

Abstract
We have previously demonstrated increased fibroblast growth factor-2 (FGF-2) expression in a lamb model of increased pulmonary blood flow secondary to congenital heart disease, which may contribute to the associated increases in pulmonary arterial muscularization. However, the mechanisms underlying these increases in FGF-2 expression remain to be identified. Initially, we found that exogenous FGF-2 increased endogenous FGF-2 promoter activity and protein levels in ovine pulmonary arterial smooth muscle cells (PASMC). Furthermore, we found that these increases in FGF-2 expression were mediated by increases in superoxide levels via NADPH oxidase activation. In addition, FGF-2-mediated increases in FGF-2 expression and PASMC proliferation were attenuated by inhibition of phosphatidylinositol 3-kinase, Akt, and NADPH oxidase. Increases in FGF-2 expression could be stimulated by other factors known to increase reactive oxygen species (ROS) signaling in PASMC (endothelin-1 and transforming growth factor-β1), whereas antioxidants attenuated these increases. Deletion constructs localized the growth factor- and ROS-sensitive region within the proximal 103 bp of the FGF-2 promoter, and sequence analysis identified a putative hypoxia response element (HRE), a DNA binding site for the ROS-sensitive transcription factor hypoxia-inducible factor-1α (HIF-1α). Stabilization of HIF-1α increased FGF-2 promoter activity, whereas mutation of the putative HRE attenuated FGF-2-induced FGF-2 promoter activity. Furthermore, FGF-2 increased HIF-1α protein levels and consensus HRE promoter activity in PASMC via antioxidant-sensitive mechanisms. Thus we conclude that FGF-2 can stimulate its own expression in PASMC via NADPH oxidase-mediated activation of ROS-sensitive transcription factors, including HIF-1α. This positive feedback mechanism may contribute to pulmonary vascular remodeling associated with increased pulmonary blood flow.
Keywords: cell signaling, proliferation, pulmonary hypertension
congenital heart disease (CHD) with increased pulmonary blood flow is often associated with the development of pulmonary hypertension (12). After birth, as pulmonary vascular resistance normally decreases, the presence of a systemic to pulmonary communication generates an increase in pulmonary blood flow. This abnormal postnatal hemodynamic state results in progressive structural and functional abnormalities of the pulmonary vascular bed (21, 23). Our animal model of pulmonary hypertension, developed by inserting an aortopulmonary vascular graft (shunt) in the late-gestational fetal lamb, may help elucidate the mechanisms involved (2, 3, 25). Postnatally, these lambs have increased pulmonary blood flow and pressure (25). In addition, at 4 wk of age they display vascular remodeling typical of pulmonary hypertension, characterized by increased medial wall thickness of the small pulmonary arteries and abnormal extension of muscle to peripheral pulmonary arteries (12, 24, 25).
Our previous studies have demonstrated that lambs with increased pulmonary blood flow display abnormal signaling by several growth factors mitogenic for vascular smooth muscle, including endothelin-1 (ET-1) (2), transforming growth factor-β1 (TGF-β1) (19), and vascular endothelial growth factor (VEGF) (18). Fibroblast growth factor-2 (FGF-2) is another mitogen for vascular smooth muscle cells (SMC) (7, 16), and our recent data suggest that dysregulated FGF-2 expression may also contribute to pulmonary vascular remodeling in these lambs (31). Lung tissue extracts displayed increased FGF-2 mRNA and protein levels relative to age-matched controls, and immunohistochemistry demonstrated increased FGF-2 protein within the pulmonary arteries (31). In addition, plasma FGF-2 protein levels were significantly higher relative to control lambs (31). Cyclic stretch and laminar shear stress stimulated the release of FGF-2 from pulmonary arterial smooth muscle cells (PASMC) and endothelial cells (PAEC) (31), suggesting that biomechanical forces resulting from increased pulmonary blood flow may contribute to elevated plasma levels. Positive feedback regulation of FGF-2 expression has been described in several tissue culture systems, including cardiomyocytes (14) and human umbilical vein endothelial cells (HUVEC) (6). From these data we hypothesize that elevated levels of circulating FGF-2 may contribute to increased FGF-2 expression and PASMC proliferation in pulmonary arteries exposed to increased blood flow. Recently, we demonstrated an association between increased pulmonary blood flow and increased oxidant stress (10). Thus, in this study, we wanted to examine potential effects of exogenous FGF-2 on endogenous FGF-2 expression in PASMC and to evaluate the role of reactive oxygen species (ROS) in this process.
MATERIALS AND METHODS
Cell culture.
Primary cultures of PASMC from 4-wk-old sheep that had not previously been surgically operated were isolated using the explant technique as described in our earlier study (30). Identity was confirmed as PASMC by immunostaining (>99% positive) with antibodies against α-smooth muscle actin, calponin, and caldesmon. This was taken as evidence that cultures were not contaminated with fibroblasts or endothelial cells. All cultures for subsequent experiments were maintained in DMEM supplemented with 10% fetal calf serum (Hyclone), antibiotics, and antimycotics (penicillin, streptomycin, and amphotericin B; Invitrogen) at 37°C in a humidified atmosphere with 5% CO2–95% air. Cells were utilized between passages 3 and 10.
For cell culture treatments, FGF-2 was obtained from Invitrogen, TGF-β1 from Sigma, and ET-1 from Calbiochem. Diethyldithiocarbamate (DETC), N-acetyl cysteine (NAC), and desferroxamine (DFO) were obtained from Sigma, and methyl viologen dichloride hydrate (paraquat) was obtained from Aldrich. Manganese(III) tetrakis (4-benzoic acid) porphyrin (MnTBAP), LY-294002, wortmannin, 1L-6-hydroxymethyl-chiro-inositol 2-(R)-2-O-methyl-3-O-octadecylcarbonate (Akt inhibitor), and apocynin were all obtained from Calbiochem.
Promoter analysis.
Plasmids containing human FGF-2 promoter DNA from −1800 to +314 bp and −103 to +314 bp (relative to the transcription start site) fused to a luciferase reporter gene were gifts from Dr. M. Stachowiak (SUNY Buffalo). A plasmid containing hypoxia response elements (HRE) fused to a luciferase reporter was a gift from Dr. Paul Schumacker (Northwestern University). Cells were cotransfected at 70% confluence with 4 μg of plasmid DNA and 0.1 μg of Renilla luciferase internal control vector (Promega) on 10-cm2 tissue culture plates using Effectine (Qiagen) according to the manufacturer's instructions. After 24 h, transfected cells were trypsinized, split onto 24-well plates, and allowed to adhere. Cells were washed in PBS and maintained in serum-free DMEM together with the appropriate growth factor or inhibitor for another 24 h. Firefly and Renilla luciferase activity of 10 μl of protein extracts was determined using the Dual-Luciferase Reporter Assay System (Promega) and a Femtomaster FB12 luminometer (Zylux). Signals were normalized to Renilla luciferase for each well.
Western blotting.
Western blot analysis was performed as previously described (4, 29). Cells were incubated with 0.05% trypsin (Invitrogen) for 15 min at 37°C to remove exogenous FGF-2, where appropriate, and lysed in Mg2+ lysis buffer (Upstate) for 5 min at room temperature. Protein extracts (20 μg) were separated on 4–20% denaturing polyacrylamide gels (Bio-Rad). Recombinant human FGF-2 (10 ng) was used as a positive control on blots to detect FGF-2. Precision Plus protein standards (5 μl; Bio-Rad) were used as molecular weight markers. All gels were electrophoretically transferred to Hybond nitrocellulose membranes (Amersham, Arlington Heights, IL). The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween (TBST). After blocking, the membranes were incubated at 4°C overnight with polyclonal antiserum raised against FGF-2 (1:200, sc-79; Santa Cruz Biotechnology) or monoclonal antiserum raised against hypoxia-inducible factor-1α (HIF-1α) (1:100, NB 100-105; Novus Biologicals), washed in TBST, and then incubated with the appropriate goat IgG-horseradish peroxidase conjugate (Pierce). After washing, chemiluminescence was used to detect the protein bands using SuperSignal West Femto Substrate (Pierce), and bands were visualized and quantified using an ImageStation (Kodak). Blots were reprobed for β-actin using a monoclonal antibody (Sigma), and intensities were determined as described above. FGF-2 and HIF-1α signals were normalized to β-actin.
Immunocytochemistry and fluorescence detection on cultured cells.
PASMC were seeded onto 96-well plates (Costar), allowed to adhere, and incubated with the appropriate treatment for 24 h as described above. Cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% IGEPAL (Sigma) for 5 min at room temperature where appropriate, blocked in 5% BSA at room temperature for 1 h, and probed with a polyclonal antibody against FGF-2 (1:200, sc-79; Santa Cruz Biotechnology) in 5% BSA at 4°C overnight. Cells were washed in PBS and probed with anti-rabbit IgG conjugated to Oregon green or rhodamine red (Molecular Probes). Nuclei were counterstained with 5 μM 4′,6-diamidino-2-phenylinodole, and fluorescence was visualized as described previously (32). Cells were imaged using a Nikon Eclipse TE-300 fluorescence microscope. Fluorescent images were captured using a CoolSnap digital camera, and the average fluorescence intensities (to correct for differences in cell number) were quantified using MetaMorph imaging software (Fryer). Briefly, the total fluorescence intensity above a constant threshold intensity was calculated for each field and divided by the total pixel number at that threshold. Total pixel number is indicative of total cell number, and dividing the total fluorescence intensity generated by every cell in a field by the total pixel number resulting from those cells yields an average fluorescence intensity that corrects for growth factor-induced cell proliferation. Three wells were used for each treatment, and three different fields were captured randomly from each well. The mean value of these nine images was calculated to generate an n value of 1. We have used this technique previously to detect changes in protein expression comparable in magnitude to those detected by Western blotting (32).
Dihydroethidium fluorescence analysis.
PASMC were seeded onto 96-well plates (Costar) and allowed to adhere for at least 18 h. Cells were then washed in PBS and incubated in serum-free DMEM containing FGF-2, apocynin, paraquat, or DETC where required. Dihydroethidium (DHE; 5 μM; Molecular Probes) was added to the media 15 min before the end of the experiment. Cells were washed with PBS and imaged using a Nikon Eclipse TE-300 fluorescence microscope. Oxidation of DHE was observed after excitation at 518 nm and emission at 605 nm. Fluorescent images were captured using a CoolSnap digital camera, and the average fluorescence intensities (to correct for differences in cell number) were quantified using MetaMorph imaging software (Fryer) as described above.
Cell proliferation assays.
PASMC at ∼2,500 cells per well were seeded onto 96-well plates (Costar) (∼25% confluence) and allowed to adhere for at least 18 h. Cells were incubated with the appropriate treatments for 72 h, since we have found previously that this time point gave easily detectable differences in viable cell number (30). This was determined using the Cell Titer 96 AQueous One Solution kit (Promega), the basis of which has been shown to be a reliable alternative to [3H]thymidine incorporation (9). The tetrazolium reagent is bioreduced to a colored product, the quantity of which is proportional to the number of metabolically active cells. Reagent (20 μl) was added directly to cells in 100 μl of medium, and following a 2-h incubation period at 37°C, the absorbance at 492 nm was read using a Labsystems Multiskan EX plate reader (Fisher).
Site-directed mutagenesis.
A 3-bp mutation was introduced into the putative HRE of the −103-bp FGF-2 promoter construct using the QuikChange II XL kit according to the manufacturer's instructions (Stratagene). The sequence TGCGTG was mutated to TGaaaG using the oligo 5′-GGACTGATGTCGCGCGCTTGaaaGTTGTGGCCGAAGCCGCCG designed from the human FGF-2 exon 1 genomic clone sequence (accession no. X04431). Briefly, mutant-strand DNA synthesis was achieved using the −103-bp construct plasmid as template, and the above oligo and its complement were extended by thermal cycling using PfuUltra DNA polymerase. Selection for nonmethylated mutant DNA was performed by digesting methylated parental DNA with DpnI. Ultracompetent XL-10 Gold cells were then transformed, and positive clones were identified by sequencing miniprep DNA. PASMC were transfected with maxiprep DNA and the Renilla luciferase control plasmid, and luciferase assays were performed as described above.
Statistical analysis.
All values were calculated and expressed as means ± SD relative to untreated samples. Comparisons between treatment groups were made by ANOVA, using the GB-STAT software program. Newman-Keuls post hoc testing was performed when differences were present between study groups. A P < 0.05 was considered statistically significant.
RESULTS
We initially determined the effects of exogenous FGF-2 on FGF-2 promoter activity. PASMC growth was induced by exogenous FGF-2 at 100 ng/ml (31), and we based our concentration range on these studies. After 24 h, exogenous FGF-2 dose-dependently stimulated luciferase activity driven by DNA from −1800 bp to +314 bp of the FGF-2 promoter (Fig. 1A). Maximal increases in luciferase activity were detected after 24 h of treatment with 100 ng/ml FGF-2, with significant increases detected at 8 and 48 h (Fig. 1B). Western blot analysis demonstrated a corresponding increase in FGF-2 protein levels in PASMC following a 24-h incubation with 100 ng/ml FGF-2 (Fig. 1, C–E). The predominant endogenous FGF-2 isoform in PASMC migrated with a slightly greater molecular mass than the exogenous human recombinant FGF-2 (Fig. 1C). Furthermore, preincubation of PASMC with trypsin before cell lysis resulted in the removal of the exogenous FGF-2 band (Fig. 1D), suggesting that this protein is associated with the extracellular matrix and does not contribute to the quantification of intracellular FGF-2 protein. We also performed immunocytochemistry using the same antibody to determine the effects of exogenous FGF-2 on FGF-2 protein levels. In agreement with the Western data, 100 ng/ml FGF-2 significantly increased FGF-2 protein in PASMC after 24 h (Fig. 1, F and G). No differences were detected between untreated and FGF-2-treated cells in the absence of cell permeabilization (Fig. 1F), suggesting that extracellular exogenous FGF-2 does not contribute to the quantification of intracellular FGF-2 in these experiments.
Fig. 1.

Exogenous fibroblast growth factor-2 (FGF-2) increases FGF-2 expression in pulmonary artery smooth muscle cells (PASMC). Luciferase activity was determined for an 1,800-bp FGF-2 promoter fragment in PASMC exposed to 0–100 ng/ml FGF-2 for 24 h (A) or to 100 ng/ml FGF-2 for 0–48 h (B). Values were normalized to an internal Renilla luciferase control. Values are means ± SD; n = 4. *P < 0.05 vs. untreated cells (UTD). C: representative Western blot depicting increased endogenous FGF-2 protein in PASMC exposed to 100 ng/ml FGF-2 for 24 h. C, 10 ng of 17-kDa human recombinant FGF-2 protein control; M, molecular mass markers with sizes indicated at right. D: representative Western blot depicting increases in endogenous FGF-2 protein in PASMC exposed to 100 ng/ml FGF-2 for 24 h. Exogenous FGF-2 was removed following digestion with trypsin. Blots were also probed for β-actin. Migration of molecular mass markers is indicated at right. E: bands from Western blots were quantified using ImageStation software, and FGF-2 signals were normalized to β-actin. Values are means ± SD; n = 6. *P < 0.05 vs. untreated cells. F: representative fluorescent images from immunocytochemistry (ICC) experiments to detect changes in FGF-2 protein in PASMC exposed to 100 ng/ml FGF-2 for 24 h. Images depicting loss of signal when cell permeabilization was omitted with the corresponding 4′,6-diamidino-2-phenylinodole (DAPI) nuclear stains are presented. G: images from ICC experiments were quantified using MetaMorph software and expressed as fold change relative to UTD. Values are means ± SD; n = 5. *P < 0.05 vs. untreated cells.
We next investigated whether FGF-2 treatment was altering superoxide levels in PASMC. Our data indicate that incubation with 100 ng/ml FGF-2 increased DHE oxidation in PASMC after 24 h (Fig. 2, A and B). Furthermore, we found that this increase was attenuated by cotreatment with the NADPH oxidase inhibitor apocynin (Fig. 2, A and B). Treatment with the CuZn-superoxide dismutase inhibitor DETC or the superoxide generator paraquat also increased DHE oxidation in PASMC (Fig. 2, A and B). Dose-response experiments demonstrated a significant increase in DHE fluorescence following a 24-h exposure to 10 ng/ml FGF-2, with a statistically insignificant increase detected at 1 ng/ml (Fig. 2C).
Fig. 2.

FGF-2 increases superoxide production in PASMC. A: fluorescence microscopy to detect changes in dihydroethidium (DHE) fluorescence. PASMC were treated with 100 ng/ml FGF-2 with or without 10 μM apocynin (Apo) for 24 h before DHE treatment. Cells were also treated with 50 μM diethylthiocarbamate (DETC) or 1 mM paraquat (Para) for 24 h to increase intracellular superoxide. B: DHE fluorescence was quantified using MetaMorph software and expressed as fold UTD. Values are means ± SD; n = 3. *P < 0.05 vs. UTD. †P < 0.05 vs. FGF-2-treated cells. C: dose-dependent changes in DHE fluorescence in PASMC exposed to 0–1,000 ng/ml FGF-2 for 24 h. Values are means ± SD; n = 3. *P < 0.05 vs. untreated cells.
The above data suggest that exogenous FGF-2 may stimulate endogenous FGF-2 expression in PASMC via a positive feedback mechanism involving ROS. We next used pharmacological inhibition to begin to identify the signaling intermediates involved. Inhibition of phosphatidylinositol 3-kinase (PI3 kinase), Akt, and NADPH oxidase all attenuated the stimulatory effects of exogenous FGF-2 on FGF-2 promoter activity (Fig. 3A). In agreement, these pharmacological inhibitors also attenuated FGF-2-mediated increases in FGF-2 protein (Fig. 3B) and PASMC number (Fig. 3C). In the same experiments, optimum growth medium containing 10% serum increased the number of PASMC to 182 ± 15% relative to serum-free medium (Fig. 3C).
Fig. 3.

Effects of pharmacological inhibition on FGF-2-induced FGF-2 expression and cell proliferation in PASMC. PASMC exposed to 0 or 100 ng/ml FGF-2 for 24 h were coincubated with 10 μM LY-294002 (LY) and 200 nM wortmannin (Wort) to inhibit phosphatidylinositol 3-kinase (PI3 kinase), 10 μM Akt inhibitor (AktI), and 10 μM Apo to inhibit NADPH oxidase. A: FGF-2 promoter activity relative to untreated cells. Values were normalized to an internal Renilla luciferase control. B: data from ICC experiments to detect changes in FGF-2 protein relative to untreated cells. C: serum-starved PASMC were treated as above. After 72 h, the number of actively respiring PASMC was determined using a methylthiazoletetrazolium-based assay. Values are means ± SD; n = 4. *P < 0.05 vs. untreated cells. †P < 0.05 vs. FGF-2-treated cells.
We next sought to establish a causal link between increased intracellular superoxide and increased FGF-2 expression in PASMC. Because we had previously shown that ET-1 and TGF-β1 stimulate NADPH oxidase-derived superoxide production in PASMC (17, 30), we investigated whether treatment with these agents would increase FGF-2 expression. After 24 h, TGF-β1 and ET-1 dose-dependently increased FGF-2 promoter activity (Fig. 4A) and increased protein expression as determined by Western blot analysis (Fig. 4, B and C) and immunohistochemistry (Fig. 4, D and E). The increases in FGF-2 protein induced by ET-1 and TGF-β1 were also blocked by the NADPH oxidase inhibitor apocynin (Fig. 4E).
Fig. 4.

Endothelin-1 (ET-1) and transforming growth factor-β1 (TGF-β1) increase FGF-2 expression in PASMC. A: luciferase activity was determined for an 1,800-bp FGF-2 promoter fragment in PASMC exposed to 100 nM ET-1 or 1 ng/ml TGF-β1 for 24 h. Values were normalized to an internal Renilla luciferase control. Values are means ± SD; n = 4. *P < 0.05 vs. untreated cells. B: representative Western blot depicting changes in FGF-2 protein in PASMC exposed to 100 nM ET-1 or 1 ng/ml TGF-β1 for 24 h. Blots were also probed for β-actin. U, untreated cells. C: bands from Western blots were quantified using ImageStation software, and FGF-2 signals were normalized to β-actin. Migration of molecular weight markers is indicated at right. Values are means ± SD; n = 4. *P < 0.05 vs. untreated cells. D: representative fluorescent images from ICC experiments to detect changes in FGF-2 protein in PASMC exposed to ET-1 or TGF-β1 for 24 h. E: cells were treated with 100 nM ET-1 or 1 ng/ml TGF-β1 and 0 or 10 μM Apo for 24 h. Images from ICC experiments were quantified using MetaMorph software and are expressed as fold change relative to untreated cells. Values are means ± SD; n = 4. *P < 0.05 vs. untreated cells. †P < 0.05 vs. growth factor-treated cells.
To further confirm the importance of ROS in mediating increases in FGF-2 expression, we exposed PASMC to DETC and paraquat. Our data indicate that DETC and paraquat increased both FGF-2 promoter activity (Fig. 5A) and FGF-2 protein levels (Fig. 5B) in PASMC after 24 h of exposure. Furthermore, the stimulatory effects of exogenous FGF-2 were attenuated by the superoxide dismutase mimetic MnTBAP (Fig. 5C) and the antioxidant NAC (Fig. 5D), although these treatments had no effect on basal promoter activity (Fig. 5, C and D). NAC also attenuated induction of FGF-2 promoter activity by both ET-1 and TGF-β1 (Fig. 5D).
Fig. 5.

Increased intracellular superoxide stimulates FGF-2 expression in PASMC. A: increased promoter activity in PASMC treated with 0–1 mM Para or with 0–50 μM DETC for 24 h. Values were normalized to an internal Renilla luciferase control. Values are means ± SD; n = 4. *P < 0.05 vs. untreated cells. B: increased FGF-2 protein in PASMC treated with 1 mM Para or 50 μM DETC for 24 h detected by ICC. Images were quantified using MetaMorph software and are expressed as fold change relative to untreated cells. Values are means ± SD; n = 4. *P < 0.05 vs. untreated cells. C: increased FGF-2 promoter activity in response to 100 ng/ml FGF-2 was attenuated by cotreatment with 100 μM manganese(III) tetrakis (4-benzoic acid) porphyrin (MnTBAP). *P < 0.05 vs. untreated cells. †P < 0.05 vs. FGF-2-treated cells. D: increased FGF-2 promoter activity in PASMC treated with 100 ng/ml FGF-2, 100 nM ET-1, or 1 ng/ml TGF-β1 (0 NAC) was attenuated by cotreatment with 1 mM N-acetyl cysteine (NAC). Values are means ± SD; n = 3. *P < 0.05 vs. untreated cells. †P < 0.05 vs. growth factor-treated cells.
Using truncated promoter fragments, we localized the FGF-2-responsive region within the proximal 103 bp of the FGF-2 promoter in PASMC (Fig. 6A). Luciferase activity of the 103-bp construct was also stimulated by DETC and paraquat (Fig. 6B). Sequence analysis identified a putative HRE binding site for the ROS-sensitive transcription factor HIF-1α within the proximal 103 bp, and treatment with 100 μM DFO, an iron-chelating stabilizer of HIF-1α protein, also stimulated luciferase activity driven by this FGF-2 promoter fragment (Fig. 6C). Mutating the putative HRE (TGCGTG to TGaaaG) within the FGF-2 promoter significantly decreased FGF-2-induced luciferase activity from the 103-bp promoter fragment, although this did not completely abolish the stimulatory effects (Fig. 6C). Conversely, DFO treatment failed to stimulate promoter activity from the HRE mutant construct (Fig. 6C). In addition, induction of promoter activity by TGF-β1 was also attenuated in the HRE mutant construct, with a statistically insignificant decrease also detected for induction by ET-1 (Fig. 6D).
Fig. 6.

FGF-2 promoter deletion constructs are responsive to FGF-2 and ROS in PASMC. A: constructs containing promoter DNA from +314 to −973, −555, or −103 bp were transfected into PASMC and treated with 0 or 100 ng/ml FGF-2 for 24 h. Values for FGF-2 treatment are expressed as fold untreated for each deletion construct. B: cells transfected with the −103-bp construct were treated with 50 μM DETC or 1 mM Para for 24 h. C and D: cells were transfected with the wild-type −103-bp construct or with a −103-bp construct containing a 3-bp mutation in the putative hypoxia response element (HRE). Cells were exposed to 100 ng/ml FGF-2 or 100 μM DFO (C) or to 100 nM ET-1 or 1 ng/ml TGF-β1 (D) for 24 h. Luciferase values were normalized to an internal Renilla luciferase control. Values are means ± SD; n = 4. *P < 0.05 vs. untreated cells. †P < 0.05 vs. wild type.
Western blot analysis demonstrated an increase in HIF-1α protein in PASMC treated with FGF-2 for 24 h, which was attenuated by cotreatment with NAC (Fig. 7, A and B). In addition, a 24-h incubation with FGF-2 increased luciferase activity driven by consensus HRE sequences in PASMC, which was also attenuated by NAC (Fig. 7C). Under the same conditions, 100 μM DFO increased luciferase activity 5.8-fold relative to untreated cells (Fig. 7C).
Fig. 7.
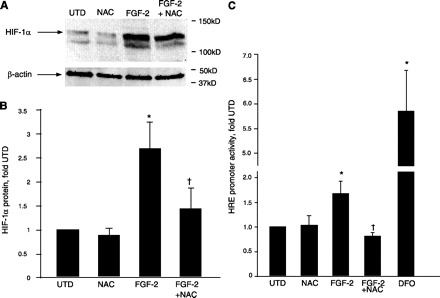
FGF-2 increases hypoxia-inducible factor-1α (HIF-1α) protein and HRE promoter activity via an antioxidant-sensitive mechanism in PASMC. A: representative Western blot showing increased HIF-1α protein in PASMC treated with 100 ng/ml FGF-2 with or without 1 mM NAC for 24 h. A protein band migrating with a lower molecular mass than HIF-1α was routinely observed using this antibody but was not used for quantification. Blots were also probed for β-actin. Migration of molecular mass markers is indicated at right. B: bands from Western blots were quantified using ImageStation software, and HIF-1α signals were normalized to β-actin. Values are means ± SD; n = 6. *P < 0.05 vs. untreated cells. †P < 0.05 vs. FGF-2-treated cells. C: PASMC were transfected with a plasmid containing a consensus HRE sequence upstream of a luciferase reporter. Cells were treated with 100 mg/ml FGF-2 with or without 1 mM NAC or with 100 μM DFO alone for 24 h. Luciferase values were normalized to an internal Renilla luciferase control. Values are means ± SD; n = 4. *P < 0.05 vs. untreated cells. †P < 0.05 vs. FGF-2-treated cells.
DISCUSSION
The excessive muscularization of the small pulmonary arteries in children with pulmonary hypertension secondary to CHD suggests that abnormal expression of growth factors mitogenic for vascular SMC may be involved in vascular remodeling. Our previous studies identified increased levels of ET-1 in plasma (33) and TGF-β1 in lung tissue (19) in a 4-wk-old lamb model of CHD with increased pulmonary blood flow. Recently, we demonstrated increased lung FGF-2 mRNA and protein in this model, with increased FGF-2 protein also evident in pulmonary arteries and in plasma (31). Since elevated FGF-2 protein levels have been detected in the urine and plasma of patients with pulmonary arterial hypertension, there is a potential role for FGF-2 in pulmonary hypertension (1). However, the mechanisms responsible for abnormal FGF-2 expression and release are unclear. The purpose of this study was to determine whether growth factors associated with pulmonary vascular remodeling potentially contribute to FGF-2 expression in PASMC, and to characterize the mechanisms involved.
Exogenous FGF-2 stimulated endogenous FGF-2 promoter activity and protein levels in PASMC isolated from 4-wk-old lambs, suggesting the potential involvement of a positive feedback mechanism. The predominant endogenous FGF-2 protein isoform in both untreated and FGF-2-stimulated cells was ∼18 kDa and could be distinguished from the 17-kDa exogenous protein on a Western blot. Digestion with trypsin before cell lysis removed the smaller band from Western blots, suggesting that the exogenous protein was associated with the extracellular matrix and not taken up by the PASMC. Furthermore, omission of cell permeabilization abolished the FGF-2-induced signal detected by immunocytochemistry, suggesting that extracellular protein was not contributing to the quantification of intracellular FGF-2 in these experiments. Since the increases in FGF-2-induced promoter activity and endogenous FGF-2 protein were comparable to increases detected by immunocytochemistry, it appears that exogenous FGF-2 does not contribute to the quantification of endogenous protein in these studies. Increases in promoter activity were detected at FGF-2 concentrations of 1 ng/ml, and our previous studies demonstrated the stimulation of PASMC proliferation at 100 pg/ml (31). We previously quantified systemic arterial FGF-2 protein concentrations to be 10 pg/ml in lambs with increased pulmonary blood flow (31), although localized pulmonary arterial concentrations may be higher in these animals due to flow-induced FGF-2 release. In addition, total lung FGF receptor 1 (FGF R1) mRNA was increased in lambs with increased pulmonary blood flow (Black SM, unpublished observations). Thus PASMC in vivo may be exposed to higher local concentrations of FGF-2 and may exhibit enhanced signaling in animals with increased pulmonary blood flow due to elevated FGF R1 expression.
Other studies have shown the potential interactions between these growth factor signaling pathways. For example, TGF-β1 was shown to upregulate FGF-2 expression in vascular SMC (5), whereas FGF-2 was found to increase ET-1 subtype A receptor (ETA) expression in PASMC (15). Our previous studies demonstrated increased expression of ETA (2) and the TGF-β1 receptor ALK-1 (19) in lambs with increased pulmonary blood flow, suggesting that PASMC isolated from these animals may also exhibit phenotypic differences in FGF-2 expression in response to growth factor treatment. Together, these data suggest that dysregulation of one pathway may perturb other signaling cascades and amplify the overall effects on smooth muscle growth in vivo.
Since ET-1 and TGF-β1 have been shown to increase superoxide production in PASMC via the activation of NADPH oxidase (17, 30), we examined the possibility that exogenous FGF-2 also stimulates this enzyme complex. FGF-2 induced superoxide production in PASMC that was attenuated by the NADPH oxidase inhibitor apocynin, thus adding FGF-2 to the family of growth factors that stimulate NADPH oxidase in PASMC. Increases in growth factor-induced FGF-2 protein were also attenuated by apocynin, suggesting that activation of the respective receptors for ET-1, TGF-β1, and FGF-2 stimulates pathways that converge on the NADPH oxidase system. Since apocynin inhibits assembly of the enzyme (28), further studies are required to determine which subunits of NADPH oxidase are affected by individual growth factors.
We have previously shown that ET-1-mediated NADPH oxidase activation in PASMC required PI3 kinase (17, 30), and the current study suggests the involvement of PI3 kinase and Akt in FGF-2 signaling. Inhibition of PI3 kinase, Akt, and NADPH oxidase also attenuated FGF-2-induced PASMC proliferation, and we have shown previously that growth factor-independent increases in ROS levels stimulate PASMC proliferation (17, 22, 30). From these data, we hypothesize that ROS are downstream mediators of several distinct FGF-2-induced pathways in PASMC, including the presently uncharacterized signaling cascade that stimulates cell proliferation and the positive feedback mechanism that increases endogenous FGF-2 expression. Recently, Schröder et al. (26) demonstrated the involvement of PI3 kinase, protein kinase C, and the NADPH oxidase subunits Rac1 and Nox1 in FGF-2-induced migration of rat aortic SMC. Together, these data suggest that common ROS-mediated FGF-2 signaling pathways may be active in vascular SMC.
Our data also show that growth factor-independent increases in ROS stimulated FGF-2 promoter activity and protein levels in PASMC, whereas antioxidants attenuated FGF-2-mediated FGF-2 promoter activity, suggesting that ROS-sensitive transcription factors play an important role in regulating the induction of FGF-2 expression. Using truncated promoter constructs, we located the FGF-2- and ROS-responsive elements within the proximal 103 bp of the FGF-2 promoter. In agreement, a previous study demonstrated that this region was responsive to FGF-2 treatment in neonatal rat cardiomyocytes (14). ET-1 and TGF-β1 also significantly increased luciferase activity from this construct, although the magnitude was less than for the 1,800-bp promoter fragment. These data suggest that additional transcription factor binding sites further upstream may be required for maximal induction of FGF-2 promoter activity by ET-1 and TGF-β1. A “growth factor-responsive element” was located in the FGF-2 promoter at −555/−513 bp in human astrocytes (20), and further studies are required to determine whether this sequence is involved in ET-1 and TGF-β1 signaling in PASMC.
Our DNA sequence analysis of the FGF-2 promoter identified a putative HRE, a consensus binding site for the transcription factor HIF-1α, located at −32 bp (5′-TGCGTG-3′). Recent studies have demonstrated the requirement for HIF-1α in FGF-2-induced human PASMC proliferation (27) and in hypoxic induction of FGF-2 mRNA in HUVEC (6). Furthermore, HIF-1α protein is stabilized by cytosolic ROS (11), suggesting that HIF-1α may be involved in the upregulation of FGF-2 promoter activity in PASMC exposed to stimuli that increase intracellular ROS. In agreement with this hypothesis, treatment with the HIF-1α-stabilizing compound DFO increased luciferase activity from the 103-bp FGF-2 promoter fragment in PASMC. In addition, FGF-2-induced increases in HIF-1α protein levels and HRE promoter activity were attenuated by the antioxidant NAC. A 3-bp mutation within the putative HRE significantly decreased, but did not abolish, FGF-2-induced FGF-2 promoter activity in PASMC, suggesting that additional ROS-sensitive transcription factors may also be involved. Candidate molecules include early growth response gene-1 (egr-1) and Sp-1, which were demonstrated to bind to the proximal 100 bp of the FGF-2 promoter in cardiomyocytes (14). Further studies are required to identify the factors responsible for maximal stimulation of ROS-induced FGF-2 promoter activity in PASMC.
Increased oxidative stress detected in the lungs of lambs with increased pulmonary blood flow was associated with upregulation of NADPH oxidase subunits and was attenuated by apocynin (10). The data presented above suggest that elevated plasma levels of FGF-2, ET-1, and TGF-β1 may all stimulate NADPH oxidase-derived superoxide in these lambs. In addition, cyclic stretch generated by increased pulmonary blood flow may also contribute to oxidative stress by activating NADPH oxidase in PASMC (17). Thus several ROS-mediated pathways are potentially involved in the increased FGF-2 expression associated with increased pulmonary blood flow (31). We showed an increase in FGF-2 promoter activity and protein levels in PASMC exposed to 24-h cyclic stretch (31), and cyclic stretch induced a transient increase in HIF-1α protein in rat aortic SMC (8). Furthermore, we have previously shown that cyclic stretch stimulated the release of TGF-β1 (17) and FGF-2 (31) from PASMC, which could potentially stimulate FGF-2 expression by activating extracellular receptors. However, a role for ROS and HIF-1α in stretch-induced FGF-2 expression in PASMC, and in pulmonary vascular remodeling associated with increased blood flow, has yet to be established.
Autocrine and paracrine signaling by FGF-2 mediates several processes, including cardioprotection and angiogenesis. In a murine model of low-flow ischemia, overexpression of FGF-2 was found to protect against myocardial dysfunction (13), whereas other studies suggested that FGF-2 regulates its own synthesis in cardiac myocytes, which may help to maintain a healthy myocardium (14). Our data suggest that FGF-2-induced FGF-2 expression involves ROS and HIF-1α in PASMC. An autocrine HIF-1α-FGF-2 amplification loop appears to be involved in hypoxia-induced angiogenesis in HUVEC (6), and dysregulation may promote tumor growth. Interestingly, hypoxia was shown to stabilize HIF-1α via the release of ROS from the mitochondria to the cytosol (11). Our data suggest that in PASMC, ROS-mediated stabilization of HIF-1α increases FGF-2 expression, which then stimulates further ROS production by NADPH oxidase and completes the loop. Although cell-specific differences likely exist, further elucidation of the complex mechanisms that regulate FGF-2 expression may identify therapeutic targets for a diverse range of diseases resulting from abnormal FGF-2 signaling.
GRANTS
This work was supported by American Heart Association (AHA) National Office Grant 0330292N (to S. Wedgwood), National Institutes of Health Grants HL60190, HL67841, HL72123, HL70061, and HD39110 (all to S. M. Black), AHA Pacific Mountain Affiliates Grant 0550133Z (to S. M. Black), and a Leducq Foundation Transatlantic Network Award (to S. M. Black).
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Bentisty JI, McLaughlin VV, Landzberg MJ, Rich JD, Newburger JW, Rich S, Folkman J. Elevated fibroblast growth factor levels in patients with pulmonary arterial hypertension. Chest 126: 1255–1261, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Black SM, Bekker JM, Johengen MJ, Parry AJ, Soifer SJ, Fineman JR. Altered regulation of the ET-1 cascade in lambs with increased pulmonary blood flow and pulmonary hypertension. Pediatr Res 47: 97–106, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Black SM, Fineman JR, Steinhorn RH, Bristow J, Soifer SJ. Increased endothelial NOS in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Heart Circ Physiol 275: H1643–H1651, 1998 [DOI] [PubMed] [Google Scholar]
- 4.Black SM, Johengen MJ, Ma ZD, Bristow J, Soifer SJ. Ventilation and oxygenation induce endothelial nitric oxide synthase gene expression in the lungs of fetal lambs. J Clin Invest 100: 1448–1458, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brogi E, Wu T, Namiki A, Isner JM. Indirect angiogenic cytokines upregulate VEGF and bFGF gene expression in vascular smooth muscle cells, whereas hypoxia upregulates VEGF expression only. Circulation 90: 649–652, 1994 [DOI] [PubMed] [Google Scholar]
- 6.Calvani M, Rapisarda A, Uranchimeg B, Shoemaker R, Melillo G. Hypoxic induction of an HIF-1α-dependent bFGF autocrine loop drives angiogenesis in human endothelial cells. Blood 107: 2705–2712, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casscells W, Lappi DA, Olwin BB, Wai C, Siegman M, Speir EH, Sasse J, Baird A. Elimination of smooth muscle cells in experimental restenosis: targeting of fibroblast growth factor receptors. Proc Natl Acad Sci USA 89: 7159–7163, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang H, Shyu K, Wang B, Kuan P. Regulation of hypoxia-inducible factor 1α by cyclical mechanical stretch in rat vascular smooth muscle cells. Clin Sci (Lond) 105: 447–456, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun 3: 207–212, 1991 [DOI] [PubMed] [Google Scholar]
- 10.Grobe A, Wells S, Benavidez E, Oishi P, Azakie A, Fineman J, Black S. Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: role of NADPH oxidase and endothelial NO synthase. Am J Physiol Lung Cell Mol Physiol 290: L1069–L1077, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Guzy R, Hoyos B, Robin E, Chen H, Liu L, Mansfield K, Simon M, Hammerling U, Schumacker P. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab: 401–408, 2005 [DOI] [PubMed]
- 12.Hoffman JI, Rudolph AM, Heymann MA. Pulmonary vascular disease with congenital heart lesions: pathologic features and causes. Circulation 64: 873–877, 1981 [DOI] [PubMed] [Google Scholar]
- 13.House S, Bolte C, Zhou M, Doetschman T, Klevitsky R, Newman G, Schultz J. Cardiac-specific overexpression of fibroblast growth factor-2 protects against myocardial dysfunction and infarction in a murine model of low-flow ischemia. Circulation 108: 3140–3148, 2003 [DOI] [PubMed] [Google Scholar]
- 14.Jimenez SK, Sheikh F, Jin Y, Detillieux KA, Dhaliwal J, Kardami E, Cattini PA. Transcriptional regulation of FGF-2 gene expression in cardiac myocytes. Cardiovasc Res 62: 548–557, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Li P, Oparil S, Sun JZ, Thompson JA, Chen YF. Fibroblast growth factor mediates hypoxia-induced endothelin-A receptor expression in lung artery smooth muscle cells. J Appl Physiol 95: 643–651, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Lindner V, Lappi DA, Baird A, Majack RA, Reidy MA. Role of basic fibroblast growth factor in vascular lesion formation. Circ Res 68: 106–113, 1991 [DOI] [PubMed] [Google Scholar]
- 17.Mata-Greenwood E, Grobe A, Kumar S, Noskina Y, Black SM. Cyclic stretch increases VEGF expression in pulmonary arterial smooth muscle cells via TGF-β1 and reactive oxygen species: a requirement for NAD(P)H oxidase. Am J Physiol Lung Cell Mol Physiol 289: L288–L298, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Mata-Greenwood E, Meyrick B, Soifer SJ, Fineman JR, Black SM. Expression of VEGF and its receptors Flt-1 and Flk-1/KDR is altered in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 285: L222–L231, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Mata-Greenwood E, Meyrick B, Steinhorn RH, Fineman JR, Black SM. Alterations in TGF-β1 expression in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 285: L209–L221, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Moffett J, Kratz E, Myers J, Stachowiak E, Florkiewicz R, Stachowiak M. Transcriptional regulation of fibroblast growth factor-2 expression in human astrocytes: implications for cell plasticity. Mol Biol Cell 9: 2269–2285, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagumo K, Yamaki S, Takahashi T. Extremely thickened media of small pulmonary arteries in fatal pulmonary hypertension with congenital heart disease–a morphometric and clinicopathological study. Jpn Circ J 64: 909–914, 2000 [DOI] [PubMed] [Google Scholar]
- 22.Patil S, Bunderson M, Wilham J, Black SM. Important role for Rac1 in regulating reactive oxygen species generation and pulmonary arterial smooth muscle cell growth. Am J Physiol Lung Cell Mol Physiol 287: L1314–L1322, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Rabinovitch M, Bothwell T, Hayakawa BN, Williams WG, Trusler GA, Rowe RD, Olley PM, Cutz E. Pulmonary artery endothelial abnormalities in patients with congenital heart defects and pulmonary hypertension. A correlation of light with scanning electron microscopy and transmission electron microscopy. Lab Invest 55: 632–653, 1986 [PubMed] [Google Scholar]
- 24.Rabinovitch M, Haworth SG, Castaneda AR, Nadas AS, Reid LM. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation 58: 1107–1122, 1978 [DOI] [PubMed] [Google Scholar]
- 25.Reddy VM, Meyrick B, Wong J, Khoor A, Liddicoat JR, Hanley FL, Fineman JR. In utero placement of aortopulmonary shunts. A model of postnatal pulmonary hypertension with increased pulmonary blood flow in lambs. Circulation 92: 606–613, 1995 [DOI] [PubMed] [Google Scholar]
- 26.Schröder K, Helmcke I, Palfi K, Krause K, Busse R, Brandes R. Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 27: 1736–1743, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Schultz K, Fanburg B, Beasley D. Hypoxia and hypoxia-inducible factor-1α promote growth factor-induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 290: H2528–H2534, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Stolk J, Hiltermann T, Dijkman J, Verhoeven A. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 11: 95–102, 1994 [DOI] [PubMed] [Google Scholar]
- 29.Wedgwood S, Bekker JM, Black SM. Shear stress regulation of endothelial NOS in fetal pulmonary arterial endothelial cells involves PKC. Am J Physiol Lung Cell Mol Physiol 281: L490–L498, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Wedgwood S, Dettman RW, Black SM. ET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen species. Am J Physiol Lung Cell Mol Physiol 281: L1058–L1067, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Wedgwood S, DeVol J, Grobe A, Benavidez E, Azakie A, Fineman J, Black S. Fibroblast growth factor-2 expression is altered in lambs with increased pulmonary blood flow and pulmonary hypertension. Pediatr Res 61: 32–36, 2007 [DOI] [PubMed] [Google Scholar]
- 32.Wedgwood S, Mitchell CJ, Fineman JR, Black SM. Developmental differences in the shear stress-induced expression of endothelial NO synthase: changing role of AP-1. Am J Physiol Lung Cell Mol Physiol 284: L650–L662, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Wong J, Reddy VM, Hendricks-Munoz K, Liddicoat JR, Gerrets R, Fineman JR. Endothelin-1 vasoactive responses in lambs with pulmonary hypertension and increased pulmonary blood flow. Am J Physiol Heart Circ Physiol 269: H1965–H1972, 1995 [DOI] [PubMed] [Google Scholar]