

Abstract
We have previously characterized a transgenic mouse model (CR2-TAg) of metastatic prostate cancer arising in the neuroendocrine (NE) cell lineage. Biomarkers of NE differentiation in this model are expressed in conventional adenocarcinoma of the prostate with NE features. To further characterize the pathways that control NE proliferation, differentiation, and survival, we established prostate NE cancer (PNEC) cell lines from CR2-TAg prostate tumors and metastases. GeneChip analyses of cell lines harvested at different passages, and as xenografted tumors, indicated that PNECs express consistent features ex vivo and in vivo and share a remarkable degree of similarity with primary CR2-TAg prostate NE tumors. PNECs express mAsh1, a basic helix–loop–helix (bHLH) transcription factor essential for NE cell differentiation in other tissues. RNA interference knockdown of mAsh1, GeneChip comparisons of treated and control cell populations, and a computational analysis of down-regulated genes identified 12 transcriptional motifs enriched in the gene set. Affected genes, including Adcy9, Hes6, Iapp1, Ndrg4, c-Myb, and Mesdc2, are enriched for a palindromic E-box motif, CAGCTG, indicating that it is a physiologically relevant mAsh1 binding site. The enrichment of a c-Myb binding site and the finding that c-Myb is down-regulated by mAsh1 RNA interference suggest that mAsh1 and c-Myb are in the same signaling pathway. Our data indicate that mAsh1 negatively regulates the cell cycle (e.g., via enhanced Cdkn2d, Bub1 expression), promotes differentiation (e.g., through effects on cAMP), and enhances survival by inhibiting apoptosis. PNEC cell lines should be generally useful for genetic and/or pharmacologic studies of the regulation of NE cell proliferation, differentiation, and tumorigenesis.
Keywords: neuroendocrine cell biology, mouse prostate neuroendocrine cancer cell lines, mAsh1, functional genomics, phylogenetic footprinting
The prostate is a tubuloaveolar gland lined with a slowly renewing epithelium composed of three cell lineages. Secretory luminal cells predominate. Lineage progenitors appear to reside in a basal cell layer underlying luminal cells. A small population of scattered neuroendocrine (NE) cells produce dendrite-like processes that extend over several cell diameters to contact members of the other two epithelial lineages. Although they export a number of neuropeptides (1–3), the precise role played by NE cells in normal prostate physiology remains unclear.
Pure NE tumors of the prostate are rare but, like most NE cell cancers, are very aggressive. Conventional adenocarcinoma of the human prostate (CaP) can display features of focal NE differentiation (NED) even when initiation does not occur in NE cells. The reported frequency of NED in CaP ranges from 30% to 100% depending on the study design and the biomarker panel used to define the phenotype (4–6). Recent findings indicate that NED correlates with poor prognosis and androgen-independent growth (7–9).
The mechanisms governing development of NED in CaP are obscure. A similar deficit in our understanding applies to the regulators of NE lineage specification and differentiation. mAsh1, the mouse homolog of the Drosophila proneural gene complex achaete–scute is one candidate regulator. mAsh1–/– mice die during the first postnatal day without discernible NE cells in their lungs and thyroid, and with severe impairment of chromaffin cell development (10–12). Their early mortality has precluded assessment of the role of this basic helix–loop–helix (bHLH) transcription factor on specification of a prostate NE cell fate because cytodifferentiation of the gland's acinar epithelium is not completed until weaning. Interestingly, the human ortholog of mAsh1 (hASH1) is expressed in at least a subset of CaP cells displaying NED (3).
Our limited understanding of NE cells, especially those that populate the prostate epithelium, is due in part to the fact that they are rare and thus difficult to harvest, in part to the absence of tools for their genetic manipulation in vivo, and in part to the lack of representative cell lines. We have developed a transgenic mouse model (CR2-TAg) where simian virus 40 large T antigen (SV40 TAg) is expressed in a subset of prostate NE cells by using transcriptional regulatory elements from the mouse cryptdin-2 gene (Defcr2). Male mice from multiple CR2-TAg pedigrees develop a stereotyped metastatic NE cell cancer (3, 13). One week after initiation of transgene expression (at 7 weeks of age), foci of transformed NE cells are evident in the acinar epithelium. These foci assume all of the known architectural phenotypes of human prostatic intraepithelial neoplasia (PIN), the presumed precursor of CaP in humans (3). Eight weeks later, the stroma of the prostate is massively infiltrated with sheets of transformed NE cells. Sixteen weeks after the onset of transgene expression, the majority of mice die with metastases to regional lymph nodes, liver, lungs, and bone (13). GeneChip studies of CR2-TAg prostates, coupled with real-time quantitative RT-PCR analyses of laser capture microdissected NE cells harvested from primary tumors and metastases, has yielded a large panel of previously unrecognized biomarkers of prostatic NE cells. A subset of these markers is also expressed in foci of NED in human CaP, including hAsh1 (3).
In this report, we describe the features of prostate NE cancer (PNEC) cell lines derived from CR2-TAg primary tumors and metastases. Global gene expression profiles of PNECs during growth ex vivo as monolayers, and in vivo as xenografts indicate that they faithfully recapitulate the features of prostate NE cell tumors. A lack of bona fide direct targets has prevented the discovery of the true DNA binding site of mAsh1, either through experimental or computational approaches. RNA interference (RNAi)-directed knockdown of mAsh1 expression in PNEC cell lines, combined with functional genomic studies and computational analyses of transcriptional regulatory elements enriched in a set of down-regulated genes, has not only disclosed likely mAsh1 binding sites but also components of the signaling network that it uses to modulate NE cell proliferation, differentiation, and survival.
Materials and Methods
Establishment of PNEC Cell Lines. Prostate tumors, plus liver and lymph node metastasis, were removed from 20- to 24-week-old male CR2-TAg mice, and cell lines were established from each as described in Supporting Materials and Methods, which is published as supporting information on the PNAS web site. Cell lines were grown at 37°C under a mixture of 5% CO2/95% air, as monolayers on poly(l-lysine)- and laminin-coated dishes in NPMM (Cambrex, East Rutherford, NJ) supplemented with 0.3% bovine pituitary extract (Cambrex) and 10% FBS (Sigma).
RNAi. RNAi Plasmids were constructed by using pSilencer 1.0-U6 (Ambion, Austin, TX) and introduced into recipient cells by using the approach outlined in Results and Discussion. Methods for comparative GeneChip studies of CR2-TAg tumors, PNEC cells, PNEC cell xenografts, and PNEC cells subjected to mAsh1 RNAi are provided below and in Supporting Materials and Methods.
Results and Discussion
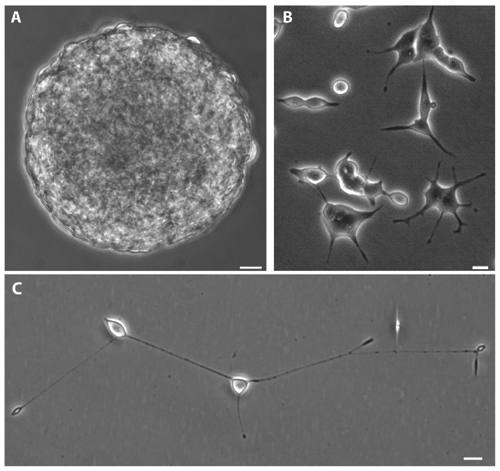
Morphologic Features and Growth Properties of PNEC Cell Lines. Cell lines were established from primary NE cell prostate tumors and liver and lymph node metastases present in 20- to 24-week-old CR2-TAg mice. These cell lines have similar morphologic phenotypes when cultured on supplemented neural progenitor medium (NPMM), irrespective of their anatomic point of origin. PNEC30 cells (derived from a primary tumor), PNEC25 cells (from a liver metastasis), and PNEC28 (lymph node metastasis) grow in suspension as multicellular aggregates that resemble the neurospheres produced when neural stem cells are cultured on noncoated surfaces (14) (e.g., see Fig. 2A, which is published as supporting information on the PNAS web site). PNEC30, PNEC28, and PNEC25 cells attach to poly(l-lysine) and laminin-coated surfaces, grow as monolayers (average doubling time, 50 h), and produce neurite-like processes (Fig. 2 B and C). Attachment is a feature shared by multiple PNEC30-derived clones, one of which (PNEC30-3) grows as a monolayer on noncoated polystyrene surfaces.
Multilabel immunohistochemistry was used to determine whether a subset of the neural/endocrine biomarkers identified in our previous functional genomics study of 16-week-old CR2-TAg prostates (3) are expressed in attached PNEC30 cells. Synaptophysin, DOPA decarboxylase [Ddc (E.C.4.1.1.28), converts l-DOPA to dopamine and 5-hydroxytryptophan to serotonin], γ-aminobutyric acid (GABA), the γ2 subunit of the GABAa receptor, and mAsh1 are all readily detectable (Fig. 3, which is published as supporting information on the PNAS web site). Studies using antibodies directed against mAsh1 and phosphohistone H3 (pSer28) disclosed that this bHLH transcription factor is expressed in a cell-cycle-dependent manner, i.e., levels are greatly diminished during M phase (Fig. 4, which is published as supporting information on the PNAS web site).
Subcutaneous injection of 6 × 106 PNEC30 cells into each flank of adult male and female BALB/cmice (n = 5 per group) produced visible tumors within 2 weeks at all sites in all recipients. Average tumor doubling time was 7.5 d irrespective of the gender of the recipient (Fig. 5A, which is published as supporting information on the PNAS web site). Histochemical and immunohistochemical studies indicated that the xenografts recapitulate features of CR2-TAg prostate tumors (Fig. 5 B and C).
Seven weeks after tail vein injection of 2 to 3 × 106 cells, >2-mm diameter mAsh1/synaptophysin-positive tumors were present in the livers of five of five BALB/c nude and two of three FVB/N recipients. One BALB/c nude mouse with liver tumors also had lesions in the femoral marrow. Tumors were not observed in their lungs.
Comparison of Global Gene Expression Profiles of PNEC Cell Lines Grown ex Vivo and as Xenografts. Mu74Av2 GeneChips, containing probe sets representing 12,483 mouse genes and ESTs, were used to obtain a more complete view of the molecular features expressed by PNECs. PNEC30 cells from passage 30 or 34 were harvested during the exponential phase of growth as attached monolayers in supplemented NPMM. Duplicate cRNA targets, independently generated from each RNA (n = 2 × 107 cells harvested at 80% confluency per dish; material from two dishes pooled/passage), were used to interrogate separate GeneChips. Comparison of these analytical duplicates yielded very few differences at a given passage (<20 transcripts with ≥2-fold change in their levels). Comparison of different passages (experimental duplicates) also produced very few differences (<20 transcripts varying ≥2-fold). These results indicate that PNECs maintain stable expression profiles.
The four PNEC30 GeneChip data sets were compared to a GeneChip profile of 24-week-old normal prostates (n = 8) where members of the NE lineage present <1% of the total epithelial cell census. Transcripts that were ≥2 fold different in the same direction, in at least three of four comparisons, were compiled. The resulting list contained 2,283 genes/ESTs, of which 1,185 were ≥2-fold higher in PNEC30 cells versus normal prostate. This list was then referenced to another list obtained from analytic duplicate GeneChip comparisons of 24-week CR2-TAg prostates, where transformed NE cells comprise >90% of the total population, versus normal littermate prostates (n = 8 per group). This second list contained 1,613 genes, of which 1,083 were ≥2-fold increased in 24-week CR2-TAg prostates. Comparison of the two lists revealed a remarkable degree of similarity in the expression profiles of PNEC30 cells and the primary 24-week CR2-TAg tumor, i.e., 1,149 genes were present in both lists, of which 753 were expressed at levels ≥2-fold higher relative to normal prostate.
We expanded our GeneChip comparison to include liver metastasis-derived PNEC25 cell monolayers, PNEC30-3 monolayers [exhibit poly(l-lysine)- and laminin-independent attachment], and PNEC30 cells grown in suspension as neurosphere-like structures. Two independent cultures were analyzed per cell line, each separated by four passages. Xenografts generated in BALB/c nude recipients by using either PNEC30 and PNEC30-3 cells were harvested 7 weeks after implantation, and their expression profiles were defined (one xenograft per mouse; five mice per PNEC line; equal-sized aliquots of RNA from a given type of xenograft were pooled). In all cases, analytical duplicate GeneChip data sets were generated and then merged into one entity and analyzed with dchip software (www.dchip.org). After normalization, model-based expression, and filtering with all four dchip default criteria, a list of 580 genes/ESTs was obtained (see Table 2, which is published as supporting information on the PNAS web site, for a list).
This 580-gene data set was used to generate hierarchical clusters with dchip (Fig. 6A, which is published as supporting information on the PNAS web site). Cells from the same PNEC line cultured at two different passages, with or without attachment, clustered together. Xenografts clustered with their PNEC cell line of origin. In addition, primary prostate tumor-derived PNEC30 lines and their xenografts more closely resembled 24-week-old CR2-TAg prostates than did the liver metastasis-derived PNEC25 line. Compared to PNEC30 cells, members PNEC25 express higher levels of mRNAs encoding proteins implicated in cell migration and metastasis, including activated leukocyte cell adhesion molecule, semaphorin3A, and thrombospondin (see Fig. 6B for dendrograms of gene clusters that exhibit differential expression in PNEC30 versus PNEC25 cells, or in xenografts versus cultured cells). These relationships were supported when a multidimensional scaling (MDS) algorithm was applied to the 580-member gene list (Fig. 6C and Table 3, which is published as supporting information on the PNAS web site).
In summary, GeneChip profiling of PNEC cell lines demonstrated that their molecular features are stably expressed in culture and that they display a high degree of similarity to NE cells in CR2-TAg prostates.
Identifying mAsh1-Regulated Genes in PNEC Cells by Using RNAi. The pathways through which mAsh1 regulates NE specification/differentiation are poorly defined, as are the cis-acting transcriptional target sequences recognized by this bHLH protein. We took advantage of the fact that PNECs represent the first available prostatic NE cell lines and used them to address these issues through RNAi knockdown of mAsh1.
PNEC cells support RNAi. We assessed the efficacy of knockdowns in the cloned PNEC30-3 cell line by using established reagents for luciferase RNAi. A plasmid that directs expression of firefly luciferase was transfected together with varying amounts of an RNAi plasmid that targets luciferase, plus a LacZ expression vector to normalize transfection efficiency. Cells cotransfected with a plasmid mixture containing the empty RNAi vector served as negative controls. Luciferase assays of cells harvested 48 h after cotransfection with the luciferase RNAi construct exhibited up to an 85% reduction in enzyme activity (Fig. 7A, which is published as supporting information on the PNAS web site).
mAsh1RNAi in transiently transfected PNEC30-3 cells. For mAsh1 RNAi, two pSilencer1.0-U6-based recombinant plasmids were constructed, each with a unique 19-bp insert that targeted distinct regions of the mRNA. PNEC30-3 monolayers were transfected at ≈50% confluency, with either one of the two plasmids, and collected 72 h later. Cells transfected with the pSilencer 1.0-U6 vector alone served as negative control. Quantitative (q) RT-PCR and Western blot analyses failed to detect knockdown of mAsh1 expression in the unfractionated PNEC30-3 population, despite multiple efforts to maximize transfection efficiency.
Our solution to this problem was to enrich for transfected cells by fluorescence-activated cell sorting (FACS). To do so, PNEC30-3 cells were cotransfected with either one of the pSilencer constructs and a GFP-expressing plasmid, harvested 72 h later, and sorted according to GFP intensity. The GFP-bright fraction was split into two aliquots, one for Western blot analysis and the other for qRT-PCR assay. mAsh1 knockdown was observed with both RNAi plasmids. pSilencer109 (mAsh1 targeting sequence, 5′-CCGGGTCAAGTTGGTCAAC) functioned better than pSilencer95 (5′-GTCAGCGGCCAAGCAGGAT) producing 2- to 3-fold reductions in mRNA and protein levels (n = 3 independent experiments; Fig. 7B and data not shown). There were no significant reductions in mAsh1 mRNA or protein levels in the GFP-negative FACS fraction or in GFP-positive cells containing the empty RNAi vector (mock-transfected PNEC30-3 cells served as the reference control). The modest decrease in mAsh1 mRNA levels observed with RNAi (Tables 4 and 5, which are published as supporting information on the PNAS web site) is consistent with previous findings that mAsh1 is negatively autoregulated (15).
GeneChip analysis of genes regulated by RNAi knockdown of mAsh1. PNEC30-3 monolayers were transiently cotransfected at 50% confluence with pSilencer109 and the GFP-expression plasmid, or with the empty pSilencer vector and GFP plasmid. GFP-positive cells were collected by FACS 72 h after transfection and RNA was extracted (n = 2 independent experiments). Two aliquots of the each RNA preparation were then used for separate cRNA syntheses (analytical duplicates). Each of the resulting cRNA targets was hybridized to a separate Mu74Av2 GeneChip. pSilencer109- and vector-transfected GeneChip data sets were compared, using the latter as baseline. Genes called “present” in at least one of the two GeneChips being compared, and “increased” or “decreased” by microarray suite 5.0 software (Affymetrix, Santa Clara, CA), were assembled into a list. Because two comparisons were generated for each experiment, four comparisons were performed in total for the two independent RNAi experiments. A final list containing 72 genes/ESTs (including mAsh1) was compiled by using the following selection criteria: (i) the gene was present in at least three of the four comparisons, and (ii) the average fold change was ≥1.3 (increased or decreased) (see Table 6, which is published as supporting information on the PNAS web site, for this list).
Real-time qRT-PCR analysis of mASH1 RNAi-associated changes in gene expression. qRT-PCR assays confirmed that the RNAi decreased expression of mAsh1 mRNA and also demonstrated that E12/E47 mRNA, which encodes the putative dimerization partners of mAsh1 required for transcription activation of its target genes (16, 17), did not change (Table 4).
Adenylyl cyclases are membrane-bound, G protein-coupled enzymes that catalyze conversion of ATP to cAMP (18). qRT-PCR verified that Adcy9 (adenylyl cyclase 9) was suppressed with mAsh1 RNAi. Levels of Adcy9 expression are normally highest in mouse brain (19). Interestingly, exposure of a non-NE prostate cancer cell line (LNCaP) to pharmacological agents that elevate cAMP levels causes reversible transdifferentiation to a NE-like phenotype (20, 21). Our in silico promoter analyses, described below, suggests that Adcy9 is a direct target of mAsh1.
qRT-PCR verified that the levels of mRNAs encoding two proteins involved in cell cycle regulation, Bub1 and Cdkn2d, fell coincident with mAsh1 RNAi. BUB family members are components of a large multiprotein kinetochore complex involved in the spindle checkpoint (22). Mutations/deletions in the human Bub1 ortholog are found in a number of cancers and cancer cell lines (23–25). Moreover, inhibition of BUB1 produces genomic instability and anchorage-independent growth of normal human fibroblasts (26). Cdkn2d (cyclin-dependent kinase inhibitor 2D) is a tumor suppressor that positively regulates the p53 pathway, leading to growth arrest and apoptosis. Because both Bub1 and Cdkn2d can be considered negative regulators of the cell cycle, these findings are consistent with our observation that mAsh1 protein levels decrease in PNECs during M phase (Fig. 4) and suggest that this bHLH transcription factor may help promote NE differentiation by suppressing proliferation. Our results are consistent with another study where forced coexpression of mAsh1 and E12 (Tcfe2a) in p19 mouse embryonal carcinoma cells caused induction of Cdkn1b (cyclin-dependent kinase inhibitor p27Kip1), withdrawal from the cell cycle, and differentiation along a neuronal pathway (27).
Hes6 was another gene confirmed by qRT-PCR to be down-regulated by mAsh1 RNAi (Table 4). Hes6 a member of the Hairy/Enhancer of split family of bHLH transcription factors and a positive regulator of mAsh1: it antagonizes the action of Hes1, which inhibits mAsh1 function during mouse embryonic development (28) and hASH1 gene transcription in small cell lung cancer (NE) cell lines (29). Moreover, Hes6 expression in Xenopus embryos is induced by Xash3, a homolog of Drosophila achaete–scute genes (30). Together, these observations suggest that there is a positive feedback loop between mAsh1 and Hes6 in prostatic NE cells.
RNAi was associated with reduced expression of a gene transcribed in endocrine cells: Iapp1 (islet amyloid polypeptide 1). IAPP was first isolated from amyloid deposits in an insulin-producing pancreatic tumor and from pancreatic islet amyloid in patients with type 2 diabetes mellitus (31, 32). Highest levels of this protein are normally encountered in pancreatic beta cells. IAPP is also expressed in endocrine cells of the normal human lung and gut, and in human NE cell tumors that arise in the GI tract and thyroid (33, 34).
The qRT-PCR study indicated that mAsh1 RNAi down-regulated a number of genes associated with neuronal differentiation/function. Ndrg4 (N-myc downstream regulated 4), was first identified based on its prominence in the early postnatal rat brain (35). Studies in PC12 cells indicate that it is involved in neurite outgrowth (36). The receptor tyrosine kinase, c-Ret, is induced in neural crest stem cells by forced expression of mAsh1 (37). c-Ret is also highly expressed in CR2-TAg tumors (data not shown). Expression of Ddc (DOPA decarboxylase), a biomarker of NE cells in normal and CR2-TAg prostates and of NED in human CaP (3), was also reduced.
qRT-PCR confirmed that expression of c-Myb was also decreased (Table 4). This transcription factor affects proliferation, differentiation, and/or apoptosis in various cellular contexts (38). Our computational analysis of mAsh1 regulatory sequences and gene targets, described below, suggests that c-Myb and mAsh1 are in the same pathway.
Finally, qRT-PCR verified the unexpected observation that mAsh1 RNAi suppressed a regulator of mesodermal development (Mesdc2; LRP5/6 chaperone) that is essential for specification of embryonic polarity (39).
Failure to obtain PNEC30-3 cells stably transfected with mAsh1 RNAi plasmids. To further define the effects of mAsh1 on PNEC cell biology, we attempted to generate cells that had stably integrated the mAsh1 RNAi plasmid into their genome. PNEC30-3 cells were cotransfected with pSilencer95 or pSilencer109, and a plasmid conferring puromycin resistance [BiCs5puroeGFP (40)]. The pSilencer 1.0-U6 RNAi vector without insert was used in lieu of pSilencer95 or pSilencer109 to generate negative controls. Puromycin-resistant clones formed in supplemented NPMM after 2–4 weeks. Clones were far more numerous, and larger, in dishes containing the pSilencer vector without insert compared to dishes containing cells exposed to pSilencer95 or pSilencer109. When 80 of the latter clones were assayed for mAsh1 knockdown by qRT-PCR and immunohistochemistry, none showed any appreciable changes in expression of the transcription factor. These clones either showed no evidence of pSilencer95 or pSilencer109 plasmid or produced a very faint PCR product derived from vector sequences. Because our multilabel immunohistochemistry studies (Fig. 4) and RNAi/GeneChip analysis indicated that mAsh1 expression negatively correlates with cell cycle progression, we thought it unlikely that RNAi knockdown would cause stably transfected cells to stop dividing. It seemed more plausible that mAsh1 deficiency could promote apoptosis in PNEC cells, e.g., c-Jun, one of the genes up-regulated in our transient transfection mAsh1 RNAi experiments (Table 4), is known to promote apoptosis in neurons (41).
Computational Analysis of mAsh1 Regulatory Targets. The genes found to be down-regulated by GeneChip/qRT-PCR studies of PNEC30-3 cells transiently transfected with mAsh1 RNAi plasmids represent potential direct targets of mAsh1. Therefore, they provided us with a gene collection for finding regulatory motifs targeted by this bHLH transcription factor (TF).
Identifying regulatory elements in mammalian genomes remains a major challenge in computational biology. A process termed “phylogenetic footprinting” has been developed to reduce search space to an evolutionarily conserved portion of queried sequence. In particular, human–rodent comparisons have proven to be a valuable resource for identifying functional motifs (42, 43).
We used a newly developed algorithm, PhyloCon (Phylogenetic Consensus), for our analysis that was specifically designed to combine phylogenetic data with coregulated genes to identify regulatory motifs (44). PhyloCon builds orthologous promoter sequences into profiles and then compares these profiles by using a new statistic, average log likelihood ratio [ALLR (44)]. The nature of ALLR and the design of the algorithm allow PhyloCon to tolerate very long sequences.
In the current study, for each gene that showed a consistent decrease in expression with mAsh1 RNAi (four of four GeneChip comparisons), we retrieved genomic sequences from Ensembl (www.ensembl.org). For most of these genes, we obtained 10 kb of sequence upstream from the transcription start site, or the translation start site if the transcription start site was undefined. If this region included a predicted ORF, we only used sequences between the ORF and the gene of interest. We also retrieved the corresponding sequences of orthologous genes from the human and rat genomes (orthology relationship based on Ensembl's definition; see www.ensembl.org/EnsMart/info).
We compiled a data set consisting of 21 orthologous groups where sequences were available from both mouse and human: 18 of these groups also contained rat sequences. The total sequence was 574 kb, or 392 kb after masking common repeats. Among all mouse sequences, 15% could be aligned confidently to orthologous human sequences with an average alignment length of 341 bp and an average identity of 68.7%, whereas 39% could be aligned confidently with orthologous rat sequences with an average alignment length of 367 bp and average identity of 80.2% (based on WU-BLAST, http://blast.wustl.edu). We applied PhyloCon to this data set and identified five significant motifs (see Table 1 and the discussion that follows below).
Table 1. Significant transcriptional motifs associated with mAsh1 regulated genes.

Sequence logos are based on ref. 54. The matrix was generated from PhyloCon predictions or selected from transfac database (Version 7.2) based on combined PBAL, PBAY, and CHN scores. The PBAL, PBAY, and CHN scores for each matrix were calculated according to formulas presented in the text. The test data set contained the list of mAsh1 RNAi-modulated genes. The background data set contained 1,000 genes randomly selected from the mouse genome. A positive score indicates relative enrichment of the corresponding site in the mAsh1-related gene set over the random gene set.
As a complementary approach, we investigated what known TF binding sites are enriched in the genes whose levels were decreased in at least three of the four GeneChip comparisons. We obtained 466 vertebrate-specific nucleotide distribution matrices for TFs from the transfac 7.2 database and used the program patser (G. Hertz and G. Stormo, unpublished; http://ural.wustl.edu) to score each potential site in the sequences against each matrix.
A scoring system that combines thermodynamics and evolutionary conservation was developed to address the significance of the specificity of a particular matrix for a promoter or set of promoters. When using an appropriate nucleotide distribution matrix to scan a sequence, patser gives each potential site a score. From a consideration of the thermodynamics of protein–DNA interactions and the statistics of the scores (45, 46), such a score is expected to be proportional to the free energy of the physical interaction. Based on this, the probability of the protein binding to a site with a score is simply
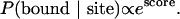 |
If a given sequence has a number of high scoring sites, then the probability of binding is the probability of the protein binding to any of these sites, or proportional to the sum of the probability of binding to each site. Because we were only interested in ranking the probabilities of binding, we could safely ignore the low-scoring sites because their overall contribution to the total probability would be small. Therefore, the probability of the protein binding to any given single sequence with i high scoring sites, each with score, scorei, is
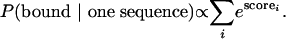 |
When more than one sequence is being considered, we are interested in two probabilities: (i) the probability of the protein binding to all of these sequences and (ii) the probability of the protein binding to any of these sequences. Such probabilities can be expressed as
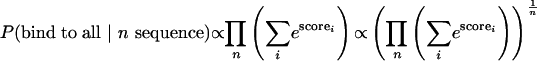 |
and
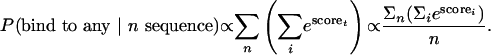 |
To include the availability of binding sites within the genome, we considered the same probability values for either all promoters in the genome, or a number of randomly selected promoters from the genome that serve as a background control data set. We took the log ratio of the estimated probability values between the data set of interest and the control data set as a discriminative score. If the control set has N sequences, then we define PBAL and PBAY as
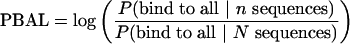 |
and
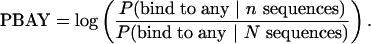 |
To take advantage of the power of comparative genomic data, we only considered the portion of mouse sequences that aligns well with orthologous human sequences. This approach may result in increased false negatives, but it may also significantly reduce false positives (T. Wang and G. Stormo, unpublished data).
In addition to PBAL and PBAY, we developed a third method that relies more on sequence conservation. We defined a term called conserved hit (CH). When patser reports a high-scoring site for a particular mouse sequence, we consider it a CH if and only if the site lies in a region that can be well aligned to a corresponding orthologous human sequence that also has a high scoring site. When there is more than one sequence to consider, we normalized CH number by sequence number. [We also considered normalizing CH number by aligned length and found that the overall performance is similar (data not shown).] Similarly, we define CHN as
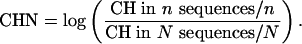 |
With these three scoring methods, we are able to assess the specificity of a given nucleotide distribution matrix for a given promoter (or set of promoters) over a random background or whole genome background. The scores are all log ratios. Therefore, any score >0 indicates a possible enrichment of the corresponding motif in the test set over the background set.
To test the discriminative power of this system, we took a muscle-specific transcription factor data set (42) and scored all 466 vertebrate specific matrices from transfac (http://ural.wustl.edu/links.html). The matrices were ranked by the sum of PBAL, PBAY, and CHN. Ten of the top 15 matrices corresponded to muscle-specific transcription factors (data not shown). We also scored these matrices on a random gene set and plotted the distribution of these three scores. The distribution of the random data set is tightly clustered around 0 (Fig. 8A, which is published as supporting information on the PNAS web site). For a group of functionally related genes, such as those in the muscle-specific data set, we expect, and indeed found, more negative scores because most of the database motifs are under-represented and there are a few significantly high-scoring motifs associated with the function of the gene group (Fig. 8B). Compared to the random data set, the broader spectrum, the negative mean, and the presence of a few high scoring motifs in our mAsh1-related gene set indicate its nonrandomness (Fig. 8C).
Seven matrices whose scores are significantly higher than others are listed in Table 1. We also calculated PBAL, PBAY, and CHN scores for the five matrices identified by PhyloCon (Table 1). For all matrices listed in Table 1, we calculated PBAL, PBAY, and CHN scores against each individual promoter (data are provided in Table 7, which is published as supporting information on the PNAS web site, and can be used to determine the likelihood of an individual promoter being regulated by a certain motif). In all experiments, 1,000 promoters chosen randomly from the mouse genome were used as a background data set.
Among the five motifs identified by PhyloCon, mAsh1.1 shows significant similarity to the binding site of SP1, a zinc finger protein that regulates expression of a large number of genes with diverse biological functions and GC-rich promoters (47). mAsh1.5 shows some similarity to the site of CAC binding protein (based on the program comparetwo; T. Wang and G. Stormo, unpublished data). Two motifs are novel: mAsh1.3 (a palindrome) and mAsh1.4 (Table 1).
Motif mAsh1.2 is a strong and conserved E-box (CAGCTG). Because many bHLH regulatory proteins are known to bind to E-box (minimally defined as “CANNTG”), mAsh1 has also been thought to bind to E-box to activate transcription of its target genes. However, the physiological binding site of mAsh1 has never been directly established. The first reported study of mAsh1 DNA binding disclosed that it forms heterooligomers with E12, and that these heterooligomer complexes interact with the Mck (muscle creatine kinase) E-box (CACCTG) in vitro (16). However, Mck is not a physiological target of mAsh1, and this interaction may be dependent on E12/E47 (16, 17). Allemann and coworkers analyzed the thermodynamics of binding of recombinant mAsh1 to DNA and found that mAsh1 binds to E-boxes with high affinity but low specificity (48). They suggested that folding of the bHLH domain of mAsh1 and DNA binding are a coupled process or “induced fit” (49). The binding affinities between mAsh1 and E-boxes with different internal sequences were largely indistinguishable in their analysis, a finding that contrasts with another group's proposal that different E-box sequences may be functionally distinct (50). The use of recombinant proteins and/or the lack of cofactors in the in vitro systems used for these biophysical studies may be a confounding factor that limits their interpretation or comparison. Genes shown by qRT-PCR to be down-regulated by mAsh1 RNAi in PNEC30-3 cells, such as Adcy9, cdkn2d, Hes6, Iapp1, Ndrg4, c-Myb, and Mesdc2, are among the those identified by PhyloCon to be enriched for the CAGCTG motif. Given that the results from our GeneChip/qRT-PCR and in silico motif analysis support one another, we propose that this E-box is a physiologically relevant binding site in mAsh1-regulated genes.
Based on our scoring system, the seven highest ranking matrices/binding sites from transfac database that are enriched in this group of mAsh1-related genes are SRF, E2F, AP2, MAZ, MAZR, E47, and CMYB (Table 1). Srf (serum response factor) controls cell growth and differentiation, as well as neuronal transmission. Srf interacts with another general transcription factor, Sp1: the Sp1 binding site was also enriched in our mAsh1 RNAi down-regulated gene set (i.e., it appeared in our PhyloCon analysis as mAsh1.1). E2F transcription factors coordinate a large group of genes involved in control of the G1/S-phase transition as well as apoptosis (51). Maz (Myc-associated zinc finger protein) is involved in neural differentiation of p19 cells (52, 53). E12/E47 can form heterodimers with mAsh1 and may assist its functions. The presence of CMYB in our list of top seven matrices (Table 1), coupled with the finding that c-Myb is one of the genes down-regulated by mAsh1 RNAi (Table 4), suggests that mAsh1 and c-Myb are in the same pathway, with c-Myb being directly regulated by mAsh1. Other genes in our data set may not be direct targets of mAsh1 but rather targets of c-Myb.
Summary. Our RNAi study indicates that mASH1 plays a role in negative regulation of the cell cycle in PNECs (e.g., it enhances expression of cdkn2d) and in promoting NE differentiation (e.g., through signaling pathways that operate via cAMP) (Fig. 1). Despite mAsh1's inhibitory effects on some regulators of cell cycle progression (Bub1 and Cdkn2d), our findings suggest that the net outcome of mAsh1 depletion may be apoptotic cell death rather than increased proliferation. This notion can be tested once an inducible system for expressing mAsh1 siRNAs is developed. Such a system could also be used in concert with PNEC cell xenografts to examine whether mAsh1 may be a potential therapeutic target for NE cell cancers and/or conventional prostate adenocarcinoma with hASH1-positive NED. Furthermore, the PNEC cell lines described in this report should be useful for genetic and pharmacologic tests of the contributions of other factors to the regulation of NE cell growth, differentiation, and tumorigenesis.
Fig. 1.

Summary of observed effects of mAsh1 RNAi in PNEC cells.
Supplementary Material
Acknowledgments
We thank Sabrina Wagoner for superb technical assistance, Tim Fleming for many helpful suggestions about cell line derivation, Joseph Ippolito for unpublished GeneChip data from CR2-TAg prostates, William Eades and Jacqueline Hughes for cell sorting, and Jeffrey Milbrandt and Gregory Hannon for luciferase RNAi plasmids. This work was supported in part by National Institutes of Health Grants DK59129 and P50-CA94056. T.W. was supported by National Institutes of Health Predoctoral Training Grant 2T32HG00045.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: bHLH, basic helix–loop–helix; CaP, conventional adenocarcinoma of the human prostate; NE, neuroendocrine; NED, NE differentiation; PNEC, prostate NE cancer; RNAi, RNA interference.
References
- 1.di Sant'Agnese, P. A. (1986) Arch. Pathol. Lab. Med. 110, 412–415. [PubMed] [Google Scholar]
- 2.Abrahamsson, P. A., Wadstrom, L. B., Alumets, J., Falkmer, S. & Grimelius, L. (1986) Pathol. Res. Pract. 181, 675–683. [DOI] [PubMed] [Google Scholar]
- 3.Hu, Y., Ippolito, J. E., Garabedian, E. M., Humphrey, P. A. & Gordon, J. I. (2002) J. Biol. Chem. 277, 44462–44474. [DOI] [PubMed] [Google Scholar]
- 4.di Sant'Agnese, P. A. (1992) Cancer 70, 254–268. [DOI] [PubMed] [Google Scholar]
- 5.Abrahamsson, P. A., Falkmer, S., Falt, K. & Grimelius, L. (1989) Pathol. Res. Pract. 185, 373–380. [DOI] [PubMed] [Google Scholar]
- 6.Aprikian, A. G., Cordon-Cardo, C., Fair, W. R. & Reuter, V. E. (1993) Cancer 71, 3952–3965. [DOI] [PubMed] [Google Scholar]
- 7.Krijnen, J. L., Bogdanowicz, J. F., Seldenrijk, C. A., Mulder, P. G. & van der Kwast, T. H. (1997) J. Urol. 158, 171–174. [DOI] [PubMed] [Google Scholar]
- 8.Theodorescu, D., Broder, S. R., Boyd, J. C., Mills, S. E. & Frierson, H. F., Jr. (1997) Cancer 80, 2109–2119. [PubMed] [Google Scholar]
- 9.Abrahamsson, P. A. (1999) Prostate 39, 135–148. [DOI] [PubMed] [Google Scholar]
- 10.Lanigan, T. M., DeRaad, S. K. & Russo, A. F. (1998) J. Neurobiol. 34, 126–134. [PubMed] [Google Scholar]
- 11.Borges, M., Linnoila, R. I., van de Velde, H. J., Chen, H., Nelkin, B. D., Mabry, M., Baylin, S. B. & Ball, D. W. (1997) Nature 386, 852–855. [DOI] [PubMed] [Google Scholar]
- 12.Huber, K., Bruhl, B., Guillemot, F., Olson, E. N., Ernsberger, U. & Unsicker, K. (2002) Development (Cambridge, U.K.) 129, 4729–4738. [DOI] [PubMed] [Google Scholar]
- 13.Garabedian, E. M., Humphrey, P. A. & Gordon, J. I. (1998) Proc. Natl. Acad. Sci. USA 95, 15382–15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiss, S., Dunne, C., Hewson, J., Wohl, C., Wheatley, M., Peterson, A. C. & Reynolds, B. A. (1996) J. Neurosci. 16, 7599–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meredith, A. & Johnson, J. E. (2000) Dev. Biol. 222, 336–346. [DOI] [PubMed] [Google Scholar]
- 16.Johnson, J. E., Birren, S. J., Saito, T. & Anderson, D. J. (1992) Proc. Natl. Acad. Sci. USA 89, 3596–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Persson, P., Jogi, A., Grynfeld, A., Pahlman, S. & Axelson, H. (2000) Biochem. Biophys. Res. Commun. 274, 22–31. [DOI] [PubMed] [Google Scholar]
- 18.Taussig, R. & Gilman, A. G. (1995) J. Biol. Chem. 270, 1–4. [DOI] [PubMed] [Google Scholar]
- 19.Premont, R. T., Matsuoka, I., Mattei, M. G., Pouille, Y., Defer, N. & Hanoune, J. (1996) J. Biol. Chem. 271, 13900–13907. [DOI] [PubMed] [Google Scholar]
- 20.Bang, Y. J., Pirnia, F., Fang, W. G., Kang, W. K., Sartor, O., Whitesell, L., Ha, M. J., Tsokos, M., Sheahan, M. D., Nguyen, P., et al. (1994) Proc. Natl. Acad. Sci. USA 91, 5330–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cox, M. E., Deeble, P. D., Lakhani, S. & Parsons, S. J. (1999) Cancer Res. 59, 3821–3830. [PubMed] [Google Scholar]
- 22.Roberts, B. T., Farr, K. A. & Hoyt, M. A. (1994) Mol. Cell. Biol. 14, 8282–8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cahill, D. P., Lengauer, C., Yu, J., Riggins, G. J., Willson, J. K., Markowitz, S. D., Kinzler, K. W. & Vogelstein, B. (1998) Nature 392, 300–303. [DOI] [PubMed] [Google Scholar]
- 24.Ru, H. Y., Chen, R. L., Lu, W. C. & Chen, J. H. (2002) Oncogene 21, 4673–4679. [DOI] [PubMed] [Google Scholar]
- 25.Hempen, P. M., Kurpad, H., Calhoun, E. S., Abraham, S. & Kern, S. E. (2003) Hum. Mutat. 21, 445–450. [DOI] [PubMed] [Google Scholar]
- 26.Musio, A., Montagna, C., Zambroni, D., Indino, E., Barbieri, O., Citti, L., Villa, A., Ried, T. & Vezzoni, P. (2003) Cancer Res. 63, 2855–2863. [PubMed] [Google Scholar]
- 27.Farah, M. H., Olson, J. M., Sucic, H. B., Hume, R. I., Tapscott, S. J. & Turner, D. L. (2000) Development (Cambridge, U.K.) 127, 693–702. [DOI] [PubMed] [Google Scholar]
- 28.Sasai, Y., Kageyama, R., Tagawa, Y., Shigemoto, R. & Nakanishi, S. (1992) Genes Dev. 6, 2620–2634. [DOI] [PubMed] [Google Scholar]
- 29.Chen, H., Thiagalingam, A., Chopra, H., Borges, M. W., Feder, J. N., Nelkin, B. D., Baylin, S. B. & Ball, D. W. (1997) Proc. Natl. Acad. Sci. USA 94, 5355–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koyano-Nakagawa, N., Kim, J., Anderson, D. & Kintner, C. (2000) Development (Cambridge, U.K.) 127, 4203–4216. [DOI] [PubMed] [Google Scholar]
- 31.Westermark, P., Wernstedt, C., Wilander, E. & Sletten, K. (1986) Biochem. Biophys. Res. Commun. 140, 827–831. [DOI] [PubMed] [Google Scholar]
- 32.Clark, A., Cooper, G. J., Lewis, C. E., Morris, J. F., Willis, A. C., Reid, K. B. & Turner, R. C. (1987) Lancet 2, 231–234. [DOI] [PubMed] [Google Scholar]
- 33.Alevizaki, M., Dai, K., Grigorakis, S. I., Legon, S. & Souvatzoglou, A. (1994) Clin. Endocrinol. (Oxford) 41, 21–26. [DOI] [PubMed] [Google Scholar]
- 34.Stridsberg, M., Eriksson, B., Lundqvist, G., Skogseid, B., Wilander, E. & Oberg, K. (1995) Regul. Pept. 55, 119–131. [DOI] [PubMed] [Google Scholar]
- 35.Zhou, R. H., Kokame, K., Tsukamoto, Y., Yutani, C., Kato, H. & Miyata, T. (2001) Genomics 73, 86–97. [DOI] [PubMed] [Google Scholar]
- 36.Ohki, T., Hongo, S., Nakada, N., Maeda, A. & Takeda, M. (2002) Brain Res. Dev. Brain Res. 135, 55–63. [DOI] [PubMed] [Google Scholar]
- 37.Lo, L., Tiveron, M. C. & Anderson, D. J. (1998) Development (Cambridge, U.K.) 125, 609–620. [DOI] [PubMed] [Google Scholar]
- 38.Oh, I. H. & Reddy, E. P. (1999) Oncogene 18, 3017–3033. [DOI] [PubMed] [Google Scholar]
- 39.Hsieh, J. C., Lee, L., Zhang, L., Wefer, S., Brown, K., DeRossi, C., Wines, M. E., Rosenquist, T. & Holdener, B. C. (2003) Cell 112, 355–367. [DOI] [PubMed] [Google Scholar]
- 40.Yu, J. Y., Taylor, J., DeRuiter, S. L., Vojtek, A. B. & Turner, D. L. (2003) Mol. Ther. 7, 228–236. [DOI] [PubMed] [Google Scholar]
- 41.Ham, J., Eilers, A., Whitfield, J., Neame, S. J. & Shah, B. (2000) Biochem. Pharmacol. 60, 1015–1021. [DOI] [PubMed] [Google Scholar]
- 42.Wasserman, W. W., Palumbo, M., Thompson, W., Fickett, J. W. & Lawrence, C. E. (2000) Nat. Genet. 26, 225–228. [DOI] [PubMed] [Google Scholar]
- 43.Lenhard, B., Sandelin, A., Mendoza, L., Engstrom, P., Jareborg, N. & Wasserman, W. W. (2003) J. Biol. 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang, T. & Stormo, G. D. (2003) Bioinformatics 19, 2369–2380. [DOI] [PubMed] [Google Scholar]
- 45.Stormo, G. D. & Fields, D. S. (1998) Trends Biochem. Sci. 23, 109–113. [DOI] [PubMed] [Google Scholar]
- 46.Stormo, G. D. (2000) Bioinformatics 16, 16–23. [DOI] [PubMed] [Google Scholar]
- 47.Kaczynski, J., Cook, T. & Urrutia, R. (2003) Genome Biol. 4, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meierhan, D., el-Ariss, C., Neuenschwander, M., Sieber, M., Stackhouse, J. F. & Allemann, R. K. (1995) Biochemistry 34, 11026–11036. [DOI] [PubMed] [Google Scholar]
- 49.Kunne, A., Sieber, M., Meierhans, D. & Allemann, R. K. (1998) Biochemistry 37, 4217–4223. [DOI] [PubMed] [Google Scholar]
- 50.Yutzey, K. E. & Konieczny, S. F. (1992) Nucleic Acids Res. 20, 5105–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.La Thangue, N. B. (2003) Nat. Cell Biol. 5, 587–589. [DOI] [PubMed] [Google Scholar]
- 52.Okamoto, S., Sherman, K., Bai, G. & Lipton, S. A. (2002) Brain Res. Mol. Brain Res. 107, 89–96. [DOI] [PubMed] [Google Scholar]
- 53.Komatsu, M., Li, H. O., Tsutsui, H., Itakura, K., Matsumura, M. & Yokoyama, K. K. (1997) Oncogene 15, 1123–1131. [DOI] [PubMed] [Google Scholar]
- 54.Schneider, T. D. & Stephens, R. M. (1990) Nucleic Acids Res. 18, 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}