google Scholar]
- 176. Dy J, Guan H, Sampath-Kumar R, Richardson BS, Yang K. Placental 11beta-hydroxysteroid dehydrogenase type 2 is reduced in pregnancies complicated with idiopathic intrauterine growth Restriction: evidence that this is associated with an attenuated ratio of cortisone to cortisol in the umbilical artery. Placenta 29: 193–200, 2008 [DOI] [PubMed] [Google Scholar]
- 177. Dzyakanchuk AA, Balazs Z, Nashev LG, Amrein KE, Odermatt A. 11β-Hydroxysteroid dehydrogenase 1 reductase activity is dependent on a high ratio of NADPH/NADP(+) and is stimulated by extracellular glucose. Mol Cell Endocrinol 301: 137–141, 2009 [DOI] [PubMed] [Google Scholar]
- 178. Edgerton DS, Basu R, Ramnanan CJ, Farmer TD, Neal D, Scott M, Jacobson P, Rizza RA, Cherrington AD. Effect of 11 beta-hydroxysteroid dehydrogenase-1 inhibition on hepatic glucose metabolism in the conscious dog. Am J Physiol Endocrinol Metab 298: E1019–E1026, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Edwards C. Sixty years after hench–corticosteroids and chronic inflammatory disease. J Clin Endocrinol Metab 97: 1443–1451, 2012 [DOI] [PubMed] [Google Scholar]
- 180. Edwards CR, Benediktsson R, Lindsay RS, Seckl JR. Dysfunction of placental glucocorticoid barrier: a link between the fetal environment and adult hypertension? Lancet 341: 355–357, 1993 [DOI] [PubMed] [Google Scholar]
- 181. Edwards CR, Stewart PM, Burt D, Brett L, McIntyre MA, Sutanto WS, de Kloet ER, Monder C. Localisation of 11 beta-hydroxysteroid dehydrogenase–tissue specific protector of the mineralocorticoid receptor. Lancet 2: 986–989, 1988 [DOI] [PubMed] [Google Scholar]
- 182. Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, Smith AV, Tobin MD, Verwoert GC, Hwang SJ, Pihur V, Vollenweider P, O'Reilly PF, Amin N, Bragg-Gresham JL, Teumer A, Glazer NL, Launer L, Zhao JH, Aulchenko Y, Heath S, Sober S, Parsa A, Luan J, Arora P, Dehghan A, Zhang F, Lucas G, Hicks AA, Jackson AU, Peden JF, Tanaka T, Wild SH, Rudan I, Igl W, Milaneschi Y, Parker AN, Fava C, Chambers JC, Fox ER, Kumari M, Go MJ, van der Harst P, Kao WH, Sjogren M, Vinay DG, Alexander M, Tabara Y, Shaw-Hawkins S, Whincup PH, Liu Y, Shi G, Kuusisto J, Tayo B, Seielstad M, Sim X, Nguyen KD, Lehtimaki T, Matullo G, Wu Y, Gaunt TR, Onland-Moret NC, Cooper MN, Platou CG, Org E, Hardy R, Dahgam S, Palmen J, Vitart V, Braund PS, Kuznetsova T, Uiterwaal CS, Adeyemo A, Palmas W, Campbell H, Ludwig B, Tomaszewski M, Tzoulaki I, Palmer ND, Aspelund T, Garcia M, Chang YP, O'Connell JR, Steinle NI, Grobbee DE, Arking DE, Kardia SL, Morrison AC, Hernandez D, Najjar S, McArdle WL, Hadley D, Brown MJ, Connell JM, Hingorani AD, Day IN, Lawlor DA, Beilby JP, Lawrence RW, Clarke R. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478: 103–109, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Engeli S, Bohnke J, Feldpausch M, Gorzelniak K, Heintze U, Janke J, Luft FC, Sharma AM. Regulation of 11beta-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes Res 12: 9–17, 2004 [DOI] [PubMed] [Google Scholar]
- 184. Ergang P, Leden P, Bryndova J, Zbankova S, Miksik I, Kment M, Pacha J. Glucocorticoid availability in colonic inflammation of rat. Dig Dis Sci 53: 2160–2167, 2007 [DOI] [PubMed] [Google Scholar]
- 185. Ergang P, Leden P, Vagnerova K, Klusonova P, Miksik I, Jurcovicova J, Kment M, Pacha J. Local metabolism of glucocorticoids and its role in rat adjuvant arthritis. Mol Cell Endocrinol 323: 155–160, 2010 [DOI] [PubMed] [Google Scholar]
- 186. Ergang P, Vytáčková K, Svec J, Bryndová J, Mikšík I, Pácha J. Upregulation of 11β-hydroxysteroid dehydrogenase 1 in lymphoid organs during inflammation in the rat. J Steroid Biochem Mol Biol 126: 19–25, 2011 [DOI] [PubMed] [Google Scholar]
- 187. Eriksson JG, Forsen T, Tuomilehto J, Winter PD, Osmond C, Barker DJ. Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ 318: 427–431, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Escher G, Galli I, Vishwanath BS, Frey BM, Frey FJ. Tumor necrosis factor a and interleukin 1b enhance the cortisone/cortisol shuttle. J Exp Med 186: 189–198, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Escher G, Nawrocki A, Staub T, Vishwanath BS, Frey BM, Reichen J, Frey FJ. Down-regulation of hepatic and renal 11 beta-hydroxysteroid dehydrogenase in rats with liver cirrhosis. Gastroenterology 114: 175–184, 1998 [DOI] [PubMed] [Google Scholar]
- 190. Esteves CL, Kelly V, Begay V, Man TY, Morton NM, Leutz A, Seckl JR, Chapman KE. Regulation of adipocyte 11beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1) by CCAAT/enhancer-binding protein (C/EBP) beta isoforms, LIP and LAP. PLoS One 7: e37953, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Esteves CL, Kelly V, Man TM, Seckl JR, Chapman KE. Dietary regulation of 11 beta-HSD1 in adipose tissue is mediated by the C/EBP beta isoforms LAP and LIP. Endocr Rev 31: S1659–S1659, 2010 [Google Scholar]
- 192. Evagelatou M, Peterson SL, Cooke BA. Leukocytes modulate 11beta-hydroxysteroid dehydrogenase (11beta-HSD) activity in human granulosa-lutein cell cultures. Mol Cell Endocrinol 133: 81–88, 1997 [DOI] [PubMed] [Google Scholar]
- 193. Evans LC, Holmes MC, Mullins JJ, Bailey MA. Evaluation of salt appetite in brain specific 11β hydroxysteroid dehydrogenase type 2 knockout (11βHSD2−/−) mice. FASEB J 26: 1069.7, 2012 [Google Scholar]
- 194. Fan Z, Du H, Zhang M, Meng Z, Chen L, Liu Y. Direct regulation of glucose and not insulin on hepatic hexose-6-phosphate dehydrogenase and 11β-hydroxysteroid dehydrogenase type 1. Mol Cell Endocrinol 333: 62–69, 2011. press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Feig PU, Shah S, Hermanowski-Vosatka A, Plotkin D, Springer MS, Donahue S, Thach C, Klein EJ, Lai E, Kaufman KD. Effects of an 11 beta-hydroxysteroid dehydrogenase type 1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome. Diabetes Obesity Metab 13: 498–504, 2011 [DOI] [PubMed] [Google Scholar]
- 196. Ferguson SE, Pallikaros Z, Michael AE, Cooke BA. The effects of different culture media, glucose, pyridine nucleotides and adenosine on the activity of 11beta-hydroxysteroid dehydrogenase in rat Leydig cells. Mol Cell Endocrinol 158: 37–44, 1999 [DOI] [PubMed] [Google Scholar]
- 197. Fernandino JI, Hattori RS, Kishii A, Strussmann CA, Somoza GM. The cortisol and androgen pathways cross talk in high temperature-induced masculinization: the 11beta-hydroxysteroid dehydrogenase as a key enzyme. Endocrinology 153: 6003–6011, 2012 [DOI] [PubMed] [Google Scholar]
- 198. Ferrari P. The role of 11beta-hydroxysteroid dehydrogenase type 2 in human hypertension. Biochim Biophys Acta 1802: 1178–1187, 2010 [DOI] [PubMed] [Google Scholar]
- 199. Finney RSH, Somers GF. The anti-inflammatory activity of glycyrretinic acid and derivatives. J Pharm Pharmacol 10: 613–620, 1958 [DOI] [PubMed] [Google Scholar]
- 200. Fowden AL, Li J, Forhead AJ. Glucocorticoids and the preparation for life after birth: are there long-term consequences of the life insurance? Proc Nutr Soc 57: 113–122, 1998 [DOI] [PubMed] [Google Scholar]
- 201. Franks PW, Knowler WC, Nair S, Koska J, Lee YH, Lindsay RS, Walker BR, Looker HC, Permana PA, Tataranni PA, Hanson RL. Interaction between an 11βHSD1 gene variant and birth era modifies the risk of hypertension in Pima Indians. Hypertension 44: 681–688, 2004 [DOI] [PubMed] [Google Scholar]
- 202. Freeman AI, Munn HL, Lyons V, Dammermann A, Seckl JR, Chapman KE. Glucocorticoid down-regulation of rat glucocorticoid receptor does not involve differential promoter regulation. J Endocrinol 183: 365–374, 2004 [DOI] [PubMed] [Google Scholar]
- 203. Freeman L, Hewison M, Hughes SV, Evans KN, Hardie D, Means TK, Chakraverty R. Expression of 11β-hydroxysteroid dehydrogenase type 1 permits regulation of glucocorticoid bioavailability by human dendritic cells. Blood 106: 2042–2049, 2005 [DOI] [PubMed] [Google Scholar]
- 204. Frick C, Atanasov AG, Arnold P, Ozols J, Odermatt A. Appropriate function of 11beta-hydroxysteroid dehydrogenase type 1 in the endoplasmic reticulum lumen is dependent on its N-terminal region sharing similar topological determinants with 50-kDa esterase. J Biol Chem 279: 31131–31138, 2004 [DOI] [PubMed] [Google Scholar]
- 205. Friso S, Pizzolo F, Choi SW, Guarini P, Castagna A, Ravagnani V, Carletto A, Pattini P, Corrocher R, Olivieri O. Epigenetic control of 11 beta-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis 199: 323–327, 2008 [DOI] [PubMed] [Google Scholar]
- 206. Fruchter O, Zoumakis E, Alesci S, De Martino M, Chrousos G, Hochberg Z. Intracrine modulation of gene expression by intracellular generation of active glucocorticoids. Steroids 71: 1001–1006, 2006 [DOI] [PubMed] [Google Scholar]
- 207. Fujisawa Y, Nakagawa Y, Ren-Shan L, Ohzeki T. Streptozotocin-induced diabetes in the pregnant rat reduces 11 beta-hydroxysteroid dehydrogenase type 2 expression in placenta and fetal kidney. Life Sci 75: 2797–2805, 2004 [DOI] [PubMed] [Google Scholar]
- 208. Funder JW. Glucocorticoid and mineralocorticoid receptors: biology and clinical relevance. Annu Rev Med 48: 231–240, 1997 [DOI] [PubMed] [Google Scholar]
- 209. Funder JW. Minireview: aldosterone and mineralocorticoid receptors: past, present, and future. Endocrinology 151: 5098–5102, 2010 [DOI] [PubMed] [Google Scholar]
- 210. Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science 242: 583–585, 1988 [DOI] [PubMed] [Google Scholar]
- 211. Fuxe K, Wikstrom AC, Okret S, Agnati LF, Harfstrand A, Yu ZY, Granholm L, Zoli M, Vale W, Gustafsson JA. Mapping of glucocorticoid receptor immunoreactive neurons in the rat tel- and diencephalon using a monoclonal antibody against rat liver glucocorticoid receptor. Endocrinology 117: 1803–1812, 1985 [DOI] [PubMed] [Google Scholar]
- 212. Gambineri A, Tomassoni F, Munarini A, Stimson R, Mioni R, Pagotto U, Chapman K, Andrew R, Mantovani V, Pasquali R, Walker BR. Combination of polymorphisms in HSD11B1 associate with in vivo 11β-HSD1 activity and metabolic syndrome in women with and without polycystic ovary syndrome. Eur J Endocrinol 165: 283–292, 2011 [DOI] [PubMed] [Google Scholar]
- 213. Gambineri A, Vicennati V, Genghini S, Tomassoni F, Pagotto U, Pasquali R, Walker BR. Genetic variation in 11β-hydroxysteroid dehydrogenase type 1 predicts adrenal hyperandrogenism among lean women with polycystic ovary syndrome. J Clin Endocrinol Metab 91: 2295–2302, 2006 [DOI] [PubMed] [Google Scholar]
- 214. Gao HB, Ge RS, Lakshmi V, Marandici A, Hardy MP. Hormonal regulation of oxidative and reductive activities of 11 beta- hydroxysteroid dehydrogenase in rat Leydig cells. Endocrinology 138: 156–161, 1997 [DOI] [PubMed] [Google Scholar]
- 215. Garcia-Bunuel L, Mc DD, Jr, Burch HB, Jones EM, Touhill E. Oxidized and reduced pyridine nucleotide levels and enzyme activities in brain and liver of niacin deficient rats. J Neurochem 9: 589–594, 1962 [DOI] [PubMed] [Google Scholar]
- 216. Gatford KL, Wintour EM, de Blasio MJ, Owens JA, Dodic M. Differential timing for programming of glucose homoeostasis, sensitivity to insulin and blood pressure by in utero exposure to dexamethasone in sheep. Clin Sci 98: 553–560, 2000 [DOI] [PubMed] [Google Scholar]
- 217. Gathercole LL, Morgan SA, Bujalska IJ, Hauton D, Stewart PM, Tomlinson JW. Regulation of lipogenesis by glucocorticoids and insulin in human adipose tissue. PLoS ONE 6: e26223, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Ge RS, Hardy DO, Catterall JF, Hardy MP. Developmental changes in glucocorticoid receptor and 11 beta-hydroxysteroid dehydrogenase oxidative and reductive activities in rat Leydig cells. Endocrinology 138: 5089–5095, 1997 [DOI] [PubMed] [Google Scholar]
- 219. Geerling JC, Engeland WC, Kawata M, Loewy AD. Aldosterone target neurons in the nucleus tractus solitarius drive sodium appetite. J Neurosci 26: 411–417, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Geerling JC, Kawata M, Loewy AD. Aldosterone-sensitive neurons in the rat central nervous system. J Comp Neurol 494: 515–527, 2006 [DOI] [PubMed] [Google Scholar]
- 221. Geerling JC, Loewy AD. 11Beta-hydroxysteroid dehydrogenase 2 vs transgene: discrepant loci of expression in the adult brain. Am J Physiol Renal Physiol 293: F440–F443, 2007 [DOI] [PubMed] [Google Scholar]
- 222. Geerling JC, Loewy AD. Aldosterone in the brain. Am J Physiol Renal Physiol 297: F559–F576, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Geerling JC, Loewy AD. Aldosterone-sensitive neurons in the nucleus of the solitary tract: bidirectional connections with the central nucleus of the amygdala. J Comp Neurol 497: 646–657, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Geerling JC, Loewy AD. Aldosterone-sensitive neurons in the nucleus of the solitary tract: efferent projections. J Comp Neurol 497: 223–250, 2006 [DOI] [PubMed] [Google Scholar]
- 225. Gelding SV, Taylor NF, Wood PJ, Noonan K, Weaver JU, Wood DF, Monson JP. The effect of growth hormone replacement therapy on cortisol-cortisone interconversion in hypopituitary adults: evidence for growth hormone modulation of extrarenal 11 beta-hydroxysteroid dehydrogenase activity. Clin Endocrinol 48: 153–162, 1998 [DOI] [PubMed] [Google Scholar]
- 226. Gelernter-Yaniv L, Feng N, Sebring NG, Hochberg Z, Yanovski JA. Associations between a polymorphism in the 11 beta hydroxysteroid dehydrogenase type I gene and body composition. Int J Obes Relat Metab Disord 27: 983–986, 2003 [DOI] [PubMed] [Google Scholar]
- 227. Gennari-Moser C, Khankin EV, Schuller S, Escher G, Frey BM, Portmann CB, Baumann MU, Lehmann AD, Surbek D, Karumanchi SA, Frey FJ, Mohaupt MG. Regulation of placental growth by aldosterone and cortisol. Endocrinology 152: 263–271, 2011 [DOI] [PubMed] [Google Scholar]
- 228. Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeold A, Bianco AC. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev 29: 898–938, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Giannopoulos G, Jackson K, Tulchinsky D. Glucocorticoid metabolism in human-placenta, decidua, myometrium and fetal membranes. J Steroid Biochem Mol Biol 17: 371–374, 1982 [DOI] [PubMed] [Google Scholar]
- 230. Giles KM, Ross K, Rossi AG, Hotchin NA, Haslett C, Dransfield I. Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J Immunol 167: 976–986, 2001 [DOI] [PubMed] [Google Scholar]
- 231. Gilmour JS, Coutinho AE, Cailhier JF, Man TY, Clay M, Thomas G, Harris HJ, Mullins JJ, Seckl JR, Savill JS, Chapman KE. Local amplification of glucocorticoids by 11β-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J Immunol 176: 7605–7611, 2006 [DOI] [PubMed] [Google Scholar]
- 232. Glock GE, McLean P. Levels of oxidized and reduced diphosphopyridine nucleotide and triphosphopyridine nucleotide in animal tissues. Biochem J 61: 388–390, 1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Gluckman PD, Hanson MA, Beedle AS. Early life events and their consequences for later disease: a life history and evolutionary perspective. Am J Hum Biol 19: 1–19, 2007 [DOI] [PubMed] [Google Scholar]
- 234. Goedecke JH, Wake DJ, Levitt NS, Lambert EV, Collins MR, Morton NM, Andrew R, Seckl JR, Walker BR. Glucocorticoid metabolism within superficial subcutaneous rather than visceral adipose tissue is associated with features of the metabolic syndrome in South African women. Clin Endocrinol 65: 81–87, 2006 [DOI] [PubMed] [Google Scholar]
- 235. Goldberg GS, Moreno AP, Bechberger JF, Hearn SS, Shivers RR, MacPhee DJ, Zhang YC, Naus CC. Evidence that disruption of connexon particle arrangements in gap junction plaques is associated with inhibition of gap junctional communication by a glycyrrhetinic acid derivative. Exp Cell Res 222: 48–53, 1996 [DOI] [PubMed] [Google Scholar]
- 236. Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol Endocrinol Metab 262: E96–E99, 1992 [DOI] [PubMed] [Google Scholar]
- 237. Gomez-Sanchez EP, Fort CM, Gomez-Sanchez CE. Intracerebroventricular infusion of RU28318 blocks aldosterone-salt hypertension. Am J Physiol Endocrinol Metab 258: E482–E484, 1990 [DOI] [PubMed] [Google Scholar]
- 238. Gomez-Sanchez EP, Ganjam V, Chen YJ, Cox DL, Zhou MY, Thanigaraj S, Gomez-Sanchez CE. The sheep kidney contains a novel unidirectional, high affinity NADP+-dependent 11 beta-hydroxysteroid dehydrogenase (11 beta-HSD-3). Steroids 62: 444–450, 1997 [DOI] [PubMed] [Google Scholar]
- 239. Gomez-Sanchez EP, Ganjam V, Chen YJ, Liu Y, Zhou MY, Toroslu C, Romero DG, Hughson MD, de Rodriguez A, Gomez-Sanchez CE. Regulation of 11β-hydroxysteroid dehydrogenase enzymes in the rat kidney by estradiol. Am J Physiol Endocrinol Metab 285: E272–E279, 2003 [DOI] [PubMed] [Google Scholar]
- 240. Gomez-Sanchez EP, Gomez-Sanchez CE. Central hypertensinogenic effects of glycyrrhizic acid and carbenoxolone. Am J Physiol Endocrinol Metab 263: E1125–E1130, 1992 [DOI] [PubMed] [Google Scholar]
- 241. Gomez-Sanchez EP, Romero DG, de Rodriguez AF, Warden MP, Krozowski Z, Gomez-Sanchez CE. Hexose-6-phosphate dehydrogenase and 11beta-hydroxysteroid dehydrogenase-1 tissue distribution in the rat. Endocrinology 149: 525–533, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Gomez-Sanchez EP, Venkataraman MT, Thwaites D, Fort C. ICV infusion of corticosterone antagonizes ICV-aldosterone hypertension. Am J Physiol Endocrinol Metab 258: E649–E653, 1990 [DOI] [PubMed] [Google Scholar]
- 243. Gottfried-Blackmore A, Sierra A, McEwen BS, Ge R, Bulloch K. Microglia express functional 11 beta-hydroxysteroid dehydrogenase type 1. Glia 58: 1257–1266, 2010 [DOI] [PubMed] [Google Scholar]
- 244. Gout J, Tirard J, Thevenon C, Riou JP, Begeot M, Naville D. CCAAT/enhancer-binding proteins (C/EBPs) regulate the basal and cAMP-induced transcription of the human 11β-hydroxysteroid dehydrogenase encoding gene in adipose cells. Biochimie 88: 1115–1124, 2006 [DOI] [PubMed] [Google Scholar]
- 245. Green H, Kehinde O. Sublines of mouse 3T3 cells that accumulate lipid. Cell 1: 113–116, 1974 [Google Scholar]
- 246. Green H, Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell 3: 127–133, 1974 [DOI] [PubMed] [Google Scholar]
- 247. Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer's disease. J Neurosci 26: 9047–9056, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Gremlich S, Roduit R, Thorens B. Dexamethasone induces posttranslational degradation of GLUT2 and inhibition of insulin secretion in isolated pancreatic beta cells–comparison with the effects of fatty acids. J Biol Chem 272: 3216–3222, 1997 [DOI] [PubMed] [Google Scholar]
- 249. Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J, Feldman RD. GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension 57: 442–451, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Grosser BI. 11-beta-Hydroxysteroid metabolism by mouse brain and glioma 261. J Neurochem 13: 475–478, 1966 [DOI] [PubMed] [Google Scholar]
- 251. Grosser BI, Axelrod LR. Conversion of cortisol to cortisol acetate, cortisone acetate and cortisone by the developing primate brain. Steroids 11: 827–836, 1968 [DOI] [PubMed] [Google Scholar]
- 252. Guan X, Wilson S, Schlender KK, Ruch RJ. Gap-junction disassembly and connexin 43 dephosphorylation induced by 18 beta-glycyrrhetinic acid. Mol Carcinog 16: 157–164, 1996 [DOI] [PubMed] [Google Scholar]
- 253. Guo C, Ricchiuti V, Lian BQ, Yao TM, Coutinho P, Romero JR, Li J, Williams GH, Adler GK. Mineralocorticoid receptor blockade reverses obesity-related changes in expression of adiponectin, peroxisome proliferator-activated receptor-gamma, and proinflammatory adipokines. Circulation 117: 2253–2261, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Guo Y, Martinez-Williams C, Gilbert KA, Rannels DE. Inhibition of gap junction communication in alveolar epithelial cells by 18alpha-glycyrrhetinic acid. Am J Physiol Lung Cell Mol Physiol 276: L1018–L1026, 1999 [DOI] [PubMed] [Google Scholar]
- 255. Guzman C, Cabrera R, Cardenas M, Larrea F, Nathanielsz PW, Zambrano E. Protein restriction during fetal and neonatal development in the rat alters reproductive function and accelerates reproductive ageing in female progeny. J Physiol 572: 97–108, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Haas CS, Creighton CJ, Pi X, Maine I, Koch AE, Haines GK, Ling S, Chinnaiyan AM, Holoshitz J. Identification of genes modulated in rheumatoid arthritis using complementary DNA microarray analysis of lymphoblastoid B cell lines from disease-discordant monozygotic twins. Arthritis Rheum 54: 2047–2060, 2006 [DOI] [PubMed] [Google Scholar]
- 257. Hadoke PW, Christy C, Kotelevtsev YV, Williams BC, Kenyon CJ, Seckl JR, Mullins JJ, Walker BR. Endothelial cell dysfunction in mice after transgenic knockout of type 2, but not type 1, 11β-hydroxysteroid dehydrogenase. Circulation 104: 2832–2837, 2001 [DOI] [PubMed] [Google Scholar]
- 258. Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 29: 4467–4483, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Hammami MM, Siiteri PK. Regulation of 11 beta-hydroxysteroid dehydrogenase activity in human skin fibroblasts: enzymatic modulation of glucocorticoid action. J Clin Endocrinol Metab 73: 326–334, 1991 [DOI] [PubMed] [Google Scholar]
- 260. Handoko K, Yang K, Strutt B, Khalil W, Killinger D. Insulin attenuates the stimulatory effects of tumor necrosis factor alpha on 11beta-hydroxysteroid dehydrogenase 1 in human adipose stromal cells. J Steroid Biochem Mol Biol 72: 163–168, 2000 [DOI] [PubMed] [Google Scholar]
- 261. Hardy DB, Pereria LE, Yang K. Prostaglandins and leukotriene B4 are potent inhibitors of 11beta-hydroxysteroid dehydrogenase type 2 activity in human choriocarcinoma JEG-3 cells. Biol Reprod 61: 40–45, 1999 [DOI] [PubMed] [Google Scholar]
- 262. Hardy DB, Yang K. The expression of 11 beta-hydroxysteroid dehydrogenase type 2 is induced during trophoblast differentiation: effects of hypoxia. J Clin Endocrinol Metab 87: 3696–3701, 2002 [DOI] [PubMed] [Google Scholar]
- 263. Hardy RS, Filer A, Cooper MS, Parsonage G, Raza K, Hardie DL, Rabbitt EH, Stewart PM, Buckley CD, Hewison M. Differential expression, function and response to inflammatory stimuli of 11β-hydroxysteroid dehydrogenase type 1 in human fibroblasts: a mechanism for tissue-specific regulation of inflammation. Arthritis Res Ther 8: R108, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Hardy RS, Rabbitt EH, Filer A, Emery P, Hewison M, Stewart PM, Gittoes N, Buckley CD, Raza K, Cooper MS. Local and systemic glucocorticoid metabolism in inflammatory arthritis. Ann Rheum Dis 67: 1204–1210, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Harris HJ, Kotelevtsev Y, Mullins JJ, Seckl JR, Holmes MC. Intracellular regeneration of glucocorticoids by 11beta-hydroxysteroid dehydrogenase (11beta-HSD)-1 plays a key role in regulation of the hypothalamic-pituitary-adrenal axis: analysis of 11beta-HSD-1-deficient mice. Endocrinology 142: 114–120, 2001 [DOI] [PubMed] [Google Scholar]
- 266. He J, Gu D, Kelly TN, Hixson JE, Rao DC, Jaquish CE, Chen J, Zhao Q, Gu C, Huang J, Shimmin LC, Chen JC, Mu J, Ji X, Liu DP, Whelton PK. Genetic variants in the renin-angiotensin-aldosterone system and blood pressure responses to potassium intake. J Hypertens 29: 1719–1730, 2011 [DOI] [PubMed] [Google Scholar]
- 267. Head GA, Obeyesekere VR, Jones ME, Simpson ER, Krozowski ZS. Aromatase-deficient (ArKO) mice have reduced blood pressure and baroreflex sensitivity. Endocrinology 145: 4286–4291, 2004 [DOI] [PubMed] [Google Scholar]
- 268. Heidbrink C, Hausler SF, Buttmann M, Ossadnik M, Strik HM, Keller A, Buck D, Verbraak E, van Meurs M, Krockenberger M, Mehling M, Mittelbronn M, Laman JD, Wiendl H, Wischhusen J. Reduced cortisol levels in cerebrospinal fluid and differential distribution of 11beta-hydroxysteroid dehydrogenases in multiple sclerosis: implications for lesion pathogenesis. Brain Behav Immun 24: 975–984, 2010 [DOI] [PubMed] [Google Scholar]
- 269. Heine VM, Rowitch DH. Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11betaHSD2-dependent mechanism. J Clin Invest 119: 267–277, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Heiniger CD, Kostadinova RM, Rochat MK, Serra A, Ferrari P, Dick B, Frey BM, Frey FJ. Hypoxia causes down-regulation of 11 beta-hydroxysteroid dehydrogenase type 2 by induction of Egr-1. FASEB J 17: 917–919, 2003 [DOI] [PubMed] [Google Scholar]
- 271. Heiniger CD, Rochat MK, Frey FJ, Frey BM. TNF-alpha enhances intracellular glucocorticoid availability. FEBS Lett 507: 351–356, 2001 [DOI] [PubMed] [Google Scholar]
- 272. Hellman L, Bradlow HL, Zumoff B, Gallagher TF. The influence of thyroid hormone on hydrocortisone production and metabolism. J Clin Endocrinol Metab 21: 1231–1247, 1961 [DOI] [PubMed] [Google Scholar]
- 273. Hench PS, Kendall EC, Slocumb CH, Polley HF. Effects of cortisone acetate and pituitary ACTH on rheumatoid arthritis, rheumatic fever and certain other conditions: a study in clinical physiology. Arch Internal Med 85: 545–666, 1950 [DOI] [PubMed] [Google Scholar]
- 274. Hench PS, Kendall EC, Slocumb CH, Polley HF. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone-compound-E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis–preliminary report. Proc Staff Meetings Mayo Clinic 24: 181–197, 1949 [PubMed] [Google Scholar]
- 275. Hench PS, Slocumb CH, Barnes AR, Smith HL, Polley HF, Kendall EC. The effects of the adrenal cortical hormone 17-hydroxy-11-dehydrocorticosterone (compound-E) on the acute phase of rheumatic fever–preliminary report. Proc Staff Meetings Mayo Clinic 24: 277–297, 1949 [PubMed] [Google Scholar]
- 276. Hennebert O, Chalbot S, Alran S, Morfin R. Dehydroepiandrosterone 7alpha-hydroxylation in human tissues: possible interference with type 1 11beta-hydroxysteroid dehydrogenase-mediated processes. J Steroid Biochem Mol Biol 104: 326–333, 2007 [DOI] [PubMed] [Google Scholar]
- 277. Hennebert O, Le Mee S, Pernelle C, Morfin R. 5Alpha-androstane-3beta,7alpha,17beta-triol and 5alpha-androstane-3beta,7beta,17beta-triol as substrates for the human 11beta-hydroxysteroid dehydrogenase type 1. Steroids 72: 855–864, 2007 [DOI] [PubMed] [Google Scholar]
- 278. Hennebert O, Pernelle C, Ferroud C, Morfin R. 7Alpha- and 7beta-hydroxy-epiandrosterone as substrates and inhibitors for the human 11beta-hydroxysteroid dehydrogenase type 1. J Steroid Biochem Mol Biol 105: 159–165, 2007 [DOI] [PubMed] [Google Scholar]
- 279. Hennebold JD, Mu HH, Poynter ME, Chen XP, Daynes RA. Active catabolism of glucocorticoids by 11β-hydroxysteroid dehydrogenase in vivo is a necessary requirement for natural resistance to infection with Listeria monocytogenes. Int Immunol 9: 105–115, 1997 [DOI] [PubMed] [Google Scholar]
- 280. Hennebold JD, Ryu SY, Mu HH, Galbraith A, Daynes RA. 11β-Hydroxysteroid dehydrogenase modulation of glucocorticoid activities in lymphoid organs. Am J Physiol Regul Integr Comp Physiol 270: R1296–R1306, 1996 [DOI] [PubMed] [Google Scholar]
- 281. Henschkowski J, Stuck AE, Frey BM, Gillmann G, Dick B, Frey FJ, Mohaupt MG. Age-dependent decrease in 11beta-hydroxysteroid dehydrogenase type 2 (11beta-HSD2) activity in hypertensive patients. Am J Hypertens 21: 644–649, 2008 [DOI] [PubMed] [Google Scholar]
- 282. Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, Le Grand CB, Li Z, Metzger JM, Mundt SS, Noonan H, Nunes CN, Olson SH, Pikounis B, Ren N, Robertson N, Schaeffer JM, Shah K, Springer MS, Strack AM, Strowski M, Wu K, Wu T, Xiao J, Zhang BB, Wright SD, Thieringer R. 11beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med 202: 517–527, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Hermanowski-Vosatka A, Gerhold D, Mundt SS, Loving VA, Lu M, Chen Y, Elbrecht A, Wu M, Doebber T, Kelly L, Milot D, Guo Q, Wang PR, Ippolito M, Chao YS, Wright SD, Thieringer R. PPARalpha agonists reduce 11beta-hydroxysteroid dehydrogenase type 1 in the liver. Biochem Biophys Res Commun 279: 330–336, 2000 [DOI] [PubMed] [Google Scholar]
- 284. Heuer H, Visser TJ. Minireview: pathophysiological importance of thyroid hormone transporters. Endocrinology 150: 1078–1083, 2009 [DOI] [PubMed] [Google Scholar]
- 285. Hirasawa G, Sasano H, Suzuki T, Takeyama T, Muramatu Y, Fukushima K, Hiwatashi N, Toyota T, Nagura H, Krozowski ZS. 11β-Hydroxysteroid dehydrogenase type 2 and mineralocorticoid receptor in human fetal development. J Clin Endocrinol Metab 84: 1453–1458, 1999 [DOI] [PubMed] [Google Scholar]
- 286. Hollander JL, Brown EM, Jessar RA, Brown CY. Hydrocortisone and cortisone injected into arthritic joints: comparative effects of and use of hydrocortisone as a local antiarthritic agent. JAMA 147: 1629–1635, 1951 [DOI] [PubMed] [Google Scholar]
- 287. Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318: 635–641, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Holmes MC, Abrahamsen CT, French KL, Paterson JM, Mullins JJ, Seckl JR. The mother or the fetus? 11Beta-hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J Neurosci 26: 3840–3844, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Holmes MC, Carter RN, Noble J, Chitnis S, Dutia A, Paterson JM, Mullins JJ, Seckl JR, Yau JL. 11Beta-hydroxysteroid dehydrogenase type 1 expression is increased in the aged mouse hippocampus and parietal cortex and causes memory impairments. J Neurosci 30: 6916–6920, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Holmes MC, Sangra M, French KL, Whittle IR, Paterson J, Mullins JJ, Seckl JR. 11beta-Hydroxysteroid dehydrogenase type 2 protects the neonatal cerebellum from deleterious effects of glucocorticoids. Neuroscience 137: 865–873, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Holmes MC, Seckl JR. The role of 11beta-hydroxysteroid dehydrogenases in the brain. Mol Cell Endocrinol 248: 9–14, 2006 [DOI] [PubMed] [Google Scholar]
- 292. Holmes MC, Yau JLW, French KL, Seckl JR. The effect of adrenalectomy on 5-hydroxytryptamine and corticosteroid receptor subtype messenger RNA expression in rat hippocampus. Neuroscience 64: 327–337, 1995 [DOI] [PubMed] [Google Scholar]
- 293. Homan A, Guan H, Hardy DB, Gratton RJ, Yang K. Hypoxia blocks 11beta-hydroxysteroid dehydrogenase type 2 induction in human trophoblast cells during differentiation by a time-dependent mechanism that involves both translation and transcription. Placenta 27: 832–840, 2006 [DOI] [PubMed] [Google Scholar]
- 294. Horigome H, Horigome A, Homma M, Hirano T, Oka K. Glycyrrhetinic acid-induced apoptosis in thymocytes: impact of 11β-hydroxysteroid dehydrogenase inhibition. Am J Physiol Endocrinol Metab 277: E624–E630, 1999 [DOI] [PubMed] [Google Scholar]
- 295. Hosfield DJ, Wu Y, Skene RJ, Hilgers M, Jennings A, Snell GP, Aertgeerts K. Conformational flexibility in crystal structures of human 11β-hydroxysteroid dehydrogenase type I provide insights into glucocorticoid interconversion and enzyme regulation. J Biol Chem 280: 4639–4648, 2005 [DOI] [PubMed] [Google Scholar]
- 296. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, Shimomura I. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56: 901–911, 2007 [DOI] [PubMed] [Google Scholar]
- 297. Hotamisligil GS. Inflammation and metabolic disorders. Nature 444: 860–867, 2006 [DOI] [PubMed] [Google Scholar]
- 298. Hu A, Fatma S, Cao J, Grunstein JS, Nino G, Grumbach Y, Grunstein MM. Th2 cytokine-induced upregulation of 11beta-hydroxysteroid dehydrogenase-1 facilitates glucocorticoid suppression of proasthmatic airway smooth muscle function. Am J Physiol Lung Cell Mol Physiol 296: L790–L803, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Huang C, Wan B, Gao B, Hexige S, Yu L. Isolation and characterization of novel human short-chain dehydrogenase/reductase SCDR10B which is highly expressed in the brain and acts as hydroxysteroid dehydrogenase. Acta Biochim Pol 56: 279–289, 2009 [PubMed] [Google Scholar]
- 300. Huang ST, Vo KC, Lyell DJ, Faessen GH, Tulac S, Tibshirani R, Giaccia AJ, Giudice LC. Developmental response to hypoxia. FASEB J 18: 1348–1365, 2004 [DOI] [PubMed] [Google Scholar]
- 301. Huang WL, Beazley LD, Quinlivan JA, Evans SF, Newnham JP, Dunlop SA. Effect of corticosteroids on brain growth in fetal sheep. Obstetrics Gynecol 94: 213–218, 1999 [DOI] [PubMed] [Google Scholar]
- 302. Huang WL, Harper CG, Evans SF, Newnham JP, Dunlop SA. Repeated prenatal corticosteroid administration delays astrocyte and capillary tight junction maturation in fetal sheep. Int J Dev Neurosci 19: 487–493, 2001 [DOI] [PubMed] [Google Scholar]
- 303. Hughes KA, Reynolds RM, Andrew R, Critchley HO, Walker BR. Glucocorticoids turn over slowly in human adipose tissue in vivo. J Clin Endocrinol Metab 95: 4696–4702, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Hult M, Elleby B, Shafqat N, Svensson S, Rane A, Jornvall H, Abrahmsen L, Oppermann U. Human and rodent type 1 11beta-hydroxysteroid dehydrogenases are 7beta-hydroxycholesterol dehydrogenases involved in oxysterol metabolism. Cell Mol Life Sci 61: 992–999, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Hult M, Jornvall H, Oppermann UC. Selective inhibition of human type 1 11beta-hydroxysteroid dehydrogenase by synthetic steroids and xenobiotics. FEBS Lett 441: 25–28, 1998 [DOI] [PubMed] [Google Scholar]
- 306. Hult M, Nobel CS, Abrahmsen L, Nicoll-Griffith DA, Jornvall H, Oppermann UC. Novel enzymological profiles of human 11beta-hydroxysteroid dehydrogenase type 1. Chem Biol Interact 130–132: 805–814, 2001 [DOI] [PubMed] [Google Scholar]
- 307. Hundertmark S, Buhler H, Fromm M, Kruner-Gareis B, Kruner M, Ragosch V, Kuhlmann K, Seckl JR. Ontogeny of 11beta-hydroxysteroid dehydrogenase: activity in the placenta, kidney, colon of fetal rats and rabbits. Horm Metab Res 33: 78–83, 2001 [DOI] [PubMed] [Google Scholar]
- 308. Hundertmark S, Buhler H, Rudolf M, Weitzel HK, Ragosch V. Inhibition of 11β-hydroxysteroid dehydrogenase activity enhances the antiproliferative effect of glucocorticosteroids on MCF-7 and ZR-75– J Endocrinol 155: 171–180, 1997 [DOI] [PubMed] [Google Scholar]
- 309. Hundertmark S, Dill A, Buhler H, Stevens P, Looman K, Ragosch V, Seckl JR, Lipka C. 11β-Hydroxysteroid dehydrogenase type 1: a new regulator of fetal lung maturation. Horm Metab Res 34: 537–544, 2002 [DOI] [PubMed] [Google Scholar]
- 310. Hundertmark S, Dill A, Ebert A, Zimmermann B, Kotelevtsev YV, Mullins JJ, Seckl JR. Foetal lung maturation in 11beta-hydroxysteroid dehydrogenase type 1 knockout mice. Horm Metab Res 34: 545–549, 2002 [DOI] [PubMed] [Google Scholar]
- 311. Ichikawa Y, Yoshida K, Kawagoe M, Saito E, Abe Y, Arikawa K, Homma M. Altered equilibrium between cortisol and cortisone in plasma in thyroid dysfunction and inflammatory diseases. Metabolism 26: 989–997, 1977 [DOI] [PubMed] [Google Scholar]
- 312. Ignatova ID, Kostadinova RM, Goldring CE, Nawrocki AR, Frey FJ, Frey BM. Tumor necrosis factor-α upregulates 11β-hydroxysteroid dehydrogenase type 1 expression by CCAAT/enhancer binding protein-β in HepG2 cells. Am J Physiol Endocrinol Metab 296: E367–E377, 2009 [DOI] [PubMed] [Google Scholar]
- 313. Iki K, Miyamori I, Hatakeyama H, Yoneda T, Takeda Y, Takeda R, Dai QL. The activities of 5-beta-reductase and 11-beta-hydroxysteroid dehydrogenase in essential-hypertension. Steroids 59: 656–660, 1994 [DOI] [PubMed] [Google Scholar]
- 314. Iqbal J, Macdonald L, Yau C, Walker B, Hadoke P. Effect of intracellular glucocorticoid metabolism on neointimal proliferation in a mouse model of wire angioplasty. Heart 97: 1–1, 2011. 21071559 [Google Scholar]
- 315. Jamieson A, Wallace AM, Andrew R, Nunez BS, Walker BR, Fraser R, White PC, Connell JM. Apparent cortisone reductase deficiency: a functional defect in 11β-hydroxysteroid dehydrogenase type 1. J Clin Endocrinol Metab 84: 3570–3574, 1999 [DOI] [PubMed] [Google Scholar]
- 316. Jamieson PM, Chapman KE, Edwards CR, Seckl JR. 11 Beta-hydroxysteroid dehydrogenase is an exclusive 11 beta-reductase in primary cultures of rat hepatocytes: effect of physicochemical and hormonal manipulations. Endocrinology 136: 4754–4761, 1995 [DOI] [PubMed] [Google Scholar]
- 317. Jamieson PM, Chapman KE, Seckl JR. Tissue- and temporal-specific regulation of 11beta-hydroxysteroid dehydrogenase type 1 by glucocorticoids in vivo. J Steroid Biochem Mol Biol 68: 245–250, 1999 [DOI] [PubMed] [Google Scholar]
- 318. Jamieson PM, Nyirenda MJ, Walker BR, Chapman KE, Seckl JR. Interactions between oestradiol and glucocorticoid regulatory effects on liver-specific glucocorticoid genes: possible evidence for a role of hepatic 11β-hydroxysteroid dehydrogenase type 1. J Endocrinol 160: 103–109, 1999 [DOI] [PubMed] [Google Scholar]
- 319. Jamieson PM, Walker BR, Chapman KE, Andrew R, Rossiter S, Seckl JR. 11β-Hydroxysteroid dehydrogenase type 1 is a predominant 11β-reductase in the intact perfused rat liver. J Endocrinol 165: 685–692, 2000 [DOI] [PubMed] [Google Scholar]
- 320. Janiak PC, Lewis SJ, Brody MJ. Role of central mineralocorticoid binding sites in development of hypertension. Am J Physiol Regul Integr Comp Physiol 259: R1025–R1034, 1990 [DOI] [PubMed] [Google Scholar]
- 321. Jenkins JS. The metabolism of cortisol by human extra-hepatic tissues. J Endocrinol 34: 51–56, 1966 [DOI] [PubMed] [Google Scholar]
- 322. Jensen E, Wood CE, Keller-Wood M. Reduction of maternal adrenal steroids results in increased VEGF protein without increased eNOS in the ovine placenta. Placenta 28: 658–667, 2007 [DOI] [PubMed] [Google Scholar]
- 323. Jensen EC, Gallaher BW, Breier BH, Harding JE. The effect of a chronic maternal cortisol infusion on the late-gestation fetal sheep. J Endocrinol 174: 27–36, 2002 [DOI] [PubMed] [Google Scholar]
- 324. Jiang JQ, Wang DS, Senthilkumaran B, Kobayashi T, Kobayashi HK, Yamaguchi A, Ge W, Young G, Nagahama Y. Isolation, characterization and expression of 11beta-hydroxysteroid dehydrogenase type 2 cDNAs from the testes of Japanese eel (Anguilla japonica) and Nile tilapia (Oreochromis niloticus). J Mol Endocrinol 31: 305–315, 2003 [DOI] [PubMed] [Google Scholar]
- 325. Joels M, de Kloet ER. Control of neuronal excitability by corticosteroid hormones. Trends Neurosci 15: 25–30, 1992 [DOI] [PubMed] [Google Scholar]
- 326. Joganathan V, Al-Hakami A, Rauz S, Stewart PM, Wallace GR, Bujalska IJ. Local cortisol generation by human macrophage subsets by 11β-hydroxysteroid dehydrogenase type 1 enzyme and its role in ocular immune privilege. Endocrine Abstr 15: OC30, 2008 [Google Scholar]
- 327. Johansson A, Andrew R, Forsberg H, Cederquist K, Walker BR, Olsson T. Glucocorticoid metabolism and adrenocortical reactivity to ACTH in myotonic dystrophy. J Clin Endocrinol Metab 86: 4276–4283, 2001 [DOI] [PubMed] [Google Scholar]
- 328. Johnstone JF, Bocking AD, Unlugedik E, Challis JR. The effects of chorioamnionitis and betamethasone on 11beta hydroxysteroid dehydrogenase types 1 and 2 and the glucocorticoid receptor in preterm human placenta. J Soc Gynecol Investig 12: 238–245, 2005 [DOI] [PubMed] [Google Scholar]
- 329. Jonas KC, Chandras C, Abayasekara DR, Michael AE. Role for prostaglandins in the regulation of type 1 11β-hydroxysteroid dehydrogenase in human granulosa-lutein cells. Endocrinology 147: 5865–5872, 2006 [DOI] [PubMed] [Google Scholar]
- 330. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet 21: 163–167, 1999 [DOI] [PubMed] [Google Scholar]
- 331. Jornvall H, Woenckhaus C, Sch9attle E, Jeck R. Modification of alcohol dehydrogenases with two NAD+-analogues containing reactive substituents on the functional side of the molecule. FEBS Lett 54: 297–301, 1975 [DOI] [PubMed] [Google Scholar]
- 332. Julan L, Guan H, van Beek JP, Yang K. Peroxisome proliferator-activated receptor delta suppresses 11beta-hydroxysteroid dehydrogenase type 2 gene expression in human placental trophoblast cells. Endocrinology 146: 1482–1490, 2005 [DOI] [PubMed] [Google Scholar]
- 333. Justesen J, Mosekilde L, Holmes M, Stenderup K, Gasser J, Mullins JJ, Seckl JR, Kassem M. Mice deficient in 11β-hydroxysteroid dehydrogenase type 1 lack bone marrow adipocytes, but maintain normal bone formation. Endocrinology 145: 1916–1925, 2004 [DOI] [PubMed] [Google Scholar]
- 334. Kajantie E, Dunkel L, Turpeinen U, Stenman UH, Wood PJ, Nuutila M, Andersson S. Placental 11 beta-hydroxysteroid dehydrogenase-2 and fetal cortisol/cortisone shuttle in small preterm infants. J Clin Endocrinol Metab 88: 493–500, 2003 [DOI] [PubMed] [Google Scholar]
- 335. Kapoor A, Kostaki A, Janus C, Matthews SG. The effects of prenatal stress on learning in adult offspring is dependent on the timing of the stressor. Behav Brain Res 197: 144–149, 2009 [DOI] [PubMed] [Google Scholar]
- 336. Kardon T, Senesi S, Marcolongo P, Legeza B, Banhegyi G, Mandl J, Fulceri R, Benedetti A. Maintenance of luminal NADPH in the endoplasmic reticulum promotes the survival of human neutrophil granulocytes. FEBS Lett 582: 1809–1815, 2008 [DOI] [PubMed] [Google Scholar]
- 337. Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci USA 102: 19204–19207, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Kaur K, Hardy R, Ahasan MM, Eijken M, van Leeuwen JP, Filer A, Thomas AM, Raza K, Buckley CD, Stewart PM, Rabbitt EH, Hewison M, Cooper MS. Synergistic induction of local glucocorticoid generation by inflammatory cytokines and glucocorticoids: implications for inflammation associated bone loss. Ann Rheumatic Dis 69: 1185–1190, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Kavanagh K, Wu X, Svensson S, Elleby B, Von Delft F, Debreczeni JE, Sharma S, Bray J, Edwards A, Arrowsmith C, Sundstrom M, Abrahmsen L, Oppermann U. Structure of human 11β-hydroxysteroid dehydrogenase in complex with NADP and carbenoxolone. PDB 2BEL, 2006. 10.2210/pdb2bel/pdb [DOI] [Google Scholar]
- 340. Kenneke JF, Mazur CS, Ritger SE, Sack TJ. Mechanistic investigation of the noncytochrome P450-mediated metabolism of triadimefon to triadimenol in hepatic microsomes. Chem Res Toxicol 21: 1997–2004, 2008 [DOI] [PubMed] [Google Scholar]
- 341. Kereszturi É, Kálmán FS, Kardon T, Csala M, Bánhegyi G. Decreased prereceptorial glucocorticoid activating capacity in starvation due to an oxidative shift of pyridine nucleotides in the endoplasmic reticulum. FEBS Lett 584: 4703–4708, 2010 [DOI] [PubMed] [Google Scholar]
- 342. Kershaw EE, Morton NM, Dhillon H, Ramage L, Seckl JR, Flier JS. Adipocyte-specific glucocorticoid inactivation protects against diet-induced obesity. Diabetes 54: 1023–1031, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Kerzner LS, Stonestreet BS, Wu KY, Sadowska G, Malee MP. Antenatal dexamethasone: effect on ovine placental 11beta-hydroxysteroid dehydrogenase type 2 expression and fetal growth. Pediatr Res 52: 706–712, 2002 [DOI] [PubMed] [Google Scholar]
- 344. Khorram NM, Magee TR, Wang C, Desai M, Ross M, Khorram O. Maternal undernutrition programs offspring adrenal expression of steroidogenic enzymes. Reprod Sci 18: 931–940, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Kim JH, Jittiwat J, Ong WY, Farooqui AA, Jenner AM. Changes in cholesterol biosynthetic and transport pathways after excitotoxicity. J Neurochem 112: 34–41, 2010 [DOI] [PubMed] [Google Scholar]
- 346. Kipari TMJ, Hadoke PWF, Man T, White C, Walker BR, Savill JS, Chapman KE, Seckl JR. 11β-Hydroxysteroid dehydrogenase type 1 deficiency in bone marrow-derived cells reduces atherosclerosis. FASEB J 27: 1519–1531, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Kipari TMJ, Hadoke PWF, Man T, White C, Walker BR, Savill JS, Chapman KE, Seckl JR. Deficiency of 11 beta-hydroxysteroid dehydrogenase type 1 reduces systemic inflammation and inflammatory cell infiltration in atherosclerotic lesions of ApoE−/− mice. Endocr Rev 31: S8–S8, 2010 [Google Scholar]
- 348. Kitanaka S, Tanae A, Hibi I. Apparent mineralocorticoid excess due to 11 beta-hydroxysteroid dehydrogenase deficiency: a possible cause of intrauterine growth retardation. Clin Endocrinol 44: 353–359, 1996 [DOI] [PubMed] [Google Scholar]
- 349. Kitraki E, Kittas C, Stylianopoulou F. Glucocorticoid receptor gene expression during rat embryogenesis. An in situ hybridization study. Differentiation 62: 21–31, 1997 [DOI] [PubMed] [Google Scholar]
- 350. Klemcke HG, Christenson RK. Porcine placental 11 beta-hydroxysteroid dehydrogenase activity. Biol Reprod 55: 217–223, 1996 [DOI] [PubMed] [Google Scholar]
- 351. Klusonova P, Kucka M, Miksik I, Bryndova J, Pacha J. Chicken 11beta-hydroxysteroid dehydrogenase type 2: partial cloning and tissue distribution. Steroids 73: 348–355, 2008 [DOI] [PubMed] [Google Scholar]
- 352. Klusonova P, Rehakova L, Borchert G, Vagnerova K, Neckar J, Ergang P, Miksik I, Kolar F, Pacha J. Chronic intermittent hypoxia induces 11beta-hydroxysteroid dehydrogenase in rat heart. Endocrinology 150: 4270–4277, 2009 [DOI] [PubMed] [Google Scholar]
- 353. Koerner DR. 11Beta-hydroxysteroid dehydrogenase of lung and testis. Endocrinology 79: 935–938, 1966 [DOI] [PubMed] [Google Scholar]
- 354. Koerner DR, Hellman L. Effect of thyroxine administration on the 11-beta-hydroxysteroid dehydrogenases in rat liver and kidney. Endocrinology 75: 592–601, 1964 [DOI] [PubMed] [Google Scholar]
- 355. Koistinen HA, Forsgren M, Wallberg-Henriksson H, Zierath JR. Insulin action on expression of novel adipose genes in healthy and type 2 diabetic subjects. Obesity Res 12: 25–31, 2004 [DOI] [PubMed] [Google Scholar]
- 356. Kojoma M, Hayashi S, Shibata T, Yamamoto Y, Sekizaki H. Variation of glycyrrhizin and liquiritin contents within a population of 5-year-old licorice (Glycyrrhiza uralensis) plants cultivated under the same conditions. Biol Pharm Bull 34: 1334–1337, 2011 [DOI] [PubMed] [Google Scholar]
- 357. Konopelska S, Kienitz T, Hughes B, Pirlich M, Bauditz J, Lochs H, Strasburger CJ, Stewart PM, Quinkler M. Hepatic 11beta-HSD1 mRNA expression in fatty liver and nonalcoholic steatohepatitis. Clin Endocrinol 70: 554–560, 2009 [DOI] [PubMed] [Google Scholar]
- 358. Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science 242: 715–723, 1988 [DOI] [PubMed] [Google Scholar]
- 359. Korbonits M, Bujalska I, Shimojo M, Nobes J, Jordan S, Grossman AB, Stewart PM. Expression of 11 beta-hydroxysteroid dehydrogenase isoenzymes in the human pituitary: induction of the type 2 enzyme in corticotropinomas and other pituitary tumors. J Clin Endocrinol Metab 86: 2728–2733, 2001 [DOI] [PubMed] [Google Scholar]
- 360. Korte SM. Corticosteroids in relation to fear, anxiety and psychopathology. Neurosci Biobehav 25: 117–142, 2001 [DOI] [PubMed] [Google Scholar]
- 361. Kossintseva I, Wong S, Johnstone E, Guilbert L, Olson DM, Mitchell BF. Proinflammatory cytokines inhibit human placental 11beta-hydroxysteroid dehydrogenase type 2 activity through Ca2+ and cAMP pathways. Am J Physiol Endocrinol Metab 290: E282–E288, 2006 [DOI] [PubMed] [Google Scholar]
- 362. Kostadinova RM, Nawrocki AR, Frey FJ, Frey BM. Tumor necrosis factor alpha and phorbol 12-myristate-13-acetate down-regulate human 11beta-hydroxysteroid dehydrogenase type 2 through p50/p50 NF-kappaB homodimers and Egr-1. FASEB J 19: 650–652, 2005 [DOI] [PubMed] [Google Scholar]
- 363. Kotelevtsev Y, Brown RW, Fleming S, Kenyon C, Edwards CR, Seckl JR, Mullins JJ. Hypertension in mice lacking 11beta-hydroxysteroid dehydrogenase type 2. J Clin Invest 103: 683–689, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, Best R, Brown R, Edwards CR, Seckl JR, Mullins JJ. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA 94: 14924–14929, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Koyama K, Krozowski Z. Modulation of 11 beta-hydroxysteroid dehydrogenase type 2 activity in Ishikawa cells is associated with changes in cellular proliferation. Mol Cell Endocrinol 183: 165–170, 2001 [DOI] [PubMed] [Google Scholar]
- 366. Koyama K, Myles K, Smith R, Krozowski Z. Expression of the 11beta-hydroxysteroid dehydrogenase type II enzyme in breast tumors and modulation of activity and cell growth in PMC42 cells. J Steroid Biochem Mol Biol 76: 153–159, 2001 [DOI] [PubMed] [Google Scholar]
- 367. Kratschmar DV, Vuorinen A, Da Cunha T, Wolber G, Classen-Houben D, Doblhoff O, Schuster D, Odermatt A. Characterization of activity and binding mode of glycyrrhetinic acid derivatives inhibiting 11β-hydroxysteroid dehydrogenase type 2. J Steroid Biochem Mol Biol 125; 129–142, 2011 [DOI] [PubMed] [Google Scholar]
- 368. Krozowski Z, MaGuire JA, Stein-Oakley AN, Dowling J, Smith RE, Andrews RK. Immunohistochemical localization of the 11 beta-hydroxysteroid dehydrogenase type II enzyme in human kidney and placenta. J Clin Endocrinol Metab 80: 2203–2209, 1995 [DOI] [PubMed] [Google Scholar]
- 369. Krozowski Z, Stuchbery S, White P, Monder C, Funder JW. Characterization of 11 beta-hydroxysteroid dehydrogenase gene-expression: identification of multiple unique forms of messenger-ribonucleic-acid in the rat-kidney. Endocrinology 127: 3009–3013, 1990 [DOI] [PubMed] [Google Scholar]
- 370. Krozowski ZS, Funder JW. Renal mineralocorticoid receptors and hippocampal corticosterone-binding species have identical intrinsic steroid specificity. Proc Natl Acad Sci USA 80: 6056–6060, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Ku YH, Koo BK, Kwak SH, Cho YM, Shin HD, Lee HK, Kim Y, Choi JW, Oh B, Park KS. Regulatory effect of common promoter polymorphisms on the expression of the 11beta-hydroxysteroid dehydrogenase type 1 gene. Horm Res 72: 25–32, 2009 [DOI] [PubMed] [Google Scholar]
- 372. Kulstad JJ, McMillan PJ, Leverenz JB, Cook DG, Green PS, Peskind ER, Wilkinson CW, Farris W, Mehta PD, Craft S. Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J Neuropathol Exp Neurolo 64: 139–146, 2005 [DOI] [PubMed] [Google Scholar]
- 373. Kusakabe M, Nakamura I, Young G. 11Beta-hydroxysteroid dehydrogenase complementary deoxyribonucleic acid in rainbow trout: cloning, sites of expression, and seasonal changes in gonads. Endocrinology 144: 2534–2545, 2003 [DOI] [PubMed] [Google Scholar]
- 374. La Fleur SE, Houshyar H, Roy M, Dallman MF. Choice of lard, but not total lard calories, damps adrenocorticotropin responses to restraint. Endocrinology 146: 2193–2199, 2005 [DOI] [PubMed] [Google Scholar]
- 375. La Fleur SE, Manalo SL, Roy M, Houshyar H, Dallman MF. Hepatic vagotomy alters limbic and hypothalamic neuropeptide responses to insulin-dependent diabetes and voluntary lard ingestion. Eur J Neurosci 21: 2733–2742, 2005 [DOI] [PubMed] [Google Scholar]
- 376. LaBorde JB, Hansen DK, Young JF, Sheehan DM, Holson RR. Prenatal dexamethasone exposure in rats: effects of dose, age at exposure, and drug-induced hypophagia on malformations and fetal organ weights. Fundam Appl Toxicol 19: 545–554, 1992 [DOI] [PubMed] [Google Scholar]
- 377. Lakshmi V, Monder C. Evidence for independent 11-oxidase and 11-reductase activities of 11 beta-hydroxysteroid dehydrogenase: enzyme latency, phase transitions, and lipid requirements. Endocrinology 116: 552–560, 1985 [DOI] [PubMed] [Google Scholar]
- 378. Lakshmi V, Monder C. Extraction of 11-beta-hydroxysteroid dehydrogenase from rat-liver microsomes by detergents. J Steroid Biochem Mol Biol 22: 331–340, 1985 [DOI] [PubMed] [Google Scholar]
- 379. Lakshmi V, Monder C. Purification and characterization of the corticosteroid 11 beta-dehydrogenase component of the rat liver 11 beta-hydroxysteroid dehydrogenase complex. Endocrinology 123: 2390–2398, 1988 [DOI] [PubMed] [Google Scholar]
- 380. Lakshmi V, Sakai RR, McEwen BS, Monder C. Regional distribution of 11 beta-hydroxysteroid dehydrogenase in rat brain. Endocrinology 128: 1741–1748, 1991 [DOI] [PubMed] [Google Scholar]
- 381. Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion: an in vitro study of dexamethasone effects in mouse islets. J Clin Invest 99: 414–423, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Landfield PW, Baskin RK, Pitler TA. Brain aging correlates: retardation by hormonal-pharmacological treatments. Science 214: 581–584, 1981 [DOI] [PubMed] [Google Scholar]
- 383. Landfield PW, Thibault O, Mazzanti ML, Porter NM, Kerr DS. Mechanisms of neuronal death in brain aging and Alzheimer's disease: role of endocrine-mediated calcium dyshomeostasis. J Neurobiol 23: 1247–1260, 1992 [DOI] [PubMed] [Google Scholar]
- 384. Langley-Evans SC, Philips G, Benediktsson R, Gardner D, Edwards CRW, Jackson AA, Seckl JR. Maternal dietary protein restriction, placental glucocorticoid metabolism and the programming of hypertension. Placenta 17: 169–172, 1996 [DOI] [PubMed] [Google Scholar]
- 385. Langlois DA, Matthews SG, Yu M, Yang K. Differential expression of 11 beta-hydroxysteroid dehydrogenase 1 and 2 in the developing ovine fetal liver and kidney. J Endocrinol 147: 405–411, 1995 [DOI] [PubMed] [Google Scholar]
- 386. Lathe R. Steroid and sterol 7-hydroxylation: ancient pathways. Steroids 67: 967–977, 2002 [DOI] [PubMed] [Google Scholar]
- 387. Laugero KD, Bell ME, Bhatnagar S, Soriano L, Dallman MF. Sucrose ingestion normalizes central expression of corticotropin-releasing-factor messenger ribonucleic acid and energy balance in adrenalectomized rats: a glucocorticoid-metabolic-brain axis? Endocrinology 142: 2796–2804, 2001 [DOI] [PubMed] [Google Scholar]
- 388. Lavery GG, Ronconi V, Draper N, Rabbitt EH, Lyons V, Chapman KE, Walker EA, McTernan CL, Giacchetti G, Mantero F, Seckl JR, Edwards CR, Connell JM, Hewison M, Stewart PM. Late-onset apparent mineralocorticoid excess caused by novel compound heterozygous mutations in the HSD11B2 gene. Hypertension 42: 123–129, 2003 [DOI] [PubMed] [Google Scholar]
- 389. Lavery GG, Walker EA, Draper N, Jeyasuria P, Marcos J, Shackleton CH, Parker KL, White PC, Stewart PM. Hexose-6-phosphate dehydrogenase knock-out mice lack 11 beta-hydroxysteroid dehydrogenase type 1-mediated glucocorticoid generation. J Biol Chem 281: 6546–6551, 2006 [DOI] [PubMed] [Google Scholar]
- 390. Lavery GG, Walker EA, Tiganescu A, Ride JP, Shackleton CH, Tomlinson JW, Connell JM, Ray DW, Biason-Lauber A, Malunowicz EM, Arlt W, Stewart PM. Steroid biomarkers and genetic studies reveal inactivating mutations in hexose-6-phosphate dehydrogenase in patients with cortisone reductase deficiency. J Clin Endocrinol Metab 93: 3827–3832, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Lavery GG, Walker EA, Turan N, Rogoff D, Ryder JW, Shelton JM, Richardson JA, Falciani F, White PC, Stewart PM, Parker KL, McMillan DR. Deletion of hexose-6-phosphate dehydrogenase activates the unfolded protein response pathway and induces skeletal myopathy. J Biol Chem 283: 8453–8461, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Lavery GG, Zielinska AE, Gathercole LL, Hughes B, Semjonous N, Guest P, Saqib K, Sherlock M, Reynolds G, Morgan SA, Tomlinson JW, Walker EA, Rabbitt EH, Stewart PM. Lack of significant metabolic abnormalities in mice with liver-specific disruption of 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology 153: 3236–3248, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Lawson AJ, Walker EA, Lavery GG, Bujalska IJ, Hughes B, Arlt W, Stewart PM, Ride JP. Cortisone-reductase deficiency associated with heterozygous mutations in 11b-hydroxysteroid dehydrogenase type 1. Proc Natl Acad Sci USA 108: 4111–4116, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Lax ER, Ghraf R, Schriefers H. The hormonal regulation of hepatic microsomal 11beta-hydroxysteroid dehydrogenase activity in the rat. Acta Endocrinol 89: 352–358, 1978 [DOI] [PubMed] [Google Scholar]
- 395. Leckie CM, Chapman KE, Edwards CRW, Seckl JR. LLC-PK1 cells model 11β-hydroxysteroid dehydrogenase type 2 regulation of glucocorticoid access to renal mineralocorticoid receptors. Endocrinology 136: 5561–5569, 1995 [DOI] [PubMed] [Google Scholar]
- 396. Leckie CM, Welberg LA, Seckl JR. 11beta-hydroxysteroid dehydrogenase is a predominant reductase in intact rat Leydig cells. J Endocrinol 159: 233–238, 1998 [DOI] [PubMed] [Google Scholar]
- 397. Lee MJ, Fried SK, Mundt SS, Wang Y, Sullivan S, Stefanni A, Daugherty BL, Hermanowski-Vosatka A. Depot-specific regulation of the conversion of cortisone to cortisol in human adipose tissue. Obesity 16: 1178–1185, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Lesage J, Blondeau B, Grino M, Breant B, Dupouy JP. Maternal undernutrition during late gestation induces fetal overexposure to glucocorticoids and intrauterine growth retardation, and disturbs the hypothalamo-pituitary adrenal axis in the newborn rat. Endocrinology 142: 1692–1702, 2001 [DOI] [PubMed] [Google Scholar]
- 399. Li A, Tedde R, Krozowski ZS, Pala A, Li KX, Shackleton CH, Mantero F, Palermo M, Stewart PM. Molecular basis for hypertension in the “type II variant” of apparent mineralocorticoid excess. Am J Hum Genet 63: 370–379, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Li JN, Ge YC, Yang Z, Guo CM, Duan T, Myatt L, Guan H, Yang K, Sun K. The Sp1 transcription factor is crucial for the expression of 11β-hydroxysteroid dehydrogenase type 2 in human placental trophoblasts. J Clin Endocrinol Metab 2010–2852, 2011 [DOI] [PubMed] [Google Scholar]
- 401. Li KXZ, Smith RE, Ferrari P, Funder JW, Krozowski ZS. Rat 11 beta-hydroxysteroid dehydrogenase type 2 enzyme is expressed at low levels in the placenta and is modulated by adrenal steroids in the kidney. Mol Cell Endocrinol 120: 67–75, 1996 [DOI] [PubMed] [Google Scholar]
- 402. Li RS, Nakagawa Y, Liu YJ, Fujisawa Y, Sai S, Nakanishi T, Chapman KE, Seckl JR, Ohzeki T. Growth hormone inhibits the 11 beta-hydroxysteroid dehydrogenase type 1 gene promoter activity via insulin-like growth factor I in HepG2 cells. Horm Metab Res 40: 286–288, 2008 [DOI] [PubMed] [Google Scholar]
- 403. Li W, Gao L, Wang Y, Duan T, Myatt L, Sun K. Enhancement of cortisol-induced 11beta-hydroxysteroid dehydrogenase type 1 expression by interleukin 1beta in cultured human chorionic trophoblast cells. Endocrinology 147: 2490–2495, 2006 [DOI] [PubMed] [Google Scholar]
- 404. Liedtke WB, McKinley MJ, Walker LL, Zhang H, Pfenning AR, Drago J, Hochendoner SJ, Hilton DL, Lawrence AJ, Denton DA. Relation of addiction genes to hypothalamic gene changes subserving genesis and gratification of a classic instinct, sodium appetite. Proc Natl Acad Sci USA 108: 12509–12514, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Liggins GC. The role of cortisol in preparing the fetus for birth. Reprod Fertil Dev 6: 141–150, 1994 [DOI] [PubMed] [Google Scholar]
- 406. Lightman SL, Conway-Campbell BL. The crucial role of pulsatile activity of the HPA axis for continuous dynamic equilibration. Nature Rev Neurosci 11: 710–718, 2010 [DOI] [PubMed] [Google Scholar]
- 407. Lim HY, Muller N, Herold MJ, van den Brandt J, Reichardt HM. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology 122: 47–53, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Lindsay RS, Lindsay RM, Edwards CR, Seckl JR. Inhibition of 11-beta-hydroxysteroid dehydrogenase in pregnant rats and the programming of blood pressure in the offspring. Hypertension 27: 1200–1204, 1996 [DOI] [PubMed] [Google Scholar]
- 409. Liu Y, Singh RJ, Usa K, Netzel BC, Liang M. Renal medullary 11 beta-hydroxysteroid dehydrogenase type 1 in Dahl salt-sensitive hypertension. Physiol Genomics 36: 52–58, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Liu YJ, Nakagawa Y, Nasuda K, Saegusa H, Igarashi Y. Effect of growth hormone, insulin and dexamethasone on 11β-hydroxysteroid dehydrogenase activity on a primary culture of rat hepatocytes. Life Sci 59: 227–234, 1996 [DOI] [PubMed] [Google Scholar]
- 411. Liu YJ, Nakagawa Y, Ohzeki T. Gene expression of 11beta-hydroxysteroid dehydrogenase type 1 and type 2 in the kidneys of insulin-dependent diabetic rats. Hypertension 31: 885–889, 1998 [DOI] [PubMed] [Google Scholar]
- 412. Liu YQ, Cousin JM, Hughes J, VanDamme J, Seckl JR, Haslett C, Dransfield I, Savill J, Rossi AG. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol 162: 3639–3646, 1999 [PubMed] [Google Scholar]
- 413. Livingstone DE, Walker BR. Is 11beta-hydroxysteroid dehydrogenase type 1 a therapeutic target? Effects of carbenoxolone in lean and obese Zucker rats. J Pharmacol Exp Ther 305: 167–172, 2003 [DOI] [PubMed] [Google Scholar]
- 414. Llorente R, Gallardo ML, Berzal AL, Prada C, Garcia-Segura LM, Viveros MP. Early maternal deprivation in rats induces gender-dependent effects on developing hippocampal and cerebellar cells. Int J Dev Neurosci 27: 233–241, 2009 [DOI] [PubMed] [Google Scholar]
- 415. Lloyd DJ, Helmering J, Cordover D, Bowsman M, Chen M, Hale C, Fordstrom P, Zhou M, Wang M, Kaufman SA, Veniant MM. Antidiabetic effects of 11beta-HSD1 inhibition in a mouse model of combined diabetes, dyslipidaemia and atherosclerosis. Diabetes Obes Metab 11: 688–699, 2009 [DOI] [PubMed] [Google Scholar]
- 416. Lombes M, Kenouch S, Souque A, Farman N, Rafestin-Oblin ME. The mineralocorticoid receptor discriminates aldosterone from glucocorticoids independently of the 11 beta-hydroxysteroid dehydrogenase. Endocrinology 135: 834–840, 1994 [DOI] [PubMed] [Google Scholar]
- 417. Lovati E, Ferrari P, Dick B, Jostarndt K, Frey BM, Frey FJ, Schorr U, Sharma AM. Molecular basis of human salt sensitivity: the role of the 11beta-hydroxysteroid dehydrogenase type 2. J Clin Endocrinol Metab 84: 3745–3749, 1999 [DOI] [PubMed] [Google Scholar]
- 418. Low SC, Assaad SN, Rajan V, Chapman KE, Edwards CR, Seckl JR. Regulation of 11 beta-hydroxysteroid dehydrogenase by sex steroids in vivo: further evidence for the existence of a second dehydrogenase in rat kidney. J Endocrinol 139: 27–35, 1993 [DOI] [PubMed] [Google Scholar]
- 419. Low SC, Chapman KE, Edwards CR, Wells T, Robinson IC, Seckl JR. Sexual dimorphism of hepatic 11 beta-hydroxysteroid dehydrogenase in the rat: the role of growth hormone patterns. J Endocrinol 143: 541–548, 1994 [DOI] [PubMed] [Google Scholar]
- 420. Low SC, Chapman KE, Edwards CRW, Seckl JR. “Liver-type” 11b-hydroxysteroid dehydrogenase cDNA encodes reductase not dehydrogenase activity in intact mammalian COS-7 cells. J Mol Endocrinol 13: 167–174, 1994 [DOI] [PubMed] [Google Scholar]
- 421. Lu L, Zhao G, Luu-The V, Ouellet J, Fan Z, Labrie F, Pelletier G. Expression of 11β-hydroxysteroid dehydrogenase type 1 in breast cancer and adjacent non-malignant tissue. An immunocytochemical study. Pathol Oncol Res: 1–6-6, 2011 [DOI] [PubMed] [Google Scholar]
- 422. Lucassen PJ, Bosch OJ, Jousma E, Kromer SA, Andrew R, Seckl JR, Neumann ID. Prenatal stress reduces postnatal neurogenesis in rats selectively bred for high, but not low, anxiety: possible key role of placental 11beta-hydroxysteroid dehydrogenase type 2. Eur J Neurosci 29: 97–103, 2009 [DOI] [PubMed] [Google Scholar]
- 423. Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP, Thakur M, McEwen BS, Hauger RL, Meaney MJ. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci 1: 69–73, 1998 [DOI] [PubMed] [Google Scholar]
- 424. Ma X, Lian QQ, Dong Q, Ge RS. Environmental inhibitors of 11β-hydroxysteroid dehydrogenase type 2. Toxicology 285: 83–89, 2011 [DOI] [PubMed] [Google Scholar]
- 425. MacLullich AM, Ferguson KJ, Reid LM, Deary IJ, Starr JM, Wardlaw JM, Walker BR, Andrew R, Seckl JR. 11Beta-hydroxysteroid dehydrogenase type 1, brain atrophy and cognitive decline. Neurobiol Aging 33: e201–208, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Man TY, Michailidou Z, Gokcel A, Ramage L, Chapman KE, Kenyon CJ, Seckl JR, Morton NM. Dietary manipulation reveals an unexpected inverse relationship between fat mass and adipose 11beta-hydroxysteroid dehydrogenase type 1. Am J Physiol Endocrinol Metab 300: E1076–E1084, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Manary MJ, Muglia LJ, Vogt SK, Yarasheski KE. Cortisol and its action on the glucocorticoid receptor in malnutrition and acute infection. Metabolism 55: 550–554, 2006 [DOI] [PubMed] [Google Scholar]
- 428. Mandyam CD, Crawford EF, Eisch AJ, Rivier CL, Richardson HN. Stress experienced in utero reduces sexual dichotomies in neurogenesis, microenvironment, and cell death in the adult rat hippocampus. Dev Neurobiol 68: 575–589, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Manning JR, Bailey MA, Soares DC, Dunbar DR, Mullins JJ. In Silico structure-function analysis of pathological variation in the HSD11B2 gene sequence. Physiol Genomics 42: 319–330, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Margittai E, Banhegyi G. Isocitrate dehydrogenase: a NADPH-generating enzyme in the lumen of the endoplasmic reticulum. Arch Biochem Biophys 471: 184–190, 2008 [DOI] [PubMed] [Google Scholar]
- 431. Marik PE. Critical illness-related corticosteroid insufficiency. Chest 135: 181–193, 2009 [DOI] [PubMed] [Google Scholar]
- 432. Martinerie L, Pussard E, Meduri G, Delezoide AL, Boileau P, Lombes M. Lack of Renal 11 beta-hydroxysteroid dehydrogenase type 2 at birth, a targeted temporal window for neonatal glucocorticoid action in human and mice. PLoS ONE 7: e31949, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177: 7303–7311, 2006 [DOI] [PubMed] [Google Scholar]
- 434. Marzolla V, Armani A, Zennaro MC, Cinti F, Mammi C, Fabbri A, Rosano GMC, Caprio M. The role of the mineralocorticoid receptor in adipocyte biology and fat metabolism. Mol Cell Endocrinol 350: 281–288, 2012 [DOI] [PubMed] [Google Scholar]
- 435. Maser E. 11Beta-hydroxysteroid dehydrogenase responsible for carbonyl reduction of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in mouse lung microsomes. Cancer Res 58: 2996–3003, 1998 [PubMed] [Google Scholar]
- 436. Maser E, Bannenberg G. 11β-Hydroxysteroid dehydrogenase mediates reductive metabolism of xenobiotic carbonyl compounds. Biochem Pharmacol 47: 1805–1812, 1994 [DOI] [PubMed] [Google Scholar]
- 437. Maser E, Bannenberg G. The purification of 11β-hydroxysteroid dehydrogenase from mouse liver microsomes. J Steroid Biochem Mol Biol 48: 257–263, 1994 [DOI] [PubMed] [Google Scholar]
- 438. Maser E, Friebertshauser J, Volker B. Purification, characterization and NNK carbonyl reductase activities of 11beta-hydroxysteroid dehydrogenase type 1 from human liver: enzyme cooperativity and significance in the detoxification of a tobacco-derived carcinogen. Chem Biol Interact 143–144: 435–448, 2003 [DOI] [PubMed] [Google Scholar]
- 439. Maser E, Netter KJ. Purification and properties of a metyrapone-reducing enzyme from mouse liver microsomes: this ketone is reduced by an aldehyde reductase. Biochem Pharmacol 38: 3049–3054, 1989 [DOI] [PubMed] [Google Scholar]
- 440. Maser E, Oppermann UC. Role of type-1 11β-hydroxysteroid dehydrogenase in detoxification processes. Eur J Biochem 249: 365–369, 1997 [DOI] [PubMed] [Google Scholar]
- 441. Maser E, Richter E, Friebertshauser J. The identification of 11β-hydroxysteroid dehydrogenase as carbonyl reductase of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Eur J Biochem 238: 484–489, 1996 [DOI] [PubMed] [Google Scholar]
- 442. Maser E, Volker B, Friebertshauser J. 11β-Hydroxysteroid dehydrogenase type 1 from human liver: dimerization and enzyme cooperativity support its postulated role as glucocorticoid reductase. Biochemistry 41: 2459–2465, 2002 [DOI] [PubMed] [Google Scholar]
- 443. Maser E, Wsol V, Martin HJ. 11Beta-hydroxysteroid dehydrogenase type 1: purification from human liver and characterization as carbonyl reductase of xenobiotics. Mol Cell Endocrinol 248: 34–37, 2006 [DOI] [PubMed] [Google Scholar]
- 444. Mason HL, Hoehn WM, McKenzie BF, Kendall EC. Chemical studies of the suprarenal cortex III. The structures of compounds A, B, and H. J Biol Chem 120: 719–741, 1937 [Google Scholar]
- 445. Mason HL, Myers CS, Kendall EC. The chemistry of crystalline substances isolated from the suprarenal gland. J Biol Chem 114: 613–631, 1936 [Google Scholar]
- 446. Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science 294: 2166–2170, 2001 [DOI] [PubMed] [Google Scholar]
- 447. Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, Shinyama H, Sharp MG, Fleming S, Mullins JJ, Seckl JR, Flier JS. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest 112: 83–90, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. McCalla CO, Nacharaju VL, Muneyyirci-Delale O, Glasgow S, Feldman JG. Placental 11 beta-hydroxysteroid dehydrogenase activity in normotensive and pre-eclamptic pregnancies. Steroids 63: 511–515, 1998 [DOI] [PubMed] [Google Scholar]
- 449. McCormick KL, Wang X, Mick GJ. Modification of microsomal 11beta-HSD1 activity by cytosolic compounds: glutathione and hexose phosphoesters. J Steroid Biochem Mol Biol 111: 18–23, 2008 [DOI] [PubMed] [Google Scholar]
- 450. McDonald ATJ, Crocker JFS, Digout SC, McCarthy SC, Blecher SR, Cole DEC. Glucocorticoid-induced polycystic kidney-disease: a threshold trait. Kidney Int 37: 901–908, 1990 [DOI] [PubMed] [Google Scholar]
- 451. McDonald SE, Henderson TA, Gomez-Sanchez CE, Critchley HO, Mason JI. 11Beta-hydroxysteroid dehydrogenases in human endometrium. Mol Cell Endocrinol 248: 72–78, 2006 [DOI] [PubMed] [Google Scholar]
- 452. McEwen BS, Biron CA, Brunson KW, Bulloch K, Chambers WH, Dhabhar FS, Goldfarb RH, Kitson RP, Miller AH, Spencer RL, Weiss JM. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Rev 23: 79–133, 1997 [DOI] [PubMed] [Google Scholar]
- 453. McSweeney SJ. 11β-Hydroxysteroid Dehydrogenase Type 1: A New Therapeutic Target Post-Myocardial Infarction? (PhD thesis) Edinburgh: Univ. of Edinburgh, 2010 [Google Scholar]
- 454. McSweeney SJ, Hadoke PW, Kozak AM, Small GR, Khaled H, Walker BR, Gray GA. Improved heart function follows enhanced inflammatory cell recruitment and angiogenesis in 11β-HSD1-deficient mice post-MI. Cardiovasc Res 88: 159–167, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455. McTernan CL, Draper N, Nicholson H, Chalder SM, Driver P, Hewison M, Kilby MD, Stewart PM. Reduced placental 11beta-hydroxysteroid dehydrogenase type 2 mRNA levels in human pregnancies complicated by intrauterine growth restriction: an analysis of possible mechanisms. J Clin Endocrinol Metab 86: 4979–4983, 2001 [DOI] [PubMed] [Google Scholar]
- 456. Meaney MJ, Aitken DH, van Berkel C, Bhatnagar S, Sapolsky RM. Effect of neonatal handling on age-related impairments associated with the hippocampus. Science 239: 766–768, 1988 [DOI] [PubMed] [Google Scholar]
- 457. Meaney MJ, Diorio J, Francis D, Widdowson J, LaPlante P, Caldji C, Sharma S, Seckl JR, Plotsky PM. Early environmental regulation of forebrain glucocorticoid receptor gene expression: implications for adrenocortical responses to stress. Dev Neurosci 18: 49–72, 1996 [DOI] [PubMed] [Google Scholar]
- 458. Meaney MJ, O'Donnell D, Rowe W, Tannenbaum B, Steverman A, Walker M, Nair NP, Lupien S. Individual differences in hypothalamic-pituitary-adrenal activity in later life and hippocampal aging. Exp Gerontol 30: 229–251, 1995 [DOI] [PubMed] [Google Scholar]
- 459. Meas T, Carreira E, Wang Y, Rauh M, Poitou C, Clement K, Dotsch J, Levy-Marchal C. 11β-Hydroxysteroid dehydrogenase type 1 of the subcutaneous adipose tissue is dysregulated but not associated with metabolic disorders in adults born small for gestational age. J Clin Endocrinol Metab 95: 3949–3954, 2010 [DOI] [PubMed] [Google Scholar]
- 460. Meijer OC, de Lange EC, Breimer DD, de Boer AG, Workel JO, de Kloet ER. Penetration of dexamethasone into brain glucocorticoid targets is enhanced in mdr1A P-glycoprotein knockout mice. Endocrinology 139: 1789–1793, 1998 [DOI] [PubMed] [Google Scholar]
- 461. Meites J. Role of hypothalamic catecholamines in aging processes. Acta Endocrinol 125 Suppl 1: 98–103, 1991 [PubMed] [Google Scholar]
- 462. Mercer W, Obeyesekere V, Smith R, Krozowski Z. Characterization of 11β-HSD1B gene expression and enzymatic activity. Mol Cell Endocrinol 92: 247–251, 1993 [DOI] [PubMed] [Google Scholar]
- 463. Meyer JS. Early adrenalectomy stimulates subsequent growth and development of the rat brain. Exp Neurol 82: 432–446, 1983 [DOI] [PubMed] [Google Scholar]
- 464. Michael AE, Gregory L, Walker SM, Antoniw JW, Shaw RW, Edwards CRW, Cooke BA. Ovarian 11-beta-hydroxysteroid dehydrogenase: potential predictor of conception by in-vitro fertilization and embryo-transfer. Lancet 342: 711–712, 1993 [DOI] [PubMed] [Google Scholar]
- 465. Michailidou Z, Coll AP, Kenyon CJ, Morton NM, O'Rahilly S, Seckl JR, Chapman KE. Peripheral mechanisms contributing to the glucocorticoid hypersensitivity in proopiomelanocortin null mice treated with corticosterone. J Endocrinol 194: 161–170, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466. Michailidou Z, Jensen MD, Dumesic DA, Chapman KE, Seckl JR, Walker BR, Morton NM. Omental 11beta-hydroxysteroid dehydrogenase 1 correlates with fat cell size independently of obesity. Obesity 15: 1155–1163, 2007 [DOI] [PubMed] [Google Scholar]
- 467. Michailidou Z, Turban S, Miller E, Zou X, Schrader J, Ratcliffe PJ, Hadoke PW, Walker BR, Iredale JP, Morton NM, Seckl JR. Increased angiogenesis protects against adipose hypoxia and fibrosis in metabolic disease-resistant 11beta-hydroxysteroid dehydrogenase type 1 (HSD1)-deficient mice. J Biol Chem 287: 4188–4197, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Mihailidou AS, Loan Le TY, Mardini M, Funder JW. Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension 54: 1306–1312, 2009 [DOI] [PubMed] [Google Scholar]
- 469. Mirescu C, Peters JD, Gould E. Early life experience alters response of adult neurogenesis to stress. Nat Neurosci 7: 841–846, 2004 [DOI] [PubMed] [Google Scholar]
- 470. Miura T, Yamauchi K, Takahashi H, Nagahama Y. Hormonal induction of all stages of spermatogenesis in vitro in the male Japanese eel (Anguilla japonica). Proc Natl Acad Sci USA 88: 5774–5778, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Miyabayashi T, Palfreyman MT, Sluder AE, Slack F, Sengupta P. Expression and function of members of a divergent nuclear receptor family in Caenorhabditis elegans. Dev Biol 215: 314–331, 1999 [DOI] [PubMed] [Google Scholar]
- 472. Mlinar B, Marc J, Jensterle M, Bokal EV, Jerin A, Pfeifer M. Expression of 11 beta-hydroxysteroid dehydrogenase type 1 in visceral and subcutaneous adipose tissues of patients with polycystic ovary syndrome is associated with adiposity. J Steroid Biochem Mol Biol 123: 127–132, 2011 [DOI] [PubMed] [Google Scholar]
- 473. Moguilewsky M, Raynaud JP. Evidence for a specific mineralocorticoid receptor in rat pituitary and brain. J Steroid Biochem Mol Biol 12: 309–314, 1980 [DOI] [PubMed] [Google Scholar]
- 474. Mohler EG, Browman KE, Roderwald VA, Cronin EA, Markosyan S, Scott Bitner R, Strakhova MI, Drescher KU, Hornberger W, Rohde JJ, Brune ME, Jacobson PB, Rueter LE. Acute inhibition of 11beta-hydroxysteroid dehydrogenase type-1 improves memory in rodent models of cognition. J Neurosci 31: 5406–5413, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475. Moisan MP, Edwards CR, Seckl JR. Differential promoter usage by the rat 11 beta-hydroxysteroid dehydrogenase gene. Mol Endocrinol 6: 1082–1087, 1992 [DOI] [PubMed] [Google Scholar]
- 476. Moisan MP, Edwards CR, Seckl JR. Ontogeny of 11 beta-hydroxysteroid dehydrogenase in rat brain and kidney. Endocrinology 130: 400–404, 1992 [DOI] [PubMed] [Google Scholar]
- 477. Moisan MP, Seckl JR, Brett LP, Monder C, Agarwal AK, White PC, Edwards CR. 11Beta-hydroxysteroid dehydrogenase messenger ribonucleic acid expression, bioactivity and immunoreactivity in rat cerebellum. J Neuroendocrinol 2: 853–858, 1990 [DOI] [PubMed] [Google Scholar]
- 478. Moisan MP, Seckl JR, Edwards CR. 11Beta-hydroxysteroid dehydrogenase bioactivity and messenger RNA expression in rat forebrain: localization in hypothalamus, hippocampus, and cortex. Endocrinology 127: 1450–1455, 1990 [DOI] [PubMed] [Google Scholar]
- 479. Monder C. Heterogeneity of 11-Beta-Hydroxysteroid Dehydrogenase in Rat-Tissues. J Steroid Biochem Mol Biol 40: 533–536, 1991 [DOI] [PubMed] [Google Scholar]
- 480. Monder C, Lakshmi V. Corticosteroid 11β-dehydrogenase of rat tissues: immunological studies. Endocrinology 126: 2435–2443, 1990 [DOI] [PubMed] [Google Scholar]
- 481. Monder C, Lakshmi V, Miroff Y. Kinetic studies on rat liver 11β-hydroxysteroid dehydrogenase. Biochim Biophys Acta 1115: 23–29, 1991 [DOI] [PubMed] [Google Scholar]
- 482. Monder C, Miroff Y, Marandici A, Hardy MP. 11β-Hydroxysteroid dehydrogenase alleviates glucocorticoid-mediated inhibition of steroidogenesis in rat Leydig cells. Endocrinology 134: 1199–1204, 1994 [DOI] [PubMed] [Google Scholar]
- 483. Monder C, Shackleton CH, Bradlow HL, New MI, Stoner E, Iohan F, Lakshmi V. The syndrome of apparent mineralocorticoid excess: its association with 11 beta-dehydrogenase and 5 beta-reductase deficiency and some consequences for corticosteroid metabolism. J Clin Endocrinol Metab 63: 550–557, 1986 [DOI] [PubMed] [Google Scholar]
- 484. Monsivais D, Bray JD, Su E, Pavone ME, Dyson MT, Navarro A, Kakinuma T, Bulun SE. Activated glucocorticoid and eicosanoid pathways in endometriosis. Fertil Steril 98: 117–125, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485. Montano MM, Wang MH, Even MD, vom Saal FS. Serum corticosterone in fetal mice: sex differences, circadian changes, and effect of maternal stress. Physiol Behav 50: 323–329, 1991 [DOI] [PubMed] [Google Scholar]
- 486. Moore CCD, Mellon SH, Murai J, Siiteri PK, Miller WL. Structure and function of the hepatic form of 11β-hydroxysteroid dehydrogenase in the squirrel monkey, an animal model of glucocorticoid resistance. Endocrinology 133: 368–375, 1993 [DOI] [PubMed] [Google Scholar]
- 487. Moore JS, Monson JP, Kaltsas G, Putignano P, Wood PJ, Sheppard MC, Besser GM, Taylor NF, Stewart PM. Modulation of 11beta-hydroxysteroid dehydrogenase isozymes by growth hormone and insulin-like growth factor: in vivo and in vitro studies. J Clin Endocrinol Metab 84: 4172–4177, 1999 [DOI] [PubMed] [Google Scholar]
- 488. Moore XL, Hoong I, Cole TJ. Expression of the 11beta-hydroxysteroid dehydrogenase 2 gene in the mouse. Kidney Int 57: 1307–1312, 2000 [DOI] [PubMed] [Google Scholar]
- 489. Morales MA, Carvajal CA, Ortiz E, Mosso LM, Artigas RA, Owen GI, Fardella CE. Possible pathogenetic role of 11β-hydroxysteroid dehydrogenase type 1 (11βHSD1) gene polymorphisms in arterial hypertension. Rev Med Chil 136: 701–710, 2008 [PubMed] [Google Scholar]
- 490. Morgan SA, Sherlock M, Gathercole LL, Lavery GG, Lenaghan C, Bujalska IJ, Laber D, Yu A, Convey G, Mayers R, Hegyi K, Sethi JK, Stewart PM, Smith DM, Tomlinson JW. 11β-Hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes 58: 2506–2515, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491. Morineau G, Sulmont V, Salomon R, Fiquet-Kempf B, Jeunemaitre X, Nicod J, Ferrari P. Apparent mineralocorticoid excess: report of six new cases and extensive personal experience. J Am Soc Nephrol 17: 3176–3184, 2006 [DOI] [PubMed] [Google Scholar]
- 492. Morris MJ, Na ES, Johnson AK. Mineralocorticoid receptor antagonism prevents hedonic deficits induced by a chronic sodium appetite. Behav Neurosci 124: 211–224, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Morton NM, Holmes MC, Fievet C, Staels B, Tailleux A, Mullins JJ, Seckl JR. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem 276: 41293–41300, 2001 [DOI] [PubMed] [Google Scholar]
- 494. Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C, Walker BR, Flier JS, Mullins JJ, Seckl JR. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 53: 931–938, 2004 [DOI] [PubMed] [Google Scholar]
- 495. Morton NM, Ramage L, Seckl JR. Down-regulation of adipose 11β-hydroxysteroid dehydrogenase type 1 by high-fat feeding in mice: a potential adaptive mechanism counteracting metabolic disease. Endocrinology 145: 2707–2712, 2004 [DOI] [PubMed] [Google Scholar]
- 496. Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci 28: 9055–9065, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497. Muller C, Pompon D, Urban P, Morfin R. Inter-conversion of 7alpha- and 7beta-hydroxy-dehydroepiandrosterone by the human 11beta-hydroxysteroid dehydrogenase type 1. J Steroid Biochem Mol Biol 99: 215–222, 2006 [DOI] [PubMed] [Google Scholar]
- 498. Mune T, Rogerson FM, Nikkila H, Agarwal AK, White PC. Human hypertension caused by mutations in the kidney isozyme of 11 beta-hydroxysteroid dehydrogenase. Nat Genet 10: 394–399, 1995 [DOI] [PubMed] [Google Scholar]
- 499. Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol 175: 6257–6263, 2005 [DOI] [PubMed] [Google Scholar]
- 500. Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmuhl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OF, Spengler D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci 12: 1559–1566, 2009 [DOI] [PubMed] [Google Scholar]
- 501. Murotsuki J, Gagnon R, Pu X, Yang K. Chronic hypoxemia selectively down-regulates 11beta-hydroxysteroid dehydrogenase type 2 gene expression in the fetal sheep kidney. Biol Reprod 58: 234–239, 1998 [DOI] [PubMed] [Google Scholar]
- 502. Murphy BE. Cortisol production and inactivation by the human lung during gestation and infancy. J Clin Endocrinol Metab 47: 243–248, 1978 [DOI] [PubMed] [Google Scholar]
- 503. Murphy BE. Ontogeny of cortisol-cortisone interconversion in human tissues: a role for cortisone in human fetal development. J Steroid Biochem 14: 811–817, 1981 [DOI] [PubMed] [Google Scholar]
- 504. Murphy VE, Clifton VL. Alterations in human placental 11beta-hydroxysteroid dehydrogenase type 1 and 2 with gestational age and labour. Placenta 24: 739–744, 2003 [DOI] [PubMed] [Google Scholar]
- 505. Murphy VE, Zakar T, Smith R, Giles WB, Gibson PG, Clifton VL. Reduced 11beta-hydroxysteroid dehydrogenase type 2 activity is associated with decreased birth weight centile in pregnancies complicated by asthma. J Clin Endocrinol Metab 87: 1660–1668, 2002 [DOI] [PubMed] [Google Scholar]
- 506. Myatt L, Sun K. Role of fetal membranes in signaling of fetal maturation and parturition. Int J Dev Biol 54: 543–553, 2009 [DOI] [PubMed] [Google Scholar]
- 507. Mziaut H, Korza G, Hand AR, Gerard C, Ozols J. Targeting proteins to the lumen of endoplasmic reticulum using N-terminal domains of 11beta-hydroxysteroid dehydrogenase and the 50-kDa esterase. J Biol Chem 274: 14122–14129, 1999 [DOI] [PubMed] [Google Scholar]
- 508. Nair S, Lee YH, Lindsay RS, Walker BR, Tataranni PA, Bogardus C, Baier LJ, Permana PA. 11β-Hydroxysteroid dehydrogenase Type 1: genetic polymorphisms are associated with Type 2 diabetes in Pima Indians independently of obesity and expression in adipocyte and muscle. Diabetologia 47: 1088–1095, 2004 [DOI] [PubMed] [Google Scholar]
- 509. Nair SM, Werkman TR, Craig J, Finnell R, Joels M, Eberwine JH. Corticosteroid regulation of ion channel conductances and mRNA levels in individual hippocampal CA1 neurons. J Neurosci 18: 2685–2696, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510. Napolitano A, Voice MW, Edwards CRW, Seckl JR, Chapman KE. 11β-Hydroxysteroid dehydrogenase 1 in adipocytes: expression is differentiation-dependent and hormonally regulated. J Steroid Biochem Mol Biol 64: 251–260, 1998 [DOI] [PubMed] [Google Scholar]
- 511. Naray-Fejes-Toth A, Fejes-Toth G. Subcellular localization of the type 2 11beta-hydroxysteroid dehydrogenase. A green fluorescent protein study. J Biol Chem 271: 15436–15442, 1996 [DOI] [PubMed] [Google Scholar]
- 512. Náray-Fejes-Tóth A, Fejes-Tóth G. Expression cloning of the aldosterone target cell-specific 11β-hydroxysteroid dehydrogenase from rabbit collecting duct cells. Endocrinology 136: 2579–2586, 1995 [DOI] [PubMed] [Google Scholar]
- 513. Náray-Fejes-Toth A, Rusvai E, Denault DL, Stgermain DL, Fejes-Toth G. Expression and characterization of a new species of 11-beta-hydroxysteroid dehydrogenase in Xenopus oocytes. Am J Physiol Renal Fluid Electrolyte Physiol 265: F896–F900, 1993 [DOI] [PubMed] [Google Scholar]
- 514. Náray-Fejes-Toth A, Rusvai E, Fejes-Toth G. Is the renal type iii corticosteroid-binding site the collecting duct-specific isoform of 11β-hydroxysteroid dehydrogenase. Endocrinology 134: 1671–1675, 1994 [DOI] [PubMed] [Google Scholar]
- 515. Nashev LG, Chandsawangbhuwana C, Balazs Z, Atanasov AG, Dick B, Frey FJ, Baker ME, Odermatt A. Hexose-6-phosphate dehydrogenase modulates 11beta-hydroxysteroid dehydrogenase type 1-dependent metabolism of 7-keto- and 7beta-hydroxy-neurosteroids. PLoS One 2: e561, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Nawrocki AR, Goldring CE, Kostadinova RM, Frey FJ, Frey BM. In vivo footprinting of the human 11beta-hydroxysteroid dehydrogenase type 2 promoter: evidence for cell-specific regulation by Sp1 and Sp3. J Biol Chem 277: 14647–14656, 2002 [DOI] [PubMed] [Google Scholar]
- 517. Newnham JP, Jobe AH. Should we be prescribing repeated courses of antenatal corticosteroids? Semin Fetal Neonatal Med 14: 157–163, 2009 [DOI] [PubMed] [Google Scholar]
- 518. Nicholas TE, Johnson RG, Lugg MA, Kim PA. Pulmonary phospholipid biosynthesis and the ability of the fetal rabbit lung to reduce cortisone to cortisol during the final ten days of gestation. Life Sci 22: 1517–1524, 1978 [DOI] [PubMed] [Google Scholar]
- 519. Nomura S, Fujitaka M, Sakura N, Ueda K. Circadian rhythms in plasma cortisone and cortisol and the cortisone/cortisol ratio. Clin Chim Acta 266: 83–91, 1997 [DOI] [PubMed] [Google Scholar]
- 520. Nunez BS, Rogerson FM, Mune T, Igarashi Y, Nakagawa Y, Phillipov G, Moudgil A, Travis LB, Palermo M, Shackleton C, White PC. Mutants of 11beta-hydroxysteroid dehydrogenase (11-HSD2) with partial activity: improved correlations between genotype and biochemical phenotype in apparent mineralocorticoid excess. Hypertension 34: 638–642, 1999 [DOI] [PubMed] [Google Scholar]
- 521. Nuotio-Antar AM, Hachey DL, Hasty AH. Carbenoxolone treatment attenuates symptoms of metabolic syndrome and atherogenesis in obese, hyperlipidemic mice. Am J Physiol Endocrinol Metab 293: E1517–E1528, 2007 [DOI] [PubMed] [Google Scholar]
- 522. Nyirenda MJ, Carter R, Tang JI, de Vries A, Schlumbohm C, Hillier SG, Streit F, Oellerich M, Armstrong VW, Fuchs E, Seckl JR. Prenatal programming of metabolic syndrome in the common marmoset is associated with increased expression of 11beta-hydroxysteroid dehydrogenase type 1. Diabetes 58: 2873–2879, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523. Nyirenda MJ, Dean S, Lyons V, Chapman KE, Seckl JR. Prenatal programming of hepatocyte nuclear factor 4alpha in the rat: a key mechanism in the “foetal origins of hyperglycaemia”? Diabetologia 49: 1412–1420, 2006 [DOI] [PubMed] [Google Scholar]
- 524. Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest 101: 2174–2181, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. O'Regan D, Kenyon CJ, Seckl JR, Holmes MC. Glucocorticoid exposure in late gestation in the rat permanently programs gender-specific differences in adult cardiovascular and metabolic physiology. Am J Physiol Endocrinol Metab 287: E863–E870, 2004 [DOI] [PubMed] [Google Scholar]
- 526. Obeid J, White PC. Tyr-179 and Lys-183 are essential for enzymatic-activity of 11-beta-hydroxysteroid dehydrogenase. Biochem Biophys Res Commun 188: 222–227, 1992 [DOI] [PubMed] [Google Scholar]
- 527. Obeyesekere VR, Li KX, Ferrari P, Krozowski Z. Truncation of the N- and C-terminal regions of the human 11beta-hydroxysteroid dehydrogenase type 2 enzyme and effects on solubility and bidirectional enzyme activity. Mol Cell Endocrinol 131: 173–182, 1997 [DOI] [PubMed] [Google Scholar]
- 528. Obeyesekere VR, Trzeciak WH, Li KXZ, Krozowski ZS. Serines at the active site of 11 beta-hydroxysteroid dehydrogenase type I determine the rate of catalysis. Biochem Biophys Res Commun 250: 469–473, 1998 [DOI] [PubMed] [Google Scholar]
- 529. Odermatt A, Arnold P, Frey FJ. The intracellular localization of the mineralocorticoid receptor is regulated by 11beta -hydroxysteroid dehydrogenase type 2. J Biol Chem 276: 28484–28492, 2001 [DOI] [PubMed] [Google Scholar]
- 530. Odermatt A, Arnold P, Stauffer A, Frey BM, Frey FJ. The N-terminal anchor sequences of 11β-hydroxysteroid dehydrogenases determine their orientation in the endoplasmic reticulum membrane. J Biol Chem 274: 28762–28770, 1999 [DOI] [PubMed] [Google Scholar]
- 531. Odermatt A, Da Cunha T, Penno CA, Chandsawangbhuwana C, Reichert C, Wolf A, Dong M, Baker ME. Hepatic reduction of the secondary bile acid 7-oxolithocholic acid is mediated by 11beta-hydroxysteroid dehydrogenase 1. Biochem J 436: 621–629, 2011 [DOI] [PubMed] [Google Scholar]
- 532. Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, Fraenkel E, Bell GI, Young RA. Control of pancreas and liver gene expression by HNF transcription factors. Science 303: 1378–1381, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533. Ogg D, Elleby B, Norstrom C, Stefansson K, Abrahmsen L, Oppermann U, Svensson S. The crystal structure of guinea pig 11beta-hydroxysteroid dehydrogenase type 1 provides a model for enzyme-lipid bilayer interactions. J Biol Chem 280: 3789–3794, 2005 [DOI] [PubMed] [Google Scholar]
- 534. Oki K, Gomez-Sanchez EP, Gomez-Sanchez CE. Role of mineralocorticoid action in the brain in salt-sensitive hypertension. Clin Exp Pharmacol Physiol 39: 90–95, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535. Olsen N, Sokka T, Seehorn CL, Kraft B, Maas K, Moore J, Aune TM. A gene expression signature for recent onset rheumatoid arthritis in peripheral blood mononuclear cells. Ann Rheum Dis 63: 1387–1392, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Olza J, Gil-Campos M, Leis R, Ruperez AI, Tojo R, Canete R, Gil A, Aguilera CM. A gene variant of 11[beta]-hydroxysteroid dehydrogenase type 1 is associated with obesity in children. Int J Obes 36: 1558–1563, 2012 [DOI] [PubMed] [Google Scholar]
- 537. Oppermann U. Carbonyl reductases: the complex relationships of mammalian carbonyl- and quinone-reducing enzymes and their role in physiology. Annu Rev Pharmacol Toxicol 47: 293–322, 2007 [DOI] [PubMed] [Google Scholar]
- 538. Orsida BE, Krozowski ZS, Walters EH. Clinical relevance of airway 11beta-hydroxysteroid dehydrogenase type II enzyme in asthma. Am J Respir Crit Care Med 165: 1010–1014, 2002 [DOI] [PubMed] [Google Scholar]
- 539. Osinski PA. Steroid 11beta-ol dehydrogenase in human placenta. Nature 187: 777, 1960 [DOI] [PubMed] [Google Scholar]
- 540. Ostreicher I, Almeida JR, CAmpean V, Rauh M, Plank K, Dotsch J. Changes in 11β-hydroxysteroid dehydrogenase type 2 expression in a low-protein rat model of intrauterine growth restriction. Nephrol Dialysis Transplant 25: 3195–3203, 2010 [DOI] [PubMed] [Google Scholar]
- 541. Ozols J. Lumenal orientation and posttranslational modifications of the liver microsomal 11β-hydroxysteroid dehydrogenase. J Biol Chem 270: 2305–2312, 1995 [DOI] [PubMed] [Google Scholar]
- 542. Palermo M, Delitala G, Mantero F, Stewart PM, Shackleton CH. Congenital deficiency of 11beta-hydroxysteroid dehydrogenase (apparent mineralocorticoid excess syndrome): diagnostic value of urinary free cortisol and cortisone. J Endocrinol Invest 24: 17–23, 2001 [DOI] [PubMed] [Google Scholar]
- 543. Pardi DS, Loftus EV, Jr., Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis Gastroenterology 124: 889–893, 2003 [DOI] [PubMed] [Google Scholar]
- 544. Pasqualini JR, Lowy J, Albepart T, Wiqvist N, Diczfalusy E. Studies on the metabolism of corticosteroids in the human foeto-placental unit. 3 Role of 21-hydroxypregnenolone in the biosynthesis of corticosteroids Acta Endocrinol (Copenh) 63: 11–20, 1970 [DOI] [PubMed] [Google Scholar]
- 545. Pasquarette MM, Stewart PM, Ricketts ML, Imaishi K, Mason JI. Regulation of 11 beta-hydroxysteroid dehydrogenase type 2 activity and mRNA in human choriocarcinoma cells. J Mol Endocrinol 16: 269–275, 1996 [DOI] [PubMed] [Google Scholar]
- 546. Patel FA, Sun K, Challis JR. Local modulation by 11beta-hydroxysteroid dehydrogenase of glucocorticoid effects on the activity of 15-hydroxyprostaglandin dehydrogenase in human chorion and placental trophoblast cells. J Clin Endocrinol Metab 84: 395–400, 1999 [DOI] [PubMed] [Google Scholar]
- 547. Paterson JM, Holmes MC, Kenyon CJ, Carter R, Mullins JJ, Seckl JR. Liver-selective transgene rescue of hypothalamic-pituitary-adrenal axis dysfunction in 11beta-hydroxysteroid dehydrogenase type 1-deficient mice. Endocrinology 148: 961–966, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548. Paterson JM, Morton NM, Fievet C, Kenyon CJ, Holmes MC, Staels B, Seckl JR, Mullins JJ. Metabolic syndrome without obesity: Hepatic overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci USA 101: 7088–7093, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549. Paulmyer-Lacroix O, Boullu S, Oliver C, Alessi MC, Grino M. Expression of the mRNA coding for 11β-hydroxysteroid dehydrogenase type 1 in adipose tissue from obese patients: an in situ hybridization study. J Clin Endocrinol Metab 87: 2701–2705, 2002 [DOI] [PubMed] [Google Scholar]
- 550. Paulsen SK, Pedersen SB, Fisker S, Richelsen B. 11β-HSD type 1 expression in human adipose tissue: impact of gender, obesity, and fat localization. Obesity 15: 1954–1960, 2007 [DOI] [PubMed] [Google Scholar]
- 551. Pavlides C, Ogawa S, Kimura A, McEwen BS. Role of adrenal steroid mineralocorticoid and glucocorticoid receptors in long-term potentiation in the CA1 field of hippocampal slices. Brain Res 738: 229–235, 1996 [DOI] [PubMed] [Google Scholar]
- 552. Payne VA, Au WS, Gray SL, Nora ED, Rahman SM, Sanders R, Hadaschik D, Friedman JE, O'Rahilly S, Rochford JJ. Sequential regulation of diacylglycerol acyltransferase 2 expression by CAAT/enhancer-binding protein beta (C/EBPβ) and C/EBPα during adipogenesis. J Biol Chem 282: 21005–21014, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553. Pepe GJ, Albrecht ED. Comparison of cortisol cortisone interconversion in vitro by the human and baboon placenta. Steroids 44: 229–240, 1984 [DOI] [PubMed] [Google Scholar]
- 554. Pepe GJ, Babischkin JS, Burch MG, Leavitt MG, Albrecht ED. Developmental increase in expression of the messenger ribonucleic acid and protein levels of 11beta-hydroxysteroid dehydrogenase types 1 and 2 in the baboon placenta. Endocrinology 137: 5678–5684, 1996 [DOI] [PubMed] [Google Scholar]
- 555. Pepe GJ, Burch MG, Albrecht ED. Estrogen regulates 11 beta-hydroxysteroid dehydrogenase-1 and -2 localization in placental syncytiotrophoblast in the second half of primate pregnancy. Endocrinology 142: 4496–4503, 2001 [DOI] [PubMed] [Google Scholar]
- 556. Pepe GJ, Davies WA, Dong KW, Luo H, Albrecht ED. Cloning of the 11 beta-hydroxysteroid dehydrogenase (11 beta-HSD)-2 gene in the baboon: effects of estradiol on promoter activity of 11 beta-HSD-1 and -2 in placental JEG-3 cells. Biochim Biophys Acta 1444: 101–110, 1999 [DOI] [PubMed] [Google Scholar]
- 557. Persson B, Kallberg Y, Bray JE, Bruford E, Dellaporta SL, Favia AD, Duarte RG, Jornvall H, Kavanagh KL, Kedishvili N, Kisiela M, Maser E, Mindnich R, Orchard S, Penning TM, Thornton JM, Adamski J, Oppermann U. The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem Biol Interact 178: 94–98, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558. Phillips DM, Lakshmi V, Monder C. Corticosteroid 11-beta-dehydrogenase in rat testis. Endocrinology 125: 209–216, 1989 [DOI] [PubMed] [Google Scholar]
- 559. Poletto R, Steibel JP, Siegford JM, Zanella AJ. Effects of early weaning and social isolation on the expression of glucocorticoid and mineralocorticoid receptor and 11beta-hydroxysteroid dehydrogenase 1 and 2 mRNAs in the frontal cortex and hippocampus of piglets. Brain Res 1067: 36–42, 2006 [DOI] [PubMed] [Google Scholar]
- 560. Pryce CR, Aubert Y, Maier C, Pearce PC, Fuchs E. The developmental impact of prenatal stress, prenatal dexamethasone and postnatal social stress on physiology, behaviour and neuroanatomy of primate offspring: studies in rhesus macaque and common marmoset. Psychopharmacology 214: 33–53, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561. Qin W, Rudolph AE, Bond BR, Rocha R, Blomme EA, Goellner JJ, Funder JW, McMahon EG. Transgenic model of aldosterone-driven cardiac hypertrophy and heart failure. Circ Res 93: 69–76, 2003 [DOI] [PubMed] [Google Scholar]
- 562. Quinkler M, Kosmale B, Bahr V, Oelkers W, Diederich S. Evidence for isoforms of 11 beta-hydroxysteroid dehydrogenase in the liver and kidney of the guinea pig. J Endocrinol 153: 291–298, 1997 [DOI] [PubMed] [Google Scholar]
- 563. Rabbitt EH, Ayuk J, Boelaert K, Sheppard MC, Hewison M, Stewart PM, Gittoes NJ. Abnormal expression of 11 beta-hydroxysteroid dehydrogenase type 2 in human pituitary adenomas: a prereceptor determinant of pituitary cell proliferation. Oncogene 22: 1663–1667, 2003 [DOI] [PubMed] [Google Scholar]
- 564. Rabbitt EH, Gittoes NJ, Stewart PM, Hewison M. 11Beta-hydroxysteroid dehydrogenases, cell proliferation and malignancy. J Steroid Biochem Mol Biol 85: 415–421, 2003 [DOI] [PubMed] [Google Scholar]
- 565. Rabbitt EH, Lavery GG, Walker EA, Cooper MS, Stewart PM, Hewison M. Prereceptor regulation of glucocorticoid action by 11beta-hydroxysteroid dehydrogenase: a novel determinant of cell proliferation. FASEB J 16: 36–44, 2002 [DOI] [PubMed] [Google Scholar]
- 566. Rae M, Mohamad A, Price D, Hadoke PW, Walker BR, Mason JI, Hillier SG, Critchley HO. Cortisol inactivation by 11beta-hydroxysteroid dehydrogenase-2 may enhance endometrial angiogenesis via reduced thrombospondin-1 in heavy menstruation. J Clin Endocrinol Metab 94: 1443–1450, 2009 [DOI] [PubMed] [Google Scholar]
- 567. Rae MT, Niven D, Ross A, Forster T, Lathe R, Critchley HO, Ghazal P, Hillier SG. Steroid signalling in human ovarian surface epithelial cells: the response to interleukin-1alpha determined by microarray analysis. J Endocrinol 183: 19–28, 2004 [DOI] [PubMed] [Google Scholar]
- 568. Rahman TJ, Mayosi BM, Hall D, Avery PJ, Stewart PM, Connell JM, Watkins H, Keavney B. Common variation at the 11-beta hydroxysteroid dehydrogenase type 1 gene is associated with left ventricular mass. Circ Cardiovasc Genet 4: 156–162, 2011 [DOI] [PubMed] [Google Scholar]
- 569. Raikkonen K, Pesonen AK, Heinonen K, Lahti J, Komsi N, Eriksson JG, Seckl JR, Jarvenpaa AL, Strandberg TE. Maternal licorice consumption and detrimental cognitive and psychiatric outcomes in children. Am J Epidemiol 170: 1137–1146, 2009 [DOI] [PubMed] [Google Scholar]
- 570. Raikkonen K, Seckl JR, Heinonen K, Pyhala R, Feldt K, Jones A, Pesonen AK, Phillips DI, Lahti J, Jarvenpaa AL, Eriksson JG, Matthews KA, Strandberg TE, Kajantie E. Maternal prenatal licorice consumption alters hypothalamic-pituitary-adrenocortical axis function in children. Psychoneuroendocrinology 35: 1587–1593, 2010 [DOI] [PubMed] [Google Scholar]
- 571. Rajan V, Chapman KE, Lyons V, Jamieson P, Mullins JJ, Edwards CRW, Seckl JR. Cloning sequencing and tissue-distribution of mouse 11β-hydroxysteroid dehydrogenase-1 cDNA. J Steroid Biochem Mol Biol 52: 141–147, 1995 [DOI] [PubMed] [Google Scholar]
- 572. Rajan V, Edwards CR, Seckl JR. 11beta-Hydroxysteroid dehydrogenase in cultured hippocampal cells reactivates inert 11-dehydrocorticosterone, potentiating neurotoxicity. J Neurosci 16: 65–70, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573. Rasheeda MK, Kagawa H, Kirubagaran R, Dutta-Gupta A, Senthilkumaran B. Cloning, expression and enzyme activity analysis of testicular 11beta-hydroxysteroid dehydrogenase during seasonal cycle and after hCG induction in air-breathing catfish Clarias gariepinus. J Steroid Biochem Mol Biol 120: 1–10, 2010 [DOI] [PubMed] [Google Scholar]
- 574. Rask E, Olsson T, Soderberg S, Andrew R, Livingstone DE, Johnson O, Walker BR. Tissue-specific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab 86: 1418–1421, 2001 [DOI] [PubMed] [Google Scholar]
- 575. Rask E, Walker BR, Soderberg S, Livingstone DE, Eliasson M, Johnson O, Andrew R, Olsson T. Tissue-specific changes in peripheral cortisol metabolism in obese women: increased adipose 11β-hydroxysteroid dehydrogenase type 1 activity. J Clin Endocrinol Metab 87: 3330–3336, 2002 [DOI] [PubMed] [Google Scholar]
- 576. Rauz S, Cheung CM, Wood PJ, Coca-Prados M, Walker EA, Murray PI, Stewart PM. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 lowers intraocular pressure in patients with ocular hypertension. Q J Med 96: 481–490, 2003 [DOI] [PubMed] [Google Scholar]
- 577. Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, Myers RM. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 19: 2163–2171, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578. Reichstein T. On components of kidney lobes. X. On knowledge of cortico-sterones. Helv Chim Acta 20: 953–969, 1937 [Google Scholar]
- 579. Reichstein T. The properties of the adrenal cortex. VI. Segregative methods, plus isolation of the substances F a H and J. Helv Chim Acta 19: 1107–1126, 1936 [Google Scholar]
- 580. Reul JMHM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdissection and differential occupation. Endocrinology 117: 2505–2511, 1985 [DOI] [PubMed] [Google Scholar]
- 581. Ricketts ML, Verhaeg JM, Bujalska I, Howie AJ, Rainey WE, Stewart PM. Immunohistochemical localization of type 1 11beta-hydroxysteroid dehydrogenase in human tissues. J Clin Endocrinol Metab 83: 1325–1335, 1998 [DOI] [PubMed] [Google Scholar]
- 582. Robinzon B, Michael KK, Ripp SL, Winters SJ, Prough RA. Glucocorticoids inhibit interconversion of 7-hydroxy and 7-oxo metabolites of dehydroepiandrosterone: a role for 11beta-hydroxysteroid dehydrogenases? Arch Biochem Biophys 412: 251–258, 2003 [DOI] [PubMed] [Google Scholar]
- 583. Robson AC, Leckie CM, Seckl JR, Holmes MC. 11 Beta-hydroxysteroid dehydrogenase type 2 in the postnatal and adult rat brain. Brain Res 61: 1–10, 1998 [DOI] [PubMed] [Google Scholar]
- 584. Rodin A, Thakkar H, Taylor N, Clayton R. Hyperandrogenism in polycystic ovary syndrome. Evidence of dysregulation of 11 beta-hydroxysteroid dehydrogenase. N Engl J Med 330: 460–465, 1994 [DOI] [PubMed] [Google Scholar]
- 585. Rodrigues SM, LeDoux JE, Sapolsky RM. The influence of stress hormones on fear circuitry. Annu Rev Neurosci 32: 289–313, 2009 [DOI] [PubMed] [Google Scholar]
- 586. Rog-Zielinska EA, Thomson A, Moran C, Brownstein DG, Kenyon CJ, Michailidou Z, Szumska D, Bhattacharya S, Richardson J, Owen E, Watt A, Forrester LM, Holmes MC, Chapman KE. Impaired cardiac function in GR−/− fetal mice. Endocrine Abstr 25: OC5.4, 2011 [Google Scholar]
- 587. Rogerson FM, Kayes KM, White PC. Variation in placental type 2 11beta-hydroxysteroid dehydrogenase activity is not related to birth weight or placental weight. Mol Cell Endocrinol 128: 103–109, 1997 [DOI] [PubMed] [Google Scholar]
- 588. Roland BL, Funder JW. Localization of 11 beta-hydroxysteroid dehydrogenase type 2 in rat tissues: In situ studies. Endocrinology 137: 1123–1128, 1996 [DOI] [PubMed] [Google Scholar]
- 589. Roland BL, Krozowski ZS, Funder JW. Glucocorticoid receptor, mineralocorticoid receptors, 11 beta-hydroxysteroid dehydrogenase-1 and -2 expression in rat brain and kidney: in situ studies. Mol Cell Endocrinol 111: R1–7, 1995 [DOI] [PubMed] [Google Scholar]
- 590. Roland BL, Li KX, Funder JW. Hybridization histochemical localization of 11 beta-hydroxysteroid dehydrogenase type 2 in rat brain. Endocrinology 136: 4697–4700, 1995 [DOI] [PubMed] [Google Scholar]
- 591. Romero DG, Zhou M, Gomez-Sanchez CE. Cloning and expression of the bovine 11beta-hydroxysteroid dehydrogenase type-2. J Steroid Biochem Mol Biol 72: 231–237, 2000 [DOI] [PubMed] [Google Scholar]
- 592. Rook G, Baker R, Walker B, Honour J, Jessop D, Hernandez-Pando R, Arriaga K, Shaw R, Zumla A, Lightman S. Local regulation of glucocorticoid activity in sites of inflammation. Insights from the study of tuberculosis. Ann NY Acad Sci 917: 913–922, 2000 [DOI] [PubMed] [Google Scholar]
- 593. Rose AJ, Diaz MB, Reimann A, Klement J, Walcher T, Krones-Herzig A, Strobel O, Werner J, Peters A, Kleyman A, Tuckermann JP, Vegiopoulos A, Herzig S. Molecular control of systemic bile acid homeostasis by the liver glucocorticoid receptor. Cell Metab 14: 123–130, 2011 [DOI] [PubMed] [Google Scholar]
- 594. Rosenstock J, Banarer S, Fonseca VA, Inzucchi SE, Sun W, Yao W, Hollis G, Flores R, Levy R, Williams WV, Seckl JR, Huber R. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 33: 1516–1522, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595. Rosmond R, Dallman MF, Bjorntorp P. Stress-related cortisol secretion in men: relationships with abdominal obesity and endocrine, metabolic and hemodynamic abnormalities. J Clin Endocrinol Metab 83: 1853–1859, 1998 [DOI] [PubMed] [Google Scholar]
- 596. Rowland NE, Fregly MJ. Sodium appetite: species and strain differences and role of renin-angiotensin-aldosterone system. Appetite 11: 143–178, 1988 [DOI] [PubMed] [Google Scholar]
- 597. Ruan H, Hacohen N, Golub TR, Van Parijs L, Lodish HF. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3–L1 adipocytes: nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes 51: 1319–1336, 2002 [DOI] [PubMed] [Google Scholar]
- 598. Ruan H, Miles PD, Ladd CM, Ross K, Golub TR, Olefsky JM, Lodish HF. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: implications for insulin resistance. Diabetes 51: 3176–3188, 2002 [DOI] [PubMed] [Google Scholar]
- 599. Rubis B, Krozowski Z, Trzeciak WH. Arginine vasopressin stimulates 11beta-hydroxysteroid dehydrogenase type 2 expression in the mineralocorticosteroid target cells. Mol Cell Endocrinol 256: 17–22, 2006 [DOI] [PubMed] [Google Scholar]
- 600. Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, Nerlov C. A CREB-C/EBPβ cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci USA 106: 17475–17480, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601. Rundle SE, Funder JW, Lakshmi V, Monder C. The intrarenal localisation of mineralocorticoid receptors and 11β-dehydrogenase: immunocytochemical studies. Endocrinology 125: 1700–1704, 1989 [DOI] [PubMed] [Google Scholar]
- 602. Rusvai E, Naray-Fejes-Toth A. A new isoform of 11 beta-hydroxysteroid dehydrogenase in aldosterone target cells. J Biol Chem 268: 10717–10720, 1993 [PubMed] [Google Scholar]
- 603. Sahni-Arya B, Flynn MJ, Bergeron L, Salyan ME, Pedicord DL, Golla R, Ma Z, Wang H, Seethala R, Wu SC, Li JJ, Nayeem A, Gates C, Hamann LG, Gordon DA, Blat Y. Cofactor-specific modulation of 11beta-hydroxysteroid dehydrogenase 1 inhibitor potency. Biochim Biophys Acta 1774: 1184–1191, 2007 [DOI] [PubMed] [Google Scholar]
- 604. Sai S, Esteves CL, Kelly V, Michailidou Z, Anderson K, Coll AP, Nakagawa Y, Ohzeki T, Seckl JR, Chapman KE. Glucocorticoid regulation of the promoter of 11beta-hydroxysteroid dehydrogenase type 1 is indirect and requires CCAAT/enhancer-binding protein-beta. Mol Endocrinol 22: 2049–2060, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605. Sai S, Nakagawa Y, Sakaguchi K, Okada S, Takahashi H, Hongo T, Seckl JR, Chapman KE, Ohzeki T. Differential regulation of 11β-hydroxysteroid dehydrogenase-1 by dexamethasone in glucocorticoid-sensitive and -resistant childhood lymphoblastic leukemia. Leuk Res 33: 1696–1698, 2009 [DOI] [PubMed] [Google Scholar]
- 606. Sai S, Nakagawa Y, Yamaguchi R, Suzuki M, Sakaguchi K, Okada S, Seckl JR, Ohzeki T, Chapman KE. Expression of 11β-hydroxysteroid dehydrogenase 2 contributes to glucocorticoid resistance in lymphoblastic leukemia cell lines. Leuk Res 35: 1644–1648, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607. Sakai RR, Lakshmi V, Monder C, McEwen BS. Immunocytochemical Localization of 11beta-hydroxysteroid dehydrogenase in hippocampus and other brain-regions of the rat. J Neuroendocrinol 4: 101–106, 1992 [DOI] [PubMed] [Google Scholar]
- 608. Sampath-Kumar R, Matthews SG, Yang K. 11beta-hydroxysteroid dehydrogenase type 2 is the predominant isozyme in the guinea pig placenta: decreases in messenger ribonucleic acid and activity at term. Biol Reprod 59: 1378–1384, 1998 [DOI] [PubMed] [Google Scholar]
- 609. Sampath-Kumar R, Yu M, Khalil MW, Yang K. Metyrapone is a competitive inhibitor of 11beta-hydroxysteroid dehydrogenase type 1 reductase. J Steroid Biochem Mol Biol 62: 195–199, 1997 [DOI] [PubMed] [Google Scholar]
- 610. Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, Piersma AH, Ozanne SE, Twinn DF, Remacle C, Rowlerson A, Poston L, Taylor PD. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 51: 383–392, 2008 [DOI] [PubMed] [Google Scholar]
- 611. San Millan JL, Botella-Carretero JI, Alvarez-Blasco F, Luque-Ramirez M, Sancho J, Moghetti P, Escobar-Morreale HF. A study of the hexose-6-phosphate dehydrogenase gene R453Q and 11beta-hydroxysteroid dehydrogenase type 1 gene 83557insA polymorphisms in the polycystic ovary syndrome. J Clin Endocrinol Metab 90: 4157–4162, 2005 [DOI] [PubMed] [Google Scholar]
- 612. Sandeep TC, Andrew R, Homer NZ, Andrews RC, Smith K, Walker BR. Increased in vivo regeneration of cortisol in adipose tissue in human obesity and effects of the 11β-hydroxysteroid dehydrogenase type 1 inhibitor carbenoxolone. Diabetes 54: 872–879, 2005 [DOI] [PubMed] [Google Scholar]
- 613. Sandeep TC, Yau JL, MacLullich AM, Noble J, Deary IJ, Walker BR, Seckl JR. 11Beta-hydroxysteroid dehydrogenase inhibition improves cognitive function in healthy elderly men and type 2 diabetics. Proc Natl Acad Sci USA 101: 6734–6739, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614. Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev 7: 284–301, 1986 [DOI] [PubMed] [Google Scholar]
- 615. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 21: 55–89, 2000 [DOI] [PubMed] [Google Scholar]
- 616. Sarkar S, Tsai SW, Nguyen TT, Plevyak M, Padbury JF, Rubin LP. Inhibition of placental 11beta-hydroxysteroid dehydrogenase type 2 by catecholamines via alpha-adrenergic signaling. Am J Physiol Regul Integr Comp Physiol 281: R1966–R1974, 2001 [DOI] [PubMed] [Google Scholar]
- 617. Schipper L, Spee B, Rothuizen J, Woutersen-van Nijnanten F, Fink-Gremmels J. Characterisation of 11 beta-hydroxysteroid dehydrogenases in feline kidney and liver. Biochim Biophys Acta 1688: 68–77, 2004 [DOI] [PubMed] [Google Scholar]
- 618. Schmidt M, Weidler C, Naumann H, Anders S, Scholmerich J, Straub RH. Reduced capacity for the reactivation of glucocorticoids in rheumatoid arthritis synovial cells: possible role of the sympathetic nervous system? Arthritis Rheum 52: 1711–1720, 2005 [DOI] [PubMed] [Google Scholar]
- 619. Schoof E, Girstl M, Frobenius W, Kirschbaum M, Dorr HG, Rascher W, Dotsch J. Decreased gene expression of 11beta-hydroxysteroid dehydrogenase type 2 and 15-hydroxyprostaglandin dehydrogenase in human placenta of patients with preeclampsia. J Clin Endocrinol Metab 86: 1313–1317, 2001 [DOI] [PubMed] [Google Scholar]
- 620. Schweizer RA, Atanasov AG, Frey BM, Odermatt A. A rapid screening assay for inhibitors of 11beta-hydroxysteroid dehydrogenases (11beta-HSD): flavanone selectively inhibits 11beta-HSD1 reductase activity. Mol Cell Endocrinol 212: 41–49, 2003 [DOI] [PubMed] [Google Scholar]
- 621. Schweizer RA, Zurcher M, Balazs Z, Dick B, Odermatt A. Rapid hepatic metabolism of 7-ketocholesterol by 11beta-hydroxysteroid dehydrogenase type 1: species-specific differences between the rat, human, and hamster enzyme. J Biol Chem 279: 18415–18424, 2004 [DOI] [PubMed] [Google Scholar]
- 622. Seckl JR. 11beta-Hydroxysteroid dehydrogenase in the brain: a novel regulator of glucocorticoid action? Front Neuroendocrinol 18: 49–99, 1997 [DOI] [PubMed] [Google Scholar]
- 623. Seckl JR. Glucocorticoids, developmental “programming” and the risk of affective dysfunction. Prog Brain Res 167: 17–34, 2008 [DOI] [PubMed] [Google Scholar]
- 624. Seckl JR, Dow RC, Low SC, Edwards CR, Fink G. The 11 beta-hydroxysteroid dehydrogenase inhibitor glycyrrhetinic acid affects corticosteroid feedback regulation of hypothalamic corticotrophin-releasing peptides in rats. J Endocrinol 136: 471–477, 1993 [DOI] [PubMed] [Google Scholar]
- 625. Seckl JR, Holmes MC. Mechanisms of disease: glucocorticoids, their placental metabolism and fetal “programming” of adult pathophysiology. Nat Clin Pract Endocrinol Metab 3: 479–488, 2007 [DOI] [PubMed] [Google Scholar]
- 626. Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology 142: 1371–1376, 2001 [DOI] [PubMed] [Google Scholar]
- 627. Semjonous NM, Sherlock M, Jeyasuria P, Parker KL, Walker EA, Stewart PM, Lavery GG. Hexose-6-phosphate dehydrogenase contributes to skeletal muscle homeostasis independent of 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology 152: 93–102, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628. Senesi S, Legeza B, Balazs Z, Csala M, Marcolongo P, Kereszturi E, Szelenyi P, Egger C, Fulceri R, Mandl J, Giunti R, Odermatt A, Banhegyi G, Benedetti A. Contribution of fructose-6-phosphate to glucocorticoid activation in the endoplasmic reticulum: possible implication in the metabolic syndrome. Endocrinology %R 101210/en2010–0614: en.2010–0614, 2010 [DOI] [PubMed] [Google Scholar]
- 629. Sequeira SM, Geerling JC, Loewy AD. Local inputs to aldosterone-sensitive neurons of the nucleus tractus solitarius. Neuroscience 141: 1995–2005, 2006 [DOI] [PubMed] [Google Scholar]
- 630. Sesti-Costa R, Baccan GC, Chedraoui-Silva S, Mantovani B. Effects of acute cold stress on phagocytosis of apoptotic cells: the role of corticosterone. Neuroimmunomodulation 17: 79–87, 2011 [DOI] [PubMed] [Google Scholar]
- 631. Shackleton CH, Honour JW, Dillon MJ, Chantler C, Jones RW. Hypertension in a four-year-old child: gas chromatographic and mass spectrometric evidence for deficient hepatic metabolism of steroids. J Clin Endocrinol Metab 50: 786–702, 1980 [DOI] [PubMed] [Google Scholar]
- 632. Shackleton CH, Rodriguez J, Arteaga E, Lopez JM, Winter JS. Congenital 11 beta-hydroxysteroid dehydrogenase deficiency associated with juvenile hypertension: corticosteroid metabolite profiles of four patients and their families. Clin Endocrinol 22: 701–712, 1985 [DOI] [PubMed] [Google Scholar]
- 633. Shah S, Hermanowski-Vosatka A, Gibson K, Ruck RA, Jia G, Zhang J, Hwang PMT, Ryan NW, Langdon RB, Feig PU. Efficacy and safety of the selective 11 beta-HSD-1 inhibitors MK-0736 and MK-0916 in overweight and obese patients with hypertension. J Am Soc Hyperten 5: 166–176, 2011 [DOI] [PubMed] [Google Scholar]
- 634. Shams M, Kilby MD, Somerset DA, Howie AJ, Gupta A, Wood PJ, Afnan M, Stewart PM. 11Beta-hydroxysteroid dehydrogenase type 2 in human pregnancy and reduced expression in intrauterine growth restriction. Hum Reprod 13: 799–804, 1998 [DOI] [PubMed] [Google Scholar]
- 635. Sharma A, Guan H, Yang K. The p38 mitogen-activated protein kinase regulates 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) expression in human trophoblast cells through modulation of 11β-HSD2 messenger ribonucleic acid stability. Endocrinology 150: 4278–4286, 2009 [DOI] [PubMed] [Google Scholar]
- 636. Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA 93: 3908–3913, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637. Sheppard K, Funder JW. Mineralocorticoid specificity of renal type 1 receptors: in-vivo binding studies. Am J Physiol Endocrinol Metab 252: E224–E229, 1987 [DOI] [PubMed] [Google Scholar]
- 638. Sheppard KE, Autelitano DJ. 11Beta-hydroxysteroid dehydrogenase 1 transforms 11-dehydrocorticosterone into transcriptionally active glucocorticoid in neonatal rat heart. Endocrinology 143: 198–204, 2002 [DOI] [PubMed] [Google Scholar]
- 639. Shih DQ, Bussen M, Sehayek E, Ananthanarayanan M, Shneider BL, Suchy FJ, Shefer S, Bollileni JS, Gonzalez FJ, Breslow JL, Stoffel M. Hepatocyte nuclear factor-1α is an essential regulator of bile acid and plasma cholesterol metabolism. Nat Genet 27: 375–382, 2001 [DOI] [PubMed] [Google Scholar]
- 640. Shimojo M, Ricketts ML, Petrelli MD, Moradi P, Johnson GD, Bradwell AR, Hewison M, Howie AJ, Stewart PM. Immunodetection of 11 beta-hydroxysteroid dehydrogenase type 2 in human mineralocorticoid target tissues: evidence for nuclear localization. Endocrinology 138: 1305–1311, 1997 [DOI] [PubMed] [Google Scholar]
- 641. Shin JW, Geerling JC, Loewy AD. Vagal innervation of the aldosterone-sensitive HSD2 neurons in the NTS. Brain Res 1249: 135–147, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642. Shoener JA, Baig R, Page KC. Prenatal exposure to dexamethasone alters hippocampal drive on hypothalamic-pituitary-adrenal axis activity in adult male rats. Am J Physiol Regul Integr Comp Physiol 290: R1366–R1373, 2006 [DOI] [PubMed] [Google Scholar]
- 643. Slavin BG, Ong JM, Kern PA. Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. J Lipid Res 35: 1535–1541, 1994 [PubMed] [Google Scholar]
- 644. Sloboda DM, Moss TJ, Li S, Matthews SG, Challis JR, Newnham JP. Expression of glucocorticoid receptor, mineralocorticoid receptor, and 11beta-hydroxysteroid dehydrogenase 1 and 2 in the fetal and postnatal ovine hippocampus: ontogeny and effects of prenatal glucocorticoid exposure. J Endocrinol 197: 213–220, 2008 [DOI] [PubMed] [Google Scholar]
- 645. Slotkin TA, McCook EC, Ritchie JC, Carroll BJ, Seidler FJ. Serotonin transporter expression in rat brain regions and blood platelets: aging and glucocorticoid effects. Biol Psychiatry 41: 172–183, 1997 [DOI] [PubMed] [Google Scholar]
- 646. Small GR, Hadoke PW, Sharif I, Dover AR, Armour D, Kenyon CJ, Gray GA, Walker BR. Preventing local regeneration of glucocorticoids by 11β-hydroxysteroid dehydrogenase type 1 enhances angiogenesis. Proc Natl Acad Sci USA 102: 12165–12170, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647. Smink JJ, Begay V, Schoenmaker T, Sterneck E, de Vries TJ, Leutz A. Transcription factor C/EBPbeta isoform ratio regulates osteoclastogenesis through MafB. EMBO J 28: 1769–1781, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648. Smit P, Dekker MJ, de Jong FJ, van den Beld AW, Koper JW, Pols HA, Brinkmann AO, de Jong FH, Breteler MM, Lamberts SW. Lack of association of the 11beta-hydroxysteroid dehydrogenase type 1 gene 83,557insA and hexose-6-phosphate dehydrogenase gene R453Q polymorphisms with body composition, adrenal androgen production, blood pressure, glucose metabolism, and dementia. J Clin Endocrinol Metab 92: 359–362, 2007 [DOI] [PubMed] [Google Scholar]
- 649. Smith BT. The role of pulmonary corticosteroid 11-reductase activity in lung maturation in the fetal rat. Pediatr Res 12: 12–14, 1978 [DOI] [PubMed] [Google Scholar]
- 650. Smith BT, Tanswell AK, Minshall D, Bogues WN, Vreeken E. Influence of corticosteroids on glycogen content and steroid 11-reductase activity in lung and liver of the fetal and newborn rat. Biol Neonate 42: 201–207, 1982 [DOI] [PubMed] [Google Scholar]
- 651. Smith JT, Waddell BJ. Increased fetal glucocorticoid exposure delays puberty onset in postnatal life. Endocrinology 141: 2422–2428, 2000 [DOI] [PubMed] [Google Scholar]
- 652. Smith RE, Li KX, Andrews RK, Krozowski Z. Immunohistochemical and molecular characterization of the rat 11 beta-hydroxysteroid dehydrogenase type II enzyme. Endocrinology 138: 540–547, 1997 [DOI] [PubMed] [Google Scholar]
- 653. Smith RE, Little PJ, Maguire JA, Stein-Oakley AN, Krozowski ZS. Vascular localization of the 11 beta-hydroxysteroid dehydrogenase type II enzyme. Clin Exp Pharmacol Physiol 23: 549–551, 1996 [DOI] [PubMed] [Google Scholar]
- 654. Smith RE, Maguire JA, Stein-Oakley AN, Sasano H, Takahashi K, Fukushima K, Krozowski ZS. Localization of 11 beta-hydroxysteroid dehydrogenase type II in human epithelial tissues. J Clin Endocrinol Metab 81: 3244–3248, 1996 [DOI] [PubMed] [Google Scholar]
- 655. Smoak KA, Cidlowski JA. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev 125: 697–706, 2004 [DOI] [PubMed] [Google Scholar]
- 656. Soehnlein O, Lindbom L, Weber C. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood 114: 4613–4623, 2009 [DOI] [PubMed] [Google Scholar]
- 657. Soldan M, Nagel G, Losekam M, Ernst M, Maser E. Interindividual variability in the expression and NNK carbonyl reductase activity of 11beta-hydroxysteroid dehydrogenase 1 in human lung. Cancer Lett 145: 49–56, 1999 [DOI] [PubMed] [Google Scholar]
- 658. Song DJ, Lorenzo B, Reidenberg MM. Inhibition of 11-Beta-hydroxysteroid dehydrogenase by gossypol and bioflavonoids. J Lab Clin Med 120: 792–797, 1992 [PubMed] [Google Scholar]
- 659. Song Z, Gong Y, Liu H, Ren Q, Sun X. Glycyrrhizin could reduce ocular hypertension induced by triamcinolone acetonide in rabbits. Mol Vis 17: 2056–2064, 2011 [PMC free article] [PubMed] [Google Scholar]
- 660. Sooy K, Webster SP, Noble J, Binnie M, Walker BR, Seckl JR, Yau JL. Partial deficiency or short-term inhibition of 11beta-hydroxysteroid dehydrogenase type 1 improves cognitive function in aging mice. J Neurosci 30: 13867–13872, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661. Soro A, Ingram MC, Tonolo G, Glorioso N, Fraser R. Evidence of coexisting changes in 11 beta-hydroxysteroid dehydrogenase and 5 beta-reductase activity in subjects with untreated essential hypertension. Hypertension 25: 67–70, 1995 [DOI] [PubMed] [Google Scholar]
- 662. Speirs HJ, Seckl JR, Brown RW. Ontogeny of glucocorticoid receptor and 11beta-hydroxysteroid dehydrogenase type-1 gene expression identifies potential critical periods of glucocorticoid susceptibility during development. J Endocrinol 181: 105–116, 2004 [DOI] [PubMed] [Google Scholar]
- 663. Staab CA, Stegk JP, Haenisch S, Neiss E, Kobsch K, Ebert B, Cascorbi I, Maser E. Analysis of alternative promoter usage in expression of HSD11B1 including the development of a transcript-specific quantitative real-time PCR method. Chem Biol Interact 191: 104–112, 2011 [DOI] [PubMed] [Google Scholar]
- 664. Stegk JP, Ebert B, Martin HJ, Maser E. Expression profiles of human 11beta-hydroxysteroid dehydrogenases type 1 and type 2 in inflammatory bowel diseases. Mol Cell Endocrinol 301: 104–108, 2009 [DOI] [PubMed] [Google Scholar]
- 665. Steiger M, Reichstein T. Desoxy-corticosterone (21-oxy-progesterone) from Delta(5)-3-oxy-ethyl-cholic acid (XII. Communication on components of kidney lobes). Helvetica Chim Acta 20: 1164–1179, 1937 [Google Scholar]
- 666. Stein MB, Koverola C, Hanna C, Torchia MG, McClarty B. Hippocampal volume in women victimized by childhood sexual abuse. Psychol Med 27: 951–959, 1997 [DOI] [PubMed] [Google Scholar]
- 667. Stewart JD, Sienko AE, Gonzalez CL, Christensen HD, Rayburn WF. Placebo-controlled comparison between a single dose and a multidose of betamethasone in accelerating lung maturation of mice offspring. Am J Obstet Gynecol 179: 1241–1247, 1998 [DOI] [PubMed] [Google Scholar]
- 668. Stewart PM, Corrie JE, Shackleton CH, Edwards CR. Syndrome of apparent mineralocorticoid excess. A defect in the cortisol-cortisone shuttle. J Clin Invest 82: 340–349, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669. Stewart PM, Murry BA, Mason JI. Type 2 11 beta-hydroxysteroid dehydrogenase in human fetal tissues. J Clin Endocrinol Metab 78: 1529–1532, 1994 [DOI] [PubMed] [Google Scholar]
- 670. Stewart PM, Rogerson FM, Mason JI. Type 2 11 beta-hydroxysteroid dehydrogenase messenger ribonucleic acid and activity in human placenta and fetal membranes: its relationship to birth weight and putative role in fetal adrenal steroidogenesis. J Clin Endocrinol Metab 80: 885–890, 1995 [DOI] [PubMed] [Google Scholar]
- 671. Stewart PM, Wallace AM, Atherden SM, Shearing CH, Edwards CR. Mineralocorticoid activity of carbenoxolone: contrasting effects of carbenoxolone and licorice on 11 beta-hydroxysteroid dehydrogenase activity in man. Clin Sci 78: 49–54, 1990 [DOI] [PubMed] [Google Scholar]
- 672. Stewart PM, Wallace AM, Valentino R, Burt D, Shackleton CH, Edwards CR. Mineralocorticoid activity of licorice: 11-beta-hydroxysteroid dehydrogenase deficiency comes of age. Lancet 2: 821–824, 1987 [DOI] [PubMed] [Google Scholar]
- 673. Stewart PM, Whorwood CB, Mason JI. Type 2 11 beta-hydroxysteroid dehydrogenase in foetal and adult life. J Steroid Biochem Mol Biol 55: 465–471, 1995 [DOI] [PubMed] [Google Scholar]
- 674. Stimson RH, Andersson J, Andrew R, Redhead DN, Karpe F, Hayes PC, Olsson T, Walker BR. Cortisol release from adipose tissue by 11beta-hydroxysteroid dehydrogenase type 1 in humans. Diabetes 58: 46–53, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 675. Stimson RH, Andrew R, McAvoy NC, Tripathi D, Hayes PC, Walker BR. Increased whole-body and sustained liver cortisol regeneration by 11 beta-hydroxysteroid dehydrogenase type 1 in obese men with type 2 diabetes provides a target for enzyme inhibition. Diabetes 60: 720–725, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676. Stocker C, O'Dowd J, Morton NM, Wargent E, Sennitt MV, Hislop D, Glund S, Seckl JR, Arch JR, Cawthorne MA. Modulation of susceptibility to weight gain and insulin resistance in low birthweight rats by treatment of their mothers with leptin during pregnancy and lactation. Int J Obes Relat Metab Disord 28: 129–136, 2004 [DOI] [PubMed] [Google Scholar]
- 677. Stojadinovic O, Lee B, Vouthounis C, Vukelic S, Pastar I, Blumenberg M, Brem H, Tomic-Canic M. Novel genomic effects of glucocorticoids in epidermal keratinocytes: inhibition of apoptosis, interferon-gamma pathway, and wound healing along with promotion of terminal differentiation. J Biol Chem 282: 4021–4034, 2007 [DOI] [PubMed] [Google Scholar]
- 678. Stokes J, Noble J, Brett L, Phillips C, Seckl JR, O'Brien C, Andrew R. Distribution of glucocorticoid and mineralocorticoid receptors and 11beta-hydroxysteroid dehydrogenases in human and rat ocular tissues. Invest Ophthalmol Vis Sci 41: 1629–1638, 2000 [PubMed] [Google Scholar]
- 679. Stulnig TM, Oppermann U, Steffensen KR, Schuster GU, Gustafsson JA. Liver X receptors downregulate 11beta-hydroxysteroid dehydrogenase type 1 expression and activity. Diabetes 51: 2426–2433, 2002 [DOI] [PubMed] [Google Scholar]
- 680. Sun K, He P, Yang K. Intracrine induction of 11beta-hydroxysteroid dehydrogenase type 1 expression by glucocorticoid potentiates prostaglandin production in the human chorionic trophoblast. Biol Reprod 67: 1450–1455, 2002 [DOI] [PubMed] [Google Scholar]
- 681. Sun K, Myatt L. Enhancement of glucocorticoid-induced 11β-hydroxysteroid dehydrogenase type 1 expression by proinflammatory cytokines in cultured human amnion fibroblasts. Endocrinology 144: 5568–5577, 2003 [DOI] [PubMed] [Google Scholar]
- 682. Sun K, Yang K, Challis JR. Differential expression of 11 beta-hydroxysteroid dehydrogenase types 1 and 2 in human placenta and fetal membranes. J Clin Endocrinol Metab 82: 300–305, 1997 [DOI] [PubMed] [Google Scholar]
- 683. Sun K, Yang K, Challis JR. Differential regulation of 11 beta-hydroxysteroid dehydrogenase type 1 and 2 by nitric oxide in cultured human placental trophoblast and chorionic cell preparation. Endocrinology 138: 4912–4920, 1997 [DOI] [PubMed] [Google Scholar]
- 684. Sun K, Yang K, Challis JR. Regulation of 11beta-hydroxysteroid dehydrogenase type 2 by progesterone, estrogen, and the cyclic adenosine 5′-monophosphate pathway in cultured human placental and chorionic trophoblasts. Biol Reprod 58: 1379–1384, 1998 [DOI] [PubMed] [Google Scholar]
- 685. Suzuki S, Koyama K, Darnel A, Ishibashi H, Kobayashi S, Kubo H, Suzuki T, Sasano H, Krozowski ZS. Dexamethasone upregulates 11beta-hydroxysteroid dehydrogenase type 2 in BEAS-2B cells. Am J Respir Crit Care Med 167: 1244–1249, 2003 [DOI] [PubMed] [Google Scholar]
- 686. Suzuki S, Tsubochi H, Ishibashi H, Suzuki T, Kondo T, Sasano H. Increased expression of 11 beta-hydroxysteroid dehydrogenase type 2 in the lungs of patients with acute respiratory distress syndrome. Pathol Int 53: 751–756, 2003 [DOI] [PubMed] [Google Scholar]
- 687. Suzuki T, Sasano H, Suzuki S, Hirasawa G, Takeyama J, Muramatsu Y, Date F, Nagura H, Krozowski ZS. 11Beta-hydroxysteroid dehydrogenase type 2 in human lung: possible regulator of mineralocorticoid action. J Clin Endocrinol Metab 83: 4022–4025, 1998 [DOI] [PubMed] [Google Scholar]
- 688. Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev 4: 141–194, 2005 [DOI] [PubMed] [Google Scholar]
- 689. Swali A, Walker EA, Lavery GG, Tomlinson JW, Stewart PM. 11 beta-hydroxysteroid dehydrogenase type 1 regulates insulin and glucagon secretion in pancreatic islets. Diabetologia 51: 2003–2011, 2008 [DOI] [PubMed] [Google Scholar]
- 690. Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, Hobbs HH, Dobbins RL. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab 288: E462–E468, 2005 [DOI] [PubMed] [Google Scholar]
- 691. Takahashi H, Yoshika M, Komiyama Y, Nishimura M. The central mechanism underlying hypertension: a review of the roles of sodium ions, epithelial sodium channels, the renin-angiotensin-aldosterone system, oxidative stress and endogenous digitalis in the brain. Hypertens Res 34: 1147–1160, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692. Takahashi KI, Fukushima K, Sasano H, Sasaki I, Matsuno S, Krozowski ZS, Nagura H. Type II 11beta-hydroxysteroid dehydrogenase expression in human colonic epithelial cells of inflammatory bowel disease. Dig Dis Sci 44: 2516–2522, 1999 [DOI] [PubMed] [Google Scholar]
- 693. Takahashi T, Hori SH. Intramembraneous localization of rat liver microsomal hexose-6-phosphate dehydrogenase and membrane permeability to its substrates. Biochim Biophys Acta 524: 262–276, 1978 [DOI] [PubMed] [Google Scholar]
- 694. Tang JI, Kenyon CJ, Seckl JR, Nyirenda MJ. Prenatal overexposure to glucocorticoids programs renal 11beta-hydroxysteroid dehydrogenase type 2 expression and salt-sensitive hypertension in the rat. J Hypertens 29: 282–289, 2011 [DOI] [PubMed] [Google Scholar]
- 695. Tannin GM, Agarwal AK, Monder C, New MI, White PC. The human gene for 11 beta-hydroxysteroid dehydrogenase. Structure, tissue distribution, and chromosomal localization. J Biol Chem 266: 16653–16658, 1991 [PubMed] [Google Scholar]
- 696. Tao Y, Shi JM, Zhang YX, Gao L, Zhan FH. Expression of 11beta-hydroxysteroid dehydrogenase type 2 in lymphoblastic cells and its relationship with glucocorticoid sensitivity. Zhongguo Shi Yan Xue Ye Xue Za Zhi 19: 109–113, 2011 [PubMed] [Google Scholar]
- 697. Teelucksingh S, Benediktsson R, Lindsay RS, Burt D, Seckl JR, Edwards CRW, Nan CL, Kelly R. Licorice. Lancet 337: 1549, 1991 [DOI] [PubMed] [Google Scholar]
- 698. Ter-Minassian M, Asomaning K, Zhao Y, Chen F, Su L, Carmella SG, Lin X, Hecht SS, Christiani DC. Genetic variability in the metabolism of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) to 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL). Int J Cancer 130: 1338–1346, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699. Terao M, Murota H, Kimura A, Kato A, Ishikawa A, Igawa K, Miyoshi E, Katayama I. 11b-Hydroxysteroid dehydrogenase-1 is a novel regulator of skin homeostasis and a candidate target for promoting tissue repair. PLoS ONE 6: e25039, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700. Tetsuka M, Haines LC, Milne M, Simpson GE, Hillier SG. Regulation of 11b-hydroxysteroid dehydrogenase type 1 gene expression by LH and interleukin-1b in cultured rat granulosa cells. J Endocrinol 163: 417–423, 1999 [DOI] [PubMed] [Google Scholar]
- 701. Tetsuka M, Thomas FJ, Thomas MJ, Anderson RA, Mason JI, Hillier SG. Differential expression of messenger ribonucleic acids encoding 11b-hydroxysteroid dehydrogenase types 1 and 2 in human granulosa cells. J Clin Endocrinol Metab 82: 2006–2009, 1997 [PubMed] [Google Scholar]
- 702. Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, Nguyen HT, Fischer JD, Matsen ME, Wisse BE, Morton GJ, Horvath TL, Baskin DG, Tsch√∂p MH, Schwartz MW. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122: 153–162, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703. Thieringer R, Le Grand CB, Carbin L, Cai TQ, Wong B, Wright SD, Hermanowski-Vosatka A. 11β-Hydroxysteroid dehydrogenase type 1 is induced in human monocytes upon differentiation to macrophages. J Immunol 167: 30–35, 2001 [DOI] [PubMed] [Google Scholar]
- 704. Thomas MP, Potter BVL. Crystal structures of 11b-hydroxysteroid dehydrogenase type 1 and their use in drug discovery. Future Med Chem 3: 367–390, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705. Thompson A, Han VK, Yang K. Differential expression of 11beta-hydroxysteroid dehydrogenase types 1 and 2 mRNA and glucocorticoid receptor protein during mouse embryonic development. J Steroid Biochem Mol Biol 88: 367–375, 2004 [DOI] [PubMed] [Google Scholar]
- 706. Thompson A, Han VK, Yang K. Spatial and temporal patterns of expression of 11β-hydroxysteroid dehydrogenase types 1 and 2 messenger RNA and glucocorticoid receptor protein in the murine placenta and uterus during late pregnancy. Biol Reprod 67: 1708–1718, 2002 [DOI] [PubMed] [Google Scholar]
- 707. Tiganescu A, Walker EA, Hardy RS, Mayes AE, Stewart PM. Localization, age- and site-dependent expression, and regulation of 11beta-hydroxysteroid dehydrogenase type 1 in skin. J Invest Dermatol 131: 30–36, 2011 [DOI] [PubMed] [Google Scholar]
- 708. Tomlinson JJ, Boudreau A, Wu D, Atlas E, Hache RJ. Modulation of early human preadipocyte differentiation by glucocorticoids. Endocrinology 147: 5284–5293, 2006 [DOI] [PubMed] [Google Scholar]
- 709. Tomlinson JW, Draper N, Mackie J, Johnson AP, Holder G, Wood P, Stewart PM. Absence of Cushingoid phenotype in a patient with Cushing's disease due to defective cortisone to cortisol conversion. J Clin Endocrinol Metab 87: 57–62, 2002 [DOI] [PubMed] [Google Scholar]
- 710. Tomlinson JW, Durrani OM, Bujalska IJ, Gathercole LL, Tomlins PJ, Reuser TTQ, Rose GE, Curnow SJ, Stewart PM, Walker EA, Rauz S. The Role of 11b-hydroxysteroid dehydrogenase 1 in adipogenesis in thyroid-associated ophthalmopathy. J Clin Endocrinol Metab 95: 398–406, 2011 [DOI] [PubMed] [Google Scholar]
- 711. Tomlinson JW, Moore J, Cooper MS, Bujalska I, Shahmanesh M, Burt C, Strain A, Hewison M, Stewart PM. Regulation of expression of 11β-hydroxysteroid dehydrogenase type 1 in adipose tissue: tissue-specific induction by cytokines. Endocrinology 142: 1982–1989, 2001 [DOI] [PubMed] [Google Scholar]
- 712. Tomlinson JW, Sherlock M, Hughes B, Hughes SV, Kilvington F, Bartlett W, Courtney R, Rejto P, Carley W, Stewart PM. Inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 activity in vivo limits glucocorticoid exposure to human adipose tissue and decreases lipolysis. J Clin Endocrinol Metab 92: 857–864, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 713. Tremblay J, Hardy DB, Pereira LE, Yang K. Retinoic acid stimulates the expression of 11beta-hydroxysteroid dehydrogenase type 2 in human choriocarcinoma JEG-3 cells. Biol Reprod 60: 541–545, 1999 [DOI] [PubMed] [Google Scholar]
- 714. Tsigelny I, Baker ME. Structures important in mammalian 11 beta- and 17 beta-hydroxysteroid dehydrogenases. J Steroid Biochem Mol Biol 55: 589–600, 1995 [DOI] [PubMed] [Google Scholar]
- 715. Tsigelny I, Baker ME. Structures stabilizing the dimer interface on human 11 beta-hydroxysteroid dehydrogenase types 1 and 2 and human 15-hydroxyprostaglandin dehydrogenase and their homologs. Biochem Biophys Res Commun 217: 859–868, 1995 [DOI] [PubMed] [Google Scholar]
- 716. Turban S, Liu X, Ramage L, Webster S, Walker B, Dunbar D, Mullins J, Seckl J, Morton N. Optimal elevation of β-cell 11β-hydroxysteroid dehydrogenase type 1 is a compensatory mechanism that prevents high fat diet-induced β-cell failure. Diabetes 61: 642–652, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717. Tye LM, Burton AF. Variation in the pattern of metabolism of corticosteroids in fetal mouse tissues. Life Sci 26: 35–39, 1980 [DOI] [PubMed] [Google Scholar]
- 718. Tzschoppe A, Struwe E, Blessing H, Fahlbusch F, Liebhaber G, Dorr HG, Rauh M, Rascher W, Goecke TW, Schild RL, Schleussner E, Scheler C, Hubler A, Dahlem P, Dotsch J. Placental 11beta-HSD2 gene expression at birth is inversely correlated with growth velocity in the first year of life after intrauterine growth restriction. Pediatr Res 65: 647–653, 2009 [DOI] [PubMed] [Google Scholar]
- 719. Ulick S, Levine LS, Gunczler P, Zanconato G, Ramirez LC, Rauh W, Rosler A, Bradlow HL, New MI. A syndrome of apparent mineralocorticoid excess associated with defects in the peripheral metabolism of cortisol. J Clin Endocrinol Metab 49: 757–764, 1979 [DOI] [PubMed] [Google Scholar]
- 720. Ulick S, Ramirez LC, New MI. An abnormality in steroid reductive metabolism in a hypertensive syndrome. J Clin Endocrinol Metab 44: 799–802, 1977 [DOI] [PubMed] [Google Scholar]
- 721. Uno H, Lohmiller L, Thieme C, Kemnitz JW, Engle MJ, Roecker EB, Farrell PM. Brain damage induced by prenatal exposure to dexamethasone in fetal rhesus macaques. I. Hippocampus. Brain Res 53: 157–167, 1990 [DOI] [PubMed] [Google Scholar]
- 722. Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz Gn, Lumeng CN, Mortensen RM. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest 120: 3350–3364, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723. Vachharajani V. Influence of obesity on sepsis. Pathophysiology 15: 123–134, 2008 [DOI] [PubMed] [Google Scholar]
- 724. Vagnerova K, Kverka M, Klusonova P, Ergang P, Miksik I, Tlaskalova-Hogenova H, Pacha J. Intestinal inflammation modulates expression of 11β-hydroxysteroid dehydrogenase in murine gut. J Endocrinol 191: 497–503, 2006 [DOI] [PubMed] [Google Scholar]
- 725. Vagnerova K, Loukotova J, Ergang P, Musilkova J, Miksik I, Pacha J. Peroxisome proliferator-activated receptor-gamma stimulates 11beta-hydroxysteroid dehydrogenase type 1 in rat vascular smooth muscle cells. Steroids 76: 577–581, 2011 [DOI] [PubMed] [Google Scholar]
- 726. Van Beek JP, Guan H, Julan L, Yang K. Glucocorticoids stimulate the expression of 11beta-hydroxysteroid dehydrogenase type 2 in cultured human placental trophoblast cells. J Clin Endocrinol Metab 89: 5614–5621, 2004 [DOI] [PubMed] [Google Scholar]
- 727. Van den Berg E, Kloppenborg RP, Kessels RPC, Kappelle LJ, Biessels GJ. Type 2 diabetes mellitus, hypertension, dyslipidemia and obesity: a systematic comparison of their impact on cognition. Biochim Biophys Acta 1792: 470–481, 2009 [DOI] [PubMed] [Google Scholar]
- 728. Van Gaalen M. A-918446 and A-801195 show preclinical promise for the treatment of AD (Abstract). 13th Int Conf Alzheimer's Dis Relat Disord (ICAD): P3–301, 2010 [Google Scholar]
- 729. Van Put DJ, Van Hove CE, De Meyer GR, Wuyts F, Herman AG, Bult H. Dexamethasone influences intimal thickening and vascular reactivity in the rabbit collared carotid artery. Eur J Pharmacol 294: 753–761, 1995 [DOI] [PubMed] [Google Scholar]
- 730. Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem 279: 47066–47075, 2004 [DOI] [PubMed] [Google Scholar]
- 731. Voice MW, Seckl JR, Edwards CR, Chapman KE. 11 Beta-hydroxysteroid dehydrogenase type 1 expression in 2S FAZA hepatoma cells is hormonally regulated: a model system for the study of hepatic glucocorticoid metabolism. Biochem J 317: 621–625, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 732. Vukelic S, Stojadinovic O, Pastar I, Rabach M, Krzyzanowska A, Lebrun E, Davis SC, Resnik S, Brem H, Tomic-Canic M. Cortisol synthesis in epidermis is induced by IL-1 and tissue injury. J Biol Chem 286: 10265–10275, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733. Wachter R, Masarik L, Burzle M, Mallik A, von Mandach U. Differential expression and activity of 11beta-hydroxysteroid dehydrogenase in human placenta and fetal membranes from pregnancies with intrauterine growth restriction. Fetal Diagn Ther 25: 328–335, 2009 [DOI] [PubMed] [Google Scholar]
- 734. Waddell BJ, Benediktsson R, Brown RW, Seckl JR. Tissue-specific messenger ribonucleic acid expression of 11beta-hydroxysteroid dehydrogenase types 1 and 2 and the glucocorticoid receptor within rat placenta suggests exquisite local control of glucocorticoid action. Endocrinology 139: 1517–1523, 1998 [DOI] [PubMed] [Google Scholar]
- 735. Waddell BJ, Benediktsson R, Seckl JR. 11β-Hydroxysteroid dehydrogenase type 2 in the rat corpus luteum: induction of messenger ribonucleic acid expression and bioactivity coincident with luteal regression. Endocrinology 137: 5386–5391, 1996 [DOI] [PubMed] [Google Scholar]
- 736. Waddell BJ, Burton PJ. Full induction of rat myometrial 11beta-hydroxysteroid dehydrogenase type 1 in late pregnancy is dependent on intrauterine occupancy. Biol Reprod 62: 1005–1009, 2000 [DOI] [PubMed] [Google Scholar]
- 737. Waddell BJ, Hisheh S, Dharmarajan AM, Burton PJ. Apoptosis in rat placenta is zone-dependent and stimulated by glucocorticoids. Biol Reprod 63: 1913–1917, 2000 [DOI] [PubMed] [Google Scholar]
- 738. Waddell BJ, Hisheh S, Krozowski ZS, Burton PJ. Localization of 11beta-hydroxysteroid dehydrogenase types 1 and 2 in the male reproductive tract. Endocrinology 144: 3101–3106, 2003 [DOI] [PubMed] [Google Scholar]
- 739. Waites GM, Wang C, Griffin PD. Gossypol: reasons for its failure to be accepted as a safe, reversible male antifertility drug. Int J Androl 21: 8–12, 1998 [DOI] [PubMed] [Google Scholar]
- 740. Wake DJ, Homer NZ, Andrew R, Walker BR. Acute in vivo regulation of 11β-hydroxysteroid dehydrogenase type 1 activity by insulin and intralipid infusions in humans. J Clin Endocrinol Metab 91: 4682–4688, 2006 [DOI] [PubMed] [Google Scholar]
- 741. Wake DJ, Stimson RH, Tan GD, Homer NZM, Andrew R, Karpe F, Walker BR. Effects of peroxisome proliferator-activated receptor-α and -γ agonists on 11β-hydroxysteroid dehydrogenase type 1 in subcutaneous adipose tissue in men. J Clin Endocrinol Metab 92: 1848–1856, 2007 [DOI] [PubMed] [Google Scholar]
- 742. Walker BR, Andrew R. Tissue production of cortisol by 11beta-hydroxysteroid dehydrogenase type 1 and metabolic disease. Ann NY Acad Sci 1083: 165–184, 2006 [DOI] [PubMed] [Google Scholar]
- 743. Walker BR, Campbell JC, Fraser R, Stewart PM, Edwards CRW. Mineralocorticoid excess and inhibition of 11β-hydroxysteroid dehydrogenase in patients with ectopic ACTH syndrome. Clin Endocrinol 37: 483–492, 1992 [DOI] [PubMed] [Google Scholar]
- 744. Walker BR, Connacher AA, Lindsay RM, Webb DJ, Edwards CR. Carbenoxolone increases hepatic insulin sensitivity in man: a novel role for 11-oxosteroid reductase in enhancing glucocorticoid receptor activation. J Clin Endocrinol Metab 80: 3155–3159, 1995 [DOI] [PubMed] [Google Scholar]
- 745. Walker BR, Seckl Cortisol Metabolism JR. In: International Textbook of Obesity, edited by Björntorp P. Chichester, UK: Wiley, 2001, p. 241–268 [Google Scholar]
- 746. Walker BR, Stewart PM, Shackleton CHL, Padfield PL, Edwards CRW. Deficient inactivation of cortisol by 11b-hydroxysteroid dehydrogenase in essential hypertension. Clin Endocrinol 39: 221–227, 1993 [DOI] [PubMed] [Google Scholar]
- 747. Walker BR, Yau JL, Brett LP, Seckl JR, Monder C, Williams BC, Edwards CRW. 11-Beta-hydroxysteroid dehydrogenase in vascular smooth-muscle and heart: implications for cardiovascular-responses to glucocorticoids. Endocrinology 129: 3305–3312, 1991 [DOI] [PubMed] [Google Scholar]
- 748. Walker EA, Ahmed A, Lavery GG, Tomlinson JW, Kim SY, Cooper MS, Ride JP, Hughes BA, Shackleton CH, McKiernan P, Elias E, Chou JY, Stewart PM. 11beta-Hydroxysteroid dehydrogenase type 1 regulation by intracellular glucose 6-phosphate provides evidence for a novel link between glucose metabolism and hypothalamo-pituitary-adrenal axis function. J Biol Chem 282: 27030–27036, 2007 [DOI] [PubMed] [Google Scholar]
- 749. Wamil M, Andrew R, Chapman KE, Street J, Morton NM, Seckl JR. 7-Oxysterols modulate glucocorticoid activity in adipocytes through competition for 11beta-hydroxysteroid dehydrogenase type. Endocrinology 149: 5909–5918, 2008 [DOI] [PubMed] [Google Scholar]
- 750. Wamil M, Battle JH, Turban S, Kipari T, Seguret D, Peixoto RD, Nelson YB, Nowakowska D, Ferenbach D, Ramage L, Chapman KE, Hughes J, Dunbar DR, Seckl JR, Morton NM. Novel fat depot-specific mechanisms underlie resistance to visceral obesity and inflammation in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 60: 1158–1167, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 751. Wamil M, Seckl JR. Inhibition of 11β-hydroxysteroid dehydrogenase type 1 as a promising therapeutic target. Drug Discovery Today 12: 504–520, 2007 [DOI] [PubMed] [Google Scholar]
- 752. Wan ZK, Chenail E, Xiang J, Li HQ, Ipek M, Bard J, Svenson K, Mansour TS, Xu X, Tian X, Suri V, Hahm S, Xing Y, Johnson CE, Li X, Qadri A, Panza D, Perreault M, Tobin JF, Saiah E. Efficacious 11 beta-hydroxysteroid dehydrogenase type i inhibitors in the diet-induced obesity mouse model. J Med Chem 52: 5449–5461, 2009 [DOI] [PubMed] [Google Scholar]
- 753. Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23: 5293–5300, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 754. Watson B, Jr, Bergman SM, Myracle A, Callen DF, Acton RT, Warnock DG. Genetic association of 11 beta-hydroxysteroid dehydrogenase type 2 (HSD11B2) flanking microsatellites with essential hypertension in blacks. Hypertension 28: 478–482, 1996 [DOI] [PubMed] [Google Scholar]
- 755. Weaver I, Cervoni N, Champagne F, D'Alessio A, Sharma S, Seckl J, Dymov S, Szyf M, Meaney M. Epigenetic programming by maternal behavior. Nature Neurosci 7: 847–854, 2004 [DOI] [PubMed] [Google Scholar]
- 756. Weaver IC, Szyf M, Meaney MJ. From maternal care to gene expression: DNA methylation and the maternal programming of stress responses. Endocr Res 28: 699, 2002 [DOI] [PubMed] [Google Scholar]
- 757. Weber B, Lewicka S, Deuschle M, Colla M, Vecsei P, Heuser I. Increased diurnal plasma concentrations of cortisone in depressed patients. J Clin Endocrinol Metab 85: 1133–1136, 2000 [DOI] [PubMed] [Google Scholar]
- 758. Wei L, MacDonald TM, Walker BR. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann Internal Med 141: 764–770, 2004 [DOI] [PubMed] [Google Scholar]
- 759. Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O'Brien CA, Thostenson J, Roberson PK, Boskey AL, Clemens TL, Manolagas SC. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in 21-month-old mice. Aging Cell 9: 147–161, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 760. Welberg LA, Seckl JR, Holmes MC. Inhibition of 11beta-hydroxysteroid dehydrogenase, the foeto-placental barrier to maternal glucocorticoids, permanently programs amygdala GR mRNA expression and anxiety-like behaviour in the offspring. Eur J Neurosci 12: 1047–1054, 2000 [DOI] [PubMed] [Google Scholar]
- 761. Welberg LA, Seckl JR, Holmes MC. Prenatal glucocorticoid programming of brain corticosteroid receptors and corticotrophin-releasing hormone: possible implications for behaviour. Neuroscience 104: 71–79, 2001 [DOI] [PubMed] [Google Scholar]
- 762. Westerbacka J, Yki-Jarvinen H, Vehkavaara S, Hakkinen AM, Andrew R, Wake DJ, Seckl JR, Walker BR. Body fat distribution and cortisol metabolism in healthy men: enhanced 5β-reductase and lower cortisol/cortisone metabolite ratios in men with fatty liver. J Clin Endocrinol Metab 88: 4924–4931, 2003 [DOI] [PubMed] [Google Scholar]
- 763. Wethmar K, Begay V, Smink JJ, Zaragoza K, Wiesenthal V, Dorken B, Calkhoven CF, Leutz A. C/EBPbetaDeltauORF mice–a genetic model for uORF-mediated translational control in mammals. Genes Dev 24: 15–20, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 764. White PC, Agarwal AK, Li A, Nikkila H, Pratt JH, Caulfield M, Clark A, McTernan C, Stewart PM. Possible association but no linkage of the HSD11B2 gene encoding the kidney isozyme of 11beta-hydroxysteroid dehydrogenase to hypertension in Black people. Clin Endocrinol 55: 249–252, 2001 [DOI] [PubMed] [Google Scholar]
- 765. White PC, Agarwal AK, Nunez BS, Giacchetti G, Mantero F, Stewart PM. Genotype-phenotype correlations of mutations and polymorphisms in HSD11B2, the gene encoding the kidney isozyme of 11beta-hydroxysteroid dehydrogenase. Endocr Res 26: 771–780, 2000 [DOI] [PubMed] [Google Scholar]
- 766. Whorwood CB, Barber PC, Gregory J, Sheppard MC, Stewart PM. 11β-Hydroxysteroid dehydrogenase and corticosteroid hormone receptors in the rat colon. Am J Physiol Endocrinol Metab 264: E951–E957, 1993 [DOI] [PubMed] [Google Scholar]
- 767. Whorwood CB, Donovan SJ, Flanagan D, Phillips DI, Byrne CD. Increased glucocorticoid receptor expression in human skeletal muscle cells may contribute to the pathogenesis of the metabolic syndrome. Diabetes 51: 1066–1075, 2002 [DOI] [PubMed] [Google Scholar]
- 768. Whorwood CB, Donovan SJ, Wood PJ, Phillips DI. Regulation of glucocorticoid receptor alpha and beta isoforms and type I 11beta-hydroxysteroid dehydrogenase expression in human skeletal muscle cells: a key role in the pathogenesis of insulin resistance? J Clin Endocrinol Metab 86: 2296–2308, 2001 [DOI] [PubMed] [Google Scholar]
- 769. Whorwood CB, Firth KM, Budge H, Symonds ME. Maternal undernutrition during early to midgestation programs tissue-specific alterations in the expression of the glucocorticoid receptor, 11β-hydroxysteroid dehydrogenase isoforms, and type 1 angiotensin II receptor in neonatal sheep. Endocrinology 142: 2854–2864, 2001 [DOI] [PubMed] [Google Scholar]
- 770. Whorwood CB, Ricketts ML, Stewart PM. Epithelial cell localization of type 2 11 beta-hydroxysteroid dehydrogenase in rat and human colon. Endocrinology 135: 2533–2541, 1994 [DOI] [PubMed] [Google Scholar]
- 771. Whorwood CB, Sheppard M, Stewart P. Tissue specific effects of thyroid hormone on 11β-hydroxysteroid dehydrogenase gene expression. J Steroid Biochem Mol Biol 46: 539–547, 1993 [DOI] [PubMed] [Google Scholar]
- 772. Wilber AA, Wellman CL. Neonatal maternal separation alters the development of glucocorticoid receptor expression in the interpositus nucleus of the cerebellum. Int J Dev Neurosci 27: 649–654, 2009 [DOI] [PubMed] [Google Scholar]
- 773. Wilckens T. Glucocorticoids and immune function: physiological relevance and pathogenic potential of hormonal dysfunction. Trends Pharmacol Sci 16: 193–197, 1995 [DOI] [PubMed] [Google Scholar]
- 774. Wilckens T, De Rijk R. Glucocorticoids and immune function: unknown dimensions and new frontiers. Immunol Today 18: 418–424, 1997 [DOI] [PubMed] [Google Scholar]
- 775. Williams LJ, Lyons V, MacLeod I, Rajan V, Darlington GJ, Poli V, Seckl JR, Chapman KE. C/EBP regulates hepatic transcription of 11beta-hydroxysteroid dehydrogenase type 1. A novel mechanism for cross-talk between the C/EBP and glucocorticoid signaling pathways. J Biol Chem 275: 30232–30239, 2000 [DOI] [PubMed] [Google Scholar]
- 776. Wilson RC, Dave-Sharma S, Wei JQ, Obeyesekere VR, Li K, Ferrari P, Krozowski ZS, Shackleton CH, Bradlow L, Wiens T, New MI. A genetic defect resulting in mild low-renin hypertension. Proc Natl Acad Sci USA 95: 10200–10205, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 777. Wilson RC, Krozowski ZS, Li K, Obeyesekere VR, Razzaghyazar M, Harbison MD, Wei JQ, Shackleton CHL, Funder JW, New MI. A mutation in the hsd11b2 gene in a family with apparent mineralocorticoid excess. J Clin Endocrinol Metab 80: 2263–2266, 1995 [DOI] [PubMed] [Google Scholar]
- 778. Wu X, Lukacik P, Kavanagh KL, Oppermann U. SDR-type human hydroxysteroid dehydrogenases involved in steroid hormone activation. Mol Cell Endocrinol 265–266: 71–76, 2007 [DOI] [PubMed] [Google Scholar]
- 779. Wyrwoll CS, Holmes MC. Prenatal excess glucocorticoid exposure and adult affective disorders: a role for serotonergic and catecholamine pathways. Neuroendocrinology 95: 47–55, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780. Wyrwoll CS, Holmes MC, Seckl JR. 11beta-hydroxysteroid dehydrogenases and the brain: from zero to hero, a decade of progress. Front Neuroendocrinol 32: 265–286, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 781. Wyrwoll CS, Mark PJ, Mori TA, Puddey IB, Waddell BJ. Prevention of programmed hyperleptinemia and hypertension by postnatal dietary omega-3 fatty acids. Endocrinology 147: 599–606, 2006 [DOI] [PubMed] [Google Scholar]
- 782. Wyrwoll CS, Mark PJ, Waddell BJ. Developmental programming of renal glucocorticoid sensitivity and the renin-angiotensin system. Hypertension 50: 579–584, 2007 [DOI] [PubMed] [Google Scholar]
- 783. Wyrwoll CS, Seckl JR, Holmes MC. Altered placental function of 11beta-hydroxysteroid dehydrogenase 2 knockout mice. Endocrinology 150: 1287–1293, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784. Yang K, Julan L, Rubio F, Sharma A, Guan H. Cadmium reduces 11 beta-hydroxysteroid dehydrogenase type 2 activity and expression in human placental trophoblast cells. Am J Physiol Endocrinol Metab 290: E135–E142, 2006 [DOI] [PubMed] [Google Scholar]
- 785. Yang K, Matthews SG, Challis JRG. Developmental and glucocorticoid regulation of pituitary 11-beta-hydroxysteroid dehydrogenase-1 gene-expression in the ovine fetus and lamb. J Mol Endocrinol 14: 109–116, 1995 [DOI] [PubMed] [Google Scholar]
- 786. Yang K, Shearman K, Asano H, Richardson BS. Effects of hypoxemia on 11 beta-hydroxysteroid dehydrogenase types 1 and 2 gene expression in preterm fetal sheep. J Soc Gynecol Invest 4: 124–129, 1997 [PubMed] [Google Scholar]
- 787. Yang K, Smith CL, Dales D, Hammond GL, Challis JR. Cloning of an ovine 11 beta-hydroxysteroid dehydrogenase complementary deoxyribonucleic acid: tissue and temporal distribution of its messenger ribonucleic acid during fetal and neonatal development. Endocrinology 131: 2120–2126, 1992 [DOI] [PubMed] [Google Scholar]
- 788. Yang X, Smith U. Adipose tissue distribution and risk of metabolic disease: does thiazolidinedione-induced adipose tissue redistribution provide a clue to the answer? Diabetologia 50: 1127–1139, 2007 [DOI] [PubMed] [Google Scholar]
- 789. Yang Z, Guo C, Zhu P, Li W, Myatt L, Sun K. Role of glucocorticoid receptor and CCAAT/enhancer-binding protein-α in the feed-forward induction of 11β-hydroxysteroid dehydrogenase type 1 expression by cortisol in human amnion fibroblasts. J Endocrinol 195: 241–253, 2007 [DOI] [PubMed] [Google Scholar]
- 790. Yang Z, Zhu X, Guo C, Sun K. Stimulation of 11β-HSD1 expression by IL-1β via a C/EBP binding site in human fetal lung fibroblasts. Endocrine 36: 404–411, 2009 [DOI] [PubMed] [Google Scholar]
- 791. Yau JL, McNair KM, Noble J, Brownstein D, Hibberd C, Morton N, Mullins JJ, Morris RG, Cobb S, Seckl JR. Enhanced hippocampal long-term potentiation and spatial learning in aged 11beta-hydroxysteroid dehydrogenase type 1 knock-out mice. J Neurosci 27: 10487–10496, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 792. Yau JL, Noble J, Graham M, Seckl JR. Central administration of a cytochrome P450–7B product 7 alpha-hydroxypregnenolone improves spatial memory retention in cognitively impaired aged rats. J Neurosci 26: 11034–11040, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 793. Yau JL, Noble J, Hibberd C, Rowe WB, Meaney MJ, Morris RG, Seckl JR. Chronic treatment with the antidepressant amitriptyline prevents impairments in water maze learning in aging rats. J Neurosci 22: 1436–1442, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 794. Yau JL, Noble J, Kenyon CJ, Hibberd C, Kotelevtsev Y, Mullins JJ, Seckl JR. Lack of tissue glucocorticoid reactivation in 11beta-hydroxysteroid dehydrogenase type 1 knockout mice ameliorates age-related learning impairments. Proc Natl Acad Sci USA 98: 4716–4721, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 795. Yau JL, Noble J, Seckl JR. 11Beta-hydroxysteroid dehydrogenase type 1 deficiency prevents memory deficits with aging by switching from glucocorticoid receptor to mineralocorticoid receptor-mediated cognitive control. J Neurosci 31: 4188–4193, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 796. Yeager MP, Guyre PM, Munck AU. Glucocorticoid regulation of the inflammatory response to injury. Acta Anaesthesiol Scand 48: 799–813, 2004 [DOI] [PubMed] [Google Scholar]
- 797. Yehuda R, Bierer LM, Andrew R, Schmeidler J, Seckl JR. Enduring effects of severe developmental adversity, including nutritional deprivation, on cortisol metabolism in aging Holocaust survivors. J Psychiatr Res 43: 877–883, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 798. Yong PY, Harlow C, Thong KJ, Hillier SG. Regulation of 11β-hydroxysteroid dehydrogenase type 1 gene expression in human ovarian surface epithelial cells by interleukin-1. Hum Reprod 17: 2300–2306, 2002 [DOI] [PubMed] [Google Scholar]
- 799. Yu L, Romero DG, Gomez-Sanchez CE, Gomez-Sanchez EP. Steroidogenic enzyme gene expression in the human brain. Mol Cell Endocrinol 190: 9–17, 2002 [DOI] [PubMed] [Google Scholar]
- 800. Yuan JM, Koh WP, Murphy SE, Fan Y, Wang R, Carmella SG, Han S, Wickham K, Gao YT, Yu MC, Hecht SS. Urinary levels of tobacco-specific nitrosamine metabolites in relation to lung cancer development in two prospective cohorts of cigarette smokers. Cancer Res 69: 2990–2995, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 801. Zbankova S, Bryndova J, Kment M, Pacha J. Expression of 11beta-hydroxysteroid dehydrogenase types 1 and 2 in colorectal cancer. Cancer Lett 210: 95–100, 2004 [DOI] [PubMed] [Google Scholar]
- 802. Zbankova S, Bryndova J, Leden P, Kment M, Svec A, Pacha J. 11β-Hydroxysteroid dehydrogenase 1 and 2 expression in colon from patients with ulcerative colitis. J Gastroenterol Hepatol 22: 1019–1023, 2007 [DOI] [PubMed] [Google Scholar]
- 803. Zennaro MC, Le Menuet D, Viengchareun S, Walker F, Ricquier D, Lombes M. Hibernoma development in transgenic mice identifies brown adipose tissue as a novel target of aldosterone action. J Clin Invest 101: 1254–1260, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 804. Zhang J, Osslund TD, Plant MH, Clogston CL, Nybo RE, Xiong F, Delaney JM, Jordan SR. Crystal structure of murine 11beta-hydroxysteroid dehydrogenase 1: an important therapeutic target for diabetes. Biochemistry 44: 6948–6957, 2005 [DOI] [PubMed] [Google Scholar]
- 805. Zhang M, Lv XY, Li J, Xu ZG, Chen L. Alteration of 11 beta-hydroxysteroid dehydrogenase type 1 in skeletal muscle in a rat model of type 2 diabetes. Mol Cell Biochem 324: 147–155, 2009 [DOI] [PubMed] [Google Scholar]
- 806. Zhang MZ, Xu J, Yao B, Yin H, Cai Q, Shrubsole MJ, Chen X, Kon V, Zheng W, Pozzi A, Harris RC. Inhibition of 11beta-hydroxysteroid dehydrogenase type II selectively blocks the tumor COX-2 pathway and suppresses colon carcinogenesis in mice and humans. J Clin Invest 119: 876–885, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 807. Zhang N, Truong-Tran QA, Tancowny B, Harris KE, Schleimer RP. Glucocorticoids enhance or spare innate immunity: effects in airway epithelium are mediated by CCAAT/enhancer binding proteins. J Immunol 179: 578–589, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 808. Zhang TY, Daynes RA. Macrophages from 11β-hydroxysteroid dehydrogenase type 1-deficient mice exhibit an increased sensitivity to lipopolysaccharide stimulation due to TGF-β-mediated up-regulation of SHIP1 expression. J Immunol 179: 6325–6335, 2007 [DOI] [PubMed] [Google Scholar]
- 809. Zhang TY, Ding X, Daynes RA. The expression of 11β-hydroxysteroid dehydrogenase type I by lymphocytes provides a novel means for intracrine regulation of glucocorticoid activities. J Immunol 174: 879–889, 2005 [DOI] [PubMed] [Google Scholar]
- 810. Zhang ZH, Kang YM, Yu Y, Wei SG, Schmidt TJ, Johnson AK, Felder RB. 11Beta-hydroxysteroid dehydrogenase type 2 activity in hypothalamic paraventricular nucleus modulates sympathetic excitation. Hypertension 48: 127–133, 2006 [DOI] [PubMed] [Google Scholar]
- 811. Zhou HY, Hu GX, Lian QQ, Morris D, Ge RS. The metabolism of steroids, toxins and drugs by 11b-hydroxysteroid dehydrogenase 1. Toxicology 292: 1–12, 2012 [DOI] [PubMed] [Google Scholar]
- 812. Zhou MY, Gomez-Sanchez EP, Cox DL, Cosby D, Gomez-Sanchez CE. Cloning, expression, and tissue distribution of the rat nicotinamide adenine dinucleotide-dependent 11 beta-hydroxysteroid dehydrogenase. Endocrinology 136: 3729–3734, 1995 [DOI] [PubMed] [Google Scholar]
- 813. Zumoff B, Bradlow HL, Levin J, Fukushima DK. Influence of thyroid function on the in vivo cortisol in equilibrium cortisone equilibrium in man. J Steroid Biochem 18: 437–440, 1983 [DOI] [PubMed] [Google Scholar]