

Abstract
A decline in energy is common in aging, and the restoration of mitochondrial bioenergetics may offer a common approach for the treatment of numerous age-associated diseases. Cardiolipin is a unique phospholipid that is exclusively expressed on the inner mitochondrial membrane where it plays an important structural role in cristae formation and the organization of the respiratory complexes into supercomplexes for optimal oxidative phosphorylation. The interaction between cardiolipin and cytochrome c determines whether cytochrome c acts as an electron carrier or peroxidase. Cardiolipin peroxidation and depletion have been reported in a variety of pathological conditions associated with energy deficiency, and cardiolipin has been identified as a target for drug development. This review focuses on the discovery and development of the first cardiolipin-protective compound as a therapeutic agent. SS-31 is a member of the Szeto-Schiller (SS) peptides known to selectively target the inner mitochondrial membrane. SS-31 binds selectively to cardiolipin via electrostatic and hydrophobic interactions. By interacting with cardiolipin, SS-31 prevents cardiolipin from converting cytochrome c into a peroxidase while protecting its electron carrying function. As a result, SS-31 protects the structure of mitochondrial cristae and promotes oxidative phosphorylation. SS-31 represents a new class of compounds that can recharge the cellular powerhouse and restore bioenergetics. Extensive animal studies have shown that targeting such a fundamental mechanism can benefit highly complex diseases that share a common pathogenesis of bioenergetics failure. This review summarizes the mechanisms of action and therapeutic potential of SS-31 and provides an update of its clinical development programme.
LINKED ARTICLES
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: bendavia, cytochrome c, cytochrome c peroxidase, mitochondria cristae, mitochondrial permeability transition, oxidative stress, reactive oxygen species, SS-31, Szeto-Schiller peptides
Introduction
Defects in energy metabolism represent a common thread among many age-associated complex diseases. A decline in bioenergetics underlies the general frailty of old age and a broad spectrum of metabolic and degenerative diseases. A wealth of research converges on the mitochondrion as the central player in cellular aging (Bratic and Trifunovic, 2010; Lee and Wei, 2012; Bratic and Larsson, 2013). Mitochondria produce about 90% of cellular energy, but they are also the major source of intracellular reactive oxygen species (ROS) and play a central role in the initiation and execution of apoptosis. As energy output declines, the most energetic tissues are preferentially affected, resulting in degenerative changes in the CNS, heart, kidney and muscle. Age-related decline in mitochondrial bioenergetics has been observed in these tissues and is associated with a decline in function in both experimental animals and humans (Short et al., 2005; Judge and Leeuwenburgh, 2007; Figueiredo et al., 2009). The universality of bioenergetic failure in age-associated complex diseases has led to the idea that restoration of mitochondrial bioenergetics may present a common approach to the treatment of disorders as diverse as heart failure, chronic kidney disease, skeletal muscle weakness and neurodegenerative diseases.
As mitochondrial oxidative stress has long been considered the primary aetiology behind aging and mitochondrial dysfunction, the majority of drug discovery efforts to date have centred on compounds that reduce mitochondrial ROS. Mitochondria-targeted antioxidants that are based on delivery of known redox agents to the mitochondrial matrix by conjugation to delocalized cations [such as triphenylphosphonium ion (TPP+)] have been reviewed extensively (Skulachev et al., 2009; Smith et al., 2012). Although these compounds can reduce mitochondrial ROS, recent studies suggest they inhibit mitochondrial bioenergetics (Fink et al., 2012; Reily et al., 2013).
More recently, cardiolipin, the phospholipid that is uniquely expressed on the inner mitochondrial membrane (IMM), has been identified as a potential drug target because of its central role in the structural formation of cristae membranes and organization of the respiratory components of the electron transport chain (ETC), as well as serving as the platform for initiation of apoptosis (Schlame and Ren, 2009; Schug and Gottlieb, 2009; Sorice et al., 2009; Paradies et al., 2010). Changes in cardiolipin not only alter fluidity and folding of the IMM, but can profoundly alter the organization and function of respiratory complexes. Cardiolipin peroxidation and loss of cardiolipin has been associated with aging and several metabolic and degenerative diseases (Han et al., 2005; Chicco and Sparagna, 2007; Shi, 2010; Claypool and Koehler, 2012). Recent lipidomic studies have identified cardiolipin as a target for drug development for traumatic brain injury, Parkinson's Disease, cancer, diabetes and the metabolic syndrome (Bayir et al., 2007; Gross and Han, 2007; Han et al., 2007; Kiebish et al., 2008; Ji et al., 2012; Tyurina et al., 2013). This review will focus on the discovery and development of the first class of compounds that targets cardiolipin on the IMM and optimizes cristae architecture, improves mitochondrial bioenergetics and reduces ROS generation. The Szeto-Schiller (SS) peptides have been evaluated in numerous preclinical disease models involving bioenergetics failure, and the lead compound (Bendavia™) is currently in Phase II clinical trials for several clinical indications.
Mitochondrial bioenergetics declines with age
Aging is known to result in biochemical and functional alterations in many components of the mitochondrial ETC, resulting in reduced efficiency of electron transport, inhibition of oxidative phosphorylation (OXPHOS), and increased ROS generation (Lesnefsky and Hoppel, 2006). Catalytic activity of complexes I, III and IV all decline with age in liver, brain, heart and skeletal muscle (Lenaz et al., 1997; Paradies et al., 1997; Fannin et al., 1999; Lesnefsky et al., 2001a; Moghaddas et al., 2003; Boveris and Navarro, 2008; Petrosillo et al., 2008). These defects in respiratory complex activity are reflected in reduced state 3 respiration and coupled respiration in isolated mitochondria (Kim et al., 1988a,b1988b; Paradies and Ruggiero, 1990; Short et al., 2005; Boveris and Navarro, 2008; Figueiredo et al., 2009; O'Toole et al., 2010). A significant decrease in coupled respiration and ATP synthesis was also found in skeletal muscles from aged mice and humans (Marcinek et al., 2005; Conley et al., 2013).
Loss of efficiency in electron transport is expected to increase electron leak and ROS generation in aging. There is substantial disagreement, however, about whether mitochondrial ROS production increases with age (Muscari et al., 1990; Drew et al., 2003; Moghaddas et al., 2003; Suh et al., 2003; Judge et al., 2005; Sen et al., 2007). These discrepancies may be caused by variation in endogenous antioxidant capacity in different tissues and cell types (Suh et al., 2003; Judge et al., 2005). Despite the lack of consistent evidence showing increased mitochondrial ROS production, there is good evidence that the mitochondrial glutathione redox status (GSH:GSSG) shifts progressively towards oxidation with age (de la Asuncion et al., 1996; Rebrin et al., 2003; Suh et al., 2003) and there is a significant decrease in mitochondrial GSH levels in brain, skeletal muscle and liver of senescent animals (Rebrin and Sohal, 2004; Perluigi et al., 2010), indicating mitochondrial oxidative stress.
Aging leads to alterations in lipid composition of the IMM
Lipids interact dynamically to form transient arrangements and influence the fluidity of lipid membranes. Lipids also interact with proteins and modulate the structural organization of proteins on the membrane, allowing for multimeric protein complexes and higher order supercomplexes (Bogdanov et al., 2008). Since the ETC resides on the IMM, changes in lipid composition of the IMM can profoundly affect mitochondrial bioenergetics. The age-associated decline in OXPHOS may result from alterations in lipid composition of the IMM (Gomez and Hagen, 2012). The defect in OXPHOS involves all respiratory complexes, including a reduction in cytochrome c (cyt c). Interestingly, the decrease in complex IV activity in cardiac mitochondria could be reversed by the addition of exogenous phospholipid liposomes, suggesting that the defect in OXPHOS may occur secondary to a defect in the phospholipid environment of complex IV (Lesnefsky and Hoppel, 2003).
The IMM contains predominantly phosphatidylcholine, phosphatidylethanolamine and cardiolipin. While phosphatidylcholine and phosphatidylethanolamine are found on other membranes, cardiolipin is only expressed on the IMM (Osman et al., 2011). Cardiolipin is synthesized from phosphatidylglycerol by cardiolipin synthase on the inner face of the IMM, followed by a transacylation reaction to achieve the desirable acyl side chains (Schlame et al., 2000). The acyl chains can vary but are predominantly comprised of linoleic acid (18:2) in the heart. Unlike other phospholipids, cardiolipin is a lipid dimer with two phosphate head groups and four acyl side chains, thus giving it a conical structure (Figure 1). To a lesser extent, phosphatidylethanolamine also has a cone shape. As a result, these two phospholipids do not form bilayers. On a membrane with other lipids, they exert a lateral pressure that modulates membrane curvature (Frey and Mannella, 2000; Osman et al., 2011) (Figure 1). Thus cardiolipin is particularly important for cristae formation, and deficiency in cardiolipin results in loss of cristae membranes (Acehan et al., 2007) (Figure 2). Cardiolipin also plays an important role in maintaining inner membrane fluidity and osmotic stability, and decreases the energy required to create folds or cristae in the IMM (Shibata et al., 1994; Nichols-Smith et al., 2004).
Figure 1.
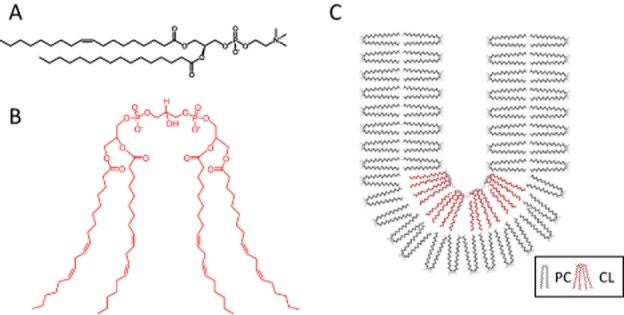
Cardiolipin promotes curvature in lipid membranes due to its unique conical structure. (A) Chemical structure of phosphatidylcholine (PC). (B) Chemical structure of cardiolipin (CL). (C) Cardiolipin exerts lateral pressure in a lipid bilayer to induce a negative curvature.
Figure 2.
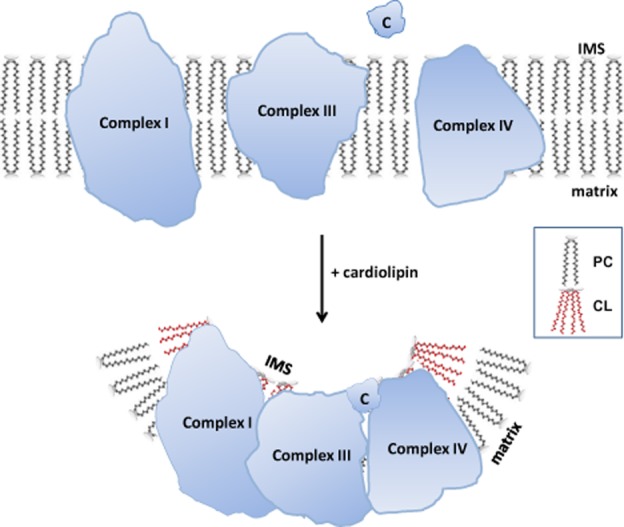
Cardiolipin is important for cristae structure and supercomplex formation on the inner mitochondrial membrane (IMM). The protein complexes of the electron transport chain reside on cristae membranes and cardiolipin (CL) provides curvatures on the IMM to increase surface area for the respiratory complexes. Cardiolipin also helps to organize the respiratory complexes into supercomplexes to facilitate electron transfer among the redox partners. Lastly, cardiolipin anchors the highly cationic cyt c (C) via electrostatic interaction to bring it in close proximity to Complex III and Complex IV for efficient electron transfer. IMS: intermembrane space.
Cardiolipin also helps to organize the respiratory complexes into supercomplexes to facilitate optimal electron transfer among the redox partners (Zhang et al., 2002; Pfeiffer et al., 2003; Mileykovskaya and Dowhan, 2009; Kiebish et al., 2012; Bazan et al., 2013) (Figure 2). Many of the respiratory complexes and carrier proteins require cardiolipin for optimal assembly and function (Fry and Green, 1981; Hoch, 1992; Mileykovskaya et al., 2005; Zhang et al., 2005; Chicco and Sparagna, 2007; Wittig and Schagger, 2009; Schwall et al., 2012). Disruption of supercomplex formation can enhance ROS generation from complex I (Maranzana et al., 2013). Cardiolipin also plays a role in anchoring cyt c to the IMM and facilitates electron transfer from complex III to complex IV (Rytomaa and Kinnunen, 1994, 1995).
A decline in cardiolipin content with age has been reported in mitochondria from brain, liver and heart (Vorbeck et al., 1982; Paradies et al., 1997; Hagen et al., 1998; Sen et al., 2007). This loss of cardiolipin may be due to changes in cardiolipin synthase activity, alterations in cardiolipin remodelling or cardiolipin peroxidation. The loss of cardiolipin can explain the decreased mitochondrial membrane fluidity reported in aged rats (Lee et al., 1999). It is also consistent with the findings that age-related decline in complex IV activity can be restored by exogenously added cardiolipin or cyt c (Paradies et al., 1997; O'Toole et al., 2010; Petrosillo et al., 2013).
Cardiolipin peroxidation, mitochondrial permeability transition and cell death
In addition to its important role in maintaining cell viability, cardioloipin can also play an important role in cell death, especially when it is oxidized. Cardiolipin is particularly vulnerable to oxidative damage because of its high content of unsaturated fatty acids and its location near the site of ROS production (Paradies et al., 2011). Cardiolipin can be oxidized by mitochondrial H2O2, but this oxidation is greatly enhanced in the presence of cyt c (Kagan et al., 2005; Basova et al., 2007; Wiswedel et al., 2010). Interaction of cyt c with cardiolipin promotes cyt c unfolding and dramatically enhances the protein's peroxidase activity. Native cyt c has a compact tertiary structure with its haem iron coordinated to Met80 and His18. Because of its hexacoordinated iron, native cyt c has very low peroxidase activity. Studies with cyt c and cardiolipin liposomes have reported substantial unfolding of cyt c that can disrupt the Met80 ligation and exposes the haem iron to H2O2 (Hanske et al., 2012; Muenzner et al., 2013). Other investigators have proposed that one or two acyl chains of cardiolipin can penetrate deep into the hydrophobic core of cyt c and loosen the Met80-Fe axial bond (Kalanxhi and Wallace, 2007; Sinibaldi et al., 2008; Sinibaldi et al., 2010).
The oxidation of cardiolipin produces kinks in the acyl chain that disturbs cardiolipin microdomains on the IMM and causes the loss of curvature (Figure 3). Cardiolipin peroxidation also disrupts supercomplexes and causes cyt c to be detached from the IMM. All of this results in inhibition of mitochondrial respiration and sets the stage for apoptosis (Gonzalvez and Gottlieb, 2007; Schug and Gottlieb, 2009). Oxidized cardiolipin synergizes with Ca2+ to induce opening of the mitochondrial permeability transition (MPT) pore (Petrosillo et al., 2007). The identity of the MPT pore remains elusive, but it is believed to be a non-specific pore which allows free passage of any molecules <1.5 kDa (Halestrap, 2009). The voltage-dependent anion channel, the adenine nucleotide translocator and the F0,F1-ATP synthase have all been postulated to be part of the MPT pore (Bernardi, 2013). Mitochondrial permeability transition disrupts the permeability barrier of the IMM and leads to collapse of the mitochondrial potential and uncoupling of oxidative phosphorylation (Halestrap, 2009). In addition, MPT causes the release of cyt c and other proapoptotic proteins into the cytosol where they trigger the caspase cascade and cell death by apoptosis (Shidoji et al., 1999; Jiang and Wang, 2004; Ott et al., 2007). Severe depletion of ATP will result in cell death by necrosis.
Figure 3.
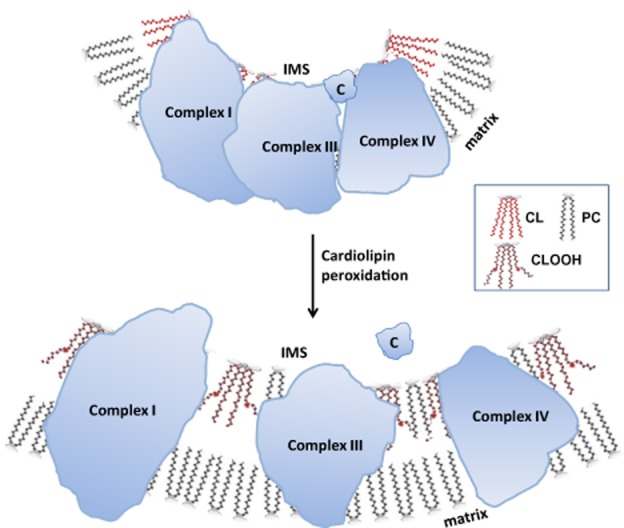
Cardiolipin peroxidation destabilizes cardiolipin microdomains on the inner mitochondrial membrane (IMM) and disrupts supercomplexes. Cardiolipin (CL) is particularly vulnerable to oxidative damage because of its high content of unsaturated fatty acids. Peroxidation of the acyl chains alters the structure of cardiolipin (CLOOH) and prevents cardiolipin from aggregating into microdomains or rafts on the IMM. The breakdown of cardiolipin rafts abolishes cristae curvatures and disrupts the organization of respiratory complexes into higher order supercomplexes. Peroxidation of cardiolipin also reduces its affinity for cyt c (C) and sets the stage for cyt c release into the cytosol and apoptosis. IMS: intermembrane space.
Cardiolipin as a target for drug development
Cardiolipin peroxidation and depletion have been reported in a variety of pathological conditions associated with energy deficiency, including skeletal muscle weakness, heart failure, neurodegenerative diseases, diabetes and ischaemia-reperfusion (IR) injury. Compounds that can inhibit cardiolipin peroxidation and preserve cardiolipin may potentially be beneficial for these diseases.
Attempts at designing molecules to inhibit cardiolipin peroxidation have been very limited. The mitochondria-targeted electron scavenger XJB-5–131 (4-amino-TEMPO conjugated to hemigramacidin S) was reported to inhibit cardiolipin peroxidation, reduce apoptotic neuronal cell death and improve behaviour in a rat traumatic brain injury model (Bayir et al., 2007). This compound may inhibit cardiolipin peroxidation by reducing mitochondrial H2O2, but there is no evidence that it directly inhibits cyt c peroxidase activity. A mitochondria-targeted inhibitor of cyt c peroxidase was shown to inhibit cardiolipin peroxidation in vitro and protect against radiation injury (Atkinson et al., 2011). The imidazole-substituted fatty acid was designed to replace Met80 as the sixth coordinate on the haem iron, and TPP+ was used to deliver it to mitochondria. It is unclear whether locking the haem iron with imidazole affects the electron carrying capacity of cyt c, and TPP+ has been shown to inhibit mitochondrial bioenergetics (Fink et al., 2012; Reily et al., 2013). The imidazole approach may also have off-target effects due to potential interaction with other haem proteins. A third approach used a poorly peroxidizable TPP+ conjugated octadecanoic acid to remodel the endogenous pool of cardiolipin to reduce cardiolipin peroxidation (Tyurina et al., 2012). This approach has only been investigated in cultured cells, and it is not known what effect this modified cardiolipin may have on mitochondrial respiration. It is important that any attempt at inhibiting cyt c peroxidase activity does not destroy the vital function of cyt c as an electron carrier.
Discovery of a new class of small molecules that target cardiolipin
The SS peptides represent a unique class of mitochondria-targeted compounds and their chance discovery was described in a recent review (Szeto and Schiller, 2011). These are synthetic tetrapeptides having an alternating aromatic-cationic motif, among which SS-31 (D-Arg-2′6′-dimethylTyr-Lys-Phe-NH2) is the most extensively studied (Figure 4A). Despite being very water soluble, the SS peptides are remarkably cell permeable, and they are readily taken up and act on all cell types, including endothelial cells, renal and intestinal epithelial cells, myotubes, cardiomyocytes, macrophages and neurons (Zhao et al., 2003; 2005; 2004; Cho et al., 2007b; Han et al., 2009; Zhu et al., 2010; 2011; Andersson et al., 2011; Calkins et al., 2011; Li et al., 2011; Dai et al., 2011a; Gilliam et al., 2012; Kloner et al., 2012; Birk et al., 2013b). The mechanism of cellular uptake for these polar peptides is consistent with diffusion as their uptake is linear and independent of receptor-or transporter-mediated processes (Zhao et al., 2003). Confocal microscopy with fluorescent-labelled analogues suggested that these compounds are localized to mitochondria (Zhao et al., 2004). In addition to fluorescent labels, the intracellular distribution of these peptides can be visualized using biotinylated analogues (Birk et al., 2013a2013b). Using isolated mitochondria, uptake of the SS peptides was found to be rapid and independent of mitochondrial potential, and fractionation studies revealed that the peptides are concentrated on the IMM rather than in the mitochondrial matrix (Zhao et al., 2004; 2005,).
Figure 4.
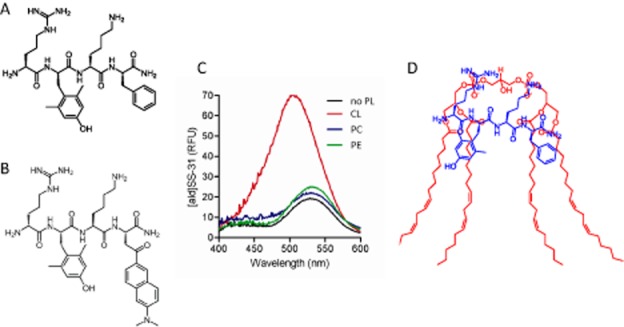
SS-31 selectively binds to cardiolipin. (A) Chemical structure of SS-31 (D-Arg-dimethylTyr-Lys-Phe-NH2). (B) Chemical structure of [ald]SS-31. The Phe4 in SS-31 is replaced by aladan (ald), a polarity-sensitive fluorescent amino acid. (C) Fluorescence emission spectra of [ald]SS-31 in the absence of phospholipids (no PL), or in the presence of cardiolipin (CL), phosphatidylcholine (PC) or phosphatidylethanolamine (PE). Addition of cardiolipin caused a shift of the emission maximum (λmax) from 530 to 500 nm. (D) Proposed model showing the interaction of SS-31 with cardiolipin. Electrostatic interaction between the two cationic moieties of SS-31 (Arg and Lys) and the phosphate head groups of cardiolipin aligns the aromatic residues (dimethyl-Tyr and Phe) within the hydrophobic acyl chain region of cardiolipin.
It was recently discovered that SS-31 selectively binds to cardiolipin via both electrostatic and hydrophobic interactions (Birk et al., 2013b). Aladan (Ald), a polarity-sensitive fluorescent amino acid whose emission maximum (λmax) undergoes a blueshift in a hydrophobic environment (Cohen et al., 2002), was used to probe whether the highly water-soluble SS-31 can interact with lipids. Ald was incorporated into the peptide sequence of SS-31 by substituting Ald for Phe (Figure 4B). Only anionic phospholipids (phosphatidylserine and cardiolipin) caused a blueshift in the λmax of [ald]SS-31, whereas the zwitterionic phospholipids (phosphatidylcholine and phosphatidylethanolamine) had no effect (Figure 4C). As phosphatidylserine is negligible in the IMM, cardiolipin is the primary target for SS-31 in the IMM. These results led us to propose that electrostatic interaction between the two basic amino acids on SS-31 and the phosphate head groups of cardiolipin aligns the aromatic residues within the hydrophobic acyl chain region. The interaction between SS-31 and cardiolipin occurs at 1:1 molar ratio (Figure 4D). The insertion of the aromatic groups into the lipid environment has now been confirmed by nuclear magnetic resonance (Birk et al., 2013a).
Interaction of SS-31 with cardiolipin changes cyt c activity
Cyt c plays major roles in mitochondrial respiration and in apoptosis, and this delicate balance between life and death is regulated by its interaction with cardiolipin. Electrostatic interaction between cyt c and cardiolipin brings cyt c in close proximity to the respiratory complexes for optimal electron transfer. However, hydrophobic interaction between cyt c and cardiolipin can result in unfolding of the tertiary structure of cyt c and converts this electron carrier into a peroxidase (Figure 5). Thus cyt c is Janus-faced with two contrasting functions, one promoting life and one promoting death. We have found that the interaction of SS-31 with cardiolipin favours the electron carrier over the peroxidase in cyt c.
Figure 5.
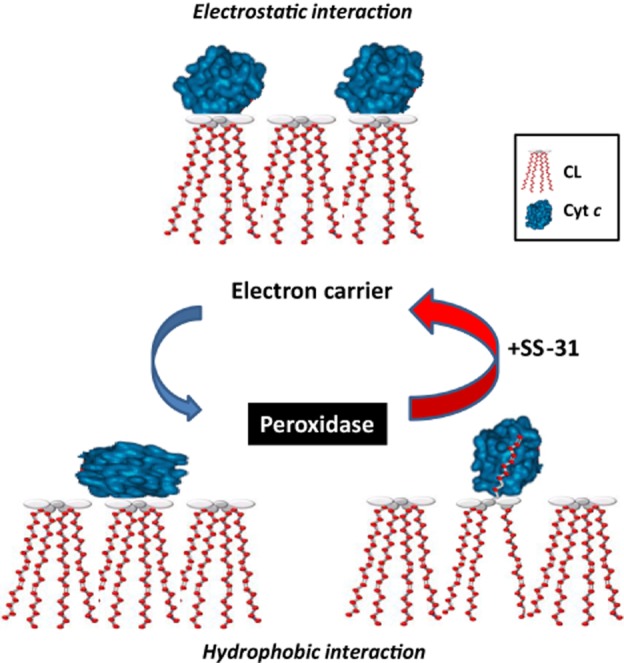
Interaction of SS-31 with cardiolipin favours the electron carrier over the peroxidase in cyt c. Electrostatic interaction between cyt c and cardiolipin brings cyt c in close proximity to the respiratory complexes for optimal electron transfer. However, hydrophobic interaction between cyt c and cardiolipin results in unfolding of the tertiary structure of cyt c and converts this electron carrier into a peroxidase by disrupting the Met80-haem ligation. The SS-31/cardiolipin complex localizes to within angstroms of the haem group to protect the Met80-haem coordination and inhibits peroxidase activity while improving π−π* interaction to promote electron transfer and ATP synthesis.
SS-31 potently inhibited cardiolipin-induced cyt c peroxidase activity in vitro as well as in isolated mitochondria, indicating that SS-31 can inhibit peroxidase activity of endogenous cyt c in mitochondria (Birk et al., 2013b). Interaction of SS-31 with cardiolipin had no effect on the binding of cyt c to cardiolipin. When cyt c was added to the [ald]SS-31/cardiolipin complex, the fluorescent signal was dramatically quenched, indicating that the peptide/cardiolipin complex is localized within angstroms of the haem which is a large resonance acceptor (Birk et al., 2013b). Structural studies with circular dichroism revealed that SS-31 effectively prevented the unfolding of cyt c by cardiolipin and protected the Met80-haem ligation, thus preventing any peroxidase activity (Birk et al., 2013b).
Importantly, SS-31 appears to have no inhibitory effect on the electron carrier function of cyt c. By disrupting the Met80-Fe ligand, cardiolipin lowers the redox potential of cyt c and inhibits its reduction by glutathione or ascorbate (Basova et al., 2007). SS-31 itself has no effect on cyt c reduction by glutathione or ascorbate, but it was able to prevent the inhibition caused by cardiolipin at a 1:1 peptide : cardiolipin ratio (Birk et al., 2013a2013b). Furthermore, addition of SS-31 to isolated mitochondria significantly increased state 3 respiration initiated by substrates for complex I or II, or when cyt c was directly reduced by N,N,N′,N′-tetramethyl-p-phenylenediamine/ascorbate, suggesting that protecting cardiolipin can improve the activity of each component of the ETC (Birk et al., 2013a2013b). Importantly, SS-31 had no effect on state 4 respiration indicating that SS-31 does not promote electron flux by uncoupling mitochondria. SS-31 promotes the efficiency of coupled respiration by increasing P/O ratio and this was confirmed by increase in the rate of ATP synthesis (Birk et al., 2013a2013b). Thus, SS-31 targets cardiolipin and interacts with cyt c to protect the Met80-haem bond and inhibits peroxidase activity while improving π-π* interaction to promote electron transfer and ATP synthesis. By modulating the interaction between cardiolipin and cyt c, SS-31 favours cyt c as an electron carrier and minimizes its role as a peroxidase (Figure 5).
SS-31 protects mitochondrial cristae, promotes ATP synthesis and inhibits mitochondrial permeability transition
During ischaemia, the rapid drop in ATP leads to elevation in cytosolic calcium followed by increase in mitochondrial calcium. Calcium induces mitochondrial ROS and cardiolipin peroxidation (Lesnefsky et al., 2001b; Petrosillo et al., 2004; Chen et al., 2008; Paradies et al., 2009). We found that calcium can directly stimulate cyt c peroxidase activity in vitro, and SS-31 can completely prevent this peroxidase activity with EC50 less than 1 μM (Birk et al., 2013b). By targeting mitochondrial cardiolipin and inhibiting cardiolipin peroxidation, SS-31 protects mitochondrial cristae and facilitates ATP recovery in ischaemic tissues (Birk et al., 2013b).
Interruption of renal blood flow for 45 min in the rat resulted in dramatic swelling of tubular cell mitochondria and loss of cristae membranes (Figure 6A). Some mitochondria remained swollen in early reperfusion, while others showed disrupted outer mitochondrial membranes and spilling of matrix materials into the cytoplasm, consistent with MPT (Figure 6C). In contrast, cristae membranes remained intact in the SS-31 treated kidneys at the end of ischaemia (Figure 6B) and after 5 min reperfusion (Figure 6D). Mitochondrial permeability transition led to sustained inhibition of mitochondrial respiration; with ATP levels down by more than 30% even 1 h after return of blood flow (Szeto et al., 2011). Mitochondria treated with SS-31 showed improved mitochondrial respiration and complete recovery of ATP after 1 h. By accelerating ATP recovery after ischaemia, SS-31 protected epithelial cells from apoptotic and necrotic cell death. These studies demonstrate that by binding to cardiolipin and inhibiting cardiolipin peroxidation, SS-31 protected the structure of the mitochondrial cristae during ischaemia, inhibited MPT pore opening and accelerated ATP recovery immediate upon reperfusion.
Figure 6.
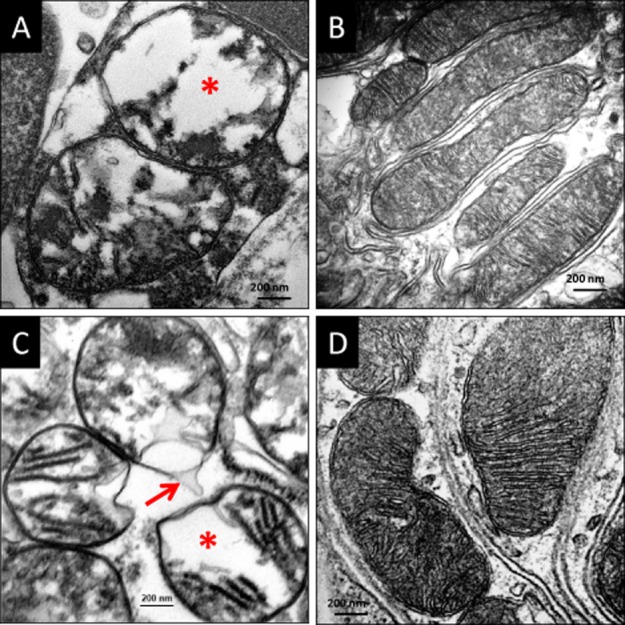
SS-31 protects mitochondrial cristae during ischaemia and prevents mitochondrial permeability transition. Rats were treated with saline or SS-31 (2 mg kg−1) before occlusion of renal blood flow for 45 min. Kidney sections were obtained either at the end of ischaemia or after 5 min reperfusion and examined by transmission electron microscopy. (A) Representative electron microscopic image of proximal tubular cells from saline-treated animal at the end of ischaemia shows swollen mitochondria with loss of cristae and matrix material (*). (B) Mitochondria from SS-31 treated kidneys looked normal and were elongated with finely stacked cristae membranes even after 45 min ischaemia. (C) Mitochondria from saline-treated animals remained swollen (*) after 5 min reperfusion and some of them showed breaks in the outer mitochondrial membrane and loss of matrix contents into the cytosol (arrow), consistent with mitochondrial permeability transition. (D) Mitochondria from SS-31 treated sections were normal with many cristae stacks. All images are taken at 80 000 × magnification.
Therapeutic potential of SS-31 in age-associated diseases
Numerous animal studies have demonstrated the remarkable efficacy of SS-31 in diverse diseases associated with bioenergetic failure (see Table 1). The effective dose(s) of SS-31 in the different disease models are summarized in Table 2007. Minimal effective dose in rodents is 0.1–0.5 mg kg−1 given subcutaneously (s.c.) or in the peritoneum (i.p.), and dose-dependent responses were observed between 0.1 and 5 mg kg−1 (Yang et al., 2009; Szeto et al., 2011). Plasma levels of SS-31 are dose-proportional over these dose ranges (Szeto and Schiller, 2011). In general, doses are lower for larger animal models (dogs, sheep and pigs). It is beyond the scope of this review to present all published reports, and some of these studies have been reviewed previously (Szeto, 2008a,b2008b; Szeto and Schiller, 2011). This review will concentrate on the most recent findings and those preclinical studies that have provided the rationale for clinical trials with SS-31. In addition to studies described in detail below, SS-31 has been reported to be beneficial for chronic kidney disease, metabolic syndrome, neurodegenerative diseases and drug-induced mitochondrial toxicity (Petri et al., 2006; Mizuguchi et al., 2008; Anderson et al., 2009; Yang et al., 2009; Duan et al., 2013; Huang et al., 2013; Toyama et al., 2013). It should be pointed out that SS-31 is also referred to as MTP-131 or Bendavia in the literature. The nomenclature used in the original paper will be used in this review.
Table 1.
Summary of preclinical studies with SS-31
| Ischaemia-reperfusion models | Species | Dose | Route | Time of administration | I/R duration | Major findings | Reference |
|---|---|---|---|---|---|---|---|
| Isolated perfused hearts | Guinea pig | 1 nM | perfusate | Starting 20 min before ischaemia and throughout reperfusion | 30′ I/60′ R | preserved contractile force | Szeto (2008b) |
| preserved ATP content | |||||||
| ↓ infarct area | |||||||
| ↓ lipid peroxidation | |||||||
| Isolated perfused hearts | Guinea pig | 1 nM | perfusate | Starting before ischaemia and throughout reperfusion | 20′ I/2h R | ↓ infarct area | Kloner et al. (2012) |
| Isolated perfused hearts | Guinea pig | 1 nM | perfusate | Starting at onset of reperfusion | 20′ I/2h R | ↓ infarct area | Kloner et al. (2012) |
| Isolated perfused hearts | Diabetic rats | 1 nM | perfusate | Starting at onset of reperfusion | 20′ I/2h R | ↓ infarct area | Sloan et al. (2012) |
| Left anterior descending coronary artery ligation | Rat | 3 mg kg−1 | i.p. | 30 min before ligation 5 min before reperfusion | 60′ I/60′R | ↓ infarct area, | Cho et al. (2007a) |
| ↓ arrhythmias | |||||||
| ↓ lipid peroxidation | |||||||
| Left anterior descending coronary artery ligation | Sheep | 0.05 mg kg−1 h−1 | i.v. | Starting 30 min before reperfusion | 60′ I/3h R | ↓ infarct area | Kloner et al. (2012) |
| Left anterior descending coronary artery ligation | Rabbit | 0.05–0.1 mg kg−1 h−1 | i.v. | Starting either 10 min before or at reperfusion | 30′ I/3h R | ↓ no-reflow | Kloner et al. (2012) |
| Middle cerebral artery occlusion | Mice | 2 or 5 mg kg−1 | s.c. | immediately after reperfusion, or at 6, 24 or 48 h after reperfusion | 30′ I/72h R | ↓ infarct volume | Cho et al. (2007a) |
| ↓ glutathione depletion | |||||||
| Renal artery occlusion | Rats | 0.5, 2 or 5 mg kg−1 | s.c. | 30 min before ischaemia 5 min before reperfusion, 2 h after reperfusion | 45′ I/24h R | protected mitochondrial structure after ischaemia | Szeto et al. (2011) |
| ↑ ATP synthesis, ↑ GSH | |||||||
| ↓ lipid peroxidation, | |||||||
| ↓ tubular apoptosis and necrosis, improved kidney function | |||||||
| Renal artery occlusion | Rats | 2 mg kg−1 | s.c. | 30 min before ischaemia 5 min before reperfusion | 30′ I/24h R | Protected renal mitochondria and ATP synthesis | Birk et al. (2013b) |
| ↓ tubular apoptosis | |||||||
| ↓ renal dysfunction | |||||||
| Islet transplantation | Donor mice | 3 mg kg−1 | s.c. | 24 h before islet harvest | ↑ islet yield | Thomas et al. (2007) | |
| Islets | 1 nM | All isolation reagents | |||||
| Recipient mice | 3 mg kg−1 | s.c. | bid x 10 days | ↓ islet apoptosis, | |||
| ↑ post-transplantation islet graft function | |||||||
| Renal artery stenosis | Pigs | 0.05 mg kg−1 h−1 | i.v. | Continuous administration for 4 h during renal angioplasty | 4 weeks | ↑ renal blood flow and GFR | Eirin et al. (2012) |
| ↓ microvascular rarefaction, ↓oxidative stress, tubular injury and fibrosis | |||||||
| Heart Failure models | Species | Dose | Route | Time of administration | Duration of study | Major findings | Reference |
| Ang II hypertensive cardiomyopathy | Mice | 3 mg kg−1 day-1 | osmotic pump | Concurrent with angiotensin exposure | 4 weeks | Prevented left ventricular hypertrophy | Dai et al. (2011b) |
| Mitigated diastolic dysfunction | |||||||
| Prevented cardiac fibrosis | |||||||
| Gαq overexpression | Mice | 3 mg kg−1 day−1 | osmotic pump | age 12–16 weeks | 4 weeks | Ameliorated diastolic dysfunction and cardiac hypertrophy | Dai et al. (2011b) |
| ↑ systolic function and myocardial performance | |||||||
| Thoracic aortic constriction | Mice | 3 mg kg−1 day−1 | osmotic pump | At time of aortic constriction | 4 weeks | Protected cardiac mitochondria | Dai et al. (2013) |
| Ameliorated cardiac enlargement and systolic dysfunction | |||||||
| Prevented cardiac fibrosis | |||||||
| Chronic ischaemia heart failure | Dogs | 0.05 mg kg−1 h−1 | i.v. | Ejection fraction ∼30% | 2 h | ↑ ejection fraction, stroke volume, cardiac output | Sabbah et al. (2012) |
| Chronic ischaemia heart failure | Dogs | 0.5 mg kg−1 | s.c. | Daily injection when ejection fraction ∼30% | 3 months | ↑ myocardial ATP synthesis | Sabbah et al. (2013) |
| ↑ systolic function | |||||||
| Kidney Failure models | Species | Dose | Route | Time of administration | Duration of study | Major findings | Reference |
| Obstructive renal fibrosis | Rats | 1 or 3 mg kg−1 | i.p. | Daily starting 1 day before obstruction | 14 days | ↓ interstitial fibrosis | Mizuguchi et al. (2008) |
| ↓ tubular apoptosis | |||||||
| ↓ macrophage infiltration | |||||||
| ↑ tubular proliferation | |||||||
| Skeletal muscle models | Species | Dose | Route | Time of administration | Duration of study | Major findings | Reference |
| Mechanical ventilation diaphragm weakness | Rats | 3 mg kg−1, followed by 0.15 mg kg−1 every 3 h | s.c. | Onset of mechanical ventilation | 12 h | Prevented ↑ mitochondrial H2O2 and lipid and protein oxidation | Powers et al. (2011) |
| Prevented diaphragmatic contractile dysfunction and atrophy | |||||||
| Immobilization-induced hindlimb muscle atrophy | Mice | 1.5 mg kg−1 | s.c. | Daily starting at onset of casting | 14 days | Prevented ↓ in mitochondrial respiration and ↑ mitochondrial H2O2 | Min et al. (2011) |
| Mitigated hindlimb muscle atrophy | |||||||
| Immobilization-induced hindlimb muscle atrophy | Rats | 3 mg kg−1 | s.c. | Daily starting at onset of casting | 7 days | Prevented ↓ in mitochondrial respiration and ↑ mitochondrial H2O2 | Talbert et al. (2013) |
| Prevented hindlimb muscle atrophy | |||||||
| Age-related muscle weakness | Aged mice | 3 mg kg−1 | s.c. | Daily starting at age 27 months | 1 h or 8 days | One dose rapidly reversed muscle energy deficits and improved fatigue resistance eight daily doses improved whole body endurance | Siegel et al. (2013) |
| Burn-induced muscle loss | Mice | 5 mg kg−1 | s.c. | Post-burn days 1,3 and 7 | 7 days | Prevented muscle apoptosis | Lee et al. (2011) |
| ↑ glucose tolerance | |||||||
| ↓ oxidized proteins | |||||||
| Metabolic studies | Species | Dose | Route | Time of administration | Duration of study | Major findings | Reference |
| Insulin resistance induced by high fat diet | Rats | 5 mg kg−1 | i.p. | Daily concurrent with onset of high fat diet | 6 weeks | ↓ mitochondrial H2O2 | Anderson et al. (2009) |
| Prevented insulin resistance | |||||||
| Burn-induced insulin resistance | Rats | 4 mg kg−1 | i.p. | Immediately after burn injury and at 12 and 24 h | 24 h | Ameliorated burn-induced insulin resistance | Carter et al. (2011) |
| Burn-induced hypermetabolism | Rats | 2 mg kg−1 followed by 4 mg kg−1 day−1 | i.v. osmotic pump | immediately after burn injury | 7 days | ↓ O2 consumption and CO2 production | Yo et al. (2013) |
| ↓ UCP1 expression in brown adipose tissue | |||||||
| Burn-induced skeletal muscle bioenergetics | Rats | 3 mg kg−1 | i.p. | 30 min pre-burn and immediately post-burn, or immediately post-burn only | 7 days | ↑ ATP synthesis rate at 6 h | Righi et al. (2013) |
| ↑ mitochondrial redox status restore mitochondrial coupling | |||||||
| Diabetic retinopathy | Diabetic rats | 3 mg kg−1 | s.c. | Daily starting 2 weeks after induction of diabetes | 4 months | Protected retinal ganglion cells and capillary basement membranes protected blood-retinal barrier | Huang et al. (2013) |
| Neurodegenerative models | Species | Dose | Route | Time of administration | Duration of study | Major findings | Reference |
| Amyotrophic lateral sclerosis (SOD1 mutant) | Mice | 5 mg kg−1 | i.p. | Starting at 30 days of age | till death | ↑ survival and motor performance | Petri et al. (2006) |
| Parkinson's Disease (MPTP model) | Mice | 0.1–10 mg kg−1 | i.p. | 30 min before first MPTP dose, and 1 and 12 h after 3rd MPTP dose | 7 days | Dose-dependent protection of dopaminergic neurons and dopamine and metabolite levels | Yang et al. (2009) |
| Drug-induced toxicity models | Species | Dose | Route | Time of administration | Duration of study | Major findings | Reference |
| Contrast dye (diatrizoate) nephropathy | Rats | 3 mg kg−1 | i.p. | 24 h and 0.5 h before, and 24 h after contrast dye | 48 h | ↓ renal dysfunction | Duan et al. (2013) |
| ↓ tubular injury | |||||||
| ↓ renal oxidative stress | |||||||
| Oxaliplatin neuropathic pain | Rats | 10 mg kg−1 | s.c. | 4 days after oxaliplatin administration | 2 h | Reverse cold hypersensitivity but not mechanical allodynia | Toyama et al. (2013) |
| Oxaliplatin neuropathic pain | Rats | 5 mg kg−1day−1 | osmotic pump | Continuous treatment concomitant with 3 weekly oxaliplatin injections | 4 weeks | Ameliorated cold hypersensitivity and mechanical allodynia | Toyama et al. (2013) |
Efficacy of SS-31 in IR injury
In addition to acute renal ischaemia, SS-31 is effective against myocardial ischaemia and cerebral ischaemia. Treatment with SS-31 prior to coronary artery ligation significantly reduced infarct size, decreased severity of arrhythmias and reduced lipid peroxidation in rats (Cho et al., 2007a). SS-31 also reduced infarct size in a mouse model of cerebral ischaemia and attenuated glutathione depletion even when administered at the onset of reperfusion (Cho et al., 2007b).
A recent paper reported the results of a collaborative study from three independent laboratories on the effects of Bendavia in acute cardiac IR injury (Kloner et al., 2012). Bendavia administered after the onset of ischaemia demonstrated cardioprotective effects in several IR models. Bendavia reduced infarct size in rabbits and sheep after coronary artery ligation, attenuated the extent of no-reflow in rabbits and reduced infarct size in isolated perfused guinea pig hearts. In cultured myocytes, Bendavia blunted bursts of intracellular ROS after hypoxia challenge, maintained mitochondrial potential and reduced cell death during reoxygenation. Importantly, the investigators reported that Bendavia is rapidly taken up by the myocardium even after ischaemia, consistent with our early report that mitochondrial uptake of SS-31 is potential-independent (Zhao et al., 2004). The encouraging results of this study provided the rationale for testing Bendavia in a clinical trial for acute myocardial infarction with percutaneous coronary intervention (see below Clinical development of Bendavia).
Mortality from myocardial ischaemia is increased in diabetics, and animal models also show increased IR injury in diabetic hearts. A recent study found that cardiac mitochondria from diabetic hearts displayed significantly greater sensitivity to MPT pore opening (Sloan et al., 2012). Diabetic animals treated with MTP-131 showed improved resistance to MPT pore opening. Diabetic hearts also showed increased infarct size after ischaemia ex vivo, and treatment with MTP-131 prior to reperfusion reduced infarct size in both non-diabetic and diabetic hearts (Sloan et al., 2012).
Bendavia has even been shown to reduce reperfusion injury after chronic ischaemia. Atherosclerotic renal artery stenosis results in chronic renal ischaemia and hypertension. Percutaneous transluminal angioplasty and stenting corrects blood pressure but does not improve renal function, and this is believed to result from reperfusion injury in a chronically ischaemic organ. In a porcine model of renal artery stenosis, a short infusion of Bendavia at the time of angioplasty resulted in significant improvement in renal blood flow and glomerular filtration rate 4 weeks later (Eirin et al., 2012). Mitochondrial biogenesis was restored, and microvascular rarefaction, apoptosis, oxidative stress, tubular injury and fibrosis all decreased. The potential of Bendavia in improving kidney outcomes in percutaneous renal angioplasty is currently being evaluated in a clinical trial (see below Clinical development of Bendavia).
Efficacy of SS-31 in organ transplantation
IR injury is also a challenge in transplantation. Islet cell transplantation is currently limited by islet cell loss during the isolation procedure and after transplantation. The use of SS-31 in the isolation procedure helped preserve mitochondrial potential, reduced islet cell apoptosis and increased islet cell yield (Thomas et al., 2007). The investigators suggested that SS-31 may present a novel approach to optimize islet isolation, reduce the need for multiple pancreata to achieve insulin independence and increase the pool of eligible organ donors.
IR injury is also a major cause of delayed graft function and chronic graft dysfunction following renal transplantation. Unlike tubular epithelial cells that can regenerate after ischaemic injury, the endothelial cell lacks comparable regenerative potential, and peritubular capillary dropout has been observed following acute renal ischaemia (Basile et al., 2001; Zhu et al., 2004). The loss of peritubular capillaries results in persistent tissue hypoxia and chronic inflammation, resulting in release of profibrotic cytokines such as TGFβ and renal fibrosis. In a model of renal IR injury in the rat, administration of SS-31 at the time of ischaemia significantly reduced endothelial damage and ameliorated microvascular dropout, chronic inflammation and fibrosis (S. Liu and H. Szeto, unpublished). These results suggest that SS-31 may be beneficial in minimizing the risk of progression to chronic kidney disease following acute kidney injury. SS-31 may also improve graft survival in renal transplantation. SS-31 has also been shown to prevent obstructive renal fibrosis (Mizuguchi et al., 2008).
Efficacy of SS-31 in heart failure
Heart failure is a major cause of mortality in the developed world and is most commonly the result of either ischaemic heart disease or chronic hypertension. The failing heart has been described as an energy-starved engine (Neubauer, 2007). The heart consumes 20–30 times its own weight in ATP every day, and 90% of the ATP is derived from mitochondrial oxidative phosphorylation. Heart failure is a mismatch between supply and demand of ATP. This mismatch may result from decreased oxygen and substrate delivery to the myocardium or from increased workload to the myocardium following hypertension. Current treatments for heart failure all rely on ‘energy sparing’ by decreasing workload – beta blockers for slowing heart rate, and ACE inhibitors and aldosterone antagonists to decrease pressure overload. However, there is no convincing evidence that they are effective for heart failure with preserved ejection fraction.
There is ample evidence that mitochondrial bioenergetics is compromised in heart failure, and considerable amount of experimental and clinical data suggest that targeting cardiac metabolism may be beneficial in treating heart failure (Rosca and Hoppel, 2010; Fillmore and Lopaschuk, 2013). There is significant decrease in myocardial ATP and phosphocreatine content in human heart failure (Beer et al., 2002). During the progression of heart failure, there is a shift in substrate preference from fatty acids to glucose, with up-regulation of enzymes involved in glycolytic pathways (Ardehali et al., 2012). The shift towards glucose metabolism improves myocardial contractile efficiency by increasing the ratio of ATP production to oxygen consumption. This has led to strategies that decrease fatty acid metabolism and/or increase glucose oxidation, and some of them are in early clinical trials (Jaswal et al., 2011; Ardehali et al., 2012; Fillmore and Lopaschuk, 2013).
Cardiac mitochondria in failing hearts have structural abnormalities and loss of cristae (Sabbah et al., 1992; Karamanlidis et al., 2011). There is decrease in activity of the individual respiratory complexes and in formation of supercomplexes, resulting in OXPHOS inhibition (Rosca et al., 2008; Lenaz and Genova, 2012; Fillmore and Lopaschuk, 2013). The loss of supercomplexes may be related to the loss of cardiolipin in both human and experimental heart failure with alterations in cardiolipin biosynthesis and remodelling (Sparagna et al., 2007; Saini-Chohan et al., 2009). These findings have led to the suggestion that dietary fatty acid intake may promote cardiolipin biosynthesis and improve mitochondrial bioenergetics and cardiac function. SS-31 represents a novel strategy to enhance mitochondrial bioenergetics in the failing heart by protecting cardiolipin to facilitate electron transfer. SS-31 has been evaluated in several experimental heart failure models.
Hypertensive heart failure model
The first reported study was a model of hypertensive cardiomyopathy in mice induced by angiotensin (Dai et al., 2011a). Continuous administration of angiotensin for 4 weeks significantly increased both systolic and diastolic pressure. After 4 weeks, echocardiography showed twofold increase in left ventricular (LV) mass and 35% decline in diastolic function, with no change in fractional shortening. Simultaneous administration of SS-31 ameliorated the cardiac hypertrophy and diastolic dysfunction without reducing the pressor response to angiotensin. SS-31 also attenuated cardiac fibrosis from tissue remodelling. The non-mitochondrial targeted antioxidant, N-acetylcysteine, had no protective effect in this model, but induction of mitochondrial catalase 2 weeks before angiotensin infusion prevented cardiomyopathy (Dai et al., 2011a,b2011b). The results from this study support the use of SS-31 in hypertensive diastolic heart failure. There is currently no approved drug therapy for heart failure with preserved ejection fraction.
Pressure-overload heart failure model
SS-31 was subsequently evaluated in the classic transverse aortic constriction (TAC) model whereby the sudden increase in end-systolic pressure results in cardiac hypertrophy with progression to overt heart failure. In mice, TAC caused more than twofold increase in LV mass and significant dilation of the LV within 4 weeks (Dai et al., 2013). There was also a significant decline in systolic function after TAC with 50% reduction in fractional shortening. Continuous delivery of SS-31 by osmotic mini pump over the 4 weeks completely ameliorated the cardiac hypertrophy and systolic failure. Myocardial fibrosis was significantly increased in this model, and this was also completely prevented by SS-31 treatment. SS-31 also abolished the metabolic switch from fatty acid oxidation to glucose metabolism. Thus the mechanism of action of SS-31 in the failing heart is entirely different from that of metabolic modulators which tend to further inhibit fatty acid metabolism.
Ultrastructural studies showed 30% reduction in mitochondrial cristae and a significant increase in mitochondrial oxidative damage that were both mitigated by SS-31. The major involvement of mitochondrial dysfunction in pressure-overload heart failure was further confirmed by examination of the cardiac proteome. Of the 538 proteins significantly changed after TAC, 30% were mitochondrial proteins, 25% were involved in metabolism and less than 10% represented regulatory proteins of the cytoskeleton. Majority of mitochondrial proteins declined in abundance after TAC, and 84% of them were attenuated by SS-31. The major pathways affected in heart failure induced by TAC were mitochondrial dysfunction/oxidative phosphorylation and citrate cycle, and these were abolished by SS-31 treatment. These results with the TAC model suggest that SS-31 may also be beneficial in heart failure with reduced ejection fraction, and clearly support a mitochondrial mechanism of action.
Myocardial ischaemia heart failure model
SS-31 was recently evaluated in the canine myocardial infarct model induced by repeated intracoronary embolizations with microspheres. This model manifests many of the sequelae of human heart failure including marked depression of LV systolic and diastolic function, LV dilatation and hypertrophy, reduced cardiac output and elevation of systemic vascular resistance. Sustained depression of cardiac function is seen in this model after embolization is discontinued, and this model has been used for evaluation of a number of experimental therapeutic agents.
In the first study, dogs with advanced heart failure received either Bendavia (0.05 mg kg−1 h−1) or saline for 2 h. Bendavia significantly increased ejection fraction, stroke volume, cardiac output and LV contractility index (dP/dt) without increasing heart rate or decreasing systemic pressure or systemic vascular resistance (Sabbah et al., 2012). These findings suggest that the improvement of LV function is likely to be the result of improved cardiac energetics, and Bendavia may be helpful in the treatment of patients with acute heart failure syndromes.
In the second study, dogs with advanced heart failure were randomized to 3 months therapy with subcutaneous injection of Bendavia (0.5 mg kg−1) or saline once daily for 3 months. Bendavia treatment significantly improved ejection fraction and reduced LV end-diastolic pressure compared with the saline group (Sabbah et al., 2013). Bendavia significantly improved mitochondrial state 3 respiration, increased mitochondrial potential, increased ATP synthesis and decreased MPT pore opening. Besides increasing coupling efficiency (P/O ratio) (Birk et al., 2013a2013b), Bendavia also protects the microvasculature after acute and chronic ischaemic injury and increases oxygen delivery to tissues (Eirin et al., 2012; Kloner et al., 2012). Thus Bendavia can increase ATP production with the same amount of oxygen available, and in addition, increase oxygen delivery to the failing heart. These studies using different animal models confirm that Bendavia improves myocardial function in advanced heart failure by promoting mitochondrial bioenergetics and suggest that Bendavia may be beneficial for both systolic and diastolic failure.
Efficacy of SS-31 in skeletal muscle aging
Aging is associated with loss of skeletal muscle mass and contractile dysfunction. There is evidence of impaired skeletal muscle mitochondrial bioenergetic function with age in humans and mice (Marcinek et al., 2005; Amara et al., 2007; Figueiredo et al., 2009). Mitochondrial coupling efficiency and maximal ATP production in skeletal muscle of 30-month-old mice was significantly lower compared to 7-month-old mice (Marcinek et al., 2005). This was associated with higher resting ADP and decreased energy charge (ATP/ADP). Studies with isolated mitochondria revealed increased number of mitochondria but significantly reduced state 3 mitochondrial respiration and increased oxidative damage (Figueiredo et al., 2009). This age-associated decline in skeletal muscle function and exercise intolerance results in significant health care costs, and there is no pharmacological treatment to rapidly reverse these mitochondrial deficits.
Siegel et al. (2013) recently reported that a single treatment with SS-31 restored in vivo mitochondrial energetics to ‘young’ levels in aged mice after only 1 h, while there was no effect in young mice. Age-related declines in resting and maximal mitochondrial ATP production, coupling of oxidative phosphorylation and cell energy state (phosphocreatine/ATP) were all rapidly reversed after SS-31 treatment. These effects of SS-31 in aged muscle were associated with a more reduced glutathione redox status and lower mitochondrial H2O2 emission. Skeletal muscle of aged mice were more fatigue-resistant after a single SS-31 treatment, and 8 days of SS-31 treatment led to increased whole-body endurance as measured by treadmill running. This rapid improvement in mitochondrial energetic cannot be due to repairing or replacing damaged mitochondria. Rather, it suggests that SS-31 can rapidly improve mitochondrial respiration by increasing the efficiency of the ETC, and this may be related to the ability of SS-31 to improve cardiolipin function and promote fluidity and the formation of supercomplexes on the IMM.
Efficacy of SS-31 in disuse muscle atrophy
Skeletal muscle weakness and atrophy commonly occur during prolonged periods of inactivity due to limb immobilization or prolonged bed rest, and is especially a problem in the aging population. Similarly, prolonged mechanical ventilation is associated with diaphragmatic weakness resulting from myofibre atrophy and contractile dysfunction that result in difficulty weaning off the machine. Muscle atrophy occurs as a result of reduction in protein synthesis and increased proteolysis, with proteolysis playing a more prominent role (Shanely et al., 2002; 2004,). In terms of the proteolytic systems, calpain, caspase-3, ubiquitin-proteosome and autophagy systems are all involved in protein breakdown during disuse muscle atrophy (Powers et al., 2012). ROS are important activators of key proteases in skeletal muscle, and recent evidence suggests that mitochondria are a dominant source of ROS production during inactivity-induced muscle atrophy (Powers et al., 2011). Disuse atrophy has been associated with loss of mitochondria, mitochondrial swelling, increased mitochondrial ROS production and impaired mitochondrial respiratory function (Muller et al., 2007; Powers et al., 2012). Mechanisms that have been proposed for increased mitochondrial ROS production in disuse atrophy include increased mitochondrial uptake of Ca2+, and increased fatty acid hydroperoxides (Bhattacharya et al., 2009; 2011,). Free fatty acids can also decrease mitochondrial respiration and increase H2O2 production (Bhattacharya et al., 2011). The role of cardiolipin peroxidation in muscle atrophy has not been investigated, but swollen mitochondria lacking cristae membranes were observed in the rat soleus muscle after hindlimb suspension (Powers et al., 2012). Cardiolipin peroxidation could account for induction of the intrinsic apoptotic pathway. Recent studies have shown that mitochondrial oxidative stress is a requisite step towards the activation of muscle proteolysis. By promoting mitochondria respiration and reducing ROS generation, SS-31 has been shown to be effective in preventing disuse atrophy in animal models.
Mechanical ventilation induced diaphragmatic atrophy
Diaphragmatic atrophy and contractile dysfunction can be seen after 12 h mechanical ventilation in rats. Mechanical ventilation significantly increased mitochondrial state 4 respiration, mitochondrial H2O2 release and oxidative damage to proteins and lipids that were completely blocked by SS-31 treatment during mechanical ventilation (Powers et al., 2011). As a result, diaphragmatic contractile dysfunction and fibre atrophy were also prevented by SS-31 treatment. Mitochondrial oxidative stress appears to be a required upstream signal for the activation of all key proteases, as SS-31 prevented activation of calpain and caspase-3, as well as 20S proteasome activity in the diaphragm. These results have significant clinical implications and suggest that SS-31 may have therapeutic potential in protecting the diaphragm and reduce weaning problems in patients after prolonged mechanical ventilation (Levine et al., 2011).
Immobilization induced skeletal muscle atrophy
Similar protection was observed with SS-31 in hindlimb muscles following immobilization in mice and rats (Min et al., 2011; Talbert et al., 2013). Fourteen days of immobilization by cast in mice resulted in significant muscle atrophy, along with muscle oxidative damage, and activation of calpain and caspase-3 (Min et al., 2011). Permeabilized muscle fibres from the soleus muscle revealed significant decrease in state 3 respiration and significantly increased mitochondrial H2O2 production in both soleus and plantaris muscles. Once-daily injection of SS-31 prevented mitochondrial respiratory inhibition and H2O2 production, and consequently mitigated protease activation and abolished atrophy of both soleus and plantaris muscles. Importantly, administration of SS-31 to normal mice had no effect on mitochondrial respiration or ROS production.
When rats were subjected to casting for 7 days, significant atrophy was observed in both the soleus and plantaris muscle (Talbert et al., 2013). Casting caused significant reduction in state 3 mitochondrial respiration and increased mitochondrial H2O2 production. Immobilization was associated with activation of calpain and caspase-3 activity, as well as an increase in the proteasome system, with up-regulation of muscle-specific E3 ligases. Autophagy signalling was also increased, suggesting that all four proteolytic systems are involved in skeletal muscle atrophy. Daily SS-31 treatment prevented the decrease in state 3 respiration and increase in H2O2 production, and abolished the activation of all four proteolytic systems. SS-31 also prevented the down-regulation of anabolic signalling molecules during immobilization, such as the Akt/mTOR pathway. Consequently, SS-31 treatment prevented immobilization-induced atrophy. Thus mitochondrial oxidative stress appears to play a requisite role in inhibiting protein synthesis and activating proteolytic systems to result in skeletal muscle atrophy.
Previous research has shown that PGC-1α, a co-activator of the PPARγ, plays a major role in mitochondrial biogenesis and oxidative metabolism (Lin et al., 2005). PGC-1α expression in skeletal muscle is down-regulated in muscle atrophy from denervation and fasting, and overexpression of PGC-1α is sufficient to attenuate the muscle atrophy (Sandri et al., 2006). However, SS-31 did not prevent the down-regulation of PGC-1α during immobilization, suggesting that the protective effect of SS-31 is not mediated by PGC-1α-induced mitochondrial biogenesis.
Skeletal muscle wasting induced by burn trauma
Severe burn injury causes a major systemic catabolic response that is associated with delayed rehabilitation and results in increased morbidity and mortality. This catabolic response is associated with mitochondrial dysfunction, with decreased OXPHOS and increased mitochondrial ROS, in skeletal muscle (Padfield et al., 2005; Khan et al., 2008). Treatment of mice with a single dose of SS-31 immediately after burn injury increased ATP synthesis rate fivefold, reduced oxidative and endoplasmic reticulum stress, and prevented apoptosis in skeletal muscle (Lee et al., 2011; Righi et al., 2013). SS-31 also reduced insulin resistance in burn injury and prevented hypermetabolism by decreasing uncoupling protein 1 in brown fat (Carter et al., 2011; Yo et al., 2013).
The results from these experimental animal models are very encouraging and suggest that SS-31 may not only be beneficial for mechanical ventilation and burn injury, but also to prevent muscle deconditioning from prolonged bed rest, especially in the aged population.
Clinical development of Bendavia
Animal studies carried out by a large number of independent investigators support the use of SS-31 for a number of major diseases with large unmet needs, including acute coronary syndrome, acute kidney injury, stroke, heart failure, sarcopenia, cachexia, neurodegenerative diseases and many others. Given all of the very promising preclinical efficacy data obtained to date, and its excellent pharmacokinetic profile, SS-31 entered into clinical development with a for-profit commercial sponsor (Stealth Peptides Inc., Newton, MA, USA) in 2010 using a clinical formulation named Bendavia.
Pre-Investigational New Drug pharmacokinetic studies
Despite being a peptide molecule, SS-31 has excellent ‘drug-like’ properties. It is a water-soluble compound and very stable in solution. The incorporation of D-Arg and amidation of the C-terminus greatly enhanced the stability of SS-31 against peptidase degradation. The in vitro and in vivo pharmacokinetics of SS-31 in many animal species have already been summarized in a previous review (Szeto and Schiller, 2011) and will only be described briefly here.
SS-31 is rapidly absorbed after s.c. administration, with peak plasma levels detected within 15 min. Consistent with its very polar property; the apparent volume of distribution of SS-31 is very small, being only 40% of total body water. Distribution of 125I-SS-31 to kidney, heart, liver, skeletal muscle and lung occurred within 30 min after s.c administration, with highest concentrations being found in the kidney (Siegel et al., 2013; Birk et al., 2013b). Being a highly water-soluble compound, there was little distribution to adipose tissue (Birk et al., 2013b). Brain concentrations are low compared to plasma, but its excellent efficacy in protecting striatal dopamine neurons in a Parkinson's disease model, indicates that SS-31 can readily cross the blood-brain barrier (Yang et al., 2009). In the isolated perfused guinea pig heart, approximately 25% of the dose in the perfusate was extracted per minute (Kloner et al., 2012). SS-31 is entirely excreted by the kidneys in non-clinical and clinical studies, accounting for 100% of the peptide (parent and metabolites) in urine. The elimination half-life is ∼2 h in rat, dog and monkey. This is sufficient for once-daily dosing to achieve pharmacological efficacy in these species. Most importantly, plasma levels and drug exposure (area under the plasma level curve) are dose-proportional within the dose range used in preclinical efficacy studies (Szeto and Schiller, 2011).
Phase I studies
Several phase I studies have assessed the safety, tolerability and pharmacokinetics of Bendavia in healthy male and female subjects with intravenous and oral dosing. Bendavia was reported to be well tolerated as an intravenous infusion over a wide dose range (0.01 mg kg−1 h−1 to 0.25 mg kg−1 h−1 over 4 h), achieving effective plasma levels even with the lowest dose. The oral formulation of Bendavia provides plasma concentrations shown to be cardioprotective in many models of acute and chronic cardiac diseases (see Table 2007). Oral Bendavia appeared to be safe and was well tolerated with no serious adverse effects across a broad dose range with highly predictable pharmacokinetics.
Phase II studies
Based on the extensive data demonstrating the effectiveness of Bendavia in preclinical models of myocardial IR injury (Cho et al., 2007a; Kloner et al., 2012), the first multinational phase II clinical study with Bendavia is focused on IR and microvascular injuries for patients experiencing acute ST-segment elevation myocardial infarction (STEMI). The rationale and design of the EMBRACE-STEMI study has been published (Chakrabarti et al., 2013). This is a randomized, double-blind, placebo-controlled trial enrolling patients with a first-time anterior STEMI undergoing primary percutaneous coronary intervention. Patients are randomized to receive Bendavia at 0.05 mg kg−1 h−1 or placebo as an intravenous infusion. This dose is based on pharmacokinetic/pharmacodynamic relationship in several animal models examining the ability of Bendavia to affect IR injury (see Table 2007), and human pharmacokinetic data from Phase I studies (Chakrabarti et al., 2013). The elimination half-life (∼4 h) is similar in humans and preclinical models and does not suggest retention by tissues. The clearance of Bendavia from isolated perfused guinea pig hearts was also found to have a similar half-time (Kloner et al., 2012). The primary end point is infarct size measured by creatine kinase release and cardiac magnetic resonance imaging with gadolinium enhancement. Patient enrolment into the EMBRACE-STEMI trial began in June 2012 and the plan is to enrol 300 patients across 40 sites within the United States and Europe. Bendavia has the potential to fill an unmet need for patients having large heart attacks or experiencing poor revascularization due to microvascular no-reflow.
A second phase II study is for treatment of acute kidney injury and renal microvascular dysfunction in hypertension. This study was initiated based on the remarkable effectiveness of Bendavia in improving renal microvascular blood flow and glomerular filtration rate after angioplasty in pigs with atherosclerotic renal artery stenosis (Eirin et al., 2012). Previous research has shown that angioplasty alone fails to reverse structural and functional deterioration in stenotic kidneys (Eirin et al., 2011). This clinical study is also supported by other animal studies showing the effectiveness of Bendavia in preventing acute ischaemia kidney injury (Szeto et al., 2011; Birk et al., 2013b). The phase II clinical study is intended to assess Bendavia's improvement of renal function in patients with hypertension and severe unilateral renal artery stenosis after treatment with angioplasty. This is a randomized, placebo-controlled, single-centre study, and patients will receive either Bendavia (0.05 mg kg−1 h−1) or saline for a maximum duration of 4 h. The primary outcome measure will be glomerular filtration rate at 8 weeks after angioplasty. Secondary outcome measures will include renal volume, regional renal blood flow, renal oxygenation and a number of inflammatory and oxidative biomarkers.
With chronic oral dosing, a third phase II clinical study is planned for the use of Bendavia in treatment of congestive heart failure. Bendavia offers a completely innovative approach to treating heart failure by improving mitochondrial bioenergetics for the energy-starved heart, and extensive preclinical data support such a clinical study (Dai et al., 2011a; 2013; Sabbah et al., 2012; 2013,).
Concluding remarks
SS-31 provides an entirely novel approach to the treatment of diseases that at first glance appear to be totally unrelated. Common to all of them, however, is the loss of cellular energy that accounts for their failure to function properly. SS-31 is the first compound that targets a specific phospholipid on the IMM, cardiolipin, to alter membrane properties and modify the activity of the ETC protein complexes to improve mitochondrial bioenergetics. In aging or diseased tissues, SS-31 serves to recharge the powerhouse of the cells. Research has shown that targeting such a fundamental mechanism can benefit highly complex diseases with multiple morbidities, such as the cardiorenal syndrome, which share a common pathogenesis of energy failure. Importantly, SS-31 has no effect on normal mitochondria which accounts for its excellent safety profile. The ongoing clinical studies with Bendavia will allow this idea to be validated and hopefully bring relief to patients suffering from these chronic diseases.
Current drug discovery efforts tend to focus on protein targets and signalling pathways for an individual disease condition, and target identification is driven by genomics and proteomics. The emerging field of lipidomics is bringing attention to lipids as drug targets. Changes in lipid composition of a membrane cannot only alter fluidity and folding of a membrane, but can profoundly change the organization and function of numerous proteins on the membrane. The complexity of the lipidome equals or exceeds that of the proteome and provides enormous opportunities for drug development.
Acknowledgments
The author's research referenced here was supported by the National Institute of Health and the Research Program in Mitochondrial Therapeutics at Weill Cornell Medical College. I am most grateful to all of my collaborators who have contributed so much to the understanding of the SS peptides. Special thanks to the following members of the Szeto laboratory for their creativity and devotion to the study of the SS peptides: Alexander V. Birk, Shaoyi Liu, Yi Soong, Wesley M. Chao and William C. Mills.
Glossary
- Cyt c
cytochrome c
- ETC
electron transport chain
- IMM
inner mitochondrial membrane
- IR
ischaemia-reperfusion
- LV
left ventricle
- MPT
mitochondrial permeability transition
- OXPHOS
oxidative phosphorylation
- ROS
reactive oxygen species
- SS
Szeto-Schiller
- TAC
transverse aortic constriction
- TPP+
triphenylphosphonium ion
Conflict of interest
The SS peptides described in this article have been licensed for commercial research and development to Stealth Peptides Inc, a clinical stage biopharmaceutical company, in which H.H.S and the Cornell Research Foundation have financial interests. H.H.S. is the scientific founder of Stealth Peptides International. The Research Program in Mitochondrial Therapeutics was established with a gift from Stealth Peptides International.
References
- Acehan D, Xu Y, Stokes DL, Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab Invest. 2007;87:40–48. doi: 10.1038/labinvest.3700480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara CE, Shankland EG, Jubrias SA, Marcinek DJ, Kushmerick MJ, Conley KE. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc Natl Acad Sci U S A. 2007;104:1057–1062. doi: 10.1073/pnas.0610131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DC, Fauconnier J, Yamada T, Lacampagne A, Zhang SJ, Katz A, et al. Mitochondrial production of reactive oxygen species contributes to the beta-adrenergic stimulation of mouse cardiomycytes. J Physiol. 2011;589(Pt 7):1791–1801. doi: 10.1113/jphysiol.2010.202838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardehali H, Sabbah HN, Burke MA, Sarma S, Liu PP, Cleland JG, et al. Targeting myocardial substrate metabolism in heart failure: potential for new therapies. Eur J Heart Fail. 2012;14:120–129. doi: 10.1093/eurjhf/hfr173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuncion de la JG, Millan A, Pla R, Bruseghini L, Esteras A, Pallardo FV, et al. Mitochondrial glutathione oxidation correlates with age-associated oxidative damage to mitochondrial DNA. FASEB J. 1996;10:333–338. doi: 10.1096/fasebj.10.2.8641567. [DOI] [PubMed] [Google Scholar]
- Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol. 2001;281:F887–F899. doi: 10.1152/ajprenal.2001.281.5.F887. [DOI] [PubMed] [Google Scholar]
- Basova LV, Kurnikov IV, Wang L, Ritov VB, Belikova NA, Vlasova II, et al. Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry. 2007;46:3423–3434. doi: 10.1021/bi061854k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayir H, Tyurin VA, Tyurina YY, Viner R, Ritov V, Amoscato AA, et al. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann Neurol. 2007;62:154–169. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- Bazan S, Mileykovskaya E, Mallampalli VK, Heacock P, Sparagna GC, Dowhan W. Cardiolipin-dependent reconstitution of respiratory supercomplexes from purified Saccharomyces cerevisiae complexes III and IV. J Biol Chem. 2013;288:401–411. doi: 10.1074/jbc.M112.425876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C, Kostler H, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40:1267–1274. doi: 10.1016/s0735-1097(02)02160-5. [DOI] [PubMed] [Google Scholar]
- Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol. 2013;4:95. doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Muller FL, Liu Y, Sabia M, Liang H, Song W, et al. Denervation induces cytosolic phospholipase A2-mediated fatty acid hydroperoxide generation by muscle mitochondria. J Biol Chem. 2009;284:46–55. doi: 10.1074/jbc.M806311200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Lustgarten M, Shi Y, Liu Y, Jang YC, Pulliam D, et al. Increased mitochondrial matrix-directed superoxide production by fatty acid hydroperoxides in skeletal muscle mitochondria. Free Radic Biol Med. 2011;50:592–601. doi: 10.1016/j.freeradbiomed.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk AV, Chao WM, Bracken WC, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2013a doi: 10.1111/bph.12468. AID 12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013b;24:1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Mileykovskaya E, Dowhan W. Lipids in the assembly of membrane proteins and organization of protein supercomplexes: implications for lipid-linked disorders. Subcell Biochem. 2008;49:197–239. doi: 10.1007/978-1-4020-8831-5_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris A, Navarro A. Brain mitochondrial dysfunction in aging. IUBMB Life. 2008;60:308–314. doi: 10.1002/iub.46. [DOI] [PubMed] [Google Scholar]
- Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123:951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratic I, Trifunovic A. Mitochondrial energy metabolism and ageing. Biochim Biophys Acta. 2010;1797:961–967. doi: 10.1016/j.bbabio.2010.01.004. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter EA, Bonab AA, Goverman J, Paul K, Yerxa J, Tompkins RG, et al. Evaluation of the antioxidant peptide SS31 for treatment of burn-induced insulin resistance. Int J Mol Med. 2011;28:589–594. doi: 10.3892/ijmm.2011.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti AK, Feeney K, Abueg C, Brown DA, Czyz E, Tendera M, et al. Rationale and design of the EMBRACE STEMI study: a phase 2a, randomized, double-blind, placebo-controlled trial to evaluate the safety, tolerability and efficacy of intravenous Bendavia on reperfusion injury in patients treated with standard therapy including primary percutaneous coronary intervention and stenting for ST-segment elevation myocardial infarction. Am Heart J. 2013;165:509–514. doi: 10.1016/j.ahj.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–C466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. 2007;292:C33–C44. doi: 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, et al. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007a;18:215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007b;282:4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends Biochem Sci. 2012;37:32–41. doi: 10.1016/j.tibs.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen BE, McAnaney TB, Park ES, Jan YN, Boxer SG, Jan LY. Probing protein electrostatics with a synthetic fluorescent amino acid. Science. 2002;296:1700–1703. doi: 10.1126/science.1069346. [DOI] [PubMed] [Google Scholar]
- Conley KE, Jubrias SA, Cress ME, Esselman P. Exercise efficiency is reduced by mitochondrial uncoupling in the elderly. Exp Physiol. 2013;98:768–777. doi: 10.1113/expphysiol.2012.067314. [DOI] [PubMed] [Google Scholar]
- Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, et al. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011a;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. 2011b;108:837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L, et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail. 2013;6:1067–1076. doi: 10.1161/CIRCHEARTFAILURE.113.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew B, Phaneuf S, Dirks A, Selman C, Gredilla R, Lezza A, et al. Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am J Physiol Regul Integr Comp Physiol. 2003;284:R474–R480. doi: 10.1152/ajpregu.00455.2002. [DOI] [PubMed] [Google Scholar]
- Duan SB, Yang SK, Zhou QY, Pan P, Zhang H, Liu F, et al. Mitochondria-targeted peptides prevent on contrast-induced acute kidney injury in the rats with hypercholesterolemia. Ren Fail. 2013;35:1124–1129. doi: 10.3109/0886022X.2013.815107. [DOI] [PubMed] [Google Scholar]
- Eirin A, Zhu XY, Urbieta-Caceres VH, Grande JP, Lerman A, Textor SC, et al. Persistent kidney dysfunction in swine renal artery stenosis correlates with outer cortical microvascular remodeling. Am J Physiol Renal Physiol. 2011;300:F1394–F1401. doi: 10.1152/ajprenal.00697.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eirin A, Li Z, Zhang X, Krier JD, Woollard JR, Zhu XY, et al. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension. 2012;60:1242–1249. doi: 10.1161/HYPERTENSIONAHA.112.199919. [DOI] [PubMed] [Google Scholar]
- Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan MO, Hoppel CL. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 1999;372:399–407. doi: 10.1006/abbi.1999.1508. [DOI] [PubMed] [Google Scholar]
- Figueiredo PA, Powers SK, Ferreira RM, Appell HJ, Duarte JA. Aging impairs skeletal muscle mitochondrial bioenergetic function. J Gerontol A Biol Sci Med Sci. 2009;64:21–33. doi: 10.1093/gerona/gln048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillmore N, Lopaschuk GD. Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochim Biophys Acta. 2013;1833:857–865. doi: 10.1016/j.bbamcr.2012.08.014. [DOI] [PubMed] [Google Scholar]
- Fink BD, Herlein JA, Yorek MA, Fenner AM, Kerns RJ, Sivitz WI. Bioenergetic effects of mitochondrial-targeted coenzyme Q analogs in endothelial cells. J Pharmacol Exp Ther. 2012;342:709–719. doi: 10.1124/jpet.112.195586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci. 2000;25:319–324. doi: 10.1016/s0968-0004(00)01609-1. [DOI] [PubMed] [Google Scholar]
- Fry M, Green DE. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J Biol Chem. 1981;256:1874–1880. [PubMed] [Google Scholar]
- Gilliam LA, Moylan JS, Patterson EW, Smith JD, Wilson AS, Rabbani Z, et al. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am J Physiol Cell Physiol. 2012;302:C195–C202. doi: 10.1152/ajpcell.00217.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LA, Hagen TM. Age-related decline in mitochondrial bioenergetics: does supercomplex destabilization determine lower oxidative capacity and higher superoxide production? Semin Cell Dev Biol. 2012;23:758–767. doi: 10.1016/j.semcdb.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalvez F, Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis. 2007;12:877–885. doi: 10.1007/s10495-007-0718-8. [DOI] [PubMed] [Google Scholar]
- Gross RW, Han X. Lipidomics in diabetes and the metabolic syndrome. Methods Enzymol. 2007;433:73–90. doi: 10.1016/S0076-6879(07)33004-8. [DOI] [PubMed] [Google Scholar]
- Hagen TM, Ingersoll RT, Wehr CM, Lykkesfeldt J, Vinarsky V, Bartholomew JC, et al. Acetyl-L-carnitine fed to old rats partially restores mitochondrial function and ambulatory activity. Proc Natl Acad Sci U S A. 1998;95:9562–9566. doi: 10.1073/pnas.95.16.9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Han X, Yang J, Cheng H, Yang K, Abendschein DR, Gross RW. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry. 2005;44:16684–16694. doi: 10.1021/bi051908a. [DOI] [PubMed] [Google Scholar]
- Han X, Yang J, Yang K, Zhao Z, Abendschein DR, Gross RW. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry. 2007;46:6417–6428. doi: 10.1021/bi7004015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Varadharaj S, Giedt RJ, Zweier JL, Szeto HH, Alevriadou BR. Mitochondria-derived reactive oxygen species mediate heme oxygenase-1 expression in sheared endothelial cells. J Pharmacol Exp Ther. 2009;329:94–101. doi: 10.1124/jpet.108.145557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanske J, Toffey JR, Morenz AM, Bonilla AJ, Schiavoni KH, Pletneva EV. Conformational properties of cardiolipin-bound cytochrome c. Proc Natl Acad Sci U S A. 2012;109:125–130. doi: 10.1073/pnas.1112312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch FL. Cardiolipins and biomembrane function. Biochim Biophys Acta. 1992;1113:71–133. doi: 10.1016/0304-4157(92)90035-9. [DOI] [PubMed] [Google Scholar]
- Huang J, Li X, Li M, Li J, Xiao W, Ma W, et al. Mitochondria-targeted antioxidant peptide SS31 protects the retinas of diabetic rats. Curr Mol Med. 2013;13:935–945. doi: 10.2174/15665240113139990049. [DOI] [PubMed] [Google Scholar]
- Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation-a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta. 2011;1813:1333–1350. doi: 10.1016/j.bbamcr.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci. 2012;15:1407–1413. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- Judge S, Leeuwenburgh C. Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol. 2007;292:C1983–C1992. doi: 10.1152/ajpcell.00285.2006. [DOI] [PubMed] [Google Scholar]
- Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–421. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- Kalanxhi E, Wallace CJ. Cytochrome c impaled: investigation of the extended lipid anchorage of a soluble protein to mitochondrial membrane models. Biochem J. 2007;407:179–187. doi: 10.1042/BJ20070459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamanlidis G, Bautista-Hernandez V, Fynn-Thompson F, Del Nido P, Tian R. Impaired mitochondrial biogenesis precedes heart failure in right ventricular hypertrophy in congenital heart disease. Circ Heart Fail. 2011;4:707–713. doi: 10.1161/CIRCHEARTFAILURE.111.961474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Mupparaju SP, Mintzopoulos D, Kesarwani M, Righi V, Rahme LG, et al. Burn trauma in skeletal muscle results in oxidative stress as assessed by in vivo electron paramagnetic resonance. Mol Med Rep. 2008;1:813–819. doi: 10.3892/mmr-00000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebish MA, Han X, Cheng H, Chuang JH, Seyfried TN. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: lipidomic evidence supporting the Warburg theory of cancer. J Lipid Res. 2008;49:2545–2556. doi: 10.1194/jlr.M800319-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebish MA, Yang K, Sims HF, Jenkins CM, Liu X, Mancuso DJ, et al. Myocardial regulation of lipidomic flux by cardiolipin synthase: setting the beat for bioenergetic efficiency. J Biol Chem. 2012;287:25086–25097. doi: 10.1074/jbc.M112.340521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Shrago E, Elson CE. Age-related changes in respiration coupled to phosphorylation. II. Cardiac mitochondria. Mech Ageing Dev. 1988a;46:279–290. doi: 10.1016/0047-6374(88)90130-3. [DOI] [PubMed] [Google Scholar]
- Kim JH, Woldgiorgis G, Elson CE, Shrago E. Age-related changes in respiration coupled to phosphorylation. I. Hepatic mitochondria. Mech Ageing Dev. 1988b;46:263–277. doi: 10.1016/0047-6374(88)90129-7. [DOI] [PubMed] [Google Scholar]
- Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, Koomalsingh KJ, et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective Peptide. J Am Heart Assoc. 2012;1:e001644. doi: 10.1161/JAHA.112.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Wei YH. Mitochondria and aging. Adv Exp Med Biol. 2012;942:311–327. doi: 10.1007/978-94-007-2869-1_14. [DOI] [PubMed] [Google Scholar]
- Lee HY, Kaneki M, Andreas J, Tompkins RG, Martyn JA. Novel mitochondria-targeted antioxidant peptide ameliorates burn-induced apoptosis and endoplasmic reticulum stress in the skeletal muscle of mice. Shock. 2011;36:580–585. doi: 10.1097/SHK.0b013e3182366872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Yu BP, Herlihy JT. Modulation of cardiac mitochondrial membrane fluidity by age and calorie intake. Free Radic Biol Med. 1999;26:260–265. doi: 10.1016/s0891-5849(98)00195-6. [DOI] [PubMed] [Google Scholar]
- Lenaz G, Genova ML. Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Adv Exp Med Biol. 2012;748:107–144. doi: 10.1007/978-1-4614-3573-0_5. [DOI] [PubMed] [Google Scholar]
- Lenaz G, Bovina C, Castelluccio C, Fato R, Formiggini G, Genova ML, et al. Mitochondrial complex I defects in aging. Mol Cell Biochem. 1997;174:329–333. [PubMed] [Google Scholar]
- Lesnefsky EJ, Hoppel CL. Ischemia-reperfusion injury in the aged heart: role of mitochondria. Arch Biochem Biophys. 2003;420:287–297. doi: 10.1016/j.abb.2003.09.046. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Hoppel CL. Oxidative phosphorylation and aging. Ageing Res Rev. 2006;5:402–433. doi: 10.1016/j.arr.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Gudz TI, Moghaddas S, Migita CT, Ikeda-Saito M, Turkaly PJ, et al. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol. 2001a;33:37–47. doi: 10.1006/jmcc.2000.1273. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol. 2001b;280:H2770–H2778. doi: 10.1152/ajpheart.2001.280.6.H2770. [DOI] [PubMed] [Google Scholar]
- Levine S, Budak MT, Dierov J, Singhal S. Inactivity-induced diaphragm dysfunction and mitochondria-targeted antioxidants: new concepts in critical care medicine. Crit Care Med. 2011;39:1844–1845. doi: 10.1097/CCM.0b013e31821e85ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen X, Xiao W, Ma W, Li T, Huang J, et al. Mitochondria-targeted antioxidant peptide SS31 attenuates high glucose-induced injury on human retinal endothelial cells. Biochem Biophys Res Commun. 2011;404:349–356. doi: 10.1016/j.bbrc.2010.11.122. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Maranzana E, Barbero G, Falasca AI, Lenaz G, Genova ML. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid Redox Signal. 2013;19:1469–1480. doi: 10.1089/ars.2012.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinek DJ, Schenkman KA, Ciesielski WA, Lee D, Conley KE. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J Physiol. 2005;569(Pt 2):467–473. doi: 10.1113/jphysiol.2005.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskaya E, Dowhan W. Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta. 2009;1788:2084–2091. doi: 10.1016/j.bbamem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskaya E, Zhang M, Dowhan W. Cardiolipin in energy transducing membranes. Biochemistry (Mosc) 2005;70:154–158. doi: 10.1007/s10541-005-0095-2. [DOI] [PubMed] [Google Scholar]
- Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol. 2011;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi Y, Chen J, Seshan SV, Poppas DP, Szeto HH, Felsen D. A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2008;295:F1545–F1553. doi: 10.1152/ajprenal.00395.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddas S, Hoppel CL, Lesnefsky EJ. Aging defect at the QO site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 2003;414:59–66. doi: 10.1016/s0003-9861(03)00166-8. [DOI] [PubMed] [Google Scholar]
- Muenzner J, Toffey JR, Hong Y, Pletneva EV. Becoming a peroxidase: cardiolipin-induced unfolding of cytochrome c. J Phys Chem B. 2013;117:12878–12886. doi: 10.1021/jp402104r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A, et al. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1159–R1168. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- Muscari C, Caldarera CM, Guarnieri C. Age-dependent production of mitochondrial hydrogen peroxide, lipid peroxides and fluorescent pigments in the rat heart. Basic Res Cardiol. 1990;85:172–178. doi: 10.1007/BF01906970. [DOI] [PubMed] [Google Scholar]
- Neubauer S. The failing heart-an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- Nichols-Smith S, Teh SY, Kuhl TL. Thermodynamic and mechanical properties of model mitochondrial membranes. Biochim Biophys Acta. 2004;1663:82–88. doi: 10.1016/j.bbamem.2004.02.002. [DOI] [PubMed] [Google Scholar]
- O'Toole JF, Patel HV, Naples CJ, Fujioka H, Hoppel CL. Decreased cytochrome c mediates an age-related decline of oxidative phosphorylation in rat kidney mitochondria. Biochem J. 2010;427:105–112. doi: 10.1042/BJ20091373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Zhivotovsky B, Orrenius S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007;14:1243–1247. doi: 10.1038/sj.cdd.4402135. [DOI] [PubMed] [Google Scholar]
- Padfield KE, Astrakas LG, Zhang Q, Gopalan S, Dai G, Mindrinos MN, et al. Burn injury causes mitochondrial dysfunction in skeletal muscle. Proc Natl Acad Sci U S A. 2005;102:5368–5373. doi: 10.1073/pnas.0501211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradies G, Ruggiero FM. Age-related changes in the activity of the pyruvate carrier and in the lipid composition in rat-heart mitochondria. Biochim Biophys Acta. 1990;1016:207–212. doi: 10.1016/0005-2728(90)90060-h. [DOI] [PubMed] [Google Scholar]
- Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: role of cardiolipin. FEBS Lett. 1997;406:136–138. doi: 10.1016/s0014-5793(97)00264-0. [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium. 2009;45:643–650. doi: 10.1016/j.ceca.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic Biol Med. 2010;48:1286–1295. doi: 10.1016/j.freeradbiomed.2010.02.020. [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Mitochondrial dysfunction in brain aging: role of oxidative stress and cardiolipin. Neurochem Int. 2011;58:447–457. doi: 10.1016/j.neuint.2010.12.016. [DOI] [PubMed] [Google Scholar]
- Perluigi M, Di Domenico F, Giorgi A, Schinina ME, Coccia R, Cini C, et al. Redox proteomics in aging rat brain: involvement of mitochondrial reduced glutathione status and mitochondrial protein oxidation in the aging process. J Neurosci Res. 2010;88:3498–3507. doi: 10.1002/jnr.22500. [DOI] [PubMed] [Google Scholar]
- Petri S, Kiaei M, Damiano M, Hiller A, Wille E, Manfredi G, et al. Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;98:1141–1148. doi: 10.1111/j.1471-4159.2006.04018.x. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Ruggiero FM, Pistolese M, Paradies G. Ca2+-induced reactive oxygen species production promotes cytochrome c release from rat liver mitochondria via mitochondrial permeability transition (MPT)-dependent and MPT-independent mechanisms: role of cardiolipin. J Biol Chem. 2004;279:53103–53108. doi: 10.1074/jbc.M407500200. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G. Synergistic effect of Ca2+ and peroxidized cardiolipin in the induction of permeability transition and cytochrome c release in rat heart mitochondria. Ital J Biochem. 2007;56:307–309. [PubMed] [Google Scholar]
- Petrosillo G, Matera M, Casanova G, Ruggiero FM, Paradies G. Mitochondrial dysfunction in rat brain with aging Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem Int. 2008;53:126–131. doi: 10.1016/j.neuint.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, De Benedictis V, Ruggiero FM, Paradies G. Decline in cytochrome c oxidase activity in rat-brain mitochondria with aging. Role of peroxidized cardiolipin and beneficial effect of melatonin. J Bioenerg Biomembr. 2013;45:431–440. doi: 10.1007/s10863-013-9505-0. [DOI] [PubMed] [Google Scholar]
- Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, et al. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873–52880. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, et al. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit Care Med. 2011;39:1749–1759. doi: 10.1097/CCM.0b013e3182190b62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Wiggs MP, Duarte JA, Zergeroglu AM, Demirel HA. Mitochondrial signaling contributes to disuse muscle atrophy. Am J Physiol Endocrinol Metab. 2012;303:E31–E39. doi: 10.1152/ajpendo.00609.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebrin I, Sohal RS. Comparison of thiol redox state of mitochondria and homogenates of various tissues between two strains of mice with different longevities. Exp Gerontol. 2004;39:1513–1519. doi: 10.1016/j.exger.2004.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebrin I, Kamzalov S, Sohal RS. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic Biol Med. 2003;35:626–635. doi: 10.1016/s0891-5849(03)00388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reily C, Mitchell T, Chacko BK, Benavides G, Murphy MP, Darley-Usmar V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013;1:86–93. doi: 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Righi V, Constantinou C, Mintzopoulos D, Khan N, Mupparaju SP, Rahme LG, et al. Mitochondria-targeted antioxidant promotes recovery of skeletal muscle mitochondrial function after burn trauma assessed by in vivo 31P nuclear magnetic resonance and electron paramagnetic resonance spectroscopy. FASEB J. 2013;27:2521–2530. doi: 10.1096/fj.12-220764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca MG, Hoppel CL. Mitochondria in heart failure. Cardiovasc Res. 2010;88:40–50. doi: 10.1093/cvr/cvq240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80:30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytomaa M, Kinnunen PK. Evidence for two distinct acidic phospholipid-binding sites in cytochrome c. J Biol Chem. 1994;269:1770–1774. [PubMed] [Google Scholar]
- Rytomaa M, Kinnunen PK. Reversibility of the binding of cytochrome c to liposomes. Implications for lipid-protein interactions. J Biol Chem. 1995;270:3197–3202. doi: 10.1074/jbc.270.7.3197. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Sharov V, Riddle JM, Kono T, Lesch M, Goldstein S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol. 1992;24:1333–1347. doi: 10.1016/0022-2828(92)93098-5. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Wang M, Zhang K, Gupta RC, Rastogi S. Acute intravenous infusion of Bendavia (MTP-131), a novel mitochondria-targeting peptide, improves left ventricular systolic function in dogs with advanced heart failure. Circulation. 2012;126:A15385. doi: 10.1161/CIRCHEARTFAILURE.115.002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah HN, Wang M, Zhang K, Gupta RC, Rastogi S. Long-term therapy with Bendavia (MTP-131), a novel mitochondria-targeting peptide, increases myocardial ATP synthesis and improves left ventricular systolic function in dogs with chronic heart failure. J Am Coll Cardiol. 2013;61:E709. [Google Scholar]
- Saini-Chohan HK, Holmes MG, Chicco AJ, Taylor WA, Moore RL, McCune SA, et al. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J Lipid Res. 2009;50:1600–1608. doi: 10.1194/jlr.M800561-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, et al. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, Ren M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim Biophys Acta. 2009;1788:2080–2083. doi: 10.1016/j.bbamem.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39:257–288. doi: 10.1016/s0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- Schug ZT, Gottlieb E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta. 2009;1788:2022–2031. doi: 10.1016/j.bbamem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Schwall CT, Greenwood VL, Alder NN. The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim Biophys Acta. 2012;1817:1588–1596. doi: 10.1016/j.bbabio.2012.04.015. [DOI] [PubMed] [Google Scholar]
- Sen T, Sen N, Jana S, Khan FH, Chatterjee U, Chakrabarti S. Depolarization and cardiolipin depletion in aged rat brain mitochondria: relationship with oxidative stress and electron transport chain activity. Neurochem Int. 2007;50:719–725. doi: 10.1016/j.neuint.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, et al. Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med. 2002;166:1369–1374. doi: 10.1164/rccm.200202-088OC. [DOI] [PubMed] [Google Scholar]
- Shanely RA, Van Gammeren D, Deruisseau KC, Zergeroglu AM, McKenzie MJ, Yarasheski KE, et al. Mechanical ventilation depresses protein synthesis in the rat diaphragm. Am J Respir Crit Care Med. 2004;170:994–999. doi: 10.1164/rccm.200304-575OC. [DOI] [PubMed] [Google Scholar]
- Shi Y. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J Biomed Res. 2010;24:6–15. doi: 10.1016/S1674-8301(10)60003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Ikawa K, Shimooka T, Terada H. Significant stabilization of the phosphatidylcholine bilayer structure by incorporation of small amounts of cardiolipin. Biochim Biophys Acta. 1994;1192:71–78. doi: 10.1016/0005-2736(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Shidoji Y, Hayashi K, Komura S, Ohishi N, Yagi K. Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem Biophys Res Commun. 1999;264:343–347. doi: 10.1006/bbrc.1999.1410. [DOI] [PubMed] [Google Scholar]
- Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, et al. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell. 2013;12:763–771. doi: 10.1111/acel.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinibaldi F, Fiorucci L, Patriarca A, Lauceri R, Ferri T, Coletta M, et al. Insights into cytochrome c-cardiolipin interaction. Role played by ionic strength. Biochemistry. 2008;47:6928–6935. doi: 10.1021/bi800048v. [DOI] [PubMed] [Google Scholar]
- Sinibaldi F, Howes BD, Piro MC, Polticelli F, Bombelli C, Ferri T, et al. Extended cardiolipin anchorage to cytochrome c: a model for protein-mitochondrial membrane binding. J Biol Inorg Chem. 2010;15:689–700. doi: 10.1007/s00775-010-0636-z. [DOI] [PubMed] [Google Scholar]
- Skulachev VP, Anisimov VN, Antonenko YN, Bakeeva LE, Chernyak BV, Erichev VP, et al. An attempt to prevent senescence: a mitochondrial approach. Biochim Biophys Acta. 2009;1787:437–461. doi: 10.1016/j.bbabio.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Sloan RC, Moukdar F, Frasier CR, Patel HD, Bostian PA, Lust RM, et al. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol. 2012;52:1009–1018. doi: 10.1016/j.yjmcc.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Smith RA, Hartley RC, Cocheme HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. 2012;33:341–352. doi: 10.1016/j.tips.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Sorice M, Manganelli V, Matarrese P, Tinari A, Misasi R, Malorni W, et al. Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett. 2009;583:2447–2450. doi: 10.1016/j.febslet.2009.07.018. [DOI] [PubMed] [Google Scholar]
- Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA, Rees ML, et al. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res. 2007;48:1559–1570. doi: 10.1194/jlr.M600551-JLR200. [DOI] [PubMed] [Google Scholar]
- Suh JH, Heath SH, Hagen TM. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic Biol Med. 2003;35:1064–1072. doi: 10.1016/s0891-5849(03)00468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH. Development of mitochondria-targeted aromatic-cationic peptides for neurodegenerative diseases. Ann N Y Acad Sci. 2008a;1147:112–121. doi: 10.1196/annals.1427.013. [DOI] [PubMed] [Google Scholar]
- Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008b;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- Szeto HH, Schiller PW. Novel therapies targeting inner mitochondrial membrane-from discovery to clinical development. Pharm Res. 2011;28:2669–2679. doi: 10.1007/s11095-011-0476-8. [DOI] [PubMed] [Google Scholar]
- Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–1052. doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert EE, Smuder AJ, Min K, Kwon OS, Szeto HH, Powers SK. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J Appl Physiol (1985) 2013;115:529–538. doi: 10.1152/japplphysiol.00471.2013. [DOI] [PubMed] [Google Scholar]
- Thomas DA, Stauffer C, Zhao K, Yang H, Sharma VK, Szeto HH, et al. Mitochondrial targeting with antioxidant peptide SS-31 prevents mitochondrial depolarization, reduces islet cell apoptosis, increases islet cell yield, and improves posttransplantation function. J Am Soc Nephrol. 2007;18:213–222. doi: 10.1681/ASN.2006080825. [DOI] [PubMed] [Google Scholar]
- Toyama S, Shimoyama N, Ishida Y, Koyasu T, Szeto HH, Shimoyama M. Characterization of Acute and Chronic Neuropathies Induced by Oxaliplatin in Mice and Differential Effects of a Novel Mitochondria-targeted Antioxidant on the Neuropathies. Anesthesiology. 2013 doi: 10.1097/01.anes.0000435634.34709.65. doi: 10.1097/01.anes.0000435634.34709.65. [DOI] [PubMed] [Google Scholar]
- Tyurina YY, Tungekar MA, Jung MY, Tyurin VA, Greenberger JS, Stoyanovsky DA, et al. Mitochondria targeting of non-peroxidizable triphenylphosphonium conjugated oleic acid protects mouse embryonic cells against apoptosis: role of cardiolipin remodeling. FEBS Lett. 2012;586:235–241. doi: 10.1016/j.febslet.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyurina YY, Winnica DE, Kapralova VI, Kapralov AA, Tyurin VA, Kagan VE. LC/MS characterization of rotenone induced cardiolipin oxidation in human lymphocytes: implications for mitochondrial dysfunction associated with Parkinson's disease. Mol Nutr Food Res. 2013;57:1410–1422. doi: 10.1002/mnfr.201200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorbeck ML, Martin AP, Long JW, Jr, Smith JM, Orr RR., Jr Aging-dependent modification of lipid composition and lipid structural order parameter of hepatic mitochondria. Arch Biochem Biophys. 1982;217:351–361. doi: 10.1016/0003-9861(82)90511-2. [DOI] [PubMed] [Google Scholar]
- Wiswedel I, Gardemann A, Storch A, Peter D, Schild L. Degradation of phospholipids by oxidative stress-exceptional significance of cardiolipin. Free Radic Res. 2010;44:135–145. doi: 10.3109/10715760903352841. [DOI] [PubMed] [Google Scholar]
- Wittig I, Schagger H. Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim Biophys Acta. 2009;1787:672–680. doi: 10.1016/j.bbabio.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid Redox Signal. 2009;11:2095–2104. doi: 10.1089/ars.2009.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yo K, Yu YM, Zhao G, Bonab AA, Aikawa N, Tompkins RG, et al. Brown adipose tissue and its modulation by a mitochondria-targeted peptide in rat burn injury-induced hypermetabolism. Am J Physiol Endocrinol Metab. 2013;304:E331–E341. doi: 10.1152/ajpendo.00098.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem. 2002;277:43553–43556. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- Zhang M, Mileykovskaya E, Dowhan W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem. 2005;280:29403–29408. doi: 10.1074/jbc.M504955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Luo G, Zhao GM, Schiller PW, Szeto HH. Transcellular transport of a highly polar 3+ net charge opioid tetrapeptide. J Pharmacol Exp Ther. 2003;304:425–432. doi: 10.1124/jpet.102.040147. [DOI] [PubMed] [Google Scholar]
- Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- Zhao K, Luo G, Giannelli S, Szeto HH. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem Pharmacol. 2005;70:1796–1806. doi: 10.1016/j.bcp.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Zhu H, Shan L, Schiller PW, Mai A, Peng T. Histone deacetylase-3 activation promotes tumor necrosis factor-alpha (TNF-alpha) expression in cardiomyocytes during lipopolysaccharide stimulation. J Biol Chem. 2010;285:9429–9436. doi: 10.1074/jbc.M109.071274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Yang Y, Wang Y, Li J, Schiller PW, Peng T. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc Res. 2011;92:75–84. doi: 10.1093/cvr/cvr145. [DOI] [PubMed] [Google Scholar]
- Zhu XY, Chade AR, Rodriguez-Porcel M, Bentley MD, Ritman EL, Lerman A, et al. Cortical microvascular remodeling in the stenotic kidney: role of increased oxidative stress. Arterioscler Thromb Vasc Biol. 2004;24:1854–1859. doi: 10.1161/01.ATV.0000142443.52606.81. [DOI] [PubMed] [Google Scholar]